

Abstract
ADP-ribosylation is a posttranslational modification involved in cellular homeostasis and stress, but has been challenging to analyze biochemically. To facilitate the detection of ADP-ribosylated proteins, we show that an alkyne-adenosine analog, N6-propargyladenosine (N6pA), is metabolically incorporated in mammalian cells and enables fluorescent detection and proteomic analysis of ADP-ribosylated proteins. Notably, our analysis of N6pA-labeled proteins up-regulated by oxidative stress revealed differential ADP-ribosylation of small GTPases. We discovered that oxidative stress induced ADP-ribosylation of Hras on Cys181 and 184 in the C-terminal hypervariable region, which are normally S-fatty-acylated. Downstream Hras signaling is impaired by ADP-ribosylation during oxidative stress, but is rescued by ADP-ribosyltransferase inhibitors. Our study demonstrates that ADP-ribosylation of small GTPases is not only mediated by bacterial toxins but is endogenously regulated in mammalian cells. N6pA provides a useful tool to characterize ADP-ribosylated proteins and their regulatory mechanisms in cells.
Graphical Abstract
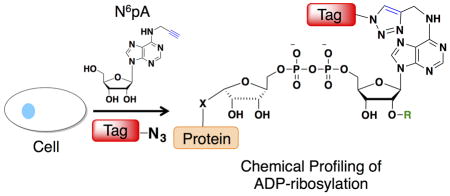
Introduction
First discovered in the nuclear extracts1, ADP-ribosylation of proteins is now known to play key roles in diverse cellular pathways in both eukaryotes and bacteria2. Proteins are ADP-ribosylated by addition of an ADP-ribose group on diverse amino acid (Arg, Asn, Cys, Glu, Asp, Lys) side chains as a monomer or elaborated into polymeric chains. The resulting mono- or poly-ADP-ribosylation can alter protein-protein interactions and affect downstream functions3,4. In mammals, DNA damage induces poly-ADP-ribosylation of several key proteins5, which can facilitate repair or activate programmed cell death6. Additionally, ADP-ribosylation has been implicated in telomere maintenance and cell division7. The discovery of additional ADP-ribosylated proteins has also suggested new roles for this posttranslational modification in translational control and cell signaling8. ADP-ribosylation is mediated by mono- and poly-ADP-ribosyltransferases (ARTDs, 18 in humans), which covalently install ADP-ribose derived from nicotinamide adenine dinucleotide (NAD) onto proteins. ADP-ribosylation is also reversible and can be removed by poly-ADP-ribose glycohydrolases (PARGs) or mono-ADP-ribose hydrolases4. While ARTD1 and 2 function in DNA repair, the activity and substrates for many of these enzymes are not well-characterized. Nonetheless, given the prominent roles of ADP-ribosylation in genome maintenance, ADP-ribosylation has emerged as a promising therapeutic target in human cancers with several ARTD inhibitors in clinical trials9,10.
ADP-ribosylation also plays a prominent role in host-pathogen interactions11. Notably, diphtheria toxin catalyzed ADP-ribosylation, which was discovered shortly after histone ADP-ribosylation, inhibits the function of mammalian elongation factor-2 (EF-2)12. Alternatively, the bacterial effector ExoS from Pseudomonas aeruginosa can ADP-ribosylated Ras and inhibit its GTPase activity13. Other secreted bacterial toxins or effectors also encode ADP-ribosyltransferase activity, but their host protein targets have not been fully characterized11,14. Despite the association with human diseases and infections, the precise functions of ADP-ribosylation in many cellular processes are still unknown due to challenges in the biochemical analysis of this complex and dynamic posttranslational modification.
The heterogeneous structures of mono- and poly-ADP-ribosylation on different protein side chains have limited the development of general methods for biochemical analysis. Radiolabeled NAD has been used to monitor ADP-ribosylation in vitro, but has low sensitivity15. Alternatively, antibodies8 and ADP-ribosylation binding reagents16 have been generated for monitoring poly-ADP-ribosylation, but have limited affinity and specificity. To exploit specific properties of ADP-ribosylated proteins, chemical methods such as borate-bead enrichment of cis-diols17 and hydrazine-labeling of hydrolyzed ribose groups18 have been developed. In addition, biotinylated and alkyne-modified NAD analogs have been synthesized for non-radioactive detection of ADP-ribosylation19–22, but are limited to in vitro studies. Despite these challenges, proteomic studies have uncovered many candidate ADP-ribosylated proteins associated with diverse cellular pathways8,23. The abundance of ADP-ribosylated proteins along with many associated regulatory enzymes and specific binding proteins suggests prominent roles for this posttranslational modification in biology, which requires more sensitive methods to detect, recover and quantify ADP-ribosylated proteins from living cells and animals.
To improve the detection of biomolecules, metabolites bearing chemical tags (i.e. azides or alkynes) have emerged as excellent chemical reporters for analyzing protein and nucleic acid modifications24. Indeed, we previously demonstrated that N6-propargyl adenosine (N6pA) can be metabolically incorporated into RNA and the poly(A) tails in mammalian cells for subsequent bioorthogonal detection25. Here, we show N6pA can also be efficiently incorporated into ADP-ribosylated proteins in mammalian cells, which enables robust fluorescent detection and enrichment of labeled proteins (Fig. 1). Proteomic analysis of N6pA-labeled proteins up-regulated by oxidative stress revealed ADP-ribosylation of small GTPases such as Hras. During H2O2-induced oxidative stress, we discovered that Hras is ADP-ribosylated on key Cys residues in C-terminal hypervariable region that is normally S-fatty-acylated and inhibits downstream cell signaling. This study demonstrates the utility of N6pA for the detection and discovery of ADP-ribosylated proteins from living cells, which should facilitate functional studies of this complex posttranslational modification in biology and human diseases.
Figure 1.

Metabolic labeling with N6pA followed by bioorthogonal labeling enables fluorescent detection and proteomic analysis of mono/poly-ADP-ribosylated proteins.
Results
N6pA labels ADP-ribosylated proteins in cells
To evaluate ADP-ribosylated protein labeling, HeLa cells were metabolically labeled with N6pA, lysed, subjected to CuI-catalyzed azide-alkyne cycloaddition (CuAAC) with azide-rhodamine (az-rho) and analyzed by in-gel fluorescence profiling. Proteins in HeLa cells were labeled with N6pA in a dose- and time-dependent manner that saturated at 1 mM after 1 hour (Supplementary Results, Supplementary Fig. 1a,b). N6pA labeled proteins in all mammalian cell lines tested (Supplementary Fig. 1c). The N6pA labeling was competed away with 5 mM adenosine, but not guanosine (Fig. 2a and Supplementary Fig. 2a). To confirm N6pA was metabolically converted to N6-propargyl NAD (N6pNAD), we performed ethanol extraction of HEK293T cells treated with DMSO or N6pA and evaluated NAD metabolites by mass spectrometry. The total ion current revealed little difference between the two runs with extracted ion current peaks for the singly charged NAD mass (m/z 664.1164) present in both DMSO and N6pA treated samples (Supplementary Fig. 3a,b). However, the singly charged N6pNAD (m/z 702.1320 Da) was only detected N6pA-treated samples at approximately 3 minutes (Supplementary Fig. 3a,b). The mass of both the NAD and N6pNAD was confirmed by high-resolution mass spectrometry and fragmentation analysis, which revealed the major ion for both peaks was nicotinamide loss (Supplementary Fig. 3c–f). These results demonstrate that mammalian cells can metabolize N6pA to N6pNAD, which is a substrate for ARTDs in vitro19, and covalently label proteins in a variety of mammalian cell types.
Figure 2.

N6pA labels ADP-ribosylated proteins in mammalian cells. a) HeLa cells were labeled with 1 mM N6pA ± 5 mM adenosine (A) or 5 mM guanosine (G) for 1 h. Cell lysates were then reacted with az-rho and analyzed by in-gel fluorescence profiling. PL = protein loading control. Fluor = fluorescence. b) HEK293T cells were transfected with H2B-GFP or the GLu3Ala ADP-ribosylation mutant, labeled 1 mM N6pA for 1 h, followed by immunopurification for GFP, and reacted with az-Rho. c) N6pA labels endogenous ARTD1 from HEK293T cells under oxidative stress. HEK293T cells were labeled 50 μM N6pA for 8 h and treated with 4 mM H2O2 for 1 h followed immunopurification for ARTD1. d) N6pA labeling of proteins is blocked by ARTD inhibitors: HeLa cells were labeled with 1 mM N6pA for 1 h and pretreated with either 10 μM olaparib (Ola) or 20 mM 3-aminobenzamide (ABA) 1 h before N6pA labeling and reacted with az-Rho. e) Wild type and ARTD1−/− MEFs were labeled with 50 μM N6pA for 8h and then treated with 4 mM H2O2 for 1 h. The cell lysates were then reacted with az-Rho. Full gels are located in Supplementary figure 2.
To evaluate the specificity to N6pA-protein labeling in mammalian cells, we evaluated known ADP-ribosylated proteins and their biochemical properties. Treatment of total cell lysates with phosphodiesterase26,27 abrogated N6pA-labeling, which showed that N6pA is indeed covalently attached to proteins by a phosphodiester linkage (Supplementary Fig. 4a). N6pA also labeled wild-type histone H2B-GFP28,29, but not its Glu3Ala mutant (Fig. 2b and Supplementary Fig. 2b), demonstrating site-specific labeling of a previously reported ADP-ribosylated protein28. Furthermore, N6pA labeled endogenous ARTD1, which was increased with H2O2-induced oxidative stress (Fig. 2c and Supplementary Fig. 2c). Notably, N6pA labeling detected low levels of ARTD1 modification in unstimulated cells, whereas anti-pan-ADP-ribose binding reagent (MABE-1016, pan-ADPr) was only reactive towards ARTD1 in H2O2-treated cells (Fig. 2c and Supplementary Fig. 2c), suggesting N6pA labeling can detect low levels of ADP-ribosylation. The specificity of N6pA labeling was further evaluated with ARTD inhibitors and knockout cells. Pretreatment of HeLa cells with ARTD inhibitors, 3-aminobenzamide or olaparib30, at their effective concentrations blocked N6pA labeling (Fig. 2d and Supplementary Fig. 2d). N6pA labeling of murine embryonic fibroblasts (MEFs) was also significantly reduced in ARTD1 (PARP1)-deficient cells under steady state conditions compared to wild-type MEFs (Fig. 2e, Supplementary Fig. 1d, and Supplementary Fig. 2e). The low levels of N6pA labeling in wild-type and H2O2-treated ARTD1−/− MEFs was still blocked by broad-spectrum ARTD inhibitors, olaparib, PJ-34, or Tiq-A, but less still sensitive to ARTD1-4 selective inhibitor ABT-88830 (Supplementary Fig. 1d). Similar results were also observed with pan-ADPr detection (Supplementary Fig. 1d). These results collectively show that ARTDs can mediate N6pA labeling of mono- and poly-ADP-ribosylated proteins in mammalian cells.
Proteomic analysis of N6pA-labeled proteins
Having established that N6pA labeled ADP-ribosylated proteins in mammalian cells, we performed proteomic analysis of HeLa cells subjected to H2O2-induced oxidative stress, as ADP-ribosylation has been reported to be up-regulated under these conditions31. Indeed, pan-ADP-ribosylation levels (pan-ADPr) were (Supplementary Fig. 2e) and N6pA-protein labeling (Supplementary Fig. 4a,b) of total cell lysates increased following H2O2-treatment (4 mM for 1 h) in the presence of serum-rich media. N6pA-protein labeling was also sensitive to phosphodiesterase treatment from H2O2-induced oxidatively stressed cells (Supplementary Fig. 4a). In H2O2-treated cells, N6pA labeling often generated smears in cell lysates reminiscent of poly-ADP-ribosylation that was also recovered after immunoprecipitation with poly-ADP-ribose-antibody (Supplementary Fig. 4c). For proteomic analysis, N6pA-labeled and H2O2-treated HeLa cell lysates were reacted with azide-biotin, affinity purified with streptavidin beads and subjected to on-bead protease digestion followed by protein identification using mass spectrometry (Supplementary Fig. 5a–c). Each sample was run in quadruplicate for label-free proteomic analysis using MaxQuant v.1.5.3.8. Correlation plots of identified peptides showed good reproducibility of the data over the four replicates (Supplementary Fig. 6a,b). A summary of the N6pA-labeled samples revealed 259 and 240 proteins enriched above the DMSO controls in untreated and H2O2-treated samples, respectively (Fig. 3a and Supplementary Data Sets 1–3). Amongst the high confidence N6pA-labeled proteins, 37 percent from the untreated samples and 35 percent from the H2O2-treated samples were identified in previous ADP-ribosylation proteomic studies using either anti-poly-ADP ribose antibody8, borate affinity17, ADP-ribose macro-domain16, alkynyl-NAD in vitro19, or ARTD1-3 alkynyl-NAD orthogonal pair22 labeling and enrichment studies (Supplementary Fig. 7a and Supplementary Data Sets 1–3). More specifically, our N6pA-enriched protein dataset overlapped 15 and 18 percent, respectively, with anti-poly-ADP ribose antibody8, which was performed in non-denaturing conditions, and ARTD1-3 alkynyl-NAD orthogonal pair22 poly-ADP-ribosylation proteomic studies (Supplementary Fig. 7a). Bioinformatic analysis of our proteomic dataset, suggests N6pA-labeled primarily labeled nuclear and cytoplasmic proteins involved in DNA and RNA processing (Supplementary Fig. 7b,c), consistent with reported functions of ADP-ribosylation3. To validate N6pA enrichment of ADP-ribosylated proteins, we analyzed DDX5, DDX17, nucleolin and filamin (Supplementary Fig. 8a,b). DDX5 was only found in the untreated samples, whereas DDX17, nucleolin and filamin were found in both untreated and H2O2-treated samples (Supplementary Fig. 8b), which correlated with our proteomic data (Supplementary Data Sets 1–3) and other ADP-ribosylation proteomic studies8,16,17,19,22. These results suggest N6pA labels known and new candidate ADP-ribosylated proteins in mammalian cells.
Figure 3.

Summary and validation of N6pA-labeled proteins. a) The scatter plot of N6pA-labeled proteins in both H2O2-treated and untreated cells. Protein hits were annotated by Maxquant analysis of recovered peptides. The x-axis is the difference of means between the H2O2-treated and untreated samples and the y-axis is the log of the probability of that difference determined by the t-test. The minimum values for a valid protein are p<0.05 and a difference of means of 2. Notable proteins are highlighted in black. b) HEK293T cells were transfected with GFP-HA-Rab7, labeled with 50 μM N6pA for 8 h and treated with 4 mM H2O2 for 1 h. GFP-HA-Rab7 was immunoprecipitated from cell lysates with antibody-conjugated beads, reacted with az-rho and analyzed in-gel fluorescence scanning. Western blots are included for protein loading control. c) N6pA labeling of FLAG-RhoA. d) N6pA labeling of HA-Hras after treatment with ADP-ribosylation inhibitors, ABT-888 (Abt, 10 μM), olaparib (Ola, 10 μM), PJ-34 (100 μM), and Tiq-A (20 μM), 2 h prior to N6pA labeling and H2O2-treatment. Full gels are located in Supplementary figure 9.
Oxidative stress induces Hras ADP-ribosylation
Our N6pA proteomic dataset revealed a number of new candidate ADP-ribosylated proteins in untreated and H2O2-treated cells (Fig. 3a and Supplementary Data Sets 1–3), including small GTPases such as Rab7a, RhoA and Hras. To validate ADP-ribosylation of these GTPases, epitope-tagged constructs were expressed in HEK293T cells, metabolically labeled with N6pA, immunoprecipitated, reacted with az-rho and visualized by in-gel fluorescence scanning. Analysis of GFP-HA-Rab7a32 confirmed N6pA labeling in control and H2O2-treated cells (Fig. 3b and Supplementary Fig. 9a). In contrast, FLAG-RhoA and HA-Hras were preferentially labeled by N6pA in H2O2-treated cells (Fig. 3c,d and Supplementary Fig. 9b,c). To further characterize the specificity of Hras ADP-ribosylation, we showed that HA-Hras N6pA-labeling was abrogated with 5 mM adenosine, but not guanosine (Supplementary Fig. 10a), sensitive to phosphodiesterase treatment (Supplementary Fig. 10b) and resistant to hydroxylamine (NH2OH) cleavage (Supplementary Fig. 10c), which hydrolyzes Glu- and Asp-linked ADP-ribosylation17 as well as S-fatty-acylation of Cys residues33 (Supplementary Fig. 10c). The enhanced modification of endogenous Hras from H2O2-treated cells was also confirmed by western blot analysis of N6pA-enriched proteins (Supplementary Fig. 5d). The apparent SDS-PAGE mobility of N6pA-labeled Hras suggests it is mono-ADP-ribosylated (Fig. 3d and Supplementary Fig. 9c). Pretreatment of HEK293T cells with the broad-spectrum ARTD inhibitors, olaparib (10 μM), PJ-34 (100 μM), or Tiq-A (10 μM) abrogated N6pA-labeling of HA-Hras (Fig. 3d and Supplementary Fig. 9c), while the ARTD1-4 selective inhibitor ABT-88830 only had a minor effect on N6pA labeling (Fig. 3d and Supplementary Fig. 9c). These results suggest Hras is ADP-ribosylated by ARTDs through an NH2OH-resistant linkage that is induced in oxidatively stressed cells.
Hras is one of four related small GTPase isoforms that regulate cell proliferation in response to growth factor signaling34. The four Ras isoforms (Hras, Nras, Kras4A, and Kras4B) share significant sequence homology except for the C-terminal hypervariable region (Fig. 4a and Supplementary Fig. 11), which are differentially lipidated to control their cellular localization and signaling properties31. All four isoforms are S-farnesylated at the canonical Cys residue of the CaaX-motif, but only Hras, Nras and Kras4A are further S-palmitoylated34 (Fig. 4a). To evaluate the generality of Hras ADP-ribosylation, we analyzed all four isoforms and found that only HA-Hras, HA-Nras, and HA-Kras4A constructs were labeled with N6pA in H2O2-treated cells (Fig. 4b and Supplementary Fig. 12a). As Kras4B has a polybasic region in hypervariable region for plasma membrane targeting instead of additional Cys residues for S-palmitoylation34, we hypothesized that ADP-ribosylation of Hras may occur on Cys residues 181 and 184. Indeed, mutation of Cys181 and 184 to Ala significantly reduced N6pA labeling of Hras (Fig. 4c and Supplementary Fig. 12b). In contrast, the Hras S-farnesylation mutant, Cys186Ala, had comparable N6pA labeling to wild type (Fig. 4c and Supplementary Fig. 12b). N6pA labeling other Hras mutants showed H2O2-induced ADP-ribosylation does not occur on Arg41 or 128, which are modified by the bacterial toxin ExoS13 and also not significantly changed in oncogenic mutants such as Gly12Val (G12V) (Supplementary Fig. 10d). Additionally, endogenous Ras immunopurified from wild-type and ARTD1−/− MEFs was labeled by N6pA, demonstrating that the ADP-ribosylation of Ras is not under ARTD1 control (Fig. 4d and Supplementary Fig. 12c). To independently verify the site-specific ADP-ribosylation, HA-Hras and the Cys mutants were expressed, immunopurified and analyzed with pan-ADP-ribose binding reagent (pan-ADPr). Only wild-type Hras and Cys186Ala mutant was detected by pan-ADPr (Fig. 4e and Supplementary Fig. 12d), confirming ADP-ribosylation of Cys181 and 184 on Hras in oxidatively stressed cells.
Figure 4.

Oxidative stress induces ADP-ribosylation of Hras at S-palmitoylation sites. a) Sequence alignment of Ras isoform C-terminal hypervariable regions. S-palmitoylated Cys are in blue and S-farnesylated sites are in green. The C-terminal hypervariable regions are italicized. b) Following transfection, HEK293T cells were labeled 50 μM N6pA for 8 h and treated with 4 mM H2O2 for 1 h. The epitope-tagged Ras isoforms were then immunoprecipitated, reacted with az-rho and analyzed by in-gel fluorescence. c) N6pA labeling of HA-Hras Cys mutants. d) Ras was immunopurified from either ARTD1+/+ or ARTD 1−/− MEFs after labeling with 50 μM N6pA for 8h, treated with 4 mM H2O2 for 1 h, and ligated with az-rho. e) HA-Hras was transfected into 293T cells, treated with 4 mM H2O2 for 1 h, and reacted with az-rho. HA-Ras isoforms were then immunoprecipitated and analyzed by western blot using anti-HA and pan-ADPr. Full gels are located in Supplementary figure 12.
ADP-ribosylation of Hras inhibits signaling under stress
ADP-ribosylation of Hras at Cys181 and 184, which are also key sites of S-palmitoylation34, suggests a switch in posttranslational modification of this small GTPase during oxidative stress. To measure endogenous Hras S-fatty-acylation levels during oxidative stress, we utilized an S-acyl-PEGylation exchange (APE) protocol developed by our laboratory (Supplementary Fig. 13a)35. The APE protocol is based on the acyl-biotin exchange protocol36, which caps free Cys residues and employs NH2OH to cleave thioesters for subsequent modification of thiols with PEG-maleimide reagents to induce a mass shift that is readily detectable by western blot (Supplementary Fig. 13a). In control HEK293T cells, APE analysis shows approximately 50 percent of endogenous Hras is di-S-fatty-acylated, with another 10 percent that is mono-S-fatty-acylated and 40 percent unmodified (Fig. 5b,c and Supplementary Fig. 13b). In contrast, endogenous Hras was not S-fatty-acylated in H2O2-treated cells compared to a control protein, calnexin (CANX)37, a dually S-palmitoylated protein that was PEGylated at similar levels in both conditions (Fig. 5b,c and Supplementary Fig. 13b). These results suggest endogenous Hras is not S-palmitoylated during oxidative stress (Fig. 5b,c and Supplementary Fig. 13b), but becomes ADP-ribosylated based on our N6pA-labeling (Fig. 3d and Supplementary Fig. 9c), enrichment (Fig. 5d and Supplementary Fig. 4c) and proteomic data (Fig. 3a). It is should also be noted that PJ-34 does not affect the levels of Hras S-fatty-acylation (Supplementary Fig. 14), suggesting ADP-ribosylation does not compete with S-fatty-acylation normally but occurs during oxidative stress.
Figure 5.

Oxidative stress induces Hras removal of S-palmitoylation. a) HEK293T cells were treated with 4 mM H2O2 for 1 h, lysed, and subjected to the APE protocol. Western blots of Hras and calnexin (Canx) after the APE protocol was performed. NH2OH-specific PEGylation is marked corresponding to mono or di S-acylation. Apo is unmodified protein. b) Bar graph depicting S-fatty-acylation levels of Hras ± H2O2-treatment measured by the APE protocol. Data is plotted as mean±s.d for 3 biological repeats. c) Quantitative analysis of endogenous Hras enrichment from N6pA-labeled cells. Data is plotted as mean±s.d for 4 biological repeats. Full gels are located in Supplementary figure 13.
We next investigated whether ADP-ribosylation affected Hras signaling during oxidative stress. To evaluate Hras signaling, we monitored the activation of downstream kinases Erk and Akt38, which are induced by cellular stress39–41, and analyzed ADP-ribosylation in parallel with N6pA labeling. For these experiments, whole cell lysates were assayed for both Erk/Akt and phosphorylated Erk/Akt by western blot, while the N6pA labeling of HA-Hras constructs was assessed by immunoprecipitation, reaction with az-rho and in-gel fluorescence (Fig. 6a, Supplementary Fig. 15, and Supplementary Fig. 16a,c). Cells were pretreated with PJ-34 2 hours before N6pA labeling and oxidative stress to evaluate pharmacological inhibition of ADP-ribosylation on Hras signaling. All samples were run in triplicate and quantified (Fig. 6b,d, Supplementary Fig. 15, Supplementary Fig. 16b,d, and Supplementary Figure 17). The N6pA labeling of Hras occurs in as little as 20 min after H2O2-treatment and increases with time, while Erk/Akt phosphorylation levels increased after 20 min and returned to baseline after 1 hour (Fig. 6a,b, Supplementary Fig. 15,and Supplementary Fig. 16). In contrast, addition of PJ-34 to H2O2-treated cells with completely abrogated N6pA labeling for all time points, while phosphorylated Erk levels increased over time and became 4-fold higher compared to control cells (Fig. 6a,b and Supplementary Fig. 15), which was similar for phosphorylated Akt levels (Supplementary Fig. 16a,b). Total Erk/Akt protein levels were not affected by H2O2 or PJ-34 treatment (Fig. 6a,b and Supplementary Fig. 16). These affects on downstream Hras signaling were also observed with lower concentrations of ARTD inhibitors 10 μM PJ-34 (Supplementary Fig. 16c,d) and olaparib (Supplementary Fig. 18). These results suggest that H2O2-induced ADP-ribosylation inhibits Hras activation of downstream signaling pathways, which can be rescued with ARTD inhibitors.
Figure 6.

Hras ADP-ribosylation and signaling during oxidative stress. a) HEK293T cells were transfected with wt HA-Hras followed by 2h of 100 μM PJ-34 treatment, 8h of N6pA labeling, 1h of 4 mM H2O2 stress. Western blots are included for protein loading control and of the whole cell lysate for both Erk and p-Erk. Western blot experiments were run in triplicate and representative data is shown. Full gels are shown in Supplementary figure 15. b) Bar graph for all three experiments showing p-Erk levels under oxidative stress with and without PJ-34 normalized against the Erk levels in the western blot for the time course data. Data is plotted as mean±s.d. c) HEK293T cells were transfected with wt HA-Hras or C181,184A followed by 2h of 10 μM PJ-34 treatment, N6pA labeling, 2h of H2O2 stress. Western blot experiments were run in triplicate and representative data is shown. Full gels are shown in Supplementary figure 17. IP=immunopurification WCL=whole cell lysate d) Bar graph for all three triplicates showing p-Erk levels under oxidative stress with and without PJ-34 normalized against the Erk levels in the western blot for the different HA-Hras constructs. Data is plotted as mean±s.d. e) HEK293T cells were transfected with vector, wt HA-Hras, G12V, or C181,184A followed by 2h of 10 μM PJ-34 treatment, N6pA labeling, 2h of H2O2 stress. Full gels are shown in Supplementary figure 19. For the bar graphs, significance was calculated for the data using a t-test and * is p < 0.05, and ** is p < 0.01.
To evaluate specific sites of Hras ADP-ribosylation during H2O2-induced oxidative stress, HEK293T cells were transfected with wt HA-Hras or the double cysteine mutant (C181A and C184A), labeled with N6pA, and treated with H2O2 for 2 h (Fig. 6c and Supplementary Fig. 17). For all HA-Hras constructs, N6pA labeling was blocked by pretreatment with PJ-34 (Fig. 6c,d and Supplementary Fig. 17). Total Erk levels appear to be unaffected by the different HA-Hras constructs, oxidative stress, or PJ-34 treatment. Interestingly, the HA-Hras double Cys mutant, which is not S-fatty-acylated or ADP-ribosylated, showed low levels of phosphorylated Erk compared to wt Hras, which is inactive in H2O2-treated cells (Fig. 6c,d and Supplementary Fig. 17). With PJ-34 treatment, phosphorylated Erk levels for wt HRas was increased 2-fold whereas no change was observed for the C181/184A HA-Hras mutant (Fig. 6c,d and Supplementary Fig. 17). The increase in phosphorylated Erk levels under these conditions is dependent upon the GTPase activity of Hras, since G12V mutant is more active than wt and C181/184A HA-Hras constructs (Fig. 6e and Supplementary Fig. 19). Collectively, these results suggest that site-specific ADP-ribosylation of key Cys residues in the C-terminal hypervariable of Hras inhibits its downstream cell signaling during oxidative stress.
Discussion
ADP-ribosylation is an important and complex posttranslational modification in biology that still requires sensitive biochemical methods for functional studies. Here we demonstrate that metabolic labeling with an alkyne-adenosine reporter, N6pA, enables sensitive fluorescence detection and enrichment of ADP-ribosylated proteins from mammalian cells. In comparison with other ADP-ribosylation methods and proteomic studies, N6pA enables detection and profiling in living cells not accessible with alkynyl-NAD analogs19,22, does not require PARG knockdown or inhibitors8,16,17 and not biased towards the modification of specific amino acids17,22. N6pA also appears to label mono- and poly-ADP-ribosylated substrates based on the proteins that are enriched (Supplementary Fig. 9a,b, Supplementary Data Sets 1–3). Although N6pA may also label poly-ADP-ribosylated substrates in cells, we primarily observe mono-ADP-ribosylation of immunoprecipitated proteins with N6pA labeling and in-gel fluorescence detection, which may depend upon the antibody, solubility and stability of mono- and poly-ADP-ribosylated proteins. These unique features of N6pA-labeling allowed us to profile ADP-ribosylated proteins in healthy and oxidatively stressed cells, which revealed known and new candidate mono- and poly-ADP-ribosylated proteins.
Most notably, our N6pA-comparative proteomic studies uncovered ADP-ribosylation of small GTPases such as RhoA and Hras in oxidatively stressed cells. Our additional results suggest oxidative stress induces mono-ADP-ribosylation of Hras at Cys residues that are normally S-palmitoylated in healthy cells and inhibits downstream Erk kinase signaling. Pharmacological inhibition of ADP-ribosylation rescues Hras signaling in oxidatively stressed cells, which may provide a mechanism by which ARTD (PARP) inhibitors may enhance cell survival in pathogenic settings42. Although, ARTD inhibitors block N6pA-labeling of Hras (Fig. 3d), we cannot exclude that these small GTPases (Hras, RhoA) may also be covalently modified by free ADP-ribose in oxidatively stressed cells. Our discovery of Hras ADP-ribosylation sites at Cys181 and 184 also adds to a growing appreciation of this modification on thiols43 and reveals an important chemical switch from S-palmitoylation during oxidative stress that inhibits downstream signaling (Supplementary Fig. 20). The ADP-ribosylation of Hras adds to the complex regulation of the Ras proteins in the hypervariable region including, phosphorylation44, and proline isomerization45 and depalmitoylation46, among other posttranslational modifications34. Our study provides the first example of endogenous ADP-ribosylation of small GTPases in mammalian cells, which has only been described during bacterial infection by ADP-ribosyltransferase toxins13. Interestingly, other studies have suggested that Hras Cys181 and 184 can also be oxidized and modified with glutathione during oxidative stress47, which suggests that these key Cys residues in the C-terminal hypervariable region of Hras are dynamically modified by S-fatty-acylation, S-glutathiolation, ADP-ribosylation and are highly sensitive to metabolic changes in cells. Our studies also provide another example of dynamic interplay between S-fatty-acylation and other posttranslational modifications48, which have been observed for example with S-palmitoylated and S-nitrosylated PSD-9549 and suggested for other proteins from PTM-selective proteomic studies50. Understanding the mechanisms that regulate the dynamic interplay between PTMs in different cell types and states will be an important area of future study.
Finally, N6pA labeling provides a complementary approach to protein-based detection reagents (macrodomain-fusions and antibodies)8,16 that may recognize specific mono- or poly-ADP-ribose structures. Moreover, N6pA derivatives could be used with specific site mapping strategies17 as well as engineered ARTDs22 to directly identify specific enzyme substrates and their sites of protein modification in cells. Adenosine chemical reporter labeling approaches therefore should provide powerful methods to discover and characterize the functions of ADP-ribosylated proteins and their regulatory mechanisms in biology and human disease.
Online Methods
N-(9-(2-(4-(6-azidohexanoyl)piperazine-1-carbonyl)phenyl)-6-(diethylamino)-3H-xanthen-3- ylidene)-N-ethylethanaminium (az-rho) was synthesized as previously reported51. N6-propargyladenosine (N6pA) was synthesized as previously reported52. After synthesis, the identity of N6-propargyladenosine was confirmed by 1H NMR in comparison with previously published spectra and by mass spectrometry.
Cell Lines and Culture
HeLa, MEF, and HEK293T cells were sourced from ATCC. The ARTD1 (PARP1)−/− MEFs were a gift from Dr. Valerie Schreiber. Cell lines were not further authenticated. Cell lines were tested for mycoplasma contamination by DAPI staining. HeLa, MEF, and HEK293T cells were cultured 10% FBS (Hyclone) in DMEM (Gibco) at 37 °C with 5% CO2 and passaged every 2–3 days at 90% confluency.
N6pA labeling and H2O2-treatment
4 × 105 cells in a 6-well plate labeled with 1 mL of fresh media containing 2 μL of 500 mM N6pA solution in DMSO or DMSO alone. The cells were incubated at 37 °C and 5% CO2 for 1 h. For drug treatment, 20 μL of 1 M 3-aminobenzamide in DMSO, 10 μL of 1 mM olaparib in DMSO, or DMSO was added to the media prior to labeling. The cells were placed in the incubator for 1 h. After 1 h, 2 μL of 500 mM N6pA solution in DMSO or DMSO control was added and the cells were placed again in the incubator for 1 h. For a 10 cm dish (Falcon) 5 mL of fresh media was used for labeling and for a 30 cm dish (Falcon), 13 mL of fresh media was used.
For N6pA labeling followed by H2O2 treatment, 4 × 105 cells in a 6-well plate were labeled with 2 mL of fresh media containing 1 μL of 100 mM N6pA solution in DMSO or DMSO alone. The cells were incubated at 37 °C and 5% CO2 for 8 h. For drug treatment, 2 μL of 10 mM olaparib in DMSO, 2 μL of 100 mM PJ-34 in DMSO, 2 μL of 10 mM ABT-888 in DMSO, 4 μL of 10 mM Tiq-A in DMSO or DMSO was added to the media prior to labeling. The cells were placed in the incubator for 2 h. After 2 h, 1 μL of 100 mM N6pA solution in DMSO or DMSO control was added and the cells were placed again in the incubator for 8 h. After 8 h, the cells were removed from the incubator and 10 μL of a 40 mM H2O2 solution in PBS or PBS was added and the cells were placed in the incubator for 1 h.
In-gel fluorescence detection
Cell lysates (50 μg - determined by Pierce BCA assay) in 45 μL 4% SDS lysis buffer (50 mM HEPES, 150 mM NaCl, pH 7.4, 250 U benzonase) were reacted with 5 μL freshly premixed click chemistry reaction cocktail [az-rho (100 μM, 10 mM stock solution in DMSO), tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (1 mM, 50 mM in water), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) (100 μM, 10 mM stock solution in DMSO) and CuSO4 (1 mM, 50 mM in water)] for a total reaction volume of 50 μL for 1 h at room temperature. The reactions were terminated by the addition of ice-cold methanol (200 μL), chloroform (75 μL), and water (150 μL) for 10 min at 20 °C and centrifuged at 15000g for 5 min at 4 °C to precipitate proteins. The protein was washed twice more with 500 μL ice-cold methanol. The supernatant from the samples was discarded. The protein pellets were air dried for 20 min, suspended in 35 μL of resuspension buffer (4% SDS, 50 mM HEPES pH 7.4, 150 mM NaCl), diluted with 12.5 μL 4× reducing SDS-loading buffer (BioRad) and 2.5 μL β-mercaptoethanol and boiled; 20 μg of protein was loaded per gel lane for separation by SDS-PAGE (4 20% Bio-Rad Stain Free Tris-HCl gel). SDS-PAGE gels were imaged on GE typhoon 5400 for fluorescent labeling. For protein loading, stain-free gels (BioRad) were imaged using Molecular Imager Gel Doc XR System (Bio Rad) with the stain-free filters.
Plasmids
Plasmids were obtained from Addgene; H2B-GFP (plasmid 11680 - deposited by Geoff Wahl) and GFP-HA-Rab7 (12605 - deposited by Richard Pagano). The HA-Kras4A and HA-Kras4B plasmids were obtained from the Phillips lab (New York University). Plasmids were also obtained from the H2B-GFP Glu3Ala mutant was generated using QuickChange Lighting mutagenesis kit following manufacturer’s directions using the primer: 5′-GCCACCATGCCAGCCCCAGCGAAGTCTGCT-3′. Hras mutants were obtained with the QuickChange II XL mutagenesis kit using the primers below. Hras mutants:
C41A: 5′-cactatagaggattcctacgcgaagcaggtggtcattgat-3′, 5′-atcaatgaccacctgcttcgcgtaggaatcctctatagtg-3′
C128A: 5′-ctgagcctgcgcagattccacagtgcgtgc-3′, 5′-gcacgcactgtggaatctgcgcaggctcag-3′
C181A and 184A: 5′-gagcacacacttggcgctcatggcgccggggccactc-3′, 5′-gagtggccccggcgccatgagcgccaagtgtgtgctc-3′
C186A: 5′-gagatcaggagagcacagccttgcagctcatgcagc-3′, 5′-gctgcatgagctgcaaggctgtgctctcctgatctc-3′
After site directed mutagenesis, all plasmids were DNA sequenced to verify their identity.
Immunoprecipitation
4 × 105 HEK293T cells in a 6-well plate were transfected with 1 μg of DNA plasmid using lipofectamine 2000 (Invitrogen) for 24 h following the manufacture instructions. The next day, the cells were labeled with N6pA, lysed in 1% Brij (50 mM HEPES, 150 mM NaCl, pH 7.4, 1 μM adenosine 5′-diphosphate (Hydroxymethyl)pyrrolidinediol (ADP-HPD)) on ice. Lysates were cleared at 15000g for 3 min. 700 μg of protein was added to protein G beads (Roche) coupled with 2 μg of anti-GFP (JL-8, clontech) for histone H2B. For the HA- and FLAG-tagged proteins, 20 μL of either anti-HA or anti-FLAG Sigma EZ-view immunopurification beads were used. The beads were rocked for 1 h at 4 °C and washed 3 times using ice-cold RIPA buffer. 50 μL of 50 mM HEPES buffer was added and on bead click chemistry was performed by adding 1 μL of 50 mM CuSO4, 1 μL of 50 mM TCEP, 2.5 μL of 2.5 mM TBTA in DMSO, and 0.5 μL of 10 mM az-rho in DMSO. The beads were mixed for 1 h and washed 3x with ice-cold RIPA buffer. 20 μL of 4% SDS loading buffer was added and the beads were boiled for 5 min and 20 μL run on an SDS-PAGE gel. ARTD1 was immunopurified using an anti-ARTD1 antibody (Santa Cruz Biotech, sc-1562) from 9 × 106 HeLa cells in a 15 cm dish using 1.5 mg of cell lysate. Poly (ADP-ribose) was immunopurified using an anti-PAR antibody (Trevigen, 4336-APC) from 9 × 106 HeLa cells in a 15 cm dish using 1.5 mg of cell lysate. Ras was immunopurified using an anti-Ras antibody (abcam 108602) from approximately 4x107 MEF cells using 10 mg of cell lysate. For the immunoblotting experiments for ADP ribose, 293T cells were transfected in a 150mm dish and the Hras constructs were immunopurified from 8 mg of cell lysate.
Immunoblotting
Proteins separated by SDS-PAGE were transferred (50 mM Tris, 40 mM glycine, 0.0375% SDS, 20% MeOH in deionized water, Bio-Rad Trans-Blot Semi-Dry Cell, 25 V, 20 min) onto a nitrocellulose membrane which was blocked with 5% nonfat dried milk in PBS for 1 h at 25 °C. The membrane was washed three times with PBST (0.1% Tween-20 in PBS), incubated with anti-HA or anti-FLAG horseradish peroxidase (1 mg/mL diluted 1:2,000 in PBST, Roche) overnight at 4 °C, and subsequently developed with ECL Western blotting detection reagents (Biorad). For proteins with primary and secondary antibodies, western blots were visualized with anti-GFP (1:1000, Clontech, JL-8), anti-calnexin (1:2000, Abcam, ab22595), anti-Erk1 (1:100, Santa Cruz Biotechnology, sc-376852), anti-p-Erk1/2 (1:100, Santa Cruz Biotechnology, sc-16982), anti-Erk1/2 (1:1000, Abcam,115799), anti-ARTD1 (1:1000, Abcam, 176491), anti-pADPr (1:500, Trevigen, 4336-BPC-100), anti-Akt (1:500, Cell Signaling, 4691p), anti-p-Akt (1:1000, Cell Signaling, 4060p), MABE1016 (Millipore, 1:100), DDX5 (1:500, Cell Signaling, 4387s), DDX17 (1:1000, Pro-Sci, 49–272), Nucleolin (1:1000, Cell Signaling, 14574s), Filamin (1:1000, Millipore, MAB1680) and anti-Ras (Millipore, 1:1000, RAS10) in PBST followed by a wash step as above. Next, the blots were treated with either goat anti-mouse-HRP conjugated secondary antibody (1:5000, Abcam, ab97023) or goat anti-rabbit-HRP conjugated secondary antibody (1:5000, Abcam, ab6013) depending on the species of the primary antibody, washed, and visualized with ECL Western blotting detection reagents.
Phosphodiesterase treatment
N6pA-labeled and H2O2-treated HEK293T cells were lysed with 1% Brij in HEPES buffer and 50 μg of lysates (measured by the BCA assay) was treated with 1 unit (based on enzymatic bis-(p-nitrophenyl) phosphate hydrolysis) of tobacco acid phosphodiesterase (Epicenter) or water, and the samples were heated to 37 °C for 0.5 h and then the lysates were reacted with az-rho, as describe above. For immunoprecipitated samples, the samples were affinity purified, washed in RIPA buffer, and then resuspended in 50 μL of HEPES buffer. To this slurry 1 unit of the tobacco acid phosphodiesterase or water was added. The slurry was shaken for 1 h at 37 °C, washed, and then reacted with az-rho.
Hydroxylamine treatment
For alk-16 labeling, HA-Hras transfected HEK293T cells in a 6-well plate were treated with 50 μM alk-16 or DMSO for 4 h. After HA-Hras transfection, N6pA labeling, oxidative stress, immunopurification, and reacted with az-rho, the EZ view beads were split in two. Half were boiled in loading buffer for 5 min. and the other half were boiled in loading buffer containing 1 M of neutralized hydroxylamine for 5 min. The samples were then separated on a SDS-PAGE gel and analyzed by in-gel fluorescence.
On-bead digestion protein identification
A 30 cm dish of HeLa cells was grown to confluency for each experiment. Cells were labeled with N6pA, treated with H2O2, and lysed with 4% SDS (50 mM HEPES, 150 mM NaCl, pH 7.4, 250 U Benzonase). 2 mg of cell lysate was reacted with azido-biotin (Click Chemistry tools) for 4 h (1 mM CuSO4, 1 mM TCEP, 200 μM TBTA, 200 μM azido-biotin) at 1 mg/ml. Protein was precipitated using methanol (8 mL), chloroform (3 mL), and water (6 mL) overnight at −20 °C and washed with methanol (5 mL) twice the following morning. Protein pellets were dried for 1 h and solubilized in 200 μL of 4% SDS buffer. The sample was diluted to 1 mg/mL with 50 mM HEPES buffer (150 mM NaCl, pH 7.4). 25 μL of streptavidin beads (Thermo) were added to the sample and the beads and solution were rocked for 1 h. The beads were washed using 3x 1% SDS (50 mM HEPES, 150 mM NaCl, pH 7.4), 3x 5 M Urea (50 mM HEPES, 150 mM NaCl, pH 7.4), and 3x PBS. Centrifugation of the beads between washing steps was carried out (2000 × g, 1 min). Samples were split and half the beads were boiled in 20 μL of 4% SDS loading buffer and separated by SDS-PAGE. The other half was suspended in 200 μL 25 mM ammonium bicarbonate, reduced with 1 mM DTT (100 mM stock) for 30 min, and then alkylated with 50 mM iodoacetamide (1 M stock) in the dark for 30 min. Then, the beads were washed with 200 μL of 25 mM ammonium bicarbonate, and suspended in 50 μL of 25 mM ammonium bicarbonate. 0.1 μg of trypsin (Promega) was added, and the samples were digested at 37 °C overnight. The supernatant was collected and dried on a speedvac and solubilized in 5% acetonitrile/1% formic acid for LC-MS/MS analysis.
Extracted tryptic peptides were desalted on a trap column following separation on a 12 cm/75 μm reversed phase C18 column (Nikkyo Technos Co., Ltd. Japan). A 180 minutes gradient increasing from 10% B to% 45% B in 133 minutes (A: 0.1% Formic Acid, B: Acetonitrile/0.1% Formic Acid) were delivered at 200 nL/min. The liquid chromatography setup (Dionex, Boston, MA, USA) was connected to an Orbitrap XL (Thermo, San Jose, CA, USA) operated in top-8-CID-mode with MS spectra measured at a resolution of 60,000@m/z 400. Acquired tandem MS spectra were extracted using Maxquant software version 1.5.3.8 and queried against the Uniprot complete human database and processed using Perseus. Peptides fulfilling a calculated 1% false discovery rate threshold were reported. Additionally, for the labeled samples vs. DMSO controls absent values were imputed at 0. For the untreated versus H2O2-treated comparison, values were imputed based on the normal distribution of values. In order to determine if a protein was a valid hit, the false discovery rate was lower than 1% and the mean difference of the control and N6pA-labeled samples had be greater than 1 with p<0.05 determined by the t-test. To determine if the hits had been previously reported, the proteomic list was compared to previously published data8,16,17,19,22.
APE protocol
Cell samples were lysed with 4% sodium dodecyl sulfate (SDS, Fischer) in TEA buffer (pH 7.3, 50 mM triethanolamine (TEA), 150 mM NaCl) containing 1x protease inhibitor cocktail (Roche), 5 mM PMSF (Sigma), and 1500 units/mL benzonase (EMD). After the samples were lysed, 5 mM EDTA (Fischer) was added. Typically, 200 μg in 92.5 μL of lysis buffer was treated with 5 μL of 200 mM neutralized TCEP for 30 minutes with nutation. N-ethylmaleimide (NEM, Sigma), 2.5 μL from freshly made 1 M stock in ethanol, was added incubated for 2 hours at room temperature. Reductive alkylation of the proteins was then terminated by methanol-chloroform-H2O precipitation (4:1.5:3) with sequential addition of methanol (400 μL), chloroform (150 μL) and distilled H2O (300 μL) (all pre-chilled on ice). The reactions were then mixed and centrifuged (Centrifuge 5417R, Eppendorf) at 15,000 g for 5 minutes at 4 °C. To pellet the precipitated proteins, the aqueous layer was removed, 500 μL of pre-chilled MeOH was added, the eppendorf tube inverted several times and centrifuged at 15,000 g for 3 minutes at 4 °C. The supernatant was then decanted, and the protein pellet washed once more with 500 μL of pre-chilled MeOH, centrifuged again and dried using a speed-vacuum (Centrivap Concentrator, Labconco) To ensure complete removal of NEM from the protein pellets, the samples were resuspended with 100 μL of TEA buffer containing 4% SDS, warmed to 37 °C for 10 minutes, briefly (~5 seconds) sonicated (Ultrasonic Cleaner, VWR) and subjected to two additional rounds of methanol-chloroform-H2O precipitations as described above.
For hydroxylamine (NH2OH) cleavage and 5 kDa mPEG-maleimide alkylation, the protein pellet was resuspended in 30 μL TEA buffer containing 4% SDS, 4 mM EDTA and treated with 90 μL of 1 M neutralized NH2OH (J.T. Baker) dissolved in TEA buffer pH 7.3. Control samples not treated with NH2OH were diluted in 90 μL TEA buffer. Samples were incubated at room temperature for 1 hour with nutation. The samples were then subjected to methanol-chloroform-H2O precipitation as described above and resuspended in 30 μL TEA buffer containing 4% SDS, 4 mM EDTA, warmed to 37 ° C for 10 minutes and briefly (~5 seconds) sonicated and treated with 90 μL TEA buffer and 1.33 mM 5 kDa methoxypolyethylene glycol-maleimide (mPEG-Mal, Sigma). Samples were incubated for 2 h at room temperature with nutation before a final methanol-chloroform-H2O precipitation. Dried protein pellets were resuspended in 100 μL 1 X Laemmli buffer (BioRad) and then heated for 5 minutes at 95 °C.
Biotin Enrichment of N6pA labeled proteins
A 10 cm dish of HEK293T cells was grown to confluency for each experiment. Cells were labeled with N6pA, treated with H2O2, and lysed with 4% SDS (50 mM HEPES, 150 mM NaCl, pH 7.4, 250 U Benzonase). 2 mg of cell lysate was reacted with azido-biotin (Click Chemistry tools) for 4 h (1 mM CuSO4, 1 mM TCEP, 200 μM TBTA, 200 μM azido-biotin) at 1 mg/ml. Protein was precipitated using methanol, chloroform, and water at −20 °C and washed with methanol (5 mL) twice the following morning. Protein pellets were dried for 1 h and solubilized in 200 μL of 4% SDS buffer. The sample was diluted to 1 mg/mL with 50 mM HEPES buffer (150 mM NaCl, pH 7.4). 25 μL of streptavidin beads (Thermo) were added to the sample and the beads and solution were rocked for 1 h. The beads were washed using 3x 1% SDS (50 mM HEPES, 150 mM NaCl, pH 7.4), 3x 5 M Urea (50 mM HEPES, 150 mM NaCl, pH 7.4), and 3x PBS. Centrifugation of the beads between washing steps was carried out (2000 × g, 1 min). The beads were then boiled in SDS-PAGE buffer and transferred to a PDVF membrane.
Western blot analysis of Erk activation
HEK293T cells in 6-well plates were transfected with 1 μg of HA-Hras per well with lipofectamine 2000 for 24 h following manufacturer instructions. Cells were treated with PJ-34 at 10 or 100 μM for 2 h or DMSO and then with 50 μM N6pA for 8 h. Cells were then oxidatively stressed with 4 mM H2O2 for 20, 60, or 240 min. Cells from 1 well were lysed with 1% Brij in 50 mM HEPES, 150 mM NaCl, pH 7.4 with 1x phosSTOP (Roche) and 1x Protease inhibitor cocktail (Roche). Protein concentration was measured by the BCA assay. 50 μg of whole cell lysate was run on an SDS-PAGE gel for each sample to measure the Erk and phosphorylated Erk levels by western blot. The remaining samples were pooled for immunoprecipitation experiments.
Cell Metabolite Extraction
293T cells were grown in a 10 cm dish were labeled with N6pA or DMSO. The following day, the media was aspirated and the cells were washed with PBS and collected with by centrifugation at 1000g for 2 min. The cells were then lysed with 500 μL of 75% ethanol/ 25% 50 mM HEPES, 150 mM NaCl pH 7.4 for 5 min at 80 °C. The extracts were then cleared by centrifugation at 16,000g for 10 min at 4 °C and the solution was pipetted off and dried in a speedvac. The extracts were then resolubilized in 20 μL of 100 mM ammonium acetate pH 9.
For the mass spectrometry, the samples were loaded onto and separated by reversed phase C18 chromatography (Acclaim 120 C18 2.1 mM ID x 150 Mm) using the following method: isocratic for 2 minutes at 5% B/95% A (A: 0.1% formic acid, B: and 0.1% formic acid /acetonitrile) followed by an increase to 60% B /40% A over 18min and hereafter further increased to 100% B over 1min. Solvents were delivered at 200 uL/min. Analytes were electrosprayed into an Orbitrap XL (Thermo, San Jose, CA, USA) using an ESI-HESI source operated at 250 °C, 30 psi sheath, and 10 psi auxiliary gas. The heated capillary was kept at 200 °C. MS spectra were measured at 60,000 resolution@400Th in the Orbitrap while MS/MS spectra (the most abundant ion per cycle) were subjected to CID and measured in the ion trap. Ion traces were extracted (window of 5ppm) using Qual Browser Thermo Excalibur 2.1, SP1.1160. Generated tandem MS spectra were manually validated.
Statisical Methods
For statistical analysis, significance was calculated for the data using an unpaired t-test with P < 0.05. All summary statistics of the results are given as mean ± s.e.m.
Supplementary Material
Acknowledgments
N.P.W. is a Leukemia and Lymphoma Society postdoctoral fellow. The authors thank Dr. Richard Pagano for GFP-HA-Rab7, Dr. Geoff Wahl for the histone H2B-GFP, Dr. Mark Phillips for the HA-Kras4A and Flag-Kras4B constructs and Dr. Valerie Schreiber for the ARTD1 (PARP1)−/− MEFs. HCH acknowledges support from NIH-NIGMS R01 GM087544 grant.
Footnotes
Competing Financial Interest Statement
The authors declare no competing financial interests.
Author Contributions
H.C.H. and N.P.W. designed the experiments, analyzed data and wrote the manuscript. H.M. helped with mass spectrometry analysis. J.P.F. performed and helped analyze the NAD metabolite mass spectrometry. N.P.W. performed all other experiments.
References
- 1.Chambon P, Weill JD, Mandel P. Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochemical and Biophysical Research Communications. 1963;11:39–43. doi: 10.1016/0006-291x(63)90024-x. [DOI] [PubMed] [Google Scholar]
- 2.Leung AKL. Poly(ADP-ribose): An organizer of cellular architecture. The Journal of Cell Biology. 2014;205:613–619. doi: 10.1083/jcb.201402114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kraus WL. PARPs and ADP-Ribosylation: 50 Years … and Counting. Molecular Cell. 2015;58:902–910. doi: 10.1016/j.molcel.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barkauskaite E, Jankevicius G, Ahel I. Structures and Mechanisms of Enzymes Employed in the Synthesis and Degradation of PARP-Dependent Protein ADP-Ribosylation. Molecular Cell. 2015;58:935–946. doi: 10.1016/j.molcel.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 5.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 6.Meder VrS, Boeglin M, de Murcia G, Schreiber Vr. PARP-1 and PARP-2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. Journal of Cell Science. 2005;118:211–222. doi: 10.1242/jcs.01606. [DOI] [PubMed] [Google Scholar]
- 7.Smith S, de Lange T. Tankyrase promotes telomere elongation in human cells. Current Biology. 2000;10:1299–1302. doi: 10.1016/s0960-9822(00)00752-1. [DOI] [PubMed] [Google Scholar]
- 8.Gagne JP, et al. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Research. 2008;36:6959–6976. doi: 10.1093/nar/gkn771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho SH, et al. PARP-14, a member of the B aggressive lymphoma family, transduces survival signals in primary B cells. Blood. 2009;113:2416–2425. doi: 10.1182/blood-2008-03-144121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 11.Westcott NP, Hang HC. Chemical reporters for exploring ADP-ribosylation and AMPylation at the host–pathogen interface. Current Opinion in Chemical Biology. 2014;23:56–62. doi: 10.1016/j.cbpa.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Honjo T, Nishizuka Y, Hayaishi O, Kato I. Diphtheria Toxin-dependent Adenosine Diphosphate Ribosylation of Aminoacyl Transferase II and Inhibition of Protein Synthesis. Journal of Biological Chemistry. 1968;243:3553–3555. [PubMed] [Google Scholar]
- 13.Ganesan AK, Vincent TS, Olson JC, Barbieri JT. Pseudomonas aeruginosa Exoenzyme S Disrupts Ras-mediated Signal Transduction by Inhibiting Guanine Nucleotide Exchange Factor-catalyzed Nucleotide Exchange. Journal of Biological Chemistry. 1999;274:21823–21829. doi: 10.1074/jbc.274.31.21823. [DOI] [PubMed] [Google Scholar]
- 14.Aktories K. Bacterial protein toxins that modify host regulatory GTPases. Nat Rev Micro. 2011;9:487–498. doi: 10.1038/nrmicro2592. [DOI] [PubMed] [Google Scholar]
- 15.Williams GT, Shall S, Ford CC. Direct radioactive labelling of poly(ADP-ribose) in developing Xenopus laevis embryos. Bioscience Reports. 1983;3:461–467. doi: 10.1007/BF01121957. [DOI] [PubMed] [Google Scholar]
- 16.Jungmichel S, et al. Proteome-wide Identification of Poly(ADP-Ribosyl)ation Targets in Different Genotoxic Stress Responses. Molecular Cell. 2013;52:272–285. doi: 10.1016/j.molcel.2013.08.026. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Wang J, Ding M, Yu Y. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat Meth. 2013;10:981–984. doi: 10.1038/nmeth.2603. [DOI] [PubMed] [Google Scholar]
- 18.Morgan RK, Cohen MS. A Clickable Aminooxy Probe for Monitoring Cellular ADP-Ribosylation. ACS Chemical Biology. 2015;8:1778–1784. doi: 10.1021/acschembio.5b00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang H, Kim JH, Frizzell KM, Kraus WL, Lin H. Clickable NAD Analogues for Labeling Substrate Proteins of Poly(ADP-ribose) Polymerases. Journal of the American Chemical Society. 2010;132:9363–9372. doi: 10.1021/ja101588r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carter-O’Connell I, et al. Identifying Family-Member-Specific Targets of Mono-ARTDs by Using a Chemical Genetics Approach. Cell Reports. 2016;14:621–631. doi: 10.1016/j.celrep.2015.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallrodt S, Buntz A, Wang Y, Zumbusch A, Marx A. Bioorthogonally Functionalized NAD+ Analogues for In-Cell Visualization of Poly(ADP-Ribose) Formation. Angewandte Chemie International Edition. 2016;14:621–631. doi: 10.1002/anie.201600464. [DOI] [PubMed] [Google Scholar]
- 22.Gibson BA, et al. Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science. 2016;353:45–50. doi: 10.1126/science.aaf7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daniels Casey M, Ong SE, Leung Anthony KL. The Promise of Proteomics for the Study of ADP-Ribosylation. Molecular Cell. 2015;58:911–924. doi: 10.1016/j.molcel.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grammel M, Hang HC. Chemical reporters for biological discovery. Nat Chem Biol. 2013;9:475–484. doi: 10.1038/nchembio.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grammel M, Hang H, Conrad NK. Chemical Reporters for Monitoring RNA Synthesis and Poly(A) Tail Dynamics. ChemBioChem. 2012;13:1112–1115. doi: 10.1002/cbic.201200091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chapman JD, Gagné JP, Poirier GG, Goodlett DR. Mapping PARP-1 Auto-ADP-ribosylation Sites by Liquid Chromatography–Tandem Mass Spectrometry. Journal of Proteome Research. 2013;12:1868–1880. doi: 10.1021/pr301219h. [DOI] [PubMed] [Google Scholar]
- 27.Daniels CM, Ong SE, Leung AKL. Phosphoproteomic Approach to Characterize Protein Mono- and Poly(ADP-ribosyl)ation Sites from Cells. Journal of Proteome Research. 2014;13:3510–3522. doi: 10.1021/pr401032q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogata N, Ueda K, Hayaishi O. ADP-ribosylation of histone H2B. Identification of glutamic acid residue 2 as the modification site. Journal of Biological Chemistry. 1980;255:7610–7615. [PubMed] [Google Scholar]
- 29.Kanda T, Sullivan KF, Wahl GM. Histone–GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Current Biology. 1998;8:377–385. doi: 10.1016/s0960-9822(98)70156-3. [DOI] [PubMed] [Google Scholar]
- 30.Wahlberg E, et al. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat Biotech. 2012;30:283–288. doi: 10.1038/nbt.2121. [DOI] [PubMed] [Google Scholar]
- 31.Leung AKL, et al. Poly(ADP-Ribose) Regulates Stress Responses and MicroRNA Activity in the Cytoplasm. Molecular cell. 2011;42:489–499. doi: 10.1016/j.molcel.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choudhury A, et al. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. The Journal of Clinical Investigation. 2002;109:1541–1550. doi: 10.1172/JCI15420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yount JS, Zhang MM, Hang HC. Current Protocols in Chemical Biology. John Wiley & Sons, Inc; 2009. [Google Scholar]
- 34.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol. 2012;13:39–51. doi: 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Percher A, et al. Mass-tag labeling reveals site-specific and endogenous levels of protein S-fatty acylation. Proceedings of the National Academy of Sciences. 2016;113:4302–4307. doi: 10.1073/pnas.1602244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wan J, Roth AF, Bailey AO, Davis NG. Palmitoylated proteins: purification and identification. Nat Protocols. 2007;2:1573–1584. doi: 10.1038/nprot.2007.225. [DOI] [PubMed] [Google Scholar]
- 37.Yount JS, et al. Palmitoylome profiling reveals S-palmitoylation–dependent antiviral activity of IFITM3. Nat Chem Biol. 2010;6:610–614. doi: 10.1038/nchembio.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol. 2011;12:104–117. doi: 10.1038/nrm3048. [DOI] [PubMed] [Google Scholar]
- 39.Wu RF, Ma Z, Liu Z, Terada LS. Nox4-Derived H2O2 Mediates Endoplasmic Reticulum Signaling through Local Ras Activation. Molecular and Cellular Biology. 2010;30:3553–3568. doi: 10.1128/MCB.01445-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choudhary S, Wang KKA, Wang HCR. Oncogenic H-Ras, FK228, and exogenous H2O2 cooperatively activated the erk pathway in selective induction of human urinary bladder cancer j82 cell death. Molecular Carcinogenesis. 2011;50:215–219. doi: 10.1002/mc.20708. [DOI] [PubMed] [Google Scholar]
- 41.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 42.Kauppinen TM, Suh SW, Berman AE, Hamby AM, Swanson RA. Inhibition of poly(ADP-ribose) polymerase suppresses inflammation and promotes recovery after ischemic injury. J Cereb Blood Flow Metab. 2009;29:820–829. doi: 10.1038/jcbfm.2009.9. [DOI] [PubMed] [Google Scholar]
- 43.West RE, Moss J, Vaughan M, Liu T, Liu TY. Pertussis toxin-catalyzed ADP-ribosylation of transducin. Cysteine 347 is the ADP-ribose acceptor site. Journal of Biological Chemistry. 1985;260:14428–14430. [PubMed] [Google Scholar]
- 44.Bivona TG, et al. PKC Regulates a Farnesyl-Electrostatic Switch on K-Ras that Promotes its Association with Bcl-Xl on Mitochondria and Induces Apoptosis. Molecular Cell. 2006;21:481–493. doi: 10.1016/j.molcel.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 45.Ahearn IM, et al. FKBP12 Binds to Acylated H-Ras and Promotes Depalmitoylation. Molecular Cell. 2011;41:173–185. doi: 10.1016/j.molcel.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goodwin JS, et al. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. The Journal of Cell Biology. 2005;170:261–272. doi: 10.1083/jcb.200502063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burgoyne JR, et al. Oxidation of HRas cysteine thiols by metabolic stress prevents palmitoylation in vivo and contributes to endothelial cell apoptosis. The FASEB Journal. 2012;26:832–841. doi: 10.1096/fj.11-189415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evangelista AM, Kohr MJ, Murphy E. S-Nitrosylation: Specificity, Occupancy, and Interaction with Other Post-Translational Modifications. Antioxidants & Redox Signaling. 2013;19:1209–1219. doi: 10.1089/ars.2012.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho GPH, et al. S-Nitrosylation and S-Palmitoylation Reciprocally Regulate Synaptic Targeting of PSD-95. Neuron. 2011;71:131–141. doi: 10.1016/j.neuron.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leonard SE, Reddie KG, Carroll KS. Mining the Thiol Proteome for Sulfenic Acid Modifications Reveals New Targets for Oxidation in Cells. ACS Chemical Biology. 2009;4:783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- 51.Nguyen T, Francis MB. Practical Synthetic Route to Functionalized Rhodamine Dyes. Organic Letters. 2003;5:3245–3248. doi: 10.1021/ol035135z. [DOI] [PubMed] [Google Scholar]
- 52.Grammel M, Luong P, Orth K, Hang HC. A Chemical Reporter for Protein AMPylation. Journal of the American Chemical Society. 2011;133:17103–17105. doi: 10.1021/ja205137d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.