

Genome-wide assessment reveals opposing patterns of vertical connectivity in two depth-generalist coral species.
Keywords: Coral reefs, deep reef refuge hypothesis, mesophotic coral ecosystems, vertical connectivity, divergent selection, RADseq, endosymbiont contamination
Abstract
Deep coral reefs (that is, mesophotic coral ecosystems) can act as refuges against major disturbances affecting shallow reefs. It has been proposed that, through the provision of coral propagules, such deep refuges may aid in shallow reef recovery; however, this “reseeding” hypothesis remains largely untested. We conducted a genome-wide assessment of two scleractinian coral species with contrasting reproductive modes, to assess the potential for connectivity between mesophotic (40 m) and shallow (12 m) depths on an isolated reef system in the Western Atlantic (Bermuda). To overcome the pervasive issue of endosymbiont contamination associated with de novo sequencing of corals, we used a novel subtraction reference approach. We have demonstrated that strong depth-associated selection has led to genome-wide divergence in the brooding species Agaricia fragilis (with divergence by depth exceeding divergence by location). Despite introgression from shallow into deep populations, a lack of first-generation migrants indicates that effective connectivity over ecological time scales is extremely limited for this species and thus precludes reseeding of shallow reefs from deep refuges. In contrast, no genetic structuring between depths (or locations) was observed for the broadcasting species Stephanocoenia intersepta, indicating substantial potential for vertical connectivity. Our findings demonstrate that vertical connectivity within the same reef system can differ greatly between species and that the reseeding potential of deep reefs in Bermuda may apply to only a small number of scleractinian species. Overall, we argue that the “deep reef refuge hypothesis” holds for individual coral species during episodic disturbances but should not be assumed as a broader ecosystem-wide phenomenon.
INTRODUCTION
Tropical coral reefs are in global decline, with many under immediate threat because of a rapidly changing climate and the accumulation of local stressors (1, 2). Even in remote areas with strong legal protection, the increase in frequency and magnitude of large-scale bleaching and storm events has severely affected coral reefs (3). Coral reef persistence increasingly depends on local areas that can offer refuge against such disturbances (4) and provide propagules to recolonize affected areas (5). Deeper sections of coral reefs (below depths of 30 to 40 m), also referred to as “mesophotic coral ecosystems,” arguably represent potentially critical ecological refuges. Although certainly not immune to disturbance (6–9), they are commonly buffered from major bleaching and storm events (10, 11) and have a near-ubiquitous presence directly adjacent to the world’s shallow coral reefs (12). Although their ability to escape disturbances has been relatively well documented, the hypothesis that deep reefs harbor reliable reproductive sources remains largely untested (11, 13–15) and this was recently highlighted as a key knowledge gap in a report by the United Nations Environment Programme (16).
The “reseed” potential of mesophotic coral reefs firstly depends on the extent of species overlap with their shallow-water counterparts. Although greater depths often confer greater protection from disturbances (11), the community similarity between shallow and deep reefs also decreases with depth, as a reflection of changing environmental conditions) (17). Given this trade-off, the “deep reef refuge” hypothesis (DRRH) pertains primarily to upper mesophotic depths (~30 to 60 m)—deep enough to escape disturbances but shallow enough to guarantee sufficient overlap in species composition (11, 18, 19). Species overlap between shallow and upper mesophotic communities can be substantial, with an estimated 25 to 40% of Caribbean coral species and ~20 to 40% of Acropora species (on the Great Barrier Reef) representing depth-generalist species (11, 20). However, initial assessments have demonstrated that depth-generalist coral species are not necessarily composed of a single panmictic population over depth (14, 21–25). The three coral species thus far assessed for shallow-mesophotic connectivity (which includes two brooding and one broadcasting species) exhibit ambiguous patterns across geographic regions, with genetic differentiation observed in certain locations but not others (14, 22, 23, 25). This geographic variation has consequently hampered a more general assessment of the DRRH as the overall role of deep reefs in shallow reef recovery remains unclear (16, 26).
To understand the reseed potential on an ecosystem-wide scale (that is, across many species), it is essential to identify the nature of barriers to vertical connectivity and how these barriers may vary across coral species with distinct life history traits. The relative roles of depth and location have remained difficult to disentangle in past assessments of vertical connectivity, often because of geographic separation of sampled shallow and mesophotic populations (14, 22, 25, 27). Nonetheless, depth-associated selection could pose an important ecological barrier to vertical connectivity (24, 28) even in the absence of physical barriers (for instance, on those reef slopes where shallow and mesophotic communities are directly adjacent). Depth-associated selection does not necessarily restrict gene flow; however, in species with localized sperm and larval dispersal (such as in most brooding corals), localized dispersal could lead to assortative mating and therefore facilitate a divergence-with-gene-flow scenario (28, 29). Similarly, assortative mating could facilitate divergence in broadcasting species if depth-associated selection is accompanied by temporal reproductive isolation, either through pleiotropic effects or through depth-related differences in spawning cues (24, 27, 30). The role of such ecological barriers to vertical connectivity remains poorly understood, and the impact of depth-associated selection on vertical connectivity has remained largely obscured because genomic sampling has been restricted to mostly to microsatellite sequencing in presumably neutral parts of the genome [but see the work of Brazeau et al. (23)].
Reduced-representation approaches to genome sequencing [for example, restriction-site associated DNA sequencing (RAD-seq)] allow for sampling of both adaptive and neutral genetic variation across the genome (31, 32). In combination with a replicated sampling design (sampling the same habitats across multiple locations in a region), such reduced-representation approaches have the potential to identify the nature of barriers (that is, ecological or physical) that might hamper cross-habitat connectivity. Nonetheless, uptake of such genotyping-by-sequencing approaches in symbiotic marine invertebrates has been slow [but see related studies (33–37)] largely due to the nonspecific nature of these methods (for example, targeting universal restriction site motifs) in light of pervasive contamination by the obligate endosymbionts. In reef-building corals, this contamination stems from the fact that they are associated with a complex consortium of eukaryotes, prokaryotes, and viruses (38), with the hosted Symbiodinium representing a significant proportion of the overall biomass (39). Endosymbiont contamination might not necessarily affect inferred genetic patterns for the host; however, this can only be ascertained when comparing it against an aposymbiotic (that is, lacking symbionts) reference (36). Contamination is of particular concern when evaluating horizontal versus vertical connectivity, given the prevalence of geographic (40) and bathymetric (41) zonation of associated Symbiodinium. Unfortunately, published genomes for scleractinian corals are still scarce (42, 43), and obtaining aposymbiotic tissue remains relatively difficult.
In this study, we test the reseeding potential of deep reefs by sampling genome-wide variation in shallow and deep population pairs of two species with contrasting reproductive modes (Agaricia fragilis and Stephanocoenia intersepta). To overcome the contamination issue, we used a novel method that generated a subtraction reference genomic library of Symbiodinium symbionts (isolated from coral hosts using fluorescence-activated cell sorting). We focused on the reef system of Bermuda, where there is extensive upper mesophotic habitat adjacent to the shallow reef (44), and whose isolated geographic setting suggests the probability that Bermudan corals depend on local recruitment sources for recovery (14, 25, 45). In addition, previous studies highlighted the potential importance of the DRRH for at least some coral species at this location, given the observed vertical connectivity for two other Bermuda corals (Montastraea cavernosa and Porites astreoides) (14, 25). Through sampling of replicate shallow and deep populations, we assess whether the vertical connectivity potential differs between the two species and how vertical connectivity might be affected by depth-associated selection. More broadly, we evaluate how these species-specific patterns affect the reseeding potential of deep reefs in Bermuda and discuss the wider implications and caveats in light of the DRRH.
RESULTS
Sample collections, skeletal morphology, and endosymbiont associations
Coral colonies from the brooding species A. fragilis and the broadcasting species S. intersepta were sampled from shallow (~12 m) and mesophotic depths (~40 m) at four different locations around Bermuda (Fig. 1A and table S1). Phenotypic characterization of two skeletal properties (corallite density and diameter; Fig. 1B) showed similar skeletal values for A. fragilis specimens from all three eastern deep populations (PD, JD, and GD; Fig. 1, A and B). In contrast, the two shallow populations and the western deep population had either a much lower corallite density (PS) or a larger diameter (GS and WD). For S. intersepta, samples from the western population (WD) were similar to those from the other deep populations (PD, JD, and GD), with, again, the shallow Princess Beach population (PS) exhibiting a much lower corallite density. Sequencing of the COX1 (mitochondrial) region of associated endosymbionts showed that A. fragilis and S. intersepta associated with distinct endosymbiont types. Further characterization of a short chloroplast minicircle locus indicated that A. fragilis associated with a single endosymbiont haplotype, while S. intersepta associated with two different Symbiodinium haplotypes (Fig. 1, C and D). However, the vast majority of S. intersepta colonies (94%) associated with a single Symbiodinium haplotype (sint_chl_a); a small proportion associated either with the alternative haplotype (sint_chl_b) or with a combination of both haplotypes (nucleotide positions ambiguous for the mutations separating the two genotypes).
Fig. 1. Sampling locations, skeletal morphology, and endosymbiont associations.

(A) Map showing sampled populations on the Bermuda reef platform. Symbols with lighter shading indicate shallow populations, and symbols with darker shading indicate deep populations. GS, “Gurnet Rock” shallow; GD, “Gurnet Rock” deep; JS, “John Smith’s Bay” shallow; JD, “John Smith’s Bay” deep; PS, “Princess Beach” shallow; PD, “Princess Beach” deep; WD, “Western Blue Cut” deep. (B) Skeletal signature of the different populations of A. fragilis (green) and S. intersepta (blue). Symbol shape corresponds to sampling locations (A), and hue indicates sampling depth [shallow (light) and deep (dark)]. (C) Endosymbiont (Symbiodinium) haplotypes associated with A. fragilis (only a single haplotype identified). (D) Endosymbiont (Symbiodinium) haplotypes associated with S. intersepta. Images of species modified from photographs by the Florida Fish and Wildlife Conservation Commission (with permission).
Contaminant filtering, missing data, and clonality
Sequencing of nextRAD libraries resulted in an average of ~1.4 million reads for each individually bar-coded sample of the two focal species (n = 213; not including failed samples). Using alignment-based clustering, we recovered 12,145 nextRAD sequence loci (hereafter referred to as “RAD loci”) for A. fragilis and 7591 for S. intersepta under initial PyRAD conditions. From the recovered RAD loci, 10% of the A. fragilis and 14% of the S. intersepta data sets were removed because they represented contaminants matching the Symbiodinium subtraction reference. Additional contaminants (~2% of RAD loci) matching non-cnidarian references (with a notable abundance of the proteobacteria Ralstonia sp.) were also removed before downstream filtering. After quality control and minimal representation filtering, we obtained 2568 biallelic single-nucleotide polymorphisms (SNPs) from 1579 RAD loci for A. fragilis and 7547 biallelic SNPs from 2187 RAD loci for S. intersepta (excluding 108 multiallelic SNPs that were included in analyses that support multiallelic data). Missing data average after filtering was 15% of SNPs for A. fragilis and 19% of SNPs for S. intersepta (Fig. 2). Although no potential clonemates were identified for S. intersepta (maximum allelic similarity, 86%), four groups of potential clones were identified for A. fragilis (allelic similarities, 94 to 98%). These represented two triplets and two pairs and always occurred within the same population (Fig. 2). In addition, a small recruit (<1.5 cm) sampled directly adjacent to an A. fragilis colony collected from a depth of 67 m was found to represent a clonemate. A single representative of each group of potential clones was kept in the data set for population-level analyses.
Fig. 2. Pairwise genetic distances between individuals of A. fragilis and S. intersepta.

Darker intensity in heat maps corresponds to greater genetic distance. Highly similar pairs are indicated in red (94 to 98% similarity, indicating potential clonemates), and the diagonal (that is, self-comparisons) is replaced with a black line. Individuals are grouped by population, and for S. intersepta, two out-group individuals from Curaçao are included. Bar graphs indicate the number of genotyped SNPs for each individual.
Genome-wide patterns of differentiation and outlier analyses
Global mean FST (fixation index, a measure of genetic differentiation between populations) varied nearly two orders of magnitude between A. fragilis (FST = 0.06998) and S. intersepta (FST = 0.00081), with many individual SNPs exhibiting high FST values for A. fragilis (Fig. 3A). From the 175 SNPs initially identified as FST outliers for A. fragilis in the overall Fdist analysis, 56 were also identified as outliers by BayeScan. From those, 25 SNPs located on 12 RAD loci were identified as potentially under depth-associated selection (outliers in at least two pairwise shallow-deep comparisons but never in within-depth pairwise comparisons). These depth-associated outliers exhibited very distinct genotype frequencies between shallow and deep populations, with a minor allele private to the deep populations (but not fixed) in nearly half of these (Fig. 3B). Depth-outlier SNPs on the same RAD locus exhibited (near-)identical patterns, and genotype frequencies are therefore given for a single representative SNP. Two of the depth-outlier RAD loci matched sections of the Acropora digitifera genome, encoding an uncharacterized protein and a phospholipase enzyme (D1-like) (42). Visual assessment of the remaining outlier SNPs (fig. S1) indicates that an additional ~13 SNPs each had a distinct allele occurring in high frequency at the Western Blue Cut population (WD). For S. intersepta, 138 outlier SNPs were identified in the Fdist analysis, 4 of which were also identified as outliers by BayeScan, but none were identified as potentially under depth-associated selection using the criterion of outliers in multiple pairwise shallow-deep comparisons (Fig. 3, C and D).
Fig. 3. Genetic differentiation and outlier SNPs.

(A) Genetic differentiation versus expected heterozygosity of all SNPs genotyped for A. fragilis. (B) Genotype frequencies of all 12 depth-associated outliers (showing one representative SNP per RAD locus). (C) Genetic differentiation versus expected heterozygosity of all SNPs genotyped for S. intersepta. (D) Genotype frequencies of all four consensus outliers (BayeScan and Fdist, indicated by a black dot) and eight additional outliers (Fdist) with the highest FST values. In the scatterplots, colors indicate the outlier category, and frequency distributions of overall FST estimates are plotted along the y axis. The dashed line indicates the minimum FST at which consensus outliers were identified (for S. intersepta). Genotype frequencies are given for shallow and deep populations, with the hue of stacks indicating genotype (ref/ref, homozygote for the reference allele; ref/alt, heterozygote; alt/alt, homozygote for the alternative allele).
Genetic structuring in A. fragilis across depths and locations
Genetic structure was identified for A. fragilis (G-statistic Monte Carlo test for all SNPs: Gobs = 56,315, P < 0.01) and supported by all approaches [Bayesian clustering, principal components analysis (PCA), and pairwise genetic distances between individuals; Figs. 2 and 4], indicating clear genetic differentiation between shallow and deep populations of A. fragilis, as well as differentiation of a deep population on the northwestern side of the island. The existence of three distinct clusters was further supported using the MedMeaK and MedMedK estimators for STRUCTURE (that is, maximum of three clusters detected; table S2). To explicitly test for divergence with depth, we categorized measures of differentiation into four groups of three pairwise population comparisons (fig. S2 and table S3): within-shallow (GS-JS, JS-PS, and PS-GS), within-deep (GD-JD, JD-PD, and PD-GD), within-deep-west (WD-GD, WD-JD, and WD-PD), and between-depths (GS-GD, JS-JD, and PS-PD). A nonparametric Kruskal-Wallis test confirmed the presence of an overall difference between groups [Kruskal-Wallis χ²(3) = 499.43, P < 2.2 × 10−16], and individual nonparametric Wilcoxon rank sum tests indicated that differentiation of the between-depths group was significantly greater than that of the within-shallow and within-deep population groups (W = 20,749,000 and 21,537,000, P < 2.2 × 10−16). However, differentiation of the between-depths group was not significantly different from that between the western (WD) and the eastern (GD, JD, and PD) deep populations. No significant differences were observed between the level of differentiation of the within-shallow and that of the within-deep populations, apart from differentiation of the western deep population (W = 17,196,000, P < 2.2 × 10−16).
Fig. 4. Genetic structuring across depths and locations.
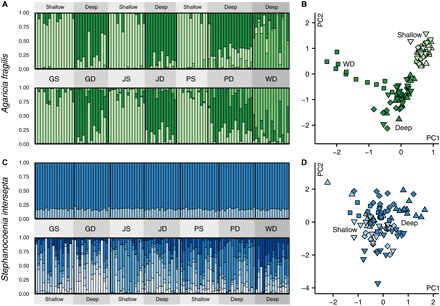
(A) STRUCTURE diagram (K = 3) for A. fragilis as inferred from the overall (top) and neutral (bottom) data set. (B) PCA for A. fragilis as inferred from the overall data set. (C) STRUCTURE diagram (K = 2) for S. intersepta as inferred from the overall data set (top) and discriminant analysis of principal components (DAPC) for the overall data set (bottom). (D) PCA for S. intersepta as inferred from the overall data set. STRUCTURE and DAPC bar graphs indicate ancestry proportions for individuals in each population. In the PCA, sampling depth of individuals is indicated by hue [shallow (light) and deep (dark)], and location is denoted by symbol shape (following Fig. 1A).
Genetic clustering for the “neutral” data set (in which any outliers identified by either Fdist or BayeScan were removed), as inferred by STRUCTURE, was very similar to the overall data set, indicating genome-wide differentiation (Fig. 4A and table S2). When considering the 83 individuals from eastern populations separately, STRUCTURE identified 26 and 9 individuals with an ancestry assignment of >0.98 to the shallow and deep cluster, respectively (K = 2). A total of 108 SNPs (in addition to the identified 25 depth-outlier SNPs) exhibited strong allelic differences (Δp ≥ 0.5) between these “purebred” groups. Plotting the genotypes of all individuals for these SNPs across the ancestry assignment gradient reveals introgression from shallow into deep individuals, which is also reflected in the overall ancestry assignments (Fig. 5). In contrast, introgression from deep into shallow appears very limited (for example, only minor assignment of shallow individuals to the deep cluster). The genetic diversity (that is, expected heterozygosity) of deep populations is also consistently higher than that of shallow populations (table S1). Despite the shallow-to-deep introgression, no migrants were observed, indicating limited migration over ecological time scales.
Fig. 5. Admixture between shallow and deep populations in A. fragilis.

SNP genotypes for the 25 outlier SNPs (indicated in red hues) (left) and 108 additional highly divergent SNPs (indicated in grayscale) (middle) for all shallow and deep individuals (eastern populations only) sorted by depth and overall ancestry assignment (right). Hues indicate genotype [following Fig. 3 (B and D)] with a white color indicating missing data. In the overall ancestry assignment, light green refers to the “shallow” cluster, and dark green corresponds to the “deep” cluster, with the horizontal solid line separating individuals originating from shallow and deep reefs. Highly divergent SNPs were selected as those with an allele frequency difference (Δp) of ≥0.5 between shallow and deep individuals with >0.98 ancestry assignment (threshold indicated by the dashed lines).
Lack of genetic structuring in S. intersepta
No genetic structuring across populations was identified for S. intersepta (G-statistic Monte Carlo test for all SNPs: Gobs = 107,590, P = 0.3). The lack of genetic structure was further confirmed by assessing pairwise genetic distances between individuals (Fig. 2), and the lack of clustering identified by PCA (Fig. 4D), DAPC, and STRUCTURE (maximum MedMeaK and MedMedK of one cluster; Fig. 4C and table S2). When running STRUCTURE for markers in the 90th to 100th percentile of overall FST (that is, most divergent SNPs), clear structuring between all seven populations was observed (K = 7), but the same structuring could be achieved when shuffling population assignments, highlighting that this apparent recovery of population delineation is an artifact of a priori population assignment (fig. S3). Genetic diversity (that is, expected heterozygosity) was slightly higher in shallow populations (table S1). Despite the lack of genetic differentiation among locations and depths, the Bermuda population was highly differentiated from the two S. intersepta samples from Curaçao (which were included as an out-group; Fig. 2). The Curaçao individuals contained 52 additional SNPs that were fixed in the Bermuda population.
Natural species densities and relative contribution to overall community structure
The species A. fragilis and S. intersepta are depth-generalists that were consistently encountered at both shallow (~12 m) and deep (~40 m) depths around Bermuda (Fig. 6A). Two additional species were also found to be common at both sampling depths: M. cavernosa [focus of a previous vertical connectivity study (25)] and Orbicella franksi. The population densities of these four depth-generalist species were highly variable across locations (Fig. 6A), but for all species other than O. franksi, population densities were higher at depth (except for A. fragilis at Gurnet Rock). In terms of the overall species composition at the three locations (Fig. 6B), the four depth-generalist species constitute 86, 87, and 88% of the community at 40 m versus 20, 16, and 16% at 12 m (19, 14, and 11% when only broadcasting species are considered).
Fig. 6. Depth-generalist species abundances and overlap in community structure.
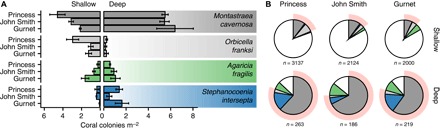
(A) Species densities of the four depth-generalist coral species common to shallow (left) and deep (right) reefs in Bermuda. (B) Proportion of the four depth-generalist species of the overall coral community on shallow and deep reefs in Bermuda (with the proportion of other species indicated in white). Shading behind pie graphs indicates the proportion of species with vertical connectivity potential: M. cavernosa (25), S. intersepta (this study), and O. franksi (based on reproductive mode and depth distribution).
DISCUSSION
The rapid decline of shallow-water coral communities has led to a growing interest in mesophotic coral ecosystems because of their potential to act as a refuge and aid in shallow reef recovery (10, 11, 19, 46). Although the ability of deep reefs to escape disturbance events is relatively well established (11), their role as reproductive sources remains largely untested (13–16). Our study demonstrates that vertical connectivity can vary greatly between species within a single reef system. Using replicated sampling of adjacent shallow and mesophotic populations and a reduced-representation genome sequencing approach (which accounts for endosymbiont contamination), we demonstrate that selection drives depth differentiation in the brooding species A. fragilis. In contrast, extensive gene flow and lack of differentiation characterize the broadcasting species S. intersepta. Overall, the reseed potential of deep reefs appears restricted to only a subset of depth-generalist coral species; although ecologically relevant to these individual species, deep-water coral reseeding should not be assumed to be a broader ecosystem-wide phenomenon.
The strong genome-wide differentiation between shallow and mesophotic populations of A. fragilis was linked to depth-associated selection for a select number of RAD loci (Fig. 3, A and B). A pattern of local-scale genetic differentiation is commonly observed among brooding species and is generally associated with the more localized dispersal of both sperm and larvae (45, 47–50). In the case of A. fragilis, geographic distance cannot account for the strong observed differentiation between shallow and deep eastern populations, because differentiation between depths within each of the three locations consistently exceeded that observed for the same depth between locations (average horizontal distances of ~1.5 km versus ~12 km, respectively). Outlier analyses identified strong depth-associated selection across a number of RAD loci (n = 12; 0.8% of those examined). In the presence of substantial gene flow, such divergent selection across opposing environments is expected to result in heterogeneous genomic divergence (or “genomic islands of divergence”) (51). Instead, for A. fragilis, the background levels of genetic differentiation were also observed to be high (with genetic structuring remaining after removing outliers), indicating that the depth-associated selection in A. fragilis has led to genome-wide “resistance” to shallow-deep gene flow.
Given the observed lack of shallow-deep migrants, immigrant inviability (that is, selection against migrants between locally adapted populations) is likely an important contributor to this resistance to gene flow in A. fragilis, reducing the chances of between-ecotype mating encounters and leading to assortative mating (52). Despite this apparent gene flow barrier, we observed introgression between populations at shallow and mesophotic depths, which may be facilitated through ecologically rare events of migration and interbreeding that were detectable with our relatively small sample sizes. Alternatively, limited introgression could be facilitated by stepping-stone populations at intermediate depths (not sampled), effectively enabling “multigenerational vertical connectivity” (15). Either way, the extent of vertical connectivity does not appear to be ecologically relevant.
Introgression between populations of A. fragilis was asymmetrical (Fig. 5), matching observations from two other studies reporting directional gene flow from shallow to deep populations in the broadcasting species M. cavernosa and the octocoral Eunicea flexuosa (24, 25). In the case of E. flexuosa, higher gamete production in shallow water (due to greater abundances and average colony sizes) was offered as a likely explanation for the asymmetric introgression (24). However, for A. fragilis, this explanation appears unlikely given that population densities at two of three locations (Fig. 6) appear lower at shallow depths. Instead, the lower population densities and the near-complete or complete fixation of the major allele in 8 of 12 outlier RAD loci at shallow depths (Fig. 3) indicate that selection against immigrants may be stronger at shallow depths (and, conversely, may not be as strong for downslope migration). Differential selective pressures could be a contributor to the observed asymmetric introgression (Fig. 5) and associated higher genetic diversity at mesophotic depths (table S1).
The observed introgression and the lack of alternatively fixed polymorphisms in A. fragilis appear to reflect a population-level rather than a species-level divergence. This is further corroborated by the observation that shallow-deep differentiation (eastern locations) is comparable to that observed between the western and eastern deep populations (fig. S2). Although distance and/or seascape resistance (for example, in relation to prevailing currents) may have played a role in the differentiation of this Western Blue Cut (WD) location, several outlier loci were identified in association with this specific population, perhaps indicative of environment-based selection (fig. S1). The western population also exhibited a phenotypic skeletal signature distinct from the other deep locations (Fig. 1B). Despite this study focusing on vertical connectivity, the identification of both neutral differentiation and outlier loci in association with this western location reiterates the broader role of environment-based selection in cross-habitat connectivity (21, 28, 53, 54).
For the gonochoric broadcasting species S. intersepta, no genetic structuring was observed across depths and locations (Figs. 2 and 4). Strong genetic differentiation was only observed for the two out-group individuals from Curaçao (Fig. 2), which is expected given their origin near the opposite end of the latitudinal distribution of this species. Eggs released by S. intersepta (as well as by M. cavernosa) have been observed to exhibit high rates of fertilization immediately upon release (55), which appears to be facilitated by intratentacular fertilization (56). Although this rapid fertilization may not directly affect larval dispersal, it would result in more localized cross-fertilization similar to that in species with a brooding reproductive mode. Assuming such localized spermcasting, the observation of a single panmictic population across the reef platform of Bermuda indicates considerable larval migration between both depths and locations. Similar genetic connectivity over depth has been inferred in Bermuda for the broadcasting species M. cavernosa (4 to 58 m depth) (25). In contrast to the brooding species A. fragilis, no RAD loci were identified to be under depth-specific selection [although, of course, only a reduced proportion of the genome was queried (57)]. In combination, the observed panmixia, apparent morphological plasticity, and association with a single endosymbiont highlight the depth-generalist nature of S. intersepta and the potential for deep populations of this species to act as a larval source for shallow reefs.
The subtraction method allowed us to successfully apply a de novo reduced-representation genome sequencing approach (in this case, nextRAD) on endosymbiont-contaminated samples. Despite our attempt to reduce the endosymbiont contamination through multiple steps of centrifugation before DNA extraction, we still found 10 to 14% of RAD loci to be of Symbiodinium origin, highlighting the necessity of further steps to identify and remove such contamination. Only a small fraction of these sequences could be identified solely by comparison with Symbiodinium reference genomes, which is not unsurprising given their potential level of divergence (particularly in the case of the Symbiodinium minutum “B1” genome). This indicates that comparison with unrelated reference genomes alone is not a viable approach to eliminating endosymbiont contamination. Thus, although genotyping-by-sequencing approaches have demonstrated their de novo potential in nonmodel organisms, population genomic assessments of endosymbiotic marine invertebrates still require either an aposymbiotic host reference [by sequencing aposymbiotic sperm or larvae; for example, see related studies (35–37)] or an endosymbiont subtraction reference (as in this study). Because obtaining aposymbiotic tissue can be difficult, particularly in brooding species, our subtraction method based on fluorescence-activated cell sorting provides an easy and cost-effective solution to this pervasive issue.
Overall, we demonstrate that coral species with similar ecological distributions can exhibit very distinct potentials for vertical connectivity at a single location. Geographic congruence in patterns of vertical connectivity in M. cavernosa and P. astreoides (despite their opposing reproductive modes) highlighted the importance of location-specific extrinsic factors in determining vertical connectivity (14). Our study focused on a single geographic region to specifically evaluate the role of depth, controlling for location-specific effects (by sampling replicate pairs of proximate shallow and mesophotic populations) and sampling genetic variation across the genome. The observed pattern for A. fragilis demonstrates how depth-associated selection can hamper vertical connectivity in the absence of physical dispersal barriers and corroborates the emerging notion that divergence by depth is prevalent in species with a brooding reproductive mode (21, 22, 29, 58). In contrast, we found that the broadcasting species S. intersepta formed a single panmictic population across the same depths and locations. Nonetheless, divergence by depth has been observed in both brooding and broadcasting species (14, 21–25), and the hypothesis that vertical connectivity is more prevalent in broadcasters than in brooders (11) cannot be evaluated until more species and locations are assessed. Regardless, these results demonstrate that the reseeding potential of deep reefs only applies to a subset of depth-generalist species.
Of the ~20 zooxanthellate scleractinian coral species described for Bermuda (59), 6 species appear to be depth-generalists occurring at both shallow and mesophotic depths (44, 60). Among these species, A. fragilis, Madracis decactis, and Scolymia cubensis are brooders, while S. intersepta, M. cavernosa, and O. franksi are broadcasters (61). Although one of the brooders, A. fragilis, occurs in reasonable abundance at both shallow and mesophotic depths, our study showed little evidence for vertical connectivity between shallow and deeper-water populations. On the basis of their reproductive mode, such vertical connectivity may be similarly hampered for M. decactis and S. cubensis. Regardless, S. cubensis was rare at both depths, and M. decactis was only common at intermediate and mesophotic depths along the southern shore (and therefore not individually reported in Fig. 6A), further indicating a limited importance of these species in a reseeding context. Similarly, P. astreoides (for which vertical connectivity was observed across 4 to 26 m depth) (14) was relatively rare at mesophotic depths (~40 m). In contrast, all three broadcasting species occurred in reasonable abundance at both shallow (12 m) and upper mesophotic (40 m) depths (Fig. 6A), and mesophotic populations of these species may represent a viable source of recruitment for the shallow reefs in this isolated reef system (although O. franksi has not been assessed). Nonetheless, although these three species constitute a considerable proportion of the diversity and composition of the upper mesophotic reef, they only represent a relatively small fraction of the diversity (~15% of species) and community (~15% of coral colonies) in shallow water (Fig. 6B).
The DRRH postulates that deep reefs (i) are protected or dampened from disturbances that affect shallow reefs and (ii) can provide a reproductive source after disturbance [sensu Bongaerts et al. (11)]. Although deep reefs can escape certain disturbances, they are not entirely immune to disturbance (6–9, 26). In fact, even their ability to escape bleaching and storm events can vary spatially and temporally, and their greater depth does not confer long-term protection against disturbances associated with climate change (9). Similarly, disturbance events can vary strongly in their impact over depth and in their impact on specific species, which can vary in thermal susceptibility or structural fragility. Thus rather than the term “refugia” (as currently used in “deep reef refugia hypothesis”), which Keppel et al. (62) classify as large geographic areas offering an escape over evolutionary time scales (for example, climate change refugia), we propose that the term “refuge(s)” is probably more appropriate (that is, “deep reef refuge hypothesis”), reflecting the role of deep reefs in providing temporal and/or spatial protection from disturbances on ecological time scales. In addition, this study confirms that the active reseeding potential is specific to only a subset of species (~15% here) and does not apply to a significant proportion of the species (diversity) that make up shallow reefs. Therefore, we conclude that the “deep reef refuge hypothesis” should be considered as an ecological concept relevant to individual species, rather than being referred to as an ecosystem-wide phenomenon.
MATERIALS AND METHODS
Study locations, specimen collections, and skeletal morphometrics
We focused on the isolated Bermuda platform in the Western Atlantic, where we targeted shallow (12 ± 2 m) and deep (40 ± 2 m) habitats at four different reef locations: Gurnet Rock, John Smith’s Bay, and Princess Beach on the southeastern side and Western Blue Cut on the northwestern side of the island (table S1). Specimens (n ≈ 30 per depth at each location) of A. fragilis (n = 208) and S. intersepta (n = 209) were collected under collection permit no. 130901b from the Department of Environmental Protection of the Bermuda Government, as part of the “XL Catlin Seaview Survey” in September 2013. Two additional samples of S. intersepta from Curaçao were included for use as a comparative out-group. Small fragments of the collected specimens were stored in salt-saturated buffer solution containing 20% dimethyl sulfoxide and 0.5 M EDTA, and for a subset of specimens, a skeletal voucher was bleached, rinsed in freshwater, and dried. For these skeletal vouchers (A. fragilis, n = 49; S. intersepta, n = 37), corallite diameter and density were assessed across five pseudoreplicates per specimen.
Separation of host fractions and DNA extraction
To counter the issue of pervasive endosymbiont contamination, we separated the tissue from the skeleton and performed multiple centrifugation steps separating out the supernatant to eliminate intact Symbiodinium cells. Genomic DNA (gDNA) was extracted using a modified version of the “Wayne’s method” (for details, see electronic notebook). Quality and yield of gDNA were assessed using gel electrophoresis and a Qubit fluorometer (Invitrogen) to select a subset of high-quality samples (n ≈ 16 per species per depth at each location) for downstream sequencing (A. fragilis, n = 110; S. intersepta, n = 113; Symbiodinium, n = 15). Selected samples were normalized (gDNA quantity) and purified using AMPure XP beads to remove potential inhibitors.
Isolation of Symbiodinium
Given that a certain degree of endosymbiont contamination remained in the “host” samples [confirmed through polymerase chain reaction (PCR) amplification with Symbiodinium-specific primers, likely due to lysed cells], we isolated individual Symbiodinium cells from the original pellet (which was, in turn, contaminated with a certain extent of host cells) to be sequenced separately as a subtraction reference. Symbiodinium isolation was carried out for aliquots of a shallow and deep specimen from each of the two species using fluorescence-activated cell sorting (BD FACSAria cell sorter) at the Queensland Brain Institute, separating out ~300,000 individual cells based on their distinct fluorescence signature (for details, see electronic notebook). In addition, Symbiodinium cells from several other coral host species were isolated from samples intended for other species-specific studies. Extractions of gDNA for the isolates were carried out as for the host.
Library preparation, sequencing, and clustering
Library preparation was carried out using the nextRAD method (Nextera-fragmented, reductively amplified DNA; SNPsaurus LLC), which uses a selective primer sequence (rather than restriction enzymes) to genotype loci consistently between samples (63). gDNA was fragmented and ligated with adapter sequences using Nextera reagent (Illumina Inc). Fragmented DNA was then PCR-amplified (73°C for 26 cycles) with one of the primers matching the adapter and extending into the gDNA using a 9–base pair (bp) selective sequence (“GTGTAGAGG”). Libraries were sequenced on three HiSeq 2500 (Illumina Inc) lanes using 100-bp single-end chemistry and following the manufacturer’s recommended protocol. Quality control, clustering, and SNP variant calling were carried out using PyRAD v3.0.6 (64) to allow for indel variation (common in marine invertebrates). After initial runs at different thresholds, we separately analyzed both species using a clustering threshold of 90%, a minimum coverage of 6, a maximum of three sites with a PHRED quality below 20, and a minimum of 10 samples in a final locus. Sequence data from the isolated Symbiodinium were filtered using PyRAD, allowing for a maximum of five sites with a PHRED quality below 20 (step 1), and reads were then clustered within samples using an 85% clustering threshold (step 3). Clusters with a coverage of ≥2 (to eliminate singletons) were reduced to a single representative sequence and used to compile a BLASTN database (n = 697,146 sequences). Parsing and analyses were carried out using custom Python (v3.4.5) scripts (available through https://github.com/pimbongaerts/radseq), unless otherwise indicated, with statistical analyses and plotting performed in R (v3.3.1) (electronic notebook available through https://github.com/pimbongaerts/bermuda-rad).
Identification of coral host loci and QC
One representative sequence was extracted for each coral RAD locus, which was then BLASTN-searched against our Symbiodinium RAD database, and two culture-based Symbiodinium genomes [Symbiodinium B1 from Shoguchi et al. (65) and Symbiodinium C1 from Chan et al., draft genome], with positive matches (maximum E-value of 10−15) removed from the coral RAD data set. An additional BLASTN comparison was carried out against the National Center for Biotechnology Information (NCBI) nonredundant nucleotide database (with the particular aim of removing any microbial contamination, if applicable), with taxonomic IDs of positive matches (maximum E-value of 10−4) extracted and classified to phylum using the NCBI Taxonomy database. RAD loci matching to non-cnidarian taxa were subsequently removed. Given an overrepresentation of SNPs toward the end of reads, RAD loci were then truncated to 75 and 80 bp for A. fragilis and S. intersepta, respectively. SNPs representing singletons/doubletons were removed (rather than filtering for a minimum allele frequency), as were SNPs that were genotyped for <50% of individuals in each population (rather than using an overall representation filter). Genetic distance/similarity (Hamming-based) was then calculated between all pairs of individuals to identify clones, with clonal groups reduced to a single representative per population. The remaining SNPs were then evaluated for significant deviations from the Hardy-Weinberg equilibrium using arlecore (v3.5.2.2) (66), with SNPs that deviated (excess or deficit) in more than five populations removed, as well as RAD loci that contained an SNP exhibiting a significant excess in at least one population (to filter out potential paralogous RAD loci).
Outliers and private alleles
Initially, outliers were identified for the overall data set and for pairwise population comparisons (considering only biallelic SNPs) using the Fdist approach (67) as implemented in Lositan (v. 1.0) (68) and BayeScan (v.2.1) (69). To conservatively identify depth-outliers, we considered those SNPs that were identified in the overall outlier analyses by both Fdist [outside the 99% confidence interval (CI); 50,000 simulations] and BayeScan (using a Bayes factor cutoff of 0.01), as well as those that were identified in at least two of three independent Fdist outlier analyses between individual shallow and deep populations within a location (but never in Fdist outlier analyses within the same depth between different locations). Genotype frequencies of outliers were calculated, and sequences of depth-outlier RAD loci were searched (BLASTN) against the NCBI nonredundant nucleotide database for potential gene region identification. To identify a subset of SNPs that was more representative of neutral genomic diversity, we compiled a separate data set (neutral), eliminating SNPs identified in one or both overall outlier analyses using more lenient cutoffs (95% CI for Fdist and a Bayes factor cutoff of 0.05 for BayeScan).
Genetic structure
To assess the genetic structure for the outlier and nonoutlier data set, we wrote a multiprocessing wrapper (“structure_mp”) for STRUCTURE (v.2.3.4) (70), which takes a Variant Call Format file and converts it into distinct replicate STRUCTURE data sets, each with a single SNP per RAD locus, which are then ran in parallel through STRUCTURE and permutated and summarized in CLUMPP (v.1.1.2) (71). Four estimators of the number of clusters (MedMeaK, MaxMeaK, MedMedK, and MaxMedK) were determined using a membership coefficient threshold of 0.5 [sensu Puechmaille (72)]. STRUCTURE runs were conducted using the admixture model with correlated allele frequencies and not considering priors (burn-in of 100,000 and 50,000 reps). Additional STRUCTURE runs were carried out for the eastern populations of A. fragilis (K = 2) for downstream ancestry analysis and for the most differentiated SNPs (90th to 100th percentile) of S. intersepta (based on overall FST calculated for the original data and when using random population assignments). Genetic structure was also assessed for both species through PCA and DAPC, both using the adegenet R package (v1.3-1) (73).
Genomic intergradation of shallow and deep individuals
Patterns of SNP ancestry were assessed for shallow and deep individuals of A. fragilis (eastern populations only) by classifying the data set into two putative “parental” groups (those with an ancestry assignment of >0.98) and an admixed group. We then extracted genotypes for the identified depth-outlier SNPs, as well as other SNPs that had a reference allele frequency differential (Δp) between parental groups of at least 0.5, and plotted for all the individuals sorted by ancestry assignment (similar to the visualization in the R package “introgress”) (74).
Population genetics statistics
To test for overall population structure in both species, we implemented Goudet’s G-statistic Monte Carlo test (75) using the “hierfstat” R package (v0.04-10) (76), with 99 permutations considering both all and the six eastern populations. Inbreeding statistics were estimated per locus among all populations (global estimates) and among all pairs of populations (using the adegenet R package). To determine whether population structure (based on Weir and Cockerham FST using the hierfstat R package) differed overall among groups of populations, we used a nonparametric Kruskal-Wallis test. To test the specific hypothesis for A. fragilis that differentiation by habitat exceeds differentiation within habitat (as expected under a scenario of divergence by ecology) (77), we compared FST values obtained from the three shallow-deep pairs against pairwise values for the three possible shallow versus shallow comparisons. For deep versus deep comparisons, we separately examined the three eastern comparisons (mirroring the shallow versus shallow comparisons) and also examined the six total possible comparisons when the western population (WD) was included. All between-habitat versus within-habitat contrasts were conducted using nonparametric Wilcoxon signed-rank tests.
Symbiodinium genotyping and community structure assessment
The associated Symbiodinium of 78 samples were sequenced for the Symbiodinium COX1 region (78) using the reverse primer under conditions outlined in Bongaerts et al. (26). Further differentiation of associated types within species was carried out on the basis of a variable region in the chloroplast minicircle recovered as a RAD locus for both species (n = 188). Coral community structure was assessed at the three eastern locations (Gurnet Rock, John Smith’s Bay, and Princess Beach) along three 30-m photographic transects at both sampling depths. The total number of scleractinian coral colonies was counted in each photograph (and standardized to surface area using the photographed transect tape), including separate counts for the depth-generalist species A. fragilis, S. intersepta, M. cavernosa, and O. franksi.
Supplementary Material
Acknowledgments
We thank D. Whillas and A. Chequer for their major efforts toward fieldwork collections. We thank K. Latijnhouwers, S. den Haan, E. Sampayo, and V. Nink for their contributions toward the Symbiodinium isolation. We thank E. Johnson and P. Etter for their input with regard to sample preparation and sequencing approach. We thank the staff at the Bermuda Institute of Ocean Sciences (particularly A. Hunter), Global Change Institute (particularly S. Naylor and D. Harris), and The Ocean Agency (particularly R. Vevers, L. Parry, and C. Bailhache) for their logistical support. Funding: This study was undertaken as part of the XL Catlin Seaview Survey, designed and undertaken by the Global Change Institute, and funded by XL Catlin in partnership with The Ocean Agency and The University of Queensland. Author contributions: P.B. and O.H.-G. conceived the study. P.B., N.E., and S.R.S. conducted the fieldwork and specimen collections. R.B. conducted the laboratory work and photographic analysis. P.B. performed the data analyses and wrote the manuscript, with contributions from C.R. All authors edited the manuscript before submission. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data and analyses are available as an electronic notebook (https://github.com/pimbongaerts/bermuda-rad), with scripts available through https://github.com/pimbongaerts/radseq. Raw Illumina sequence data can be obtained from the NCBI Sequence Read Archive (SRA) under the accession code PRJNA361144 (https://www.ncbi.nlm.nih.gov/bioproject/361144).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/2/e1602373/DC1
fig. S1. Genotype frequencies of SNP outliers for A. fragilis.
fig. S2. Genome-wide pairwise differentiation (FST) of populations.
fig. S3. Genetic structuring of S. intersepta populations based on most divergent SNPs.
table S1. Details of the sampled A. fragilis and S. intersepta populations.
table S2. Estimators of the number of STRUCTURE clusters for A. fragilis and S. intersepta.
table S3. Genome-wide pairwise differentiation (FST) of populations.
REFERENCES AND NOTES
- 1.Pandolfi J. M., Bradbury R. H., Sala E., Hughes T. P., Bjorndal K. A., Cooke R. G., McArdle D., McClenachan L., Newman M. J. H., Paredes G., Warner R. R., Jackson J. B. C., Global trajectories of the long-term decline of coral reef ecosystems. Science 301, 955–958 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Hoegh-Guldberg O., Mumby P. J., Hooten A. J., Steneck R. S., Greenfield P., Gomez E., Harvell C. D., Sale P. F., Edwards A. J., Caldeira K., Knowlton N., Eakin C. M., Iglesias-Prieto R., Muthiga N., Bradbury R. H., Dubi A., Hatziolos M. E., Coral reefs under rapid climate change and ocean acidification. Science 318, 1737–1742 (2007). [DOI] [PubMed] [Google Scholar]
- 3.De'ath G., Fabricius K. E., Sweatman H., Puotinen M., The 27-year decline of coral cover on the Great Barrier Reef and its causes. Proc. Natl. Acad. Sci. U.S.A. 109, 17995–17999 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glynn P. W., Coral reef bleaching: Facts, hypotheses and implications. Global Change Biol. 2, 495–509 (1996). [Google Scholar]
- 5.McCook L. J., Almany G. R., Berumen M. L., Day J. C., Green A. L., Jones G. P., Leis J. M., Planes S., Russ G. R., Sale P. F., Management under uncertainty: Guide-lines for incorporating connectivity into the protection of coral reefs. Coral Reefs 28, 353–366 (2009). [Google Scholar]
- 6.Thomson D. P., Bearham D., Graham F., Eagle J. V., High latitude, deeper water coral bleaching at Rottnest Island, Western Australia. Coral Reefs 30, 1107 (2011). [Google Scholar]
- 7.Bongaerts P., Muir P., Englebert N., Bridge T. C. L., Hoegh-Guldberg O., Cyclone damage at mesophotic depths on Myrmidon Reef (GBR). Coral Reefs 32, 935 (2013). [Google Scholar]
- 8.Appeldoorn R., Ballantine D., Bejarano I., Carlo M., Nemeth M., Otero E., Pagan F., Ruiz H., Schizas N., Sherman C., Weil E., Mesophotic coral ecosystems under anthropogenic stress: A case study at Ponce, Puerto Rico. Coral Reefs 35, 63–75 (2016). [Google Scholar]
- 9.Smith T. B., Gyory J., Brandt M. E., Miller W. J., Jossart J., Nemeth R. S., Caribbean mesophotic coral ecosystems are unlikely climate change refugia. Global Change Biol. 22, 2756–2765 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Lesser M. P., Slattery M., Leichter J. J., Ecology of mesophotic coral reefs. J. Exp. Mar. Biol. Ecol. 375, 1–8 (2009). [Google Scholar]
- 11.Bongaerts P., Ridgway T., Sampayo E. M., Hoegh-Guldberg O., Assessing the ‘deep reef refugia’ hypothesis: Focus on Caribbean reefs. Coral Reefs 29, 309–327 (2010). [Google Scholar]
- 12.Bridge T. C. L., Hughes T. P., Guinotte J. M., Bongaerts P., Call to protect all coral reefs. Nat. Clim. Change 3, 528–530 (2013). [Google Scholar]
- 13.Slattery M., Lesser M. P., Brazeau D., Stokes M. D., Leichter J. J., Connectivity and stability of mesophotic coral reefs. J. Exp. Mar. Biol. Ecol. 408, 32–41 (2011). [Google Scholar]
- 14.Serrano X. M., Baums I. B., Smith T. B., Jones R. J., Shearer T. L., Baker A. C., Long distance dispersal and vertical gene flow in the Caribbean brooding coral Porites astreoides. Sci. Rep. 6, 21619 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holstein D. M., Paris C. B., Vaz A. C., Smith T. B., Modeling vertical coral connectivity and mesophotic refugia. Coral Reefs 35, 23–37 (2016). [Google Scholar]
- 16.E. K. Baker, K. A. Puglise, P. T. Harris, Mesophotic Coral Ecosystems—A Lifeboat for Coral Reefs? (The United Nations Environment Programme and GRID-Arendal, Nairobi and Arenal, 2016), 98 pp. [Google Scholar]
- 17.Kahng S. E., Garcia-Sais J. R., Spalding H. L., Brokovich E., Wagner D., Weil E., Hinderstein L., Toonen R. J., Community ecology of mesophotic coral reef ecosystems. Coral Reefs 29, 255–275 (2010). [Google Scholar]
- 18.Kahng S. E., Copus J. M., Wagner D., Recent advances in the ecology of mesophotic coral ecosystems (MCEs). Curr. Opin. Environ. Sustain. 7, 72–81 (2014). [Google Scholar]
- 19.Loya Y., Eyal G., Treibitz T., Lesser M. P., Appeldoorn R., Theme section on mesophotic coral ecosystems: Advances in knowledge and future perspectives. Coral Reefs 35, 1–9 (2016). [Google Scholar]
- 20.Muir P., Wallace C., Bridge T. C. L., Bongaerts P., Diverse staghorn coral fauna on the mesophotic reefs of north-east Australia. PLOS ONE 10, e0117933 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bongaerts P., Riginos C., Ridgway T., Sampayo E. M., van Oppen M. J. H., Englebert N., Vermeulen F., Hoegh-Guldberg O., Genetic divergence across habitats in the widespread coral Seriatopora hystrix and its associated Symbiodinium. PLOS ONE 5, e10871 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Oppen M. J., Bongaerts P., Underwood J. N., Peplow L. M., Cooper T. F., The role of deep reefs in shallow reef recovery: An assessment of vertical connectivity in a brooding coral from west and east Australia. Mol. Ecol. 20, 1647–1660 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Brazeau D. A., Lesser M. P., Slattery M., Genetic structure in the coral, Montastraea cavernosa: Assessing genetic differentiation among and within mesophotic reefs. PLOS ONE 8, e65845 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prada C., Hellberg M. E., Long prereproductive selection and divergence by depth in a Caribbean candelabrum coral. Proc. Natl. Acad. Sci. U.S.A. 110, 3961–3966 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serrano X., Baums I. B., O'Reilly K., Smith T. B., Jones R. J., Shearer T. L., Nunes F. L. D., Baker A. C., Geographic differences in vertical connectivity in the Caribbean coral Montastraea cavernosa despite high levels of horizontal connectivity at shallow depths. Mol. Ecol. 23, 4226–4240 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Bongaerts P., Frade P. R., Hay K. B., Englebert N., Latijnhouwers K. R., Bak R. P. M., Vermeij M. J. A., Hoegh-Guldberg O., Deep down on a Caribbean reef: Lower mesophotic depths harbor a specialized coral-endosymbiont community. Sci. Rep. 5, 7652 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eytan R. I., Hayes M., Arbour-Reily P., Miller M., Hellberg M. E., Nuclear sequences reveal mid-range isolation of an imperilled deep-water coral population. Mol. Ecol. 18, 2375–2389 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Bongaerts P., Riginos C., Hay K. B., van Oppen M. J. H., Hoegh-Guldberg O., Dove S., Adaptive divergence in a scleractinian coral: Physiological adaptation of Seriatopora hystrix to shallow and deep reef habitats. BMC Evol. Biol. 11, 303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlon D. B., Budd A. F., Incipient speciation across a depth gradient in a scleractinian coral? Evolution 56, 2227–2242 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Gilmour J. P., Underwood J. N., Howells E. J., Gates E., Heyward A. J., Biannual spawning and temporal reproductive isolation in Acropora corals. PLOS ONE 11, e0150916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller M. R., Dunham J. P., Amores A., Cresko W. A., Johnson E. A., Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 17, 240–248 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baird N. A., Etter P. D., Atwood T. S., Currey M. C., Shiver A. L., Lewis Z. A., Selker E. U., Cresko W. A., Johnson E. A., Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLOS ONE 3, e3376 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toonen R. J., Puritz J. B., Forsman Z. H., Whitney J. L., Fernandez-Silva I., Andrews K. R., Bird C. E., ezRAD: A simplified method for genomic genotyping in non-model organisms. PeerJ 1, e203 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Combosch D. J., Vollmer S. V., Trans-pacific RAD-Seq population genomics confirms introgressive hybridization in Eastern Pacific Pocillopora corals. Mol. Phylogenet. Evol. 88, 154–162 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Dixon G. B., Davies S. W., Aglyamova G. V., Meyer E., Bay L. K., Matz M. V., Genomic determinants of coral heat tolerance across latitudes. Science 348, 1460–1462 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Barfield S., Aglyamova G. V., Matz M. V., Evolutionary origins of germline segregation in Metazoa: Evidence for a germ stem cell lineage in the coral Orbicella faveolata (Cnidaria, Anthozoa). Proc. Biol. Sci. 283, 20152128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howells E. J., Abrego D., Meyer E., Kirk N. L., Burt J. A., Host adaptation and unexpected symbiont partners enable reef-building corals to tolerate extreme temperatures. Global Change Biol. 22, 2702–2714 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Blackall L. L., Wilson B., van Oppen M. J. H., Coral—The world’s most diverse symbiotic ecosystem. Mol. Ecol. 24, 5330–5347 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Muscatine L., McCloskey L. R., Marian R. E., Estimating the daily contribution of carbon from zooxanthellae to coral animal respiration. Limnol. Oceanogr. 26, 601–611 (1981). [Google Scholar]
- 40.LaJeunesse T. C., Pettay D. T., Sampayo E. M., Phongsuwan N., Brown B., Obura D. O., Hoegh-Guldberg O., Fitt W. K., Long-standing environmental conditions, geographic isolation and host–symbiont specificity influence the relative ecological dominance and genetic diversification of coral endosymbionts in the genus Symbiodinium. J. Biogeogr. 37, 785–800 (2010). [Google Scholar]
- 41.Bongaerts P., Carmichael M., Hay K. B., Tonk L., Frade P. R., Hoegh-Guldberg O., Prevalent endosymbiont zonation shapes the depth distributions of scleractinian coral species. R. Soc. Open Sci. 2, 140297 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shinzato C., Shoguchi E., Kawashima T., Hamada M., Hisata K., Tanaka M., Fujie M., Fujiwara M., Koyanagi R., Ikuta T., Fujiyama A., Miller D. J., Satoh N., Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 476, 320–323 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Bhattacharya D., Agrawal S., Aranda M., Baumgarten S., Belcaid M., Drake J. L., Erwin D., Foret S., Gates R. D., Gruber D. F., Kamel B., Lesser M. P., Levy O., Liew Y. J., MacManes M., Mass T., Medina M., Mehr S., Meyer E., Price D. C., Putnam H. M., Qiu H., Shinzato C., Shoguchi E., Stokes A. J., Tambutté S., Tchernov D., Voolstra C. R., Wagner N., Walker C. W., Weber A. P. M., Weis V., Zelzion E., Zoccola D., Falkowski P. G., Comparative genomics explains the evolutionary success of reef-forming corals. eLife 5, e13288 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goodbody-Gringley G., Marchini C., Chequer A. D., Goffredo S., Population structure of Montastraea cavernosa on shallow versus mesophotic reefs in Bermuda. PLOS ONE 10, e0142427 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goodbody-Gringley G., Vollmer S. V., Woollacott R. M., Giribet G., Limited gene flow in the brooding coral Favia fragum (Esper, 1797). Mar. Biol. 157, 2591–2602 (2010). [Google Scholar]
- 46.Hughes T. P., Tanner J. E., Recruitment failure, life histories, and long-term decline of Caribbean corals. Ecology 81, 2250–2263 (2000). [Google Scholar]
- 47.Carlo D. B., Olson R. R., Larval dispersal distance as an explanation for adult spatial pattern in two Caribbean reef corals. J. Exp. Mar. Biol. Ecol. 173, 247–263 (1993). [Google Scholar]
- 48.Ayre D. J., Hughes T. P., Genotypic diversity and gene flow in brooding and spawning corals along the Great Barrier Reef, Australia. Evolution 54, 1590–1605 (2000). [DOI] [PubMed] [Google Scholar]
- 49.Underwood J. N., Smith L. D., van Oppen M. J. H., Gilmour J. P., Multiple scales of genetic connectivity in a brooding coral on isolated reefs following catastrophic bleaching. Mol. Ecol. 16, 771–784 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Warner P. A., Willis B. L., van Oppen M. J. H., Sperm dispersal distances estimated by parentage analysis in a brooding scleractinian coral. Mol. Ecol. 25, 1398–1415 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Nosil P., Funk D. J., Ortiz-Barrientos D., Divergent selection and heterogeneous genomic divergence. Mol. Ecol. 18, 375–402 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Nosil P., Vines T. H., Funk D. J., Reproductive isolation caused by natural selection against immigrants from divergent habitats. Evolution 59, 705–719 (2005). [PubMed] [Google Scholar]
- 53.Barshis D. J., Stillman J. H., Gates R. D., Toonen R. J., Smith L. W., Birkeland C., Protein expression and genetic structure of the coral Porites lobata in an environmentally extreme Samoan back reef: Does host genotype limit phenotypic plasticity? Mol. Ecol. 19, 1705–1720 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Kenkel C. D., Goodbody-Gringley G., Caillaud D., Davies S. W., Bartels E., Matz M. V., Evidence for a host role in thermotolerance divergence between populations of the mustard hill coral (Porites astreoides) from different reef environments. Mol. Ecol. 22, 4335–4348 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Hagman D. K., Gittings S., Vize P. D., Fertilization in broadcast-spawning corals of the Flower Garden Banks National Marine Sanctuary. Gulf Mex. Sci. 16, 180–187 (1998). [Google Scholar]
- 56.Vermeij M. J. A., Barott K. L., Johnson A. E., Marhaver K. L., Release of eggs from tentacles in a Caribbean coral. Coral Reefs 29, 411 (2010). [Google Scholar]
- 57.Hoban S., Kelley J. L., Lotterhos K. E., Antolin M. F., Bradburd G., Lowry D. B., Poss M. L., Reed L. K., Storfer A., Whitlock M. C., Finding the genomic basis of local adaptation: Pitfalls, practical solutions, and future directions. Am. Nat. 188, 379–397 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gorospe K. D., Karl S. A., Depth as an organizing force in Pocillopora damicornis: Intra-reef genetic architecture. PLOS ONE 10, e0122127 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.J. M. Locke, J. P. Bilewitch, K. A. Coates, in Coral Reefs of the United Kingdom Overseas Territories, C.R.C. Sheppard, Ed. (Springer, 2013), pp. 189–200. [Google Scholar]
- 60.Fricke H., Meischner D., Depth limits of Bermudan scleractinian corals: A submersible survey. Mar. Biol. 88, 175–187 (1985). [Google Scholar]
- 61.Baird A. H., Guest J. R., Willis B. L., Systematic and biogeographical patterns in the reproductive biology of scleractinian corals. Annu. Rev. Ecol. Evol. Syst. 40, 551–571 (2009). [Google Scholar]
- 62.Keppel G., Van Niel K. P., Wardell-Johnson G. W., Yates C. J., Byrne M., Mucina L., Schut A. G. T., Hopper S. D., Franklin S. E., Refugia: Identifying and understanding safe havens for biodiversity under climate change. Global Ecol. Biogeogr. 21, 393–404 (2012). [Google Scholar]
- 63.Russello M. A., Waterhouse M. D., Etter P. D., Johnson E. A., From promise to practice: Pairing non-invasive sampling with genomics in conservation. PeerJ 3, e1106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eaton D. A., PyRAD: Assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics 30, 1844–1849 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Shoguchi E., Shinzato C., Kawashima T., Gyoja F., Mungpakdee S., Koyanagi R., Takeuchi T., Hisata K., Tanaka M., Fujiwara M., Hamada M., Seidi A., Fujie M., Usami T., Goto H., Yamasaki S., Arakaki N., Suzuki Y., Sugano S., Toyoda A., Kuroki Y., Fujiyama A., Medina M., Coffroth M. A., Bhattacharya D., Satoh N., Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr. Biol. 23, 1399–1408 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Excoffier L., Lischer H. E., Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010). [DOI] [PubMed] [Google Scholar]
- 67.Beaumont M. A., Nichols R. A., Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. London Ser. B. 263, 1619–1626 (1996). [Google Scholar]
- 68.Antao T., Lopes A., Lopes R. J., Beja-Pereira A., Luikart G., LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics 9, 323 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Foll M., Gaggiotti O., A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 180, 977–993 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pritchard J. K., Stephens M., Donnelly P., Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jakobsson M., Rosenberg N. A., CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (2007). [DOI] [PubMed] [Google Scholar]
- 72.Puechmaille S. J., The program structure does not reliably recover the correct population structure when sampling is uneven: Subsampling and new estimators alleviate the problem. Mol. Ecol. Resour. 16, 608–627 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Jombart T., Ahmed I., adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gompert Z., Buerkle C. A., introgress: A software package for mapping components of isolation in hybrids. Mol. Ecol. Resour. 10, 378–384 (2010). [DOI] [PubMed] [Google Scholar]
- 75.Goudet J., Raymond M., de Meeüs T., Rousset F., Testing differentiation in diploid populations. Genetics 144, 1933–1940 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Goudet J., Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Resour. 5, 184–186 (2005). [Google Scholar]
- 77.Schluter D., Evidence for ecological speciation and its alternative. Science 323, 737–741 (2009). [DOI] [PubMed] [Google Scholar]
- 78.Pochon X., Putnam H. M., Burki F., Gates R. D., Identifying and characterizing alternative molecular markers for the symbiotic and free-living dinoflagellate genus Symbiodinium. PLOS ONE 7, e29816 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/2/e1602373/DC1
fig. S1. Genotype frequencies of SNP outliers for A. fragilis.
fig. S2. Genome-wide pairwise differentiation (FST) of populations.
fig. S3. Genetic structuring of S. intersepta populations based on most divergent SNPs.
table S1. Details of the sampled A. fragilis and S. intersepta populations.
table S2. Estimators of the number of STRUCTURE clusters for A. fragilis and S. intersepta.
table S3. Genome-wide pairwise differentiation (FST) of populations.