

ABSTRACT
During fermentative growth in natural and industrial environments, Saccharomyces cerevisiae must redistribute the available nitrogen from multiple exogenous sources to amino acids in order to suitably fulfill anabolic requirements. To exhaustively explore the management of this complex resource, we developed an advanced strategy based on the reconciliation of data from a set of stable isotope tracer experiments with labeled nitrogen sources. Thus, quantifying the partitioning of the N compounds through the metabolism network during fermentation, we demonstrated that, contrary to the generally accepted view, only a limited fraction of most of the consumed amino acids is directly incorporated into proteins. Moreover, substantial catabolism of these molecules allows for efficient redistribution of nitrogen, supporting the operative de novo synthesis of proteinogenic amino acids. In contrast, catabolism of consumed amino acids plays a minor role in the formation of volatile compounds. Another important feature is that the α-keto acid precursors required for the de novo syntheses originate mainly from the catabolism of sugars, with a limited contribution from the anabolism of consumed amino acids. This work provides a comprehensive view of the intracellular fate of consumed nitrogen sources and the metabolic origin of proteinogenic amino acids, highlighting a strategy of distribution of metabolic fluxes implemented by yeast as a means of adapting to environments with changing and scarce nitrogen resources.
IMPORTANCE A current challenge for the wine industry, in view of the extensive competition in the worldwide market, is to meet consumer expectations regarding the sensory profile of the product while ensuring an efficient fermentation process. Understanding the intracellular fate of the nitrogen sources available in grape juice is essential to the achievement of these objectives, since nitrogen utilization affects both the fermentative activity of yeasts and the formation of flavor compounds. However, little is known about how the metabolism operates when nitrogen is provided as a composite mixture, as in grape must. Here we quantitatively describe the distribution through the yeast metabolic network of the N moieties and C backbones of these nitrogen sources. Knowledge about the management of a complex resource, which is devoted to improvement of the use of the scarce N nutrient for growth, will be useful for better control of the fermentation process and the sensory quality of wines.
KEYWORDS: complex nitrogen resource, metabolic network, metabolism, nitrogen, quantitative analysis, regulation, yeasts
INTRODUCTION
The management of nutrients provided by the external environment, mainly carbon and nitrogen, and their redistribution inside the cells for the formation of biosynthetic precursors through the metabolic network are essential for all living organisms. The topology of the metabolic network of the yeast Saccharomyces cerevisiae is one of the best known, and it has been described at the genomic scale (1–3). This knowledge has led to the development of yeast metabolic models, which aim at a comprehensive overview of cellular functioning (4). Accordingly, metabolic flux analysis (MFA) approaches have been implemented to quantify the activities of the different pathways in a defined metabolic network. This methodology relies on model-based interpretation of measurable data, including metabolite uptake and production rates and quantitative intracellular information obtained from isotope tracer experiments (5–8). An MFA approach based on a 13C isotopic tracer from carbon substrates was extensively and successfully applied to gain insight into the in vivo management of the pathways of central carbon metabolism (CCM) in S. cerevisiae (5, 9–13). In contrast, many experimental constraints prevented the implementation of MFA approaches focused on nitrogen metabolism, and the flux distribution through this network remains virtually unexplored. First, the multiplicity of compounds that often compose the nitrogen resource in the environment may have hindered investigations. For example, during industrial fermentations, such as fermentations of wine or beer, nitrogen is provided as a composite mixture of 18 amino acids and ammonium, while in general, the carbon source consists only of glucose/fructose or maltose, respectively (14). Then, in accordance with this diversity, the network structure of nitrogen metabolism is very complex, involving a large number of highly interconnected intermediates and reactions that contribute both to the catabolism of exogenous sources and to anabolism, for the de novo synthesis of amino acids. As a consequence, a large amount of quantitative data, unavailable until now, is required for a complete understanding of the operation of the nitrogen metabolism network.
Schematically, the central core of nitrogen metabolism comprises α-ketoglutarate derived from the CCM as well as glutamate and glutamine, together with the enzymatic system composed of glutamate dehydrogenase, glutamine synthetase, and glutamate synthase, which catalyzes their interconversion (15, 16). Combined with transaminases and deaminases, this system permits the gathering of amine groups from ammonium or other amino acids, releasing α-keto acid intermediates (17, 18). Amine groups are redistributed to carbon precursors for de novo amino acid synthesis, providing the building blocks for protein synthesis. The α-keto acids cluster in two groups. First, pyruvate, oxaloacetate, and α-ketoglutarate are redirected toward the CCM, underlying the strong interaction of nitrogen metabolism with carbon, energetic, and redox metabolisms (19–23). Other compounds, including α-ketoisocaproate (KIC), α-ketoisovalerate (KIV), α-ketobutyrate (KIB), α-ketomethylvalerate, 3-indolepyruvate, and phenylpyruvate, are subsequently assimilated through the Ehrlich pathway. This group is responsible for the formation of higher alcohols, often referred to as fusel alcohols, which can combine with acetyl coenzyme A (acetyl-CoA) to form acetate esters (24). These molecules are of great interest to the fermented beverage and food industries as essential contributors to the sensory profiles of products. Both the biomass production and the profiles of fermentation compounds are affected by the operation of nitrogen metabolism (active metabolic routes, extent, efficiency, etc.). Despite extensive knowledge of the reactions, enzymes, and genes involved in nitrogen and aroma metabolism in S. cerevisiae, no quantitative information is available on the management of a complex mixture of nitrogen sources by this yeast.
The aim of this study was to comprehensively explore the redistribution—particularly for biomass production and the formation of volatile compounds—of the available nitrogen present as a mixture of compounds during fermentation. We developed a dedicated approach that utilizes the labeling information obtained during fermentations carried out in the presence of selected labeled nitrogen sources. Analysis of the complete data set allowed us first to quantify, throughout the fermentation dynamics, the redistribution of nitrogen from the more abundant sources toward the other amino acids and, more generally, to elucidate the fate of nitrogen sources after entry into the cells. These compounds, according to their nature and availability, can be directly used for protein synthesis, stored within the cells, or further catabolized to provide nitrogen for the de novo synthesis of amino acids (Fig. 1A). The second question addressed through this study is the identification of the metabolic origins of the proteinogenic amino acids and of the main higher alcohols produced during fermentation. Proteinogenic amino acids can originate from the direct incorporation of consumed compounds; however, they can also be synthesized using nitrogen from ammonium or other amino acids and carbon backbones, derived from the CCM or interconversion between α-keto acids (Fig. 1A).
FIG 1.
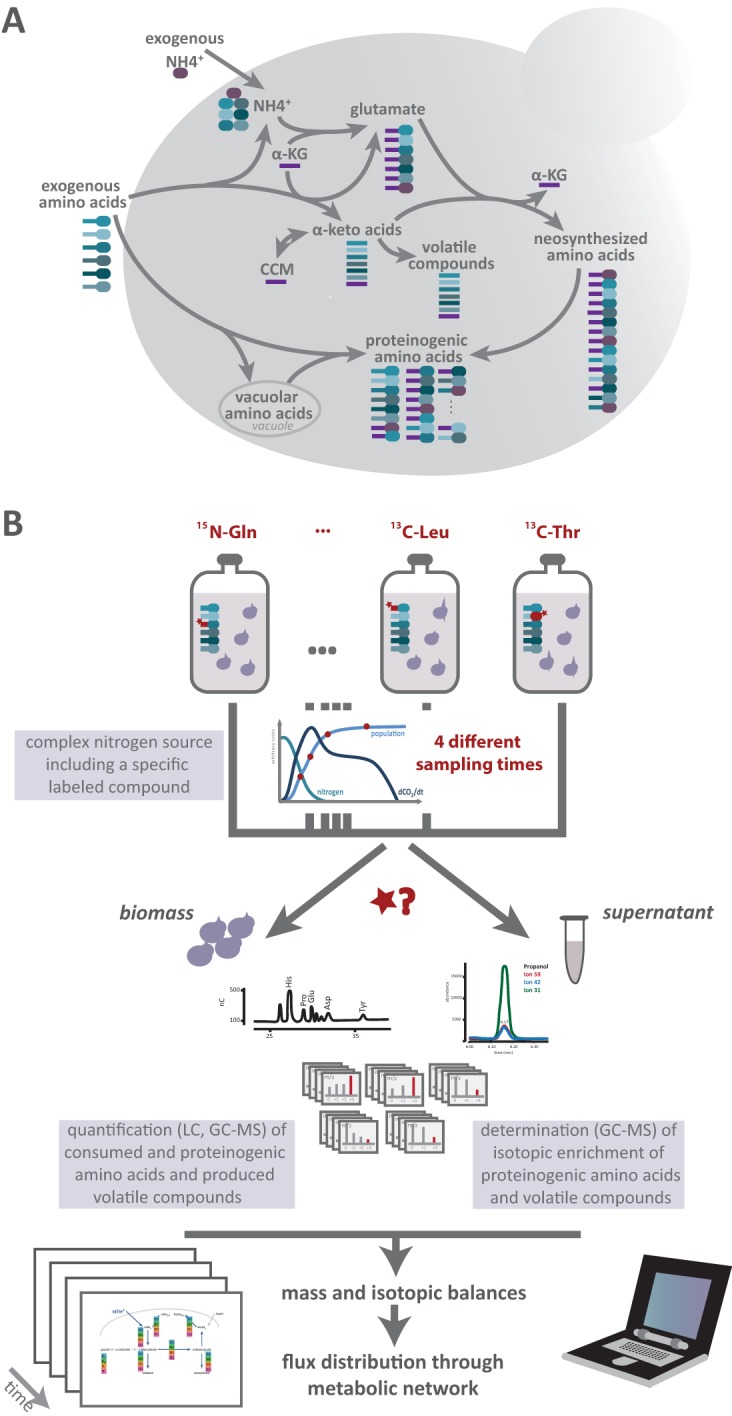
Quantitative analysis of the metabolism of a complex nitrogen resource. (A) Comprehensive outline of the management of a complex nitrogen resource by S. cerevisiae. After entering the cells, the 19 nitrogen sources can be either (i) directly used for the biosynthesis of macromolecules as building blocks, (ii) stored in vacuoles, or (iii) catabolized to provide nitrogen for the de novo synthesis of other amino acids, releasing carbon backbones as α-keto acids to be further converted into volatile compounds or redirected toward the CCM. Diverse origins can be assigned to proteinogenic amino acids, including direct incorporation of consumed amino acids and de novo synthesis. (B) Schematic overview of the design of the isotopic tracer experiment and the data analysis workflow. A set of fermentations was carried out in a synthetic medium containing a complex mixture of nitrogen sources with only one (a different one for each fermentation) in 13C- or 15N-labeled form. The production of biomass and its composition in labeled and unlabeled proteinogenic amino acids, the residual concentration of amino acids in the supernatant, and the production and isotopic enrichment of volatile compounds were measured at 4 sampling times. Mass and isotopic balances for each consumed labeled amino acid were calculated from these data and were further analyzed in an integrated way so as to assess the quantitative contributions of the different metabolic pathways involved in both the catabolism of nitrogen sources and the formation of volatile molecules. LC, liquid chromatography.
RESULTS
Experimental design for 13C and 15N isotopic labeling.
To assess the multiple origins of proteinogenic amino acids and volatile compounds, we implemented an approach based on the combination of stable isotope tracer experiments with a selected panel of 13C- and 15N-labeled amino acids (Fig. 1B). This method relied on the achievement of parallel fermentations on the same medium, which includes the nitrogen source as a chemically defined mixture of ammonium and amino acids that mimics the nitrogen composition of grape juice without oligopeptides (25).
For each fermentation, a single nitrogen compound was supplied as a 13C- or 15N-labeled molecule, while the other nitrogen compounds remained unlabeled. We focused specifically on ammonium, glutamine, and arginine, the three major nitrogen sources in the medium, as well as on amino acids derived from major intermediates of the Ehrlich pathway (leucine, isoleucine, threonine, valine). The whole set of proteinogenic amino acids was quantified (see Data Sets S2 and S3 in the supplemental material), and their isotopic enrichment was measured by quantitative mass spectrometry (26) (see Data Sets S5 and S6 in the supplemental material). Furthermore, for the 13C experiments, the formation of the most abundant volatile compounds and the incorporation of the label into these molecules were also determined (see Data Sets S4 and S6). Together with balances from the consumption of nitrogen sources (see Data Set S1), the data on the transfer of the 15N isotopic tracer from ammonium, glutamine, and arginine to the amino acids in biomass provided information on the extent of nitrogen redistribution from consumed nitrogen sources to proteinogenic amino acids and on the contribution of de novo synthesis to the fulfillment of anabolic requirements. In the same way, analysis of 13C enrichment patterns permitted the assessment of the fraction of exogenous amino acids that was directly incorporated into proteins and provided quantitative information on the further catabolism of these molecules toward the formation of volatile compounds via their α-keto acid intermediates (Fig. 1A).
High reproducibility between fermentations was found for the growth characteristics, nitrogen consumption patterns, and profiles of proteinogenic amino acids included in biomass (see Fig. S1 in the supplemental material). This demonstrated the relevance of this approach, which combined all the data generated during the set of independent fermentations to provide a quantitative and comprehensive analysis of the use of multiple nitrogen sources for the biosynthesis of proteinogenic amino acids.
When provided as a mixture of amino acids and ammonium, nitrogen sources are sequentially assimilated by S. cerevisiae (27, 28). As a consequence, the nature of the nitrogen resource available for cellular anabolism evolves continuously throughout the growth phase of alcoholic fermentation. This led us to investigate the fates of the different assimilated nitrogen sources at various stages of culture: partial (one-half or three-fourths) or complete consumption of the resource nitrogen (N½, N¾, or NT, respectively) and the end of fermentation (EF), corresponding to 16, 20, 40, and 110 h of fermentation, respectively.
Contributions of glutamine, ammonium, and arginine to de novo synthesis of amino acids.
During fermentations in synthetic medium (SM), most of the yeast assimilable nitrogen (YAN) was provided as ammonium, arginine, and glutamine; both amino acids were supplied in surplus in the medium relative to the anabolic requirements (Fig. 2A). However, these three major nitrogen sources were consumed at different rates by S. cerevisiae strain EC1118 and were sequentially exhausted in the medium (27), resulting in changes in the nature of the nitrogen compounds that could be used for biosynthesis over the growth phase (Fig. 2B; also Data Set S1). Until half of the YAN is consumed, the nitrogen sources that might be available for anabolism consist mainly of ammonium (40%), with a low contribution of arginine (11%). In contrast, during the last stages of growth (from N¾ to NT), arginine accounted for 37% of the nitrogen resource imported into the cells, while the portion provided by ammonium decreased considerably (4%).
FIG 2.

Redistribution of nitrogen from the three major nitrogen sources to proteinogenic amino acids during fermentation. Fermentations in which the total of arginine, glutamine, or ammonium was provided in 15N-labeled form were carried out, and the isotopic enrichment of each proteinogenic amino acid was measured. (A) Molar ratios of consumed to proteinogenic amino acids at different stages of fermentation. A value higher than 100% indicates that the amino acid is provided in the medium in excess of the requirements for protein synthesis. (B) Nitrogen amounts relating to the consumption of arginine, glutamine, ammonium, and other amino acids (gray). The bar on the left (T0) shows the initial amounts of the three major nitrogen sources supplied in the medium. N½, partial (one-half) nitrogen consumption; N¾, partial (three-fourths) nitrogen consumption; NT, complete nitrogen consumption; EF, end of fermentation. (C) Portion of nitrogen provided by arginine, glutamine, and ammonium recovered in proteinogenic amino acids (Ala, Gly, Val, Asp, Phe, Leu, Ile, Thr, Ser, Pro, Lys, His, Glu, and Arg). Nitrogen consumption (B) or incorporation (C) between the previous stage and the current stage is represented by dark colors, while the amount already consumed (B) or incorporated (C) at the previous stage is represented by light colors. (D) Contributions of nitrogen provided by consumed arginine, glutamine, and ammonium to the de novo synthesis of proteinogenic amino acids. The size of spheres is proportional to the isotopic enrichment of proteinogenic amino acids. The data were calculated using the averages of measured values from 2 or 8 independent biological experiments.
We first aimed to determine the origin of nitrogen for de novo amino acid synthesis and its dynamics throughout the fermentation. The redistribution of nitrogen from the major nitrogen sources to all of the proteinogenic amino acids (except for tryptophan, methionine, cysteine, and tyrosine, which were not detected by the gas chromatography [GC]-mass spectrometry [MS] method used) was investigated during cultivations in the presence of 15N-labeled NH4+, glutamine, or arginine. Analysis of the 15N enrichment of proteinogenic amino acids revealed that a substantial portion of the amino acids recovered in biomass were synthesized de novo using nitrogen provided mainly by ammonium, arginine, and glutamine, in accordance with their sequential assimilation (Fig. 2C; see also Table S2 in the supplemental material). In particular, the incorporation of nitrogen from arginine was strongly limited during the first stages of culture (N½, N¾) but accounted for as much as 13% of the total nitrogen contained in other amino acids at the end of growth (NT, EF). The fraction of nitrogen provided by ammonium varied in the opposite way (Fig. 2C; also Table S2).
Interestingly, most of the proteinogenic amino acids displayed similar patterns of incorporation of nitrogen from labeled sources (Fig. 2D). This is in agreement with the formation of an intracellular pool of nitrogen from multiple origins, likely through glutamate, with this common intermediate being further redistributed toward proteins. Combining data from 15N and 13C labeling experiments, we determined that the contribution of the three most abundant nitrogen sources (ammonium, arginine, and glutamine) to this pool was on the order of 80% (see Table S4 in the supplemental material).
Amino acid-specific patterns of incorporation of nitrogen from glutamine, ammonium, and arginine.
However, different profiles were identified in the case of amino acids exhibiting specific metabolic features (Fig. 2D). First, early in growth, the isotopic enrichment of glutamate and, to a lesser extent, aspartate from both 15N-labeled ammonium and 15N-labeled glutamine was higher than that of other proteinogenic amino acids. This is in accordance with efficient and intensive nitrogen transfer from these N sources to precursors derived from the tricarboxylic acid (TCA) pathway, α-ketoglutarate and oxaloacetate, through glutamate synthase, glutamate dehydrogenase, and aspartate aminotransferase activities.
In contrast, the labeling of lysine and histidine, amino acids that are unable to support the growth of S. cerevisiae (29), was lower than that of other amino acids during culture in the presence of [15N]ammonium, [15N]glutamine, or [15N]arginine (Fig. 2C; see also Fig. S2 in the supplemental material). In particular, at the end of the growth stage (NT), only 39% of proteinogenic histidine incorporated nitrogen from ammonium, glutamine, or arginine, a finding consistent with total de novo synthesis of this compound of ∼49%. This value, compared with the amount of histidine consumed (Fig. S2), suggested that most of the histidine consumed was recovered in proteins. Such an important contribution of direct incorporation of consumed lysine and histidine into biomass may be related, first, to the inability of S. cerevisiae to catabolize these amino acids, in contrast to other yeast species (29, 30). In addition, the synthesis of these compounds involves multiple-step pathways requiring energy, reducing power, and biological precursors, promoting the direct use of consumed histidine and lysine for protein synthesis.
Finally, proteinogenic proline exhibited its own distinctive pattern of nitrogen incorporation. During the earlier stages (N½) of fermentation, a larger fraction of nitrogen in proteinogenic proline than in other amino acids is derived from arginine (Table S2; Fig. 2D). This reflects the specificities of the metabolic network of proline, which cannot be further catabolized by yeast under anaerobiosis (31) (Fig. 3A): proline is synthesized directly from intracellular glutamate (32), and equimolar production of proline results from the catabolism of arginine (17). We also found a pronounced decrease in the labeling of proline throughout fermentations in the presence of [15N]ammonium or [15N]glutamine (Fig. 2D), which was not found for the other proteinogenic amino acids. Overall, these labeling patterns suggested that the increase in arginine catabolism during the last stage of growth limited specifically the formation of proline from glutamate. At the end of fermentation, <35% of the proline in proteins was synthesized using nitrogen from the three major sources, indicating an important contribution of proline taken from the culture medium to protein biosynthesis, even if no significant changes were observed in residual proline concentrations throughout the fermentation. This may be explained by the production of this amino acid through arginine catabolism, occurring together with its consumption.
FIG 3.
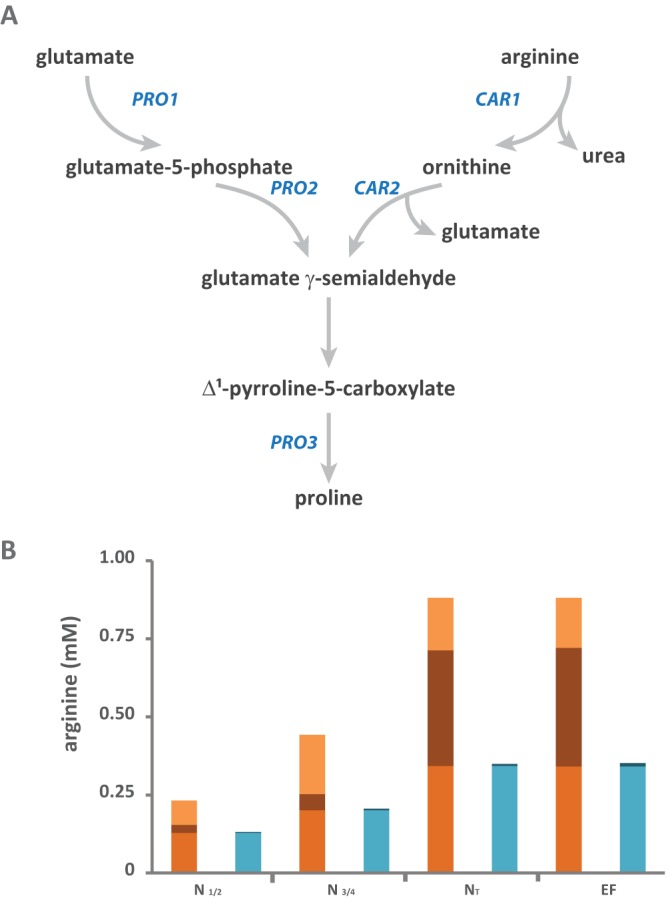
Focus on the fate of arginine during fermentation. (A) Schematic representation of the connections between the metabolic pathways involved in proline biosynthesis and those involved in arginine and glutamate degradation. (B) Isotopic labeling from 15N-labeled arginine during fermentation. Consumed arginine (orange) was mainly incorporated directly into proteins (medium orange) and was used to provide nitrogen for the de novo synthesis of other amino acids (brown) (calculated from the biomass content in proteinogenic amino acids and their isotopic enrichments). The labeled fraction of proteinogenic arginine (light blue) represents the consumed arginine directly incorporated into proteins, while the unlabeled fraction (dark blue) corresponds to de novo-synthesized arginine. The raw data and details of the calculations are provided in the supplemental material. N½, partial (one-half) nitrogen consumption; N¾, partial (three-fourths) nitrogen consumption; NT, complete nitrogen consumption; EF, end of fermentation.
Fate of arginine throughout fermentation.
Taking into account these observations and the key role of arginine in the management of a complex N resource during fermentation (33), we further focused on the fate of the nitrogen provided by arginine (Fig. 3; Table S2). We first found that the 15N isotopic enrichment of arginine in biomass was >97% for all stages of fermentation carried out in the presence of [15N]arginine. This observation showed that most of the proteinogenic arginine originated from direct incorporation of the exogenous compound, with a very limited contribution of de novo synthesis. This is consistent with the feedback inhibition by arginine of the first steps in its biosynthesis (34). Furthermore, the amount of arginine consumed greatly exceeded the amount directly incorporated into proteins at all of the fermentation stages (Fig. 2A). Accordingly, substantial redistribution of nitrogen from arginine to other amino acids was expected all along the growth phase. This was actually the case at the end of the growth phase (NT), as shown by the repartition of consumed arginine between direct incorporation into proteins (38%) and catabolism (62%) (Fig. 3B). In contrast, at the beginning of culture (N½), incorporation of the label into other amino acids was very limited, with 15N from arginine accounting for <4% of the total nitrogen in these compounds (Fig. 2C and D and 3B). As a consequence, at this stage of fermentation, recovery into proteinogenic arginine and other amino acids accounted for only 56% and 12% of nitrogen from exogenous arginine, respectively. The remaining 32% likely corresponded to the arginine stored intracellularly in the vacuole. In agreement with that notion, we recently reported that a substantial fraction of arginine, once transported into the cell, had accumulated inside the vacuole before being metabolized during the last stages of the growth phase (33). When arginine started to be substantially consumed (N¾), the isotopic enrichment of glutamate, an intermediate of the arginine assimilation pathway (17, 35), increased over that at the first stage of growth, while the level of labeling in other amino acids remained very low (Fig. 2D); at the end of the growth phase (NT), nitrogen from arginine was equally redistributed between proteinogenic amino acids. Thus, this specific transient increase in glutamate labeling is consistent with a more efficient use of arginine as an N donor during the last stages of growth (from N¾) that involves glutamate as a mediator for the transfer.
Origin of the carbon backbone of proteinogenic aliphatic amino acids.
We next investigated the provenance of the carbon skeleton of amino acids in proteins by analyzing 13C isotopic labeling data. We focused on four aliphatic amino acids, selected on the basis of differences in their order of assimilation from a complex nitrogen resource (27), as well as in the intermediates synthesized from the CCM and used as carbon precursors for their synthesis: threonine and isoleucine originate from oxaloacetate, and leucine and valine originate from pyruvate. Except for threonine, the level of consumption of the exogenous amino acids was as much as 5 times higher than their content in biomass (Fig. 2A). This substantial imbalance, observed early in the growth phase, suggested that an important fraction of proteinogenic amino acids is synthesized de novo. In accordance with this observation, the labeled fraction of aliphatic amino acids in proteins, which corresponds to the part that originates directly from the exogenous compound, did not exceed 50% throughout the fermentation (Fig. 4; see also Table S3 in the supplemental material). As a result, at the end of the growth, the direct incorporation of any consumed amino acid into proteins accounted for <20% of the total proteinogenic compound.
FIG 4.

Important contribution of de novo synthesis of proteinogenic amino acids. Tracer experiments using 13C-labeled leucine (A), isoleucine (B), valine (C), or threonine (D) during fermentation were carried out both to quantify the fates of these branched amino acids once they enter the cells and to assess the metabolic origins of their proteinogenic counterparts. Consumed amino acid (orange) was either directly incorporated into proteins (light orange) or catabolized (dark orange). Proteinogenic amino acids (blue) originated from direct incorporation of consumed amino acids (light blue) or from de novo synthesis using CCM precursors (dark blue). N½, partial (one-half) nitrogen consumption; N¾, partial (three-fourths) nitrogen consumption; NT, complete nitrogen consumption; EF, end of fermentation.
Over the growth phase, the fractions of labeled threonine and leucine—amino acids assimilated early by yeasts (27, 28)—in proteins decreased from 46 and 50% to 20 and 19%, respectively (Fig. 5D and 6A; also Table S3). Interestingly, the label incorporated into proteinogenic valine from the late consumption of valine was less important, and the proportion decreased from 23 to 15% throughout growth (Fig. 5A). Isoleucine exhibited both an intermediate profile of consumption and an intermediate range of variation of its labeling in proteins (from 34 to 19%) (Fig. 6D). These observations highlight the important involvement of de novo synthesis using the carbon skeleton from the CCM in fulfilling the amino acid requirements for growth; the level of de novo synthesis increases with the depletion of those amino acids in the medium. Moreover, delayed consumption of a nitrogen source likely results in a decrease in the contribution of its direct incorporation into the proteinogenic pool.
FIG 5.


Flux partitioning around valine and leucine metabolism. The distributions of fluxes around the α-ketoisovalerate and α-ketoisocaproate nodes, which are the main intermediates of both the catabolism and the anabolism of leucine and valine, were investigated through stable isotope tracer experiments with 13C-labeled valine and 13C-labeled leucine. (A and D) Portions of consumed (cons) valine (A) and leucine (D) recovered in proteinogenic (prot) leucine and valine. The labeled fraction (light color) corresponds to consumed valine or leucine directly incorporated into proteins, while the unlabeled fraction (dark color) represents the proteinogenic amino acids synthesized de novo from CCM precursors. (B and E) Portions of consumed valine (B) or leucine (E) converted into higher alcohols (isoamyl alcohol and isobutanol). The labeled fraction (light color) corresponds to the fraction of higher alcohol synthesized using the carbon backbone from consumed valine or leucine; the unlabeled fraction (dark color) represents the part of volatile molecules synthesized from CCM α-keto acids. Isotopic enrichments (defined as the molar ratio of the quantity of labeled compound to the total quantity of compound) are shown above each bar. The raw data and details of the calculations are provided in the supplemental material. (C and F) Partitioning of fluxes involved in the use of valine (C) or leucine (F) during fermentation. The fraction of consumed valine or leucine catabolized through a pathway, reported in the colored boxes, was assessed from the molar ratio of the amount of a labeled proteinogenic amino acid or volatile compound to the total amount of consumed amino acid. ic, intracellular.
FIG 6.


Flux partitioning around threonine and isoleucine metabolism. The flux distribution through the metabolic routes around the threonine node was investigated using stable isotope tracer experiments with 13C-labeled threonine and 13C-labeled isoleucine. (A and D) Portion of consumed (cons) threonine (A) or isoleucine (D) recovered in proteinogenic (prot) threonine, glycine, or isoleucine. The labeled fraction (light color) corresponds to consumed threonine or isoleucine directly incorporated into proteins, while the unlabeled fraction (dark color) represents the amino acids in proteins synthesized de novo from CCM precursors. (B and E) Contribution of threonine (B) or isoleucine (E) catabolism to propanol synthesis. The labeled fraction (light color) corresponds to the fraction of propanol synthesized using the carbon backbone from consumed threonine (B) or isoleucine (E), while the unlabeled fraction (dark color) represents the part of propanol synthesized from CCM via aspartate. Isotopic enrichments (defined as the molar ratio of the quantity of labeled compound to the total quantity of compound) are shown above each bar. The raw data and details of the calculations are provided in the supplemental material. (C and F) Partitioning of fluxes involved in the use of threonine (C) or isoleucine (F) during fermentation. The fraction of consumed threonine or isoleucine catabolized through a pathway, reported in colored boxes, was assessed from the molar ratio of the amount of a labeled proteinogenic amino acid or volatile compound to the amount of consumed amino acid. The fluxes indicated by light blue arrows were not quantified because of the small amounts of metabolites produced, which prevented accurate measurement of labeling incorporation.
Fates of aliphatic amino acids consumed during the growth phase.
We also attempted to accurately assess the fates of the consumed aliphatic amino acids during the growth phase. We measured the 13C isotopic enrichment of both proteinogenic amino acids and higher alcohols (volatile compounds) that may be synthesized from labeled branched amino acids through the Ehrlich pathway, namely, isobutanol, isoamyl alcohol, and propanol (24) (Fig. 5B and E and 6B and E; also Table S3). Amyl alcohol, derived from isoleucine degradation, was reasonably removed from the analysis due to insufficient formation (<1 μM [not shown]). This information allowed us to assess the quantitative distribution of the consumed aliphatic amino acids across the metabolic network involved in valine and leucine biosynthesis and degradation, on the one hand, and in threonine catabolism pathways, on the other (Fig. 5C and F and 6C and F).
The considerable imbalance between the availability of these compounds in the medium and their content in biomass (Fig. 2A) was expected to result in massive incorporation of consumed amino acids into proteins during fermentation. However, analysis of the 13C labeling pattern of proteinogenic aliphatic amino acids (Fig. 4) revealed substantial differences in the fraction of exogenous N compound directly incorporated into the proteins depending on the nature of the amino acid, and, accordingly, contrasting mechanisms of management of amino acids once they are imported into the cells were highlighted.
In the presence of [13C]leucine, the amount of labeled leucine in biomass was close to that taken from the medium by yeasts, while no isotopic enrichment was found in the other proteinogenic amino acids. Therefore, between 77% and 86% of assimilated leucine was directed toward protein synthesis. Accordingly, the contribution of further catabolism to the fate of consumed leucine was low and was limited to the formation of isoamyl alcohol (Fig. 5F).
In contrast, the portions of exogenous isoleucine, valine, and threonine directly recovered in proteins accounted for only 51, 41, and 38% of their consumption at the end of the growth phase, respectively, (Fig. 5C and F and 6F). Although the anabolic requirements for these amino acids were at least twice as high as the amount available in the medium, their direct incorporation into biomass once they entered the cells was restricted. This suggests that a portion of these consumed aliphatic amino acids was catabolized.
In agreement with this assumption, during cultures in the presence of labeled valine, we further measured 13C isotopic enrichment on leucine as well as on two higher alcohols: isobutanol and isoamyl alcohol (Fig. 5E). This was consistent with the formation of proteinogenic leucine from the carbon backbone of consumed valine, which likely involved the unidirectional conversion of α-ketoisovalerate (KIV) from valine transamination into α-ketoisocaproate (KIC) (36). In addition, the substantial fractions of KIV and KIC derived from labeled valine, accounting for 15% and 23% of the consumed valine at the end of the growth phase (NT), respectively, were reduced in higher alcohols. Overall, this quantitative analysis of the fate of consumed valine showed that its intracellular flux is allocated between direct incorporation into proteins (45%) and catabolism (55%).
In the same way, a substantial portion of the labeling provided by [13C]threonine (35% at the end of the growth phase) was detected in propanol and in proteinogenic isoleucine, demonstrating the important contribution of catabolism to the fate of consumed threonine via its α-keto acid derived through the Ehrlich pathway, α-ketobutyrate (KIB). Moreover, a lesser portion of consumed threonine (7%) was further converted into proteinogenic glycine, likely due to a threonine aldolase, Gly1p (37). It has been reported that during S. cerevisiae growth on glucose, the metabolic pathway involving phosphoglycerate dehydrogenases, encoded by SER3 and SER33, is responsible for the formation of most of the proteinogenic glycine (38). Our data clearly support the notion that the threonine pathway contributes substantially to fulfilling the anabolic requirements for glycine. Overall, 20% of the consumed threonine was not recovered in related proteinogenic amino acids or propanol. This may be explained by the conversion of this amino acid into other metabolites, each of them produced in amounts insufficient for detection by the analytical methods we used. These include propanoic acid, synthesized by the oxidation of KIB (24), as well as propyl acetate and ethyl propionate, formed by condensing acetyl-CoA with propanol and propionic acid with ethanol, respectively. Another option is the reduction of threonine into α-amino-β-ketobutyrate, since a low threonine dehydrogenase activity has been reported in S. cerevisiae (39). Finally, a portion of the glycine and isoleucine from labeled threonine was likely further converted, primarily into glutathione and amyl alcohol and its derivatives, respectively.
The catabolism of a portion of intracellular isoleucine to amyl alcohol and related compounds through the Ehrlich pathway (40) may also explain the imbalance between the amounts of consumed labeled isoleucine and the portion of labeled proteinogenic isoleucine in our experiments (Fig. 6F).
Metabolic origin of fusel alcohols.
During S. cerevisiae fermentative metabolism, volatile compounds of three related chemical classes are derived from the catabolism of α-keto acids: fusel organic acids, fusel alcohols, and their acetate-ester derivatives (24). All of these are involved in the sensory quality of fermented products. We asked, finally, what was the metabolic origin of the volatile compounds derived from three α-keto acids (KIB, KIC, and KIV), because these intermediates either are newly synthesized through the CCM or come from the transamination of consumed amino acids. This issue was addressed by quantitatively analyzing the incorporation of label into higher alcohols, the most abundant volatile compounds derived from α-keto acids.
During experiments with labeled threonine, the isotopic enrichment detected in propanol, which was the same as that measured in proteinogenic threonine (Fig. 6), decreased throughout the growth phase but remained constant during the stationary phase. This observation implies that propanol originates entirely from the catabolism of intracellular threonine, consisting of 19% consumed threonine (labeled fraction) and 81% newly synthesized threonine at the end of the growth phase. Moreover, the synthesis of propanol depended on the availability of threonine inside the cells, which explains why propanol is produced exclusively during growth, before nitrogen depletion in the medium (41).
Volatile compounds synthesized from KIV and KIC exhibited metabolic origins different from those of KIB derivatives. Thus, during fermentations carried out using [13C]valine or [13C]leucine within the complex nitrogen source (Fig. 5), we measured low levels of incorporation of label into isobutanol and isoamyl alcohol, corresponding to the fraction coming from exogenous amino acids once they entered the cells. This demonstrates the very limited contribution of the catabolism of valine and leucine consumed by yeasts to the formation of these higher alcohols. In contrast to that of propanol, the isotopic enrichment of both isobutanol and isoamyl alcohol substantially decreased after growth stopped, between the end of the growth and the end of fermentation, supporting the key role of the CCM in the formation of these compounds, which was the only way to achieve their synthesis during the stationary phase. This metabolic pattern was consistent with the dynamics of production of isoamyl alcohol and isobutanol, which occurs throughout the fermentation process (41).
DISCUSSION
In natural and industrial environments, the available nitrogen resource supporting microbial growth often consists of a mixture of amino acids and ammonium at variable concentrations (42). These conditions differ greatly from those commonly used for physiological studies, which are carried out using a unique source of nitrogen. The nitrogen metabolism of S. cerevisiae, extensively described in the literature (for a review, see reference 16), involves a large number of branched reactions and intermediates taking part in both the catabolism of exogenous nitrogen sources and the anabolism of amino acids through de novo synthesis. The activity through these different interconnected routes also results in the excretion of metabolites synthesized from intermediates of the network. In this study, we explored quantitatively how yeast manages a complex nitrogen resource to ensure its growth and redistributes exogenous nitrogen sources to fulfill its anabolic requirements throughout fermentation. Taking into account the great complexity of the nitrogen metabolic network, we developed a system-level approach based on the reconciliation of data from a series of isotopic tracer experiments. Quantification of the cometabolism of various nitrogen sources using multiply labeled elemental isotopes is currently impossible. Indeed, the analytical techniques available enable the accurate detection of the labeling pattern of proteinogenic amino acids by using a single-element isotope and possibly colabeling with two isotopic elements (43). Consequently, to address these limitations, we chose to repeat a set of fermentations under the same environmental conditions, but labeling a different selected nitrogen source each time. To broaden the analytic framework of the fate of these consumed N molecules, label incorporation was monitored not only on proteinogenic amino acids, as is usually done, but also on higher alcohols, because these two chemical classes of compounds share common metabolic precursors: α-keto acids. Further combination of the specific data provided by each 13C or 15N tracer experiment, which was made possible by the reproducible behavior of the strains between fermentations, offered a quantitative extended vision of nitrogen metabolism in the presence of multiple nitrogen sources.
Unexpected catabolism of consumed amino acids within yeast cells.
Since the amounts of amino acids supplied in the medium were substantially lower than the anabolic demand, except for glutamine and arginine (44), we expected massive direct incorporation of exogenous amino acids into biomass through the activity of aminoacyl-tRNA synthases, which catalyze the initial step of protein biogenesis (45). This mode of operation is generally assumed, particularly in model-based quantitative analyses of yeast metabolic networks (46–48). In contrast to this assumption, our data revealed, as a general rule, partial use of most of the exogenous amino acids assimilated by yeast for direct protein synthesis, at a level independent of their availability. Therefore, substantial catabolism of consumed N sources, which first involves transaminations (16, 17, 49), occurred inside the cells and was accompanied by substantial de novo synthesis of proteinogenic amino acids. Such a dual allocation of the consumed amino acids was made possible by the similar orders of magnitude of the catalytic efficiencies of aminoacyl-tRNA synthetases and transaminases (50–53). The question arises why S. cerevisiae breaks down a large portion of the consumed amino acids and, thereafter, resynthesizes amino acids to fulfill anabolic requirements. This degradation/rebuilding cycle is, at first glance, time- and energy-consuming. It is key to point out that the direct incorporation of consumed amino acids into proteins would require nutrient-sensing and signaling mechanisms, in particular, to regulate the de novo synthesis of amino acids according to both the cellular need for each amino acid and its intracellular concentration. Many protein components are involved in the operation of regulatory networks; for example, more than 20 proteins, including a large number of kinases, contribute to the sensing of intracellular glutamate/glutamine and further regulation through the retrograde response (RTG) and nitrogen catabolite repression (NCR) signaling pathways (54). Consequently, the implementation of such systems for the sensing and control of the formation of the 20 proteinogenic amino acids would be exceedingly costly in terms of protein synthesis and functioning. Another point to be considered is that S. cerevisiae is frequently found in harsh environments deprived of nitrogen sources. The ability to quickly take up and catabolize amino acids allows these yeasts to manage nitrogen scarcity in natural environments as effectively as possible, efficiently harvesting N sources and storing them in a common pool of nitrogen as glutamate, which is further redistributed for de novo biosynthesis of amino acids. This is particularly relevant for high-sugar environments, in which the accumulation of ethanol leads to inhibition of amino acid uptake (55, 56).
Different fates of exogenous amino acids inside the cells.
Intracellularly, different outcomes occurred for each amino acid. We could connect these outcomes with the order of assimilation of the amino acids, their abilities to be further catabolized, and the cell requirements for protein synthesis. A first illustration of this point is the specific behavior of exogenous arginine imported into the cells. Our results showed that arginine in proteins originated exclusively from the uptake of exogenous arginine. The absence of de novo synthesis of this compound likely resulted from the negative control exerted by cytoplasmic arginine on the ARG1, ARG3, ARG4, ARG5, ARG6, and ARG8 genes, which are responsible for arginine catabolism via the ArgR/Mcm1 complex (16, 57, 58). Then we showed the transient accumulation in vacuoles, during the first stages of growth, of a substantial fraction of the remaining consumed exogenous arginine, which was not immediately catabolized further. This storage, confirming previous observations (33, 59), is in accordance with the preferential use by S. cerevisiae of nitrogen compounds that suitably support biomass production, such as glutamine or ammonium (15, 16, 60), at the expense of poor sources, such as arginine (17, 61). Later in the fermentation, when the preferred sources became limiting, yeast remobilized the nonpreferred, stored sources to supply nitrogen for the last steps of growth.
Among the other nitrogen sources characterized, leucine was the only exogenous compound that was directed entirely toward protein synthesis. Several factors may contribute to this distinct allocation. First, although leucine can be used as a sole nitrogen source by yeast, it supports growth less efficiently than other amino acids (58), most likely because its catabolism results in the formation of α-ketoisocaproate. This intermediate cannot be metabolized further through the CCM or converted into another α-keto acid (36) and is assimilated solely through the Ehrlich pathway, resulting in the formation of isoamyl derivatives (24, 62). In accordance with this, no labeling was detected in other amino acids when [13C]leucine was provided as the labeled nitrogen source, but a small fraction of the label was recovered in isoamyl alcohol. Second, since leucine is one of the most abundant amino acids in biomass, the anabolic demand for leucine is important (44). Finally, the genes encoding the two specific transporters of leucine, BAP2 and BAP3, are under SPS control. The SPS amino-acid-sensing pathway allows the Ssy1p-mediated regulation of specific permeases, activating their transcription as soon as an extracellular substrate (leucine in case of Bap2p and Bap3p) is detected (16, 63, 64). Consequently, leucine was imported into the cells early during the fermentation process (27), while we observed the redistribution of nitrogen from exogenous compounds to the de novo synthesis of amino acids mainly during the last stages of growth.
In contrast to what we observed for leucine, only a fraction of exogenous threonine, isoleucine, valine, or glutamine consumed by yeast was directly incorporated into proteins, corresponding to <50% of the amount required for anabolism. Interestingly, only 20% of the glutamine was directly integrated into biomass after being imported into the cells, which is consistent with the extensive conversion (80%) of this key intermediate of the central nitrogen metabolism (CNM) into glutamate and then to α-ketoglutarate to provide nitrogen that can be redistributed to other amino acids (16). The released α-ketoglutarate can be easily used in different ways, entering the tricarboxylic acid route or being used as a skeleton for the de novo synthesis of some amino acids. A portion of threonine, isoleucine, and valine was likewise further catabolized inside the cells, but to a lesser extent, ranging from 50 to 60%. This could be explained by the fact that the catabolism of these amino acids provided precursors that were used through a limited number of biosynthetic pathways. Investigators using 14C tracer experiments have reported previously that the carbon backbone derived from glutamate catabolism was retrieved exclusively in amino acids of the same family: glutamine, arginine, proline, and lysine (65). This study revealed that the same pattern occurs in the use of threonine, isoleucine, and valine. The carbon skeletons derived from the transamination of these compounds were not redistributed toward all of the proteinogenic amino acids throughout the central metabolism but were found specifically in the amino acids synthesized through related metabolic pathways: isoleucine and glycine in the case of threonine and leucine in the case of valine. However, the fraction recovered in derived amino acids does not account for all of the α-keto acids originating from the transaminations of aliphatic amino acids. In yeast, α-keto acids have been reported to be the major precursors for the synthesis through the Ehrlich pathway of fusel alcohols and their acetate ester derivatives (24). These aromatic molecules are produced at approximately 10 to 70 mg/liter during alcoholic fermentation (66). We demonstrated here that the residual portion of α-keto acids from threonine, isoleucine, and valine transamination was further catabolized through the Ehrlich metabolic route.
Key role of the catabolism of amino acids in the de novo synthesis of proteinogenic compounds but not in the formation of volatile compounds.
On the basis of 15N incorporation patterns of proteinogenic amino acids, we showed that the nitrogen provided by the three main sources, ammonium, glutamine and arginine, was equally redistributed to all of the newly synthesized amino acids. Furthermore, the involvement of these sources in supplying nitrogen for de novo synthesis differed depending on the fermentation stage. For example, the contribution of arginine to supplying NH3 groups increased considerably at the end of the growth phase. This was in accordance with the order of assimilation of the nitrogen sources by yeast during growth in the presence of multiple nitrogen sources. Indeed, S. cerevisiae sequentially imports amino acids and ammonium when provided as a mixture (27, 28, 66), as a consequence of both NCR (15) and SPS (16) control. In particular, NCR inhibits the transcription of genes coding for permeases and enzymes involved in the catabolism of specific nitrogen sources, such as arginine (15, 16).
These observations can be explained by the involvement of a unique intermediate for the management of NH3 groups with a time-dependent origin. The glutamate/glutamine pair has been reported to play a major role in the CNM, collecting NH3 groups from the available nitrogen sources and then serving as amine donors for the transamination of α-keto acids (67, 68). Interestingly, during 15N-labeled arginine assimilation, isotopic enrichment occurred earlier in glutamate than in the other proteinogenic amino acids, supporting the key contribution of this compound as a mediator for nitrogen redistribution.
Such an efficient device for nitrogen reallocation emphasizes that the main role of the catabolism of most of the consumed amino acids is to supply nitrogen for the de novo synthesis of proteinogenic amino acids in accordance with anabolic requirements. This de novo synthesis, which provided approximately 60 to 80% of the demand for nearly all amino acids, was achieved with the carbon backbones of precursors (mainly α-keto acids) in addition to NH3 donors. 13C tracer experiments provided evidence that the carbon intermediates originated primarily from the catabolism of sugars through the CCM, while the catabolism of and interconversions between amino acids accounted for a limited portion of the formation of α-keto acids. In accordance with this distribution, in this study, we demonstrated the low contribution of the carbon skeletons of consumed amino acids to the production of volatile compounds derived from α-keto acids. On the basis of these data, modulation of the production of targeted fermentative aromas, a key issue in the food and beverage industries, cannot be achieved by modifying the availability of exogenous amino acids. Moreover, quantitative knowledge of the metabolic origins of α-keto acids may provide new insights regarding the regulation of the formation of fermentation products. Indeed, the formation of amino acid precursors from glucose affects not only the carbon balance but also yeast energetic and redox states through the demand for ATP, NAD, and NADPH cofactors (19–23). In turn, the redox and energetic status of the cells controls the distribution of fluxes through the CCM. As a consequence, the formation of fermentation products may be modulated depending on the extent of the synthesis of amino acid precursors from glucose.
Overall, the experimental design implemented in this study, based on a combination of 13C and 15N isotopic tracer experiments, provided a consistent and reliable quantitative data set on the distribution of fluxes through the nitrogen metabolic network. This approach offers new perspectives for study of the regulatory mechanisms controlling nitrogen metabolism, in accordance with the adaptation of yeasts to their environments, and for a better understanding of the mechanisms underlying metabolic diversity within the species S. cerevisiae. Furthermore, these results provide new insights regarding the management of multiple nitrogen resources to fulfill anabolic requirements during the fermentative growth of S. cerevisiae according to both the carbon and nitrogen central metabolisms. This knowledge will also provide clues for the design of rational strategies to improve either the kinetics of the fermentation process or the formation of targeted metabolites that contribute to the sensory quality of wines.
MATERIALS AND METHODS
Yeast strain, fermentation conditions, and sampling.
All of the experiments in this study were performed with the commercial wine yeast strain Saccharomyces cerevisiae EC1118 (Lalvin EC1118; Lallemand). Fermentations were carried out in 330-ml reactors equipped with fermentation locks to maintain anaerobiosis at 28°C with continuous magnetic stirring (150 rpm). The synthetic medium (SM) used for fermentations, which mimics grape juice (25), is characterized by a low pH (3.5) and a high glucose content (240 g glucose · liter−1), in contrast to a low availability of nitrogen (180 mg nitrogen · liter−1) as a complex mixture of inorganic and organic sources without oligopeptide addition (Data Set S1), and the presence of ergosterol (1.875 mg · liter−1), oleic acid (0.625 mg · liter−1), and Tween 80 (0.05 g · liter−1) as anaerobic growth factors. A set of fermentations was carried out in SM, each of them containing one labeled N compound among the mixture of ammonium and amino acids composing the nitrogen resource. The labeled nitrogen sources include ammonium chloride (15N [99%]), l-glutamine (15N2 [98%]), l-arginine (U-15N4 [98%]), l-valine (13C5 [97 to 98%]), l-threonine (13C4 [97 to 99%]), l-leucine (13C6 [97 to 99%]), and l-isoleucine (13C6 [97 to 99%]) from Euriso-top (Cambridge Isotope Laboratories). For each condition, duplicate cultures were carried out under the same conditions using only unlabeled molecules. The progress of fermentation was monitored by the determining the CO2 release while measuring the weight loss throughout the process (69).
Samples (2 aliquots of 6 ml each) were taken throughout the fermentation process at different stages (16 h, 20 h, 28 h, and 150 h) corresponding to a partial (one-half or three-fourths) or complete consumption of nitrogen sources and at the end of fermentation (referred as N½, N¾, NT, and EF, respectively). Cells were pelleted by centrifugation (at 2,000 × g for 5 min at 4°C) and were washed twice with distilled water. Supernatants and cell pellets were stored at −80°C for further analysis. In parallel, 10-ml samples of cultures carried out without labeled molecules were taken for dry cell weight determination.
Quantification of consumed and proteinogenic amino acids.
Dry cell weight was determined by weight difference by filtering 10 ml of the culture through preweighed nitrocellulose filters (pore size, 0.45 μm). The filter was washed twice with 50 ml distilled water and was dried at 105°C for 48 h before weighing (until no further change in weight was observed).
Residual ammonium ions in the supernatant were assayed spectrophotometrically using an Enzytec kit (catalog no. 5380; Enzytec) according to the manufacturer's instructions. Before the quantification of residual amino acids, molecules with high molecular weights were removed from supernatants by the addition of 1 volume of a 25% (wt/vol) sulfosalicylic acid solution to 4 volumes of the sample, followed by 1 h of incubation at 4°C. After centrifugation (4°C, 10 min, 3,000 × g), the sample was filtered through a 0.22-μm-pore-size nitrocellulose membrane (Millipore). Amino acid concentrations were determined using a specific amino acid analyzer (Biochrom 30; Biochrom) combining ion-exchange chromatography and spectrophotometric detection after ninhydrin revelation, as described previously (Data Set EV1 in reference 27).
The protein fraction of the biomass was quantified using a bicinchoninic acid (BCA) assay kit (BCA1; Sigma-Aldrich), according to the manufacturer's instructions, after the extraction of proteins from cellular pellets (1 to 2 mg) by incubation for 1 h in the presence of dimethyl sulfoxide (DMSO) (50%, vol/vol) at 105°C. In parallel, proteins contained in the cell pellets were precipitated by overnight incubation in the presence of trichloroacetic acid (10% [vol/vol] in acetone) at −20°C and were hydrolyzed in 6N HCl for 24 h at 105°C to determine the relative concentrations of amino acids within proteins using the Biochrom system described above. The percentage of each amino acid in proteins was further calculated from these data, dividing the measured amount of each amino acid (in milligrams per liter) by the total amount of amino acids measured in the protein extract (sum in milligrams per liter) (Data Set S2). To assess the concentration of each proteinogenic amino acid in the culture (in milligrams per liter), these percentages were then multiplied by the concentration of proteins in the culture (in milligrams per liter), that is, the product of the protein content of biomass and the dry weight (Data Set S3).
Measurement of isotopic enrichment of intracellular amino acids. (i) Biomass hydrolysis.
The isotopic enrichment of proteinogenic amino acids was determined from 1 to 2 mg dried biomass, prehydrolyzed in 1,200 μl of 6 M HCl for 16 h at 105°C. The sample, after the addition of 800 μl of distilled water, was centrifuged at 3,000 × g for 5 min to remove cellular debris. The supernatant was distributed into four 400-μl fractions that were further dried at 105°C until they reached the consistency of syrup (4 to 5 h). These fractions were further used for the derivatization of amino acids using three different agents.
Ethylchloroformate (ECF) derivatization was modified from a previously published procedure (26). The dried hydrolysate was dissolved in 200 μl of 20 mM HCl and 133 μl of pyridine-ethanol (1:4). This mixture was derivatized by adding 50 μl of ECF. The derivatives were extracted into 500 μl of dichloromethane. For analysis, the upper organic phase was collected after 4 min of centrifugation at 10,000 rpm and was injected directly into the GC-MS.
(N,N)-Dimethylformamide dimethyl acetal (DMFDMA) derivatization was modified from a previously published procedure (26). The dried hydrolysate was dissolved in 50 μl of methanol and 200 μl of acetonitrile. The mixture was derivatized by adding 300 μl of DMFDMA. The extract could be injected directly into the GC-MS.
N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA) derivatization includes the dissolution of the dried hydrolysate in 200 μl of acetonitrile and the derivatization of the compounds by adding 200 μl of BSTFA. The mixture was then incubated for 4 h at 135°C. The extract was directly analyzed in the GC-MS.
(ii) GC-MS analysis.
Samples were then analyzed with a Hewlett-Packard (Agilent Technologies, Santa Clara, CA, USA) 6890 gas chromatograph equipped with a CTC Combi PAL autosampler (AOC-5000; Shimadzu, Columbia, MD, USA) and coupled to an HP 5973 mass spectrometry detector. The instrument was controlled, and the data were analyzed, using HP G1701DA ChemStation software. The gas chromatograph was fitted with a DB-17MS column, (length, 30 m; inside diameter, 0.25 mm; film thickness, 0.15 μm) (Agilent Technologies, Santa Clara, CA, USA).
For ECF derivatives, the oven temperature was initially held at 130°C for 3 min. Thereafter, the temperature was gradually raised at a gradient of 15°C min−1 up to 260°C. This temperature, 260°C, was held for 20 min. The carrier gas was He at a flow rate of 1.2 ml · min−1. A total of 2 μl was injected with a split ratio of 3:1. The temperature of the inlet was 230°C.
For DMFDMA derivatives, the oven temperature was maintained at 60°C for 1 min, and then the temperature was increased up to 130°C (20°C min−1), followed by a second gradient of 4°C min−1 up to 260°C. This temperature, 260°C, was held for 10 min. The carrier gas was He, and the flow rate through the column was 1.2 ml · min−1. A total of 2 μl was injected with a split ratio of 3:1. The temperature of the inlet was 230°C.
For BSTFA derivatives, the oven temperature was initially held at 110°C for 1 min, and then the temperature was increased at a gradient of 2°C min−1 up to 154°C, followed by a second gradient of 5°C min−1 up to 300°C. This temperature, 300°C, was maintained for 10 min. The flow through the column was held constant at 1.2 ml He · min−1. A total of 2 μl was injected with a split ratio of 3:1. The temperature of the inlet was 275°C.
The mass spectrometer quadrupole temperature was set at 150°C. The source was set at 250°C for ECF and DMFDMA derivatives and at 300°C for BSTFA derivatives. The transfer line was held at 250°C. The MS was operated in selected ion monitoring (SIM) mode with positive-ion electron impact at 70 eV. The different ions, characteristic of an amino acid fragment, monitored in SIM runs are listed in Table S1 in the supplemental material. For each amino acid fragment, the outcome of the analysis was a cluster of intensities corresponding to its different mass isotopomers. These data were subsequently processed using IsoCor software, recently developed by P. Millard (70); they were corrected for natural labeling and were assessed for the isotopic enrichment of each amino acid, defined as the labeled fraction of a given compound (expressed as a percentage) (Data Set S4 and S5).
Quantification and isotopic enrichment of volatile compounds.
The labeled volatile compounds were extracted according to the method described by Rollero et al. (69). Samples were prepared by the addition of deuterated internal standards at 100 μg · liter−1 to 5 ml supernatant, followed by two successive extractions with 1 ml of dichloromethane. The organic extracts were further dried over anhydrous sodium sulfate and were concentrated under a nitrogen flux. The extracted volatile molecules were separated with a Hewlett-Packard (Agilent Technologies, Santa Clara, CA, USA) 6890 gas chromatograph equipped with a Phenomenex fused silica capillary column (length, 30 m; inside diameter, 0.25 mm; film thickness, 0.25 μm) (ZB-WAX; Agilent Technologies, Santa Clara, CA, USA) and with helium as the carrier gas (constant flow rate, 1.0 ml/min). The oven temperature, initially maintained at 40°C for 3 min, was increased by 4°C/min up to 220°C and was held at this temperature for 20 min. The injector and the transfer line were held at 250°C. We used an injection volume of 2 μl, and the splitter, at 10:1, was opened after 30 s. The mass spectrometer quadrupole temperature was set at 150°C, the source at 230°C, and the transfer line at 250°C. Compounds were detected using an HP 5973 mass spectrometer recording the mass spectra in SIM mode with positive-ion electron impact at 70 eV. The ion clusters used for the quantification and determination of the labeling patterns of volatile compounds, reported in Table S1 in the supplemental material, were selected on the basis of a high signal-to-noise ratio and low interference from other compounds. The concentration of each volatile molecule was quantified from the sum of intensities of the corresponding ion cluster (Data Set S6). In parallel, for each ion cluster, the intensities were subsequently corrected for natural labeling using IsoCor software (70) and were processed to assess the isotopic enrichment of volatile compounds, defined as the labeled fraction of molecules (expressed as a percentage) (Data Set S5).
Outline of calculations for flux quantification.
The experimental design of this work was based on a set of 14 fermentations carried out under the same experimental conditions (medium, operating conditions, and yeast strain). The only difference was the labeled nitrogen source used. No substantial differences were found between the fermentation kinetics obtained and the production of biomass at the different stages of the process (Fig. S1 in the supplemental material). Moreover, determination of within-subject variation coefficients revealed that the consumption profiles of the 20 nitrogen sources were similar for the 14 experiments (Fig. S1). This approach was also implemented to assess the reproducibility of the content of proteinogenic amino acids in biomass from measurements obtained during eight independent fermentations (use of four labeled sources in duplicate [Fig. S1]). According to these analyses, mean values from 14 or 8 independent measurements of the above-mentioned physiological parameters were used for further calculations, including the amounts of unlabeled/labeled amino acids and volatile compounds, and the determination of the distribution of fluxes. The labeled fraction of a proteinogenic amino acid or a volatile compound was calculated by multiplying its concentration (expressed as a millimolar concentration) by its isotopic enrichment. The difference from the total amount of the compound corresponded to the unlabeled portion. Fluxes through metabolic reactions involved in the synthesis of a target compound (a proteinogenic amino acid or a volatile molecule) from a labeled nitrogen source were quantified by dividing the labeled fraction of the compound by the total amount of consumed labeled amino acid, expressed as a millimolar concentration. Further details on the calculations are provided in Tables S2 to S4 and Data Sets S1 to S6 in the supplemental material.
Supplementary Material
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02617-16.
REFERENCES
- 1.Duarte NC, Herrgård MJ, Palsson BØ. 2004. Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model. Genome Res 14:1298–1309. doi: 10.1101/gr.2250904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herrgård MJ, Lee B-S, Portnoy V, Palsson BØ. 2006. Integrated analysis of regulatory and metabolic networks reveals novel regulatory mechanisms in Saccharomyces cerevisiae. Genome Res 16:627–635. doi: 10.1101/gr.4083206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sauer U. 2006. Metabolic networks in motion: 13C-based flux analysis. Mol Syst Biol 2:62. doi: 10.1038/msb4100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Osterlund T, Nookaew I, Nielsen J. 2012. Fifteen years of large scale metabolic modeling of yeast: developments and impacts. Biotechnol Adv 30:979–988. doi: 10.1016/j.biotechadv.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 5.Gombert AK, Moreira dos Santos M, Christensen B, Nielsen J. 2001. Network identification and flux quantification in the central metabolism of Saccharomyces cerevisiae under different conditions of glucose repression. J Bacteriol 183:1441–1451. doi: 10.1128/JB.183.4.1441-1451.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wiechert W. 2001. 13C metabolic flux analysis. Metab Eng 3:195–206. doi: 10.1006/mben.2001.0187. [DOI] [PubMed] [Google Scholar]
- 7.Fischer E, Zamboni N, Sauer U. 2004. High-throughput metabolic flux analysis based on gas chromatography-mass spectrometry derived 13C constraints. Anal Biochem 325:308–316. doi: 10.1016/j.ab.2003.10.036. [DOI] [PubMed] [Google Scholar]
- 8.Birkemeyer C, Luedemann A, Wagner C, Erban A, Kopka J. 2005. Metabolome analysis: the potential of in vivo labeling with stable isotopes for metabolite profiling. Trends Biotechnol 23:28–33. doi: 10.1016/j.tibtech.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Frick O, Wittmann C. 2005. Characterization of the metabolic shift between oxidative and fermentative growth in Saccharomyces cerevisiae by comparative 13C flux analysis. Microb Cell Fact 4:30. doi: 10.1186/1475-2859-4-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grotkjaer T, Akesson M, Christensen B, Gombert AK, Nielsen J. 2004. Impact of transamination reactions and protein turnover on labeling dynamics in. 13C-labeling experiments. Biotechnol Bioeng 86:209–216. doi: 10.1002/bit.20036. [DOI] [PubMed] [Google Scholar]
- 11.Christen S, Sauer U. 2011. Intracellular characterization of aerobic glucose metabolism in seven yeast species by 13C flux analysis and metabolomics. FEMS Yeast Res 11:263–272. doi: 10.1111/j.1567-1364.2010.00713.x. [DOI] [PubMed] [Google Scholar]
- 12.Blank LM, Kuepfer L, Sauer U. 2005. Large-scale 13C-flux analysis reveals mechanistic principles of metabolic network robustness to null mutations in yeast. Genome Biol 6:R49. doi: 10.1186/gb-2005-6-6-r49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.dos Santos MM, Gombert AK, Christensen B, Olsson L, Nielsen J. 2003. Identification of in vivo enzyme activities in the cometabolism of glucose and acetate by Saccharomyces cerevisiae by using 13C-labeled substrates. Eukaryot Cell 2:599–608. doi: 10.1128/EC.2.3.599-608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perpete P, Santos G, Bodart E, Collin S. 2005. Uptake of amino acids during beer production: the concept of a critical time value. J Am Soc Brew Chem 63:23–27. [Google Scholar]
- 15.Magasanik B, Kaiser CA. 2002. Nitrogen regulation in Saccharomyces cerevisiae. Gene 290:1–18. doi: 10.1016/S0378-1119(02)00558-9. [DOI] [PubMed] [Google Scholar]
- 16.Ljungdahl PO, Daignan-Fornier B. 2012. Regulation of amino acid, nucleotide, and phosphate metabolism in Saccharomyces cerevisiae. Genetics 190:885–929. doi: 10.1534/genetics.111.133306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper TG. 1982. Nitrogen metabolism in Saccharomyces cerevisiae, p 39–99. In Strathern JN, Jones EW, Broach JR (ed), The molecular biology of the yeast Saccharomyces: metabolism and gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 18.Jones EW, Fink GR. 1982. Regulation of amino acid and nucleotide biosynthesis in yeast, p 182–299. In Strathern JN, Jones EW, Broach JR (ed), The molecular biology of the yeast Saccharomyces: metabolism and gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 19.Reeds PJ, Wahle KW, Haggarty P. 1982. Energy costs of protein and fatty acid synthesis. Proc Nutr Soc 41:155–159. doi: 10.1079/PNS19820025. [DOI] [PubMed] [Google Scholar]
- 20.Aoyagi Y, Tasaki I, Okumura J, Muramatsu T. 1988. Energy cost of whole-body protein synthesis measured in vivo in chicks. Comp Biochem Physiol A Comp Physiol 91:765–768. doi: 10.1016/0300-9629(88)90962-0. [DOI] [PubMed] [Google Scholar]
- 21.Albers E, Larsson C, Lidén G, Niklasson C, Gustafsson L. 1996. Influence of the nitrogen source on Saccharomyces cerevisiae anaerobic growth and product formation. Appl Environ Microbiol 62:3187–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Camarasa C, Grivet J-P, Dequin S. 2003. Investigation by 13C-NMR and tricarboxylic acid (TCA) deletion mutant analysis of pathways for succinate formation in Saccharomyces cerevisiae during anaerobic fermentation. Microbiology 149:2669–2678. doi: 10.1099/mic.0.26007-0. [DOI] [PubMed] [Google Scholar]
- 23.Radler F. 1993. Yeasts—metabolism of organic acids, p 165–182. In Fleet GH. (ed), Wine microbiology and biotechnology. Harwood Academic Publishers, Chur, Switzerland. [Google Scholar]
- 24.Hazelwood LA, Daran J-M, van Maris AJA, Pronk JT, Dickinson JR. 2008. The Ehrlich pathway for fusel alcohol production: a century of research on Saccharomyces cerevisiae metabolism. Appl Environ Microbiol 74:2259–2266. doi: 10.1128/AEM.02625-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bely M, Sablayrolles JM, Barre P. 1990. Automatic detection of assimilable nitrogen deficiencies during alcoholic fermentation in oenological conditions. J Ferment Bioeng 70:246–252. doi: 10.1016/0922-338X(90)90057-4. [DOI] [Google Scholar]
- 26.Christensen B, Nielsen J. 1999. Isotopomer analysis using GC-MS. Metab Eng 1:282–290. doi: 10.1006/mben.1999.0117. [DOI] [PubMed] [Google Scholar]
- 27.Crépin L, Nidelet T, Sanchez I, Dequin S, Camarasa C. 2012. Sequential use of nitrogen compounds by Saccharomyces cerevisiae during wine fermentation: a model based on kinetic and regulation characteristics of nitrogen permeases. Appl Environ Microbiol 78:8102–8111. doi: 10.1128/AEM.02294-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiranek V, Langridge P, Henschke PA. 1995. Amino acid and ammonium utilization by Saccharomyces cerevisiae wine yeasts from a chemically defined medium. Am J Enol Vitic 46:75–83. [Google Scholar]
- 29.Brunke S, Seider K, Richter ME, Bremer-Streck S, Ramachandra S, Kiehntopf M, Brock M, Hube B. 2014. Histidine degradation via an aminotransferase increases the nutritional flexibility of Candida glabrata. Eukaryot Cell 13:758–765. doi: 10.1128/EC.00072-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhattacharjee J. 1992. Evolution of α-aminoadipate pathway for the synthesis of lysine in fungi: the catabolism of lysine, p 69–70. In Mortlock RJ. (ed), The evolution of metabolic function. CRC Press, Boca Raton, FL. [Google Scholar]
- 31.Liang X, Dickman MB, Becker DF. 2014. Proline biosynthesis is required for endoplasmic reticulum stress tolerance in Saccharomyces cerevisiae. J Biol Chem 289:27794–27806. doi: 10.1074/jbc.M114.562827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang SS, Brandriss MC. 1987. Proline utilization in Saccharomyces cerevisiae: sequence, regulation, and mitochondrial localization of the PUT1 gene product. Mol Cell Biol 7:4431–4440. doi: 10.1128/MCB.7.12.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crépin L, Sanchez I, Nidelet T, Dequin S, Camarasa C. 2014. Efficient ammonium uptake and mobilization of vacuolar arginine by Saccharomyces cerevisiae wine strains during wine fermentation. Microb Cell Fact 13:109. doi: 10.1186/s12934-014-0109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pauwels K, Abadjieva A, Hilven P, Stankiewicz A, Crabeel M. 2003. The N-acetylglutamate synthase/N-acetylglutamate kinase metabolon of Saccharomyces cerevisiae allows co-ordinated feedback regulation of the first two steps in arginine biosynthesis. Eur J Biochem 270:1014–1024. doi: 10.1046/j.1432-1033.2003.03477.x. [DOI] [PubMed] [Google Scholar]
- 35.Martin O, Brandriss MC, Schneider G, Bakalinsky AT. 2003. Improved anaerobic use of arginine by Saccharomyces cerevisiae. Appl Environ Microbiol 3:1623–1628. doi: 10.1128/AEM.69.3.1623-1628.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kohlhaw GB. 2003. Leucine biosynthesis in fungi: entering metabolism through the back door. Microbiol Mol Biol Rev 67:1–15. doi: 10.1128/MMBR.67.1.1-15.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monschau N, Stahmann KP, Sahm H, McNeil JB, Bognar AL. 1997. Identification of Saccharomyces cerevisiae GLY1 as a threonine aldolase: a key enzyme in glycine biosynthesis. FEMS Microbiol Lett 1:55–60. [DOI] [PubMed] [Google Scholar]
- 38.Albers E, Laizé V, Blomberg A, Hohmann S, Gustafsson L. 2003. Ser3p (Yer081wp) and Ser33p (Yil074cp) are phosphoglycerate dehydrogenases in Saccharomyces cerevisiae. J Biol Chem 12:10264–10272. doi: 10.1074/jbc.M211692200. [DOI] [PubMed] [Google Scholar]
- 39.Murata K, Saikusa T, Fukuda Y, Watanabe K, Inoue Y, Shimosaka M, Kimura A. 1986. Metabolism of 2-oxoaldehydes in yeasts. Possible role of glycolytic bypath as a detoxification system in l-threonine catabolism by Saccharomyces cerevisiae. Eur J Biochem 2:297–301. [DOI] [PubMed] [Google Scholar]
- 40.Dickinson JR, Harrison SJ, Dickinson JA, Hewlins MJ. 2000. An investigation of the metabolism of isoleucine to active amyl alcohol in Saccharomyces cerevisiae. J Biol Chem 15:10937–10942. doi: 10.1074/jbc.275.15.10937. [DOI] [PubMed] [Google Scholar]
- 41.Mouret JR, Camarasa C, Angenieux M, Aguera E, Perez M, Farines V, Sablayrolles JM. 2014. Kinetic analysis and gas–liquid balances of the production of fermentative aromas during winemaking fermentations: effect of assimilable nitrogen and temperature. Food Res Int 62:1–10. doi: 10.1016/j.foodres.2014.02.044. [DOI] [Google Scholar]
- 42.Bell SJ, Henschke PA. 2005. Implications of nitrogen nutrition for grapes, fermentation and wine. Aust J Grape Wine Res 11:242–295. doi: 10.1111/j.1755-0238.2005.tb00028.x. [DOI] [Google Scholar]
- 43.Blank LM, Desphande RR, Schmid A, Hayen H. 2012. Analysis of carbon and nitrogen co-metabolism in yeast by ultrahigh-resolution mass spectrometry applying 13C- and 15N-labeled substrates simultaneously. Anal Bioanal Chem 403:2291–2305. doi: 10.1007/s00216-012-6009-4. [DOI] [PubMed] [Google Scholar]
- 44.Lange HC, Heijnen JJ. 2001. Statistical reconciliation of the elemental and molecular biomass composition of Saccharomyces cerevisiae. Biotechnol Bioeng 75:334–344. doi: 10.1002/bit.10054. [DOI] [PubMed] [Google Scholar]
- 45.Ibba M, Söll D. 2000. Aminoacyl tRNA synthesis. Annu Rev Biochem 69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 46.Varela C, Pizarro F, Agosin E. 2004. Biomass content governs fermentation rate in nitrogen-deficient wine musts. Appl Environ Microbiol 6:3392–3400. doi: 10.1128/AEM.70.6.3392-3400.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vargas FA, Pizarro F, Pérez-Correa JR, Agosin E. 2011. Expanding a dynamic flux balance model of yeast fermentation to genome-scale. BMC Syst Biol 5:75. doi: 10.1186/1752-0509-5-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vázquez-Lima F, Silva P, Barreiro A, Martínez-Moreno R, Morales P, Quirós M, González R, Albiol J, Ferrer P. 2014. Use of chemostat cultures mimicking different phases of wine fermentations as a tool for quantitative physiological analysis. Microb Cell Fact 13:85. doi: 10.1186/1475-2859-13-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cooper TG. 2002. Transmitting the signal of excess nitrogen in Saccharomyces cerevisiae from the Tor proteins to the GATA factors: connecting the dots. FEMS Microbiol Rev 3:223–238. doi: 10.1111/j.1574-6976.2002.tb00612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roy H, Ibba M. 2006. Phenylalanyl-tRNA synthetase contains a dispensable RNA-binding domain that contributes to the editing of noncognate aminoacyl-tRNA. Biochemistry 45:9156–9162. doi: 10.1021/bi060549w. [DOI] [PubMed] [Google Scholar]
- 51.Islam MM, Nautiyal M, Wynn RM, Mobley JA, Chuang DT, Hutson SM. 2010. Branched-chain amino acid metabolon: interaction of glutamate dehydrogenase with the mitochondrial branched-chain aminotransferase (BCATm). J Biol Chem 2851:265–276. doi: 10.1074/jbc.M109.048777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karsten WE, Reyes ZL, Bobyk KD, Cook PF, Chooback L. 2011. Mechanism of the aromatic aminotransferase encoded by the Aro8 gene from Saccharomyces cerevisiae. Arch Biochem Biophys 516:67–74. doi: 10.1016/j.abb.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 53.Ibba M, Hong KW, Sherman JM, Sever S, Söll D. 1996. Interactions between tRNA identity nucleotides and their recognition sites in glutaminyl-tRNA synthetase determine the cognate amino acid affinity of the enzyme. Proc Natl Acad Sci U S A 93:6953–6958. doi: 10.1073/pnas.93.14.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conrad M, Schothorst J, Kankipati HN, Van Zeebroeck G, Rubio-Texeira M, Thevelein JM. 2014. Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev 38:254–299. doi: 10.1111/1574-6976.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leao C, Van Uden N. 1984. Effects of ethanol and other alkanols on the general amino acid permease of Saccharomyces cerevisiae. Biotechnol Bioeng 26:404–406. [DOI] [PubMed] [Google Scholar]
- 56.Mishra P, Prasad R. 1989. Relationship between ethanol tolerance and fatty acyl composition of Saccharomyces cerevisiae. Appl Microbiol Biotechnol 30:294–229. [Google Scholar]
- 57.Messenguy F, Dubois E. 2003. Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene 316:1–21. doi: 10.1016/S0378-1119(03)00747-9. [DOI] [PubMed] [Google Scholar]
- 58.Godard P, Urrestarazu A, Vissers S, Kontos K, Bontempi G, Van Helden J, André B. 2007. Effect of 21 different nitrogen sources on global gene expression in the yeast Saccharomyces cerevisiae. Mol Cell Biol 27:3065–3086. doi: 10.1128/MCB.01084-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kitamoto K, Yoshizawa K, Ohsumi Y, Anraku Y. 1988. Dynamic aspects of vacuolar and cytosolic amino acid pools of Saccharomyces cerevisiae. J Bacteriol 170:2683–2686. doi: 10.1128/jb.170.6.2683-2686.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Broach JR. 2012. Nutritional control of growth and development in yeast. Genetics 192:73–105. doi: 10.1534/genetics.111.135731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gutiérrez A, Chiva R, Sancho M, Beltran G, Arroyo-López FN, Guillamon JM. 2012. Nitrogen requirements of commercial wine yeast strains during fermentation of a synthetic grape must. Food Microbiol 31:25–32. doi: 10.1016/j.fm.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 62.Dickinson R, Lanterman M, Danner D, Pearson B, Sanz P, Harrison S, Hewlins M. 1997. A 13C nuclear magnetic resonance investigation of the metabolism of leucine to isoamyl alcohol in Saccharomyces cerevisiae. J Biol Chem 272:26871–26878. doi: 10.1074/jbc.272.43.26871. [DOI] [PubMed] [Google Scholar]
- 63.Iraqui I, Vissers S, Bernard F, de Craene JO, Boles E, Urrestarazu A, André B. 1999. Amino acid signaling in Saccharomyces cerevisiae: a permease-like sensor of external amino acids and F-Box protein Grr1p are required for transcriptional induction of the AGP1 gene, which encodes a broad-specificity amino acid permease. Mol Cell Biol 19:989–1001. doi: 10.1128/MCB.19.2.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gaber RF, Ottow K, Andersen HA, Kielland-Brandt MC. 2003. Constitutive and hyperresponsive signaling by mutant forms of Saccharomyces cerevisiae amino acid sensor Ssy1. Eukaryot Cell 2:922–929. doi: 10.1128/EC.2.5.922-929.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Albers E, Gustafsson L, Niklasson C, Lidén G. 1998. Distribution of 14C-labelled carbon from glucose and glutamate during anaerobic growth of Saccharomyces cerevisiae. Microbiology 144:1683–1690. doi: 10.1099/00221287-144-6-1683. [DOI] [PubMed] [Google Scholar]
- 66.Jones M, Pierce JS. 1964. Absorption of amino acids from wort by yeasts. J Inst Brew 70:307–315. doi: 10.1002/j.2050-0416.1964.tb01996.x. [DOI] [Google Scholar]
- 67.DeLuna A, Avendano A, Riego L, Gonzalez A. 2001. NADP-glutamate dehydrogenase isoenzymes of Saccharomyces cerevisiae. Purification, kinetic properties, and physiological roles. J Biol Chem 276:43775–43783. doi: 10.1074/jbc.M107986200. [DOI] [PubMed] [Google Scholar]
- 68.Swiegers JH, Bartowsky EJ, Henschke P, Pretorius I. 2005. Yeast and bacterial modulation of wine aroma and flavour. Aust J Grape Wine Res 11:139–173. doi: 10.1111/j.1755-0238.2005.tb00285.x. [DOI] [Google Scholar]
- 69.Rollero S, Bloem A, Camarasa C, Sanchez I, Ortiz-Julien A, Sablayrolles JM, Dequin S, Mouret JR. 2015. Combined effects of nutrients and temperature on the production of fermentative aromas by Saccharomyces cerevisiae during wine fermentation. Appl Microbiol Biotechnol 5:2291–2304. doi: 10.1007/s00253-014-6210-9. [DOI] [PubMed] [Google Scholar]
- 70.Millard P, Letisse F, Sokol S, Portais JC. 2012. IsoCor: correcting MS data in isotope labeling experiments. Bioinformatics 28:1294–1296. doi: 10.1093/bioinformatics/bts127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.