

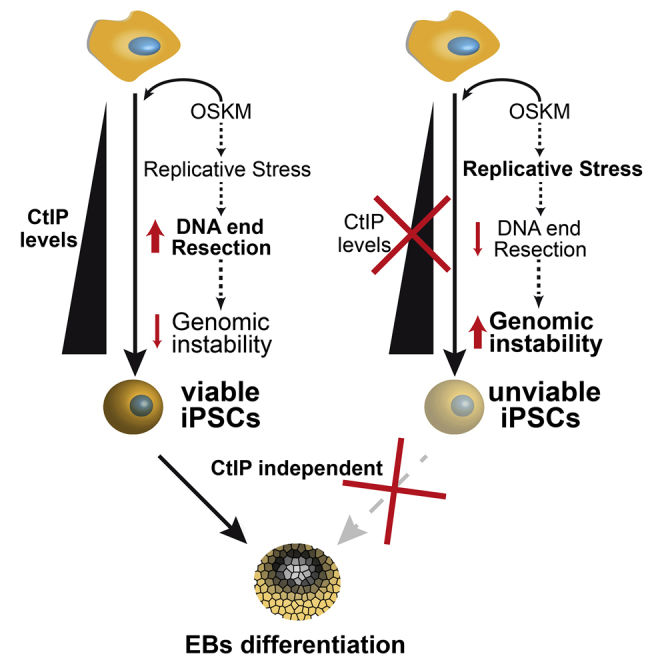
Summary
Acquired genomic instability is one of the major concerns for the clinical use of induced pluripotent stem cells (iPSCs). All reprogramming methods are accompanied by the induction of DNA damage, of which double-strand breaks are the most cytotoxic and mutagenic. Consequently, DNA repair genes seem to be relevant for accurate reprogramming to minimize the impact of such DNA damage. Here, we reveal that reprogramming is associated with high levels of DNA end resection, a critical step in homologous recombination. Moreover, the resection factor CtIP is essential for cell reprogramming and establishment of iPSCs, probably to repair reprogramming-induced DNA damage. Our data reveal a new role for DNA end resection in maintaining genomic stability during cell reprogramming, allowing DNA repair fidelity to be retained in both human and mouse iPSCs. Moreover, we demonstrate that reprogramming in a resection-defective environment has long-term consequences on stem cell self-renewal and differentiation.
Keywords: iPSC, cell reprogramming, genetic instability, DNA resection
Graphical Abstract
Highlights
-
•
DNA end resection mechanism is hyper-activated during reprogramming
-
•
CtIP protein levels increase during mouse and human cell reprogramming
-
•
Loss of CtIP impairs mouse and human reprogramming efficiency
-
•
Reprogramming without CtIP causes defects in iPSC self-renewal and differentiation
Therapeutic use of iPSC is limited due to the genomic alterations that accompany cell reprograming. Huertas, Gómez-Cabello, and colleagues show that DNA resection is essential to avoid such phenomena and that CtIP is critically required for mouse and human cell reprogramming. Indeed, reprogramming in the absence of CtIP has severe consequences for cell maintenance and differentiation.
Introduction
The ability to generate induced pluripotent stem cells (iPSCs) has been heralded to have great potential in regenerative medicine and research (Yu et al., 2007, Takahashi and Yamanaka, 2006). However, this potential is currently under debate, due to evidence that iPSCs can acquire DNA damage and genomic instability during the reprogramming process (Ruiz et al., 2015, Liang and Zhang, 2013, Gore et al., 2011, Mayshar et al., 2010). In fact, even just expressing the reprogramming factors, regardless of the methodology used to generate them, causes DNA damage, mainly by replication stress (Ruiz et al., 2015, Gonzalez et al., 2013, Tilgner et al., 2013). It is critical, however, to obtain “safe” iPSCs that are genetically identical to their parent cells for clinical use. An essential prerequisite for this is to obtain a thorough understanding about how the DNA repair machinery acts in these cells.
Several pieces of evidence suggest that pluripotent stem cells need more active DNA repair pathways than somatic differentiated cells (Rocha et al., 2013). Supporting this view, members of the DNA damage response (DDR) have been shown to prevent genomic instability in iPSCs (Hong et al., 2009, Kawamura et al., 2009, Li et al., 2009). Indeed, proteins involved in the repair of DNA double-strand breaks (DSBs), in both homologous recombination (HR) and non-homologous end joining (NHEJ), have a relevant role in reprogramming efficiency (Gonzalez et al., 2013, Ruiz et al., 2013, Tilgner et al., 2013). HR is required for an error-free repair of DSBs, using homologous sequences (normally from the sister chromatid) (Heyer et al., 2010), and for restarting replication forks stalled during replication stress (Petermann and Helleday, 2010). In contrast, NHEJ competes with HR for DSB repair in a more error-prone pathway (Gomez-Cabello et al., 2013, Huertas, 2010, Lieber, 2008). DNA end resection is a key event that regulates the DSB repair pathway choice between NHEJ and HR. This mechanism generates single-strand DNA (ssDNA) by 5′ to 3′ degradation at both sides of a break (Huertas and Jackson, 2009, Jackson and Bartek, 2009). Although resected DNA is an obligate substrate for HR, it blocks NHEJ (Heyer et al., 2010). CtIP is a major player in the decision between HR and NHEJ as it allows for ssDNA formation, precluding binding of the NHEJ machinery to DNA breaks (Huertas, 2010). DNA end resection is highly regulated by multiple signals, including cell-cycle-dependent CtIP phosphorylation (Sartori et al., 2007). Cells depleted of CtIP fail to repair DNA DSBs by HR, are sensitive to DNA damaging agents, and accumulate chromosomal aberrations in response to DNA damage (Sartori et al., 2007, Huertas and Jackson, 2009).
Here, we identified DNA end resection as one of the predominant mechanisms required for cell reprogramming in human and mouse iPSCs (miPSCs), most likely based on its function in repairing replication-induced DNA insults. In addition, we have determined that CtIP is essential for reprogramming efficiency in both organisms. Cells deficient for CtIP underwent apoptosis rather than reprogramming, probably due to an unbearable load of DNA damage during the process. Those cells that could reprogram with low levels of CtIP acquired a burden in terms of genomic mutations that compromised their long-term survival and ability to differentiate again. We suggest that CtIP has a genomic stability protector role during reprogramming. Exploiting such a role could contribute to creating genetically stable iPSCs that meet clinical safety standards for use in regenerative medicine.
Results
DNA End Resection Increases in Mouse and Human iPSCs
Cell reprogramming per se, by the expression of reprogramming factors (OCT4, SOX2, KLF4, and c-MYC), increases DNA damage and genetic instability, mainly by replication stress (Ruiz et al., 2015). Here, we used a previously reported mouse embryonic fibroblast (MEF) cell line bearing the doxycycline-inducible system of mouse genes of Klf4, Oct4, Sox2, and c-Myc to carry out the reprogramming process (Abad et al., 2013). First, we analyzed cellular levels of DNA end resection in MEFs and their corresponding iPSCs generated by doxycycline treatment. We developed a new strategy for a readout of DNA end resection based on bromodeoxyuridine (BrdU) detection by fluorescence-activated cell sorting (FACS) analysis using native conditions. In contrast to standard proliferation assays using BrdU incorporation, this assay is based on a BrdU epitope that is hidden in double-stranded DNA, and thereby unavailable to anti-BrdU antibodies under native conditions. Critically, the assay is non-responsive to DNA replication, and the epitope is only exposed after formation of ssDNA by resection. This novel method demonstrated that miPSCs had more exposed BrdU than primary MEFs not treated with doxycycline, showing that a higher amount of endogenously occurring breaks were resected in reprogrammed cells (Figure 1A). We further confirmed that this increased BrdU signal intensity was indeed due to canonical DNA end resection, as it disappeared when the key resection factor CtIP was depleted (Figure S1A).
Figure 1.

DNA End Resection Is an Essential Mechanism for Cell Reprogramming
(A) FACS analysis of BrdU exposed by DNA end resection in MEFs and their respective reprogrammed cells (miPSCs). p Values were calculated using the Kolmogorov-Smirnov test. At least three independent experiments were performed. Representative histogram is shown.
(B) Resected DNA length obtained by SMART technique in MEFs and miPSCs. Error bars indicate ±SEM of three independent experiments.
(C) Representative images of DNA fibers visualized with the anti-BrdU antibody.
(D) Same as (A) except using human foreskin fibroblasts (HFFs) and the human iPSCs (hiPSCs) derived from them. At least three independent experiments were performed. Representative histogram is shown.
(E) Same as (B) except using human cells. Error bars indicate ±SEM of three independent experiments.
(F) Same as (C) except using human cells.
(G) MEFs and miPSCs were immunoblotted to analyze the indicated proteins. At least three independent experiments were performed. A representative western blot is shown.
(H) Same as (G) except showing protein levels in HFFs and hiPSCs. At least three independent experiments were performed. A representative western blot is shown.
See also Figures S1–S3.
These results demonstrated that the DNA end resection process was activated in miPSCs in the absence of exogenous damage, most likely due to replication stress and DNA damage generated during cell reprogramming. We hypothesized that this resection activation reflects not only an increased number of breaks being processed, but also a higher processivity of the resection machinery itself. Thus, we analyzed whether the length of resected DNA was longer in miPSCs than in MEFs, using a high-resolution technique to measure the length of resected DNA in individual DNA fibers (Cruz-Garcia et al., 2014). We demonstrated that miPSCs generated significantly longer tracks of ssDNA compared with the primary differentiated parent cells (Figures 1B and 1C). A 50% increase in the median length of resected DNA was observed in pluripotent cells with respect their MEF control (Figures 1B and 1C). Again, we could demonstrate that this was caused by activation of the canonical resection machinery, as this increased length of ssDNA depended on CtIP activity (Figure S1C). Strikingly, the number of lesions and the amount of resected DNA following reprogramming to iPSCs was equivalent to that seen after treating primary cells with high doses of exogenous damage (Figures S1B and S1C), in agreement with the idea that this process represents a severe challenge for genomic integrity.
To address whether the activation of resection during cell reprogramming was evolutionarily conserved, we investigated whether DNA end processing also increases during reprogramming of primary human cells. We used four retroviral vectors bearing one of the OSKM factors (OCT4, SOX2, KLF4, or c-MYC) to generate human iPSCs (hiPSCs) from human foreskin fibroblasts (HFFs). Similar to miPSCs, hiPSCs showed both increased ssDNA-BrdU exposure, indicating a higher number of resected DSBs (Figure 1D) and longer resected tracks (Figures 1E and 1F) than the parental HFF somatic cells, despite the lack of any exogenous source of DNA damage. In fact, this effect in hiPSCs was more exacerbated than in miPSCs, as hiPSCs had up to a 3-fold gain on the length of resected DNA. All of our results point toward a hyper-activation of DNA resection during reprogramming in mouse and human cells, which likely minimizes the impact of the DNA damage caused during the process.
CtIP Levels Increase in miPSCs and hiPSCs
CtIP activates DNA end processing in HR and is known to be a major regulator of DNA end resection (Cruz-Garcia et al., 2014, Wang et al., 2013, Nakamura et al., 2010, Huertas and Jackson, 2009, Huertas et al., 2008, Sartori et al., 2007). Indeed, we observed that CtIP was required for the resection hyper-activation observed in miPSCs (Figures S1A and S1C). Congruently, we observed that CtIP expression and protein levels increased in miPSCs with respect to primary MEFs, concurrent with the expression of the pluripotency marker Nanog (Figures 1G and S2A–S2C). We confirmed by immunoblot that hiPSCs also incremented the expression and protein levels of CtIP in a concomitant manner to NANOG and OCT4 (Figures 1H, S2D, and S2E). These results clearly suggested that the hyper-active DNA resection in iPSCs requires CtIP upregulation during the reprogramming process, most likely to manage an increased load of DSBs that have to be repaired by HR. Thus, both mouse and human iPSCs showed intrinsic increased levels of CtIP and DNA end resection compared with their parent cells.
CtIP Is Required for Efficient Cell Reprogramming for Both Mouse and Human Cells
To determine the relevance of CtIP and its upregulation during cell reprogramming, we forced the induction process in cells with reduced levels of the CtIP protein. For this, we transduced MEFs with short hairpin RNA (shRNA) lentivirus against CtIP or control shRNA (shNon-Target [shNT]), which also contained the OSKM factors under doxycycline induction, and then analyzed them for reprogramming (Figure S2A). Three weeks after induction, we observed a dramatic and significant reduction of the number of colonies with stem cell-like morphology in CtIP-depleted cells compared with shNT control cells (Figure 2A). Moreover, CtIP-depleted colonies were smaller than control ones (Figure 2B). In agreement with results published previously (Chen et al., 2005), and as a control of the CtIP depletion effects in primary MEFs, we observed less proliferation in CtIP-downregulated MEFs (Figure S3A). However, this reduced proliferation was not associated with an increase in sub-G1 phase cells (Figure 2C, –Dox), even though, as expected, CtIP deficiency provoked a significant increase in DNA damage, as measured by γH2ax (Figures 2F and 2G). Interestingly, in agreement with the idea that unrepaired DNA damage during cell reprogramming triggers apoptosis as a consequence of genomic instability, we observed that reprogrammed mouse cells expressing an shRNA against CtIP showed a strong increase in sub-G1 cells 10 days after doxycycline induction (Figures 2C and 2D), compared with either reprogrammed cells expressing a control shRNA or MEFs depleted for CtIP. Further, we observed an increase of proteolytic cleavage and activation of caspase-3 in reprogrammed MEFs with short hairpin CtIP (shCtIP) at day 10 (Figure 2E), confirming that the increased levels of sub-G1 were indeed due to induced apoptosis.
Figure 2.

CtIP Deficiency Impairs Mouse Cell Reprogramming
(A) Doxycycline-inducible OSKM MEFs were transduced with shCtIP and shNT lentivirus prior to induction with doxycycline. Reprogramming efficiency was analyzed by counting the number of colonies in triplicate of each biological sample. Error bars indicate ±SEM of a biological triplicate.
(B) Representative pictures of miPSC morphology.
(C) The sub-G1 peak was quantified in MEFs (–Dox) or reprogrammed MEFs at day 10 (+Dox) by FACS analysis in cells downregulated for CtIP (black bars) or expressing a control shRNA (white bars). Error bars indicate ±SEM of three independent experiments.
(D) Representative cell-cycle plots of reprogrammed MEFs in each condition. At least three independent experiments were performed.
(E) Immunoblot of proactive and active/cleavage caspase-3 in reprogrammed MEFs at day 10 (+Dox) bearing the indicated shRNAs. A representative western blot is shown of three independent experiments.
(F) FACS quantification of cells positive for γH2ax. Other details as in (C).
(G) The indicated proteins were immunodetected in samples from OSKM-induced MEFs after 10 days of continuous doxycycline treatment. A representative western blot is shown of three independent experiments.
t Test statistical analyses in (A), (C), and (F) were performed using at least three independent experiments.
See also Figures S2 and S3.
We also detected that the amount of the DNA damage marker γH2ax increased slightly but significantly in cells containing shCtIP versus those with shNT, as shown by both FACS analysis and immunoblot (Figures 2F and 2G). Even though this difference was also observed in MEFs, it was more intense in reprogrammed cells, in agreement with CtIP playing a more critical role in repairing DNA damage during cell reprogramming. This suggests that excessive damage prompted the observed induction of apoptosis. Pluripotent status was confirmed by the observation of Nanog protein levels (Figures 2G, S2A, and S2C).
Efficient reprogramming requires functional repair pathways to favor an error-free dedifferentiation. In agreement with this, DDR was activated during reprogramming process, as shown by the increase in Chk1, γH2ax, and CtIP protein levels (Figures 2G and S2A). Reprogrammed cells that had downregulated CtIP also had increased DNA damage levels compared with reprogrammed control cells at 10 days after doxycycline induction (Figure 2G), supporting the idea that replication stress causes DNA damage and Chk1 activation during reprogramming (Ruiz et al., 2015).
Congruently, hiPSCs obtained from HFF cells by expression of OSKM factors in the presence of an shRNA against CtIP showed an increased sub-G1 peak (Figures 3A and 3B), an enrichment of the activated form of caspase-3 (Figure 3C) and elevated numbers of γH2AX-positive cells (Figure 3D), when compared with the same cells reprogramed bearing a control shRNA. In addition, expressing shCtIP constitutively during reprogramming completely blocked iPSC colony generation, while expressing control shRNA had no effects (not shown). We wondered whether this strong effect was due to cell death caused by a lack of CtIP in HFF cells (e.g., an excessive depletion of CtIP) or reflected a problem during reprogramming. Strikingly, CtIP depletion in HFF cells without OSKM expression neither changed their proliferation rate (Figure S3B) nor significantly induced DNA damage or apoptosis, measured as sub-G1 cells (Figures 3A and 3D). Thus, we conclude that the lack of iPSCs was not caused by problems in HFF cells, but by a critical role of CtIP in human cell reprogramming. To confirm this relevant role, we next generated iPSCs with limited CtIP downregulation by using an isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible shRNA (shCtIP-IPTG) and titrating IPTG concentration. Indeed, and similar to miPSCs, HFF cells partially depleted of CtIP by IPTG induction also formed fewer colonies, confirming that they had an impaired reprogramming efficiency (Figure 3E). In addition, colonies partially deficient for CtIP were smaller than control colonies (Figure 3F). Indeed, although we were able to expand several clones from the iPSCs with an IPTG-induced control shRNA (shNon-target [shNT] clones), we only successfully expanded one clone of iPSCs bearing the shCtIP-IPTG under IPTG induction (CtIP C1 clone; Figure 3G). Interestingly, we could recover CtIP expression on this clone by removing IPTG. As an additional control, we reprogrammed HFF cells containing shCtIP-IPTG to iPSCs in the absence of IPTG (C4 clone) (Figure 3G). Hence, by adding IPTG at different time points, we could compare distinct states of HFF cells after reprogramming: (1) reprogrammed in the absence of CtIP (C1 clone); (2) reprogrammed in the presence of CtIP and depleted of CtIP once pluripotency had been established (C4 clone); and (3) always with CtIP (Figure 3G). Analyzing these reprogrammed clones for chromosome number variation, we found that the C1 clone (reprogrammed without CtIP) had a significant increase in chromosomal aberrations, measured as aneuploidy, compared with those from the other two conditions (Figure 3H). This is in agreement with our results showing increased DNA damage markers when CtIP is absent during reprogramming (Figure 3D). We provide strong evidence that CtIP plays a role in avoiding the genomic instability generated specifically during cell reprogramming.
Figure 3.

CtIP Depletion Impairs Human Cell Reprogramming
(A) Percentage of cells in sub-G1 phase in HFF cells or those reprogrammed to iPSCs bearing either the control shRNA shNT (white bars) or shCtIP (black bars). Error bars indicate ±SEM of three independent experiments. p Values were calculated using the t test. ns, not significant.
(B) Representative FACS plots of cell-cycle analysis in reprogrammed HFF cells of each condition. At least three independent experiments were performed.
(C) Reprogrammed HFF cells bearing the indicated shRNAs at day 10 were immunoblotted to analyze for the proactive and active forms of caspase-3. A representative western blot is shown of three independent experiments.
(D) Percentage of positive cells for γH2AX. Other details as in (A).
(E) Reprogramming efficiency of human cells containing an IPTG-inducible shRNA against CtIP (shCtIP-IPTG) or the control shNT-IPTG was analyzed in three independent experiments. Error bars indicate ±SEM. Statistical significance was performed using a t test.
(F) Representative images of hiPSC morphology in cells harboring the inducible shCtIP-IPTG obtained in the presence or absence of IPTG, as indicated.
(G) Western blot analysis against the CtIP and OCT4 proteins in HFF cells and hiPSC clones bearing the indicated shRNAs and in the presence (+) or absence (−) of IPTG. A representative western blot is shown of three independent experiments.
(H) Analysis of chromosomes per metaphase in iPSCs harboring IPTG-inducible shCtIP or shNT and reprogrammed in the presence (+) or absence (−) of IPTG. Tukey's multiple comparison test was used with n > 75.
See also Figure S2.
Normal CtIP Levels during Reprogramming Are Required for Maintenance and Differentiation of iPSCs
Due to the difficulty of expanding iPSC colonies from MEFs or HFFs reprogrammed in the absence of CtIP, we wondered whether the genomic instability generated during reprogramming in CtIP-downregulated cells affected self-renewal and differentiation attributes. In fact, it is known that deficiency in BRCA1, another protein involved in HR and DNA end resection that interacts with CtIP, impairs maintenance of iPSCs in culture (Gonzalez et al., 2013). To deepen our understanding about the role of CtIP, we selected several miPSC clones obtained in the presence of shCtIP or a control shRNA. Taking advantage of the presence of the GFP gene on the plasmid, we analyzed the permanence of GFP cells in the colonies as a proxy for the presence of the shRNA targeting CtIP. We determined that all control shNT-harboring iPSC colonies maintained higher numbers of GFP cells than shCtIP iPSCs, suggesting that the cells that continued to grow and to maintain cell pluripotency had a tendency to lose the cassette containing GFP and the shRNA against CtIP (Figures 4A and 4B). Strikingly, this effect was specific for iPSC cells that had been reprogrammed in the presence of shCtIP and was not observed in primary MEFs with shCtIP (Figure S4). Thus, collectively, miPSC colonies reprogrammed with CtIP depletion had a natural selection favoring cells with normal CtIP expression (Figure 4C). This could explain the high CtIP levels in cells bearing shCtIP (Figure S2A). To study the kinetics of loss of shCtIP expression, we sorted GFP-positive miPSCs generated from cells containing either shNT-GFP or shCtIP-GFP and analyzed them for GFP disappearance during growth (Figure 4D). We found that GFP cells were rapidly purified from the cell population containing shCtIP-GFP compared with control cells, with a reduction in the number of GFP-positive cells of up to 50% after only three passages. We next performed a self-renewal assay using shCtIP- or shNT-miPSCs after enriching the populations of shRNA-bearing cells by cell sorting, using GFP as a marker. Again, reprogrammed cells depleted for CtIP formed less-viable colonies than control cells (Figures 4E and 4G). To differentiate whether this effect of CtIP deficiency was due to defects gained during cell reprogramming or those acquired during the pluripotent state of the reprogrammed cells, we transducted already-reprogrammed iPSCs with either an shRNA against CtIP or against a control sequence and analyzed them for self-renewal by colony formation. Interestingly, in this scenario, CtIP downregulation did not modify self-renewal capacity compared with control cells (Figures 4F and 4H). Along the same lines, the murine D3 embryonic stem cell line (ES-D3) was not affected by CtIP depletion in self-renewal experiments (Figures S5A–S5C).
Figure 4.

The Levels of CtIP During Reprogramming Affect iPSC Maintenance
(A) Percentage of GFP-positive cells in iPSC colonies isolated after MEFs reprogramming in the presence (white bars) or absence (black bars) of CtIP.
(B) Representative bright field (BF) and GFP images of miPSCs obtained by microscopy.
(C) Average percentage of GFP cells in several clones of shNT and shCtIP iPSCs. Error bars indicate ±SEM of different clones (n = 6 and n = 10). Statistical analysis was performed using a t test.
(D) Percentage of iPSCs harboring shCtIP-GFP or shNT-GFP during subsequence passages of the cells. The average and SD of five independent experiments is plotted. t test analysis for each passage is shown. ∗∗p < 0.01, ∗∗∗p < 0.005.
(E) The same amount of miPSCs reprogrammed either in the presence (white bars) or absence (black bars) of CtIP were seeded at low density and the number of each colony was measured. The relative number of colonies formed from three independent experiments is plotted. The t test was performed to compare both conditions.
(F) Same as (E) but with cells reprogrammed in the presence of CtIP, and then transduced with an shRNA against CtIP (black bars) or control shNT (white bars). Other details as in (E). ns, not significant.
(G) miPSCs reprogrammed in the presence of shCtIP or control shNT were immunoblotted to analyze the indicated proteins. A representative western blot is shown of three independent experiments.
(H) Same as in (G) but with cells transduced with an shRNA against CtIP after reprogramming.
See also Figures S4 and S5.
As self-renewal was compromised by inherited defects aroused upon cell reprogramming in the absence of CtIP, we wondered whether the differentiation process could also be jeopardized in a similar way. Despite the difficulty in expanding CtIP-deficient miPSCs, we were able to force mouse embryonic body differentiation from several clones, by removing the growth factor, leukemia inhibitory factor (LIF) for 6 days. We observed that miPSCs reprogrammed under CtIP depletion also generated embryonic bodies (EBs) smaller than their respective control cell reprogrammed in the presence of the control shNT, hence showing a clear deficiency in differentiation (Figures 5A–5C). We then tested if, as for self-renewal, this defect was a consequence of problems inherent to reprogramming in a CtIP-defective environment rather than a consequence of the loss of an active role of CtIP in stem cell differentiation. Indeed, CtIP downregulation in already-reprogrammed miPSCs (Figures 5D and 5E) or ES-D3 cells (Figures S5D–S5F) did not affect their differentiation capacity. Collectively, these data confirmed that genomic instability created during cell reprogramming under CtIP deficiency is the main cause of impairment of self-renewal and the differentiation process of miPSCs.
Figure 5.

CtIP Is Essential for Differentiation and Genomic Stability Maintenance in iPSCs
(A) Median size of embryonic bodies (EBs) generated at 6 days after spontaneous differentiation of miPSCs reprogrammed in the presence of shCtIP or control shNT. At least 300 EBs of each condition were measured. The average and SD of five independent experiments is plotted. Statistical significance was obtained by a t test.
(B) Representative images of EBs using BF and GFP filters.
(C) Western blot analysis of indicated proteins in EBs generated from miPSCs reprogrammed in the presence of shCtIP or control shNT. A representative western blot is shown of three independent experiments.
(D) Same as in (A), but with cells reprogrammed in the presence of CtIP and then transduced with shCtIP or shNT as indicated. ns, not significant.
(E) Same as in (C) but with cells transduced with shCtIP or shNT after reprogramming.
(F) Copy number variation (CNV) of genomic DNA between iPSCs obtained in the presence or absence of CtIP during cell reprogramming. Two independent experiments (the first and second assays) are shown. CNVs were detected using SurePrint G3 Unrestricted CGH (4 × 180 K) arrays. See also Figure S5.
To strengthen our hypothesis, we analyzed copy number variation (CNV) between early passes of shNT- or shCtIP-iPSC clones (two of each). We found a large difference in the number of CNV (>50 CNV) between miPSCs reprogrammed with CtIP depletion and their respective control cells with shNT (Figure 5F).
Discussion
Genomic instability is one of the biggest concerns in the potential clinical use of iPSCs (Rocha et al., 2013). The challenging field of regenerative medicine requires an in-depth understanding of the causes and consequences of genetic abnormalities that arise during the reprogramming process (Studer et al., 2015, Tabar and Studer, 2014, Buganim et al., 2013). Here, we study mouse and human reprogramming to iPSCs, to elucidate the role of DNA end resection as a relevant mechanism for avoiding genomic instability in this process. We demonstrate that expression of the CtIP protein, a key protein in DNA end resection (Sartori et al., 2007), is upregulated during the formation of iPSCs and is required for efficient reprogramming. A CtIP deficiency during reprogramming not only drastically impairs the reprogramming process but also endangers the future of the reprogrammed cells by critically limiting the maintenance of their pluripotency state and their further differentiation to EBs. All of these effects are likely caused by the genomic aberrations acquired by cells during reprogramming. These severe genomic consequences correlate with the well-established roles of CtIP in DNA resection, HR, and DSB repair pathway choice (Sartori et al., 2007, Huertas, 2010, Gomez-Cabello et al., 2013, López-Saavedra et al., 2016), rather than reflecting a novel role of CtIP in the reprogramming process. Intriguingly, and unexpectedly, the drastic effects of these CtIP roles are highly specific to the cell-reprogramming process: CtIP depletion in already-established iPSCs or ES cells does not reduce the ability of the cells to self-renew or differentiate into EBs. We hypothesize that this phenomenon is related to the load of endogenously induced DNA damage in these different situations. Cell reprogramming severely increases replication stress and therefore DNA damage (Ruiz et al., 2015) (see also Figures 1G, 2F, 2D, 3D, and S2A), while endogenous DNA damage in fibroblasts or iPSCs (under normal cell culture conditions) is low. Thus, these differences in the amount of DNA damage could explain why CtIP is essential during iPSC formation but not for maintenance of iPSCs or fibroblasts. Indeed, CtIP is essential for cell viability in cells that have been exposed to mutagens that result in high levels of DNA damage (Sartori et al., 2007, Huertas and Jackson, 2009) or chromosomal aberrations (Huertas and Jackson, 2009). Strikingly, and in agreement with this idea, our data suggest that cell reprogramming in wild-type human and mouse fibroblasts causes an increase in DNA resection that is comparable with high doses of ionizing radiation (Figure S1).
DDR and DNA repair genes have been shown to control genetic stability during cell reprogramming (Lu et al., 2016, Rocha et al., 2013, Hong et al., 2009, Kawamura et al., 2009, Li et al., 2009). Along those lines, we have now demonstrated that DNA end resection, a key process in DSB repair by HR, is hyper-activated in cells undergoing reprogramming compared with the parental somatic cells or already-differentiated cells. During reprogramming, cells not only have an increase in the amount of breaks that are resected, but also a gain in processivity, measured as the length of resected DNA. The most likely explanation for upregulation of DNA end resection is the occurrence of DNA damage during reprogramming. This idea is supported by an increase in the γH2AX marker levels after expression of reprogramming factors, as observed by us and others (Ruiz et al., 2015, Gonzalez et al., 2013, Tilgner et al., 2013). Replication stress, due to the presence of a stalled replication fork, is a major generator of DNA damage, and this is commonly resolved mainly by HR or microhomology-mediated end joining (Petermann and Helleday, 2010, Aguilera and Gomez-Gonzalez, 2008). Hence, the occurrence of DNA damage by replication stress during iPSC development could explain both the upregulation and essential role of DNA end resection in reprogramming.
Here we show that CtIP, a bona fide regulator of DNA end resection (Huertas and Jackson, 2009, Huertas et al., 2008, Sartori et al., 2007), is upregulated during cell reprogramming, and that it is essential for this process. Similarly, CHK1 has been suggested to be pertinent to generating iPSCs (Ruiz et al., 2015). CtIP is known to be a key player in maintaining genomic stability, and we reasoned that CtIP could be appropriately activated to repair DSBs generated by the presence of reprogramming factors. Selective CtIP depletion during mouse and human cell reprogramming interferes with iPSC generation and triggers apoptosis. In agreement with these data, BRCA1, which accelerates DNA resection through its interaction with CtIP (Cruz-Garcia et al., 2014), is also required for successful reprogramming (Gonzalez et al., 2013). In addition, other proteins linked to HR repair, such as BRCA2 and RAD51, are critical for cell reprogramming, at least in mice (Gonzalez et al., 2013).
We also observed that CtIP depletion during cell reprogramming in mouse and human cells hampers the growth and maintenance of their derived iPSCs. As a matter of fact, the human C1 clone obtained from cells bearing an inducible shRNA in the presence of IPTG showed the same long-term problems in viability regardless of the restoration of CtIP expression (data not shown, see Figure 3G for CtIP expression recovery). Likewise, Brca1-deficient MEFs have problems for cell reprogramming, and the derived iPSCs are unable to establish colonies (Gonzalez et al., 2013). This clearly differentiates proteins involved in early steps of HR, such as CtIP and BRCA1 in resection, from those affecting later steps, such as BRCA2 and RAD51, which are in fact dispensable for iPSC colony expansion (Gonzalez et al., 2013). We reasoned that iPSCs are defective for DNA end resection, which is critical for choosing between HR and NHEJ (Gomez-Cabello et al., 2013). As mentioned before, we suggest that both pluripotent and differentiated cells can likely repair their DNA by both NHEJ and HR, such that eliminating one of them only has a mild effect on cell viability unless an exogenous source of damage is present. However, during cell reprogramming, recombination is the only mechanism able to deal with the endogenous damage. We postulate that this is due to the nature of the DNA lesion, as replication stress will readily cause the appearance of one-ended DNA DSBs and will require recombination to restore replication. In the absence of CtIP, those breaks would be erroneously repaired, inducing the observed chromosomal abnormalities and CNV differences (Figures 3H and 5F). Indeed, this seems to be a recurrent mechanism that causes cells undergoing reprogramming to acquire a high degree of chromosomal instability. Even by analyzing cells from a single clone that differentiated in the absence of CtIP (the C1 clone), we can observe that each metaphase has a different number of chromosomes (Figure 3H). Thus, in the absence of CtIP, DSBs created by replication stress would be repaired through more mutagenic repair pathways, thereby increasing mutagenesis and chromosomal rearrangements (Bunting et al., 2010). This is consistent with an increase in DNA damage and genetic instability and would ultimately lead to apoptosis during reprogramming.
We could not expand the only clone we were able to obtain from CtIP-depleted hiPSCs (C1), but did establish a few clones from CtIP-depleted miPSCs, albeit with low efficiency. These clones showed high levels of alterations in chromosome number and CNV with respect to iPSCs generated under normal CtIP levels. These data are consistent with early embryonic lethality (E4.0) observed for CtIP knockout mice, which shows a slightly elevated apoptosis (Chen et al., 2005). Although this lethality has been previously associated with the retinoblastoma protein, our data support the idea that the resection function of CtIP is required for embryonic viability (Polato et al., 2014). Curiously, we found that established pluripotent cells, namely miPSCs and ES-D3 cells, did not require CtIP protein (at least not in the absence of an exogenous source of DNA damage). Cell reprogramming seems to be one such source of internal stress, so it is possible that other situations arise during normal embryogenesis in which CtIP, and specifically its resection activity, are essential.
Our data and those from previous reports suggest that cells from patients with deficient DDR and DNA repair pathways could not be efficiently reprogrammed and used in regenerative medicine. However, more in-depth investigations are needed to clarify which genes are specifically required to avoid genomic instability during the reprogramming process, to grant this powerful tool a future as a clinical standard procedure. Indeed, we suggest that, when studying self-renewal and differentiation of iPSCs, it is of capital importance to discriminate between the actual roles of repair proteins on those processes and the inherent, long-term consequences of genomic instability caused by reprogramming in a repair-defective environment.
Experimental Procedures
Cell Cultures
HEK293T, SNL, and C57BL/6 primary MEFs carrying a doxycycline-inducible tetracistronic cassette encoding the four murine reprogramming factors Oct4, Sox2, Klf4, and c-Myc (provided by M. Serrano) (Abad et al., 2013) were grown in DMEM (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich), 100 units/mL penicillin and 100 g/mL streptomycin (Sigma-Aldrich). HFF cells were growth in DMEM (Gibco) supplemented with 20% FBS (ATCC, LGC Promochem) and 100 units/mL penicillin (Sigma-Aldrich). miPSCs were cultured on gelatin-coated plates with DMEM/F12 + GlutaMAX (Gibco) supplemented with 20% knockout serum replacement (Gibco), 1,000 U/mL LIF (Millipore), 1% non-essential amino acids, 0.1 mM β-mercaptoethanol, and 100 units/mL penicillin (Sigma-Aldrich). hiPSCs were cultured on SNL feeders with DMEM/F12 + GlutaMAX (Gibco) supplemented with 20% knockout serum replacement (Gibco), 10 ng/mL basic fibroblast growth factor (Miltenyi Biotec), 1% non-essential amino acids, 0.1 mM β-mercaptoethanol, and 100 units/mL penicillin (Sigma-Aldrich).
Retroviral and Lentiviral Production
Retroviral and lentiviral particles were produced in HEK293T cells as described previously (Gomez-Cabello et al., 2013) using the plasmids listed in Table S1. Lentiviruses harboring shRNA vectors (Sigma) targeting human CtIP (CAG AAG GAT GAA GGA CAG TTT), mouse CtIP (GCA AGG TTT ACA AGT CAA AGT), and a non-target sequence (GCG CGA TAG CGC TAA TAA TTT) were used.
Mouse and Human iPSC Reprogramming
For miPSCs, MEFs were seeded at 1.5 × 105 cells per well of a 12-well plate. MEFs were cultured in miPSC medium supplemented with 1 μg/mL doxycycline to induce the expression of OSKM and promote reprogramming. Medium was changed every 24 hr for 21 days or until iPSCs colonies appeared. iPSCs were then expanded in 6-well gelatin-coated plates and in iPSC medium without doxycycline. For hiPSCs, HFFs with low number of passes were incubated with retroviral supernatants containing OCT4, c-MYC, SOX2, and KLF4 three times for 48 hr each time. Cells were then plated on irradiated (45 Gy) SNL feeders, the medium was changed 1 day later to iPSC medium, and cells were incubated for a further 21–30 days. For IPTG induction reprogramming, HFFs were transduced with an IPTG-inducible shCtIP lentivirus or the respective shNT lentivirus as a control. After 48 hr, iPSC regular medium with 1 mM IPTG (Sigma) was added to start the reprogramming process and maintained during the whole process. Reprogramming efficiency was calculated as the number of colonies normalized to the number of cells seeded.
Single-Molecule Analysis of Resection Tracks
iPSCs and differentiated cells (MEFs and HFFs) were seeded in 6-well plates at the required density to reach 80% confluence at the time of harvest. Cells were grown in the presence of 10 μM BrdU (GE Healthcare) for 24 hr and then harvested. Single-molecule analysis of resection tracks (SMART) was performed as described previously (Cruz-Garcia et al., 2014).
Immunoblotting
Protein extracts were prepared in Laemmli buffer (4% SDS/20% glycerol, 120 mM Tris-HCl [pH 6.8]). Proteins were resolved by SDS-PAGE, transferred to polyvinylidene fluoride (Millipore) membrane and visualized by immunoblotting. Western blot analysis used the antibodies listed in Table S2. Results were visualized and quantified using an Odyssey Infrared Imaging System (LI-COR Biosciences).
Flow-Cytometric Analysis of DNA End Resection
MEF cells, HFF cells, hiPSCs, and miPSCs were prepared for FACS analysis as follows: cells were grown in the presence of 10 μM BrdU (GE Healthcare) for 16–18 hr and then detached using Accutase (eBioscence). Cells grown in absence of BrdU were used as FACS-negative control. Cells were fixed with 4% paraformaldehyde for 10 min at 4°C, permeabilized with 0.1% Triton X-100 in PBS, washed in PBS, and then blocked with 5% FBS in PBS. After blocking, cells were incubated with an anti-BrdU mouse monoclonal antibodies (Table S2) for 1–2 hr at room temperature, and then with the appropriate secondary antibody (Table S2) for 30 min at room temperature. Additional control cells without primary antibody were used to set up FACS conditions. Cells were then washed and resuspended in PBS. Samples were analyzed with a BD FACSCalibur flow cytometer (BD Biosciences, Ref: 342975). At least 10,000 events were recorded for each sample.
Flow-Cytometric Analysis Cell Cycle
Mouse and hiPSCs were grown in 6-well plates. After 2 days, Accutase was added to remove the cells, which were then fixed with 70% ethanol at 4°C for at least 1 day. Cells were then washed and resuspended in PBS. Samples were incubated with 1 mg/mL propidium iodide and 10 mg/mL RNase (Sigma) for 20 min prior to FACS analysis. Samples were analyzed with a BD FACSCalibur flow cytometer (BD Biosciences). At least 10,000 events were recorded for each sample.
Karyotyping
hiPSCs were grown for 48 hr on gelatinized 6-well plates. At 3 hr prior to cell collection, the medium was changed and supplemented with 0.1 μg/mL demecolcine (Sigma, D7385) for 2 hr. Cells were washed, detached with Accutase, and centrifuged at 200 × g for 5 min at 4°C. iPSCs were then resuspended in hypotonic KCl solution (0.56%) and incubated at 37°C for 10 min. Cells underwent two rounds of fixation in methanol:glacial acetic acid (3:1) and centrifugation, after which they were resuspended in fixation solution and stained with DAPI. DAPI-stained chromosomes from at least 75 cells were counted for each condition.
EBs
miPSCs growing in gelatinized 6-well plates were detached with Accutase, counted, and replated onto ultra-low attachment 6-well plate with iPSC regular medium without LIF for 3–4 days. EBs were analyzed for size and number through microscopic images using Adobe Photoshop CS6 (Adobe Systems Incorporated).
Array Comparative Genomic Hybridization
For mouse array comparative genomic hybridization, CNV was detected from genomic DNA isolated from iPSC clones and hybridized to SurePrint G3 Human High-Resolution 1 × 1 M Microarrays (CNV) (Agilent Technologies) following manufacturer's instructions. CNV was identified using Agilent CytoGenomics v2.0 analysis software, following ADM-2 algorithm suggested by Agilent Technologies.
All iPSC clones were obtained at early passes after reprogramming and colony selection.
Author Contributions
D.G-C. and C.C-R. generated and characterized mouse and hiPSCs. OSKM-inducible MEFs were generated by M.A. and M.S. D.G.-C. and P.H. supervised the work and wrote the paper.
Acknowledgments
We wish to thank the cytometer unit of CABIMER for technical support, Diana Aguilar for critical reading of the manuscript, and Veronica Raker for style corrections. This work was funded by an R + D + I grant from the Spanish Ministry of Economy and Competitivity (SAF2013-43255-P) and an ERC Starting Grant (DSBRECA).
Published: January 5, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.12.009.
Contributor Information
Daniel Gómez-Cabello, Email: daniel.gomez@cabimer.es.
Pablo Huertas, Email: pablo.huertas@cabimer.es.
Accession Numbers
The accession number for the microarray data reported in this paper is GEO: GSE90888.
Supplemental Information
References
- Abad M., Mosteiro L., Pantoja C., Canamero M., Rayon T., Ors I., Grana O., Megias D., Dominguez O., Martinez D. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature. 2013;502:340–345. doi: 10.1038/nature12586. [DOI] [PubMed] [Google Scholar]
- Aguilera A., Gomez-Gonzalez B. Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008;9:204–217. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- Buganim Y., Faddah D.A., Jaenisch R. Mechanisms and models of somatic cell reprogramming. Nat. Rev. Genet. 2013;14:427–439. doi: 10.1038/nrg3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting S.F., Callen E., Wong N., Chen H.T., Polato F., Gunn A., Bothmer A., Feldhahn N., Fernandez-Capetillo O., Cao L. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.L., Liu F., Cai S., Lin X., Li A., Chen Y., Gu B., Lee E.Y., Lee W.H. Inactivation of CtIP leads to early embryonic lethality mediated by G1 restraint and to tumorigenesis by haploid insufficiency. Mol. Cell. Biol. 2005;25:3535–3542. doi: 10.1128/MCB.25.9.3535-3542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Garcia A., Lopez-Saavedra A., Huertas P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 2014;9:451–459. doi: 10.1016/j.celrep.2014.08.076. [DOI] [PubMed] [Google Scholar]
- Gomez-Cabello D., Jimeno S., Fernandez-Avila M.J., Huertas P. New tools to study DNA double-strand break repair pathway choice. PLoS One. 2013;8:e77206. doi: 10.1371/journal.pone.0077206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez F., Georgieva D., Vanoli F., Shi Z.D., Stadtfeld M., Ludwig T., Jasin M., Huangfu D. Homologous recombination DNA repair genes play a critical role in reprogramming to a pluripotent state. Cell Rep. 2013;3:651–660. doi: 10.1016/j.celrep.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore A., Li Z., Fung H.L., Young J.E., Agarwal S., Antosiewicz-Bourget J., Canto I., Giorgetti A., Israel M.A., Kiskinis E. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer W.D., Ehmsen K.T., Liu J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H., Takahashi K., Ichisaka T., Aoi T., Kanagawa O., Nakagawa M., Okita K., Yamanaka S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat. Struct. Mol. Biol. 2010;17:11–16. doi: 10.1038/nsmb.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P., Jackson S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009;284:9558–9565. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P., Cortes-Ledesma F., Sartori A.A., Aguilera A., Jackson S.P. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–692. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura T., Suzuki J., Wang Y.V., Menendez S., Morera L.B., Raya A., Wahl G.M., Izpisua Belmonte J.C. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140–1144. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Collado M., Villasante A., Strati K., Ortega S., Canamero M., Blasco M.A., Serrano M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136–1139. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G., Zhang Y. Genetic and epigenetic variations in iPSCs: potential causes and implications for application. Cell Stem Cell. 2013;13:149–159. doi: 10.1016/j.stem.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- López-Saavedra A., Gómez-Cabello D., Domínguez-Sánchez M.S., Mejías-Navarro F., Fernández-Ávila M.J., Dinant C., Martínez-Macías M.I., Bartek J., Huertas P. A genome-wide screening uncovers the role of CCAR2 as an antagonist of DNA end resection. Nat. Commun. 2016;7:12364. doi: 10.1038/ncomms12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., Li H., Baccei A., Sasaki T., Gilbert D.M., Lerou P.H. Influence of ATM-mediated DNA damage response on genomic variation in human induced pluripotent stem cells. Stem Cells Dev. 2016;25:740–747. doi: 10.1089/scd.2015.0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayshar Y., Ben-David U., Lavon N., Biancotti J.C., Yakir B., Clark A.T., Plath K., Lowry W.E., Benvenisty N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010;7:521–531. doi: 10.1016/j.stem.2010.07.017. [DOI] [PubMed] [Google Scholar]
- Nakamura K., Kogame T., Oshiumi H., Shinohara A., Sumitomo Y., Agama K., Pommier Y., Tsutsui K.M., Tsutsui K., Hartsuiker E. Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLoS Genet. 2010;6:e1000828. doi: 10.1371/journal.pgen.1000828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E., Helleday T. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010;11:683–687. doi: 10.1038/nrm2974. [DOI] [PubMed] [Google Scholar]
- Polato F., Callen E., Wong N., Faryabi R., Bunting S., Chen H.T., Kozak M., Kruhlak M.J., Reczek C.R., Lee W.H. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J. Exp. Med. 2014;211:1027–1036. doi: 10.1084/jem.20131939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha C.R., Lerner L.K., Okamoto O.K., Marchetto M.C., Menck C.F. The role of DNA repair in the pluripotency and differentiation of human stem cells. Mutat. Res. 2013;752:25–35. doi: 10.1016/j.mrrev.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Ruiz S., Gore A., Li Z., Panopoulos A.D., Montserrat N., Fung H.L., Giorgetti A., Bilic J., Batchelder E.M., Zaehres H. Analysis of protein-coding mutations in hiPSCs and their possible role during somatic cell reprogramming. Nat. Commun. 2013;4:1382. doi: 10.1038/ncomms2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz S., Lopez-Contreras A.J., Gabut M., Marion R.M., Gutierrez-Martinez P., Bua S., Ramirez O., Olalde I., Rodrigo-Perez S., Li H. Limiting replication stress during somatic cell reprogramming reduces genomic instability in induced pluripotent stem cells. Nat. Commun. 2015;6:8036. doi: 10.1038/ncomms9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori A.A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S.P. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer L., Vera E., Cornacchia D. Programming and reprogramming cellular age in the era of induced pluripotency. Cell Stem Cell. 2015;16:591–600. doi: 10.1016/j.stem.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabar V., Studer L. Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nat. Rev. Genet. 2014;15:82–92. doi: 10.1038/nrg3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tilgner K., Neganova I., Moreno-Gimeno I., Al-Aama J.Y., Burks D., Yung S., Singhapol C., Saretzki G., Evans J., Gorbunova V. A human iPSC model of Ligase IV deficiency reveals an important role for NHEJ-mediated-DSB repair in the survival and genomic stability of induced pluripotent stem cells and emerging haematopoietic progenitors. Cell Death Differ. 2013;20:1089–1100. doi: 10.1038/cdd.2013.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Shi L.Z., Wong C.C., Han X., Hwang P.Y., Truong L.N., Zhu Q., Shao Z., Chen D.J., Berns M.W. The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet. 2013;9:e1003277. doi: 10.1371/journal.pgen.1003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Vodyanik M.A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J.L., Tian S., Nie J., Jonsdottir G.A., Ruotti V., Stewart R. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.