

Abstract
Epoxyeicosatrienoic acids (EETs) are synthesized in astrocytes, and inhibitors of soluble epoxide hydrolase (sEH), which hydrolyzes EETs, reduce infarct volume in ischemic stroke. Astrocytes can release protective neurotrophic factors, such as vascular endothelial growth factor (VEGF). We found that addition of sEH inhibitors to rat cultured astrocytes immediately after oxygen-glucose deprivation (OGD) markedly increased VEGF concentration in the medium 48 h later and the effect was blocked by an EET antagonist. The sEH inhibitors increased EET concentrations to levels capable of increasing VEGF. When the sEH inhibitors were removed from the medium at 48 h, the increase in VEGF persisted for an additional 48 h. Neurons exposed to OGD and subsequently to astrocyte medium previously conditioned with OGD plus sEH inhibitors showed increased phosphorylation of their VEGF receptor-2, less TUNEL staining, and increased phosphorylation of Akt, which was blocked by a VEGF receptor-2 antagonist. Our findings indicate that sEH inhibitors, applied to cultured astrocytes after an ischemia-like insult, can increase VEGF secretion. The released VEGF then enhances Akt-enabled cell survival signaling in neurons through activation of VEGF receptor-2 leading to less neuronal cell death. These results suggest a new strategy by which astrocytes can be leveraged to support neuroprotection.
Keywords: Akt, astrocyte, epoxide hydrolase, epoxyeicosatrienoic acid, oxygen-glucose deprivation, vascular endothelial growth factor
Graphical Abstract
Inhibition of soluble epoxide hydrolase after oxygen-glucose deprivation in astrocytes stabilizes epoxyeicosatrienoic acids, which then augment the release of VEGF. Exposure of neurons to oxygen-glucose deprivation followed by the conditioned astrocyte medium increases neuronal phosphorylated Akt through VEGF receptor-2 signaling and leads to increased neuronal survival, thereby providing evidence of one way by which astrocytes can be leveraged for neuronprotection.
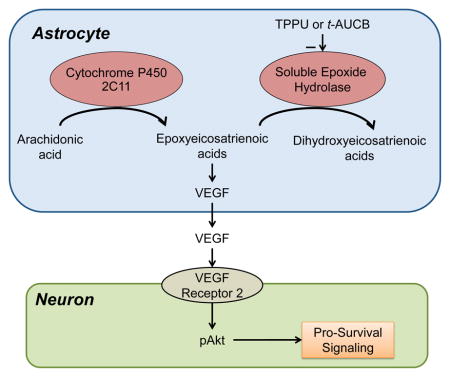
Introduction
Astrocytes protect neurons from stroke by a variety of mechanisms, such as buffering the changes in extracellular ions, importing released glutamate, synthesizing glutathione, and promoting vasodilation (Ouyang et al. 2014). Astrocytes can also serve as a source of trophic factors that protect neurons and promote neurogenesis and angiogenesis (Newton et al. 2013, Oliveira et al. 2013).
Within astrocytes, epoxyeicosatrienoic acids (EETs) derived from the epoxygenation of arachidonic acid have emerged as signaling molecules that facilitate openings of TRPV4 and calcium-activated potassium (KCa) channels (Dunn et al. 2013, Higashimori et al. 2010, Yamaura et al. 2006, Gebremedhin et al. 2003). Exposure of cultured astrocytes to hypoxia or glutamate increases the synthesis and release of EETs (Yamaura et al. 2006, Nithipatikom et al. 2001), suggesting that they may be functionally important under conditions of ischemia. EETs are hydrolyzed by soluble epoxide hydrolase (sEH) to corresponding 1,2-dihydroxyeicosatrienoic acids (DHETs) (Morisseau & Hammock 2013). In vivo, inhibition of sEH or gene deletion of sEH reduces infarct volume after transient middle cerebral artery occlusion (Shaik et al. 2013, Zhang et al. 2008). In these studies, sEH null mice have less severe reduction in intraischemic cerebral blood flow, but other mechanisms are also likely to contribute to the reduction in infarct volume. Administration of an sEH inhibitor at the start of reperfusion following transient focal ischemia also reduces infarct volume (Zhang et al. 2007), suggesting that EETs play a protective role after ischemia.
In this study, in order to identify other neuroprotective mechanisms that are independent of blood flow, we exposed primary astrocyte cultures to oxygen-glucose deprivation (OGD) and then treated them with sEH inhibitors after reoxygenation. We focused on administration after reoxygenation because treatment after reperfusion is more clinically relevant, and oxygen-dependent formation of epoxides is more likely to occur after reoxygenation. We also investigated the effect of treating OGD-exposed primary neuronal cultures with medium from astrocytes previously conditioned with OGD and sEH inhibitors. We focused on astrocyte release of vascular endothelial growth factor (VEGF) because astrocytes release VEGF under hypoxic conditions (Sinor et al. 1998, Schmid-Brunclik et al. 2008), sEH inhibitors can promote VEGF release in other tissue (Panigrahy et al. 2013) and VEGF can exert pro-survival effects in neurons and may promote reparative mechanisms through its angiogenic effects (Sanchez et al. 2010, Shibuya 2009, Li et al. 2012). Two main hypotheses were tested. First, administration of sEH inhibitors to astrocytes after OGD increases the release of VEGF into the medium by a mechanism that requires the action of EETs. Second, medium derived from astrocytes that are treated with sEH inhibitors after OGD augments the pro-survival phosphorylation of Akt in OGD-exposed neurons. We also determined whether this augmentation requires activation of neuronal VEGF receptor-2 (VEGFR2), the primary receptor mediating neuronal protection by VEGF (Hao & Rockwell 2013). Throughout the studies, two structurally distinct sEH inhibitors were used:
1-(1-Propanoylpiperidin-4-yl)-3-[4-(trifluoromethoxy)phenyl]urea (TPPU) (Rose et al. 2010, Ulu et al. 2012) and trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB) (Hwang et al. 2007).
Materials and Methods
Animals
Timed-pregnant Sprague-Dawley rats (14 to 15 days of gestation) were purchased from Charles River (Wilmington, MA, USA) and housed at the Johns Hopkins University animal facilities. Primary cultured astrocytes were prepared from 1-day postnatal rat pups, and neurons were prepared from E15 rat embryos. All studies were performed in accordance with National Institutes of Health Guidelines for the Care and Use of Laboratory Animals, and protocols were approved by the Johns Hopkins University Animal Care and Use Committee.
Chemicals
The sEH inhibitors TPPU and t-AUCB were synthesized according to published procedures (Hwang et al. 2007, Rose et al. 2010). Both agents were dissolved in dimethyl sulfoxide (DMSO that was also used as vehicle control) as 10 mM stock solutions. They were used at final concentrations of 0.1, 1, 3, 10, 30, and 100 μM. The EET antagonist 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE) and authentic 14,15-EET were purchased from Cayman Chemical Company (Ann Arbor, MI), dissolved in DMSO, and used at a final concentration of 100 μM and 30 μM, respectively.
Astrocyte cultures
Primary cultures of astrocytes were generated as described (McCarthy & de Vellis 1980). Briefly, cerebral cortices were minced and dissociated with 0.25 mg/mL trypsin-EDTA for 15 min at 37°C and then rinsed with Dulbecco’s Modified Eagle Medium (DMEM)/Nutrient Mixture F-12 containing 10% fetal bovine serum (FBS; Invitrogen, Grand Island, NY, USA). Tissue pieces were triturated by a pipette and then passed through a 40-μm mesh strainer (BD Falcon, Franklin Lakes, NJ, USA). Isolated cells were then suspended in DMEM/F12 with 10% FBS and 1% penicillin/streptomycin, plated on 10 μg/mL poly-D-lysine–coated 75 cm2 flasks (Corning, Corning, NY, USA) at a density of 1×106 cells/cm2, and incubated at 37°C in a 95%/5% mixture of atmospheric air and CO2. At day in vitro (DIV) 8, glial cultures were shaken for 18 h at 260 rpm in a temperature-controlled incubator to dislodge oligodendrocytes and microglial cells. The remaining cells were maintained for an additional 3 days, detached with 0.25 mg/mL trypsin-EDTA, and seeded in 24-well plates. The cultured astrocytes were used for experiments at DIV 15. Immunocytochemical staining of the cell cultures with an antibody specific to glial fibrillary acidic protein (GFAP) showed that >95% of cells were GFAP-positive astrocytes. The density of confluent astrocytes was 1×105 cells/cm2.
The number of dead astrocytes was determined by staining with propidium iodide (PI, Invitrogen, Waltham, MA, USA), which enters the nuclei of dead cells and co-localizes with the nuclear stain, DAPI. Astrocytes were fixed with 4% paraformaldehyde for 30 min at room temperature after which the cells were covered with methanol for 5 min and immunostained for GFAP. Then they were washed and equilibrated briefly in 2X SSC (0.3 M NaCl, 0.03 M sodium citrate, pH 7.0) and incubated in 100 μg/mL DNase-free RNase in 2X SSC for 20 minutes at 37°C. After rinsing three times for 1 min in 2X SSC, the cells were incubate with 1uM PI at room temperature for 10 min. The working solution of 1uM PI was made by diluting the 1mg/ml (1.5 mM) stock solution 1:1500 in 2X SSC. Finally, the cells were washed three times with 2X SSC, and then observed under a fluorescence microscope.
To test the effect of sEH inhibitors on VEGF expression and secretion, we allocated primary astrocytes into three groups: normoxia (without OGD or sEH inhibitor treatment), ODG plus DMSO vehicle (OGD and then 0.1% DMSO treatment), and OGD plus sEH inhibitor (OGD and then sEH inhibitor treatment). Because induction of VEGF after ischemia is unlikely to be immediate, we extended the inhibitor treatment for 48 h after OGD to assure that the inhibitor was present during the period of potential induction.
Neuronal cultures
Primary neuronal cultures were prepared from cerebral cortices of E15 Sprague-Dawley rat embryos. Following the same dissociation protocol described for astrocyte cultures, we plated cells at a low density (1×105 cells/cm2) on poly-D-lysine–coated 12 mm coverslips for immunocytochemistry, and at a higher density (5×105 cells/cm2) in coated 6-well plates for Western blot analysis. The cells were cultured initially in DMEM/F12 containing 10% FBS. After 4 h, the medium was replaced with Neurobasal medium supplemented with B27 (NB/B27), L-glutamine, and 1% penicillin/streptomycin (Invitrogen). At DIV 4, one-half of the medium in each well was replaced, and 2 μM cytosine arabinoside was added to prevent non-neuronal proliferation. Experiments were performed on cells at DIV 8. The cultures contained >95% neurons as demonstrated by immunocytochemical staining for microtubule associated protein 2 (MAP2).
Oxygen-glucose deprivation
The OGD model was performed as previously described (Huang et al. 2008). Briefly, cells were washed twice, incubated in glucose-free DMEM (Invitrogen), and then placed in a hypoxic incubator filled with a gas mixture of 95% N2 and 5% CO2 at 37°C for the designated period (6 h for astrocytes, 1 h for neurons). After OGD, cultures were returned to standard medium and reoxygenated in a normoxic incubator with 5% CO2/95% air.
Drug administration
Vehicle (0.1% DMSO), TPPU, or t-AUCB was added to astrocytes in oxygenated, glucose-containing DMEM/F12 immediately after 6 h of OGD. TPPU and t-AUCB were used at final concentrations of 0.1, 1, 3, 10, 30, and 100 μM. The cells were left in culture for an additional 48 h, and then both cells and medium were processed for VEGF ELISA assay as described below. To test the role of EETs in TPPU/t-AUCB-induced VEGF secretion, we added the EET antagonist 14,15-EEZE (100 μM) in combination with TPPU or t-AUCB (100 μM) or authentic 14,15-EET (0.1–30 μM) during the 48 h of reoxygenation.
Liquid chromatographic-mass spectrometric (LC-MS) measurements of EETs
To determine changes in the production levels of CYP epoxygenase-derived EETs, cultured astrocytes were treated with 40 μM arachidonic acid in potassium phosphate buffer (0.1 M, pH 7.4) containing the NADPH-generating system (20 mM isocitrate and 0.1 units/ml isocitrate dehydrogenase), 1 mM EDTA, 10 mM MgCl2. The normoxia group was incubated with media containing 5.6 mM glucose for the entire course of the experiment, whereas the remaining groups underwent 6 hours of OGD. Following OGD, the media was replaced with culture media containing 5.6 mM glucose and either 0.1% DMSO or the sEH inhibitors TPPU (100 μM) and t-AUCB (100 μM). Following 24 h of reoxygenation, the media was collected and kept on ice. The samples were spiked with 2 ng deuterated 2H8-EETs as an internal standard. The samples were acidified to pH 3.5 and extracted twice with 3 ml of diethyl ether with gentle vortexing for 1 min and then centrifuged at 5000 g for 5 min. The diethyl ether layer was then transferred to a clean 10 ml glass centrifuge tube and dried under a flow of nitrogen gas. The dried samples were then reconstituted in 200 μl of acetonitrile for analysis by LC-MS in the negative ion mode. For quantitative measurements, the m/z 319 and 327 ions were used for EETs and [2H8] EETs, respectively. The standard curves were typically constructed over the range of 1–100 pg per injection. The concentrations of the four regioisomers of EETs were calculated by comparing the ratios of their peak areas to standard curves as previously described (Nithipatikom et al. 2001) and expressed as concentration per unit volume of astrocyte media.
Co-culture
For co-culture experiments, primary astrocytes were seeded at a density of 1×105 cells/cm2 on filter membrane (0.4 μm pores) tissue culture inserts in 24- and 6-well plates (Corning). These astrocytes were exposed to 6-h OGD and reoxygenated for 48 h with or without sEH inhibitor TPPU/t-AUCB (100 μM). After the OGD/reoxygenation period, the astrocyte medium was replaced with NB/B27 medium for an additional 24 h to generate conditioned medium compatible with the neuronal medium. The conditioned medium was processed for VEGF ELISA assay or co-culture as described below. Primary neurons were seeded on 12-mm coverslips in 24-well and 6-well plates as described above; at DIV 8, neurons were subjected to 1 h OGD. At the time of reoxygenation, the neuron-covered coverslips were placed under the inserts holding OGD/reoxygenation-stimulated astrocytes that had been grown for 24 h in NB/B27. The neurons were co-cultured with astrocytes on the transwell membranes for 24 h, after which cell lysates of neurons in the 6-well plates were collected for Western blot analysis, and neurons on the coverslips were fixed and processed for assessment of neuronal cell death as described below.
ELISA
Culture medium of astrocytes was collected after treatment, and cells were lysed in RIPA lysis buffer (Thermo Scientific, Waltham, MA, USA). VEGF concentrations in the cell lysate and culture medium were measured in duplicate with VEGF ELISA kits (ab100786; ab100787, Abcam, Cambridge, UK) according to the manufacturer’s instructions, and levels were detected with a microplate reader at a wavelength of 450 nm. Measurements of VEGF concentrations in the astrocyte medium included: 1) following 48-h exposure to 100 μM TPPU or t-AUCB in normoxic astrocytes not subjected to OGD, 2) after OGD and 48-h exposure to vehicle or 0.1, 1, 3, 10, 30, and 100 μM TPPU or t-AUCB, 3) after OGD and 48-h exposure to vehicle, 100 μM TPPU, 100 μM t-AUCB, or 100 μM 14,15-EEZE plus TPPU or t-AUCB, 4) in response to addition of 0.1, 1, 10, and 30 μM 14,15-EET to the astrocyte medium for 48 h after OGD, 5) evaluation of the time course of VEGF over 48 h after switching the astrocyte medium to NB/B27 following previous treatment with OGD and either vehicle or 100 μM TPPU, and 6) after switching the astrocyte medium to NB/B27 following previous treatment with OGD and vehicle, 100 μM TPPU, or 100 μM t-AUCB.
In situ labeling of fragmented DNA
Terminal deoxynucleotidyl transferase (TdT)–mediated dUTP Nick-End labeling (TUNEL) staining was performed according to the manufacturer’s instructions (In situ cell death detection kit, Roche, Mannheim, Germany). Briefly, neurons grown on coverslips and subjected to OGD as described above were fixed for 30 min in 4% paraformaldehyde and blocked with 3% H2O2 in methanol. After three washes in phosphate-buffered saline (PBS), the cells were permeabilized with 0.1% Triton X-100 for 2 min on ice, and then incubated for 60 min in a 37°C humidified chamber with TUNEL reaction buffer containing TdT enzyme and fluorescein-labeled nucleotide mixture. The reaction was terminated in PBS. In the second step, the cells were incubated with antibody against MAP2 (ab11267, 1:200, Abcam) at 4°C overnight and then with Alexa Fluor 568-conjugated goat anti-mouse IgG antibody (A-11004, 1:400, Invitrogen) for 1 h at room temperature. Coverslips were washed 3×5 min with PBS between each step. Coverslips were stained with DAPI and mounted on glass microscope slides (Vector Laboratories, Burlingame, CA, USA). Fluorescence microscopy was performed and the number of apoptotic neurons was determined by counting the MAP2/TUNEL double-stained cells in three different fields of view on each coverslip.
Western blot analysis
Neurons grown on 6-well plates and subjected to treatment described above were lysed in RIPA lysis buffer (Thermo Scientific). Protein concentrations were determined with a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific) according to the manufacturer’s instructions. Equal amounts of each protein sample (total protein extract, 30 μg) were separated by electrophoresis in 4–15% SDS-polyacrylamide gels (Mini-PROTEAN TGX precast gel, Bio-Rad, Hercules, CA, USA) and transferred to polyvinylidene fluoride membranes. After being blocked, membranes were incubated overnight at 4°C with primary antibodies against phosphorylated VEGFR-2 (Tyr 996; sc-16629-R, 1:200, Santa Cruz Biotechnology, Dallas, TX, USA), total forms of VEGFR-2 (ab131441, 1:300, Abcam), and β-actin (4967, 1:500, Cell Signaling Technology, Danvers, MA, USA). The membranes were then incubated with horseradish peroxidase-conjugated secondary antibody (1:10000, Cell Signaling Technology) for 1 h at room temperature. The blots were developed in enhanced chemiluminescence reagents (Thermo Scientific) and exposed to X-ray film. Signals were quantified with Quantity One software 4.62 (Bio-Rad).
Statistical analysis
The number of wells with each treatment was balanced within each cell culture experiment. The person performing cell counts was blinded to treatment, whereas the person performing biochemical assays was not blinded to treatment. Work by others indicated a coefficient of variation of approximately 10% in VEGF levels measured by ELISA in astrocyte media after anoxia (Schmid-Brunclik et al. 2008). Based on this coefficient of variation, we chose a sample size of 4 independent experiments to provide 80% power for detecting 30% relative changes in VEGF with P<0.05.
All data were analyzed by analysis of variance. If the F-value was significant at the 0.05 level, individual mean values were compared with the Holm-Sidak procedure for multiple comparisons. The primary comparisons were to a normoxic control group and to an OGD plus vehicle group. In cases where tests for normality and homogeneity of variance failed, a logarithmic transformation was applied to the data, which then passed these tests. Data are presented as means ± SD.
Results
Effect of sEH inhibitor on astrocyte viability after OGD
Triple-labeling with GFAP, propidium iodide (PI), and DAPI was used to identify the effect of sEH inhibitors on astrocyte viability after OGD (Fig. 1). Compared with viability in the normoxia group, OGD with vehicle caused a significant reduction in viable astrocytes (95.5 ± 0.3% vs. 71.9 ± 2.5%, P<0.01). Administration of either TPPU or t-AUCB (100 μM) after OGD led to significant improvements in cell viability (86.3 ± 1.8% and 87.3 ± 3.4%, respectively), thereby indicating that sEH inhibitors have a protective effect against OGD in astrocytes (Fig. 1).
Fig. 1.

Effect of sEH inhibitors on astrocyte viability after oxygen-glucose deprivation (OGD). Astrocytes were exposed to normoxia (A) or 6-h OGD followed by 48-h reoxygenation with DMSO (vehicle; B), 100 μM TPPU (C), or 100 μM t-AUCB (D). Astrocytes were labeled with GFAP (green), propidium iodide (red), and DAPI (blue). (E) The percentage of viable astrocytes was calculated from the percentage of DAPI-stained nuclei that were not co-stained with propidium iodide. Three different fields of view on each coverslip were counted and averaged. Data are shown as mean ± SD; n = 3 independent experiments. *P<0.05 vs. normoxia; #P<0.05 vs. OGD + vehicle. Scale bars = 50 μm on images obtained with a 20× lens.
Inhibitors of sEH help restore astrocyte release of EETs after OGD
The concentration of the four regioisomers of EETs was measured in the astrocyte medium 24 h after OGD. Relative to normoxic controls, OGD reduced the levels of all four EET regioisomers by two orders of magnitude (Fig. 2). Addition of TPPU or t-AUCB largely preserved the levels of the regioisomers after OGD, although the levels remained significantly less than those in the normoxic controls for 5,6-EET, 8,9-EET and 11,12-EET. Thus, the sEH inhibitors achieved the intended effect of increasing EETs after OGD.
Fig. 2.

Concentration of the four EET regioisomers in the astrocyte medium 24 h after normoxia or oxygen-glucose deprivation (OGD) and subsequent treatment with vehicle or 100 μM TPPU or t-AUCB. Values are plotted on a log scale as mean ± SD; n = 5 independent experiments. *P<0.05 vs. normoxia; #P<0.05 vs. OGD + vehicle.
Inhibitors of sEH increase astrocyte VEGF secretion and expression after OGD
ELISA analysis showed that OGD with vehicle led to a small, but significant, 16% increase in VEGF secreted from primary astrocytes into the media (Fig. 3A) compared with levels in the normoxia group. Administration of TPPU or t-AUCB after OGD significantly and dose-dependently increased astrocyte VEGF secretion into the media and cell protein expression (Fig. 3A, B). For example, 1 μM TPPU increased VEGF in the medium by 50%, whereas 100 μM increased VEGF by 300% (Fig. 3A). In the absence of OGD, 100 μM TPPU and t-AUCB increased VEGF in the media by 33% and 25%, respectively. These increases were significantly less than the corresponding levels attained with treatment after OGD. Thus, the sEH inhibitors exert a synergistic effect on VEGF secretion when combined with OGD and reoxygenation.
Fig. 3.

sEH inhibitor and EET increase astrocyte vascular endothelial growth factor (VEGF) secretion after oxygen-glucose deprivation (OGD). (A) ELISA analysis of VEGF secreted into the medium of astrocytes exposed to normoxia plus 48 h of 0.1% DMSO vehicle (V), 100 μM TPPU, or 100 μM t-AUCB or to 6 h OGD plus 48 h reoxygenation with V or different concentrations of TPPU and t-AUCB. (B) VEGF expression in the whole-cell lysates of astrocytes exposed to the same experimental conditions as described in (A). *P<0.05 vs. normoxia; #P<0.05 vs. OGD + vehicle. (C) VEGF concentration in medium of OGD astrocytes that were treated with or without 100 μM of TPPU or t-AUCB alone or in combination with 100 μM 14,15-EEZE during the 48 h of reoxygenation. *P<0.05 vs. OGD + vehicle; #P<0.05 vs. OGD + TPPU; †P<0.01 vs. OGD + t-AUCB. (D) VEGF concentration in medium of OGD astrocytes that were treated for 48 h with 0.1% DMSO vehicle, 0.1, 1, 10, or 30 μM 14,15-EET, 1 μM 14,15-EET plus 30 μM TPPU, or 1 μM 14,15-EET plus 100 μM TPPU. *P<0.05 vs. OGD + vehicle; #P<0.05 vs. OGD + 1 μM 14,15-EET. The data are shown as mean ± SD; n = 4 independent experiments each in A–D.
To determine whether the augmentation of VEGF release by sEH inhibitors required the actions of EETs exclusively, we treated wells of OGD astrocytes with a combination of TPPU (100 μM) or t-AUCB (100 μM) and the EET antagonist 14,15-EEZE (100 μM) during the 48 h of reoxygenation. As in the previous experiments, after 6 h of OGD, treatment with 100 μM of TPPU or t-AUCB for 48 h of reoxygenation led to corresponding increases in secreted VEGF of 260% or 120% when compared with OGD/vehicle-treated astrocytes. These increases were completely suppressed by 14,15-EEZE application (Fig. 3C), thereby indicating that the effect of the inhibitors could be attributed to the actions of EETs.
To evaluate whether administration of exogenous EETs recapitulate the effect of the sEH inhibitors, we added 14,15-EET over a concentration range of 0.1–30 μM to the astrocyte medium after OGD. Concentration-dependent increases were observed over the 1–30 μM range of 14,15-EET (Fig. 3D). Furthermore, addition of 30–100 μM TPPU to the lowest effective 14,15-EET concentration of 1 μM significantly augmented the VEGF response.
Enhanced activation of neuronal VEGF receptor-2 and neuronal survival
Because we found that the sEH inhibitors can augment astrocyte VEGF secretion and expression after OGD, we wanted to investigate whether this augmentation would subsequently protect neurons from oxygen and glucose restoration after OGD. For this reason, we did co-culture experiments. Primary neurons were subjected to 1-h OGD, which is sufficient to produce moderate levels of cell death. These cells were then exposed to conditioned medium from OGD astrocytes that had been treated with vehicle or an sEH inhibitor (100 μM TPPU or t-AUCB). However, for the medium to be compatible with neurons, the astrocytes were transferred to fresh NB/B27 medium for 24 h before exposure of the neurons to the astrocyte medium. Because the replacement medium did not contain sEH inhibitors, we determined whether the increased release of VEGF into the new astrocyte medium would be sustained. As shown in Fig. 4A, the increase in the VEGF concentration 24 h after removal of TPPU or t-AUCB from OGD-treated astrocytes (77.9 ± 10.5 or 47.8 ± 6.8 pg/ml, respectively) was significantly higher than after removal of vehicle from OGD-treated astrocytes (22.5±2.1 pg/ml, P<0.05; Fig. 4A). These values were similar to those obtained in the previous experiments in which the medium was not replaced with NB/B27 medium and the sEH inhibitors were still present. This observation indicates that astrocytes previously exposed to OGD and sEH inhibitor can continue secreting VEGF and at a high concentration despite medium replacement and sEH inhibitor removal. This finding may also indicate that these sEH inhibitors have long enzyme residence times that lengthen their effects (Lee et al. 2014). Because the OGD neurons were exposed to the astrocyte replacement medium for an additional 24 h, we further evaluated the time course of the VEGF concentration after replacing the astrocyte medium with NB/B27. We found that the VEGF concentration remained elevated for at least 48 h after removal of 100 μM TPPU compared to the control group previously treated with vehicle (Fig. 4B). This 48-h time point for astrocytes corresponds to 24 h after the neurons underwent OGD.
Fig. 4.

Increased VEGF persists after removal of sEH inhibitor. (A) VEGF measurements were made 24 h after switching the astrocyte medium from DMEM/F-12 to NB/B27. The switch in medium occurred after astrocytes were exposed to 6-h oxygen-glucose deprivation (OGD) and 48-h reoxygenation with vehicle, 100 μM TPPU, or 100 μM t-AUCB. The data show that VEGF remained elevated 24 h after removal of sEH inhibitors, corresponding to the time at which co-culture with neurons was started. Values are mean ± SD; n = 4 independent experiments; *P<0.01 vs. vehicle-treated OGD astrocytes. (B) Time course of VEGF concentration in the astrocyte medium over 48 h after switching the medium in astrocytes previously exposed to OGD and 48-h reoxygenation with vehicle or 100 μM TPPU. Values are mean ± SD; n = 3 independent experiments. *P<0.05 vs. vehicle-treated OGD + vehicle. Note that VEGF remained elevated at 48 h, corresponding to the time when co-culture with neurons ended.
Activation of VEGFR-2 is associated with phosphorylation at Tyr 996. Western blotting with an antibody selective for Tyr 996 showed that phosphorylated VEGFR-2 was significantly increased 24 h after OGD in neurons that were co-cultured with conditioned medium from TPPU- or t-AUCB-treated OGD astrocytes compared with those co-cultured with conditioned medium from vehicle-treated OGD astrocytes (Fig. 5).
Fig. 5.

Co-culturing neurons immediately after oxygen-glucose deprivation (OGD) with astrocytes that have been stimulated by OGD and an sEH inhibitor increases phosphorylation of vascular endothelial growth factor (VEGF) receptor 2 (VEGFR-2) in the neurons. Neurons were subjected to 1-h OGD and then cultured for 24 h in NB/B27 medium alone or co-cultured with the astrocytes subjected to 6-h OGD, 48-h rexoygenation with vehicle or 100 μM TPPU or t-AUCB, and then 24-h in NB/B27 medium without sEH inhibitors. Neuronal lysates in each condition were analyzed by Western blot for the levels of phosphorylated (p-) or total VEGFR-2. β-actin served as the loading control. The levels of phosphorylation were normalized to the total protein. Values are mean ± SD; n = 4 independent experiments. *P<0.01 vs. OGD neurons co-cultured with vehicle-treated OGD astrocytes.
Neuronal cell death was assessed by TUNEL staining of DAPI-demarcated nuclei (Fig. 6). As expected, compared to untreated, normoxic controls, the number of TUNEL-positive neurons increased when OGD neurons were co-cultured with astrocytes previously treated with vehicle after OGD. The increase in TUNEL-positive neurons was attenuated when the astrocytes had been treated with TPPU or t-AUCB after OGD (Fig. 6). MAP2 staining also suggested that treatment with the sEH inhibitors reduced swelling of the cell body and maintained fine processes of neurons (Fig. 6).
Fig. 6.

Co-culturing neurons after oxygen-glucose deprivation (OGD) with sEH inhibitor-treated OGD astrocytes decreases TUNEL-positive neurons and preserves MAP2-positive processes. Neurons were exposed to normoxia (A) or 1-h OGD followed by co-culture with OGD astrocytes that had been treated with DMSO (vehicle; B), 100 μM TPPU (C), or 100 μM t-AUCB (D) at reoxygenation. Neurons were labeled with MAP2 (red), TUNEL (green), and DAPI (blue). (E) The percentage of TUNEL-positive cells with DAPI-stained nuclei was taken as a measure of cell death. Three different fields of view on each coverslip were counted and averaged. The bar graph shows the percentage of neurons that were not TUNEL-positive (mean ± SD; n = 3 independent experiments). *P<0.05 vs. normoxia; #P<0.05 vs. OGD neurons co-cultured with vehicle-treated OGD astrocytes. Scale bars = 50 μm on images obtained with a 20× lens.
Phosphorylation of Akt
Because VEGFR-2 activation can increase phosphorylation of Akt, which promotes neuronal survival (Hao & Rockwell 2013), we compared the Akt phosphorylation state after exposure of neurons to OGD followed by astrocyte conditioned media. As shown in Fig. 7, the phosphorylation of Akt (p-Akt) in OGD neurons was significantly greater when the neurons were co-cultured with TPPU- or t-AUCB-treated OGD astrocytes than when co-cultured with vehicle-treated OGD astrocytes. This increase was blocked by treatment of the neurons with the VEGFR-2 antagonist SU1498. These results suggest that the neuronal pro-survival signaling of astrocyte medium previously conditioned with TPPU or t-AUCB requires VEGFR-2 activation.
Fig. 7.

TPPU/t-AUCB signaling through vascular endothelial growth factor (VEGF)/VEGF receptor-2 (VEGFR-2) leads to activation of the Akt pathway. Neurons were subjected to 1-h oxygen-glucose deprivation (OGD) and then cultured in NB/B27 alone or co-cultured with astrocytes previously stimulated with OGD plus vehicle (0.1% DMSO) or 100 μM TPPU or t-AUCB. During the 24 h of co-culture, some neurons were also treated with the VEGFR-2 antagonist SU1498 (10 μM). Neuronal lysates in each condition were analyzed by Western blotting for detection of phosphorylated Akt (p-Akt) and total Akt. β-actin served as the loading control. Levels of p-Akt were normalized to total Akt and then quantified relative to the OGD neurons without astrocyte co-culture (100%). Data are shown as mean ± SD; n = 4 independent experiments. *P<0.01 vs. OGD neurons co-cultured with vehicle-treated OGD astrocytes; #P<0.05 vs. OGD neurons co-cultured with TPPU-treated astrocytes; †P<0.05 vs. OGD neurons co-cultured with t-AUCB-treated astrocytes.
Discussion
The major findings of this study are that 1) inhibition of sEH in astrocytes after 6-h OGD increases survival of astrocytes in association with an increase in EETs levels, 2) inhibition of sEH in astrocytes after OGD produces a sustained release of VEGF, 3) this increase is blocked by an EET antagonist and recapitulated by administration of exogenous EETs, 4) medium from astrocyte culture treated with OGD and sEH inhibition augments pro-survival p-Akt signaling in neurons after OGD by a mechanism that requires VEGF receptor-2 activation, and 5) astrocyte medium conditioned by OGD and sEH inhibition improves survival of neurons subjected to OGD.
Inhibition of sEH is known to reduce infarct volume in experimental stroke (Shaik et al. 2013, Zhang et al. 2007). Astrocytes, which are more resistant than neurons to ischemic injury, may help support the viability of neurons. Application of EETs to cultured astrocytes has been reported to improve survival from OGD (Li et al. 2012, Yuan et al. 2015) and to protect astrocyte mitochondria from β-amyloid toxicity (Sarkar et al. 2014). We used 6 h of OGD to produce a stress from which the majority of astrocytes could survive. In this condition, the sEH inhibitor provided substantial reduction in astrocyte cell death. This observation supports the concept that EETs have a protective effect in astrocytes.
Previous work from our group showed that the EET antagonist 14,15-EEZE reduces acute hypoxia-induced cerebral vasodilation in vivo (Liu et al. 2015) and that hypoxia without glucose deprivation increases the formation of EETs by cultured astrocytes (Yamaura et al. 2006). Thus, we anticipated that OGD would increase the level of EETs. In contrast, we observed a profound reduction of EETs 24 h after OGD. Possible explanations for the low EET levels include the greater insult associated with combining hypoxia with glucose deprivation and with the relatively long 6-h duration of the OGD. Nevertheless, administration of TPPU or t-AUCB after OGD nearly restored the levels of all EET regioisomers. This restoration is presumably attributable to inhibition of the hydrolysis of EETs. Because EETs can open calcium-sensitive K+ channels in astrocytes (Yamaura et al. 2006) and protect astrocytes (Li et al. 2012), improved physiological function of astrocytes afforded by EETs may have helped preserve the astrocyte synthetic capacity for EETs and thereby further augment the restoration of EETs after OGD. Because we used both TPPU and t-AUCB in all experiments and obtained similar results, it is less likely that the effects of the chemicals are due to nonspecific effects but more likely that they are acting as sEH inhibitors.
Astrocytes are a source of several neurotrophic factors. A recent report found that addition of 14,15-EET to astrocytes after OGD increased BDNF mRNA approximately 20% in association with an increase in viable astrocytes; glial cell line-derived neurotrophic factor, nerve growth factor, and neurotrophin-4/5 mRNA were unaffected by 14-15-EET (Yuan et al. 2015). Thus, EETs exert some degree of specificity in regulating neurotrophic factors in astrocytes. However, this study did not examine VEGF. We focused on astrocyte release of VEGF because astrocytes release VEGF under hypoxic conditions (Sinor et al. 1998, Schmid-Brunclik et al. 2008), sEH inhibitors can promote VEGF release in other tissue (Panigrahy et al. 2013, Sander et al. 2011, Xu et al. 2013), and VEGF can exert pro-survival effects in neurons (Hao & Rockwell 2013, Sanchez et al. 2010). Here, we found that administration of sEH inhibitors without exposure to OGD produced 25–33% increases in VEGF in the astrocyte medium. These results indicate that the action of EETs on VEGF expression does not require hypoxia. However, because the VEGF gene has a hypoxic response element, we expected that VEGF would be substantially upregulated after OGD. Twenty-four hours after OGD, we detected only a modest 16% increase in VEGF protein in the cell lysate and medium of vehicle-treated astrocytes. Importantly, addition of TPPU after OGD markedly amplified this increase in VEGF as high as three-fold, and addition of t-AUCB increased VEGF by as much as two-fold. Interestingly, the magnitude of this effect on VEGF is greater than the approximately 50% augmentation of BDNF protein by 14,15-EET administration reported by others (Yuan et al. 2015). Because the magnitude of this amplification is much greater than the relative increase in astrocyte cell survival, this amplification likely reflects a strong synergistic interaction of epoxides with hypoxia on VEGF expression, rather than simply an increase in the number of surviving astrocytes. The mechanism of this interaction remains to be determined. Induction of VEGF through a peroxisome-proliferator-activated receptor-γ coactivator-1α mechanism that is independent of hypoxia inducible factors has been described (Arany et al. 2008). Whether EETs act through hypoxia inducible factors or independent of them in astrocytes remains to be determined. Nevertheless, the effect of TPPU and t-AUCB on VEGF secretion into the medium persisted 48 h after removal of the sEH inhibitors. This persistence suggests an effect on VEGF gene induction rather than an effect on VEGF transport or metabolism. The persistence effect may also be due to the fact that the inhibitors used in this study have long enzyme residence times, which could prolong their biological effect (Lee et al. 2014).
Several epoxides of long-chain fatty acids are hydrolyzed by sEH. The observations that the EET antagonist 14,15-EEZE blocked the augmentation of VEGF secretion by TPPU and t-AUCB suggest that the action of EETs, rather than other epoxides, is primarily responsible for this effect of sEH inhibition. Further support for the role of EETs is derived from the VEGF response data to different concentrations of 14,15-EET. A concentration of 0.1 μM 14,15-EET, which exceeds the concentration measured after OGD with vehicle treatment (Fig. 2), failed to increase VEGF (Fig. 3D). In contrast, concentrations in the range of 1–10 μM, which were achieved after OGD with TPPU and t-AUCB treatment, significantly increased VEGF (Fig. 3D) and further increases could be obtained by combining 1 μM 14,15-EET with 30 or 100 μM TPPU (Fig. 3D). Thus, the concentration of EETs measured with the different treatments is internally consistent with the concentration of EETs required to increase VEGF release.
In the transwell experiments, neurons were removed from OGD and, immediately upon reoxygenation, were cultured with the conditioned astrocyte medium. Exposure of the neurons to the medium from astrocytes treated with either of the sEH inhibitors increased neuronal VEGFR-2 phosphorylation and Akt phosphorylation and decreased neuronal TUNEL staining. The increased VEGFR-2 phosphorylation reflects increased receptor activation and indicates that the VEGF released by astrocytes is sufficient to activate neuronal receptors despite dilution by the relatively large extracellular space in cell culture. Phosphorylation of Akt promotes anti-apoptotic signaling, and the decrease in TUNEL signaling indicates decreased neuronal cell death. The increase in Akt phosphorylation was blocked by addition of the VEGFR-2 antagonist SU1498 to the neuronal medium after OGD. Thus, the beneficial effect of the sEH inhibitor–conditioned astrocyte medium on this pro-survival pathway in neurons appears to require activation of neuronal VEGFR-2.
A limitation of this study is that we did not investigate whether the VEGFR-2 antagonist completely blocked the beneficial effect on TUNEL staining of the astrocyte medium conditioned with sEH inhibitors. Although we did not examine whether VEGFR-1 also contributes to this neuronal protection, this possibility seems unlikely because VEGFR-2 is the primary receptor required for VEGF’s anti-apoptotic effect in neurons (Hao & Rockwell 2013). It should also be noted that we used different durations of OGD for neurons and astrocytes because of their differing vulnerability. When the neurons were reoxygenated and immediately exposed to the astrocyte conditioned medium, three days had elapsed since the astrocytes had been exposed to OGD. Our intention was to allow time for the astrocyte VEGF secretion to occur and then to wash out the sEH inhibitor so that it would not have a direct effect on neurons. We reasoned that an early intervention to manipulate EETs could have a prolonged effect on trophic factor release that would ameliorate secondary neuronal death and possibly promote neurorepair.
Despite these artificial conditions, these experiments provide a proof of concept for one of the potential mechanisms by which astrocytes can protect neurons after reoxygenation from ischemia. Astrocytes can release growth factors other than VEGF and some of these may also provide acute neuroprotection or help to restore function of the remaining neurons after an ischemic event. For example, 14,15-EET can promote expression of BDNF after OGD, and interfering with astrocyte BDNF-TrkB signaling attenuated the 14,15-EET protection of astrocytes and neurons (Yuan et al. 2015). Thus, there might be convergence of BDNF and VEGF signaling in promoting neuronal survival. In addition, our experiments do not exclude the possibility that, in vivo, EETs released from astrocytes might act directly on neurons to provide protection. Hence, other EET-dependent or -independent neuroprotective mechanisms may exist in astrocytes besides those demonstrated for astrocyte VEGF release in the present model. Therefore, promoting EET bioavailability through inhibition of sEH could be one strategy for leveraging astrocyte-based neuroprotection and possibly neurorepair.
Acknowledgments
The authors thank Claire F. Levine for her editorial assistance.
Y.Z. was supported by a Chinese Scholarship Council student fellowship grant and a grant from the National Natural Science Foundation of China (No. 81600965). R.C.K. was supported by NIH grants NS060703 and NS038684. B.D.H. and K.S.S.L. were supported by NIEHS grant R01 ES002710, Superfund Research Program grant P42 ES004699, and NIH CounterAct U54 NS079202. K.S.S.L. was also supported by NIH/NIEHS Pathway to Independence Award K99 ES024806. D.R.H. was supported by NIH grants HL033833, HL092105, and HL105997 and by a grant from the Veterans Administration Research Career Scientist Award.
Abreviations Used
- BDNF
brain-derived neurotrophic factor
- DHET
1,2-dihydroxyeicosatrienoic acid
- DIV
day in vitro
- DMEM
Dulbecco’s Modified Eagle Medium
- DMSO
dimethyl sulfoxide
- EET
epoxyeicosatrienoic acid
- 14,15-EEZE
14,15-epoxyeicosa-5(Z)-enoic acid
- FBS
fetal bovine serum
- GFAP
glial fibrillary acidic protein
- MAP2
microtubule associated protein 2
- NB/B27
Neurobasal medium supplemented with B27
- OGD
oxygen-glucose deprivation
- PBS
phosphate-buffered saline
- PI
propidium iodide
- sEH
soluble epoxide hydrolase
- SSC
saline-sodium citrate buffer
- t-AUCB
trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid
- TPPU
1-(1-propanoylpiperidin-4-yl)-3-[4-(trifluoromethoxy)phenyl]urea
- TUNEL
terminal deoxynucleotidyl transferase (TdT)–mediated dUTP Nick-End labeling
- VEGF
vascular endothelial growth factor
- VEGFR2
vascular endothelial growth factor receptor-2
Footnotes
Conflicts of Interests
B.D.H. is on University of California patents on sEH inhibitors. Other authors have no conflicts of interest to declare.
References
- Arany Z, Foo SY, Ma Y, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- Dunn KM, Hill-Eubanks DC, Liedtke WB, Nelson MT. TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci U S A. 2013;110:6157–6162. doi: 10.1073/pnas.1216514110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebremedhin D, Yamaura K, Zhang C, Bylund J, Koehler RC, Harder DR. Metabotropic glutamate receptor activation enhances the activities of two types of Ca2+-activated K+ channels in rat hippocampal astrocytes. J Neurosci. 2003;23:1678–1687. doi: 10.1523/JNEUROSCI.23-05-01678.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao T, Rockwell P. Signaling through the vascular endothelial growth factor receptor VEGFR-2 protects hippocampal neurons from mitochondrial dysfunction and oxidative stress. Free Radic Biol Med. 2013;63:421–431. doi: 10.1016/j.freeradbiomed.2013.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashimori H, Blanco VM, Tuniki VR, Falck JR, Filosa JA. Role of epoxyeicosatrienoic acids as autocrine metabolites in glutamate-mediated K+ signaling in perivascular astrocytes. Am J Physiol Cell Physiol. 2010;299:C1068–C1078. doi: 10.1152/ajpcell.00225.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XJ, Zhang WP, Li CT, Shi WZ, Fang SH, Lu YB, Chen Z, Wei EQ. Activation of CysLT receptors induces astrocyte proliferation and death after oxygen-glucose deprivation. Glia. 2008;56:27–37. doi: 10.1002/glia.20588. [DOI] [PubMed] [Google Scholar]
- Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50:3825–3840. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Liu JY, Wagner KM, et al. Optimized inhibitors of soluble epoxide hydrolase improve in vitro target residence time and in vivo efficacy. J Med Chem. 2014;57:7016–7030. doi: 10.1021/jm500694p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Xu X, Chen C, Yu X, Edin ML, Degraff LM, Lee CR, Zeldin DC, Wang DW. Cytochrome P450 2J2 is protective against global cerebral ischemia in transgenic mice. Prostaglandins Other Lipid Mediat. 2012;99:68–78. doi: 10.1016/j.prostaglandins.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Gebremedhin D, Harder DR, Koehler RC. Contribution of epoxyeicosatrienoic acids to the cerebral blood flow response to hypoxemia. J Appl Physiol. 2015 doi: 10.1152/japplphysiol.01043.2014. jap 01043 02014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. 2013;53:37–58. doi: 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton SS, Fournier NM, Duman RS. Vascular growth factors in neuropsychiatry. Cell Mol Life Sci. 2013;70:1739–1752. doi: 10.1007/s00018-013-1281-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nithipatikom K, Grall AJ, Holmes BB, Harder DR, Falck JR, Campbell WB. Liquid chromatographic-electrospray ionization-mass spectrometric analysis of cytochrome P450 metabolites of arachidonic acid. Anal Biochem. 2001;298:327–336. doi: 10.1006/abio.2001.5395. [DOI] [PubMed] [Google Scholar]
- Oliveira SL, Pillat MM, Cheffer A, Lameu C, Schwindt TT, Ulrich H. Functions of neurotrophins and growth factors in neurogenesis and brain repair. Cytometry A. 2013;83:76–89. doi: 10.1002/cyto.a.22161. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, Xu L, Yue S, Liu S, Giffard RG. Neuroprotection by astrocytes in brain ischemia: importance of microRNAs. Neurosci Lett. 2014;565:53–58. doi: 10.1016/j.neulet.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahy D, Kalish BT, Huang S, et al. Epoxyeicosanoids promote organ and tissue regeneration. Proc Natl Acad Sci USA. 2013;110:13528–13533. doi: 10.1073/pnas.1311565110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose TE, Morisseau C, Liu JY, Inceoglu B, Jones PD, Sanborn JR, Hammock BD. 1-Aryl-3-(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: structure-activity relationships, pharmacokinetics, and reduction of inflammatory pain. J Med Chem. 2010;53:7067–7075. doi: 10.1021/jm100691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez A, Wadhwani S, Grammas P. Multiple neurotrophic effects of VEGF on cultured neurons. Neuropeptides. 2010;44:323–331. doi: 10.1016/j.npep.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander AL, Jakob H, Sommer K, Sadler C, Fleming I, Marzi I, Frank J. Cytochrome P450-derived epoxyeicosatrienoic acids accelerate wound epithelialization and neovascularization in the hairless mouse ear wound model. Langenbecks Arch Surg. 2011;396:1245–1253. doi: 10.1007/s00423-011-0838-z. [DOI] [PubMed] [Google Scholar]
- Sarkar P, Zaja I, Bienengraeber M, Rarick KR, Terashvili M, Canfield S, Falck JR, Harder DR. Epoxyeicosatrienoic acids pretreatment improves amyloid beta-induced mitochondrial dysfunction in cultured rat hippocampal astrocytes. Am J Physiol Heart Circ Physiol. 2014;306:H475–484. doi: 10.1152/ajpheart.00001.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid-Brunclik N, Burgi-Taboada C, Antoniou X, Gassmann M, Ogunshola OO. Astrocyte responses to injury: VEGF simultaneously modulates cell death and proliferation. Am J Physiol Regul Integr Comp Physiol. 2008;295:R864–873. doi: 10.1152/ajpregu.00536.2007. [DOI] [PubMed] [Google Scholar]
- Shaik JS, Ahmad M, Li W, et al. Soluble epoxide hydrolase inhibitor trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid is neuroprotective in rat model of ischemic stroke. Am J Physiol Heart Circ Physiol. 2013;305:H1605–1613. doi: 10.1152/ajpheart.00471.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M. Brain angiogenesis in developmental and pathological processes: therapeutic aspects of vascular endothelial growth factor. FEBS J. 2009;276:4636–4643. doi: 10.1111/j.1742-4658.2009.07175.x. [DOI] [PubMed] [Google Scholar]
- Sinor AD, Irvin SM, Cobbs CS, Chen J, Graham SH, Greenberg DA. Hypoxic induction of vascular endothelial growth factor (VEGF) protein in astroglial cultures. Brain Res. 1998;812:289–291. doi: 10.1016/s0006-8993(98)00976-7. [DOI] [PubMed] [Google Scholar]
- Ulu A, Appt S, Morisseau C, et al. Pharmacokinetics and in vivo potency of soluble epoxide hydrolase inhibitors in cynomolgus monkeys. Br J Pharmacol. 2012;165:1401–1412. doi: 10.1111/j.1476-5381.2011.01641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu DY, Davis BB, Wang ZH, et al. A potent soluble epoxide hydrolase inhibitor, t-AUCB, acts through PPARgamma to modulate the function of endothelial progenitor cells from patients with acute myocardial infarction. Int J Cardiol. 2013;167:1298–1304. doi: 10.1016/j.ijcard.2012.03.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaura K, Gebremedhin D, Zhang C, Narayanan J, Hoefert K, Jacobs ER, Koehler RC, Harder DR. Contribution of epoxyeicosatrienoic acids to the hypoxia-induced activation of Ca2+-activated K+ channel current in cultured rat hippocampal astrocytes. Neuroscience. 2006;143:703–716. doi: 10.1016/j.neuroscience.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Yuan L, Liu J, Dong R, Zhu J, Tao C, Zheng R, Zhu S. 14,15-epoxyeicosatrienoic acid promotes production of BDNF from astrocytes and exerts neuroprotective effects during ischemic injury. Neuropathol Appl Neurobiol. 2015 doi: 10.1111/nan.12291. [DOI] [PubMed] [Google Scholar]
- Zhang W, Koerner IP, Noppens R, et al. Soluble epoxide hydrolase: a novel therapeutic target in stroke. J Cereb Blood Flow Metab. 2007;27:1931–1940. doi: 10.1038/sj.jcbfm.9600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Otsuka T, Sugo N, Ardeshiri A, Alhadid YK, Iliff JJ, Debarber AE, Koop DR, Alkayed NJ. Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke. 2008;39:2073–2078. doi: 10.1161/STROKEAHA.107.508325. [DOI] [PMC free article] [PubMed] [Google Scholar]