

Abstract
Human onchocerciasis is a serious neglected tropical disease caused by the filarial nematode Onchocerca volvulus that can lead to blindness and chronic disability. Control of the disease relies largely on mass administration of a single drug, and the development of new drugs and vaccines depends on a better knowledge of parasite biology. Here, we describe the chromosomes of O. volvulus and its Wolbachia endosymbiont. We provide the highest-quality sequence assembly for any parasitic nematode to date, giving a glimpse into the evolution of filarial parasite chromosomes and proteomes. This resource was used to investigate gene families with key functions that could be potentially exploited as targets for future drugs. Using metabolic reconstruction of the nematode and its endosymbiont, we identified enzymes that are likely to be essential for O. volvulus viability. In addition, we have generated a list of proteins that could be targeted by Federal-Drug-Agency-approved but repurposed drugs, providing starting points for anti-onchocerciasis drug development.
The filaria are a group of tissue-dwelling parasitic nematodes of vertebrates that are spread by blood-feeding arthropods. Onchocerca volvulus is the most pathogenic and is the agent of onchocerciasis (or ‘river blindness’), a leading cause of morbidity and socioeconomic loss for the world’s poorest populations1. Approximately 17 million people are still infected with O. volvulus, predominantly in Africa2. Infections are chronic and manifest clinically as debilitating skin disease and—in 1.2 million people—vision impairment or blindness. First-stage larvae, known as microfilariae (L1/mf), are produced by fertile female worms residing within onchocercomata (nodules). They migrate to the skin and other organs (for example, the anterior chamber of the eye), where they induce inflammatory reactions that are responsible for most Onchocerca-related pathology.
Onchocerciasis was identified by the World Health Organization (WHO) as a potential candidate for disease elimination through annual (or semiannual) mass drug administration (MDA) of ivermectin3, an approach that has eliminated onchocerciasis from all but two countries in the Americas4. Ivermectin is solely microfilaricidal, which means it must be given over decades, past the lifespan of the long-lived adult worms5,6. Moreover, in much of Central Africa where Loa loa is co-endemic with O. volvulus, ivermectin cannot be used due to the risk of Loa-associated irreversible neurological severe adverse events and death7. Despite its success in Latin America and small foci in Africa, elimination of onchocerciasis in Africa is unlikely to be achieved within the proposed timeframes solely through MDA with ivermectin8. Reliance on a single drug also increases the potential for the emergence of ivermectin-resistant O. volvulus5, making the development of new drugs or novel therapies imperative.
To gain better insight into this important but neglected pathogen, we generated a high-quality genome assembly of O. volvulus. Although draft whole-genome assemblies exist for other filarial nematode species9–13, we have, for the first time in any species of this group, reconstructed whole chromosomes, including a sex chromosome. We also generated a genome assembly for its obligate intracellular endosymbiont Wolbachia (wOv) and transcriptional data from eight life stages. Our analysis highlights the metabolic interplay between O. volvulus and wOv as a path to novel drug targets.
Results
O. volvulus genome structure and features
The 97 Mb nuclear genome of O. volvulus comprising three autosomes and a pair of sex chromosomes14 was assembled using a combination of sequencing, an optical map and manual improvement. Four large scaffolds (16–31 Mb) comprise 94% of the assembly and seven out of eight of their ends correspond to ends of optical maps or telomeric repeats (Fig. 1a and Supplementary Fig. 1). These scaffolds thus represent essentially complete chromosomes of O. volvulus. This is the highest-quality assembly for any parasitic nematode (Supplementary Tables 1 and 2) and only the fourth nematode species15–17 for which chromosome sequences are available. The assembly also includes the mitochondrial genome and complete wOv genome.
Figure 1. O. volvulus chromosomes.
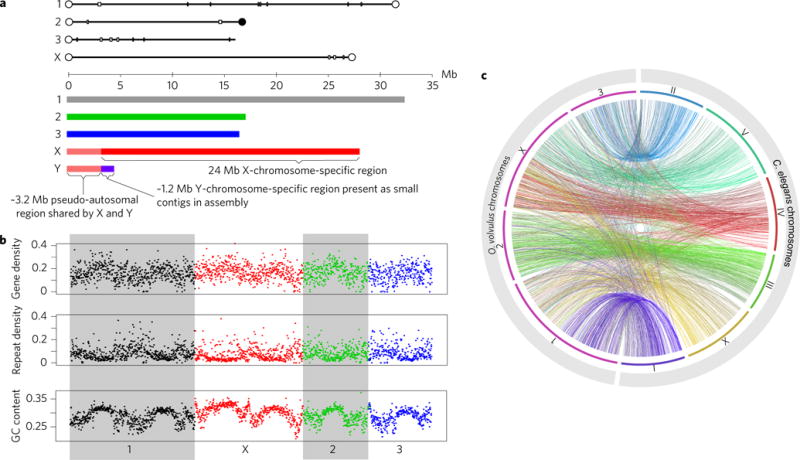
a, Lines above the axis show sizes of O. volvulus chromosomes in the assembly and the locations of potential telomeric repeats. Filled circles indicate telomere repeats present in the assembly, open circles are ends of optical map scaffolds (Supplementary Fig. 1c). Rectangular boxes/bars indicate sequence gaps of at least 50 kb. The inferred karyotype for O. volvulus is shown below the axis, based on the assembly and sequence coverage data. b, GC content, gene density and repeat density (proportion of bases in each window covered by genes/at least one annotated repeat) in non-overlapping 10 kb windows for each of the four large scaffolds. Colours and shading indicate scaffold boundaries. c, Comparison of four O. volvulus chromosomes with six C. elegans chromosomes. Links show PROmer hits with similarity greater than 70% over at least 100 amino acids, coloured according to chromosome location of C. elegans hit.
By analysing sequence data from male and female worms, we identified scaffold OM2 as the X chromosome (Supplementary Fig. 2a). For a long contiguous portion (22.2 of 25.5 Mb of the scaffold) the median depth of coverage of male sequence data was 50% that for the rest of the genome. The same region had a coverage of 75% using data from mature females, which are likely to be gravid and contain a mixture of male and female cells, and 100% using data from juvenile female worms only (Supplementary Fig. 2a and Supplementary Table 3). We propose that this region represents the X-chromosome-specific sequence, while the rest of the scaffold is a 3.2 Mb pseudo-autosomal region (PAR) shared by X and Y chromosomes and presumably still capable of chromosomal crossover. Other scaffolds lack data from juvenile females and show low coverage in adult female libraries (Supplementary Fig. 2c,d), allowing us to identify ∼1.2 Mb as the potentially Y-specific sequence. Only this small portion of the Y chromosome is present in our assembly as this chromosome is largely pseudo-autosomal (Fig. 1a) and so mostly assembled with the X chromosome. The small extent of sequence divergence between X and Y and the presence of an extensive PAR confirm that this evolved recently from an ancestral XO karyotype14 and contrasts with the situation in other nematodes that have X and Y chromosomes, where the Y is largely unique, repeat-rich and degenerate18. Furthermore, one region of the PAR adjacent to the X-specific region shows an excess of heterozygous sites missing in the juvenile female sample (Supplementary Fig. 2b). This represents a region in which X and Y have begun to diverge, but where the two chromosomes are sufficiently similar that they are still represented by the same region of the assembly. We propose that this is a region of more recent divergence between X and Y chromosomes than the X- and Y-specific regions. This suggests a process of sex chromosome evolution similar to that observed in other systems, in which recombination suppression and subsequent divergence between sex chromosomes occurs in a patchy way, leading to different ‘strata’ of divergence19.
We identified over 97% of a conserved set of eukaryotic genes20 in this assembly, with five of the six missing genes also missing from all other filarial genome assemblies, presumably reflecting ancestral gene losses from this group (Supplementary Table 4). O. volvulus shows large-scale variation in gene density, GC content and repeat density (Fig. 1b and Supplementary Table 5), establishing that these patterns are present in nematodes beyond Caenorhabditis spp. The pattern of variation on chromosomes 1 and X is more complex, with two peaks in gene density and GC. We propose that this pattern is due to the origins of these chromosomes as fusions of two ancestral chromosomes. For comparison of gene content, we have also generated sequence data for the related species O. ochengi, a filarial parasite of cattle, and produced a fragmented draft assembly of this species (Supplementary Tables 1 and 2).
Gene content
A total of 12,143 protein-coding genes were predicted in the O. volvulus genome guided by RNAseq data from eight stages of the parasite life cycle (Supplementary Tables 1 and 2). The majority (∼91%) of genes had orthologues in other nematodes, with ∼9% (1,173) being O. volvulus-specific, with little or no homology to genes annotated in other helminths (Supplementary Table 6). Predicted proteins were classified into functional categories21 (Supplementary Fig. 3a), although 44% of O. volvulus proteins have no predicted function. The distinctive biology of O. volvulus is likely to be underpinned by genes with potentially novel functions and with relatively few homologues in other helminth parasites. Of the O. volvulus-specific genes, 92% encode putative proteins of unknown function, of which 7% are potentially secreted (Supplementary Fig. 3b and Supplementary Table 6).
A total of 3,152 protein-coding genes were present on the large contiguous X-chromosome-specific region and an additional 113 on small contigs assigned to X, while only 100 genes (Supplementary Table 7) were inferred to be on Y-chromosome-specific contigs. The X-specific genes are enriched for those encoding putative G-protein coupled receptors, translational release factors, calcium-binding proteins and membrane proteins involved in cell–cell communication.
Gene synteny and gene family expansions
The free-living nematode Caenorhabditis elegans remains the most genetically tractable model nematode, so we used that species to perform comparative analyses with O. volvulus. Mapping orthologues indicated a clear correspondence between C. elegans chromosomes I, II and III with O. volvulus chromosomes 1, 3 and 2, respectively. C. elegans chromosomes IV and V orthologues match mostly the O. volvulus X chromosome, while C. elegans chromosome X orthologues are split between O. volvulus chromosomes 1 and X (Fig. 1c). Gene synteny between O. volvulus chromosomes and the ten largest O. ochengi scaffolds shows that both genomes are predominantly co-linear, but not all genes have predicted one-to-one orthologues (Supplementary Fig. 4).
We further investigated the evolutionary history of O. volvulus genes using Ensembl Compara22. This produced high support for the traditional classification of this group as found in previous molecular work23 and confirmed the close relationship between O. volvulus and O. ochengi (Fig. 2). We observed many gene duplication events of potential biological importance in the evolutionary history of O. volvulus (Supplementary Table 8 and Supplementary Data). For example, the gene family encoding the α subunit of the enzyme collagen prolyl 4-hydroxylase (C-4PHα), an important enzyme for collagen synthesis in the cuticle of nematodes, has been repeatedly duplicated in the lineage leading to O. volvulus, resulting in a paralogous family of 16 genes. Two copies are clearly truncated and may be pseudogenes, while at least 11 of these paralogues are full-length, have the conserved catalytic sites residues24 (Supplementary Fig. 5) and are predicted to be secreted. This drastic expansion probably reflects the requirement for changes in collagen composition during cuticle remodelling throughout O. volvulus development.
Figure 2. Gene family evolution and comparative genomics of O. volvulus and relatives.
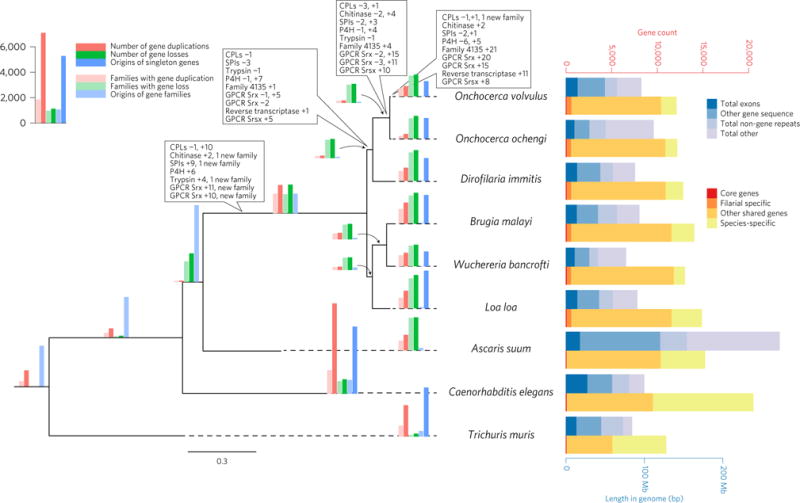
Maximum-likelihood genome phylogeny of six filarial nematode species and three outgroup species. All nodes were fully supported by 100 bootstrap replicates. The phylogeny is annotated with histograms showing the number of duplications (red) and losses (green) for individual genes (dark red or dark green); number of families (light red or light green) with one or more duplications/losses; and numbers of gene families (light blue) inferred to appear on each branch and (on terminal branches) numbers of singleton genes (dark blue) as estimated by the Ensembl Compara pipeline. Note that our data cannot reconstruct gene losses on the most basal branch of the tree. Bar charts in yellow-red summarize the evolutionary history of the genome of each species, defining genes shared among all nine nematode species, the six filarial species and genes with more complex patterns of conservation. The total heights of these bars represent the total number of protein-coding loci annotated on each genome. Boxes on branches show numbers of gene duplications (+X) and losses (−X) in five gene families of specific interest in O. volvulus: trypsin, cathepsin-L like proteases (CPL), chitinase, serine protease inhibitors (SPI) and prolyl-4 hydroxylase alpha-subunits (P4H) and in other families with many gene duplications on the branch leading to O. volvulus. Duplications are observed in two different Srx GPCR families; the reverse transcriptase gene family could be missing from some species because of differences in annotation and repeat finding methods. Family 4135 comprises weakly conserved hypothetical proteins. Stacked bar charts in blue summarize the genome of each species, with total heights representing the size of each genome assembly, divided into exons, other genic sequences (introns and UTRs; non-coding genes), annotated repeats and DNA sequence not annotated in any of these categories.
Similarly, three G-protein-coupled receptor (GPCR) gene families show a remarkable expansion in O. volvulus (Fig. 2 and Supplementary Table 8). In general, filarial genomes lack several GPCR gene families present in C. elegans, including Srj, Sra and Srb receptors9. However, unlike the other filariae, O. volvulus contains members of all other GPCR families represented in C. elegans (Fig. 3). For example, it retains the Str family involved in odorant detection, a family not found in other filariae but present in Ascaris suum, and the root-knot nematode Meloidogyne hapla, perhaps reflecting responses to specific cues in their environments9. The maintenance of Str and other families of GPCRs may suggest their importance throughout the developmental stages of O. volvulus, but could also reflect the high quality of the sequence assembly and annotation. Further underlining the importance of GPCRs to O. volvulus biology, the Srx and Srsx families are among the most duplicated genes in the O. volvulus genome, most striking of which is the expansion of the Srx family, with 35 duplications inferred to be specific to O. volvulus.
Figure 3. GPCRs.
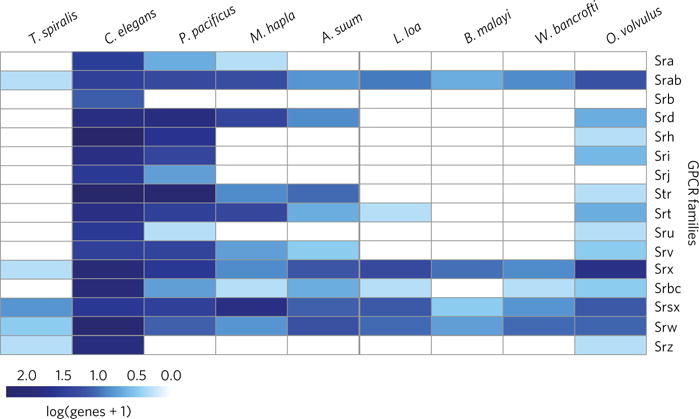
Profile of GPCR families present in nematode genomes in comparison to O. volvulus. The log of the number of genes plus one is plotted per family.
In contrast to GPCRs, the nuclear hormone receptor (NHR) gene family is particularly sparse. NHRs are important transcriptional modulators that regulate cellular differentiation, homeostasis and reproduction in nematodes and other animals25. C. elegans has over 270 NHRs—the highest number identified in any species—but the filarial nematodes have a far smaller repertoire. Brugia malayi has 50 and O. volvulus even fewer, with only 24 classical NHRs and 8 orphan receptors identified (Supplementary Table 9 and Supplementary Fig. 6). Despite this, O. volvulus has orthologues of all five C. elegans NHR genes that exclusively participate in moulting and metamorphosis26,27 (Supplementary Table 10). Interestingly, the ecdysone receptor (OVOC9104) was gained at the base of the filarial lineage. Ecdysone and related hormones regulate moulting and metamorphosis in arthropods, so it is tempting to speculate that this gain in NHRs may be an adaptation to the insect host.
Control of the neuromusculature is a principal target for many anthelmintic drugs, where they bind and activate the pentameric ligand-gated ion channels (pLGICs) that mediate fast-synaptic signalling. Like other filarial nematodes, O. volvulus encodes fewer pLGIC subunit genes (only 48 compared to 120 in C. elegans28 and other clade V nematodes29), but has retained genes encoding subunits of the glutamate- and GABA-gated chloride channels (Supplementary Fig. 7) that are putative targets for ivermectin30. In filarial nematodes, ivermectin paralyses or kills microfilariae gradually, but also suppresses microfilarial production from adult females. O. volvulus encodes an orthologue of the avr-14 ivermectin target that in B. malayi is expressed within reproductive tissue; this may explain the effect of ivermectin on microfilarial production31.
Although the emergence of drug resistance in human parasites has been less rapid than in parasitic nematodes of livestock, cases of unexpected earlier reappearance of O. volvulus microfilariae following ivermectin treatment have been reported31. The mechanisms of ivermectin resistance remain unclear, but polymorphism of P-glycoprotein (Pgp) drug transporters in other nematodes has been linked to resistance, together with evidence that Pgp inhibitors can increase drug efficacy32,33. The Pgps are members of the ATP-binding cassette (ABC) transporter family. The genome of O. volvulus contains relatively few ABC transporter orthologues, with only 10 compared to 54 in C. elegans31.
Metabolic reconstruction and identification of potential drug targets
To examine the metabolic capabilities of O. volvulus and L. loa, a filarial parasite without a Wolbachia endosymbiont, we performed genome-scale metabolic reconstructions (Supplementary Tables 11–13). The resultant networks comprise 767 reactions (378 distinct enzymes) for O. volvulus and 648 reactions (301 enzymes) for L. loa. Each share a core of 628 reactions, with the majority (100 of 139) of additional reactions in the O. volvulus reconstruction contributed by wOv. Next, we performed flux balance analysis (FBA)34,35 to investigate the impact of single reaction knockouts on parasite growth. Metabolites were made freely available across reactions, an assumption that will minimize false-positive essential reaction predictions at the expense of false-negative predictions (for details see Methods).
For O. volvulus, FBA predicted 71 essential reactions (Table 1 and Supplementary Tables 11 and 12). For L. loa, 112 reactions were predicted to be essential (70 common to O. volvulus essential reactions), including 23 transport reactions (Supplementary Table 12). Essential reactions are associated with nucleotide and lipid metabolism, energy production, biosynthesis of cofactors and transport (Table 1). Only threonine transport was essential for O. volvulus but not L. loa, which can produce its own threonine. In contrast, O. volvulus benefits from possible wOv contributions to fatty acid metabolism, haem synthesis and nucleotide metabolism, which, unlike L. loa, does not require the salvage of key metabolites in these pathways from the host. The endosymbiont also allows the conversion of lysine from aspartate (Supplementary Table 11). Finally, a single reaction predicted to be essential to both parasites is uniquely provided by wOv: NAD kinase (Enzyme Commission number: EC 2.7.1.23; Table 1 and Supplementary Table 11).
Table 1.
Overview of the metabolic pathways included in the metabolic reconstructions.
| Number of reactions in pathway
|
Number of reactions predicted essential*
|
||||||
|---|---|---|---|---|---|---|---|
| Superpathway | Metabolic pathway | L. loa | Ov and wOv | wOv only | L. loa | Ov and wOv | wOv only |
| Amino acid metabolism | Alanine, aspartate and glutamate metabolism | 14 | 15 | 3 | 1 | 1 | 0 |
| Arginine and proline metabolism | 26 | 25 | 2 | 3 | 3 | 0 | |
| Biosynthesis of amino acids | 37 | 41 | 9 | 3 | 3 | 0 | |
| Cysteine and methionine metabolism | 17 | 19 | 2 | 4 | 4 | 0 | |
| Glycine, serine and threonine metabolism | 17 | 17 | 3 | 1 | 1 | 0 | |
| Biosynthesis of other secondary metabolites | Streptomycin biosynthesis | 4 | 4 | 0 | 2 | 2 | 0 |
| Carbohydrate metabolism | Amino sugar and nucleotide sugar metabolism | 23 | 23 | 3 | 4 | 4 | 0 |
| Butanoate metabolism | 8 | 8 | 0 | 1 | 0 | 0 | |
| Inositol phosphate metabolism | 12 | 12 | 0 | 3 | 3 | 0 | |
| Pentose phosphate pathway | 17 | 18 | 0 | 2 | 2 | 0 | |
| Propanoate metabolism | 14 | 14 | 1 | 1 | 1 | 0 | |
| Pyruvate metabolism | 20 | 20 | 1 | 1 | 1 | 0 | |
| Energy metabolism | Methane metabolism | 16 | 16 | 0 | 2 | 2 | 0 |
| Lipid metabolism | Arachidonic acid metabolism | 16 | 14 | 0 | 3 | 3 | 0 |
| Biosynthesis of unsaturated fatty acids | 14 | 15 | 2 | 7 | 2 | 0 | |
| Fatty acid biosynthesis | 3 | 15 | 32 | 1 | 1 | 0 | |
| Fatty acid degradation | 32 | 32 | 0 | 19 | 0 | 0 | |
| Fatty acid elongation | 28 | 28 | 0 | 24 | 0 | 0 | |
| Fatty acid metabolism | 48 | 57 | 32 | 31 | 2 | 0 | |
| Glycerolipid metabolism | 9 | 9 | 0 | 2 | 2 | 0 | |
| Glycerophospholipid metabolism | 22 | 22 | 1 | 6 | 6 | 0 | |
| Sphingolipid metabolism | 16 | 16 | 0 | 5 | 5 | 0 | |
| Metabolism of cofactors and vitamins | Nicotinate and nicotinamide metabolism† | 11 | 12 | 1 | 1 | 0 | 1 |
| One carbon pool by folate | 10 | 10 | 6 | 1 | 0 | 0 | |
| Pantothenate and CoA biosynthesis | 11 | 11 | 1 | 3 | 3 | 0 | |
| Metabolism of other amino acids | Beta-alanine metabolism | 9 | 9 | 0 | 1 | 1 | 0 |
| Glutathione metabolism | 19 | 18 | 0 | 2 | 2 | 0 | |
| Metabolism of terpenoids and polyketides | Terpenoid backbone biosynthesis | 12 | 12 | 2 | 8 | 8 | 0 |
| Tetracycline biosynthesis | 1 | 1 | 0 | 1 | 1 | 0 | |
| Nucleotide metabolism | Purine metabolism | 53 | 63 | 9 | 7 | 3 | 0 |
| Pyrimidine metabolism | 47 | 51 | 7 | 7 | 5 | 0 | |
| Transport reactions | Extracellular transport | 43 | 43 | 0 | 23 | 18‡ | 0 |
| Total (non-redundant) number of reactions in pathways | 428 | 455 | 72 | 86 | 49 | 1 | |
| Total number of reactions in reconstructions | 777 | 796 | 100 | 112 | 70 | 1 | |
One reaction was essential either from L. loa (with or without genetic evidence); O. volvulus (Ov) and Wolbachia (wOv) with reactions in the O. volvulus reconstruction (both from gene annotation and added in pathway gap-filling) not being contributed by wOv only; or wOv with reactions in the Ov reconstruction supported by genetic evidence only from wOv.
Only a single reaction, in the nicotinate and nicotinamide pathway, is essential and uniquely provided by wOv. Transport reactions for both nematode and Wolbachia lack genetic evidence therefore making wOv only and O. volvulus and wOv transporters indistinguishable.
Note that of the 18 transport reactions essential to O. volvulus, all are essential to L. loa as well except for threonine transport. Also reported is the total number of reactions belonging to the pathways listed here, as well as the total number of reactions in the reconstructions.
Focusing on purine metabolism (Fig. 4), wOv provides an alternative route for inosine monophosphate (IMP) production in O. volvulus, a crucial precursor in purine metabolism, while L. loa depends exclusively on adenine import. It is not known whether IMP produced by wOv is available to the nematode, but given their ability to take up purines and purine nucleosides36,37, the relative susceptibility of the two species to the inhibition of adenosine monophosphate (AMP) aminohydrolase (EC 3.5.4.6) activity warrants further investigation. Furthermore, the presence of purine-nucleoside phosphorylase (EC 2.4.2.1)—enabling the conversions of guanine and adenine to deoxyguanosine and deoxyadenosine, respectively—ensures O. volvulus is robust to the deletion of ribonucleoside-diphosphate reductase (EC 1.17.4.1). Given the strikingly different pathways involved in the production of purines for the two species, any reliance by O. volvulus on this alternative pathway could be exploited to selectively target O. volvulus7, a credible prospect because inhibitors of purine-nucleoside phosphorylase already exist38.
Figure 4. Contribution of Wolbachia to O. volvulus in purine metabolism.
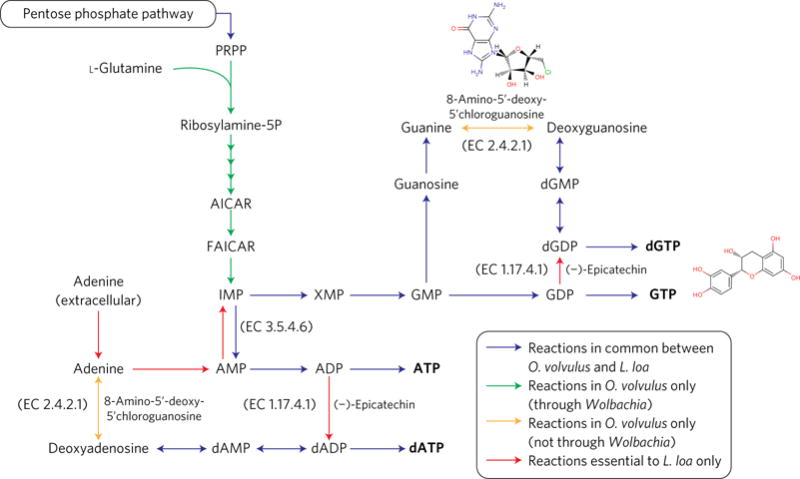
Inhibitors 8-amino-5′-deoxy-5′chloroguanosine and (−)-epicatechin (shown in the figure) have been found against the activities of purine nucleoside phosphorylase (EC 2.4.2.1) and ribonucleoside diphosphate reductase (EC 1.17.4.1)113,114. The chemical structures of the inhibitors shown were obtained from the Braunschweig Enzyme Database (BRENDA)115. Metabolites in bold represent biomass components.
Insights into possible repurposed drugs or new drug targets
We have also investigated potential O. volvulus targets of currently available Federal Drug Agency (FDA)-approved drugs. We excluded wOv loci, as repurposing of antibacterial compounds for anti-Wolbachia chemotherapy has been extensively investigated39. This strategy identified 51 O. volvulus proteins, mostly enzymes and proteins involved in ion transport and neurotransmission, that 85 drugs may target (Supplementary Table 14). We used the WHO Anatomical Therapeutic Classification (ATC) to remove from the set those drugs classed as anti-neoplastics (these are likely to have intolerable side effects for anthelmintic use), resulting in a set of 42 drugs. By screening these targets based on sequence features, we identified 16 O. volvulus proteins likely to be good drug targets (Table 2).
Table 2.
The top 16 O. volvulus targets and their predicted drugs.
| Ov gene ID | Function of family | Drugs (WHO name) | ATC level 3 classification |
|---|---|---|---|
| OVOC2713 | Ribonucleotide reductase, small chain | Fludarabine phosphate, clofarabine, gemcitabine, (hydroxyurea), gallium nitrate | Antimetabolites, other antineoplastic agents. |
| OVOC11146 | Calcineurin-like phosphoesterase | Tiagabine | Antiepileptics |
| OVOC215 | Sodium neurotransmitter symporter family | Imipramine, clomipramine, amitriptyline, nortriptyline, protriptyline, amoxapine, fluoxetine, citalopram, paroxetine, sertraline, fluvoxamine, escitalopram, trazodone, nefazodone, venlafaxine, milnacipran, duloxetine, desvenlafaxine, vilazodone, vortioxetine, (levomilnacipran, chlorphentermine) | Antidepressants (unclassified) |
| OVOC3747 | Ion transport protein | Dronedarone, (nimodipine, felodipine) | Antiarrhythmics, class I and III (selective calcium channel blockers with mainly vascular effects) |
| OVOC1986 | KQT-1 potassium channel | Dronedarone | Antiarrhythmics, class I and III |
| OVOC1111 | Receptor family ligand binding region | Baclofen, oxybate | Muscle relaxants, central acting agents, null |
| OVOC5191 | G-protein coupled GABA receptor activity | ||
| OVOC12632 OVOC12637 OVOC12635 OVOC968 |
NADH-ubiquinone oxidoreductase chains 1, 4 and 5; probable NADH dehydrogenase [ubiquinone] iron-sulfur protein 7 | Metformin | Blood glucose lowering drugs excluding insulin |
| OVOC6309 | 3-oxo-5-α-steroid 4-dehydrogenase | Dutasteride | Drugs used in benign prostatic hypertrophy |
| OVOC10592 | ERG2 and sigma1 receptor like protein | Pentazocine, (dextromethorphan) | Opiods, cough suppressants (other nervous system drugs) |
| OVOC8585 | C-terminal tandem repeated domain in type 4 procollagen | Xiaflex, (ocriplasmin) | Other drugs for disorders of the musculoskeletal system (other ophthalmologicals) |
| OVOC10695 | Glycosyl transferase family 21 | Miglustat, eliglustat | Other alimentary tract and metabolism products |
| OVOC4110 | Amidase | Acetaminophen | Other analgesics and antipyretics |
The table shows anatomical therapeutic classification groups for drugs identified as potential repurposing candidates. For drugs in parentheses, the mechanism is shown in similar style parentheses in the ‘ATC level 3 classification’ column.
In a second approach, we leveraged known mechanisms of action of particular drug classes on specific nematode protein families to prioritize from a wider set of potential targets. Among the O. volvulus protein families with at least one drug target, those comprising zinc finger C4, NHR, dopamine neurotransmitter, GPCR, serotonin receptors, GABA receptor and ion channels could be considered ‘privileged’ based on the extensive list of drugs by which they may be targeted. The genome sequence reveals the full repertoire of these families, providing a rich resource of potential leads for future drug development efforts (Supplementary Fig. 8). Finally, there is the possibility that existing antiparasitic drugs such as levamisole and morantel, which each target distinct classes of acetylcholine-gated cation channels40 (AcHR), should be revisited as antifilarial agents. Levamisole targets unc-38 and unc-63 (duplicated in O. volvulus), while morantel targets acr-26 and acr-27 (OVOC7603; a distinct class of AcHR found only in parasitic nematodes). Importantly, these targets are all conserved between O. volvulus and B. malayi41,42. The availability of the complete genome sequence will enhance the opportunity to investigate the mechanisms of action of existing classes of anthelminthics.
wOv and lateral gene transfer
In addition to the filarial genome, we assembled the complete 956 kb genome of wOv (Supplementary Table 15). The O. volvulus genome was examined for evidence of lateral gene transfers (LGTs) from wOv to its host. These are termed nuclear Wolbachia transfers or nuwts. In total, 531 putative nuwts were identified. Interestingly, one LGT event (OVOC_OM2 22,596,036–22,595,692) was found to have a best match to Midichloria mitochondrii, the endosymbiont of tick mitochondria (Supplementary Table 16). Seven nuwts were larger than 1 kb (maximum of 8.1 kb), four of which were confirmed by amplification, cloning and sequence verification of the Wolbachia–nematode junction (Supplementary Table 17). In a gene-based analysis aimed at detecting more divergent nuwts, a majority (98%) of the 576 regions that contain nuwt open reading frames were fragmented, but four were full-length, potentially functional genes, with relatively few overall mutations, suggesting they may be full-length merely because they are recent transfers (Supplementary Table 16). Overall, nuwts appear to be largely nonfunctional in this genome.
Insights into host–parasite interactions
O. volvulus, like the other filarial parasites, interacts with its definitive human host and its intermediate arthropod host (Simulium spp.) during its life cycle. These parasites must have their own innate immune system to protect them from microbial pathogens, but they also are thought to have evolved mechanisms to subvert both human and insect host defence mechanisms. Although molecules such as immunoglobulins or Toll-like receptors (TLRs) are absent in all filarial nematodes, O. volvulus and other filariae sequenced to date have homologues of some downstream proteins from the TLR signalling pathway (Supplementary Table 18). The innate immune system encoded by the O. volvulus genome also includes C-type lectins, galectins, jacalins and scavenger receptors. Similar to the other filariae, O. volvulus does not appear to produce the antibacterial peptides seen in C. elegans (and in other non-filarial nematodes), although it is possible that different peptides are produced by filariae.
Analysis of the putative proteome of O. volvulus identified a number of human cytokine and chemokine mimics and/or antagonists (Supplementary Table 18). In addition, the O. volvulus genome encodes 12 serine protease inhibitors (SPIs) including serpins and small SPIs, and five cysteine protease inhibitors including cystatins. Both protein families have been shown to interfere with antigen processing and presentation43 and are potentially involved in immune regulation and in parasite interference with the host immune response44. The O. volvulus genome also encodes proteins with sequences similar to those of human autoantigens, some of which have been implicated in inducing cross-reactive antibodies that have been connected with the pathogenesis of posterior eye disease45 and the nodding syndrome46.
Discussion
The O. volvulus genome assembly represents the highest-quality genomic data available for any non-model nematode species. It will be a critical resource for research on other filarial nematodes that reside in different niches within the host, for which only draft genomes are available. Comparative analysis with other nematode genomes has confirmed the recent evolution of an XY karyotype suggesting ancient chromosomal fusions led to the formation of the Onchocerca chromosomes. It should be possible to confirm this hypothesis when chromosome-scale assemblies for filarial nematodes without these fusions become available.
The genome data constitute an invaluable and comprehensive resource for the development of new and urgently needed interventions against onchocerciasis and other filariases. In particular, we describe the orthologues and paralogues of known or suspected targets of existing anthelmintic compounds and identify targets of other licensed compounds that could show activity against O. volvulus. Our analysis of the targets of these drugs highlights several protein families that could guide further drug discovery in Onchocerca. Finally, we performed metabolic reconstructions of O. volvulus in conjunction with its Wolbachia. By investigating in silico the impact of single reaction knockouts on parasite growth, we identified enzymes likely to be essential to O. volvulus viability. While an ideal method would model the multicellular nature of O. volvulus and treat Wolbachia and Onchocerca metabolisms as two separate compartments with transport processes between them, we believe our approach sets a basis for future methodologies to understanding filarial metabolism and approaches the current state of the art in C. elegans metabolic models47. By comparing O. volvulus and L. loa, which does not harbour Wolbachia, we also identified a unique O. volvulus target and those that are compensated by symbiosis. Developing additional therapeutic strategies seems likely to be vital in achieving timely elimination of onchocerciasis8, and these targets are now important candidates for experimental testing and validation.
Methods
Parasite material for genome sequencing
All O. volvulus parasite material used for genome sequencing was collected in the research facility at the Tropical Medicine Research Station, Kumba, Cameroon. Written informed consent was obtained. In cases of illiteracy, a literate witness signed and a thumbprint was made by the participant. Institutional Review Board (IRB) approvals were obtained from both the New York Blood Center and from the Tropical Medicine Research Station, Kumba (Protocols 321 and 01, respectively). The individuals who consented to participate in the study were born or had resided for more than ten years in villages around Kumba. They were confirmed to have microfilariae in their skin snips and clinical symptoms of disease, such as dermatitis, nodules and ocular lesions. None of the subjects had received ivermectin treatment before the study. The adult worm samples were obtained as part of a nodulectomy campaign conducted in villages surrounding Kumba in 1996–1998 and in 2006. Nodules were excised under sterile conditions and were treated with collagenase overnight, following the protocol of Schulz-Key and colleagues48. Briefly, cleaned individual nodules were immersed in 0.5% collagenase (Sigma grade IV) in RPMI 1640 containing 10% FCS + 200 units of penicillin and 200 μg ml−1 streptomycin. The flat tubes containing the nodule were then placed in a rocking water bath and incubated at 35 °C until the tissue was completely digested. Once digested, the liberated worms were unravelled from residual tissue with mounted needles under a dissecting scope and then washed in several changes of RPMI. Individual female worms were snap-frozen in Eppendorf tubes with liquid nitrogen. They were then stored and shipped in liquid nitrogen and, upon arrival in New York, stored at −80 °C until shipment on dry ice to the Wellcome Trust Sanger Institute.
Freshly dissected O. volvulus L3s were also cryopreserved according to the method described by Cupp and colleagues49 and were shipped to the New York Blood Center in liquid nitrogen and, upon arrival in New York, were stored in liquid nitrogen. To collect sexed juvenile adult worms (40 days in culture), thawed and washed L3s were cultured for 14 days in the presence of peripheral blood mononuclear cells (PBMCs) (1.5 × 105 per well of a 96-well plate) and then over a monolayer of human dermal fibroblasts (105 per well of a 24-well plate) (S. Lustigman, unpublished data). On day 40, they were separated into juvenile female and male worms based on their size (males being much smaller than females) and the morphology of the posterior end50. They were then frozen individually, stored at −80 °C until shipment on dry ice to the Wellcome Trust Sanger Institute.
Cows that had grazed in northern Cameroon, where O. ochengi is highly endemic, were brought to abattoirs located in Douala, Cameroon. Subcutaneous nodules containing adult O. ochengi worms were identified on the umbilical skin of slaughtered infected cows. Adult worm masses containing one viable adult female and zero to several adult males were then carefully recovered from the purchased skins by dissection of the nodule with a sterile razor blade and then snap-frozen in liquid nitrogen. The material was transported to the USA in liquid nitrogen and, upon arrival in New York, stored at −80 °C.
Parasite material for transcriptomics
All parasite material was prepared in the Tropical Medicine Research Station, Kumba, Cameroon, between 1993 and 1999, except for female worm samples, which were from both Cameroon and Ecuador, where samples were collected as part of a previous study51. L3 were obtained from Simulium damnosum flies 7 days after infection with skin microfilariae, as described previously52,53. To obtain moulting larvae, freshly dissected L3s were cultured in vitro in groups of ten larvae in 96-well plates containing a 1:1 mixture of Iscove’s modified Dulbecco’s medium and NCTC-135, 20% fetal calf serum and antibiotic-antimycotic solution (Life Technologies) for 3 days at 37 °C. Larvae were collected immediately after dissection (L3) or after 1, 2 or 3 days in culture, washed with Tris-EDTA buffer and then snap-frozen in liquid nitrogen. Ultrastructural examination by electron microscopy confirmed that these cultured larvae had started the moulting process, as the separation between the cuticle of L3 and the newly synthesized cuticle of L4 was evident in some of the cross-sections52. Nodular and skin microfilariae were purified as described previously54,55 and the adult worms were excised from collagenase-treated nodules as previously described49. Parasites were stored in trizol until RNA isolation.
Optical mapping
Snap-frozen L3s were thawed and used to make agarose plugs using the CHEF Genomic DNA Plug Kit (Bio-Rad). Approximately 3 × 103 L3s were spun at 1,000g for 5 min, resuspended in 32 μl cell suspension buffer and incubated in a 50 °C water bath; 53 μl of 2% clean-cut agarose, melted at 50 °C was added to the L3 and mixed gently before being transferred to a plug mould and stored at 4 °C. The plug was incubated in 100 μl proteinase K and 2.5 μl proteinase K reaction buffer at 50 °C for 1 day, before being washed five times, 1 h per wash, with gentle agitation, in 1× Wash buffer. The plug was stored at 4 °C until use. For optical mapping, DNA molecules were stretched and immobilized along microfluidic channels before digestion with the restriction endonuclease SpeI, yielding a set of restriction fragments ordered by their location in the genome. The fragments were fluorescently stained and visualized to determine the fragment sizes. Assembling overlapping fragment patterns of single-molecule restriction maps produced an optical map of the genome consisting of six large optical contigs with a total size of 92.54 Mb.
Genome sequencing for assembly
Whole-genome sequence libraries (Supplementary Table 19) were generated from genomic DNA extracted from a single adult female O. volvulus worm and a single adult female O. ochengi worm, both washed thoroughly in 0.1 M EDTA. DNA was extracted using the Qiagen Genomic-tip kit. The ratio of host to parasite DNA was examined by qPCR using single-copy genes as markers. PCR-free 400–550 bp paired-end Illumina libraries were produced (one library per species) using a protocol based on a previously described method56 but using Agencourt AMPure XP beads for sample clean-up and size selection. Genomic DNA was precipitated onto beads after each enzymatic stage with an equal volume of 20% polyethylene glycol 6000 and 2.5 M sodium chloride solution. Beads were not separated from the sample throughout the process until after the adapter ligation stage. Fresh beads were then used for size selection. O. volvulus genomic DNA was used to generate a 3 kb mate pair library using a modified SOLiD 5500 protocol adapted for Illumina sequencing57.
Genome resequencing of sexed juvenile female and male worms
DNA was extracted from sexed juvenile male and female worms using the Promega Wizard kit, following the manufacturer’s instructions. Then, 400 bp fragment Illumina libraries were produced (Supplementary Table 19) following the protocol used for genome sequencing (see above), but using 12 cycles of amplification.
Genome assemblies
The assembly of the genome was produced using a combination of sequencing, a de novo optical restriction map (Supplementary Fig. 1a) and extensive manual assembly improvement.
The initial assembly for O. volvulus was produced from a combination of short-fragment paired-end and mate-pair Illumina libraries. Short paired-end sequence reads were first corrected and initially assembled using SGA v0.9.7 (ref. 58). This draft assembly was then used to calculate the distribution of k-mers for all odd values of k between 41 and 81, using GenomeTools v.1.3.7 (ref. 59). The k-mer length for which the maximum number of unique k-mers were present in the SGA assembly was then used as the k-mer setting for de Bruijn graph construction in a second assembly with Velvet v1.2.03 (ref. 60). The mate-pair library was then used to further scaffold this Velvet assembly using SSPACE (ref. 61), followed by gap closing using Gapfiller (ref. 62) and IMAGE (ref. 63). A ‘bin’ assembly using the unaligned reads was incorporated into the main assembly and this assembly was scaffolded using SSPACE. The O. ochengi genome assembly was produced using the above method except that only a short-fragment paired-end (PCR-free) library was used and therefore the SSPACE scaffolding step was excluded. The O. ochengi assembly was found to be contaminated with sequences derived from the host (cow) genome. To identify contaminating contigs, PROmer (ref. 64) was used to compare the Bos taurus UMD 3.1 (ref. 65) assembly against the initial O. ochengi assembly and contigs with hits of greater than 97% identity covering more than 90% of the contig length were removed. This process removed ∼20 Mb of the O. ochengi assembly and this cleaned version was used for all comparisons reported here.
For O. volvulus, the output of the automated assembly process above produced an ‘interim’ assembly, which was improved by an extensive manual finishing effort, using GAP5 (ref. 66); scaffolds were extended, linked and, where possible, errors detected by REAPR (ref. 67) were fixed. The de novo optical map guided this process. The improved sequence scaffolds (totalling 90.9 Mb) were aligned against the optical contigs using MapSolver. Any miss-joins were resolved and new potential joins were investigated and executed where possible. Further automated gap closure was undertaken during this finishing process using IMAGE and Gapfiller, and the accuracy of the consensus sequence was improved using ICORN2 (ref. 68) with both short-fragment and mate-pair Illumina reads. Finally, REAPR was run to detect and break misassembled regions based on the mate-pair library data. The v.3 genome assembly resulting from this work was used for gene finding (Supplementary Table 2).
Although it was not possible to extend sequence data beyond the boundaries of the optical contigs, two additional joins could be made between four of the six superscaffolds, with evidence of these joins coming from read pairs joining the two ends and in one case sequence similarity between the two ends, and synteny with C. elegans and a B. malayi optical map (unpublished) supporting that, in each case, the two scaffolds being joined belong to the same chromosome. The v.4 genome assembly includes just these two additional joins, with minimum sizes for the introduced gaps (approximately 100 and 300 kb) estimated as the lengths of unjoined ‘overhangs’ in the optical mapping molecules. The final assembly thus has all four chromosome pairs represented by large contiguous scaffolds (Supplementary Table 2).
To assess the completeness of the assemblies, we ran CEGMA v2 (ref. 20), which reports the percentage of 248 highly conserved eukaryotic gene families that are present as full or partial genes in the assembly. For most eukaryotes, we would expect to see nearly 100% of CEGMA families represented by a full gene in the genome. Thus, CEGMA provides a measure of the completeness of the assembly for a species.
The Wolbachia endosymbiont genome sequence was assembled by first identifying scaffolds from the initial Velvet assembly that showed sequence similarity to published sequences of five published lambda phage clones containing Wolbachia sequence from Fenn and colleagues69 (a total of 70.8 kb). Nucmer70 identified a set of eight contigs totalling 113 kb that all had >99% similarity to the published clones over at least 500 bp and that all had very similar coverage (43–46×) in the Illumina data. These contigs also had more distant similarity to the published Brugia malayi Wolbachia sequence71. Applying a cutoff of depth between 42 and 47× coverage and PROMER similarity of at least 75% over 250 bp with the B. malayi Wolbachia sequence identified 44 contigs covering 926 kb that were putatively assigned to the O. volvulus Wolbachia sequence. Manual finishing using GAP5 then allowed us to order, extend and link these contigs to complete the Wolbachia genome assembly.
Transcriptome sequencing and analyses
High-throughput transcriptome data were generated from the RNA of O. volvulus stage-specific parasites: nodular microfilariae, skin microfilariae, L2, L3, L3D1, L3D3, adult male and adult female worms. For all larval stages and adult worms, RNA was prepared using TRIzol and lysing matrix D (1.4 mm ceramic spheres) and a Fastprep24 (MP Biomedicals). RNAseq libraries were prepared following the RNAseq protocols of the Illumina mRNAseq Sample Prep kit and the Illumina TruSeq kit (Illumina). Transcriptome libraries were sequenced on Illumina HiSeq 2000 or MiSeq machines (Supplementary Table 19).
Gene prediction
Gene predictions were conducted by various methods available in MAKER version 2.2.28 (ref. 72). The MAKER annotation pipeline consists of four general steps to generate high-quality annotations by taking into account evidence from multiple sources. First, assembled contigs are filtered against RepeatRunner73, RepBase74 and a species specific repeat library generated by Repeat Modeler (http://www.repeatmasker.org/RepeatModeler.html) using RepeatMasker (http://www.repeatmasker.org/) to identify and mask repetitive elements in the genome. Second, gene predictors Augustus 2.5.5 (ref. 75), GeneMark-ES 2.3a (self-trained)76 and SNAP 2013-02-16 (ref. 77) are employed to generate ab initio models that can use evidence within MAKER. Further species-specific gene models were provided to MAKER using comparative algorithms against the genome: genBlastG (ref. 78) output of C. elegans gene models (WormBase79) and RATT (Rapid Annotation Transfer Tool80) output of gene models from B. malayi, the taxonomically nearest reference genome. Third, a set of expressed sequence tags (ESTs), cDNAs and proteins from related organisms were aligned against the genome using BLASTN and BLASTX (ref. 81), respectively, and these alignments were further refined with respect to splice sites using Exonerate82. Finally, the EST and protein homology alignments, comparative gene models and the ab initio gene predictions were integrated and filtered by MAKER to produce a set of evidence informed gene annotations. The MAKER genome annotation pipeline was run three consecutive times. In the absence of a species-specific trained gene predictor, Augustus and SNAP were trained using CEGMA protein evidence gained from the default KOGs. The RNAseq data were mapped to the genome assembly using TopHat2 (ref. 83). The TopHat output includes the inferred positions of intron splice sites in the genomes and this information was fed into MAKER. The first run of MAKER was performed using the est2genome and protein2genome option with taxonomy-specific ESTs and cDNAs available from INSDC (ref. 84), including data from a large-scale EST project for O. volvulus85,86 and nematode protein sequences, respectively. Gene models obtained from the first run were used to train SNAP and models from the second run were used to train Augustus. With the trained models, MAKER was run a third time using a taxonomically broader protein set that included metazoan proteins from the UniProt Complete protein database87 and a subset of nematode proteomes from GeneDB (ref. 88). Finally, the MAKER gene set was filtered to remove less reliable gene models, as follows. First, MAKER gene models that were predicted based on Exonerate or BLASTX alignments and that did not overlap with any Augustus, genBlastG or RATT gene models were discarded, as they were probably due to spurious alignments. Second, MAKER gene models that encoded proteins of fewer than 30 amino acids were discarded. Third, if two different MAKER gene models overlapped in their coding sequence, the gene model with the worse score was discarded.
Because the O. ochengi genome assembly is much more fragmented than the O. volvulus assembly and there is no RNAseq evidence to guide gene finding, exons are frequently missed or complex gene models broken up in the annotation of this species.
Identification of Wolbachia insertions in O. volvulus genome
The O. volvulus Wolbachia 956 kb genome contains 785 predicted protein-coding genes. Comparison of wOv with the published Wolbachia of O. ochengi (wOo)89 revealed the two genomes to be nearly identical (99.51%) but wOv to be 1,937 bp larger, predominantly due to a small number of large indels. Approximately 4,400 single nucleotide polymorphisms (SNPs) were also identified, of which 57 variants may result in functional differences, including some that could be involved in the interaction of the symbionts with their hosts. The O. volvulus assembly was searched against the wOv genome with NUCMER v3.06 using MAXMATCH, revealing a total of 531 matches. Of those, 486 were >100 bp and 7 were >1 kbp. Subsequently, the predicted wOv proteins were used to search for regions with protein homology in the O. volvulus genome using TBLASTN as implemented in NCBI BLAST 2.2.21, with results reported in tabular format and an e-value threshold of 1e-15. The regions with matches in the O. volvulus assembly were extracted and searched against NT with BLASTN and NR with BLASTX as implemented in NCBI BLAST 2.2.21. Matches not meeting an e-value threshold of 1e-15 were discarded, as well as those without a best match to a bacterial gene/protein (which would include mitochondrial and nuclear mitochondrial sequences), yielding a list of putative nuwts in the O. volvulus genome (Supplementary Table 16). These regions were also searched with PRAZE (http://ber.sourceforge.net) against the predicted wOv proteins to identify frameshifts, nonsense mutations and truncations.
Comparative analysis of the predicted O. volvulus gene set
To help us examine the evolution of gene families and to identify the families of genes lost or expanded in O. volvulus, we ran the Ensembl Compara pipeline22. Ensembl Compara is a pipeline that clusters genes into families based on all-against-all blast scores, produces multiple alignments and phylogenies for each gene family, and predicts paralogy and orthology relationships by reconciling reconstructed gene trees with the phylogeny for the taxa included. Along with O. volvulus and O. ochengi, we included published filarial nematode genomes (Dirofilaria immitis, Wuchereria bancrofti, B. malayi and Loa loa), Ascaris suum as the only other clade III nematode species for which genome data has been published, C. elegans as the model free-living nematode species and the clade I parasitic nematode Trichuris muris as an outgroup to all of these. The phylogenetic tree of these species used as input to Compara was constructed from existing evidence on relationships between filarial species and was identical in topology to that shown in Fig. 2. Using the Compara Perl API to query our custom Ensembl Compara database, we identified gene families with multiple expansions or losses in O. volvulus relative to the other comparator species. We removed all the ‘dubious’ duplications (those with a duplication confidence score ≤0) and defined a gene family as expanded if there were at least two gene duplication events reported in O. volvulus. To help us in identifying gene families that are expanded in O. volvulus, we looked at both the gene count and the total protein length per species in these families, as the latter statistic is less sensitive to fragmented gene models.
A phylogeny of the nine nematode species was generated from 3,148 single-copy Compara gene families that have genes from at least seven species. Sequences for each gene family were aligned using Mafft v7.205 (ref. 90) in automatic (−auto) mode; these alignments were then trimmed with GBlocks v0.91b (ref. 91) with default parameters, and trimmed alignments were then concatenated to produce a single global data matrix. The phylogeny was then inferred using RAxML v8.0.24 (ref. 92), with each gene family alignment treated as a separate partition using the best-fitting model for that alignment (minimum corrected Akaike information criterion (AIC)) from the empirical amino acid substitution models available in that version of RAxML. The preferred phylogeny was estimated with 10 random addition-sequence replicates and support for splits on the tree estimated using 100 bootstrap replicates.
Functional annotation of genes and gene families
Gene ontology (GO) terms were assigned to genes by transferring GO terms from C. elegans orthologues based on the Ensembl Compara approach for transferring GO terms to orthologues in vertebrate species22, but modified for improved accuracy in transferring GO terms across phyla. Manually curated GO annotations were downloaded from the GO Consortium website93 and, for a particular predicted protein in the present study, the manually curated GO terms were obtained for its C. elegans orthologues and then transferred to our predicted protein. GO terms of the three possible types (molecular function, cellular component and biological process) were assigned to predicted proteins in this way. Additional GO terms were identified using InterproScan94. We developed two pipelines to annotate the Compara gene families: one for the assignment of GO terms to genes and another to assign product description to the gene families.
Chemogenomics screening
A computational target-based approach was used to screen FDA95-approved drugs across all World Health Organization Anatomical Therapeutic Classes (WHO ATC)96 to identify drugs with potential for use as anthelmintics. For protein target identification, we built a Compara database containing all the proteins in release WBPS1 of WormBase ParaSite together with proteins from the 2010-07 Ensembl gene build for the human reference genome assembly GRCh37. An initial broad stroke approach identified as a potential drug target any O. volvulus protein belonging to the same gene family in this database as a human protein annotated as a drug target in ChEMBL. A more stringent approach identified O. volvulus proteins as targets only if they were one-to-one orthologues of a human target protein in the same Compara database. Further filtering of the identified proteins was based on molecular weight, number of transmembrane domains, number of disulfide bonds and life-cycle stage. We identified approved drugs with annotated targets in ChEMBL for targets with O. volvulus homologues identified by both the stringent one–one orthologue ‘individual’ protein approach and the more comprehensive approach using Compara families. Both drug sets were filtered on mode of administration (that is, oral, topical and parenteral). Drugs administered solely via an intravenous route were removed from the set. Further filtering was carried out to restrict the drug set to those with a single protein target, thus excluding drugs targeting protein complexes. We created a drug–protein family interaction network to represent family-level drug identification using the network visualization tool Cytoscape97. The edges (lines) connect nodes (circles) representing drugs and their protein family targets, respectively (Supplementary Fig. 8). This allowed protein families to be identified as having at least one drug target and those with particularly high numbers to be highlighted as potentially the most ‘druggable’ targets.
Ion channels and other anthelmintic drug targets
Nucleotide sequences for selected ion-channel genes were aligned as codons using MAFFT (ref. 98) and regions with uncertainty caused by high levels of sequence divergence and differing sequence length were removed. Maximum-likelihood phylogenies were inferred using PhyML (v20120412)99 with branch node significance determined from the Shimodaira–Hasegawa (SH) statistics and from 100 bootstrap replicates.
Metabolic reconstruction and FBA
For each organism, an initial set of EC predictions was obtained from several methods: (1) DETECT v2.0 (cutoff ILS ≥0.9, ≥5 positive hits100), (2) BLASTP81 (e-value 1e-10 against SWISSPROT101 enzymes), (3) PRIAM102 enzyme rel. Feb-2014 (minimum probability >0.5, profile coverage >70%, check catalytic - TRUE), (4) KAAS103, (5) EFICAz104 and (6) EC assignments from BRENDA105. From these assignments, a set of high-confidence predictions was derived as follows from BRENDA, DETECT and reactions identified by both PRIAM and KAAS. Reaction assignments to metabolic pathways and pathway hole filling were performed using Pathway Tools v18.0 (ref. 106). Those predictions with support from either EFICAz, PRIAM or BLASTP were used to augment the high-confidence set of predictions. In addition, novel Pathway Tool predictions to genes that were previously unannotated were also incorporated.
From our draft networks, we completed our metabolic reconstruction, starting with our high-confidence sets of enzymes for both O. volvulus and L. loa. In the case of O. volvulus, we also added 100 enzymes (ECs) unique to its Wolbachia endosymbiont. We used KEGG as a guide to find the reactions catalysed by individual enzymes. Reactions that use macromolecules to derive certain metabolites, specifically those reactions that contain the same compound (such as DNA) on both sides of the equation, were considered uninformative and filtered. We further modified some reactions from their original KEGG formulation as they contained a glycan identified in KEGG as equivalent to a compound also present in the reconstruction. Reaction directionalities were defined with reference to KEGG, with reactions marked as either reversible (lower bound −1,000 and upper bound 1,000 mmol (gDW h)−1) or irreversible (with lower bound 0 and upper bound 1,000 mmol (gDW h)−1 or lower bound −1,000 and upper bound 0 mmol (gDW h)−1 depending on directionality). We added a non-growth-associated maintenance (NGAM) equation, accounting for the organism’s needs outside of growth, to be at least 5 mmol (gDW h)−1. We allowed for glucose uptake at a maximum of 10 mmol (gDW h)−1 and initially allowed for diffusion of water, oxygen, carbon dioxide, ammonia, diphosphate, phosphate and ethanol. We also allowed, by default, the transport of all amino acids into the system. Model biomass was defined with reference to a previous metabolic reconstruction for the parasites Toxoplasma gondii107 and Leishmania major108 (Supplementary Table 13).
Pathway gap-filling for both O. volvulus and L. loa was first performed to ensure production of biomass components and subsequently to complete metabolic pathways missing a limited number of reactions. In this process, we identified from the literature additional metabolites as possibly important for growth and added these as part of the biomass (biomass components 46–49, Supplementary Table 13)109–111. We also added as part of the biomass such important cofactors as NADH, NADP+ and FAD (biomass components 50–52). In all, we added 68 KEGG reactions for O. volvulus and 50 for L. loa. In addition, two reactions previously added for gap-filling purposes for O. volvulus were found to have some evidence through reciprocal BLAST against L. loa (R04230 and R04231 corresponding to EC 6.3.2.5). To enable arachidonic acid metabolism from linoleic acid, two reactions (common to both reconstructions and with gene evidence: R03814_1 (1.14.19.3) and R00390_1 (6.2.1.3)) were modified from their KEGG formulations, while one reaction (common to both reconstructions: N00001) was custom-made. Further modifications involve changing the directionalities of R01665 (2.7.4.14) and R01664 (3.1.3.5) to ensure CMP was not a dead end; R00086 (ATP phosphohydrolase; linked to many ECs) was modified to allow proton export. For both models, we ensured that the same utility reactions were added so that the models were comparable. Thus, in all, each reconstruction consists of 63 utility reactions, namely 43 transport reactions, 8 sinks, 4 demand reactions and 8 diffusion reactions (a diffusion reaction was added when allowing for the movement of bicarbonate). Moreover, both reconstructions contain six irreversible reactions to ensure that specific metabolites (hexanoyl-CoA, octanoyl-CoA, decanoyl-CoA, lauroyl-CoA, tetradecanoyl-CoA and palmitoyl-CoA, with KEGG IDs C05270, C01944, C05274, C01832, C02593 and C00154, respectively) are considered as contenders for the more general compound acyl-CoA (KEGG ID: C00040) involved in such pathways as glycerophospholipid metabolism. Note that we predict L. loa to be a threonine autotroph through pathway gap filling. In particular, the presence of the enzyme EC 1.1.1.103 (present in L. loa only) specializing in glycine, serine and threonine metabolism suggests the possible production of threonine from glycine. Consequently, the reaction catalysed by EC 2.3.1.29 (R00371) was added to complete the circuit.
Shared between O. volvulus and L. loa are 628 reactions and 250 distinct EC numbers. Most of the additional reactions present in the O. volvulus reconstruction (139 reactions) are contributed by wOv (100 reactions), none of which are encoded in the L. loa genome.
FBA34,35 was used to simulate growth of the worms. A naive objective function was set as maximizing yield (biomass production) for both nematodes. The COBRA Toolbox (version 1.3.4) was used in conjunction with MATLAB to perform simulations112. Single-reaction knockout experiments were performed by blocking the flow through individual reactions (that is, setting the upper and lower bounds as zero) and then measuring the yield through the biomass function. Growth ratios were found by dividing the yield following the block by the yield in an unrestrained model (that is, with the reaction unblocked). Reactions were defined as essential if their growth ratio was predicted to be less than 0.1 upon removal of that reaction. Further details of the metabolic reconstruction are provided in Supplementary Table 13.
In these analyses no attempt was made to partition reactions due to the challenge of accurately assigning reactions to distinct intracellular compartments (for example, mitochondria, golgi and wOv), as well as determining the specific small-molecule transporter activities that allow metabolites to move between compartments. Instead, metabolites were assumed to be freely available across reactions, an assumption, as previously mentioned, that is expected to minimize false-positive essential reaction predictions at the expense of increasing the likelihood false-negative predictions.
Supplementary Material
Acknowledgments
The O. volvulus and O. ochengi genome sequencing was funded by the Wellcome Trust through the core funding of the Wellcome Trust Sanger Institute, WTSI (grant no. 098051). The unpublished RNAseq data used in this study were produced by the parasite genomics group at the Wellcome Trust Sanger Institute in collaboration with the laboratory of T.B. Nutman and S. Lustigman and are available from the sequence data archives under BioProject PBJEB2965. The authors thank C. Griffiths, N. Park, L. Shirley and DNA Pipelines at WTSI for generating the libraries, J. Keane, M. Aslett and A. Page of WTSI pathogen informatics for informatics support and for providing the pipeline to annotate the Wolbachia genome, N. Tricoche of the New York Blood Center (NYBC) for the generation of O. volvulus D40 juvenile female and male worms, and J. Liu and A. Contreras from the NYBC for preparing all the DNA and RNA samples used in these studies. J.C.D.H. was supported by NIAID (U19 AI110820) as well as an NIH Director’s New Innovator Award (DP2-OD007372). J.P., N.N. and L.S.S. were supported by the Canadian Institutes of Health Research (MOP# 84556) and the Natural Sciences and Engineering Research Council of Canada (RGPIN-2014-06664). Additional computing resources were provided through Compute Canada by the University of Toronto SciNet HPC Consortium. S.L. was supported in part by intramural funding from the NYBC, The Edna McConnell Clark Foundation and NIH/NIAID (R01 AI42328). The authors thank the people of the villages around Kumba, Marumba I, Marumba II, Boa Bakundu, Bombanda and Bombele, Cameroon, who were part of the field studies that enabled the collection of O. volvulus parasite material. The authors also thank P. Enyong and other personnel at the Tropical Medicine Research Station, Kumba, for their assistance in collecting the O. volvulus and O. ochengi parasite material.
Footnotes
Data availability. Sequence data have been deposited in the European Nucleotide Archive (ENA). Assemblies and annotation are available at WormBase and WormBase-ParaSite (http://parasite.wormbase.org/Onchocerca_volvulus_prjeb513/Info/Index/ and http://parasite.wormbase.org/Onchocerca_ochengi_prjeb1204/Info/Index/). All have been submitted to GenBank under BioProjects PRJEB513 (O. volvulus) and PRJEB1204 (O. ochengi). RNAseq data are available under BioProject PRJEB2965 (O. volvulus). All accession numbers are available in Supplementary Table 19. The wOv version described here differs slightly from the GenBank version and is available from ftp://ftp.sanger.ac.uk/pub/project/pathogens/Onchocerca/volvulus/).
The Supplementary Tables provide information that supports the data presented. Supplementary Data are also provided for all the Ensembl Compara analyses.
Author contributions
All authors read and approved the manuscript. N.H., T.K., J.T., A.T., O.L., E.S., M.B.R., M.D., I.J.T. and S.B. carried out sample preparation, sequencing, genome assembly and/or gene finding and annotation. S.R.D., J.C.D.H and D.V. analysed Wolbachia sequence data. J.P., N.N. and L.S.S. performed metabolic reconstructions and constraints-based analyses. P.M. performed the chemogenomics screening. R.B., B.H., A.G., A.M., T.R.U., S.B. and J.M.C.R. performed data analyses. J.A.C., T.B.N., E.G., M.B. and S.L. analysed data and wrote the manuscript.
Additional information
Supplementary information is available for this paper.
Competing interests
The authors declare no competing financial interests.
References
- 1.Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386:743–800. doi: 10.1016/S0140-6736(15)60692-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hotez PJ, et al. The Global Burden of Disease Study 2010: interpretation and implications for the neglected tropical diseases. PLoS Negl Trop Dis. 2014;8:e2865. doi: 10.1371/journal.pntd.0002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brady MA, Hooper PJ, Ottesen EA. Projected benefits from integrating NTD programs in sub-Saharan Africa. Trends Parasitol. 2006;22:285–291. doi: 10.1016/j.pt.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention (CDC) Progress toward elimination of onchocerciasis in the Americas—1993–2012. Morb Mortal Wkly Rep. 2013;62:405–408. [PMC free article] [PubMed] [Google Scholar]
- 5.Turner HC, et al. Uncertainty surrounding projections of the long-term impact of ivermectin treatment on human onchocerciasis. PLoS Negl Trop Dis. 2013;7:e2169. doi: 10.1371/journal.pntd.0002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner HC, et al. Reaching the London declaration on neglected tropical diseases goals for onchocerciasis: an economic evaluation of increasing the frequency of ivermectin treatment in Africa. Clin Infect Dis. 2014;59:923–932. doi: 10.1093/cid/ciu467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelly-Hope LA, Cano J, Stanton MC, Bockarie MJ, Molyneux DH. Innovative tools for assessing risks for severe adverse events in areas of overlapping Loa loa and other filarial distributions: the application of microstratification mapping. Parasit Vectors. 2014;7:307. doi: 10.1186/1756-3305-7-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stolk WA, Walker M, Coffeng LE, Basáñez MG, de Vlas SJ. Required duration of mass ivermectin treatment for onchocerciasis elimination in Africa: a comparative modelling analysis. Parasit Vectors. 2015;8:552. doi: 10.1186/s13071-015-1159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desjardins CA, et al. Genomics of Loa loa, a Wolbachia-free filarial parasite of humans. Nat Genet. 2013;45:495–500. doi: 10.1038/ng.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lau YL, et al. Draft genome of Brugia pahangi: high similarity between B. pahangi and B. malayi. Parasit. Vectors. 2015;8:451. doi: 10.1186/s13071-015-1064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghedin E, et al. Draft genome of the filarial nematode parasite Brugia malayi. Science. 2007;317:1756–1760. doi: 10.1126/science.1145406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Godel C, et al. The genome of the heartworm, Dirofilaria immitis, reveals drug and vaccine targets. FASEB J. 2012;26:4650–4661. doi: 10.1096/fj.12-205096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tallon LJ, et al. Single molecule sequencing and genome assembly of a clinical specimen of Loa loa, the causative agent of loiasis. BMC Genomics. 2014;15:788. doi: 10.1186/1471-2164-15-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Post R. The chromosomes of the Filariae. Filaria J. 2005;4:10. doi: 10.1186/1475-2883-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- 16.Hillier LW, et al. Comparison of C. elegans and C. briggsae genome sequences reveals extensive conservation of chromosome organization and synteny. PLoS Biol. 2007;5:e167. doi: 10.1371/journal.pbio.0050167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunt VL, et al. The genomic basis of parasitism in the Strongyloides clade of nematodes. Nat Genet. 2016;48:299–307. doi: 10.1038/ng.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foth BJ, et al. Whipworm genome and dual-species transcriptome analyses provide molecular insights into an intimate host-parasite interaction. Nat Genet. 2014;46:693–700. doi: 10.1038/ng.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bellott DW, et al. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature. 2014;508:494–499. doi: 10.1038/nature13206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 21.Bennuru S, et al. Stage-specific proteomic expression patterns of the human filarial parasite Brugia malayi and its endosymbiont Wolbachia. Proc Natl Acad Sci USA. 2011;108:9649–9654. doi: 10.1073/pnas.1011481108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vilella AJ, et al. Ensemblcompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lefoulon E, et al. Shaking the tree: multi-locus sequence typing usurps current onchocercid (filarial nematode) phylogeny. PLoS Negl Trop Dis. 2015;9:e0004233. doi: 10.1371/journal.pntd.0004233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Myllyharju J, Kivirikko KI. Characterization of the iron- and 2-oxoglutarate-binding sites of human prolyl 4-hydroxylase. EMBO J. 1997;16:1173–1180. doi: 10.1093/emboj/16.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vogeler S, Galloway TS, Lyons BP, Bean TP. The nuclear receptor gene family in the Pacific oyster, Crassostrea gigas, contains a novel subfamily group. BMC Genomics. 2014;15:369. doi: 10.1186/1471-2164-15-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antebi A. Nuclear hormone receptors in. C elegans WormBook. 2006:1–13. doi: 10.1895/wormbook.1.64.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tzertzinis G, et al. Molecular evidence for a functional ecdysone signaling system in Brugia malayi. PLoS Negl Trop Dis. 2010;4:e625. doi: 10.1371/journal.pntd.0000625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones AK, Davis P, Hodgkin J, Sattelle DB. The nicotinic acetylcholine receptor gene family of the nematode Caenorhabditis elegans: an update on nomenclature. Invert Neurosci. 2007;7:129–131. doi: 10.1007/s10158-007-0049-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williamson SM, Walsh TK, Wolstenholme AJ. The cys-loop ligandgated ion channel gene family of Brugia malayi and Trichinella spiralis: a comparison with Caenorhabditis elegans. Invert Neurosci. 2007;7:219–226. doi: 10.1007/s10158-007-0056-0. [DOI] [PubMed] [Google Scholar]
- 30.Dent JA, Smith MM, Vassilatis DK, Avery L. The genetics of ivermectin resistance in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2000;97:2674–2679. doi: 10.1073/pnas.97.6.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li BW, Rush AC, Weil GJ. High level expression of a glutamate-gated chloride channel gene in reproductive tissues of Brugia malayi may explain the sterilizing effect of ivermectin on filarial worms. Int J Parasitol Drugs Drug Resist. 2014;4:71–76. doi: 10.1016/j.ijpddr.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ardelli BF. Transport proteins of the ABC systems superfamily and their role in drug action and resistance in nematodes. Parasitol Int. 2013;62:639–646. doi: 10.1016/j.parint.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 33.Kotze AC, et al. Recent advances in candidate-gene and whole-genome approaches to the discovery of anthelmintic resistance markers and the description of drug/receptor interactions. Int J Parasitol Drugs Drug Resist. 2014;4:164–184. doi: 10.1016/j.ijpddr.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Orth JD, Thiele I, Palsson BO. What is flux balance analysis? Nat Biotechnol. 2010;28:245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Varma A, Palsson BO. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl Environ Microbiol. 1994;60:3724–3731. doi: 10.1128/aem.60.10.3724-3731.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen SN, Howells RE. Brugia pahangi: uptake and incorporation of adenosine and thymidine. Exp Parasitol. 1979;47:209–221. doi: 10.1016/0014-4894(79)90074-2. [DOI] [PubMed] [Google Scholar]
- 37.Chen SN, Howells RE. Brugia pahangi: uptake and incorporation of nuclei acid precursors by microfilariae and macrofilariae in vitro. Exp Parasitol. 1981;51:296–306. doi: 10.1016/0014-4894(81)90117-x. [DOI] [PubMed] [Google Scholar]
- 38.Rejman D, et al. N-phosphonocarbonylpyrrolidine derivatives of guanine: a new class of bi-substrate inhibitors of human purine nucleoside phosphorylase. J Med Chem. 2012;55:1612–1621. doi: 10.1021/jm201409u. [DOI] [PubMed] [Google Scholar]
- 39.Taylor MJ, Hoerauf A, Townson S, Slatko BE, Ward SA. Anti-Wolbachia drug discovery and development: safe macrofilaricides for onchocerciasis and lymphatic filariasis. Parasitology. 2014;141:119–127. doi: 10.1017/S0031182013001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boulin T, et al. Eight genes are required for functional reconstitution of the Caenorhabditis elegans levamisole-sensitive acetylcholine receptor. Proc Natl Acad Sci USA. 2008;105:18590. doi: 10.1073/pnas.0806933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Courtot E, et al. Functional characterization of a novel class of morantel-sensitive acetylcholine receptors in nematodes. PLoS Pathogens. 2015;11:e1005267. doi: 10.1371/journal.ppat.1005267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mostafa E, et al. Transient effects of levamisole on Brugia malayi microfilariae. Invert Neurosci. 2015;15:5. doi: 10.1007/s10158-015-0181-0. [DOI] [PubMed] [Google Scholar]
- 43.Gregory WF, Maizels RM. Cystatins from filarial parasites: evolution, adaptation and function in the host-parasite relationship. Int J Biochem Cell Biol. 2008;40:1389–1398. doi: 10.1016/j.biocel.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 44.Molehin AJ, Gobert GN, McManus DP. Serine protease inhibitors of parasitic helminths. Parasitology. 2012;139:681–695. doi: 10.1017/S0031182011002435. [DOI] [PubMed] [Google Scholar]
- 45.Cooper PJ, Guderian RH, Proano R, Taylor DW. Absence of cellular responses to a putative autoantigen in onchocercal chorioretinopathy. Cellular autoimmunity in onchocercal chorioretinopathy. Invest Ophthalmol Vis Sci. 1996;37:405–412. [PubMed] [Google Scholar]
- 46.Dowell SF, et al. Nodding syndrome. Emerg Infect Dis. 2013;19:1374–1384. doi: 10.3201/eid1909.130401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gebauer J, et al. A genome-scale database and reconstruction of Caenorhabditis elegans metabolism. Cell Syst. 2016;2:312–322. doi: 10.1016/j.cels.2016.04.017. [DOI] [PubMed] [Google Scholar]
- 48.Schulz-Key H, Albiez EJ, Buttner DW. Isolation of living adult Onchocerca volvulus from nodules. Tropenmed Parasitol. 1977;28:428–430. [PubMed] [Google Scholar]
- 49.Cupp EW, Sauerbrey M, Richards F. Elimination of human onchocerciasis: history of progress and current feasibility using ivermectin (Mectizan®) monotherapy. Acta Trop. 2011;120(Suppl 1):S100–S108. doi: 10.1016/j.actatropica.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 50.Abraham D, et al. Survival and development of larval Onchocerca volvulus in diffusion chambers implanted in primate and rodent hosts. J Parasitol. 1993;79:571–582. [PubMed] [Google Scholar]
- 51.Zimmerman PA, et al. Polymerase chain reaction-based diagnosis of Onchocerca volvulus infection: improved detection of patients with onchocerciasis. J Infect Dis. 1994;169:686–689. doi: 10.1093/infdis/169.3.686. [DOI] [PubMed] [Google Scholar]
- 52.Lustigman S, Huima T, Brotman B, Miller K, Prince AM. Onchocerca volvulus: biochemical and morphological characteristics of the surface of third -and fourth-stage larvae. Exp Parasitol. 1990;71:489–495. doi: 10.1016/0014-4894(90)90075-n. [DOI] [PubMed] [Google Scholar]
- 53.Miller KM, Hotze C, Brotman B, Prince AM. An economical procedure for screening of hybridoma supernatants for surface reactive antibodies to filarial larvae. Trop Med Parasitol. 1990;41:221–222. [PubMed] [Google Scholar]
- 54.Ham PJ, Townson S. Improved development of Brugia microfilariae following cryopreservation in liquid nitrogen using a technique suitable for field conditions. Trans R Soc Trop Med Hyg. 1986;80:150–153. doi: 10.1016/0035-9203(86)90217-8. [DOI] [PubMed] [Google Scholar]
- 55.Ham PJ, Townson S, James ER, Bianco AE. An improved technique for the cryopreservation of Onchocerca microfilariae. Parasitology. 1981;83:139–146. doi: 10.1017/s0031182000050113. [DOI] [PubMed] [Google Scholar]
- 56.Kozarewa I, et al. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat Methods. 2009;6:291–295. doi: 10.1038/nmeth.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park N, et al. An improved approach to mate-paired library preparation for Illumina sequencing. Methods in Next Generation Sequencing. 2013;1:10–20. [Google Scholar]
- 58.Simpson JT, Durbin R. Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 2012;22:549–556. doi: 10.1101/gr.126953.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurtz S, Narechania A, Stein JC, Ware D. A new method to compute K-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genomics. 2008;9:517. doi: 10.1186/1471-2164-9-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 62.Nadalin F, Vezzi F, Policriti A. Gapfiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics. 2012;13(Suppl 14):S8. doi: 10.1186/1471-2105-13-S14-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsai IJ, Otto TD, Berriman M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010;11:R41. doi: 10.1186/gb-2010-11-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30:2478–2483. doi: 10.1093/nar/30.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zimin AV, et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009;10:R42. doi: 10.1186/gb-2009-10-4-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bonfield JK, Whitwham A. Gap5—editing the billion fragment sequence assembly. Bioinformatics. 2010;26:1699–1703. doi: 10.1093/bioinformatics/btq268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hunt M, et al. REAPR: a universal tool for genome assembly evaluation. Genome Biol. 2013;14:R47. doi: 10.1186/gb-2013-14-5-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Otto TD, Sanders M, Berriman M, Newbold C. Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology. Bioinformatics. 2010;26:1704–1707. doi: 10.1093/bioinformatics/btq269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fenn K, et al. Phylogenetic relationships of the Wolbachia of nematodes and arthropods. PLoS Pathogens. 2006;2:e94. doi: 10.1371/journal.ppat.0020094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Foster J, et al. The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 2005;3:e121. doi: 10.1371/journal.pbio.0030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics. 2011;12:491. doi: 10.1186/1471-2105-12-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith CD, et al. Improved repeat identification and masking in Dipterans. Gene. 2007;389:1–9. doi: 10.1016/j.gene.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics. 2006;7:474. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stanke M, et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:W435–W439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18:1979–1990. doi: 10.1101/gr.081612.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.She R, et al. Genblastg: using BLAST searches to build homologous gene models. Bioinformatics. 2011;27:2141–2143. doi: 10.1093/bioinformatics/btr342. [DOI] [PubMed] [Google Scholar]
- 79.Yook K, et al. Wormbase 2012: more genomes, more data, new website. Nucleic Acids Res. 2012;40:D735–D741. doi: 10.1093/nar/gkr954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Otto TD, Dillon GP, Degrave WS, Berriman M. RATT: Rapid Annotation Transfer Tool. Nucleic Acids Res. 2011;39:e57. doi: 10.1093/nar/gkq1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim D, et al. Tophat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nakamura Y, Cochrane G, Karsch-Mizrachi I. The International Nucleotide Sequence Database Collaboration. Nucleic Acids Res. 2013;41:D21–D24. doi: 10.1093/nar/gks1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lizotte-Waniewski M, et al. Identification of potential vaccine and drug target candidates by expressed sequence tag analysis and immunoscreening of Onchocerca volvulus larval cDNA libraries. Infect Immun. 2000;68:3491–3501. doi: 10.1128/iai.68.6.3491-3501.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Williams SA, Laney SJ, Lizotte-Waniewskia M, Bierwerta LA, Unnasch TR. The river blindness genome project. Trends Parasitol. 2002;18:86–90. [Google Scholar]
- 87.UniProt, Consortium. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Logan-Klumpler FJ, et al. GeneDB—an annotation database for pathogens. Nucleic Acids Res. 2012;40:D98–108. doi: 10.1093/nar/gkr1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Darby AC, et al. Analysis of gene expression from the Wolbachia genome of a filarial nematode supports both metabolic and defensive roles within the symbiosis. Genome Res. 2012;22:2467–2477. doi: 10.1101/gr.138420.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 92.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and postanalysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jones P, et al. Interproscan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.US FDA. Approved Drug Products with Therapeutic Equivalence Evaluations. 33rd. US Department of Health and Human Services; 2013. [Google Scholar]
- 96.WHO Collaborating Centre for Drug Statistics Methodology. Guidelines for ATC Classification and DDD Assignment 2015. 19th. World Health Organization; 2015. [Google Scholar]
- 97.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 100.Hung SS, Wasmuth J, Sanford C, Parkinson J. DETECT—a density estimation tool for enzyme classification and its application to Plasmodium falciparum. Bioinformatics. 2010;26:1690–1698. doi: 10.1093/bioinformatics/btq266. [DOI] [PubMed] [Google Scholar]
- 101.UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Claudel-Renard C, Chevalet C, Faraut T, Kahn D. Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res. 2003;31:6633–6639. doi: 10.1093/nar/gkg847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tian W, Arakaki AK, Skolnick J. EFICAz: a comprehensive approach for accurate genome-scale enzyme function inference. Nucleic Acids Res. 2004;32:6226–6239. doi: 10.1093/nar/gkh956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schomburg I, et al. BRENDA: a resource for enzyme data and metabolic information. Trends Biochem Sci. 2002;27:54–56. doi: 10.1016/s0968-0004(01)02027-8. [DOI] [PubMed] [Google Scholar]
- 106.Karp PD, et al. Pathway Tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief Bioinform. 2010;11:40–79. doi: 10.1093/bib/bbp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Song C, et al. Metabolic reconstruction identifies strain-specific regulation of virulence in Toxoplasma gondii. Mol Syst Biol. 2013;9:708. doi: 10.1038/msb.2013.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chavali AK, Whittemore JD, Eddy JA, Williams KT, Papin JA. Systems analysis of metabolism in the pathogenic trypanosomatid Leishmania major. Mol Syst Biol. 2008;4:177. doi: 10.1038/msb.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Foster JM, Zhang Y, Kumar S, Carlow CK. Parasitic nematodes have two distinct chitin synthases. Mol Biochem Parasitol. 2005;142:126–132. doi: 10.1016/j.molbiopara.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 110.Rojo-Arreola L, et al. Chemical and genetic validation of the statin drug target to treat the helminth disease, schistosomiasis. PLoS ONE. 2014;9:e87594. doi: 10.1371/journal.pone.0087594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wittich RM, Walter RD. Putrescine N-acetyltransferase in Onchocerca volvulus and Ascaris suum, an enzyme which is involved in polyamine degradation and release of N-acetylputrescine. Mol Biochem Parasitol. 1990;38:13–17. doi: 10.1016/0166-6851(90)90199-v. [DOI] [PubMed] [Google Scholar]
- 112.Becker SA, et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nat Protoc. 2007;2:727–738. doi: 10.1038/nprot.2007.99. [DOI] [PubMed] [Google Scholar]
- 113.Daddona PE, et al. Expression of human malaria parasite purine nucleoside phosphorylase in host enzyme-deficient erythrocyte culture. Enzyme characterization and identification of novel inhibitors. J Biol Chem. 1986;261:11667–11673. [PubMed] [Google Scholar]
- 114.Schroeder P, et al. Loss of the tyrosyl radical in mouse ribonucleotide reductase by (−)-epicatechin. Biochem Biophys Res Commun. 2005;326:614–617. doi: 10.1016/j.bbrc.2004.11.089. [DOI] [PubMed] [Google Scholar]
- 115.Chang A, et al. BRENDA in 2015: exciting developments in its 25th year of existence. Nucleic Acids Res. 2015;43:D439–D446. doi: 10.1093/nar/gku1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.