

Abstract
Heritable disorders that feature high bone mass (HBM) are rare. The etiology is typically a mutation(s) within a gene that regulates the differentiation and function of osteoblasts (OBs) or osteoclasts (OCs). Nevertheless, the molecular basis is unknown for approximately one-fifth of such entities.
NF-κB signaling is a key regulator of bone remodeling and acts by enhancing OC survival while impairing OB maturation and function. The NF-κB transcription complex comprises five subunits. In mice, deletion of the p50 and p52 subunits together causes osteopetrosis (OPT). In humans, however, mutations within the genes that encode the NF-κB complex, including the Rela/p65 subunit, have not been reported.
We describe a neonate who died suddenly and unexpectedly and was found at post-mortem to have HBM documented radiographically and by skeletal histopathology. Serum was not available for study. Radiographic changes resembled malignant OPT, but histopathological investigation showed morphologically normal OCs and evidence of intact bone resorption excluding OPT. Furthermore, mutation analysis was negative for eight genes associated with OPT or HBM. Instead, accelerated bone formation appeared to account for the HBM. Subsequently, trio-based whole exome sequencing revealed a heterozygous, de novo, missense mutation (c.1534_1535delinsAG, p.Asp512Ser) in exon 11 of RELA encoding Rela/p65. The mutation was then verified using bi-directional Sanger sequencing. Lipopolysaccharide stimulation of patient fibroblasts elicited impaired NF-κB responses compared to healthy control fibroblasts. Five unrelated patients with unexplained HBM did not show a RELA defect.
Ours is apparently the first report of a mutation within the NF-κB complex in humans. The missense change is associated with neonatal osteosclerosis from in utero increased OB function rather than failed OC action. These findings demonstrate the importance of the Rela/p65 subunit within the NF-κB pathway for human skeletal homeostasis, and represent a new genetic cause of HBM.
Keywords: osteoblast, osteoclast, osteopetrosis, osteosclerosis, trio-exome sequencing
II) Introduction
High bone mass (HBM) is a feature of many phenotypically diverse conditions, including rare monogenic diseases.[1] The clinical spectrum of heritable HBM ranges broadly from autosomal dominant (AD) typically benign findings exemplified by the low-density lipoprotein receptor-related protein 5 (LRP5) type of endosteal hyperostosis,[2] to autosomal recessive (AR) life-threatening types of osteopetrosis.[3] Investigation of the molecular basis of Mendelian HBM disorders has revealed genes that regulate the cells responsible for bone apposition and for bone resorption.[4] For example, the HBM of LRP5 activation results from increased osteoblast (OB) activity,[4] whereas the HBM of the osteopetroses results from failed osteoclast (OC) formation or function.[5]
Among the genes that regulate skeletal remodeling are those involved in the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway.[6] NF-κB signaling, besides being linked to cancer and inflammation,[7] functions in bone turnover.[8] Activation of NF-κB is mediated via two different pathways; i.e., the “classical” and “alternative” pathway.[9,10] Upstream activation of the classical NF-κB pathway is mediated by IκB kinase complex (IKK), that includes IKKβ and the NF-κB essential modulator (NEMO), whereas the key control node of the alternative pathway is NF-κB Inducing Kinase (NIK).[11] Downstream, the NF-κB transcription complex consists of homo- and heterodimers of five subunits: v-rel avian reticuloendotheliosis viral oncogene homolog A (RelA or p65), RelB, c-Rel, p50, and p52.[12]
Mice globally deprived of NF-κB activation due to combined deletion of both the p50 and p52 subunits of the NF-κB transcription complex lack OCs and develop osteopetrosis.[13] Mice without IKKβ in the OC lineage also develop osteopetrosis.[14] Additionally, mice lacking either p65/RelA or RelB, or those with inhibition of NEMO, show reduced bone loss in several models of pathological osteolysis.[15–17] RelB-deficient mice also exhibit transient increases in bone formation, and their mesenchymal stem cells show more rapid repair of a cortical bone defect.[18] Inhibition of the classical NF-κB pathway specifically in OBs, via expression of a dominant negative NEMO, leads to enhanced bone formation.[19] Conversely, constitutive activation of IKKβ impairs osteogenesis.[20] Thus, in mouse models, the NF-κB pathway controls the function of both OCs and OBs.
Despite this overwhelming evidence derived from mouse models for a role of NF-κB in bone cells, few human disorders involving bone have been associated with mutations in this pathway. Mutations in the NEMO gene can cause osteopetrosis with ectodermal dysplasia and immunodeficiency.[21] Activating mutations in an inhibitor of classical NF-κB, IκBα cause a similar skin abnormality and immunodeficiency, but a bone phenotype was not reported.[22] In OCs, the RANKL/RANK signalling pathway potently induces both NF-κB pathways.[23] Loss-of-function of either the cytokine RANKL or the receptor RANK causes especially rare “OC-poor” forms of human osteopetrosis,[24, 25] although all the consequences cannot be ascribed to loss of NF-κB signalling. To our knowledge, there are no reports in humans of mutations within the genes that encode the NF-κB complex itself.
We investigated a neonate who died suddenly and unexpectedly and was found to have HBM apparently due to enhanced bone formation. Trio exome sequencing disclosed a heterozygous de novo missense mutation in RELA encoding the Rela/p65 component of NF-κB. Studies of the patient’s fibroblasts revealed impaired NF-κB activation.
III) Patient and Methods
A) Patient
Our study subject (“patient”), a boy, was the first child born to non-consanguineous healthy parents. The mother and father were 33 and 35 years old, respectively. She had taken as recommended during her pregnancy iron, folic acid, and vitamin D supplements. Neither parent reported within three generations in their families premature deaths, cardiovascular diseases, bone disorders, or any other life-threatening conditions.
The pregnancy of 38 weeks gestation was uneventful with spontaneous onset of labor. However, after twelve hours, the cardiotocograph showed two episodes with decelerations and bradycardia, and a small amount of green amniotic fluid was observed. Consequently, caesarean section was performed.
Apgar scores were eight after one minute, and ten after both two and five minutes. Birth weight (3200 g) and length (50 cm) were both at the 50th centile, and the head circumference was 34 cm (30th centile). Routine new-born physical examination was normal. He was breastfeed and body weight increased by 115 g when discharged from hospital at three days-of-life.
Seven days after birth, respirations stopped suddenly. Cardiopulmonary resuscitation was unsuccessful and death was declared upon arrival at our hospital. The parents reported that their baby had been sleeping more than anticipated, but otherwise seemed well. There was no history to suggest hypocalcaemia or respiratory distress.
Thirteen months later and at term, the parents had an apparently healthy baby girl with normal birth weight, length, and head circumference.
B) Post-mortem Studies
According to the Danish Health Law sudden, unexpected deaths must be reported to the police, who, as would be expected in such a case, requested a forensic autopsy at the Institute of Forensic Medicine, University of Southern Denmark. As is routine in sudden unexplained infant deaths, a full post-mortem radiographic skeletal survey of the boy was performed at Odense University Hospital, Denmark. Serum was not available for study. Biopsy of the Achilles tendon was used to harvest fibroblasts. Bone from a femur was fixed in formalin, decalcified, and then examined for histopathology.
C) Genetic studies
1) Genetic screening
After signed informed consent from the parents, the boy’s genomic DNA was obtained from his blood and eight osteopetrosis genes were tested by sequencing and by copy number variation analysis at a commercial laboratory (Centogene, Rostock, Germany). Then, DNA in peripheral blood leukocytes was obtained from the parents and their baby girl.
2) Trio-based whole-exome sequencing
When mutation analysis of the boy for osteopetrosis proved negative, we searched elsewhere for causal mutations using a trio-based whole-exome sequencing approach. DNA from the patient and the parents was subjected to exome capture using SureSelect Human All Exon V5 (Agilent Technologies, Glostrup, Denmark) and sequenced on the Illumina HiSeq 2000 platform by Oxford Gene Technology (Oxfordshire, United Kingdom). A mean coverage of 62–67× was obtained with 93–94 per cent of targeted bases covered with minimum 20x coverage. Raw reads were processed using the Burrows-Wheeler Alignment tool (BWA-MEM) v. 0.7.12[26] and the GATK Best Practice pipeline v. 3.3–0 was used for variant calling.[27] Annotation of variants was performed using Annovar.[28] Because both parents were healthy, an X-linked recessive, AR, or de novo AD transmission pattern was possible. Only rare coding variants and splicing variants were considered (minor allele frequency < 1%).
3) Sanger sequencing
The variant identified in the boy was then validated using Sanger sequencing. Exon 11 of RELA (RefSeq: NM_021975.3) was analyzed by bi-directional sequencing using the BigDye® Terminator v.3.1 cycle sequencing kit and an ABI3730XL capillary sequencer (Applied Biosystems, Naerum, Denmark). Primers used were RelAe11F: CCGGCCCCTCCCCAAGTC and RelAe11B: CACACCCCACCAGAATCCGTAAGT.
4) Protein sequence analysis
The three-dimensional organization of the altered p65 was evaluated by structure prediction using HHpred and visualizing in Protean using DNAStar v. 12.0 (DNASTAR, Madison, WI, USA).[29]
5) Other High Bone Mass Patients
To explore if five of our patients in St. Louis, USA who persisted to have unexplained HBM[30–32] harboured mutations in RELA, we performed Sanger sequencing using primers and annealing temperatures that we developed to amplify and sequence all 11 coding exons and adjacent mRNA splice sites. Some amplicons required a G-C-rich protocol (AccuPrime GC-Rich, Invitrogen, Walthan, MA, USA) (Supplementary Appendix, Table 1). Amplicons were sequenced in both directions using an ABI3130 (Applied Biosystems, Foster City, CA, USA) and analyzed using Sequencher software (Gene Codes Corp, Ann Arbor, MI, USA).
D) Cell studies
1 1) NF-κB reporter assay
2 Fibroblasts from the patient and normal controls were seeded at 12000/cm2 in a 96-well plate and infected with pLenti-NFκB-Luc reporter (Qiagen) (MOI=5). The following day, cells were treated with or without lipopolysaccharides (LPS) (1μg/ml) for 3 hours. The luciferase assay was performed by luciferase assay system (Promega) and normalized against the protein concentration measured by Pierce™ Coomassie (Bradford) Protein Assay Kit.
3 2) Gene expression analysis
Fibroblasts from the patient and normal controls were seeded in 24-well plates and then treated with or without LPS (1μg/ml) for 20 hours. Total RNA was isolated by GenElute mammalian total RNA miniprep kit (Sigma-Aldrich) and 1μg total RNAs were reverse transcribed by iScript™ cDNA synthesis kit (Bio-Rad). Real-time RT-PCR was performed using fast SYBR Green master mix (Applied Biosystem) on a StepOnePlus™ system (Applied Biosystem) according to the manufacturer’s protocol. Primers are listed in Supplementary Appendix, Table 2.
IV) Results
A) Post-mortem Findings
At the autopsy, there were no dysmorphological findings and no macroscopic pathological features were noted. A normal brain structure was observed without signs of increased intracranial pressure or incarceration. Histological studies revealed small hemorrhagic infarcts in periventricular areas, possibly birth labor-related. The bone marrow was normal and without signs of infection. Except for small areas of erythropoietic tissue in the liver, no microscopic abnormalities were observed in other organs including the thymus and spleen. Although HBM was clearly present, it was not extreme and the cause of death remained unknown.
B) Radiographic Findings
The skeleton was shaped normally (i.e., without errors of bone modeling), but diffusely osteosclerotic and with long bone cortices that were slightly thickened with less well-defined inner margins. The medullary cavities were osteosclerotic presumably with woven bone. The diffuse osteosclerosis was more than seen in normal newborns.
Specifically, the skull was osteosclerotic most evident at the base, but including the orbital roofs, sphenoid bones, basiocciput, and occiput (Figure 1A). The maxilla and mandible were osteosclerotic, and the frontal and occipital bones were thickened. The cervical bodies and neural arches were sclerotic.
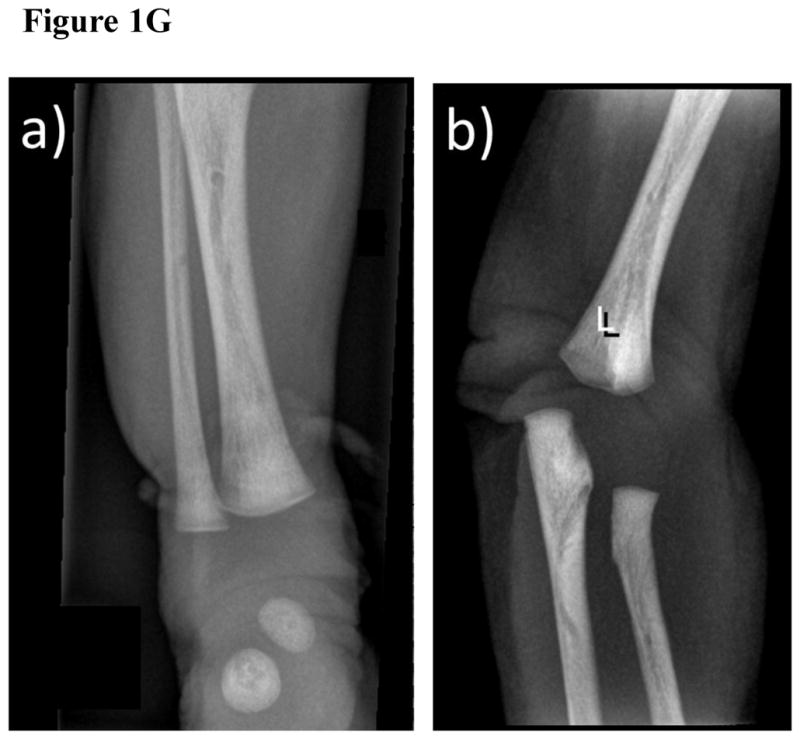
Figure 1. Radiological investigations.


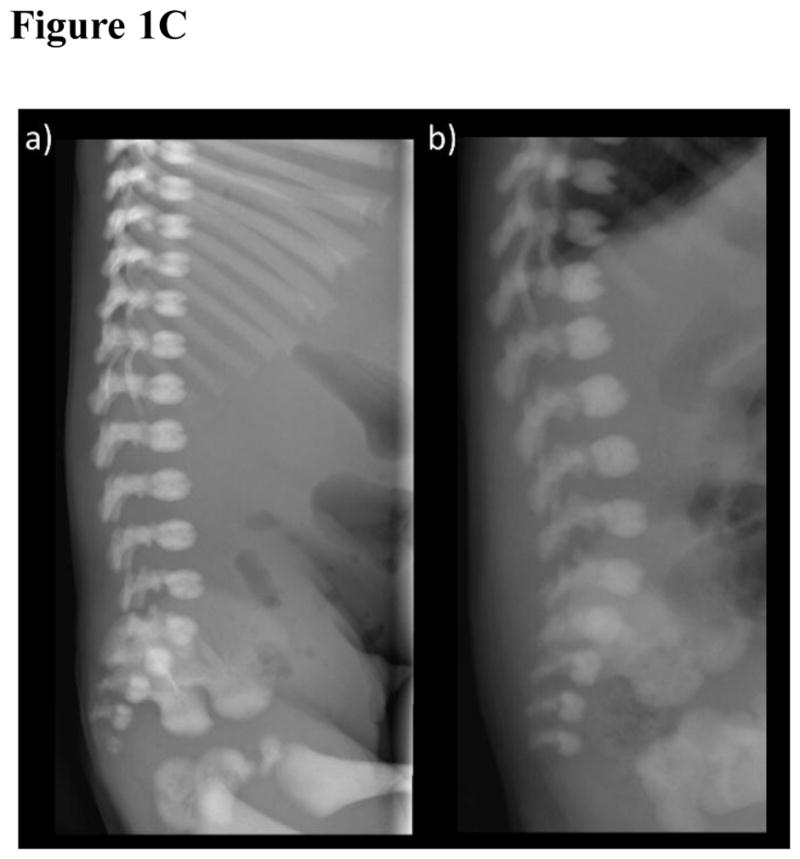



- The patient exhibits osteosclerosis greatest at the base including the orbital roofs. The cervical vertebral bodies and neural arches are sclerotic.
- Similar findings are seen in a 20-day-old infant with malignant osteopetrosis due to compound heterozygous TCIRG1 mutations.
- The patient has sclerotic ribs, clavicles, and scapulae. The lungs are airless and the thorax bell-shaped.
- The bones are sclerotic in this infant with malignant osteopetrosis.
- The patient shows diffuse osteosclerosis of the vertebral bodies and neural arches.
- Similar findings are noted in the infant with malignant osteopetrosis.
- The pelvic bones of the patient, including the proximal femora, show osteosclerosis with thickened inner pelvic margins.
- In this infant with malignant osteopetrosis, the proximal ends of the femora are irregular.
- The normal appearance of the left femur of a healthy 7 day-old neonate.
- In the patient, there is diffuse osteosclerosis with cortical thickening best seen in the midshaft. The provisional zones of calcification are sharp with normal-appearing physeal lines. There is minimal linear hypomineralization in the distal metaphysis.
- In the infant with malignant osteopetrosis, there is irregularity at the bone ends and wide physeal lines (“osteopetrorickets”), diffuse osteosclerosis, and poorly defined medullary cortices.
(F) Radiographs of the Right Knee
AP (a) and Lateral (b) views of the patient showing diffuse osteosclerosis, which is slightly patchy in the metaphysis, and normal physeal lines without irregularity. There is a small band of metaphyseal demineralization.
(c) In the infant with malignant osteopetrosis, in addition to the osteosclerosis the bone ends are irregular and the physeal lines are wide.
(G) Anteroposterior Radiograph of the Patient’s Right Ankle (A) and Left Elbow (B)
In addition to the overall osteosclerosis, there are areas of patchy increased osteosclerosis and cortical thickening.
The thorax was bell-shaped and the ribs diffusely osteosclerotic, and having faint medullary cavities (Figure 1B). The same sclerosis was seen in the ribs, clavicles and scapulae. The postmortem lungs were airless.
The spine had normally-shaped neural arches and vertebral bodies that were diffusedly osteosclerotic. The distal lumbar interpediculate distances flared normally. The odontoid was correctly developed. Vertebral bodies (Figure 1C) were slightly less dense in their mid portions with vascular channels present anteriorly, but not unduly prominent. Slight rounding was noted of the upper anterior surfaces. There was no “bone-in-bone” or “rugger jersey” appearance to suggest an osteopetrosis.
The pelvis was normally formed, having an overall increase in sclerosis with thickened inner pelvic margins (Figure 1D). A faint bone-in-bone appearance involved the iliac wings. The hips were seated, and the proximal femora quite sclerotic.
Long and short tubular bones were osteosclerotic, but normally modeled (Figure 1E, F). The proximal and distal thirds had greater osteosclerosis than normal newborn bones, and there was a poorly defined linear sclerotic pattern. There was a small band of metaphyseal demineralization on the right knee. Mottled irregular areas of sclerosis and thickened trabeculae were especially about the ankles and elbows (Figure 1G). In the middle third, cortices were thicker with not well-defined inner margins. Provisional zones of calcification were sharp, and not serrated or irregular as sometimes found in infantile (malignant) osteopetrosis.[33] A narrow band of hypomineralization was adjacent to the provisional zones of calcification. The medullary cavities, presumably containing woven bone, were osteosclerotic. The ossified femoral and tibial epiphyses were slightly osteosclerotic.
The hands and feet showed an overall increased osteosclerosis, more evident in the metacarpals and tarsals. The ends of the proximal phalanges of the hands, feet, and metacarpals had greater sclerosis than the mid-portions. The short tubular bones had medullary cavities. Cortices were thickened and irregular, especially in the metacarpals.
Overall, the skeletal appearance simulated infantile osteopetrosis, but medullary cavities were present in the long bones and ribs, and there were no irregularities in the bone ends to indicate “osteopetrorickets” that sometimes develops in severe osteopetrosis.[33]
C) Skeletal Histopathology
Bone harvested from the distal femur metaphysis was osteosclerotic and lacked clear distinction between the cortex and trabeculae (Figure 2A). Picrosirius red staining observed with polarization indicated a mixture of lamellar and woven bone throughout the specimen, suggesting increased and abnormal bone formation (Figure 2B). Additionally, osteocytes in many areas were disorganized and closely spaced, as usually seen in high-turnover woven bone (Figure 2C). In contrast to this evidence of increased bone formation, findings for a defect in OC-mediated bone resorption were largely lacking. Using toluidine blue staining, cartilage entrapped within bone (likely primary spongiosa) was identified in some areas of the specimen, within 3 mm of the periosteum (Figure 2D). However, the specimen did not contain any growth plate, and therefore cartilage distance from a growth plate was unknown. In a neonate, the observed cartilage may be normal. Additionally, the specimen showed abundant OCs associated with scalloping of the bone surface, indicating active bone resorption (Figure 2E). The bone marrow was normal, and showed normal hematopoietic elements and no fibrosis (Figure 2F). Overall, the histological appearance was more consistent with increased bone formation rather than OC failure and osteopetrosis.
Figure 2. Histology of Decalcified Femur Specimen.

(A) A low-power image of the H&E stain shows dense bone from cortex to cortex (top to bottom).
(B) Polarized image of picrosirius red stain in the center of the specimen demonstrates lamellar bone (appears solid red) admixed with woven bone (hatched red).
(C) A slightly magnified view of the area in B, stained with H&E, shows an irregular arrangement of osteocytes, with the more tightly grouped cells corresponding to areas of woven bone, and sparsely cellular areas with lamellar bone.
(D) Toluidine stained section near the periosteal surface (just out of the field to the right) shows some retained cartilaginous matrix (purple).
(E) Numerous morphologically normal osteoclasts are identified in the specimen on H&E stains, associated with scalloped bone surfaces indicative of active resorption.
(F) The small amount of bone marrow in the specimen shows normal hematopoietic elements, without marrow fibrosis.
Scale bars: A = 2 mm; B,D = 500 μm; C,F = 200 μm; E = 100 μm.
D) Genetic studies
Mutation analysis revealed no pathogenic defect in eight genes associated with osteopetrosis; i.e., chloride channel 7 (CLCN7), T-cell immune regulator 1 (TCIRG1), osteopetrosis-associated transmembrane protein 1 (OSTM1), pleckstrin homology domain-containing protein, family M, member 1 (PLEKHM1), sorting nexin 10 (SNX10), tumour necrosis factor receptor superfamily, member 11 A (TNFRSF11A), tumor necrosis factor ligand superfamily, member 11 (TNFSF11), and carbonic anhydrase II (CA2).[5] Nor was there a mutation in LRP5 that causes HBM in Worth-type endosteal hyperostosis,[34] in the recent past often erroneously considered an osteopetrosis (i.e., “autosomal dominant osteopetrosis, type 1”).[35]
Subsequent mutation analysis applying exome sequencing variant filtering identified six genes (CADPS2, FAT1, MKI67, ZFHX3, ZNF469, MUC16) containing compound heterozygote variants, and five hemizygous variants (MAP3K15, FAM47B, GPKOW, ATRX, SLITRK4) fulfilling X-linked recessive models. However, comprehensive review of published literature revealed no plausible link to osteopetrosis, or to other bone-related phenotypes. In particular, none of these genes are located up- or downstream of the NF-κB pathway. (Supplementary Appendix, Table 3). In contrast, filtering for de novo variants revealed one sporadic heterozygote variant (c.1534_1535delinsAG, numbering according to NM_021975.3, in RELA located on chromosome 11q13 (Figure 3). The p65 protein is known to have at least four isoforms all coded within the same gene resulting from alternative splicing, the predominant one being 551 amino acids long. This mutation predicted replacement of aspartate with serine at amino acid position 512 (p.Asp512Ser) within the C-terminal part of the protein.[36] Neither the specific mutation nor any other missense change at amino acid position 512 has been registered in the dbSNP database, Exome Variant Server (EVS), or The Exome Aggregation Consortium (ExAC) database.
Figure 3. Exome Sequencing. Alignment Reads of Exon 11 of RELA.

Visualization of aligned reads in exon 11 of RELA showing the de novo mutation c.1534_1535delinsAG (g.65421970_65421971delinsCT) detected as heterozygous in the patient and homozygous wildtype in his parents. Mutation reads are highlighted by yellow (C) and dark blue colors (T).
The patient’s mutation was not present in his parents or sister, and was validated with Sanger sequencing. (Figure 4). Evolutionary alignment for the RELA mutation was evaluated (Figure 5). None of our additional five patients with idiopathic HBM had an exon or splice-site RELA mutation.
Figure 4. RELA Mutation.

(A) Electropherograms of forward and reverse DNA sequence show the variant of the patient and the forward DNA sequence of his parents. The c.1534_1535delinsAG variant is indicated with an arrow.
(B) Normal and proband cDNA sequence (as codons) and amino acid sequence show the identified mutation RELA p.Asp512Ser (the altered nucleotide and amino acid changes are in bold and underlined).
Figure 5. Evolutionary Alignment For Patient’s RELA Mutation.

Alignment of the C-terminal part of RelA using Clustal W. Amino acids conserved relative to the human sequence are shaded grey. Boxed amino acids indicate TA1 and TA2, respectively. The arrow shows the position of Asp512Ser.
E) Cell studies
The effect of the patient’s RELA mutation on transduction of NF-κB signaling was examined using a NF-κB luciferase reporter assay. LPS induced higher levels of NF-κB activity in the control fibroblasts, whereas the patient’s fibroblasts did not respond to LPS stimulation. Consistent with the results from the luciferase assay, gene expression analysis on NF-κB target genes including IL8[37] and CD98[38] revealed that LPS did not elicit biological responses in the patient’s fibroblasts (Figure 7).
Figure 7. Patient RELA Mutation Response to NF-κB Activation in Fibroblasts.

(A) Cells were seeded at at 12000/cm2 in 96-well plate, infected with pLenti-NFκB-Luc reporter (Qiagen) (MOI=5), treated the following day without lipopolysaccharides (LPS) as control (Ctrl) or with 1μg/ml LPS for 3 hours, and then processed in a luciferase reporter assay. Data shown are mean+SD of five replicates.
(B) Fibroblasts were seeded in a 24-well plate, treated without LPS as control (Ctrl) or with 1μg/ml LPS for 20 hours, and then processed for real-time RT-PCR. The gene expression was normalized against GAPDH. Data shown are mean+SD of three replicates.
*P<0.05, **P<0.01 compared to Ctrl.
V) Discussion
Sequence analysis revealed that our patient carried a single (heterozygous), de novo, missense (p.Asp512Ser) variant in RELA. LPS stimulation test of his fibroblasts showed impaired NF-κB responses, indicating that the RELA mutation was pathogenic. To our knowledge, this is the first report of a mutation within the NF-κB complex in humans. Upon radiographic and then histological assessment of his skeleton, the generalized osteosclerosis appeared to be from excessive bone formation. Bone resorption seemed to be intact as evidenced by numerically and morphologically normal OCs associated with eroded bone surfaces. Despite his HBM, there was no accumulation of primary spongiosa, impairment of the bone marrow, compression of nerves, or fractures that would indicate poor-quality bone and osteopetrosis. Although he died at one week-of-life, a benign form of HBM, rather than infantile osteopetrosis seemed present. Molecular defects both up- and downstream of NF-κB are understood to alter skeletal homeostasis, but to date mutation of any of the five NF-κB subunits constituting the transcriptional complex has not been described in any Mendelian disorder.
Mice lacking RELA are embryonically lethal, in part explained by liver apoptosis.[39] Post-mortem assessment of our patient did not reveal any major hepatic abnormalities, and the cause of his death remains unknown. Chimeric mice engrafted with Rela−/− bone marrow cells are viable, but the number of OCs is reduced because the osteoclastogenic response to RANKL injection is diminished.[16] Although animal studies have established the pivotal role of p65 in impeding RANKL-induced OC apoptosis, thus allowing for OC differentiation,[16] and in inhibiting OB activity and differentiation,[8] knowledge concerning p65 function in humans is limited.
Our patient’s RELA variant predicts a polar but uncharged serine rather than a negatively charged aspartate in p65. Therefore, this substitution may have functional consequences due to their differing side groups. The NF-κB transcription factors join in various combinations of homo- and hetero-dimers, and have a structurally conserved N-terminal rel homology region (RHR) responsible for DNA binding and containing the dimerization domain (DD).[40] At its C-terminal, p65 has a trans-activating domain (TAD) essential for control of p65-regulated genes. The TAD is largely unstructured,[36] but this intrinsic domain (ID) is a common feature of eukaryotic TADs.[41] The ID confers functional flexibility, increasing the number of possible intermolecular protein–protein interactions. Numerous kinases can phosphorylate the TAD of p65. Commonly phosphorylated residues in p65 TAD are T435, S468, T505, S529 and S536.[36] Phosphorylation at any of these sites influences the ability of p65 to bind to downstream regulatory elements, contributing to either activation or inactivation of the target.[41–44] The TAD can be further subdivided in two alpha-helical regions, TA1 and TA2, which are anticipated to work in union. TA1 and TA2 are interspersed by linker-rich-in-proline residues. With our patient’s genetic variant S512, a serine replaces an aspartate within this linker (Figure 5 and 6). This newly inserted serine may be a phosphorylation site, and the rigid proline-rich region could stabilize the positioning of a phosphorylated serine to the same surface -- a hypothesis proposed for TA1.[45] Hence, our patient’s replacement amino acid having a different side group might alter the protein’s conformation thus impairing p65 action by an AD “loss-of-function” effect. Our unpublished data suggest, however, that partial loss of p65 function is unlikely to affect OCs, consistent with our observations in this patient.
Figure 6. The Three-Dimensional Structure of RelA.

The missense variant replaces aspartate with serine (pink coloured amino acid marked with black arrow) located in linker between TA1 (blue alpha helix) TA2 (grey alpha helix).
TNFα is among several factors that activate the NF-κB-mediated inhibition of OB differentiation via p65.[46] p65 mediates this inhibition by blocking the Sma and Mad-related protein (SMAD) induction of Runx-related transcription factor 2 (Runx2) induced DNA transcription.[23, 47] Additionally, p65 down-regulates OB activity by blocking c-Jun N-terminal kinases (JNK) stimulated Fra-1 expression.[19] Studies in vivo demonstrated that loss of TNFα stimulated NF-κB activity[48] increased OB differentiation, as did the blockade of NF-κB at the level of IκBαDN upstream of p65.[49] Furthermore, in the mature OB, inhibition of the classical NF-κB pathway resulted in higher bone mass in young mice,[19] whereas inhibition of NF-κB increased OB surface area, bone formation rate, trabecular bone volume, and reduced the number of OCs in ovariectomized mice.[19, 50] By contrast, overexpression of p65/p50, or a “gain-of-function” model, reduced OB differentiation.[48] These studies would favour our patient’s p.Asp512Ser RELA mutation having a dominant negative effect on p65-mediated inhibition of OB differentiation. Additionally, reduced p65 inhibition of JNK activity would promote Fra1 expression and increase OB activities, and thereby be consistent with our patient’s findings of excessive bone formation with normal OB morphology.
Having hundreds of target genes, the ubiquitous NF-κB signalling pathway potentially influences inflammation responses, autoimmunity, and cancer processes.[51, 52] Consequently, the patient’s RELA variant may have altered NF-κB signaling in tissues other than bone e.g. caused impairment of the immune response. The Toll-like receptor (TLR) is expressed in immature immune cells. When microbial pathogens bind to TLR, this activates the NF-κB-induced pro-inflammatory cytokine response.[53] In our patient, the autopsy revealed no macro- or microscopic signs of infection, sepsis, inflammation, or other pathological processes. Furthermore, additional blood cultures did not expose any evidence of infection.
Our investigation has limitations. In vitro tetracycline labelling was not possible for a dynamic quantitation of our patient’s bone formation rate. His serum or plasma was not available for biochemical studies to support accelerated bone formation. Although the RELA mutation seems pathogenetic, further functional analyses including additional cell or animal studies must provide evidence for HBM from this alteration in RELA.
In conclusion, we report the first mutation within the human NF-κB complex. HBM in the affected neonate occurred without bone marrow impairment or fractures. Radiographic and static histopathological findings of the skeleton indicated the HBM was from increased bone formation rather than a type of osteopetrosis from impaired numbers or function of OCs. Further studies to explore this mutation’s effects on NF-κB signalling as well as OB and OC differentiation and function are planned. This p.Asp512Ser mutation of p65 seems to explain a new HBM disorder.
Supplementary Material
Acknowledgments
Supported by Shriners Hospitals for Children, The Hypophosphatasia Research Fund at The Barnes-Jewish Hospital Foundation, the National Institute of Diabetes and Digestive and Kidney Diseases (DK067145) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (AR052705) of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Margaret Huskey and Shenghui Duan helped with RELA sequencing.
Footnotes
Author contributions:
Study initiator and coordinator: ALF
Study conduct: ALF, MF
Data collection: ALF, PJTK, HDS, CE, SM, WQ
Data analysis and interpretation: ALF, ML, KB, MF, SM, WQ, MK, WHM, MPW
Drafting manuscript: ALF, MF, DVN, MPW
Revising manuscript: DVN, MPW
Approving final version of manuscript: All authors.
Presented in part at the European Calcified Tissue Society/International Bone and Mineral Society (ECTS/IBMS) Joint Meeting 2015, April 25 - 28, 2015, Rotterdam, The Netherlands (The abstract will have a citation but we do not know where and when yet), and to be presented at the American Society for Bone and Mineral Research 2015 Annual Meeting, October 9 – 12, 2015, Seattle, Washington, USA.
Disclosures: None
Contributor Information
Martin J. Larsen, Email: Martin.Larsen@rsyd.dk.
Klaus Brusgaard, Email: Klaus.Brusgaard@rsyd.dk.
Deborah V. Novack, Email: Novack@wustl.edu.
Peter Juel Thiis Knudsen, Email: pthiis@health.sdu.dk.
Henrik Daa Schrøder, Email: Henrik.Daa.Schroeder@rsyd.dk.
Weimin Qiu, Email: wqiu@health.sdu.dk.
Christina Eckhardt, Email: Maria-Christina.Eckhardt@rsyd.dk.
William H. McAlister, Email: mcalisterw@mir.wustl.edu.
Moustapha Kassem, Email: mkassem@health.sdu.dk.
Steven Mumm, Email: smumm@dom.wustl.edu.
Morten Frost, Email: frostnielsen@yahoo.com.
Michael P. Whyte, Email: mwhyte@shrinenet.org.
VII) References
- 1.Whyte MP. Primer On Metabolic Bone Diseases and Disorders of Mineral Metabolism. American Society for Bone and Mineral Research, Wiley-Blackwell; 2013. Sclerosing Bone Disorders; pp. 769–785. [Google Scholar]
- 2.Rickels MR, Zhang X, Mumm S, Whyte MP. Oropharyngeal skeletal disease accompanying high bone mass and novel LRP5 mutation. J Bone Miner Res. 2005;20:878–85. doi: 10.1359/JBMR.041223. [DOI] [PubMed] [Google Scholar]
- 3.Del Fattore A, Cappariello A, Teti A. Genetics, pathogenesis and complications of osteopetrosis. Bone. 2008;42:19–29. doi: 10.1016/j.bone.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 4.de Vernejoul MC, Kornak U. Heritable sclerosing bone disorders: presentation and new molecular mechanisms. Ann N Y Acad Sci. 2010;1192:269–77. doi: 10.1111/j.1749-6632.2009.05244.x. [DOI] [PubMed] [Google Scholar]
- 5.Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol. 2013;9:522–36. doi: 10.1038/nrendo.2013.137. [DOI] [PubMed] [Google Scholar]
- 6.Crockett JC, Mellis DJ, Scott DI, Helfrich MH. New knowledge on critical osteoclast formation and activation pathways from study of rare genetic diseases of osteoclasts: focus on the RANK/RANKL axis. Osteoporos Int. 2011;22:1–20. doi: 10.1007/s00198-010-1272-8. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin AS. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 8.Novack DV. Role of NF-κB in the skeleton. Cell Res. 2011;21:169–82. doi: 10.1038/cr.2010.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene. 1999;18:6867–74. doi: 10.1038/sj.onc.1203219. [DOI] [PubMed] [Google Scholar]
- 10.Schreiber S, Nikolaus S, Hampe J. Activation of nuclear factor kappa B inflammatory bowel disease. Gut. 1998;42:477–84. doi: 10.1136/gut.42.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun SC. Non-canonical NF-κB signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh G, van Duyne G, Ghosh S, Sigler PB. Structure of NF-kappa B p50 homodimer bound to a kappa B site. Nature. 1995;373:303–10. doi: 10.1038/373303a0. [DOI] [PubMed] [Google Scholar]
- 13.Iotsova V, Caamaño J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997;3:1285–9. doi: 10.1038/nm1197-1285. [DOI] [PubMed] [Google Scholar]
- 14.Otero JE, Dai S, Foglia D, Alhawagri M, Vacher J, Pasparakis M, Abu-Amer Y. Defective osteoclastogenesis by IKKbeta-null precursors is a result of receptor activator of NF-kappaB ligand (RANKL)-induced JNK-dependent apoptosis and impaired differentiation. J Biol Chem. 2008;283:24546–53. doi: 10.1074/jbc.M800434200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaira S, Johnson T, Hirbe AC, Alhawagri M, Anwisye I, Sammut B, O’Neal J, Zou W, Weilbaecher KN, Faccio R, Novack DV. RelB is the NF-kappaB subunit downstream of NIK responsible for osteoclast differentiation. Proc Natl Acad Sci U S A. 2008;105:3897–902. doi: 10.1073/pnas.0708576105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaira S, Alhawagri M, Anwisye I, Kitaura H, Faccio R, Novack DV. RelA/p65 promotes osteoclast differentiation by blocking a RANKL-induced apoptotic JNK pathway in mice. J Clin Invest. 2008;118:2088–97. doi: 10.1172/JCI33392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clohisy JC, Yamanaka Y, Faccio R, Abu-Amer Y. Inhibition of IKK activation, through sequestering NEMO, blocks PMMA-induced osteoclastogenesis and calvarial inflammatory osteolysis. J Orthop Res. 2006;24:1358–65. doi: 10.1002/jor.20184. [DOI] [PubMed] [Google Scholar]
- 18.Yao Z, Li Y, Yin X, Dong Y, Xing L, Boyce BF. NF-κB RelB negatively regulates osteoblast differentiation and bone formation. J Bone Miner Res. 2014;29:866–77. doi: 10.1002/jbmr.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang J, Wang Z, Tang E, Fan Z, McCauley L, Franceschi R, Guan K, Krebsbach PH, Wang CY. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med. 2009;15:682–9. doi: 10.1038/nm.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swarnkar G, Zhang K, Mbalaviele G, Long F, Abu-Amer Y. Constitutive activation of IKK2/NF-κB impairs osteogenesis and skeletal development. PLoS One. 2014;9:e91421. doi: 10.1371/journal.pone.0091421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dupuis-Girod S, Corradini N, Hadj-Rabia S, Fournet JC, Faivre L, Le Deist F, Durand P, Döffinger R, Smahi A, Israel A, Courtois G, Brousse N, Blanche S, Munnich A, Fischer A, Casanova JL, Bodemer C. Osteopetrosis, lymphedema, anhidrotic ectodermal dysplasia, and immunodeficiency in a boy and incontinentia pigmenti in his mother. Pediatrics. 2002;109:e97. doi: 10.1542/peds.109.6.e97. [DOI] [PubMed] [Google Scholar]
- 22.Lopez-Granados E, Keenan JE, Kinney MC, Leo H, Jain N, Ma CA, Quinones R, Gelfand EW, Jain A. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum Mutat. 2008;29:861–8. doi: 10.1002/humu.20740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, Armstrong A, Shen V, Bain S, Cosman D, Anderson D, Morrissey PJ, Peschon JJ, Schuh J. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–24. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sobacchi C, Frattini A, Guerrini MM, Abinun M, Pangrazio A, Susani L, Bredius R, Mancini G, Cant A, Bishop N, Grabowski P, Del Fattore A, Messina C, Errigo G, Coxon FP, Scott DI, Teti A, Rogers MJ, Vezzoni P, Villa A, Helfrich MH. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet. 2007;39:960–2. doi: 10.1038/ng2076. [DOI] [PubMed] [Google Scholar]
- 25.Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A, Moratto D, Mazzolari E, Clayton-Smith J, Orchard P, Coxon FP, Helfrich MH, Crockett JC, Mellis D, Vellodi A, Tezcan I, Notarangelo LD, Rogers MJ, Vezzoni P, Villa A, Frattini A. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet. 2008;83:64–76. doi: 10.1016/j.ajhg.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H. Aligning sequence reads, clone sequences and assembly contains with BWA-MEM. Oxford Universtity Press; 2014. pp. 1–3. Available from: http://arxiv.org/abs/1303.3997. 2013. [Google Scholar]
- 27.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;11:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biegert A, Mayer C, Remmert M, Söding J, Lupas AN. The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res. 2006;34:W335–9. doi: 10.1093/nar/gkl217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whyte MP, Madson KL, Mumm S, McAlister WH, Novack DV, Blair JC, Helliwell TR, Stolina M, Abernethy LJ, Shaw NJ. Rapid skeletal turnover in a radiographic mimic of osteopetrosis. J Bone Miner Res. 2014;29:2601–9. doi: 10.1002/jbmr.2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tebben P, Wenkert D, Novack D, DiMeglio L, Sedighi H, McAlister W, Mumm S, Whyte M. A new pediatric sclerosing bone disorder featuring fragility fractures. J Bone Miner Res. 2006;21(Suppl 1):S432. [Google Scholar]
- 32.Eddy M, Gannon R, McAlister W, Green W, Whyte M. Polycystic osteosclerosis with hypercalcemia: A new disorder. J Bone Miner Res. 1999;14(Suppl 1):S447. [Google Scholar]
- 33.Kaplan FS, August CS, Fallon MD, Gannon F, Haddad JG. Osteopetrorickets. The paradox of plenty. Pathophysiology and treatment. Clin Orthop Relat Res. 1993:64–78. [PubMed] [Google Scholar]
- 34.Van Wesenbeeck L, Cleiren E, Gram J, Beals RK, Bénichou O, Scopelliti D, Key L, Renton T, Bartels C, Gong Y, Warman ML, De Vernejoul MC, Bollerslev J, Van Hul W. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet. 2003;72:763–71. doi: 10.1086/368277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bollerslev J, Henriksen K, Nielsen MF, Brixen K, Van Hul W. Autosomal dominant osteopetrosis revisited: lessons from recent studies. Eur J Endocrinol. 2013;169:R39–57. doi: 10.1530/EJE-13-0136. [DOI] [PubMed] [Google Scholar]
- 36.Schmitz ML, dos Santos Silva MA, Altmann H, Czisch M, Holak TA, Baeuerle PA. Structural and functional analysis of the NF-kappa B p65 C terminus. An acidic and modular transactivation domain with the potential to adopt an alpha-helical conformation. J Biol Chem. 1994;269:25613–20. [PubMed] [Google Scholar]
- 37.Kunsch C, Rosen CA. NF-kappa B subunit-specific regulation of the interleukin-8 promoter. Mol Cell Biol. 1993;13:6137–46. doi: 10.1128/mcb.13.10.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan Y, Dalmasso G, Sitaraman S, Merlin D. Characterization of the human intestinal CD98 promoter and its regulation by interferon-gamma. Am J Physiol Gastrointest Liver Physiol. 2007;292:G535–45. doi: 10.1152/ajpgi.00385.2006. [DOI] [PubMed] [Google Scholar]
- 39.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–70. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 40.Huxford T, Ghosh G. A structural guide to proteins of the NF-kappaB signaling module. Cold Spring Harb Perspect Biol. 2009;1:a000075. doi: 10.1101/cshperspect.a000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milanovic M, Kracht M, Schmitz ML. The cytokine-induced conformational switch of nuclear factor κB p65 is mediated by p65 phosphorylation. Biochem J. 2014;457:401–13. doi: 10.1042/BJ20130780. [DOI] [PubMed] [Google Scholar]
- 42.Hu J, Nakano H, Sakurai H, Colburn NH. Insufficient p65 phosphorylation at S536 specifically contributes to the lack of NF-kappaB activation and transformation in resistant JB6 cells. Carcinogenesis. 2004;25:1991–2003. doi: 10.1093/carcin/bgh198. [DOI] [PubMed] [Google Scholar]
- 43.Buss H, Handschick K, Jurrmann N, Pekkonen P, Beuerlein K, Müller H, Wait R, Saklatvala J, Ojala PM, Schmitz ML, Naumann M, Kracht M. Cyclin-dependent kinase 6 phosphorylates NF-κB P65 at serine 536 and contributes to the regulation of inflammatory gene expression. PLoS One. 2012;7:e51847. doi: 10.1371/journal.pone.0051847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, Nanes MS. Inhibition of osteoblast differentiation by tumor necrosis factor-alpha. Endocrinology. 2000;141:3956–64. doi: 10.1210/endo.141.11.7739. [DOI] [PubMed] [Google Scholar]
- 45.Schmitz ML, Baeuerle PA. The p65 subunit is responsible for the strong transcription activating potential of NF-kappa B. EMBO J. 1991;10:3805–17. doi: 10.1002/j.1460-2075.1991.tb04950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mattioli I, Geng H, Sebald A, Hodel M, Bucher C, Kracht M, Schmitz ML. Inducible phosphorylation of NF-kappa B p65 at serine 468 by T cell costimulation is mediated by IKK epsilon. J Biol Chem. 2006;281:6175–83. doi: 10.1074/jbc.M508045200. [DOI] [PubMed] [Google Scholar]
- 47.Yamazaki M, Fukushima H, Shin M, Katagiri T, Doi T, Takahashi T, Jimi E. Tumor necrosis factor alpha represses bone morphogenetic protein (BMP) signaling by interfering with the DNA binding of Smads through the activation of NF-kappaB. J Biol Chem. 2009;284:35987–95. doi: 10.1074/jbc.M109.070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Y, Li A, Strait K, Zhang H, Nanes MS, Weitzmann MN. Endogenous TNFalpha lowers maximum peak bone mass and inhibits osteoblastic Smad activation through NF-kappaB. J Bone Miner Res. 2007;22:646–55. doi: 10.1359/jbmr.070121. [DOI] [PubMed] [Google Scholar]
- 49.Eliseev RA, Schwarz EM, Zuscik MJ, O’Keefe RJ, Drissi H, Rosier RN. Smad7 mediates inhibition of Saos2 osteosarcoma cell differentiation by NFkappaB. Exp Cell Res. 2006;312:40–50. doi: 10.1016/j.yexcr.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 50.Alles N, Soysa NS, Hayashi J, Khan M, Shimoda A, Shimokawa H, Ritzeler O, Akiyoshi K, Aoki K, Ohya K. Suppression of NF-kappaB increases bone formation and ameliorates osteopenia in ovariectomized mice. Endocrinology. 2010;151:4626–34. doi: 10.1210/en.2010-0399. [DOI] [PubMed] [Google Scholar]
- 51.Brown KD, Claudio E, Siebenlist U. The roles of the classical and alternative nuclear factor-kappaB pathways: potential implications for autoimmunity and rheumatoid arthritis. Arthritis Res Ther. 2008;10:212. doi: 10.1186/ar2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Escárcega RO, Fuentes-Alexandro S, García-Carrasco M, Gatica A, Zamora A. The transcription factor nuclear factor-kappa B and cancer. Clin Oncol (R Coll Radiol) 2007;19:154–61. doi: 10.1016/j.clon.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 53.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.