

Abstract
The interface between implanted devices and their host tissue is complex and is often optimized for maximal integration and cell adhesion. However, this also gives a surface suitable for bacterial colonization. We have developed a novel method of modifying the surface at the material–tissue interface with an antimicrobial peptide (AMP) coating to allow cell attachment while inhibiting bacterial colonization. The technology reported here is a dual AMP coating. The dual coating consists of AMPs covalently bonded to the hydroxyapatite surface, followed by deposition of electrostatically bound AMPs. The dual approach gives an efficacious coating which is stable for over 12 months and can prevent colonization of the surface by both Gram-positive and Gram-negative bacteria.
Keywords: surface engineering, antimicrobial, device–tissue interface
1. Background
Infection is recognized as one of the leading complications of bone replacement implant operations within the field of orthopaedics, with 2.8% of patients contracting a surgical site infection [1]. While acute infections occur immediately, it is common for some infections, particularly those in prosthetic joints, to be detected over 1 year after implantation of the device. These infections are often a complex and challenging complication to prevent or treat [2].
The bacteria most commonly associated with infection in orthopaedic implants are Gram-positive bacteria—coagulase-negative Staphylococcus epidermidis, Staphylococcus aureus (both meticillin sensitive and resistant) and Enterococcus spp., which are responsible for 75% of all implant infections [3]—and Gram-negative bacteria—Pseudomonas aeruginosa. Current clinical practice for treating infection around an implant site is an extended course of antibiotics. However, systemic and oral administration of antibiotics often results in low availability at the site of infection [4]. Bacteria can also colonize the implant, forming a biofilm on the surface resulting in infection [5]. This type of infection is difficult to treat as the dense extracellular matrix associated with the biofilm acts as a barrier against detergents and antibiotics, with further complications presented by the rise of multi-resistant bacteria such as meticillin-resistant S. aureus [6]. Well-established infection around the implant results in necrosis of the surrounding tissue, often requiring surgical debridement. Extensive tissue necrosis breaks down the bone–implant interface, causing loosening of the implant and loss of limb function, which then requires surgical retrieval and replacement of the implant [7,8]. Implant retrieval not only incurs significant financial costs and patient discomfort but also results in further bone tissue loss, meaning that implant replacement can only be performed a limited number of times over the lifetime of the patient.
The use of titanium for the manufacture of bone replacement implants is well established as a result of its high strength and stiffness properties, along with its corrosion resistance and excellent biocompatibility in bone replacement applications [9]. Hydroxyapatite (HA) is a mineral with similar chemical composition to the mineral fraction of human bone and is used as a coating on metal implants to promote osseointegration [10,11]. Currently, HA coatings are being used to coat cement-less implants and to promote bone ingrowth [12]. Many reports suggest that HA coatings on metal implants precipitate faster than bone fixation, resulting in a reduction of pain and recovery time for implant patients [13,14]. Self-cleaning surfaces are well established in the natural world to prevent bacterial adhesion [15].
There is an array of technologies that can be used to deposit coatings on surfaces. These include the deposition of patterned HA [16,17], inorganic surfaces binding peptides [18] and the incorporation of silver nanoparticles [19]. Numerous strategies for incorporating antimicrobial agents with orthopaedic implants have been investigated, including: (i) coating titanium with polymers loaded with antibiotics for sustained release [20,21], (ii) deposition of antimicrobial ion coatings such as Ag or Cu [22,23], and (iii) altering the topography of the surface [24].
These approaches have also been exploited for incorporating antimicrobials into HA (and other calcium phosphate-based) coatings. Silver ions have been shown to display high antimicrobial efficiency and are easily incorporated into materials such as HA through an ion exchange process [25–27]. Antimicrobial agents have been added onto the implant surface using dip/spray coating, which relies on adsorption of the agent onto the implant surface. However, adsorbed molecules are often weakly bound to the implant surface and can be rapidly desorbed under physiological conditions. The leaching of the antimicrobial agent from the surface into the surrounding area following implantation may provide relatively short-term potency in the local tissue area [19]. However, rapid desorption of the antimicrobial agent correspondingly reduces the antimicrobial potency at the implant surface. Studies on titanium have shown that it is possible to use the strong association between the titanium oxide passivation layer and catechol chemistry to give stable binding of an antibiotic to the surface [21]. However, this chemistry is not directly translatable to HA. Also the inclusion of antibiotics such as cefotaxime is not desirable in a clinical setting as cefotaxime-resistant species are already established [28]. If an infection occurs from these bacteria, the coating would then be ineffectual.
Antimicrobial peptides (AMPs) have been identified as promising active agents against bacterial and fungal microorganisms due to their broad spectrum of activity coupled with low toxicity. However, clinical applications and commercial development of these compounds is still very limited [29]. Difficulties in their production, poor optimization of their properties and efficacy, together with high manufacturing costs have contributed to the slow transfer from research to clinical practice and development of commercial products [30]. The rise of antibiotic-resistant bacteria is stimulating significant interest in applying AMPs to biomedical devices and translating them to clinical use. The main sectors of application of AMPs are mainly those of prevention and control of infections in humans and animals, while other opportunities can also be foreseen such as food preservation systems and utilization in agricultural and environmental protection.
Antimicrobial coatings have many uses in medicine and include coatings for implants [31], catheters [32] and dressings for wounds [33]. There are several AMP compounds that are undergoing clinical trials in combined antimicrobial and immune-modulatory functions. However, at present no drugs based on AMPs have been approved [34]. Owing to their chemical nature (peptides), oral and intravenous administration of AMPs poses problems due to the possible reduction or neutralization of the active ingredient or induction of an allergic reaction [35]. On the other hand, AMP-derived drugs appear very promising compounds for topical formulations; for example, for the treatment of skin burns/lesions, open wounds and the protection of implanted devices ranging from catheters to contact lenses, stents and artificial tissue substitute applications. Natural methods of combating infection in the body include defence proteins such as defensins. Defensins are small proteins purified from granulocytes that display high levels of antimicrobial activity [36]. Defensins counteract bacterial invasion of the body through direct and indirect pathways. The direct pathway involves attacking the invading pathogen by inducing pore formation in the membrane of the cell, leading to cell lysis [37]. Novel synthetic peptides based on plant defensins have been designed and shown to exhibit good levels of antimicrobial activity [38]. Scudiero et al. [39] used human β-defensins as a model for novel synthetic peptides and demonstrated good antimicrobial activity. However, this activity was reliant on the localized ion concentration, although the synthetic analogues were not as susceptible to this effect as the natural defensin.
In this study, we demonstrate an alternative method for imparting antimicrobial activity to orthopaedic implants. The method uses two mechanisms in tandem for binding a defensin-based AMP to a HA surface with the aim of both providing a robust long-term antimicrobial coating of the implant and facilitating partial release of the antimicrobial payload into the surrounding tissue. This uses two different methodologies: a covalent attachment of the peptide (cAMP) that gives a ‘permanent’ coating and an electrostatic attachment of the peptide (eAMP) that is released from this short-term surface coating into the area surrounding the implant, sterilizing the tissue around the implant. Employing both coatings simultaneously on the same surface gives a dual coating (dAMP) that has allowed us to create a multi-functional antimicrobial surface, which can sterilize the surgical site and prevent colonization of the implant for an extended period of time.
2. Material and methods
2.1. Materials
All reagents were purchased from Sigma (Poole, UK), unless otherwise stated. Peptides were purchased from Genscript (Piscataway, NJ, USA). Bacteria were obtained as isolates from patients in the Queen Elizabeth Hospital, Birmingham, UK.
2.2. Hydroxyapatite synthesis
HA (for both electrostatic and covalent studies) was prepared using the following steps.
Aqueous solutions of calcium nitrate 1 M (Fisher Scientific UK Ltd, Loughborough, UK) and diammonium hydrogen phosphate dibasic 0.6 M were prepared and the pH adjusted to 10 through dropwise addition of 37–39% ammonium hydroxide. The phosphate salt solution was added dropwise to the calcium salt solution using a burette while stirring and maintaining a pH of 10 through further addition of ammonium hydroxide. After ageing overnight under stirring, the nanocrystals were washed by five cycles of centrifugation at 4000 r.p.m. and resuspension with deionized water before filtering. The typical yield of solid product was approximately 30 g, with 1 g resuspended at 1 mg ml–1 in deionized water adjusted to pH 7 and dried overnight in an oven at 60°C to give HA as a powder.
HA pellets for electrostatic studies were prepared using dried HA powder mixed with deionized water (830 µl for each gram of HA) to form a thick paste. The paste was put into a homemade polytetrafluoroethylene mould and incubated at 60°C overnight before sintering at 1100°C for 6 h with a temperature ramp rate of 5°C min−1.
Thiol-functionalized hydroxyapatite (tHA) was prepared for covalent HA studies. The thiol functionalization solution consisted of 3% (v/v) (3-mercaptopropyl) trimethoxysilane (MPTS) stock solution, 3% (v/v) ultra-pure deionized water and 94% (v/v) methanol. The water–methanol mixture was prepared before adding the MPTS. The pH was adjusted slowly to 4 by addition of 1 M hydrochloric acid and left to hydrolyse for 4 h. The hydrolysed MPTS solution was then pH adjusted to 7 by addition of ammonium hydroxide before adding the cut HA pellets and leaving it to react overnight while on a flat shaker plate running at 300 r.p.m. (ThermoScientific Inc., Waltham, MA, USA).
No more than 30 pellets were placed in each reaction bottle at a time to ensure adequate separation and coating of the pellets. The pellets were carefully washed five times in 20% methanol before drying in an oven for 2 h at 60°C.
2.3. eAMP coating of HA
The moulded HA pellets which had been prepared as described above were incubated in eAMP (5(6)-carboxyfluorescein (5(6)-CF))–RRRRRRGALAGRRRRRRGALAGEEEEEEE (0.9 mM) in phosphate-buffered saline (PBS) for 30 min. The coated pellets were then removed from the solution and washed five times with PBS and stored at room temperature before being used for studies.
2.4. cAMP coating of hydroxyapatite
tHA pellets which had been prepared as described above were incubated in N,N-dimethylformamide (DMF) (10 ml), succinimidyl trans-4-(maleimidylmethyl)cyclohexane-1-carboxylate (SMCC) (0.2 M) and the cAMP (5(6)-CF)–RRRRRRGALAGRRRRRRGALAG (0.9 mM) for 12 h with shaking. The covalently coated pellets were removed from the solution and washed five times with DMF and five times with PBS before being used in the studies.
2.5. dAMP coating of thiol–hydroxyapatite
tHA pellets which had been prepared as described above were incubated in DMF (10 ml), SMCC (0.2 M) and the cAMP (5(6)-CF)--RRRRRRGALAGRRRRRRGALAG (0.9 mM) for 12 h with shaking. The covalently coated pellets were removed from the solution and washed five times with DMF and five times with PBS. The pellets were then incubated in eAMP (5(6)-CF)–RRRRRRGALAGRRRRRRGALAGEEEEEEE (0.9 mM) in PBS for 30 min. The coated pellets were then removed from the solution and washed five times with PBS and stored at room temperature before being used for studies. Control pellets were uncoated HA which had been washed in DMF (5 × 1 ml) and PBS (5 × 1 ml).
2.6. AMP coating of hydroxyapatite
As a control for release studies HA pellets were coated with (5(6)-CF)–RRRRRRGALAGRRRRRRGALAG. The peptide was added to PBS (10 ml). HA pellets were added to the solution and incubated for 12 h with shaking. The pellets were removed from solution and washed five times with PBS.
2.7. Quantification of peptide on hydroxyapatite
Pellets were coated as described above. The amount of peptide remaining in the reaction vessel and the washes was quantified using fluorescence of the 5(6)-CF tag. This was then used to calculate the remaining peptide which had been immobilized on the surface.
2.8. Release of AMP from eAMP and cAMP surfaces
eAMP-, cAMP- and AMP-coated pellets were incubated in PBS (1 ml) at 37°C and the solution was changed at each time point. The solutions were stored in the dark at 4°C and the fluorescence of the 5(6)-CF tag was monitored at excitation/emission 495/525 nm on a Wallac 1420 plate reader (Perkin Elmer, London, UK). Percentage values were calculated from the known concentration immobilized on the surface.
2.9. Stability of dAMP coatings
HA pellets were coated as described above. Twelve pellets were split into four treatment groups of three samples: (1) HA, (2) eAMP–HA, (3) tHA, and (4) cAMP–tHA. The surface of the pellets was imaged immediately following the coating procedure to ensure that the coating was present. The pellets were then incubated for 12 months at room temperature (20–24°C) in simulated body fluid prepared according to the literature [40].
At the 7 day, 14 day, 1 month and 12 month time points, the pellets were removed from the simulated body fluid solution, dried and viewed with an Axioplan-2 fluorescence microscope and images of their surfaces obtained with Axiovision software (both Carl Zeiss Ltd, Cambridge, UK) to detect residual fluorescent dAMP. Peptide on the surface was quantified using established methods [41]. Briefly, each of the pellets was imaged on nine different areas of the surface (total 27 images per treatment). The levels of the peptide on the surface were quantified using Image J (National Institutes of Health). All images were taken at the same exposure, and the percentage of fluorescent pixels above a standardized background threshold was calculated using Image J.
2.10. Proteolytic stability
cAMP-coated pellets were coated as described and the levels of fluorescence on the surfaces quantified. The pellets were incubated in trypsin–ethylenediaminetetraacetic acid (1 ml, 0.02 M) or human leucocyte elastase (1 ml, 0.6 µg ml−1; Merck Millipore, Nottingham, UK) for 2 h. The pellets were removed from the solution and the surfaces washed three times in PBS (1 ml). The pellets were air dried in the dark and imaged and quantified as previously described.
2.11. Scanning electron microscopy and energy-dispersive X-ray microscopy
2.11.1. Spectroscopy of surfaces
Surfaces were prepared as previously described for fluorescence microscopy. The samples were studied on a Philips XL30 FEG scanning electron microscope. Imaging was carried out in triplicate and the images shown are representative of each surface. After scanning electron microscopy (SEM) samples were submitted for energy-dispersive X-ray spectroscopy (EDX). Samples were run in triplicate and spectrum sites were chosen at random across each surface. The data presented are the averages of all spectra.
2.11.2. Antimicrobial efficacy testing of AMP on laboratory strains of bacteria
The minimum inhibitory concentration (MIC) of the prepared AMP against various bacteria was determined by the broth dilution method according to the recommendations of the British Society of Antimicrobial Chemotherapy [42]. Briefly, serial dilutions of peptide solutions were prepared at 3.7 to 0.002 mM concentrations made up in lysogeny (LB) broth and tested against S. aureus and S. epidermidis; the lowest concentration required to inhibit bacterial growth was used.
2.12. Antimicrobial resistance studies
Bacterial resistance studies were then carried out by incubating each bacterium in LB broth supplemented with 50% of the concentration of the peptide required to obtain an MIC concentration. Incubation was carried out at 37°C for 12 h and the MIC test was repeated on the pre-treated bacteria as described above.
Further resistance studies were carried out by incubating each bacterium with 0.003 mM AMP and then doubling the concentration of AMP every 24 h until half the MIC value was reached. At each time point, bacteria were sampled using the MIC assay and the MIC noted. These studies allowed us to establish how easily resistance might be developed to the AMP peptide.
2.12.1. Antimicrobial tests of AMP surfaces
Antimicrobial-coated HA surfaces were tested by incubating cAMP–tHA/dAMP–tHA and uncoated pellets were inoculated with 10 µl of approximately 109 colony-forming units (cfu) per millilitre of coagulase-negative S. epidermidis, S. aureus and P. aeruginosa. Pellets were incubated for 24 h at 37°C. The optical density of LB broth around the pellet was measured at 600 nm using a spectrophotometer (Jenway, Stone, UK). The pellet surface was then swabbed and plated out on LB agar. The plates were incubated at 37°C for 24 h and the number of visible colonies on the plate counted.
2.12.2. Effect of serum incubation on antimicrobial efficacy
dAMP pellets were prepared as previously described. The pellets were incubated in fetal calf serum (1 ml) for 4 h. The pellets were washed with PBS and then the antimicrobial efficacy was tested against S. aureus as described above.
2.12.3. Long-term antimicrobial studies
HA and dAMP–tHA-coated pellets were incubated in LB broth (1 ml) and inoculated with 10 µl of approximately 109 cfu ml−1 of coagulase-negative S. epidermidis. At 7, 14 and 21 days, the broth was tested for optical density to determine bacterial number and the surfaces swabbed and plated out on LB agar. The agar plates were incubated at 37°C overnight and the colonies counted.
2.13. Toxicity studies of AMP and dAMP with mammalian cells
Toxicity studies were carried out using osteoblast precursor cells (MC3T3). Cells were detached from a confluent flask and seeded at 50 000 cells per well. The medium (Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS; 10% v/v), penicillin/streptomycin (1% v/v)) was supplemented with 37 µg of peptide per well. At each time point, the cells were detached from the plate using trypsin (200 µl). The cells were counted using a haemocytometer.
dAMP–tHA was placed in a 12-well plate and seeded with 50 000 cells and covered with medium (1 ml). Samples were prepared separately for resazurin and cell counts. Samples for resazurin had the medium removed and were washed with PBS. The pellets were resuspended in fresh medium (1 ml) with resazurin (20 µl, 15 mg ml−1) and incubated for 2 h at 37°C. Aliquots (200 µl) of the medium were removed and added to a 96-well plate. The absorbance was read at 570 nm on a Wallac Victor 3 1420 multilabel counter. Samples for cell number had the medium removed and the pellet incubated in trypsin (200 µl) for 5 min at 37°C. The cell suspension was added to a haemocytometer and counted.
Toxicity studies were carried out using SAOS-2 cells (osteosarcoma cells, ECACC; used at passages 10–14).
Cells were detached from a confluent flask and seeded at 5000 cells per well. The medium (DMEM, FBS (10% v/v), penicillin/streptomycin (1% v/v)) was supplemented with 37 µg of peptide per well. At each time point, the cells were detached from the plate using trypsin (200 µl). The cells were counted using a haemocytometer.
Electrostatic and covalently coated HA pellets were prepared as previously described. SAOS-2 cells were seeded onto dAMP-coated and -uncoated HA pellets at a density of 5000 cells per pellet in 500 µl of McCoys 5a medium supplemented with 10% FBS and 1% penicillin/streptomycin and the medium changed every 3–4 days. At each sample time point, the HA pellets with their attached SAOS-2 cells were removed from the medium and gently washed with PBS (3 × 1 ml). The cells growing on the HA pellets were then tested for metabolic activity using alamarBlue following established protocols [33]. Briefly, alamarBlue dye (10 µl) was added to the medium surrounding the cell-coated pellets and incubated for 2 h and then the absorbance of the medium was read at 570 nm on a Glomax multi-detection system (Promega, Southampton, UK). The cell-bearing HA pellets were washed in PBS to remove any dead cells, then placed in sterile water and frozen at –80°C. The samples were subjected to a three-cycle freeze–thaw process to lyse the cells and release their DNA into solution, and the DNA content of each sample was measured using Hoechst 33342 dye to assess SAOS-2 cell survival [43].
2.14. Statistics
All statistical analyses were carried out using SPSS 17.0 (IBM SPSS Inc., Chicago, IL, USA), and data are presented as the mean ± s.e.m. The Shapiro–Wilk test was used to ensure that all data were normally distributed before parametric testing using a one-way ANOVA with a Tukey post hoc test or a Mann–Whitney U-test. The statistical significance threshold was p < 0.05.
3. Results
3.1. Peptide design and synthesis
AMPs were designed using sequence matching from human α- and β-defensins. Amino acid residue properties including positive/negative charges and hydrophilic/hydrophobic patterns were compared across the different defensins and condensed into a small peptide sequence pattern which was then synthesized using solid-phase peptide synthesis (figure 1). All peptides were additionally tagged with 5(6)-CF to allow visualization of the peptide on attachment surfaces and peptide quantification. The individual peptide sequences generated provided a toolkit of activity (antimicrobial activity, surface binding activity or both antimicrobial and surface binding activity) which allowed a mix-and-match approach to building candidate peptides for binding to specific surfaces (e.g. the RRRRRRGALAGRRRRRRGALAGGGGEEEEEEE peptide sequence gave a broad-spectrum high activity AMP which binds to HA electrostatically).
Figure 1.
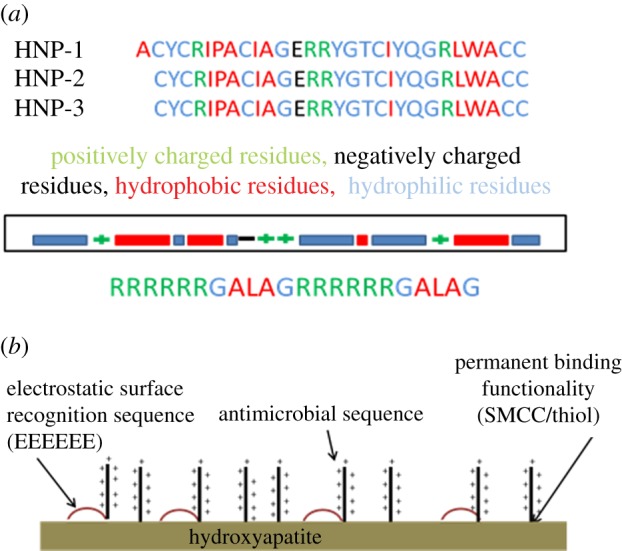
(a) The sequence of human defensins α1, 2 and 3 with the nature of the amino acids highlighted. The main patterns are then shown pictorially. The sequences of the first- and second-generation peptides are shown with the nature of the amino acids indicated by colour. (b) The attachment of the peptide to the HA through both electrostatic binding and covalent linkages.
The cAMP coating was characterized using EDX (table 1). These data showed that there is a significant increase in the presence of Si between HA (0 ± 0) and tHA (0.2 ± 0.08) (p = 0.002). This was mirrored by the increase in sulfur with HA (0 ± 0), tHA (0.37 ± 0.10) (p = 0.000). The increase in sulfur can be attributed to the sulfhydryl groups of MPTS, demonstrating thiol modification of the surface. This was further supported by Raman spectroscopy (see the electronic supplementary material) that showed a peak at 2572 cm–1, which corresponds to the S–H stretching mode, as well as 2890 and 2923 cm–1, which are the CH2 vibrations of the propyl chain of the MPTS. The cAMP–tHA was characterized by EDX. The spectra showed a significant increase in the amount of carbon present on the surface between tHA (8.78 ± 0.44) and the cAMP–tHA (25.95 ± 4.76) (p < 0.000; table 1). The increase in carbon is consistent with the incorporation of the peptide onto the surface. The amount of peptide incorporated onto the surface was determined to be 0.54 mol cm−2 (cAMP) and 1.78 mol cm−2 (eAMP). The calculations also showed a 1 : 1.2 cAMP to eAMP ratio in dAMP-coated pellets.
Table 1.
Elemental composition (% wt) of the substrate and functionalized substrate as determined through EDX measurements. Values shown are the averages ±s.d. of five readings taken at different points across the surface of each pellet.
| Ca | P | C | O | Si | S | |
|---|---|---|---|---|---|---|
| HA | 29.39 ± 0.64 | 16.74 ± 0.74 | 6.93 ± 0.35 | 46.94 ± 0.44 | 0 ± 0 | 0 ± 0 |
| HA–thiol modified | 31.62 ± 0.93 | 16.25 ± 0.43 | 8.78 ± 0.44 | 42.40 ± 1.13 | 0.20 ± 0.08 | 0.37 ± 0.10 |
| HA–dAMP | 19.50 ± 2.72 | 11.94 ± 1.28 | 25.95 ± 4.76 | 41.99 ± 1.76 | 0.24 ± 0.04 | 0.39 ± 0.04 |
Raman spectroscopy was also used to examine the composition of the HA to determine whether tricalcium phosphate (TCP) was present (electronic supplementary material). The spectra showed HA peaks with slight peak broadening, suggesting a small amount of TCP was present in the pellets.
Surfaces with either eAMP or cAMP coating were imaged for surface coverage using fluorescence microscopy (figure 2a–f), which showed that AMP was successfully bound to the surface with good coverage immediately following both covalent and electrostatic attachment. The coated HA pellets were incubated in simulated body fluid for one month with regular changes of fluid before fluorescent reimaging to visualize residual AMP (both eAMP and cAMP), and this showed that at this time point all of the electrostatically bound peptide was removed from the HA surface (figure 2a–c). However, with covalent functionalization there was still coverage of the cAMP on the tHA surface following the incubation period (figure 2d–f).
Figure 2.

Fluorescence microscopy images of: (a) HA; (b) HA coated with electrostatic coating, imaged immediately after washing following coating; (c) HA coated with electrostatic coating, after samples had been stored in simulated body fluid for one month with changes every 7 days; (d) thiol-modified HA; (e) thiol-modified HA with covalent coating, imaged immediately after washing; (f) thiol-modified HA with covalent coating, after samples had been stored in simulated body fluid for one month with changes every 7 days before. All images are representative of all sample surfaces. (g) Quantification of the fluorescence on the covalently coated, thiol-modified HA over time, showing the average surface pixel count, n = 3, and error bars show the standard error of the mean. (h) Quantification of the fluorescence on the covalently coated, thiol-modified HA after treatment with proteases, n = 3, and error bars show the standard error of the mean. (i) Release from the eAMP, cAMP and AMP coatings from the surface over 96 h, n = 3, error bars show the standard error of the mean.
Incorporation of the binding sequences into the AMP structure allowed the electrostatic binding to HA surfaces to give a sustained peptide release profile over 24 h. The release profile showed that 100% of the eAMP was released over 96 h (figure 2i). This is significantly higher than cAMP samples, which showed no release in the same time period, while the AMP coating with no specific attachment method to the HA showed 100% release at 8 h. The long-term stability of the coating was examined after incubation in PBS for 12 months (figure 2g). This showed no significant differences in the levels of fluorescence immediately after coating (1.3 × 106 ± 7.2 × 104) compared with after incubation for 12 months (1.2 × 106 ± 3.8 × 104). The surfaces were also shown to be resistant to proteolysis from both human leucocyte elastase (1.3 × 106 ± 2.3 × 104) and trypsin (1.3 × 106 ± 5.6 × 104) (figure 2h). All the coated samples showed significantly higher fluorescence than the uncoated samples (83 ± 85).
The surfaces were also characterized by SEM (electronic supplementary material, figure S2a–c). This showed that there were no differences in HA structure between HA, tHA and dAMP–tHA. The images show crystalline structures consistent with HA.
3.2. Antimicrobial efficiency of antimicrobial peptide sequences in solution
The AMP sequence (RRRRRRGALAGRRRRRRGALAG) was tested in solution against a broad spectrum of bacterial species including clinical isolates (table 2). The peptide was effective against all the species. The peptide showed effectiveness at slightly lower concentrations for Gram-positive bacteria (S. aureus, both meticillin-sensitive and -resistant, S. epidermidis and Enterococcus spp.), with MICs ranging between 0.08 and 0.52 mM, than with Gram-negative bacteria (P. aeruginosa, Klebsiella pneumoniae, Escherichia coli and Enterobacter cloacae), which had MICs between 0.67 and 0.95 mM.
Table 2.
MIC of AMP against clinically isolated bacteria.
| bacterium | MIC (mM) ± s.e.m. |
|---|---|
| P. aeruginosa | 0.87 ± 0.19 |
| K. pneumoniae | 0.95 ± 0.13 |
| E. coli | 0.67 ± 0.18 |
| E. cloacae | 0.74 ± 0.18 |
| S. aureus (meticillin-sensitive) | 0.37 ± 0.03 |
| S. aureus (meticillin-resistant) | 0.35 ± 0.07 |
| S. epidermidis (coagulase-negative) | 0.08 ± 0.03 |
| Enterococcus spp. | 0.52 ± 0.31 |
AMP was tested against bacteria commonly found in implant infections (S. aureus and S. epidermidis) to try to ascertain its ability to generate resistant bacteria. When the bacteria were preconditioned in two different ways to the peptide to generate resistance, no resistance was observed. However, the pre-treatment of the bacteria with peptide did affect the MIC value for the peptide in each strain. Incubation overnight of the bacteria in half the MIC of the peptide did reduce the MIC concentration in S. epidermidis (table 3). Consecutive incubation of the bacteria with increasing concentrations of peptide resulted in the MIC decreasing at each concentration for both S. aureus and S. epidermidis (figure 3).
Table 3.
The effect of peptide pre-incubation with bacteria on MIC.
| bacterium | MIC (naive bacteria) mM | MIC (preconditioned 24 h) mM |
|---|---|---|
| S. aureus (NCTC 8532) | 0.9 | 0.9 |
| S. epidermidis (C8—clinical isolate) | 0.9 | 0.2 |
Figure 3.

MIC for S. aureus and S. epidermidis after incubation in increasing concentrations of AMP, shown as a percentage of the initial MIC required to inhibit growth in naive bacteria.
3.3. Antimicrobial efficacy of the AMP coating against clinical isolates
eAMP-, cAMP- and dAMP-coated and -uncoated HA were tested to determine the efficacy of the surface against clinical isolates commonly found in orthopaedic infections. The surfaces were trialled against S. aureus, S. epidermidis and P. aeruginosa, which are the most commonly found bacteria in orthopaedic infections (figure 4). The data demonstrated that both the cAMP and dAMP could inhibit the growth of bacteria on the HA surfaces (figure 4a) while the eAMP coating alone could not significantly reduce bacterial colonization of the surface. Swabs of both cAMP and dAMP surfaces showed no colony-forming units against any of the bacteria tested (figure 4a). However, the cAMP coating could only reduce the levels of planktonic bacteria in solution around the surface, while the eAMP and dAMP coatings could successfully inhibit planktonic growth of the bacteria in solution and bacterial colonization on the surface (figure 4b).
Figure 4.

(a) The number of colony-forming units found on cAMP–tHA, dAMP–tHA or tHA after incubation with S. aureus, S. epidermidis and P. aeruginosa. Error bars show standard error of the mean, n = 6, ***p < 0.000. (b) The absorbance of the bacteria in broth surrounding the surface at 600 nm for S. epidermidis, S. aureus and P. aeruginosa in the solution surrounding a cAMP-tHA/eAMP-tHA/dAMP-tHA or tHA. Error bars show standard error of the mean, n = 6, **p < 0.001, *p < 0.05.
3.4. Efficacy of the coating after incubation in serum
Following incubation in serum the efficacy of the dAMP at inhibiting growth of bacteria in solution was not significantly different from dAMP-coated HA which had not been exposed to serum. Both surfaces were significantly lower than uncoated HA (p < 0.001; figure 5a). However, following incubation in serum the efficacy of inhibiting bacteria on the surface was slightly, although not significantly, reduced when compared with dAMP-coated HA which had not been exposed to serum (figure 5b). Both dAMP-coated HA and dAMP-coated HA that had been incubated in serum had significantly lower bacterial colonization than uncoated HA (p < 0.001).
Figure 5.

(a) The number of colony-forming units of S. aureus found on dAMP–tHA or tHA with and without a 4 h incubation in serum. Error bars show standard error of the mean, n = 6, ***p < 0.000. (b) The absorbance of the bacteria in broth surrounding the surface at 600 nm of dAMP–tHA or tHA with and without a 4 h incubation in serum. Error bars show standard error of the mean, n = 6, ***p < 0.001, *p < 0.05.
3.5. Long-term antimicrobial efficacy against S. epidermidis
dAMP–tHA pellets were incubated with S. epidermidis over three weeks. The optical density of the solution surrounding the pellets was measured each week. This showed that HA surfaces had a significantly higher number of bacteria with optical densities of 0.12 ± 0.02 at week 1, increasing to 0.27 ± 0.04 at week 2 and 0.35 ± 0.06 at week 3 than dAMP–tHA surfaces at week 1 (0.02 ± 0.01), week 2 (0.05 ± 0.02) and week 3 (0.09 ± 0.02) (figure 6a). After three weeks, the surfaces were removed from the LB broth and swabs of the surfaces were plated to show that the dAMP–tHA surfaces still had not been colonized by the bacteria in this time compared with HA surfaces, which showed high bacterial levels (5.8 × 104 ± 5.3 × 104; figure 6b).
Figure 6.

(a) The survival of S. epidermidis in the solution surrounding the dAMP–tHA pellet at 1, 2 and 3 weeks compared with HA measured using optical density. (b) Survival of S. epidermidis on the surface of the dAMP–tHA pellet compared with the HA pellet after three weeks in culture. Error bars show standard error of the mean, n = 6.
3.6. Toxicity of the peptide in culture with mammalian cells
The toxicity of the peptide alone was tested in both osteoblast precursor cells and osteosarcoma cells (figure 7a,b). The peptide demonstrated no toxicity in either cell type over a 7 day period. In osteoblast precursor cells, the cell number increased steadily over one week from 1.7 × 105 ± 2.5 × 105 (control) and 2.0 × 105 ± 1.5 × 105 (peptide) at day 1 to 1.7 × 106 ± 1.7 × 105 (control) and 1.7 × 106 ± 1.2 × 105 (peptide) as the cells reach confluency at day 7. In osteosarcoma cells, the cell number increased from 9895 ± 96 (control) and 9391 ± 113 (peptide) at day 1 to day 3 29 880 ± 598 (control) and 23 149 ± 620 (peptide). The cell number plateaued over the following 4 days with final readings of 23 119 ± 495 (control) and 24 718 ± 2166 (peptide). Both growth curves match the control samples of cells grown without peptide present in the cell medium with no statistical differences between the number of cells with peptide or without peptide (figure 7).
Figure 7.

(a) Osteoblast precursor cells grown on tissue culture plastic with and without AMP in the cell media throughout growth. (b) Osteosarcoma cells grown on tissue culture plastic with and without AMP in the cell media, n = 6, error bars show standard error of the mean.
3.7. Toxicity of the dAMP coating on HA to mammalian cells
Osteoblast precursor cells were cultured on dAMP–tHA and HA for 14 days (figure 8a,b). In both treated and untreated HA, the cell number increased up to day 7, 1.6 × 106 ± 1.8 × 105 (peptide coated) and 1.6 × 106 ± 2.2 × 105, and then plateaued. Although the cell number appears to decrease at day 14, there was no significant difference between day 9 peptide coated (1.4 × 106 ± 1.5 × 105), control (1.5 × 106 ± 7.9 × 105) and day 14 peptide coated (1.1 × 106 ± 6.2 × 105), control (1.0 × 106 ± 4.6 × 105). The cell number followed the same growth pattern on both dAMP–tHA and HA with no significant differences between the two groups. Over the 14 days, the osteosarcoma cell number increased up to day 3 peptide coated (2.2 × 104 ± 395), control (2.9 × 104 ± 1713) and then plateaued with no significant differences from days 3–14, peptide coated (2.3 × 104 ± 158), control (2.6 × 104 ± 1133) (figure 8). There were no significant differences between the dAMP–tHA and HA groups, indicating that the dAMP did not demonstrate toxicity to the cells when bound onto a surface.
Figure 8.

(a) Osteoblast precursor cells grown on dAMP–tHA pellets compared with osteoblast precursor cells grown on HA pellets. (b) Osteosarcoma cells grown on dAMP–tHA pellets compared with HA pellets. Error bars show standard error of the mean, n = 6.
4. Discussion
As infection is established as a major complication of orthopaedic surgery, there have been multiple attempts to manipulate the host–device interface by the addition of antimicrobial agents [17,18]. While there has been a good deal of success, the main barrier to clinical translation has been the lack of ability to tailor implant material chemistry at a molecular scale and a reliance on incorporating agents with a secondary material phase onto the implant surface. An implant formed from titanium with a polymer surface coating, which is then covalently functionalized, changes the properties of the implant for the host tissue and correspondingly the host tissue response to the device.
By binding peptides to the surfaces of material such as HA directly, we aim to leave as much of the original material ‘visible’ to the host tissue as possible and therefore retain the osseointegrative properties of the implant. The peptide coating has a surface density of 1 molecule nm−1; this suggests the cells will still be able to attach to the HA. While previous studies have demonstrated that the idea of dual coating is possible on different surfaces, all of the coatings rely on the deposition of polymers which mask the surface completely, changing the material surface properties exposed to the in vivo environment [44]. In this study, we are tailoring the surface chemistry of the implant and tethering the AMPs directly onto the surface in order to inhibit bacterial growth. The SEM images support that the macroscale of the material is unchanged and the cell growth experiments have shown that the nanoscale changes have not affected the cell response to the material.
The defensin sequence synthesized, based on human defensin sequences, showed comparable levels of antimicrobial efficacy to human defensins [45]. While these values can vary depending on the environment, this comparison crucially demonstrates that the efficiency of the antimicrobial sequences is unchanged by the shorter peptide length and simplification of the sequence. One real potential of the peptide sequence presented here is the apparent inability of the bacteria to develop resistance in the studies carried out. This is believed to be due to the proposed mechanism of action of the peptide. AMPs are known to insert into bacterial cells causing pore formation [46]. This destabilizes the membrane and triggers cell lysis and death [47]. The positively charged AMP will associate strongly with the negative charges found in the bacterial cell wall [48]. This allows the hydrophobic regions of the peptide to induce pore formation. The bacteria will struggle to develop resistance to this as it would demand either removal or complete restructuring of the bacterial cell wall. With antimicrobial resistance becoming a global problem, developing antimicrobial surfaces for clinical applications, against which bacteria struggle to present resistance, is of great interest to the clinical community [49]. While further studies are of course required, this presents a promising outlook for the AMP. Long-term studies into antimicrobial-coated implants have demonstrated that after 5 years the implants are colonized and the bacteria are resistant to the chosen antibiotic [50]. Using a broad spectrum AMP (that the bacteria cannot rapidly develop resistance to) ensures that colonization of the surface will not occur.
Mikos and co-workers [51] demonstrated that short sequence polyglutamic acid residues can bind strongly to the surface of HA. Our work here has demonstrated that the inclusion of the binder sequence does not hinder the efficacy of the peptide and ensures that the solution around the surface can be sterilized in the short term. In the long-term studies, the efficacy of the surface at sterilizing the solution surrounding it was reduced as the electrostatically bound peptide was removed over sequential broth changes.
The stability of the cAMP to long-term incubation supports the spectroscopic data that the peptide has formed a covalent bond with the surface which will not allow the peptide to desorb from the surface. The apparent stability of the peptides to proteoloytic degradation is due to steric hindrance. As the peptides are grafted directly to the surface without linker spacer units, the HA surface provides a large amount of steric hindrance around the peptides. The high coverage of peptide across the surface also provides steric bulk, preventing access to the peptide chain. This prevents enzymes such as proteases reaching the regions on the chain they can activate, and gives the AMP coating its high stability profile.
The stability of the covalent functionality of the dAMP coating also supports translation of this work. The coating is shown to be stable for up to 12 months in simulated body fluid with motion and change of the solution surrounding the surface. This suggests that the coating will retain stability in the body and give a long-lasting coating on the implant, preventing biofilm formation without displaying cytotoxicity. The long-term microbiological efficacy study demonstrated not only that the surface maintained its efficacy in optimum bacterial growth conditions, but also that the dAMP surface still significantly reduced the number of planktonic bacteria in solution despite the eAMP coating dissociation and removal in the initial broth changes. This demonstrates that the covalent bond form of the peptide is effective at maintaining surface sterilization and may reduce the number of bacteria overall in a closed system by eliminating bacteria when they come into contact with the surface.
Previous work on the surface chemistry of HA by Williams et al. [52] has shown that it is possible to alter the surface chemistry of the HA to include thiol residues without significantly altering the material properties. This has allowed us to use the thiol groups to act as tethering sites to covalently link the AMPs. This gives permanent functionalization with the peptide without compromising the bulk properties of the material, as evidenced by the SEM images. From a biological perspective, cell proliferation and metabolic activity studies showed the AMP coating had no cytotoxic effects.
The results we present here have shown that thiol modification has occurred (both EDX and Raman data, table 1 and electronic supplementary material, figure S1). The EDX data also demonstrate that we can develop this technique further and attach the AMPs to the free thiol functionality. The fluorescence spectroscopy images demonstrate that this technique can be used to coat bulk material over large surface areas. This makes the case for translation of the dAMP coating into clinical application very strong.
The development of these dual surface coatings, which impart both short- and long-term antimicrobial functionalities, is vitally important for orthopaedic infection as it can occur at the time of surgery or later. This results in a need for a dual coating that can sterilize the area surrounding the implant at the time of surgery and give long-lasting functionality [53,54]. The data presented here demonstrate that, although both cAMP and eAMP are efficacious, neither alone can inhibit both the planktonic and surface growth of bacteria. The dAMP coating can inhibit both.
In summary, in the fight to treat infection following implantation of a prosthesis, there are several important needs which must be met within the host–device interface: (1) short-term delivery of antimicrobial agents to sterilize the host tissue and device to prevent colonization by bacteria, (2) long-term stable antimicrobial coatings to prevent bacterial colonization of the implant surface, and (3) no alteration of the material properties of the implant device which would compromise the ability of the implant to integrate with native bone tissue. To the best of our knowledge, these needs are not met by the current state of the art in the field. There are many studies showing coating of orthopaedic devices by dip/spray coating or incorporation of antimicrobial agents during production and these give excellent materials [55,56]. However, these materials only provide short-term antimicrobial activity (less than 12 months), failing to tackle the whole problem. There has been further work on coating devices with polymers, which can then have antimicrobial agents ‘permanently’ attached, but this method changes the properties of the implant material due to the inclusion of the polymer material. We have addressed this problem by incorporating the dual antimicrobial surface coating which facilitates short-term release as well as a long-term stable antimicrobial presence at the material surface.
5. Conclusion
We have demonstrated the manufacture of a novel dual-system approach to prevent antimicrobial infections which uses two different binding mechanisms for the same antimicrobial sequence. The electrostatically released peptide inhibits bacterial growth in solution while the covalently bonded peptide inhibits adherence and biofilm formation. Both together can inhibit the colonization of bacteria at the interface of an engineered surface with tissue, while maintaining cell growth. The peptides can be tailored to interact with a surface and maintain good efficacy against a broad spectrum of bacteria and show strong antimicrobial activity against clinical isolates of S. aureus, S. epidermidis and P. aeruginosa (the bacteria most commonly found in joint infections). By using our mix-and-match system combining multiple fragments with different properties (surface binding and antimicrobial activity) without compromising the individual functions, the dAMP coating gives short-term sterilization of the neighbouring tissue, followed by long-term sustained antimicrobial activity of the surface. With this work established, further work is needed to test the peptide coatings in a preclinical animal infection model to move them towards a clinical environment.
Supplementary Material
Authors' contributions
The study was designed, carried out and the manuscript written by F.d.C.; L.T., R.L.W., O.A., M.R.B., F.H. and E.H. carried out experimental work; B.O., J.G., M.W. and A.F.A.P. designed the study and reviewed the manuscript; A.S., L.G., R.A.H.S., A.B. and A.L. reviewed the manuscript.
Competing interests
We declare we have no competing interests.
Funding
This work was funded by the NIHR Surgical Reconstruction and Microbiology Research Centre and the University of Birmingham (RDF-2016). Student support was also provided through the Frank Ker Bequest.
References
- 1.Whitehouse JD, Friedman ND, Kirkland KB, Richardson WJ, Sexton DJ. 2002. The impact of surgical-site infections following orthopaedic surgery at a community hospital and a university hospital: adverse quality of life, excess length of stay and extra cost. Infect. Control Hosp. Epidimiol. 23, 183–189. ( 10.1086/502033) [DOI] [PubMed] [Google Scholar]
- 2.Noroozi H, Kazemi A, Fadee R, Alavi S, Mohammadzadeh M. 2010. Microbiological assessment of nonsurgical traumatic wound infections and surgical site infections in hospitalised patients. Iran. J. Clin. Infect. Disease 5, 80–83. [Google Scholar]
- 3.Philips JE, Crane TP, Noy M, Elliott TSJ, Grimer RJ. 2006. The incidence of deep prosthetic infections in a specialist orthopaedic hospital. J. Bone Joint Surg. 88-B, 943–948. ( 10.1302/0301-620X.88B7.17150) [DOI] [PubMed] [Google Scholar]
- 4.Song Z, Borgwardt L, Hoiby N, Wu H, Sorensen TS, Borgwardt A. 2013. Prosthesis infections after orthopaedic joint replacement: the possible role of bacterial biofilms. Orthop. Rev. 5, e14 ( 10.4081/or.2013.e14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Funao H, et al. 2016. A novel hydroxyapatite film coated with ionic silver via inositol hexaphosphate chelation prevents implant-associated infection. Sci. Rep. 6, 23238 ( 10.1038/srep23238) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zapotoczna M, O'Neill E, O'Gara JP. 2016. Untangling the diverse and redundant mechanisms of Staphylococcus aureus biofilm formation. PLoS Pathog. 12, e1005671 ( 10.1371/journal.ppat.1005671) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moran E, Byren I, Atkins BL. 2010. The diagnosis and management of prosthetic joint infections. J. Antimicrob. Chemother. 65, 45–54. ( 10.1093/jac/dkq305) [DOI] [PubMed] [Google Scholar]
- 8.Uesenko M, Windhager R, Kontekakis A, Hanstein T, Kuehn K-D. 2016. Risk factors for periprosthetic joint infections following primary total hip arthroplasty. Univ. J. Med. Sci. 4, 38–44. ( 10.13189/ujmsj.2016.040105) [DOI] [Google Scholar]
- 9.Geetha M, Singh AK, Asokamani R, Gogia AK. 2009. Ti based biomaterials, the ultimate choice for orthopaedic implants—a review. Prog. Mater Sci. 54, 397–425. ( 10.1016/j.pmatsci.2008.06.004) [DOI] [Google Scholar]
- 10.l-Sanabani JS, Madfa AA, Al-Sanabani FA. 2013. Application of calcium phosphate materials in dentistry. Int. J. Biomater. 2013, 876132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shamsul JB, Nurhidayah AZ, Ruzaidi CM. 2007. Characterization of cobalt-chromium-HAP biomaterial for biomedical application. J. Appl. Sci. Res. 3, 1544–1553. [Google Scholar]
- 12.Vahabzadeh S, Roy M, Bandyopadhyay A, Bose S. 2015. Phase stability and biological property evaluation of plasma sprayed hydroxyapatite coatings for orthopaedic and dental applications. Acta Biomater. 17, 47–55. ( 10.1016/j.actbio.2015.01.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chambers B, St Clair SF, Froimson MI. 2007. Hydroxyapatite-coated tapered cementless femoral components in total hip arthroplasty. J. Arthroplasty 22(4 Suppl. 1), 71–74. ( 10.1016/j.arth.2007.01.019) [DOI] [PubMed] [Google Scholar]
- 14.Ferreira A, Aslanian T, Dalin T, Picaud J. 2016. Ceramic bearings with bilayer coating in cementless total hip arthroplasty. A safe solution. A retrospective study of one hundred and twenty six cases with more than ten years follow-up. Int. Orthop. 1, 1–7. ( 10.1007/s00264-016-3271-7) [DOI] [PubMed] [Google Scholar]
- 15.Xu Q, Zhang W, Dong C, Sreeprassad TS, Xia Z. 2016. Biomimetic self-cleaning surfaces: synthesis, mechanism and application. J. R. Soc. Interface 13, 1–12. ( 10.1098/rsif.2016.0300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nithyanandan A, Suntharavathanan M, Huang J, Rehman S, Draper E, Edirisinghe M. 2015. Bioinspired electrohydrodynamic ceramic patterning of curved metallic substrates. Bioinsp. Biomim. Nanobiomater. 4, 59–67. ( 10.1680/bbn.14.00020) [DOI] [Google Scholar]
- 17.Boccaccini AR, Keim S, Ma R, Li Y, Zhitomirsky I. 2010. Electrophoretic deposition of biomaterials. J. R. Soc. Interface 7, 581–613. ( 10.1098/rsif.2010.0156.focus) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang S, Karaca BT, VanOosten SK, Yuca E, Mahalingam S, Edirisinghe M, Tamerler C. 2015. Coupling infusion and gyration for the nanoscale assembly of functional polymer nanofibers integrated with genetically engineered proteins. Macromol. Rapid Commun. 36, 1322–1328. ( 10.1002/marc.201500174) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu Z, Mahalingam S, Rohn JL, Ren G, Edirisinghe M. 2015. Physico-chemical and antibacterial characteristics of pressure spun nylon nanofibers embedded with functional silver nanoparticles. Mater. Sci. Eng. C 56, 195–204. ( 10.1016/j.msec.2015.06.003) [DOI] [PubMed] [Google Scholar]
- 20.Taha M, Chai F, Blanchemain N, Neut C, Goube M, Maton M, Martel B, Hildebrand HF. 2014. Evaluation of sorption capacity of antibiotic and antibacterial properties of a cyclodextrin-polymer functionalised hydroxyapatite-coated titanium hip prosthesis. Int. J. Pharm. 477, 380–389. ( 10.1016/j.ijpharm.2014.10.026) [DOI] [PubMed] [Google Scholar]
- 21.He S, Zhou P, Wang L, Xiong X, Zhang Y, Deng Y, Wei S. 2014. Antibiotic-decorated titanium with enhanced antibacterial activity through adhesive polydopamine for dental/bone implant. J. R. Soc. Interface 11, 20140169 ( 10.1098/rsif.2014.0169) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lischer S, Korner E, Balaza DJ, Shen D, Wick P, Grieder K, Haas D, Heuberger M, Hegemann D. 2011. Antibacterial burst-release from minimal Ag-containing plasma polymer coatings. J. R. Soc. Interface 8, 1019–1030. ( 10.1098/rsif.2010.0596) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cattalini JP, Hoppe A, Pishbin F, Roether J, Boccaccini AR, Lucangioli S, Mourino V. 2015. Novel nanocomposite biomaterials with controlled copper/calcium release capability for bone tissue engineering multifunctional scaffolds. J. R. Soc. Interface 12, 20150509 ( 10.1098/rsif.2015.0509) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nowlin K, Boseman A, Covell A, LaJeunesse D. 2015. Adhesion-dependent rupturing of Saccharomyces cerevisiae on biological antimicrobial nanostructured surfaces. J. R. Soc. Interface 12, 20140999 ( 10.1098/rsif.2014.0999) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Díaz M, Barba F, Miranda M, Guitian F, Torrecillas R, Moya JS. 2009. Synthesis and antimicrobial activity of a silver-hydroxyapatite nanocomposite. J. Nanomater. 2009, 498505 ( 10.1155/2009/498505) [DOI] [Google Scholar]
- 26.Lu X, et al. 2011. Nano-Ag-loaded hydroxyapatite coatings on titanium surfaces by electrochemical deposition. J. R. Soc. Interface 8, 529–539. ( 10.1098/rsif.2010.0366) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Behra R, Sigg L, Clift MJD, Herzog F, Minghetti M, Johnston B, Petri-Fink A, Rothen-Rutishauser B. 2013. Bioavailability of silver nanoparticles and ions: from a chemical and biochemical perspective. J. R. Soc. Interface 10, 20130396 ( 10.1098/rsif.2013.0396) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsueh P-R, Chen W-H, Luh K-T. 2005. Relationships between antimicrobial use and antimicrobial resistance in gram-negative bacteria causing nosocomial infections from 1991–2003 at a university hospital in Taiwan. Int. J. Antimicrob. Agents 26, 463–472. ( 10.1016/j.ijantimicag.2005.08.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen TL, Haney EF, Vogel HJ. 2011. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 29, 464–472. ( 10.1016/j.tibtech.2011.05.001) [DOI] [PubMed] [Google Scholar]
- 30.Wimley WC, Hristova K. 2011. Antimicrobial peptides; successes, challenges and unanswered questions. J. Membr. Biol. 239, 27–34. ( 10.1007/s00232-011-9343-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang BG, Myers DE, Wallace GG, Brandt M, Choong P. 2014. Bioactive coatings for orthopaedic implants—recent trends in development of implant coatings. Int. J. Mol. Sci. 15, 11 878–11 892. ( 10.3390/ijms150711878) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mishra B, Basu A, Chua RRY, Saravanan R, Tambyah PA, Ho B, Chang MW, Leong SSJ. 2014. Site specific immobilization of a potent antimicrobial peptide onto silicone catheters: evaluation against urinary tract infection pathogens. J. Mater. Chem. B 2, 1706–1716. ( 10.1039/c3tb21300e) [DOI] [PubMed] [Google Scholar]
- 33.Veldhuizen EJA, Schneider VAF, Agustiandari H, van Dijk A, Tjeerdsma-van Bokhoven JLM, Bikker FJ, Haagsman HP. 2014. Antimicrobial and immunomodulatory activities of PR-39 derived peptides. PLoS ONE 9, 1–7. ( 10.1371/journal.pone.0095939) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steinstraesser L, Kraneburg U, Jacobsen F, Al-Benna S. 2011. Host defence peptides and their antimicrobial-immunomodulatory duality. Immunobiology 216, 322–333. ( 10.1016/j.imbio.2010.07.003) [DOI] [PubMed] [Google Scholar]
- 35.Huby RDJ, Dearman RJ, Kimber I. 2000. Why are some proteins allergens? Toxicol. Sci. 55, 235–246. ( 10.1093/toxsci/55.2.235) [DOI] [PubMed] [Google Scholar]
- 36.Chairatana P, Chu H, Castillo PA, Shen B, Bevins CL, Nolan EM. 2016. Proteolysis triggers self-assembly and unmasks innate immune function of a human defensin peptide. Chem. Sci. 7, 1738–1752. ( 10.1039/C5SC04194E) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chileveru HR, Lim SA, Chairatana P, Wommack AJ, Chiang I-L, Nolan EM. 2015. Visualising attack of Escherichia coli by the antimicrobial peptide human defensin 5. Biochemistry 54, 1767–1777. ( 10.1021/bi501483q) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rigano MM, et al. 2012. A novel synthetic peptide from a tomato defensin exhibits antibacterial activities against Helicobacter pylori. J. Pept. Sci. 18, 755–762. ( 10.1002/psc.2462) [DOI] [PubMed] [Google Scholar]
- 39.Scudiero O, et al. 2010. Novel synthetic, salt-resistant analogs of human beta-defensins 1 and 3 endowed with enhanced antimicrobial activity. Antimicrob. Agents Chemother. 54, 2312–2322. ( 10.1128/AAC.01550-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marques MRC, Loebenberg R, Almukainzi M. 2011. Simulated biological fluids with possible application in dissolution testing. Dissolution Technol. 18, 9–23. ( 10.14227/DT180311P15) [DOI] [Google Scholar]
- 41.Kelly M, et al. 2015. Peptide aptamers: novel coatings for orthopaedic implants. Mater. Sci. Eng. C 54, 84–93. ( 10.1016/j.msec.2015.04.021) [DOI] [PubMed] [Google Scholar]
- 42.Andrews JM. 2001. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 48, 5–16. ( 10.1093/jac/48.suppl_1.5) [DOI] [PubMed] [Google Scholar]
- 43.Ciapetti G, et al. 2003. Osteoblast growth and function in porous poly-ε-caprolactone matrices for bone repair: a preliminary study. Biomaterials 24, 3815–3824. ( 10.1016/S0142-9612(03)00263-1) [DOI] [PubMed] [Google Scholar]
- 44.Yang CC, Lin CC, Liao JW, Yen SK. 2013. Vancomycin-chitosan composite deposited on post-porous hydroxyapatite coated Ti6Al4 V. Mater. Sci. Eng. C 33, 2203–2212. ( 10.1016/j.msec.2013.01.038) [DOI] [PubMed] [Google Scholar]
- 45.Dhople V, Krukemeyer A, Ramamoorthy A. 2006. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim. Biophys. Acta Biomembr. 158, 1499–1512. ( 10.1016/j.bbamem.2006.07.007) [DOI] [PubMed] [Google Scholar]
- 46.Brogden KA. 2005. Antimicrobial peptides: pore formers of metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250. ( 10.1038/nrmicro1098) [DOI] [PubMed] [Google Scholar]
- 47.Bechinger B, Lohner K. 2006. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochem. Biophys. Acta Biomembr. 1758, 1529–1539. ( 10.1016/j.bbamem.2006.07.001) [DOI] [PubMed] [Google Scholar]
- 48.Hancock REW. 2001. Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. 1, 156–164. ( 10.1016/S1473-3099(01)00092-5) [DOI] [PubMed] [Google Scholar]
- 49.Kilgus DJ, Howe DJ, Strang A. 2002. Results of periprosthetic hip and knee infections caused by resistant bacteria. Clin. Orthop. Relat. Res. 404, 116–124. ( 10.1097/00003086-200211000-00021) [DOI] [PubMed] [Google Scholar]
- 50.Berbari EF, Hanssen AD, Duffy MC, Steckelberg JM, Ilstrup DM, Harmsen WS, Osmon DR. 1998. Risk factors for prosthetic joint infection: case-control study. Clin. Infect. Dis. 27, 1247–1254. ( 10.1086/514991) [DOI] [PubMed] [Google Scholar]
- 51.Murphy MB, Hartgerink JD, Goepferich A, Mikos AG. 2007. Synthesis and in vitro hydroxyapatite binding of peptides conjugated to calcium-binding moieties. Biomacromolecules 8, 2237–2243. ( 10.1021/bm070121s) [DOI] [PubMed] [Google Scholar]
- 52.Williams RL, Hadley MJ, Jiang PJ, Mendes PM, Rappoport JZ, Grover LM. 2013. Thiol modification of silicon-substituted hydroxyapatite nanocrystals facilitates fluorescent labelling and visualization of cellular internalization. J. Mater. Chem. B 1, 4370–4378. ( 10.1039/c3tb20775g) [DOI] [PubMed] [Google Scholar]
- 53.Tang S, Tian B, Ke QF, Zhu Z-A, Guo YP. 2014. Gentamicin-loaded carbonated hydroxyapatite coatings with hierarchically porous structures: drug delivery properties, bactericidial properties and biocompatibility. RSC Adv. 4, 41500 ( 10.1039/C4RA05493H) [DOI] [Google Scholar]
- 54.Neut D, de Belt H, van Horn JR, van der Mei HC, Busscher HJ. 2003. Residual gentamicin-release from antiobiotic loaded polymethylmethacrylate beads after 5 years of implantation. Biomaterials 24, 1829–1831. ( 10.1016/S0142-9612(02)00614-2) [DOI] [PubMed] [Google Scholar]
- 55.Kim HW, Knowles JC, Kim HE. 2005. Porous scaffolds of gelatin-hydroxyapatite nanocomposites obtained by biomimetic approach: characterization and antibiotic drug release. J. Biomed. Mater. Res. B Appl. Biomater. 74B, 686–699. ( 10.1002/jbm.b.30236) [DOI] [PubMed] [Google Scholar]
- 56.Stigter M, de Groot K, Layrolle P. 2002. Incorporation of tobramycin into biomimetic hydroxyapatite coating on titanium. Biomaterials 23, 4143–4153. ( 10.1016/S0142-9612(02)00157-6) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.