

Abstract
Nonsense-mediated RNA decay (NMD) selectively degrades mutated and aberrantly processed transcripts that contain premature termination codons (PTC). Cellular NMD activity is typically assessed using exogenous PTC-containing reporters. We overcame some inherently problematic aspects of assaying endogenous targets and developed a broadly applicable strategy to reliably and easily monitor changes in cellular NMD activity. Our new method was genetically validated for distinguishing NMD regulation from transcriptional control and alternative splicing regulation, and unexpectedly disclosed a different sensitivity of NMD targets to NMD inhibition. Applying this robust method for screening, we identified NMD-inhibiting stressors but also found that NMD inactivation was not universal to cellular stresses. The high sensitivity and broad dynamic range of our method revealed a strong correlation between NMD inhibition, endoplasmic reticulum (ER) stress, and polysome disassembly upon thapsigargin treatment in a temporal and dose-dependent manner. We found little evidence of calcium signaling mediating thapsigargin-induced NMD inhibition. Instead, we discovered that of the three unfolded protein response (UPR) pathways activated by thapsigargin, mainly protein kinase RNA-like endoplasmic reticulum kinase (PERK) was required for NMD inhibition. Finally, we showed that ER stress compounded TDP-43 depletion in the up-regulation of NMD isoforms that had been implicated in the pathogenic mechanisms of amyotrophic lateral sclerosis and frontotemporal dementia, and that the additive effect of ER stress was completely blocked by PERK deficiency.
Keywords: NMD, cellular stress, alternative splicing, Psd-95, Ptbp1, Ptbp2, Hnrnpl, Srsf11, Tra2b, Upf2, PERK, IREα, ATF6α, Tdp-43, ALS, FTD, UPR, cryptic splicing
INTRODUCTION
Nonsense-mediated RNA decay (NMD) is a post-transcriptional quality control mechanism that ensures the fidelity of cellular gene expression by selectively degrading aberrant transcripts containing a premature stop codon (PTC) (Chang et al. 2007; Rebbapragada and Lykke-Andersen 2009; Popp and Maquat 2013; Lykke-Andersen and Jensen 2015). NMD is also an essential gene regulation mechanism that quantitatively determines the abundance of physiologic transcripts with NMD features (Nott et al. 2003; Rehwinkel et al. 2005; Chan et al. 2007; Isken and Maquat 2008; Chen 2010; Wang et al. 2011; Yepiskoposyan et al. 2011; Tani et al. 2012; Zheng and Black 2013; Lykke-Andersen et al. 2014; Peccarelli and Kebaara 2014). Most strikingly, snoRNA host genes and some alternatively spliced NMD-sensitive isoforms are consistently repressed by NMD in various cell types and organs (Weischenfeldt et al. 2008; Thoren et al. 2010; Lykke-Andersen et al. 2014; Lykke-Andersen and Jensen 2015). The coupling of alternative splicing and NMD (AS-NMD) is widely used by splicing regulators and some RNA binding proteins (RBP) to maintain their own homeostatic expression (Boutz et al. 2007; Lareau et al. 2007; Ni et al. 2007; Spellman et al. 2007; Saltzman et al. 2008). AS-NMD is also harnessed to control the expression of non-RBP genes during development (Zheng et al. 2012; Zheng 2016a). NMD regulation of endogenous genes is integrated into a variety of physiological settings including the fine-tuning of the unfolded protein response (UPR) (Maquat and Gong 2009; Sakaki et al. 2012; Lou et al. 2014; Oren et al. 2014; Karam et al. 2015). Despite extensive studies on the molecular mechanisms of target recognition and decay, less is known about the modulation of NMD activity. Several studies showed that NMD was inhibited as an adaptive response to hypoxia and calcium signaling (Gardner 2008; Nickless et al. 2014) or in the tumor microenvironment (Wang et al. 2011).
Given the essential role of NMD in disease pathogenesis and gene expression regulation, methods that precisely detect changes in cellular NMD activity are of great interest and can facilitate the analysis of NMD controls in response to extracellular stimuli or during development. Traditional methods rely on a pair of plasmid reporters, with one containing a PTC and the other lacking a PTC. The two contrasting reporters are separately delivered into the cultured cells, usually along with a third plasmid to control cell-to-cell variation, and the reporter transcripts are assayed individually. The reporter pair is engineered to normalize the impact of transcription and other regulatory mechanisms affecting transcript abundance in order to isolate NMD regulation. Variables inherent to a general reporter gene approach, including the degree of overexpression, transfection methods, the choice of the reporters and cell density, affect the robustness of the assay and have to be painstakingly controlled to enhance the signal-to-noise ratio. Cell lines stably expressing a reporter gene can mitigate variation induced by transient transfection, but they introduce new variables, such as integration loci and copy numbers, which unpredictably affect reporter readout (Zheng et al. 2013; Zheng 2016b).
Exogenous reporter assays are invaluable in characterizing molecular mechanisms of NMD. Their application has unfortunately been restricted to transfectable cells. There is an urgent need for a broadly applicable method to track cellular NMD activity where transfection of reporters is difficult, e.g., in primary cells, tissue organs and animals. Ideally this new method would also be simpler, faster, and less expensive than exogenous reporter assays.
To address these needs, we aimed to develop an assay based on endogenous NMD targets. Assaying endogenous targets does not have the limitations associated with exogenous reporters and does not require secondary validations. Hard-to-transfect samples and postmortem tissues also become assessable. Furthermore, multiple endogenous targets can be examined in parallel to improve the robustness of the assay, whereas the reporter approach usually deals with one exogenous target at a time. The advantages of directly assaying endogenous targets are summarized in Table 1.
TABLE 1.
Comparison between our method and traditional NMD reporter methods
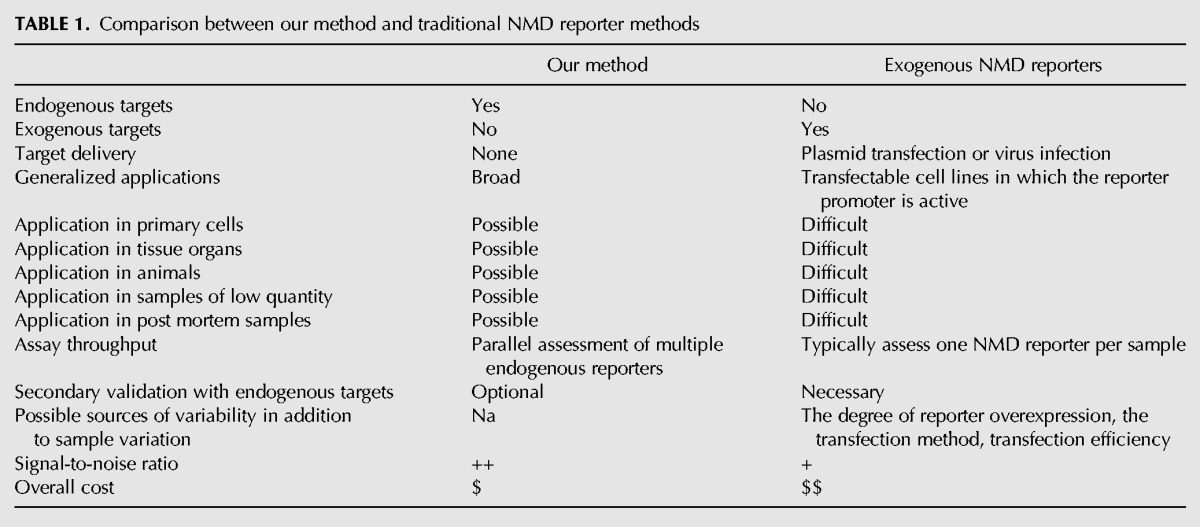
Monitoring NMD activity through endogenous NMD targets is, however, inherently challenging. It is hard to discern direct NMD targets. Although many genes have altered expression levels in NMD-deficient cells, some of the changes observed upon NMD deficiency may instead result from transcriptional regulation as an indirect consequence of NMD inhibition. It is also unclear whether their responses to NMD regulation depend on the cell type or cellular context. All endogenous NMD targets, direct or indirect, are under constant transcriptional regulation and subject to myriad possibilities of multifactorial regulation; hence, changes in their expression levels alone are not robust predictors of cellular NMD efficiency. These endogenous NMD targets are not suitable for unbiased screening of NMD modulators expecting high specificity and low false discovery, because transcriptional modulators would constitute a great proportion of the false positives. When a change in cellular NMD activity is an unknown and a change in the abundance of an NMD target is detected, researchers need a robust method to distinguish the pertinent regulatory processes and either confidently claim a change or avoid falsely reporting a change in NMD activity.
RESULTS
Development of a quantitative assay for monitoring changes in cellular NMD activity
To more effectively distinguish NMD regulation from transcriptional control, we designed our assay based on isoform-centric quantitation instead of gene-centric quantitation; thus, the analysis incorporated the non-NMD isoform of the same gene, which became one major advantage of our assay. A transcript with a stop codon >50 nt upstream of an exon–exon junction is consistently selected by NMD for degradation (Lykke-Andersen et al. 2000; Maquat et al. 2010). Such an RNA structure can result from alternative RNA splicing shifting the reading frame and creating a PTC. Because transcriptional regulation should equally affect the NMD and non-NMD isoforms of a given gene, the effects of transcriptional regulation can be better separated from those of NMD regulation. We focused on alternative isoforms that include or skip a small cassette exon to minimize the sequence difference between the two isoforms. We deliberately excluded alternative 5′ UTR, alternative 3′ UTR, and intron retention to avoid complications reflecting differential translation efficiency or miRNA targeting.
In our method, we measure the individual abundance of NMD-sensitive isoforms and that of their non-NMD counterparts via quantitative real-time PCR (qPCR). The NMD isoforms are first examined for differential expression between a treatment and a control condition. An increase in a NMD isoform may be due to NMD inhibition, transcriptional activation or a change in alternative splicing favoring the NMD isoform. These three scenarios can be distinguished by examining the expression of the non-NMD counterparts. Genuine NMD regulation, transcriptional activation and alternative splicing regulation would lead to no change, up-regulation, and down-regulation of the NMD-insensitive isoforms, respectively (Supplemental Fig. S1A). Similarly, a decrease in a NMD isoform can be interpreted as a result of enhanced cellular NMD activity if the non-NMD isoform exhibits no change (Supplemental Fig. S1B).
One technical difficulty of our method is designing RT-qPCR primers specific to the cassette exon-skipping isoform, whose entire sequence is present in the inclusion isoform. To specifically detect an inclusion isoform, we designed a primer entirely annealing to the cassette exon (exon B in Fig. 1A). For the exclusion isoform, junction primers annealing to the exon-skipping junction appear to be the only possible choices. This can be either a reverse primer (Fig. 1A) with its 5′ portion reverse complementary to the downstream constitutive exon (exon C) and its 3′ portion to the upstream constitutive exon (exon A) or a forward primer (not shown) with its 5′ and 3′ portions matching the upstream and downstream exons, respectively. The challenge is herein illustrated with reverse primers but also applies to forward primers. Many exon-skipping junction primers are able to amplify the inclusion isoforms (Supplemental Fig. S2). The longer the 3′ portion of the junction primer annealing to exon A, the easier the primer amplifies the exon-inclusion transcripts (Supplemental Fig. S2B). As we have observed, some exon-skipping junction primers with 3′ portions as short as 6 nt can still anneal to the inclusion isoform at 55°C–60°C, albeit at a lower efficiency than the exclusion isoform. On the other hand, a long 5′ portion and a short 3′ portion increase the possibility of the primer annealing to the exon B–exon C junction because of the general sequence similarity around 5′ splice sites (Supplemental Fig. S2C,D). We therefore identified NMD-associated cassette exons containing a 3′ end unlike that of its upstream exon. We then screened exon-skipping junction primers for one that amplified only the exclusion isoform and not the inclusion isoform. All primer pairs were tested and confirmed for their specificity, RT-qPCR efficiency, and linear dynamic ranges (Supplemental Table S1). Finally, two stably expressed housekeeping genes, Gapdh and Sdha, were used as internal controls for normalization by geometric averaging.
FIGURE 1.

A new quantitative method, distinct from alternative splicing assays, distinguishes NMD activity from alternative splicing and transcriptional regulation. (A) Schematics of the RT-qPCR method specifically detecting alternative splicing isoforms. The NMD isoform can be either the inclusion or exclusion isoform, while the other isoform is designated as the non-NMD isoform. The inclusion isoform is detected by primer F1 and isoform-specific primer R1. The exclusion isoform is detected by F2 and isoform-specific junction primer R2. Primers F3 and R3 are commonly used in alternative splicing assays to detect both inclusion and exclusion isoforms. (B) Conventional splicing assay to derive expression ratios of the Psd-95 NMD isoform relative to the non-NMD isoform in N2a cells depleted of UPF1 using two independent siRNAs or N2a cells overexpressing PTBP1. The ratios by themselves do not distinguish NMD regulation from alternative splicing regulation. Representative digital gel images are shown in the lower panel. A one-way ANOVA test was used to determine significant ratio changes between different samples, followed by a Tukey's multiple comparison test. N = 3. (C) RT-qPCR analysis of the Upf1 deficient cells as in B but using different primers specific to the Psd-95 non-NMD isoform and Psd-95 NMD (NPsd-95) isoform along with validation of the siUpf1 knockdown efficiency. (D) RT-qPCR analysis of the PTBP1-overexpressing cells as in B but using different primers specific to the Psd-95 isoforms described in C. Western blots (right panel) of the samples transfected with GFP and Flag-PTBP1 plasmids. Arrow: Flag-PTBP1; arrowheads: endogenous PTBP1 proteins that are down-regulated by PTBP1 overexpression. (E) Responses of the two Psd-95 isoforms to double knockdown of PTBP1 and PTBP2 in N2a cells assayed by RT-qPCR. Right panel shows the Western blots verifying the knockdown efficiencies. (F) RT-qPCR analysis of the two Psd-95 isoforms 4 h after 4 µg/µL actinomycin D treatment. (C–F) A two-way ANOVA test followed by Dunnett's multiple comparison tests was used to determine significant expression changes between samples. (*) P < 0.05. Error bars represent mean ± SEM.
Our method is different from an alternative splicing assay that simultaneously amplifies both isoforms in one RT-PCR, with primers in the flanking constitutive exons, then resolves the two isoforms by electrophoresis and derives an expression ratio between the inclusion and exclusion isoforms. Such an assay has been used to confirm AS-NMD targets but has not been used on its own to monitor NMD activity, because it cannot definitively distinguish NMD regulation from alternative splicing regulation. For example, the splicing assay could not discriminate between NMD regulation and alternative splicing regulation of postsynaptic density protein 95 (Psd-95, Dlg4). Psd-95 is transcribed in many cells including embryonic stem cells (Zheng 2016a). Both polypyrimidine tract binding proteins, PTBP1 and PTBP2, inhibit exon 18 of Psd-95, leading to a frameshift of the transcripts, which are then targeted by NMD (Zheng et al. 2012). The inclusion of exon 18 yields the non-NMD isoform. Thus, RNAi-mediated knockdown of the NMD factor Upf1 increased the exon 18-skipping NMD isoform, and PTBP1 overexpression promoted exon 18 skipping (Fig. 1B). Under these two experimental scenarios, the exclusion to inclusion isoform ratios derived from the alternative splicing assay could be very similar, and it is not possible to differentiate NMD regulation from alternative splicing regulation.
In contrast, our method effectively discriminated NMD regulation from alternative splicing regulation. When differential expression of the NMD isoform is detected, the expression of the non-NMD isoform is also scrutinized to determine whether and how the two isoforms are differentially regulated. In cells deprived of Upf1, the NMD isoform but not the non-NMD isoform exhibited significant changes (Fig. 1C). In cells overexpressing PTBP1, the non-NMD isoform significantly decreased and the NMD isoform increased (Fig. 1D). Our assay also accurately reported the response of Psd-95 to the loss of PTBPs. Both PTBP1 and PTBP2 promote the NMD isoform of Psd-95. As expected, RNAi-mediated knockdown of both PTBP1 and PTBP2 significantly decreased the level of the NMD isoform (Fig. 1E). Our assay attributed this change to alternative splicing regulation because of the concomitant increase in the non-NMD isoform (Fig. 1E).
To test whether our assay could discern transcriptional regulation as proposed, we applied actinomycin D to N2a cells to inhibit transcription of these AS-NMD targets. We used U6 snRNA as the internal control for qPCR. As expected, both NMD and non-NMD isoforms of Psd-95 were suppressed, which our method correctly assigned to transcriptional inhibition rather than NMD regulation (Fig. 1F). In summary, our assay precisely disclosed the distinct responses of the NMD and non-NMD isoforms of Psd-95 to transcriptional regulation, alternative splicing regulation, and NMD regulation.
Application of the new quantitative assay in animal samples
One advantage of assessing endogenous NMD targets over exogenous PTC reporters is the applicability in hard-to-transfect cells, tissue organs, postmortem samples, and samples of low quantity (Table 1). Using alternative splicing assays alone to distinguish NMD regulation from alternative splicing regulation in these kinds of samples is challenging. For example, the molar ratio between the Psd-95 exclusion and inclusion isoforms increased from 0.21 in control wild-type cortices to 1.7 in conditional knockout cortices of Upf2 (Upf2loxP/loxP; Emx1-cre), another key NMD factor, at embryonic day 17 (Fig. 2A, left panel). This ratio decreased to 0.11 and 0.02 in Ptbp2+/− and Ptbp2−/− cortices of the same age, respectively (Fig. 2A, right panel). Without prior knowledge of the sample identification, it would be impossible simply based on these numbers to attribute the observed ratio changes to either NMD regulation or alternative splicing regulation.
FIGURE 2.

The new method distinguishes NMD activity from alternative splicing regulation in animal samples. (A) Alternative splicing assay of the Psd-95 NMD and non-NMD isoforms in Upf2 conditional knockout cortices (left panel) and Ptbp2 null cortices (right panel). (Lower left panel) Western blot of UPF2 in Upf2loxP/lox and Upf2loxP/loxP; Emx1-cre cortices. The Upf2 conditional knockout cortices produce truncated UPF2 proteins (ΔUPF2). (Lower right panel) Genotyping of Ptbp2 animals. Statistics were calculated using a two-tailed unpaired Student's t-test for the Upf2 knockout samples (N = 2) and a one-way ANOVA test followed by Tukey's multiple comparison test for the Ptbp2 mutant samples (Ptbp2+/+ and Ptbp2+/−, N = 2; Ptbp2−/−, N = 4). (B,C) RT-qPCR analysis of the samples shown in A for individual expression of the Psd-95 non-NMD isoform and Psd-95 NMD (NPsd-95) isoform. (D–G) Expression levels of both NMD and non-NMD isoforms of Ptbp2, Tra2b, Hnrnpl, and Srsf11 in Upf2 knockout cortices relative to wild type. (B–G) A two-way ANOVA test followed by Dunnett's multiple comparison tests was used to determine significant expression changes between samples. (*) P < 0.05; (ns) not significant. Error bars represent mean ± SEM.
In contrast, without relying on the ratio analysis, our method effectively discriminated between NMD and alternative splicing regulation. In the conditional Upf2loxP/loxP; Emx1-cre cortices, our assay detected no change in the exon 18-inclusion isoform but an eightfold increase in the skipping (NMD) isoform (Fig. 2B). Our method therefore ascribed these changes to NMD regulation. Consistent with PTBP2 inhibiting exon 18 splicing and as shown by our assay, the levels of the inclusion isoform in the Ptbp2−/− cortices increased twofold relative to wild-type, while the exclusion isoform decreased significantly (Fig. 2C). Our method correctly attributed these changes to alternative splicing regulation. Thus, our method can be used on its own to effectively monitor cellular NMD activity in hard-to-transfect cells and organs as well as postmortem tissues.
To further improve the robustness and specificity of our assay, we added other known AS-NMD targets including heterogeneous nuclear ribonucleoprotein L (Hnrnpl), serine/arginine-rich splicing factor 11 (Srsf11 or Sfrs11), transformer 2 beta (Tra2b), and polypyrimidine tract binding protein 2 (Ptbp2, nPTB, or brPTB). The NMD transcript isoforms of these genes are as follows: Hnrnpl including exon 6, Srsf11 including exon 4, Tra2b including exon 2, and Ptbp2 excluding exon 10 (Stoilov et al. 2004; Boutz et al. 2007; Spellman et al. 2007; Saltzman et al. 2008). These are all small cassette exons that moderately distinguish the two isoforms at the sequence level. Monitoring different genes that are targeted by NMD upon either exon inclusion or exon skipping was intended to exclude false positives affecting splicing and to confirm consistent NMD regulatory patterns. These genes were included in the final assay also because RT-qPCR primers specific to their exclusion isoforms were successfully identified. To allow further improvement and standardization of the method by the community, we followed the guidelines of the international Real-time PCR Data Markup Language (RDML) consortium and provided detailed information about our qPCR assays (Supplemental Table S1 and Materials and Methods). When tested in the conditional Upf2loxP/loxP; Emx1-cre cortices, our methods showed that the non-NMD isoform levels of these genes barely changed. In contrast, the NMD isoforms were up-regulated significantly by seven- to 50-fold (Fig. 2D–G). These data confirmed NMD regulation of these genes and validated our method of including these additional reporters. These five genes encode very different proteins and have been studied in various cell lines and tissues. They are also widely transcribed and are not known to be transcriptionally coupled, making them suitable for broadly monitoring NMD activity.
Thapsigargin specifically enhances the endogenous NMD targets
Because our method can be used on its own to infer NMD regulation with high confidence, one of its applications is unbiased screening for changes in cellular NMD activity. We screened a small panel of widely used pharmacological inhibitors at various concentrations for their effects on NMD. The expression of both the NMD and non-NMD isoforms of the endogenous reporter genes were simultaneously measured before and after drug treatment. When the levels of all the NMD isoforms were altered in the same direction, the expression of the NMD and non-NMD isoforms under the treatment and control conditions was then subjected to ANOVA analysis to determine whether the two isoforms were differentially regulated. We were interested in drugs that affected the NMD isoforms across the board and minimally affected the non-NMD counterparts. Many chemicals, including protein kinase A inhibitor KT5720, Cam Kinase II inhibitor KN-93, PKC inhibitor GF109203X, and Na+/K+-ATPase inhibitor ouabain, did not show any effect on NMD activity (Supplemental Figs. S3, S4). However, thapsigargin had potent activity in blocking cellular NMD.
The abundance of the NMD isoform transcripts increased as early as 1 h after thapsigargin treatment and continued to increase gradually (Fig. 3). At 5 h, the NMD isoform levels were enhanced by three- to 10-fold. In contrast, the non-NMD isoform levels barely changed for Srsf11, Tra2b, and Hnrnpl and increased slightly to around 1.8-fold for Ptbp2 and Psd-95. Although transcription and alternative splicing regulation might have modestly contributed to the changes in Ptbp2 and Psd-95, the dramatic increase in the levels of the NMD isoforms for all five genes could not be attributed solely to transcriptional stimulation or splicing changes. Rather, the changes were consistent with attenuation of the decay pathway specific to these isoforms, i.e., NMD.
FIGURE 3.

Thapsigargin specifically enhances the endogenous NMD targets. (A–E) Temporal expression of NMD isoforms (red lines) and non-NMD isoforms (blue lines) of Srsf11, Ptbp2, Tra2b, Hnrnpl, and Psd-95 upon 0.2 µM thapsigargin treatment. Expression levels are normalized to DMSO-treated samples. A two-way ANOVA test was used to determine significant difference of expression between the two isoforms. (#) P < 0.05. Dunnett's multiple comparison tests were used to determine significant expression changes of individual isoforms over time relative to DMSO treatment. (*) P < 0.05. N = 3. Error bars represent mean ± SEM.
The new method reveals a strong correlation between ER stress, polysome disassembly, and NMD inhibition in a thapsigargin dose-dependent manner
Thapsigargin is a noncompetitive inhibitor of the sarco/endoplasmic reticulum Ca2+ ATPase. Thapsigargin treatment increases intracellular Ca2+ concentration, which stimulates various Ca2+ dependent signaling pathways. In fact, Nickless et al. (2014) recently reported that intracellular calcium inhibited NMD after administration of thapsigargin. Thapsigargin also induces ER stress. Some cellular stresses including hypoxia inhibit NMD via phosphorylation of eukaryotic initiation factor 2α (eIF2α) (Gardner 2008; Wang et al. 2011), although the exact mechanism underlying NMD inhibition by eIF2α phosphorylation remains to be determined (Karam et al. 2015). The diverging mechanisms reported by these studies were both based on the exogenous NMD reporter approach and gene-centric validation. We therefore investigated which of the proposed mechanisms our independent new method would support. We first tested whether some core NMD factors were affected at 5 h after thapsigargin treatment and found no changes in their expression (Supplemental Fig. 5). A previous study reported a 1.5- to 2.5-fold increase in Upf1 mRNA in HeLa cells 4 d after chronic depletion of various NMD factors (Yepiskoposyan et al. 2011). The apparent contrast may be due to the difference in the time scale of the treatment.
We examined whether an increase in intracellular Ca2+ was sufficient to inhibit NMD. We applied a wide dose range of ouabain, a cardiac glycoside also used by Nickless et al. (2014) and measured the abundance of both NMD and non-NMD isoforms at various time-points. However, no clear changes in cellular NMD efficiency were observed (Supplemental Fig. S4). Even at concentrations as high as 200 or 400 µM that clearly stimulated the immediate early genes c-fos and Pip92, the NMD isoforms were not substantially enhanced (Supplemental Fig. S4). To further test whether increased intracellular Ca2+ was sufficient to inhibit NMD, we treated the cells with ionomycin, a Ca2+ ionophore, as another method of raising the cytoplasmic Ca2+ concentration. Cells were responsive to ionomycin, with increased expression of the immediately early genes c-Jun and Pip92 (Supplemental Fig. S6A). However, ionomycin treatment in a range of 1 to 100 µM did not specifically alter the levels of the NMD isoforms (Supplemental Fig. S6B). Ionomycin had no effect on Ptbp2 expression but equally repressed both NMD and non-NMD isoforms of Psd-95, suggesting that ionomycin inhibited Psd-95 transcription. Taken together, the results of ouabain and ionomycin treatment suggested that enhanced cytosolic Ca2+ signaling was not sufficient to inhibit NMD in N2a cells.
We investigated another consequence of thapsigargin treatment, ER stress, for its possible association with NMD inhibition. We first examined cellular stress levels by measuring the splicing activity of X-box binding protein 1 (Xbp1), because thapsigargin-induced NMD inhibition occurred as early as 1 h post-treatment and Xbp1 splicing responds rapidly to ER stress. As a part of the unfolded protein response (UPR) triggered by ER stress, the serine/threonine protein kinase/endoribonuclease inositol-requiring enzyme 1 (IRE1α) oligomerizes and activates its ribonuclease activity through trans-autophosphorylation (Samali et al. 2010; Rubio et al. 2011; Hetz 2012; van Schadewijk et al. 2012). Activated IRE1α excises a 26-nt intron of Xbp1 mRNA, resulting in a shorter isoform (Xbp1s) that encodes a potent transcriptional activator for the expression of chaperones. RT-PCR with primers flanking the 26-nt intron detected Xbp1 splicing at 1 h after thapsigargin treatment (Fig. 4A). The intron excision was continuously enhanced thereafter, mirroring the kinetics of NMD inhibition. IRE1α was reported to be targeted by NMD (Oren et al. 2014; Karam et al. 2015), and we also observed that thapsigargin enhanced Ire1α expression (Supplemental Fig. S7A). However, Ire1α expression remained unchanged at 1 h when Xbp1s was first detected, suggesting that the initial Xbp1 splicing was due to oligomerization-induced activation of IRE1α protein. Subsequent Ire1α induction may potentiate Xbp1 intron excision. In summary, NMD inhibition and Xbp1 intron excision happened almost simultaneously in the early phase of thapsigargin treatment.
FIGURE 4.

Dose-dependent correlation between ER stress, polysome disassembly, and NMD inhibition upon thapsigargin treatment. (A) Time course analysis of Xbp1 splicing upon application of 0.2 µM thapsigargin in N2a cells. N = 3. (B) Thapsigargin dose-dependent Xbp1 splicing in N2a cells at 5 h after treatment. N = 3. (C) Thapsigargin dose-dependent expression of the NMD (red lines) and non-NMD (blue lines) isoforms of Srsf11, Ptbp2, Tra2b, Hnrnpl, and Psd-95 in N2a cells at 5 h after treatment. A two-way ANOVA test was used to determine the significant difference between the two isoforms. (#) P < 0.05. Dunnett's multiple comparison tests were used to determine significant expression changes of individual isoforms after thapsigargin treatment in comparison to DMSO treatment. (*) P < 0.05. N = 3. Error bars represent mean ± SEM. (D) Polysome fractionation graphs of N2a cells at 5 h after treatment with DMSO, 0.01 µM, 0.02 µM, 0.05 µM, 0.1 µM, and 0.2 µM thapsigargin (TG). 40S, 60S, 80S, disome (black arrow) and polysome are labeled accordingly. In each graph, a red line is drawn from the disome peak to the peak of the eight-ribosome fraction.
To further evaluate the association between ER stress and NMD inhibition, we titrated the doses of thapsigargin and reexamined NMD activity and ER stress levels. Cells were treated with thapsigargin at concentrations of 0.002, 0.005, 0.01, 0.02, 0.05, 0.1, and 0.2 µM and harvested at 5 h post-treatment for analysis. Xbp1 intron excision and Ire1α induction occurred at 0.01 µM and intensified with increasing concentration (Fig. 4B; Supplemental Fig. S7B). NMD inhibition, collectively and consistently demonstrated by the expression of Srsf11, Ptbp2, Tra2b, Hnrnpl, and Psd-95, was observed at a concentration as low as 0.02 µM and escalated in a dose-dependent manner (Fig. 4C). Xbp1s splicing and NMD repression exhibited the same kinetics, and both plateaued at 0.1 µM and above (Fig. 4B,C). Concentrations above 0.2 µM did not further increase NMD inhibition (data not shown). This analysis indicated that Xbp1 intron excision had a higher sensitivity than NMD inhibition to low concentrations of thapsigargin treatment. Nevertheless, these data support a strong positive correlation between ER stress and NMD inhibition.
In addition to altering gene expression via the IRE1-Xbp1s signaling pathway to retain homeostasis, ER stress also reduces global protein synthesis (Harding et al. 1999; Ron 2002; Wek et al. 2006). The dose of thapsigargin inhibiting NMD was as low as 20 nM, whereas in the literature thapsigargin has been widely used at 1 µM to induce ER stress. To assess whether the NMD-inhibiting thapsigargin doses also repressed translation, we performed ribosome fractionation using sucrose density-gradient centrifugation and examined polysome integrity as an indicator of translational activity. Control cells treated with DMSO showed a typical polysome profile consisting of peaks of individual ribosome subunits (40S and 60S), monosomes (80S), and polysomes (2, 3, 4, 5, and ≥6 ribosomes). Under the culture conditions used, the heights of the polysome peaks steadily increased with the number of ribosomes (Fig. 4D). Thapsigargin effectively reduced the heights of the polysome peaks. The heavier polysomes (≥4 ribosomes) were affected the most and further collapsed with increasing doses of thapsigargin. Polysome disintegration was also accompanied by increasing optical density values of 80S. To quantify the dose effect of thapsigargin, we drew a line from the peak of the disome to the peak of the polysome consisting of roughly eight ribosomes based on relative elution time. We measured the angle of this line relative to a horizontal line through the disome peak and used the angle as an indicator of polysome integrity. This angle was 30 for DMSO-treated cells and decreased to 0, −18, −20, −30, and −40 in response to increasing thapsigargin concentrations of 0.01, 0.02, 0.05, 0.1, and 0.2 µM, respectively. Interestingly, 0.01 µM thapsigargin induced noticeable disintegration of heavy polysomes but little NMD repression. These data showed that polysome disassembly was positively associated with and also appeared to lead NMD inhibition.
Since NMD is translation dependent, the isoforms responsive to NMD inhibition were presumably undergoing translation. Without isoform-specific antibodies and proven assays to detect the protein isoforms translated from these five NMD transcripts, we examined their mRNA abundance on the polysome fractions. We note that association with polysomes is not a proof but a proxy of active translation. Using RT-qPCR, we quantified the expression levels of the NMD isoforms relative to Sdha, a relatively abundant housekeeping gene. As shown in Supplemental Figure S8, both the NMD and non-NMD isoforms were readily detectable on the polysome fractions of DMSO-treated N2a cells. The relative Ct values (ΔCt) for the NMD isoforms of Psd-95, Ptbp2, Hnrnpl, Tra2b, and Srsf11 were 0.44, 0.94, 2.94, 1.19, and 3.27, respectively, while those of the non-NMD counterparts were 0.42, −1.30, −0.78, −2.12, and −2.17, respectively. These data suggested that their RNA copy numbers were roughly within one order of magnitude from that of Sdha. We also examined their abundance in the polysome fractions of N2a cells at 5 h after treatment of 0.2 µM thapsigargin. The ΔCt values were still within (−3.3, 3.8), suggesting that their representation on the polysome was still within one order of magnitude from that of Sdha. It should be noted that the collapsing polysomes yielded lower amounts of RNA, and the ΔCt values became less indicative of translational activity. In summary, these data supported that the NMD isoforms were undergoing translation at the onset of thapsigargin treatment and could therefore readily respond to NMD inhibition.
Thapsigargin inhibits NMD by activating the PERK pathway
Since the degree of NMD inhibition was strongly correlated with the extent of ER stress measured by Xbp1 splicing and polysome disassembly, we next checked which signaling pathway directly led to NMD inhibition. ER stress stimulates three branches of the unfolded protein response (UPR): the IRE1, activating transcription factor 6α (ATF6α), and protein kinase RNA-like endoplasmic reticulum kinase (PERK) pathways (Yoshida et al. 1998, 2001; Harding et al. 1999; Calfon et al. 2002; Hetz 2012; Han et al. 2013). These three stress sensors bind to chaperone protein BIP and are quiescent under non-stress conditions. Under ER stress, misfolded proteins sequester BIP from continually interacting with these three proteins. Upon release from BIP binding, PERK is activated via autophosphorylation, similar to IRE1α. ATF6α is activated by intramembrane proteolysis and translocates into the nucleus to induce the transcription of chaperones, such as Bip. To test which pathway mediates NMD activity, we knocked down the stress sensors by RNAi for 48 h before thapsigargin treatment and examined whether the thapsigargin-inhibited NMD activity could be restored.
We found that depletion of only PERK completely prevented thapsigargin from repressing NMD. Two independent siRNAs reduced the endogenous Perk transcripts to around 20% (Fig. 5A). PERK knockdown did not significantly affect the steady-state levels of either the NMD or non-NMD isoforms of Psd-95, Ptbp2, and Tra2b (Fig. 5B–D). Thapsigargin application in control siRNA-pretreated cells induced only the NMD isoforms of these genes along with Xbp1s as previously demonstrated (Fig. 5B–E) and also interestingly increased Perk transcript levels (Fig. 5A). In siPerk-pretreated cells, none of the NMD isoforms were stimulated by thapsigargin (Fig. 5B–D).
FIGURE 5.

PERK is necessary for thapsigargin-induced NMD inhibition. Expression levels of Perk (A); the NMD (red) and non-NMD (blue) isoforms of Psd-95 (B), Ptbp2 (C), and Tra2b (D); Xbp1s (E); and Bip (F) after siPerk knockdown and thapsigargin treatment. Control siRNA and two different siRNAs targeting Perk were transfected into N2a cells for 48 h before thapsigargin application. Note that the thapsigargin effect on the NMD isoforms was completely blocked by siPerk transfection (B–D). Expression levels of the NMD (red) and non-NMD (blue) isoforms of Psd-95 (G), Ptbp2 (H), and Tra2b (I) as well as Xbp1s (J) and Bip (K) in N2a cells treated with DMSO, thapsigargin, or thapsigargin plus PERK inhibitor GSK2606414 at various concentrations. A concentration of 0.1 µM GSK2606414 or above was sufficient to revert the effect of thapsigargin on the NMD isoforms (G–I). (L) Western blots of phosphorylated eIF2α (P-eIF2α) and total eIF2α in cells treated with DMSO, DMEM, 0.2 µM thapsigargin (TG), and 0.3 µM GSK2606414 (GSK) plus TG. (M) Quantification of L using the intensity ratio of P-eIF2α over total eIF2α. N = 2. (N) Schematics of the proposed mechanism of thapsigargin and other stressors leading to NMD inhibition. A one-way ANOVA test was used for A, E, F, J, and K. A two-way ANOVA test followed by Dunnett's multiple comparison tests was used for B, C, D, G, H, and I. (*) P < 0.05; (ns) not significant. N = 3. Error bars represent mean ± SEM.
Depletion of Ire1α or Atf6 alone was not sufficient to fully rescue thapsigargin-induced inhibition of NMD activity. Both Ire1α and Atf6α were up-regulated by thapsigargin (Supplemental Figs S7A, S9A). RNAi treatment efficiently reduced thapsigargin-increased Ire1α and Atf6α (Supplemental Figs S7C, S9A). Depletion of Ire1α dampened thapsigargin-enhanced NMD isoforms by about twofold but did not revert their levels to those of pre-thapsigargin treatment (Supplemental Fig. S7D–F). Reduction of Atf6α did not alter the effect of thapsigargin on the NMD isoforms (Supplemental Fig. S9B–E). These experiments were corroborated by the Perk knockdown cells, in which thapsigargin continued to activate IREα and ATF6α but no longer inhibited NMD. Xbp1s levels, which are indicative of IRE1α activity, were still six- to eightfold higher in the thapsigargin-treated siPerk cells than in DMSO-treated cells (Fig. 5E). Similarly, Bip transcript levels, which are widely used as a reporter of ATF6α activity, were unchanged by control siRNA, Perk siRNAs or DMSO treatment and remained high after thapsigargin treatment regardless of siPerk application (Fig. 5F). Therefore, activation of IRE1α and ATF6α by thapsigargin was not sufficient to inhibit NMD.
Because RNAi-mediated depletion could not resolve whether it was the physical scaffold or enzymatic activity of PERK that was essential for NMD inhibition, we tested a small molecule inhibitor that inhibits only the enzymatic activity of PERK. GSK2606414 is a potent selective PERK inhibitor (Axten et al. 2012), and we applied it along with thapsigargin treatment. Application of GSK2606414, similar to siPerk alone, did not affect NMD activity in DMSO-treated cells. In thapsigargin-treated cells, however, GSK2606414 effectively reversed the up-regulation of the NMD isoforms in a dose-dependent manner (Fig. 5G–I). GSK2606414 phenocopied Perk siRNA treatment in attenuating thapsigargin-induced Xbp1s but to a level still well above that in DMSO-treated cells (Fig. 5J). Like RNAi depletion of Perk, GSK2606414 did not down-regulate Bip transcripts (Fig. 5K). These data further show that of the three UPR branches only PERK activity was required for thapsigargin to inhibit NMD.
PERK activation phosphorylates eIF2α at serine 51, leading to attenuation of protein synthesis (Harding et al. 1999; Wek et al. 2006). To test whether eIF2α was part of the signaling cascade, we performed quantitative immunoblots on total eIF2α and phosphor-eIF2α (Ser51) in cells treated with thapsigargin and PERK inhibitor GSK2606414. As expected, DMSO did not alter the level of phosphorylation. We observed an increase in phosphor-eIF2α at 1.5 h after thapsigargin treatment, which was completely diminished by GSK2606414 (Fig. 5L,M). Meanwhile, total eIF2α levels remained constant across all conditions. These data supported the notion that thapsigargin inhibits NMD via PERK activation and subsequent attenuation of global protein translation (Fig. 5N).
NMD inhibition is not ubiquitous under various cellular stresses
We then investigated whether NMD inhibition was ubiquitous under various cellular stresses. We did not observe changes in NMD isoform levels in cells subjected to high temperatures (up to 45°C for 2 h) or serum deprivation (as low as 0% fetal bovine serum for up to 24 h). However, NMD inhibition was observed after culturing N2a cells in L-glutamine-free media (Supplemental Fig. S10). The NMD isoform levels of Psd-95, Ptbp2, and Tra2b were not apparently altered within the first 6 h of switching to L-glutamine-free media, possibly due to residual cellular glutamine sustaining cell metabolism. These NMD isoforms were clearly up-regulated at 12 h and further enhanced at 15 h. These data show that induced NMD inhibition was not limited to stress caused by thapsigargin but also not ubiquitous under all cellular stresses.
ER stress enhances TDP-43-controlled NMD isoforms through PERK
Given the critical roles of NMD in physiology and pathogenesis, modulation of NMD activity may impact NMD-associated events. To show the relevance of NMD inhibition by ER stress, we turned to recent discoveries implicating NMD regulation in the pathogenic mechanisms of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (Barmada et al. 2015; Ling et al. 2015). RNA binding protein TDP-43 is commonly found in the cytoplasmic inclusion bodies of ALS and FTD, and its genetic mutations are linked to familial ALS and FTD (Arai et al. 2006; Neumann et al. 2006; Guo et al. 2011; Polymenidou et al. 2012; Ling et al. 2013). Increased cryptic splicing in TDP-43-deficient cells has been proposed as one of the pathogenic mechanisms (Ling et al. 2015). Many of these cryptic isoforms are presumably subject to NMD regulation. We therefore reasoned that ER stress might aggravate TDP-43 deficiency in the up-regulation of these NMD isoforms.
To investigate the possible compounding effect of ER stress on TDP-43-mediated NMD isoforms, we examined the expression of TDP-43-repressed cryptic exons with and without thapsigargin treatment. We designed specific PCR primers flanking the previously reported cryptic exons of A230046K03Rik, Mib1, and Usp15 and measured the ratios of the NMD isoforms to the normal isoforms. The splicing of cryptic exons was not detected in cells treated with control siRNA or DMSO but was drastically boosted by RNAi-mediated depletion of Tdp-43 (Fig. 6). Subsequent thapsigargin treatment further increased the level of the NMD isoforms in TDP-43-deficient cells but had no effect in the mock-transfected or control siRNA-transfected cells.
FIGURE 6.

Thapsigargin enhances TDP-43-repressed NMD isoforms through PERK. The expression levels of Tdp-43 (A) and Perk (B) in N2a cells transfected with control siRNA, siTdp43, and/or siPerk and subsequently treated with DMSO or 0.2 µM thapsigargin. These cells were also assayed for the expression of the normal and NMD isoforms of A230046K03Rik (C), Mib1 (D), and Ups15 (E). Representative digital gels are shown in the lower panels. Arrows point to the NMD and normal isoforms. The ratio of the NMD isoform relative to the normal isoform was quantified for each gene under each experimental condition (upper panels). In siTdp43 cells, thapsigargin further increased the ratios. In siTdp43 and siPerk double-knockdown cells, thapsigargin had no effect on the ratios compared to DMSO. (ns) P ≥ 0.05; (*) P < 0.05 (two-way ANOVA followed by Dunnett's multiple comparison tests). All error bars represent mean ± SEM. N = 3.
To test whether the thapsigargin activity was due to PERK-mediated NMD inhibition, we knocked down Perk prior to the drug treatment. As shown by two independent Perk siRNAs, loss of PERK did not cause cryptic splicing on its own nor interfere with the activity of TDP-43 in DMSO-treated cells (Fig. 6). However, PERK knockdown completely eliminated thapsigargin's additive effects to TDP-43 depletion, resulting in similar isoform ratios between the DMSO and thapsigargin treatments. These results confirmed that ER stress exacerbated the up-regulation of cryptically spliced NMD isoforms through PERK activation in TDP-43-deficient cells.
DISCUSSION
In this study, we devised a new method to precisely monitor changes in cellular NMD activity through individual quantitative measurement of a panel of endogenous NMD isoforms and their corresponding non-NMD isoforms. Because the method can be used on its own to assess changes in cellular NMD activity with high specificity and sensitivity, it can facilitate analysis of NMD controls by cellular pathways in response to stimuli or during development and is particularly suitable for unbiased screening of NMD modulators. The assay was designed to distinguish NMD regulation from transcriptional regulation and alternative splicing control, which also affect the steady-state levels of NMD substrates. Furthermore, we included five different NMD isoforms to enhance the robustness of this new method. These five isoforms responded consistently to the changes in NMD activity, albeit to varying degrees, indicating that fewer may suffice. Using this new method as an independent assay to traditional PTC reporters and gene-centric quantitation, we demonstrated that thapsigargin-induced NMD inhibition occurred via PERK activation rather than Ca2+ signaling. We further showed that NMD inhibition was not universal under all cellular stresses.
Our new method is both sensitive and quantitative thanks to the high sensitivity and broad dynamic range of RT-qPCR. This is essential for determining the efficiency and dynamics of NMD activity regulated by cellular pathways or during development, for analyzing the kinetics of drug response and for dissecting the underlying molecular mechanisms. While the repressive effect of thapsigargin on NMD started to plateau at a concentration of 0.2 µM and above, NMD inhibition was already detected at treatment levels as low as 0.02 µM. At this low concentration, the NMD isoform up-regulation was only ∼20% of the maximal up-regulation but was nevertheless consistently detected by our method (Fig. 4). Similarly, in the time course experiment, 20%–30% of full inhibition (typically achieved at 5 h post-treatment) was readily detectable at 1 h post-treatment (Fig. 3).
Our method indicated that different NMD targets have varying sensitivity to NMD inactivation. For example, the NMD isoform of Hnrnpl was up-regulated by about 60-fold in the conditional Upf2 knockout cortices, whereas the Psd-95 NMD isoform increased by about eightfold (Fig. 2B,F). Surprisingly, the ranking of these tested genes regarding their sensitivity to NMD inhibition was relatively preserved between the Upf2 knockout and thapsigargin experiments, with Hnrnpl and Tra2b exhibiting much higher sensitivity and Psd-95 and Srsf11 displaying lower sensitivity. The varying sensitivity of these NMD targets does not appear to be associated with the 3′ UTR length or the length between the PTC and the downstream exon–exon junction (Supplemental Table S2). For example, the 3′ UTR of the Hnrnpl and Psd-95 NMD isoforms are similar in length and both are much shorter than others.
Among the three UPR branches, only the PERK signaling pathway but not IRE1α or ATF6α was required for thapsigargin-induced NMD inhibition. Our results demonstrated the necessity of PERK activation as well as a strong correlation between polysome disassembly and NMD inhibition. The findings are therefore consistent with previous reports and support the hypothesis that NMD inhibition is specific to PERK-eIF2α-translation repression signaling (Chiu et al. 2004; Gardner 2008, 2010; Hu et al. 2010; Wang et al. 2011).
Our studies did not detect a role of intracellular Ca2+ signaling in NMD inhibition. Downstream from thapsigargin treatment, PERK inhibition via a specific drug inhibitor or siRNA depletion did not reverse the increase in cytosolic Ca2+ but did completely restore NMD activity. Furthermore, other chemicals increasing intracellular Ca2+ failed to attenuate NMD. Therefore, increased intracellular Ca2+ was not enough to inhibit NMD in our system, contradicting a recent study reporting calcium's sufficient role (Nickless et al. 2014). One possible explanation is the type of cells used for the analysis. We used mouse N2a cells, whereas Nickless et al. used human osteosarcoma cells (U2OS). Additionally, Nickless et al. used exogenous fluorescent mini-gene reporters to measure NMD activity, whereas we used endogenous NMD reporters.
With the new method, we also found that glutamine deprivation inhibited NMD, while heat shock or serum withdrawal did not. This result was consistent with a previous study reporting NMD attenuation upon deprivation of all amino acids (Mendell et al. 2004). Although the mechanism of NMD inhibition by amino acid starvation remains unclear, it is probably due to translation interference. Amino acid starvation activates general control nonderepressible 2 (GCN2), which can phosphorylate eIF2α (Dever et al. 1992) to slow down translation (Pain 1994).
Our studies showed an adverse effect of ER stress on TDP-43 proteinopathy enhancing TDP-43-repressed NMD substrates. ER stress-stimulating episodes could thus restrain TDP-43 defective cells from clearing up these unwanted transcripts in a timely manner and could even possibly accelerate progressive loss of neurons in ALS and FTD patients. We also showed that PERK inhibition completely blocked the undesirable effect of ER stress, providing a possible target of therapeutics to ameliorate the ill influence of ER stress on TDP-43 deficiency.
As a proof-of-principle analysis, our study demonstrated the utility of a new quantitative method for accurately monitoring changes in cellular NMD activities. The high sensitivity and broad dynamic range of our method allow for the detection of continual changes in NMD activity during development or in response to environmental changes. This presents a new avenue for exploring endogenous NMD modulation and subsequent applications in the study of genetic diseases. Our method can also be applied to monitor PERK activity and translation inhibition, which are tightly linked to NMD inhibition. Our method, however, cannot distinguish translation repression and NMD inhibition. In the present analysis, we only tested a small panel of widely used small molecule inhibitors to demonstrate the potential of our assay for unbiased screening. With the aid of cataloged libraries, high-throughput robotic liquid handling systems and next-generation sequencing, our new method can be adapted to screen larger libraries.
MATERIALS AND METHODS
Cell cultures and treatments
Neuro-2a (N2a) cells were maintained in N2a complete media consisting of L-glutamine-free Dulbecco's modified Eagle's medium (DMEM), 10% FBS and 1× GlutaMAX at 37°C. For the application of actinomycin D (Sigma, cat. no. A1410), thapsigargin (VWR, cat. no. 89161-410), KT5720 (Tocris Bioscience, cat. no. 1288100U), KN-93 (Fisher Scientific, cat. no. NC0362431), GF109203X (R&D, cat. no. A00061), ionomycin (Fisher Scientific, cat. no. AG-CN2-0416-M001), and ouabain octahydrate (Fisher Scientific, cat. no. 80055-364), 750,000 cells were plated on 35 mm BioLite TC plates and incubated with 2 mL of N2a complete media overnight before treatment with the drugs at the indicated concentrations. The cells were collected at 5 h post-treatment unless otherwise specified. The PERK inhibitor GSK2606414 (Thermo Fisher, cat. no. 501016108) was dissolved in DMSO and stored at −20°C. N2a cells were incubated with GSK2606414 for 1 h before thapsigargin treatment. For L-glutamine deprivation, 750,000 N2a cells were plated overnight and switched to L-glutamine-free DMEM with 10% FBS. SiRNA knockdown experiments were conducted using Lipofectamine RNAiMax and Silencer Select siRNAs (siTdp-43, cat. no. s106688 and s106686; siPerk, cat. no. s201280 and s65405; siPtbp1, cat. no. s72335 and s72336; siPtbp2, cat. no. s80148 and s80149; siUpf1, cat. no. s72879 and s72878; siIre1 cat. no. s95859; siAtf6, cat. no. s105470) according to the manufacturer's instructions. Silencer Negative Control siRNA (AM4615) was used as the siRNA control. Lipofectamine 2000 (Life Technologies) was used to transfect the Flag-Ptbp1 plasmid and control GFP plasmid into N2a cells. Cells were incubated for 48 h for optimal knockdown efficiency or overexpression before downstream treatments.
Animals
Conditional Upf2−/− (that is, Upf2loxP/loxP; Emx1-cre) mice were generated by first breeding Upf2loxP/loxP mice to Emx1-cre mice and subsequently breeding Upf2loxP/+; Emx1-cre mice to Upf2loxP/loxP mice (Zheng et al. 2012). Ptbp2−/− mice were generated by breeding Ptbp2+/− mice to Ptbp2+/− mice (Li et al. 2014). All animal procedures were approved by the Institutional Animal Care and Use Committee at UCR.
RNA extraction, cDNA synthesis, and RT-qPCR
TRIzol (Life Technologies, cat. no. 15596-018) was directly added to the cells or mouse brain tissues to extract total RNA following the TRIzol reagent standard protocol. Isolated RNA was treated with 4 units of Turbo DNase (Ambion) at 37°C for 35 min to degrade all remaining genomic DNA. After the DNase treatment, RNA was purified using phenol-chloroform (pH 4.5, VWR cat. no. 97064-744). RNA concentrations were measured using a Nanodrop 2000c (Thermo Fisher). One microgram of freshly isolated DNA-free RNA was converted to cDNA using 1 µL random hexamers (30 µM) and 200 units of Promega M-MLV reverse transcriptase (cat. no. M1705) following the Promega protocol in a 20 µL reaction. For all qPCR primers, quality control was performed for their specificity, sensitivity, melting curves, and standard curves (Supplemental Table S1). RT-qPCR experiments were conducted using a QuantStudio 6 Real-Time PCR instrument with 2× Power SYBR Green PCR master mix (Life Tech) following the Life Tech protocol. Each 10 µL reaction contained 0.3 µL cDNA, 5 µL 2× Power SYBR Green PCR master mix, 0.3 nM forward primer, and 0.3 nM reverse primer. The QuantStudio 6 RT-qPCR run program was as follows: 50°C for 2 min; 95°C for 15 sec and 60°C for 1 min, with the 95°C and 60°C steps repeated for 40 cycles; and a melting curve test from 60°C to 95°C at a 0.05°C/sec measuring rate. QuantStudio Real-Time PCR software was used for the analysis. All RT-qPCR reactions were conducted with three technical replicates along with a no template control (NTC, not amplified). Outliers were excluded when the coefficient of variation of Ct for the three technical replicates was larger than 0.3. Relative expression (fold changes) was calculated using the ΔΔCt method. For the splicing assays of the NMD exons, PCR was performed using New England Biolab Taq DNA polymerase (cat. no. M0267E). All statistical analysis was performed using GraphPad Prism 6.
Protein extraction, immunoblotting, and quantitative image analysis
Cells in six-well plates were washed twice with 1 mL cold 1× PBS and harvested with freshly prepared RIPA buffer. The BCA assay (Thermo Scientific) was performed to determine protein concentrations before loading equal amounts of the materials onto SDS-PAGE gels (4% stacking and 12% resolving) for Western blotting. For primary antibodies, we used antibodies to PTBP1-NT (1:3000), PTBP2-IS2 (1:1000), P-eIF2 (1:333, Thermo Fisher), eIF2 (1:1000, Thermo Fisher), and GAPDH (1:1500, Ambion). Specific Western blot bands were quantified using ImageQuant TL and normalized with GAPDH to derive the relative protein levels.
Quantitative analysis of cryptic exon splicing
We optimized the PCR cycle numbers for Psd-95 (28 cycles), A230046K03Rik (29 cycles), Mib1 (29 cycles), and Usp15 (29 cycles). Quantitative gel electrophoresis of PCR amplicons was conducted. Splicing of the exons was determined using the following formula. All statistical analysis was performed using GraphPad Prism 6.
Polysome fractionation
For polysome fractionation, 1.5 × 107 N2a cells in 20 mL N2a complete media were plated on 150 mm petri dishes overnight and treated with varying concentrations of thapsigargin the next day. Cycloheximide (Fisher Scientific, cat. no. 50490338) was added at a concentration of 100 µg/mL, and the cells were incubated for 10 min at 37°C before lysate collection. The cells were washed twice with 10 mL cold 1× PBS containing 100 µg/mL cycloheximide then collected in 4 mL of the same cold PBS solution. The cells were lysed with 0.5 mL lysis buffer (20 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl2, 2 mM DTT, 100 µg/mL cycloheximide, 1% Triton X-100, 50 µ/mL RNaseout, and 1× EDTA-free protease inhibitor cocktail). Roughly 400 µL (6000 optical units) of lysate was loaded onto premade sucrose gradients (60% to 15%) and balanced (within 0.5 mg) before ultracentrifugation at 4°C and 237,000g (50,000 rpm for a SW55 Ti rotor) for 1.5 h. Products were carefully removed from the ultracentrifuge and fractionated with the apparatus consisting of the gradient fractionator (Brandel SYN-202), the ISCO absorbance detector (ISCO # UA-6), and the fraction collector (R1 Fraction Collector) at 2.0 sensitivity and 150 cm/h chart speed to record absorbance data and collect fractionations.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Xuemei Chen (Howard Hughes Medical Institute, University of California, Riverside, USA) for providing the gradient fractionator and Dr. Kestrel Rogers for technical assistance with the polysome fractionation experiments. We thank Dr. Douglas Black (UCLA) for the gift of the PTBP1 and PTBP2 antibodies and Dr. Jens Lykke-Andersen (UCSD) for the UPF2 antibody. We thank Ruchira Puri for her help with RNA extraction and cDNA synthesis, and James Randolph and Angela Reinert for their assistance with the amino acid deprivation studies. We also thank members of the Zheng laboratory for their helpful discussions. This work was supported by the National Institutes of Health (grant number R00MH096807 to S.Z.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.058040.116.
REFERENCES
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. 2006. TDP-43 is a component of ubiquitin-positive τ-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351: 602–611. [DOI] [PubMed] [Google Scholar]
- Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WHH, Heerding DA, Minthorn E, Mencken T, et al. 2012. Discovery of 7-methyl-5-(1-[3-(trifluoromethyl)phenyl]acetyl-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 55: 7193–7207. [DOI] [PubMed] [Google Scholar]
- Barmada SJ, Ju S, Arjun A, Batarse A, Archbold HC, Peisach D, Li X, Zhang Y, Tank EMH, Qiu H, et al. 2015. Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proc Natl Acad Sci 112: 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Stoilov P, Li Q, Lin C-H, Chawla G, Ostrow K, Shiue L, Ares M Jr, Black DL. 2007. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev 21: 1636–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. 2002. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415: 92–96. [DOI] [PubMed] [Google Scholar]
- Chan W-K, Huang L, Gudikote JP, Chang Y-F, Imam JS, MacLean JA II, Wilkinson MF. 2007. An alternative branch of the nonsense-mediated decay pathway. EMBO J 26: 1820–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y-F, Imam JS, Wilkinson MF. 2007. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem 76: 51–74. [DOI] [PubMed] [Google Scholar]
- Chen L. 2010. A global comparison between nuclear and cytosolic transcriptomes reveals differential compartmentalization of alternative transcript isoforms. Nucleic Acids Res 38: 1086–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu S-Y, Lejeune F, Ranganathan AC, Maquat LE. 2004. The pioneer translation initiation complex is functionally distinct from but structurally overlaps with the steady-state translation initiation complex. Genes Dev 18: 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF, Hinnebusch AG. 1992. Phosphorylation of initiation factor 2 α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68: 585–596. [DOI] [PubMed] [Google Scholar]
- Gardner LB. 2008. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol Cell Biol 28: 3729–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner LB. 2010. Nonsense-mediated RNA decay regulation by cellular stress: implications for tumorigenesis. Mol Cancer Res 8: 295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Chen Y, Zhou X, Kar A, Ray P, Chen X, Rao EJ, Yang M, Ye H, Zhu L, et al. 2011. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat Struct Mol Biol 18: 822–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin Y-H, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. 2013. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15: 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. 1999. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397: 271–274. [DOI] [PubMed] [Google Scholar]
- Hetz C. 2012. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13: 89–102. [DOI] [PubMed] [Google Scholar]
- Hu W, Petzold C, Coller J, Baker KE. 2010. Nonsense-mediated mRNA decapping occurs on polyribosomes in Saccharomyces cerevisiae. Nat Struct Mol Biol 17: 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken O, Maquat LE. 2008. The multiple lives of NMD factors: balancing roles in gene and genome regulation. Nat Rev Genet 9: 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam R, Lou C-H, Kroeger H, Huang L, Lin JH, Wilkinson MF. 2015. The unfolded protein response is shaped by the NMD pathway. EMBO Rep 16: 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Brooks AN, Soergel DAW, Meng Q, Brenner SE. 2007. The coupling of alternative splicing and nonsense-mediated mRNA decay. EMBO Rep 16: 599–609. [DOI] [PubMed] [Google Scholar]
- Li Q, Zheng S, Han A, Lin CH, Stoilov P, Fu XD, Black DL. 2014. The splicing regulator PTBP2 controls a program of embryonic splicing required for neuronal maturation. Elife 3: e01201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW. 2013. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79: 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JP, Pletnikova O, Troncoso JC, Wong PC. 2015. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349: 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou CH, Shao A, Shum EY, Espinoza JL, Huang L, Karam R, Wilkinson MF. 2014. Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Rep 6: 748–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen S, Jensen TH. 2015. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol 16: 665–677. [DOI] [PubMed] [Google Scholar]
- Lykke-Andersen J, Shu MD, Steitz JA. 2000. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 103: 1121–1131. [DOI] [PubMed] [Google Scholar]
- Lykke-Andersen S, Chen Y, Ardal BR, Lilje B, Waage J, Sandelin A, Jensen TH. 2014. Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev 28: 2498–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE, Gong C. 2009. Gene expression networks: competing mRNA decay pathways in mammalian cells. Biochem Soc Trans 37: 1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE, Tarn W-Y, Isken O. 2010. The pioneer round of translation: features and functions. Cell 142: 368–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC. 2004. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet 36: 1073–1078. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133. [DOI] [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O'Brien G, Shiue L, Clark TA, Blume JE, Ares M Jr. 2007. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21: 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickless A, Jackson E, Marasa J, Nugent P, Mercer RW, Piwnica-Worms D, You Z. 2014. Intracellular calcium regulates nonsense-mediated mRNA decay. Nat Med 20: 961–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Meislin SH, Moore MJ. 2003. A quantitative analysis of intron effects on mammalian gene expression. RNA 9: 607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren YS, McClure ML, Rowe SM, Sorscher EJ, Bester AC, Manor M, Kerem E, Rivlin J, Zahdeh F, Mann M, et al. 2014. The unfolded protein response affects readthrough of premature termination codons. EMBO Mol Med 6: 685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pain VM. 1994. Translational control during amino acid starvation. Biochimie 76: 718–728. [DOI] [PubMed] [Google Scholar]
- Peccarelli M, Kebaara BW. 2014. Regulation of natural mRNAs by the nonsense-mediated mRNA decay pathway. Eukaryot Cell 13: 1126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Bennett CF, Cleveland DW, Yeo GW. 2012. Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res 1462: 3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MWL, Maquat LE. 2013. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu Rev Genet 47: 139–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbapragada I, Lykke-Andersen J. 2009. Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr Opin Cell Biol 21: 394–402. [DOI] [PubMed] [Google Scholar]
- Rehwinkel J, Letunic I, Raes J, Bork P, Izaurralde E. 2005. Nonsense-mediated mRNA decay factors act in concert to regulate common mRNA targets. RNA 11: 1530–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D. 2002. Translational control in the endoplasmic reticulum stress response. J Clin Invest 110: 1383–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio C, Pincus D, Korennykh A, Schuck S, El-Samad H, Walter P. 2011. Homeostatic adaptation to endoplasmic reticulum stress depends on Ire1 kinase activity. J Cell Biol 193: 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaki K, Yoshina S, Shen X, Han J, DeSantis MR, Xiong M, Mitani S, Kaufman RJ. 2012. RNA surveillance is required for endoplasmic reticulum homeostasis. Proc Natl Acad Sci 109: 8079–8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzman AL, Kim YK, Pan Q, Fagnani MM, Maquat LE, Blencowe BJ. 2008. Regulation of multiple core spliceosomal proteins by alternative splicing-coupled nonsense-mediated mRNA decay. Mol Cell Biol 28: 4320–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samali A, Fitzgerald U, Deegan S, Gupta S. 2010. Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int J Cell Biol 2010: 830307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman R, Llorian M, Smith CWJ. 2007. Crossregulation and functional redundancy between the splicing regulator PTB and its paralogs nPTB and ROD1. Mol Cell 27: 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoilov P, Daoud R, Nayler O, Stamm S. 2004. Human tra2-beta1 autoregulates its protein concentration by influencing alternative splicing of its pre-mRNA. Hum Mol Genet 13: 509–524. [DOI] [PubMed] [Google Scholar]
- Tani H, Imamachi N, Salam KA, Mizutani R, Ijiri K, Irie T, Yada T, Suzuki Y, Akimitsu N. 2012. Identification of hundreds of novel UPF1 target transcripts by direct determination of whole transcriptome stability. RNA Biol 9: 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoren LA, Nørgaard GA, Weischenfeldt J, Waage J, Jakobsen JS, Damgaard I, Bergström FC, Blom AM, Borup R, Bisgaard HC, et al. 2010. UPF2 is a critical regulator of liver development, function and regeneration. PLoS One 5: e11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schadewijk A, van't Wout EFA, Stolk J, Hiemstra PS. 2012. A quantitative method for detection of spliced X-box binding protein-1 (XBP1) mRNA as a measure of endoplasmic reticulum (ER) stress. Cell Stress Chaperones 17: 275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Zavadil J, Martin L, Parisi F, Friedman E, Levy D, Harding H, Ron D, Gardner LB. 2011. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol Cell Biol 31: 3670–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Mönch K, Thoren LA, Nielsen FC, Jacobsen SEW, Nerlov C, Porse BT. 2008. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev 22: 1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek RC, Jiang HY, Anthony TG. 2006. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 34: 7–11. [DOI] [PubMed] [Google Scholar]
- Yepiskoposyan H, Aeschimann F, Nilsson D, Okoniewski M, Mühlemann O. 2011. Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. RNA 17: 2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Haze K, Yanagi H, Yura T, Mori K. 1998. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins: involvement of basic leucine zipper transcription factors. J Biol Chem 273: 33741–33749. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. 2001. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891. [DOI] [PubMed] [Google Scholar]
- Zheng S. 2016a. Alternative splicing and nonsense-mediated mRNA decay enforce neural specific gene expression. Int J Dev Neurosci 55: 102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S. 2016b. IRAS: high-throughput identification of novel alternative splicing regulators. Methods Enzymol 572: 269–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Black DL. 2013. Alternative pre-mRNA splicing in neurons: growing up and extending its reach. Trends Genet 29: 442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Gray EE, Chawla G, Porse BT, O'Dell TJ, Black DL. 2012. PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2. Nat Neurosci 15: 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Damoiseaux R, Chen L, Black DL. 2013. A broadly applicable high-throughput screening strategy identifies new regulators of Dlg4 (Psd-95) alternative splicing. Genome Res 23: 998–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.