

Abstract
Ribozymes use divalent cations for structural stabilization, as catalytic cofactors, or both. Because of the prominent role of Ca2+ in intracellular signaling, engineered ribozymes with stringent Ca2+ selectivity would be important in biotechnology. The wild-type glmS ribozyme (glmSWT) requires glucosamine-6-phosphate (GlcN6P) as a catalytic cofactor. Previously, a glmS ribozyme variant with three adenosine mutations (glmSAAA) was identified, which dispenses with GlcN6P and instead uses, with little selectivity, divalent cations as cofactors for site-specific RNA cleavage. We now report a Ca2+-specific ribozyme (glmSCa) evolved from glmSAAA that is >10,000 times more active in Ca2+ than Mg2+, is inactive in even 100 mM Mg2+, and is not responsive to GlcN6P. This stringent selectivity, reminiscent of the protein nuclease from Staphylococcus, allows rapid and selective ribozyme inactivation using a Ca2+ chelator such as EGTA. Because glmSCa functions in physiologically relevant Ca2+ concentrations, it can form the basis for intracellular sensors that couple Ca2+ levels to RNA cleavage. Biochemical analysis of glmSCa reveals that it has co-opted for selective Ca2+ binding a nonspecific cation-binding site responsible for structural stabilization in glmSWT and glmSAAA. Fine-tuning of the selectivity of the cation site allows repurposing of this preexisting molecular feature.
Keywords: catalytic RNA, in vitro selection, phosphorothioate interference, SAXS, molecular exaptation
INTRODUCTION
Divalent cations stabilize the 3D architecture of ribozymes and other structured RNAs (for review, see Draper 2004; Koculi et al. 2007). In addition, alkaline earth ions directly participate in the catalytic mechanism of several ribozymes (for review, see Fedor and Williamson 2005; Ferré-D'Amaré and Scott 2010; Aufinger et al. 2011). The specific identity of the divalent cation is generally less important for electrostatic structural stabilization than for catalytic cofactor function. For instance, the Tetrahymena ribozyme can adopt a near-native fold in the presence of Ca2+, Ba2+, or Sr2+, but is inactive unless its active site is bound to Mg2+ (Grosshans and Cech 1989; Celander and Cech 1991; Koculi et al. 2007). Structure-guided engineering and in vitro evolution have been used to alter the cation specificity of several natural ribozymes. In the case of the Tetrahymena ribozyme, in vitro selection produced variants that have 300-fold increased activity in Ca2+ (Lehman and Joyce 1993; Riley and Lehman 2003). However, these also had increased activity in Mg2+. Biochemical analyses suggested that their active sites did not acquire specificity for Ca2+; instead they relaxed their cation selectivity (Cernak et al. 2008). Similarly, in vitro evolution of the ribonuclease P ribozyme for cleavage in Ca2+ produced a point mutant that acquired the ability to cleave in this cation. However, and despite a decrease of reactivity in Mg2+, the cleavage rate of the mutant in Ca2+ was only 91-fold higher than in Mg2+ (Frank and Pace 1997). Consistent with its intracellular abundance, Mg2+ is preferentially used by most natural ribozymes that depend on a divalent cation as a catalytic cofactor. Ribozymes specific for Ca2+ would have biotechnological applications, given the importance of this metal ion in intracellular signaling (for review, see Clapham 2007). Altering the cation selectivity of natural ribozymes so that they are stringently dependent on Ca2+ has remained a challenging molecular engineering problem.
The glmS ribozyme is a bacterial gene-regulatory RNA that controls the abundance of glucosamine-6-phosphate (GlcN6P), an essential precursor for cell-wall biosynthesis (for review, see Ferré-D'Amaré 2010). It is part of the mRNA for the enzyme GlcN6P synthetase. GlcN6P binds to the ribozyme and activates its self-cleavage (Winkler et al. 2004). This leads to degradation of the mRNA, thereby enabling negative-feedback regulation of GlcN6P synthesis (Collins et al. 2007). Uniquely among self-cleaving RNAs, the glmS ribozyme uses an exogenous small molecule, GlcN6P, as an obligate catalytic cofactor (Klein and Ferré-D'Amaré 2006; Viladoms and Fedor 2012). Consistent with the coenzyme function of GlcN6P, the activity of this ribozyme is largely insensitive to the identity of divalent cations. Thus, and as expected for their role solely in structural stabilization, the glmS ribozyme exhibits comparable GlcN6P-catalyzed activity in the presence of Mg2+, Ca2+, Mn2+, or even the complex ion cobalt (III) hexammine (Roth et al. 2006; Klawuhn et al. 2010). The amino ligands of the latter are kinetically inert, and therefore activity of the ribozyme in this compound is strong evidence that inner-sphere cation coordination is not required for folding or catalysis (Cowan 1993).
Previously, we examined whether RNAs closely related to the wild-type glmS ribozyme (glmSWT) could perform divalent cation-mediated catalysis without using the coenzyme GlcN6P (Lau and Ferré-D'Amaré 2013). The evolutionary ancestors of glmSWT could have been such unregulated ribozymes. Acquisition of GlcN6P-dependence by RNAs like these would have given rise to the gene-regulatory capability of the present-day glmSWT (Ferré-D'Amaré 2011). We discovered that three adenosine mutations in the ribozyme core produced a variant (termed glmSAAA) that is fully active (kobs ∼ 2.5 min−1, which is within 20-fold of glmSWT) in the presence of divalent cations alone. This mutant ribozyme was not stimulated by GlcN6P. As is the case for many engineered variants of natural ribozymes, glmSAAA has only modest cation specificity; its maximum activity in Ca2+ exceeds that in Mg2+ by less than a factor of 50 (Lau and Ferré-D'Amaré 2013).
The crystal structure of glmSAAA revealed essentially the same 3D-architecture as that of glmSWT, underscoring the high stability of this ribozyme fold, and demonstrating that this RNA structural scaffold can be functionalized to support divergent biochemical activities (GlcN6P and cation-mediated catalysis by glmSWT and glmSAAA, respectively; Lau and Ferré-D'Amaré 2013). The functional plasticity of the structurally rigid glmS ribozyme fold we uncovered suggested that other variants that exhibit a higher degree of cation selectivity than glmSAAA might exist. To test this hypothesis, we performed further in vitro selection experiments to isolate variants of the glmS ribozyme that are selectively activated by Ca2+. Such RNAs could potentially be developed into tools that link intracellular signaling by Ca2+ with RNA stability and gene expression. Unlike in the glmSAAA selection (Lau and Ferré-D'Amaré 2016a), these new experiments explicitly selected and counterselected for catalysis based on the identity of the divalent cation (Ca2+ versus Mg2+) in solution. This work has now produced a mutant of the glmS ribozyme that does not use GlcN6P for catalysis and is exquisitely selective for Ca2+. Small-angle X-ray scattering (SAXS) demonstrates that the mutant can fold equally in Ca2+ or Mg2+, excluding a primarily structural role for the cation. Biochemical analysis indicates that rather than evolving a new Ca2+-binding site, the mutant RNA co-opted a nonspecific cation-binding site that had played a structural role in both the glmSWT and glmSAAA ribozymes.
RESULTS
In vitro selection yields a highly Ca2+-selective glmS ribozyme variant
To seek variants of the glmS ribozyme that function without GlcN6P and are highly selective for specific metal ions, we performed in vitro selection using a pool of RNA sequences based on glmSAAA in which nucleotides near the active site that do not participate in forming the characteristic, triply pseudoknotted secondary structure (Klein and Ferré-D'Amaré 2006) of the ribozyme were mutagenized (Fig. 1), and enriched for cleavage in either Mg2+ or Ca2+ (Materials and Methods). Ribozymes with varying degrees of selectivity for Ca2+ were isolated after eight selection rounds (Fig. 2).
FIGURE 1.
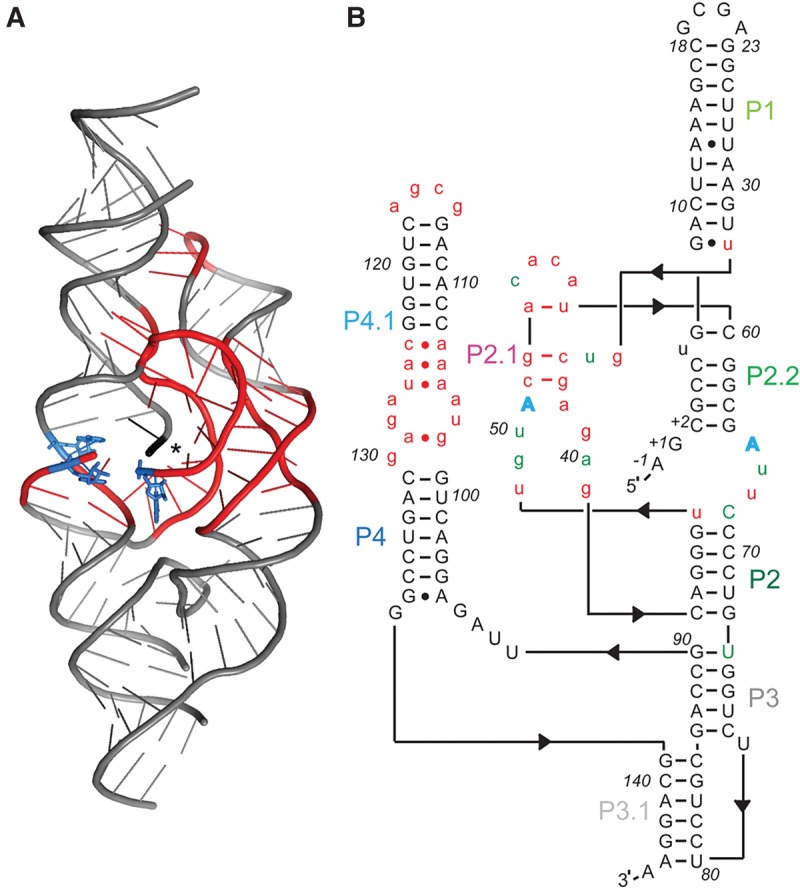
Mutagenized RNA pool based on glmSAAA for in vitro selection. (A) Cartoon depiction of the crystal structure of glmSAAA (Lau and Ferré-D'Amaré 2013). Nucleotides mutagenized for this study are in red. Blue residues denote the three adenosine mutations (relative to wild-type, A49, A51, G65) that characterize glmSAAA. (*) Location of scissile phosphate. (B) Sequence and hypothetical secondary structure of glmSCa. Nucleotides in red lower case were mutagenized by 30% relative to glmSAAA. Nucleotides in gray were constant. Two of the three adenosine mutations that characterize glmSAAA (blue) were retained in glmSCa. Nucleotides in green denote the eight mutations in glmSCa relative to glmSAAA.
FIGURE 2.
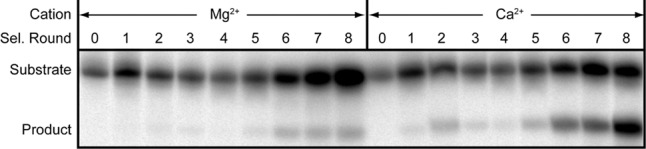
In vitro selection of cation-specific ribozymes from glmSAAA. RNAs from each of eight rounds in the Ca2+-specific selection (Pool B, Materials and Methods) were 32P-body-labeled and incubated in either 25 mM Mg2+ or 25 mM Ca2+ for 1 h. Samples were analyzed by denaturing PAGE and visualized by autoradiography.
A group of ribozymes that displayed strong discrimination against Mg2+ was chosen for further analyses. These RNAs all have in common 10 point mutations from the wild-type sequence (eight mutations relative to glmSAAA; Figs. 1B, 3), and reversion of any of these to the wild-type nucleotide reduced or eliminated Ca2+-dependent activity (not shown). A ribozyme containing just these mutations (hereafter glmSCa) displayed high selectivity for Ca2+. Its cleavage rate was at least 10,000-fold faster in 2 mM Ca2+ than in 2 mM Mg2+. This is a lower bound because this ribozyme exhibited no detectable activity in Mg2+, and therefore the estimate is limited by the sensitivity of our cleavage assay. Even in the presence of 100 mM Mg2+ (Fig. 4A), glmSCa was inactive. This Ca2+-selective ribozyme is also inactive in common monovalent and lanthanide ions and the soft divalent ion Cd2+, but exhibits residual activity in Sr2+ (Fig. 4A). Like glmSAAA (Lau and Ferré-D'Amaré 2013), glmSCa is inactive in cobalt hexammine, indicating that it requires inner-sphere cation coordination for activity.
FIGURE 3.

Alignment of sequences from three selections after nine rounds. (A) Mg2+-dependent selection, pool A. (B) Ca2+-dependent selection, pool B. (C) Ca2+-dependent selection with mutagenic PCR, pool C (Materials and Methods). Nucleotides in red were mutagenized by 30% relative to glmSAAA in the selection pool. Nucleotides in black were constant. The three adenosines that characterize glmSAAA are in cyan. The eight mutations in glmSCa (relative to glmSAAA) are boxed in green. For comparison, glmSWT and glmSAAA sequences are shown at the bottom of each panel. Paired (duplex) regions of glmSWT and glmSAAA are labeled as in Figure 1A.
FIGURE 4.
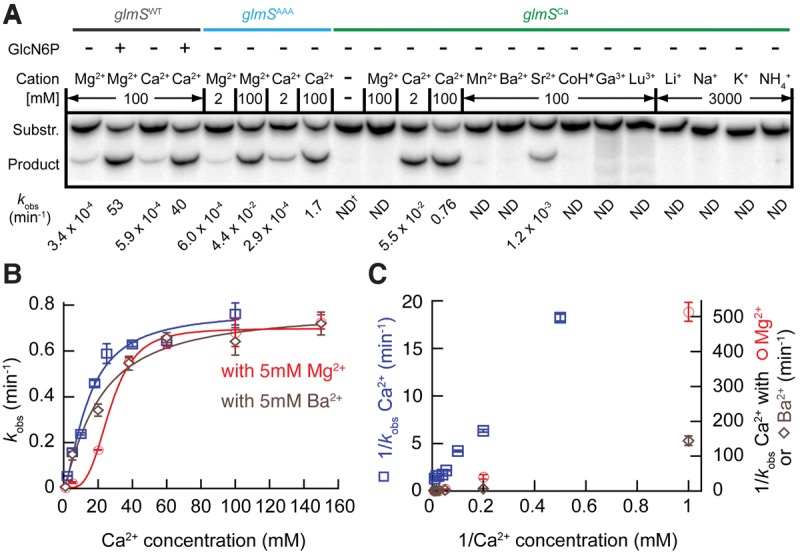
The glmSCa ribozyme is highly Ca2+-selective. (A) glmSCa activity in the presence of various metal ions. Autoradiogram of PAGE analysis of cleavage of a 5′ 32P-radiolabeled decanucleotide by different ribozymes for 30 min. Reaction rates are indicated. (*) Cobalt hexammine (CoH); (†) not determined (ND). (B) glmSCa activity in different Ca2+ concentrations (blue) and in the presence of an additional 5 mM Mg2+ (red) or 5 mM Ba2+ (brown). All reactions were in the presence of a background of 100 mM KCl. (C) Lineweaver–Burk representation of the reaction rates from B. Error bars represent standard errors of the mean of at least three independent experiments.
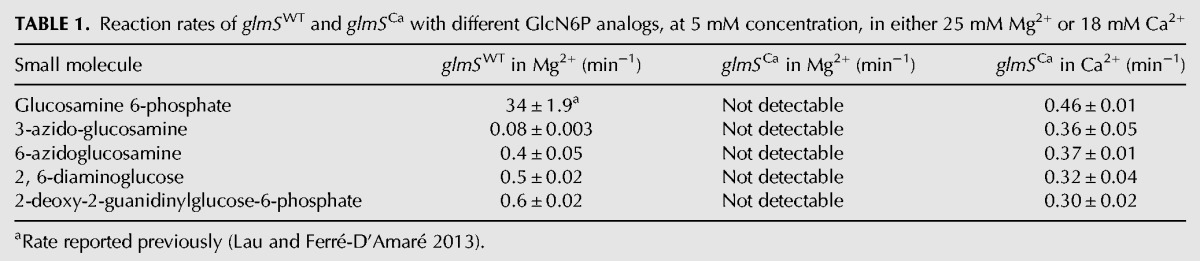
Comparison of the activity of glmSCa in the presence and absence of GlcN6P shows no stimulation by the cofactor of glmSWT (Table 1). Indeed, glmSCa is not stimulated by a number of other small molecule activators of glmSWT with vicinal amine and hydroxyl groups (McCarthy et al. 2005; Lim et al. 2006; Posakony and Ferré-D'Amaré 2013; Fei et al. 2014), nor a recently synthesized GlcN6P analog activator of glmSWT that bears a guanidinium group instead of the amine (Table 1). Thus, glmSCa, like glmSAAA from which it evolved, is a small molecule cofactor-independent ribozyme.
TABLE 1.
Reaction rates of glmSWT and glmSCa with different GlcN6P analogs, at 5 mM concentration, in either 25 mM Mg2+ or 18 mM Ca2+
Mg2+ inhibits glmSCa catalysis but not folding
Kinetic analysis shows that glmSCa attains half-maximal activity (K1/2) at 14 mM Ca2+ (in the presence of 100 mM KCl) (Fig. 4B). This is lower than for glmSWT (in saturating GlcN6P and 100 mM KCl) and glmSAAA (K1/2 = 31 and 93 mM, respectively, Lau and Ferré-D'Amaré 2013), indicating a higher affinity for the divalent cation. Addition of 5 mM Mg2+ to the Ca2+ titration of glmSCa yielded a sigmoidal relationship, and increased K1/2 by approximately twofold (Fig. 4B). Lineweaver–Burk analysis shows a nonlinear relationship in the presence of Mg2+, suggestive of mixed-type inhibition by this cation (Fig. 4C). Competition by Mg2+ with Ca2+ for inner-sphere binding to functionally important sites is further supported by analogous experiments in which a Ca2+ titration was supplemented with 5 mM Ba2+. In these, the K1/2 was increased by ∼1.5 fold, but the kinetics remained Michaelis–Menten. Presumably, the larger ionic radius of Ba2+ (Table 2) precludes it from competing efficiently for functionally important inner-sphere Ca2+ binding sites, but it can displace some diffusely RNA-associated (Draper 2004) Ca2+ ions (Fig. 4B).
TABLE 2.
Properties of select divalent cations
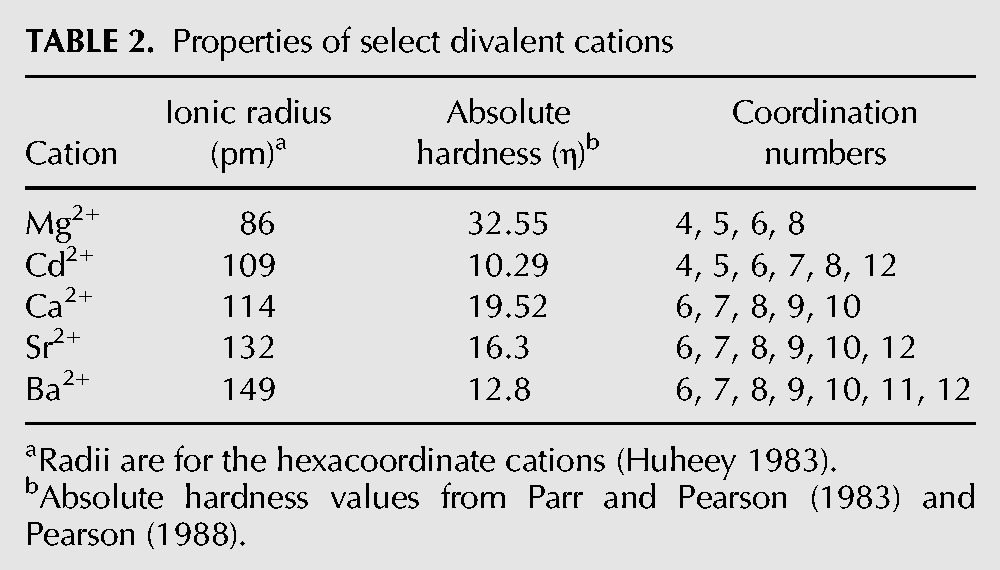
To examine whether glmSCa requires Ca2+ for folding, we compared its overall three-dimensional structure in solution in the presence of a near-physiological concentration of MgCl2 (Romani and Scarpa 1992; Grubbs 2002), or the same concentration of CaCl2 or SrCl2 (2.5 mM of the alkaline earth salt in addition to 100 mM KCl in each case, Materials and Methods) by small-angle X-ray scattering (SAXS). In the presence of Mg2+, glmSCa exhibits a radius of gyration (Rg) of 41.4 ± 1.2 Å. Unexpectedly, given the inactivity of the ribozyme in Mg2+, the shapes of its pair-probability distribution function P(r) (Fig. 5A) and Kratky plots (Fig. 5B) are indicative of a folded RNA. The Rg of the RNA in the presence of either Ca2+ or Sr2+ is identical within experimental error to that in Mg2+. Moreover, the P(r) and Kratky plots of the RNA in the three divalent cations tested are virtually identical (Fig. 5B). This analysis indicates that all three divalent cations can globally fold the RNA to a comparable extent at the same ionic strength. SAXS analysis, therefore, suggests that inactivity of glmSCa in Mg2+ reflects inhibition of the chemical step, rather than a deficit in folding. Under our experimental conditions, the Rg of the glmSAAA ribozyme (in the equivalent cis-acting form) in 2.5 mM MgCl2 is 38.5 ± 2.3 Å (not shown), suggesting that the parental ribozyme may be slightly more compact than glmSCa.
FIGURE 5.
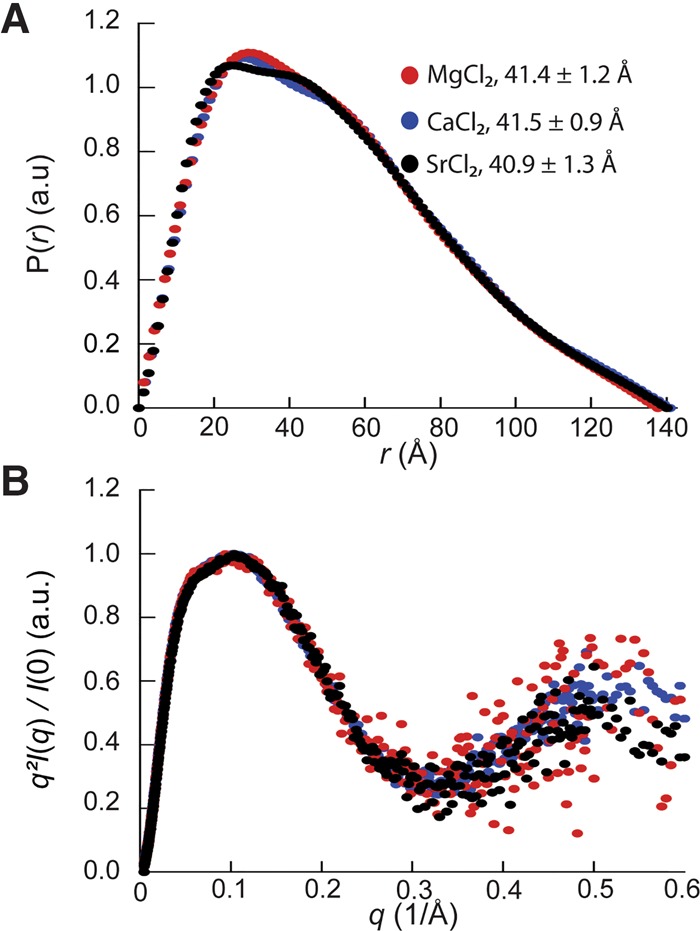
SAXS analysis indicates that glmSCa adopts the wild-type fold in a variety of divalent cations. (A) Pair-probability functions of glmSCa in the presence of 2.5 mM of either Mg2+, Ca2+, of Sr2+ (red, blue, and black, respectively) in addition to 100 mM KCl. The radii of gyration of the RNA in the three divalent cations are indicated (mean ± standard error of the mean of thirty measurements). (B) Kratky plots of glmSCa color-coded as in A.
The glmSCa ribozyme cleaves one nucleotide 3′ to glmSWT
Both glmSWT and glmSAAA cleave RNA by internal transesterification, yielding products with 2′,3′-cyclic phosphate and 5′-OH termini (Winkler et al. 2004; Lau and Ferré-D'Amaré 2013). To characterize the cleavage reaction of glmSCa, we compared its 5′ cleavage product with those of glmSWT and glmSAAA. Upon treatment of the 5′ cleavage products of the three ribozymes with acid or polynucleotide kinase, they exhibited parallel gel mobility shifts consistent with opening and removal, respectively, of a 2′,3′-cyclic phosphate (Fig. 6A). This suggests that glmSCa-catalyzed RNA cleavage also proceeds through internal transesterification.
FIGURE 6.
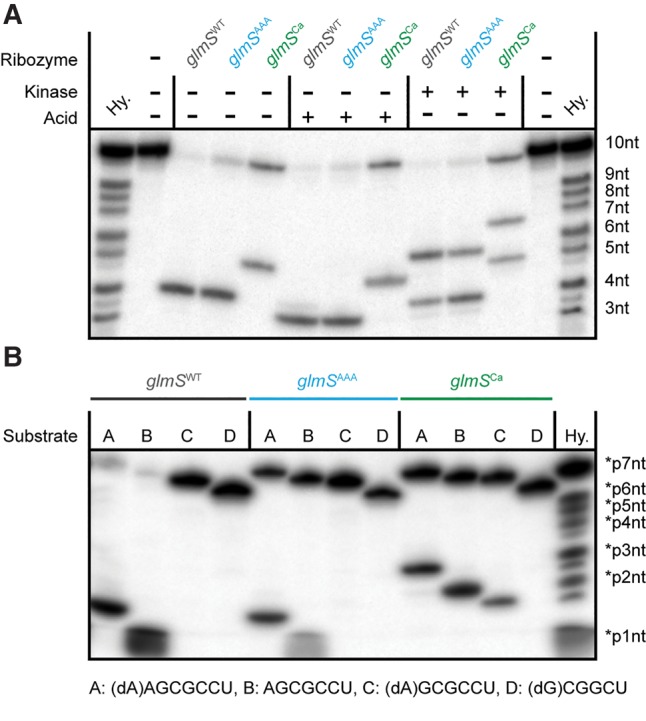
Similar to glmSWT and glmSAAA, glmSCa RNA cleavage proceeds by internal transesterification, but at a site 1 nt distal. (A) 5′ Cleavage product end-group analysis. 5′ 32P-radiolabeled substrate was cleaved in trans with the three ribozymes for 30 min and treated with acid or with phage T4 polynucleotide kinase prior to analysis by denaturing PAGE, as previously described (Winkler et al. 2004; Xiao et al. 2008; Lau and Ferré-D'Amaré 2013). (B) Mapping of glmSCa cleavage site. 5′ 32P-radiolabeled of RNA substrates of varying lengths with or without single-site 2′-deoxy substitutions were cleaved by the three ribozymes and analyzed by denaturing PAGE. (Hy.) Hydrolysis ladder.
Unexpectedly, the 5′ cleavage product of glmSCa had an electrophoretic mobility consistent with being one nucleotide longer than the products of the two other RNAs. This hinted that the cleavage site of the Ca2+-selective ribozyme shifted one nucleotide 3′ to the cleavage site of the wild type. To confirm this, we subjected oligonucleotides bearing a 2′-deoxyribose at positions −2, −1, or +1 (by convention, there is no zero position) to cleavage by the three ribozymes (Fig. 6B). Because the 2′-OH group is the nucleophile in the transesterification reaction, substitution of the reactive ribose with a deoxyribose is expected to abrogate cleavage. Consistent with previous experiments, glmSWT and glmSAAA cleavage was dependent on a 2′-OH at position −1 (Winkler et al. 2004; Lau and Ferré-D'Amaré 2013). In contrast, glmSCa could cleave a substrate with the deoxy substitution at position −1, but not at position +1. Overall, these experiments indicate that glmSCa cleaves its substrate between positions +1 and +2 by using the ribose 2′-OH of the former as the nucleophile.
Phosphorothioate interference suggests location of an essential Ca2+
To locate Ca2+ ions important for glmSCa activity, we performed phosphorothioate interference mapping. This technique exploits the altered affinity for hard cations (e.g., Mg2+ or Ca2+) resulting from substitution of nonbridging phosphate oxygen (NBPO) atoms of an RNA with sulfur (Schatz et al. 1991). We synthesized glmSCa analogs with random sulfur substitution of pro-RP NBPOs by transcription, and separated active from inactive RNAs by electrophoresis following self-cleavage. Positions within glmSCa with deleterious substitutions were identified by comparing the relative abundance of active versus inactive RNA fragments after selective phosphorothioate cleavage with iodine. In our mapping analysis of glmSCa, which provided readout between residues G3 and G90, we observed strong interferences only at residues A40 and G41 (Fig. 7).
FIGURE 7.
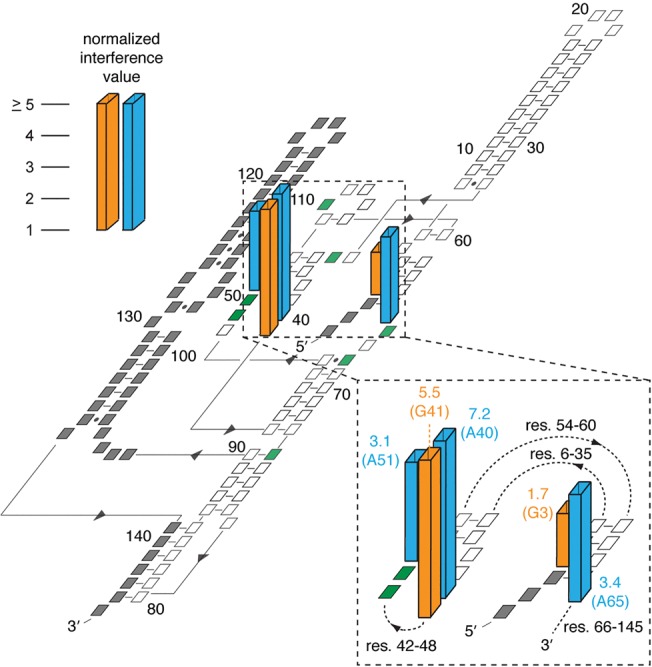
Strong phosphorothioate interferences in glmSCa. Normalized interference values (Lau and Ferré-D'Amaré 2013) are shown. Only positions with interferences >1 are shown, and interferences >5 are plotted as having the value 5.0. Interferences from AαS (adenosine phosphorothioate) and GαS are in cyan and orange, respectively. Significant interferences were not observed from CαS and UαS. (Inset) Close-up of residues with strong interferences near the active site (res., residue numbers). Residues are colored green and gray in the plane of the secondary structure to represent the eight mutations in glmSCa relative to glmSAAA, and positions that were not addressed in the phosphorothioate mapping experiment, respectively.
Previously, we identified functionally important divalent cations for glmSWT and glmSAAA by combining unbiased interference mapping and site-specific phosphorothioate substitution. We found that for both RNAs, the pro-Rp NBPO of residue C2 is important in metal ion coordination. The best ordered structural metal ion discovered in multiple crystallographic analyses of the two ribozymes (Fig. 8A) is coordinated by this oxygen atom (Klein and Ferré-D'Amaré 2006; Cochrane et al. 2007, 2009; Klein et al. 2007a,b). This hexacoordinate cation, MA, is too far from the cleavage sites of either glmSWT or glmSAAA to participate directly in catalysis. Moreover, full activity of glmSWT in cobalt hexammine (Roth et al. 2006) strongly argues against any essential direct (i.e., inner-sphere) role for this, or any other divalent cation in folding or catalysis by the wild-type ribozyme (Cowan 1993).
FIGURE 8.

glmSCa co-opts a preexisting structural cation for activity. (A) The glmSAAA active site (Lau and Ferré-D'Amaré 2013). NBPOs with strong and moderate phosphorothioate interference are in red and pink, respectively. Scissile phosphate is in black (*). Residue (−1) is modeled (Lau and Ferré-D'Amaré 2013). (B) Phosphorothioate interferences in glmSCa mapped onto the glmSAAA structure. Residues that differ between glmSWT and glmSCa are in green. (C) Effect of NBPO sulfur substitutions on glmSCa. Substitutions of G3 serve as a quality control on the assembly of phosphorothioate-containing ribozymes. Activities of unsubstituted glmSCa, pro-Rp, and pro-Sp NBPO substitutions, and diastereomeric substitutions are in gray, yellow, purple, and orange, respectively. Sr2+ activities are shown in brown. (*), (**), and (***) denote 0.05 < P < 0.01, 0.001 < P < 0.01, and P < 0.001, respectively (two-tailed t-test).
Activity of glmSAAA was also dependent on metal ion coordination by the pro-Rp NBPO of residue A38 and the pro-Sp NBPO of residue C2 (Lau and Ferré-D'Amaré 2013). Crystallographic analysis confirmed the presence of a divalent cation bound at this position (MB). Multiple lines of evidence indicated that this cation participates directly in the catalytic mechanism of glmSAAA (but not glmSWT). In contrast, we did not observe any significant interference at A38 for glmSCa, which implies that this ribozyme does not require MB for catalysis. The interference at residues A40 and G41 of glmSCa is consistent with the presence of a metal ion (MC) observed in crystal structures of glmSWT and glmSAAA (Fig. 8B). MC is too far from the active site of glmSCa to participate in catalysis, and thus probably plays a structural role in all three ribozymes.
If neither MB nor MC is involved in catalysis by glmSCa, where is the essential metal ion cofactor? Examination of crystal structures suggests that by virtue of its novel cleavage site one nucleotide 3′ to the scissile phosphates of glmSWT and glmSAAA, glmSCa may recruit MA for catalysis (Fig. 8B). We tested this by site-specific phosphorothioate substitution. Consistent with lack of involvement of MB in catalysis by glmSCa, sulfur substitution of the pro-Sp NBPO of C2 only reduced the maximal cleavage fraction to ∼80% (Fig. 8C). In contrast, substitution of the pro-Rp NBPO of C2, which coordinates MA, markedly impaired glmSCa activity, with the maximum extent of cleavage reduced to ∼9%. This impairment could not be relieved with Sr2+, which suggests a high degree of cation selectivity of the MA site in glmSCa (it could also not be rescued with the thiophilic cation Cd2+, in which the ribozyme is inactive, not shown). Substitution of both NBPOs at G3 had no effect on activity, arguing against a nonlocal, nonspecific effect of sulfur substitution. Finally, we confirmed the importance of the structural cation MC suggested by our mapping results by synthesizing glmSCa constructs containing a diastereomeric mixture of phosphorothioates at residues A40 and G41 (the internal position of the substitution precluded chromatographic resolution of the diastereomers). These had moderately impaired activity that could be partially relieved by Sr2+ (Fig. 8C). Overall, our analysis indicates that MA is essential for glmSCa activity, without precluding a role in facilitating overall ribozyme folding.
DISCUSSION
Ten point mutations distinguish glmSCa from the GlcN6P-dependent wild-type ribozyme (Figs. 1, 3). Two of these, U49A and G65A, are shared with glmSAAA. Previous analyses of glmSAAA demonstrated that while both adenosine mutations contribute to the coenzyme-independence of that ribozyme, the mutation at position 65 is the most important in abrogating the ability of the RNA to use GlcN6P as a general acid catalyst (Lau and Ferré-D'Amaré 2013). These mutations are therefore consistent with the GlcN6P-independence of glmSCa. Site-directed mutagenesis has previously demonstrated that the phylogenetically conserved guanine at position 40 is essential for the activity of both glmSWT and glmSAAA (Cochrane et al. 2007; Klein et al. 2007a; Lau and Ferré-D'Amaré 2013). Structural analysis implicated the purine base of this residue in orienting the nucleophile of the transesterification reaction (the 2′-OH of residue −1), and possibly in facilitating its deprotonation (Klein and Ferré-D'Amaré 2006; Cochrane et al. 2007; Klein et al. 2007a). Moreover, Raman crystallographic investigation of the ribozyme demonstrated that G40 contributes to lowering the pKa of the amine of GlcN6P, the general acid catalytic moiety (Gong et al. 2011). The presence of the G40A mutation in glmSCa thus explains why this ribozyme no longer cleaves its substrates between positions −1 and +1 (Fig. 6). Of the remaining seven mutations, A35U is particularly interesting, as this residue is located in close proximity to the MA site (Fig. 8), and therefore may be involved in endowing Ca2+ selectivity to glmSCa.
Since the ionic radii of Ca2+ and Mg2+ differ by only 28 pm (Table 2), definitive characterization of the exquisite cation selectivity glmSCa will require atomic-resolution structural analysis of the transition state of the ribozyme. However, none of the mutations that characterize glmSCa disrupt the secondary structure elements that define the unusually stable glmS ribozyme fold (Fig. 1B). Therefore, previously determined structures of the RNA can guide provisional interpretation of our results (Fig. 8A,B). Because glmSCa is itself overall structurally stable (indicated by the fact that it can adopt structures indistinguishable by SAXS in the presence of either Mg2+, Ca2+, or Sr2+) (Fig. 5), and because its structure is comparable in compactness to that of the wild-type and glmSAAA, the cation selectivity of glmSCa likely results from a localized reorganization of the MA cation binding site. Our biochemical data are consistent with an enlargement of this site relative to that of the wild-type, such that it cannot achieve tight binding (at least in the transition state) of Mg2+ (ionic radius = 86 pm) while being optimal for Ca2+ (ionic radius = 114 pm), tolerant for the somewhat larger Sr2+ (ionic radius = 132 pm) and unable to accommodate the much larger Ba2+ (ionic radius = 149 pm; Table 2). This expansion of the MA site could be largely the result of the mutation of residue A35, which is immediately adjacent to this cation-binding site, to the smaller uracil (Fig. 8B). The inability of Sr2+ to activate a glmSCa ribozyme containing a sulfur substitution of the MA-coordinating pro-Rp NBPO of C2, despite its ability to weakly support catalysis of the unmodified glmSCa (Fig. 4), could hint that replacement of the nonbridging phosphate oxygen with sulfur makes the MA site too small to accommodate the larger Sr2+ cation. In addition, the inability of Cd2+ to rescue the same phosphorothioate-containing RNA, despite an ionic radius (109 pm) comparable to that of Ca2+ suggests that the remaining ligands of the MA site, or the substrate atoms with which MA interacts during catalysis, have a strong preference for hard cations (Table 2).
Compared to the glmSAAA ribozyme from which it was selected, glmSCa has adopted a new catalytic strategy that relies on MA. In the wild-type ribozyme, this cation is structural, since it lies too far away from the cleavage site of that ribozyme (Klein and Ferré-D'Amaré 2006). Moreover, as glmSWT is fully active in a variety of metal ions (including cobalt hexammine), its structural cation binding sites (e.g., MA) have limited selectivity (Roth et al. 2006; Klawuhn et al. 2010). Likewise, glmSAAA has only modest cation selectivity, and functions even in high concentrations of the monovalent cation Li+. Both crystallographic and phosphorothioate interference analyses indicate that MA retains a structural function in glmSAAA (Lau and Ferré-D'Amaré 2013). The co-option of an erstwhile structural metal ion for catalysis (directly or indirectly) by glmSCa is an example of molecular exaptation, that is, the evolutionary repurposing of preexisting features for adaptation to new selective pressures (Gould and Vrba 1982). The evolution of glmSCa illustrates both the rugged fitness landscape of ribozymes, wherein a small number of mutations can have large effects on activity (Pitt and Ferré-D'Amaré 2010; Lau and Ferré-D'Amaré 2016b), and the exceptional structural stability of the triply pseudoknotted glmS fold (Hampel and Tinsley 2006; Klein and Ferré-D'Amaré 2006; Tinsley et al. 2007; Lau and Ferré-D'Amaré 2013).
In addition to demonstrating that a self-cleaving ribozyme that requires a metal ion for activity can achieve remarkable specificity for Ca2+, glmSCa is a potentially useful biotechnological tool. Like in the case of the Ca2+-dependent protein nuclease from Staphylococcus (Heins et al. 1967), RNA cleavage by glmSCa can be initiated in the presence of Ca2+, and then abruptly stopped by addition of the Ca2+-specific chelator EGTA (Fig. 9). In addition, because this ribozyme is active in modest Ca2+ concentrations (∼1 mM), it may serve as the basis for new live-cell Ca2+ sensors whose output is site-specific RNA cleavage. Organelles such as the endoplasmic reticulum, mitochondria, and nucleus contain total Ca2+ levels of ∼0.01 to 1 mM. In stimulated tumor mast cells and hyperplastic prostate epithelial cells, this level can reach as high as 1.2 and 7 mM, respectively (Tvedt et al. 1987; Chandra et al. 1994). Such Ca2+ concentrations would provide strong and selective activation of glmSCa in vivo.
FIGURE 9.
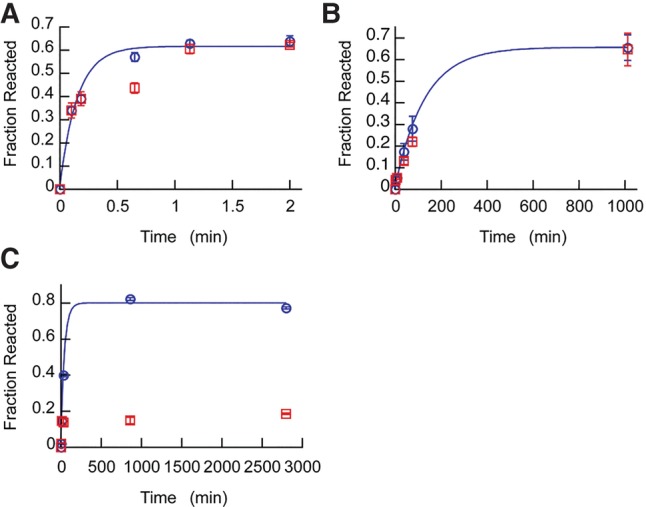
Chelation of Ca2+ by EGTA halts glmSCa, but not glmSWT or glmSAAA under physiological cation concentrations. Reaction time courses for (A) glmSWT (in the presence of 25 µM GlcN6P), (B) glmSAAA, and (C) glmSCa were initiated by the addition of both 25 mM Mg2+ and 25 mM Ca2+. After the second time point, half of the sample from each reaction was transferred into a fresh tube containing a twofold molar excess of EGTA. The reactions with (red) or without EGTA (blue) added were continued for the remaining time points. First-order rate fits are shown for reactions with no EGTA added (blue). Error bars are standard errors of the mean from three independent time courses.
MATERIALS AND METHODS
RNA pool synthesis and selection
A double-stranded mutagenized DNA pool was synthesized as described, with the exception of constant residues A49, A51, and A65 (Fig. 1; Lau and Ferré-D'Amaré 2013, 2016a). Approximately 100 nmol of DNA (∼6 × 1015 unique sequences) were used to template transcription of 32P body-labeled RNA for the first selection round. Two parallel selections were performed with 15 nmol of RNA in incubation buffers containing 50 mM HEPES-KOH (pH 7.5), and either 10 mM MgCl2 (Pool A) or 10 mM CaCl2 (Pool B). Active ribozymes were reverse-transcribed as previously described (Lau and Ferré-D'Amaré 2013). In round 2, cDNAs from Pool B were further divided into two before the second PCR step, with one (Pool B) amplified using standard PCR and the other (Pool C) using mutagenic PCR (20 mM Tris–HCl, pH 8.4, 50 mM KCl, 7 mM MgCl2, 0.5 mM MnCl2, 0.2 mM dATPs, 0.2 mM dGTPs, 1 mM dCTPs, 1 mM dTTPs) for 13 cycles (Cadwell and Joyce 1994). Starting with round 3, the divalent cation concentrations in the incubation buffers were reduced to 2 mM, and the 5′ primer was switched to 5′-TTCTAATACGACTCACTATAGGTGCACGATCTAAATACTGACCAATCTAAAGGATGCCTAGCGGCTGGACTTAAAGCCGCGAGG-3′ (T7 promoter in italics) during the second PCR step to yield a different leader sequence. A more stringent selection scheme was used starting with round 5, in which RNA pools were 5′ labeled with 6-thioguanosine 5′-monophosphate (6SGMP) and selected using an [(N-acryloylamino)phenyl]mercuric chloride (APM) gel mobility-shift assay (Igloi 1988; Lau and Ferré-D'Amaré 2013). A counter-selection step was also introduced starting with round 6 in which 6SGMP tagged RNAs were first incubated overnight in 50 mM HEPES, pH 7.5, with either 25 mM CaCl2, 25 mM MgCl2, or 25 mM MnCl2 for Pools A, B, and C, respectively. RNAs that did not self-cleave were recovered by electrophoresis through APM gels, and ribozymes specifically active in either Mg2+ or Ca2+ were selected using conditions described above.
Kinetics
Ribozyme rate measurements in different metal ion conditions were performed as previously described (Lau and Ferré-D'Amaré 2013). For determination of the inhibitory effects of Mg2+ and Ba2+ on glmSCa activity, 5 mM MgCl2 or 5 mM BaCl2 was added to cleavage reactions in varying concentrations of CaCl2 (from 2 to 150 mM). Pseudo–first-order rates were determined as previously described (Lau and Ferré-D'Amaré 2013). All cleavage rate measurements were performed on a background of 100 mM KCl.
Small-angle X-ray scattering
SAXS experiments were performed at beamline 12-ID-C of The Advanced Photon Source at Argonne National Laboratories. Unimolecular glmS ribozyme constructs were used in this study, rather than the bimolecular RNAs used by Lau and Ferré-D'Amaré (2013). The constructs correspond to those in Figure 1A, except that their first nucleotide is residue +1. RNA samples were extensively exchanged into a solution containing 20 mM HEPES-KOH pH 7.5, 100 mM KCl, and 10 μM EDTA using Millipore Amicon Ultra centrifugal concentrators (10 kDa molecular weight cutoff). The samples were then prepared for analysis on-site by heating to 85°C for 2 min, followed by buffer exchange into the buffer listed above, supplemented with 2.5 mM of the appropriate group II divalent ion. Samples were then passed through a Durapore PVDF 0.1 μm filter prior to UV/Vis analysis and SAXS data collection. Samples were allowed to equilibrate at room temperature for 30 min prior to collection. Samples, ranging from 0.7–1.5 mg/mL, were passed through a flow cell, during which they were exposed to the X-ray beam (12 keV) for 0.5 sec, followed by a 2.0 s rest time. Thirty data sets were collected per sample using a Pilatus 2M detector. Prior to averaging, each data set was examined for radiation damage and aggregation using Igor Pro (WaveMetrics). Guinier analysis was performed using Igor Pro. Indirect Fourier transformation was performed using the programs DATGNOM and GNOM (Svergun 1992; Petoukhov et al. 2007).
Phosphorothioate interference mapping and NBPO substitution experiments
Phosphorothioate interference mapping was performed using 185-nt cis-cleaving glmSCa constructs, and interference values were calculated as previously described (Lau and Ferré-D'Amaré 2013). For individual NBPO substitution experiments, RNA oligos 5′-AG*CGCCU-3′ and 5′-AGC*GCCU-3′ ([*] indicates position of diastereomeric phosphorothioate substitutions) were purchased from Dharmacon. Rp and Sp phosphorothioate isomers were resolved by reversed-phase HPLC (Frederiksen and Piccirilli 2009). Rates for NBPO substitutions at C2 or G3 were determined by incubation of HPLC-purified RNA oligos (Rp or Sp), respectively, with a 126-nt trans-acting glmSCa construct in 25 mM HEPES-KOH, pH 7.5, 5 mM CaCl2. Of note, 126-nt trans-acting glmSCa constructs containing diastereomeric sulfur substitutions at the NBPOs of A40 or G41 were synthesized by enzymatic ligation of a 104 nt glmSCa ribozyme truncated to start at position 42 (CAGGGU … CAGGAA), with RNA oligo 5′-GGCUUUAAGUUGUCGAG*AG-3′ and 5′-GGCUUUAAGUUGUCGAGA*G-3′, respectively. The ligated RNAs were purified by 8% denaturing PAGE. The ribozymes were incubated with 5′ 32P-labeled RNA oligo 5′-AGCGCCUGGACUUAAAGCC-3′ to determine the effects of A40 or G41 NBPO substitutions. The same RNA constructs and conditions were used for Sr2+ experiments, except 5 mM Ca2+ was replaced with 5 mM Sr2+.
ACKNOWLEDGMENTS
We thank J. Sellers for access to rapid-quench instrumentation, X. Zuo and the staff at beamline 12-ID-C of the Advanced Photon Source (APS), Argonne National Laboratory (ANL) for SAXS data collection support, S. Bachas, N. Baird, M. Chen, T. Dever, J. Hogg, C. Jones, J. Posakony, K. Warner, and J. Zhang for discussions, and two anonymous referees for motivating the SAXS experiments and suggesting the inclusion of Table 2. M.W.L. was a Croucher Foundation Postdoctoral Fellow. SAXS data were collected in a core facility of the Center for Cancer Research, US National Cancer Institute (NCI), allocated under an agreement between NCI and ANL (PUP-24152). Use of APS was supported by the US Department of Energy. This work was supported in part by the Intramural Program of the National Heart, Lung and Blood Institute, National Institutes of Health (NIH).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.059824.116.
REFERENCES
- Aufinger P, Grover N, Westhof E. 2011. Metal ion binding to RNA. Met Ions Life Sci 9: 1–35. [PubMed] [Google Scholar]
- Cadwell RC, Joyce GF. 1994. Mutagenic PCR. PCR Methods Appl 3: S136–S140. [DOI] [PubMed] [Google Scholar]
- Celander DW, Cech TR. 1991. Visualizing the higher order folding of a catalytic RNA molecule. Science 251: 401–407. [DOI] [PubMed] [Google Scholar]
- Cernak P, Madix RA, Kuo LY, Lehman N. 2008. Accommodation of Ca(II) ions for catalytic activity by a group I ribozyme. J Inorg Biochem 102: 1495–1506. [DOI] [PubMed] [Google Scholar]
- Chandra S, Fewtrell C, Millard PJ, Sandison DR, Webb WW, Morrison GH. 1994. Imaging of total intracellular calcium and calcium influx and efflux in individual resting and stimulated tumor mast cells using ion microscopy. J Biol Chem 269: 15186–15194. [PubMed] [Google Scholar]
- Clapham DE. 2007. Calcium signaling. Cell 131: 1047–1058. [DOI] [PubMed] [Google Scholar]
- Cochrane JC, Lipchock SV, Strobel SA. 2007. Structural investigation of the GlmS ribozyme bound to its catalytic cofactor. Chem Biol 14: 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane JC, Lipchock SV, Smith KD, Strobel SA. 2009. Structural and chemical basis for glucosamine 6-phosphate binding and activation of the glmS ribozyme. Biochemistry 48: 3239–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins JA, Irnov I, Baker S, Winkler WC. 2007. Mechanism of mRNA destabilization by the glmS ribozyme. Genes Dev 21: 3356–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan JA. 1993. Metallobiochemistry of RNA. Co(NH3)63+ as a probe for Mg2+ (aq) binding sites. J Inorg Biochem 49: 171–175. [DOI] [PubMed] [Google Scholar]
- Draper DE. 2004. A guide to ions and RNA structure. RNA 10: 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedor MJ, Williamson JR. 2005. The catalytic diversity of RNAs. Nat Rev Mol Cell Biol 6: 399–412. [DOI] [PubMed] [Google Scholar]
- Fei X, Holmes T, Diddle J, Hintz L, Delaney D, Stock A, Renner D, McDevitt M, Berkowitz DB, Soukup JK. 2014. Phosphatase-inert glucosamine 6-phosphate mimics serve as actuators of the glmS riboswitch. ACS Chem Biol 9: 2875–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré-D'Amaré AR. 2010. The glmS ribozyme: use of a small molecule coenzyme by a gene-regulatory RNA. Q Rev Biophys 43: 423–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré-D'Amaré AR. 2011. Use of a coenzyme by the glmS ribozyme-riboswitch suggests primordial expansion of RNA chemistry by small molecules. Philos Trans R Soc Lond B Biol Sci 366: 2942–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré-D'Amaré AR, Scott WG. 2010. Small self-cleaving ribozymes. Cold Spring Harbor Persp Biol 2: a003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, Pace NR. 1997. In vitro selection for altered divalent metal specificity in the RNase P RNA. Proc Nat Acad Sci 94: 14355–14360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiksen JK, Piccirilli JA. 2009. Separation of RNA phosphorothioate oligonucleotides by HPLC. Methods Enzymol 468: 289–309. [DOI] [PubMed] [Google Scholar]
- Gong B, Klein DJ, Ferré-D'Amaré AR, Carey PR. 2011. The glmS ribozyme tunes the catalytically critical pKa of its coenzyme glucosamine-6-phosphate. J Am Chem Soc 133: 14188–14191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould SJ, Vrba ES. 1982. Exaptation—a missing term in the science of form. Paleobiology 8: 4–15. [Google Scholar]
- Grosshans CA, Cech TR. 1989. Metal ion requirements for sequence-specific endoribonuclease activity of the Tetrahymena ribozyme. Biochemistry 28: 6888–6894. [DOI] [PubMed] [Google Scholar]
- Grubbs RD. 2002. Intracellular magnesium and magnesium buffering. Biometals 15: 251–259. [DOI] [PubMed] [Google Scholar]
- Hampel KJ, Tinsley MM. 2006. Evidence for preorganization of the glmS ribozyme ligand binding pocket. Biochemistry 45: 7861–7871. [DOI] [PubMed] [Google Scholar]
- Heins JN, Suriano JR, Taniuchi H, Anfinsen CB. 1967. Characterization of a nuclease produced by Staphylococcus aureus. J Biol Chem 242: 1016–1020. [PubMed] [Google Scholar]
- Huheey JE. 1983. Inorganic chemistry principles of structure and reactivity. Harper International, Cambridge, MA. [Google Scholar]
- Igloi GL. 1988. Interaction of tRNAs and of phosphorothioate-substituted nucleic acids with an organomercurial. Probing the chemical environment of thiolated residues by affinity electrophoresis. Biochemistry 27: 3842–3849. [DOI] [PubMed] [Google Scholar]
- Klawuhn K, Jansen JA, Souchek J, Soukup GA, Soukup JK. 2010. Analysis of metal ion dependence in glmS ribozyme self-cleavage and coenzyme binding. Chembiochem 11: 2567–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DJ, Ferré-D'Amaré AR. 2006. Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science 313: 1752–1756. [DOI] [PubMed] [Google Scholar]
- Klein DJ, Been MD, Ferré-D'Amaré AR. 2007a. Essential role of an active-site guanine in glmS ribozyme catalysis. J Am Chem Soc 129: 14858–14859. [DOI] [PubMed] [Google Scholar]
- Klein DJ, Wilkinson SR, Been MD, Ferré-D'Amaré AR. 2007b. Requirement of helix P2.2 and nucleotide G1 for positioning of the cleavage site and cofactor of the glmS ribozyme. J Mol Biol 373: 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koculi E, Hyeon C, Thirumalai D, Woodson SA. 2007. Charge density of divalent metal cations determines RNA stability. J Am Chem Soc 129: 2676–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau MWL, Ferré-D'Amaré AR. 2013. An in vitro evolved glmS ribozyme has the wild-type fold but loses coenzyme dependence. Nat Chem Biol 9: 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau MWL, Ferré-D'Amaré AR. 2016a. In vitro evolution of coenzyme-independent variants from the glmS ribozyme structural scaffold. Methods 106: 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau MWL, Ferré-D'Amaré AR. 2016b. Many activities, one structure: functional plasticity of ribozyme folds. Molecules 21: E1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman N, Joyce GF. 1993. Evolution in vitro of an RNA enzyme with altered metal dependence. Nature 361: 182–185. [DOI] [PubMed] [Google Scholar]
- Lim J, Grove BC, Roth A, Breaker RR. 2006. Characteristics of ligand recognition by a glmS self-cleaving ribozyme. Angew Chem Int Ed Engl 45: 6689–6693. [DOI] [PubMed] [Google Scholar]
- McCarthy TJ, Plog MA, Floy SA, Jansen JA, Soukup JK, Soukup GA. 2005. Ligand requirements for glmS ribozyme self-cleavage. Chem Biol 12: 1221–1226. [DOI] [PubMed] [Google Scholar]
- Parr RG, Pearson RG. 1983. Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105: 7512–7516. [Google Scholar]
- Pearson RG. 1988. Absolute electronegativity and hardness: application to inorganic chemistry. Inorg Chem 27: 734–740. [Google Scholar]
- Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. 2007. ATSAS 2.1—towards automated and web-supported small-angle scattering data analysis. J Appl Crystallogr 40: s223–s228. [Google Scholar]
- Pitt JN, Ferré-D'Amaré AR. 2010. Rapid construction of empirical RNA fitness landscapes. Science 330: 376–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posakony JJ, Ferré-D'Amaré AR. 2013. Glucosamine and glucosamine-6-phosphate derivatives: catalytic cofactor analogues for the glmS ribozyme. J Org Chem 78: 4730–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley CA, Lehman N. 2003. Expanded divalent metal-ion tolerance of evolved ligase ribozymes. Biochimie 85: 683–689. [DOI] [PubMed] [Google Scholar]
- Romani A, Scarpa A. 1992. Regulation of cell magnesium. Arch Biochem Biophys 298: 1–12. [DOI] [PubMed] [Google Scholar]
- Roth A, Nahvi A, Lee M, Jona I, Breaker RR. 2006. Characteristics of the glmS ribozyme suggest only structural roles for divalent metal ions. RNA 12: 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz D, Leberman R, Eckstein F. 1991. Interaction of Escherichia coli tRNASer with its cognate aminoacyl-tRNA synthetase as determined by footprinting with phosphorothioate-containing tRNA transcripts. Proc Natl Acad Sci 88: 6132–6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr 25: 495–503. [Google Scholar]
- Tinsley RA, Furchak JR, Walter NG. 2007. Trans-acting glmS catalytic riboswitch: locked and loaded. RNA 13: 468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tvedt KE, Kopstad G, Haugen OA, Halgunset J. 1987. Subcellular concentrations of calcium, zinc, and magnesium in benign nodular hyperplasia of the human prostate: X-ray microanalysis of freeze-dried cryosections. Cancer Res 47: 323–328. [PubMed] [Google Scholar]
- Viladoms J, Fedor MJ. 2012. The glmS ribozyme cofactor is a general acid-base catalyst. J Am Chem Soc 134: 19043–19049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. 2004. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 428: 281–286. [DOI] [PubMed] [Google Scholar]
- Xiao H, Murakami H, Suga H, Ferré-D'Amaré AR. 2008. Structural basis of specific tRNA aminoacylation by a small in vitro selected ribozyme. Nature 454: 358–361. [DOI] [PubMed] [Google Scholar]