

Abstract
HIV-1 particle assembly, which occurs at the plasma membrane (PM) of cells, is driven by the viral polyprotein Gag. Gag recognizes phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2], a PM-specific phospholipid, via the highly basic region (HBR) in its N-terminal matrix (MA) domain. The HBR is also known to bind to RNA. We have previously shown, using an in vitro liposome binding assay, that RNA inhibits Gag binding to membranes that lack PI(4,5)P2. If this RNA block is removed by RNase treatment, Gag can bind nonspecifically to other negatively charged membranes. In an effort to identify the RNA species that confer this inhibition of Gag membrane binding, we have tested the impact of purified RNAs on Gag interactions with negatively charged liposomes lacking PI(4,5)P2. We found that some tRNA species and RNAs containing stem–loop 1 of the psi region in the 5′ untranslated region of the HIV-1 genome impose inhibition of Gag binding to membranes lacking PI(4,5)P2. In contrast, a specific subset of tRNAs, as well as an RNA sequence previously selected in vitro for MA binding, failed to suppress Gag–membrane interactions. Furthermore, switching the identity of charged residues in the HBR did not diminish the susceptibility of Gag–liposome binding for each of the RNAs tested, while deletion of most of the NC domain abrogates the inhibition of membrane binding mediated by the RNAs that are inhibitory to WT Gag–liposome binding. These results support a model in which NC facilitates binding of RNA to MA and thereby promotes RNA-based inhibition of Gag–membrane binding.
Keywords: Gag, retrovirus assembly, HIV-1, membrane binding, tRNA, viral RNA
INTRODUCTION
Each of the four major domains of the Gag polyprotein of HIV-1 plays an important role in viral particle assembly. The matrix domain (MA) drives binding of Gag to the plasma membrane (PM), the capsid domain (CA) makes critical interprotein interactions with other Gag molecules to drive multimerization, the nucleocapsid domain (NC) packages a dimer of genomic RNA (gRNA), and the p6 domain recruits cellular factors that pinch off nascent viral particles from the PM (for review, see Balasubramaniam and Freed 2011; Sundquist and Krausslich 2012).
Accumulating evidence supports the notion that specific interactions between the MA domain and the head group of phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2], an acidic phospholipid enriched in the inner leaflet of the PM, drive HIV-1 assembly to the PM over other intracellular membranes (Ono et al. 2004; Saad et al. 2006; Shkriabai et al. 2006; Chan et al. 2008; Chukkapalli et al. 2008; Alfadhli et al. 2009; Anraku et al. 2010; Monde et al. 2011; Gerber et al. 2015; Barros et al. 2016; Mercredi et al. 2016). Studies using various structural, biophysical, or biochemical approaches have shown that recognition of the PI(4,5)P2 headgroup requires a group of positively charged amino acids in MA known as the highly basic region (HBR) (Saad et al. 2006; Shkriabai et al. 2006; Chukkapalli et al. 2008, 2010; Alfadhli et al. 2011; Mercredi et al. 2016). We have previously demonstrated, using an in vitro liposome flotation assay, that despite the presence of significant positive charge in the HBR, Gag does not bind efficiently to negatively charged membranes unless PI(4,5)P2 is present in the liposomes (Chukkapalli et al. 2008; Inlora et al. 2011). This is in a stark contrast to the MA domain of HTLV-1 Gag, which has a similar total positive charge and efficiently binds negatively charged membranes in the absence of PI(4,5)P2 (Inlora et al. 2011, 2014).
The HBR region also binds to RNA (Cimarelli and Luban 1999; Purohit et al. 2001; Hearps et al. 2008; Jones et al. 2011). Notably, treatment of Gag with RNase prior to the liposome flotation assay revealed that in the absence of RNA, HIV-1 Gag can bind significantly to negatively charged liposomes even in the absence of PI(4,5)P2 (Chukkapalli et al. 2010; Inlora et al. 2011, 2014; Dick et al. 2013). This response to RNase treatment was observed not only with Gag synthesized in rabbit reticulocyte lysates, which has been routinely used for this assay, but also with Gag recovered from the cytosol of human cells expressing HIV-1 molecular clones (Chukkapalli et al. 2013). Furthermore, addition of RNase to cell homogenates has also been shown to increase Gag association to cell membranes (Kutluay et al. 2014). MA-bound, but not NC-bound, RNA is likely to mediate the suppression of Gag interactions with membranes lacking PI(4,5)P2, since the response to RNase treatment can still be observed when most of the NC domain has been deleted (Chukkapalli et al. 2010; Inlora et al. 2014; Olety et al. 2015). These results suggest that MA-bound RNA acts as a chaperone of Gag–membrane interactions, possibly by screening the electrostatic charges of the HBR from negatively charged membranes unless there is specific recognition of the PI(4,5)P2 headgroup.
When the charged residues of the HBR are swapped (HBR switch mutant: R to K and vice versa), HIV-1 Gag no longer specifically recognizes PI(4,5)P2-containing liposomes (Llewellyn et al. 2013). However, its ability to bind to PI(4,5)P2-lacking liposomes is still enhanced upon RNase treatment, suggesting that RNA binding to MA does not rely on the exact arrangement of basic residues in HBR. Interestingly, while HTLV-1 MA is insensitive to RNA-mediated suppression, introduction of a basic residue, which is predicted to increase the size of the surface-exposed basic patch, renders HTLV-1 MA susceptible to the RNA block (Inlora et al. 2014). Thus the presence of clustered charge in the HBR, rather than a specific amino acid sequence, is likely to be required for RNA-mediated suppression of HIV-1 Gag–membrane interactions.
Since specific MA residues do not appear to be essential for RNA binding, it is possible that the MA–RNA interaction might not depend on a specific RNA sequence or structure. A recent crosslinking-immunoprecipitation sequencing (CLIP-seq) study in HEK293T cells demonstrated that over 90% of MA-bound RNAs are a subset of tRNAs (Kutluay et al. 2014). However, it remains to be determined whether the predominance of these tRNAs is due to specific molecular interactions between tRNAs and MA, or whether tRNA binding reflects nonspecific electrostatic interactions with abundant or accessible RNA molecules. To determine whether the RNA-mediated inhibition of Gag interactions with non-PI(4,5)P2 acidic lipids requires a specific RNA binding partner for MA, we extended our previous liposome flotation assay to study the role of specific RNAs in Gag–membrane interactions. Following RNase treatment of Gag and subsequent inactivation of the RNase, we found that only some RNAs inhibit Gag binding to negatively charged liposomes. This result indicates the presence of specific determinants in RNAs for suppression of MA-mediated membrane binding. Moreover, we observed that this suppression takes place only when NC is present, suggesting a role for NC in the MA–RNA interaction.
RESULTS
Yeast total tRNA inhibits Gag interactions with acidic liposomes in a dose-dependent manner
To monitor the effects of specific RNA species on Gag–membrane interactions, we used an extension of an in vitro liposome flotation assay we previously developed (Chukkapalli et al. 2013). Briefly, full-length, myristoylated Gag was produced via an in vitro transcription/translation reaction in rabbit reticulocyte lysate. Following protein production, RNA was removed by treatment with RNase A, which was subsequently fully deactivated by the inhibitor RNasin. Chemically synthesized or in vitro transcribed, gel-purified RNA was then added back to the reaction and incubated with Gag prior to the addition of liposomes. Liposome binding was monitored by sucrose gradient membrane flotation as described previously (Chukkapalli et al. 2008). Liposomes with a 2:1 ratio of neutral phosphatidylcholine (PC) and acidic phosphatidylserine (PS) were prepared by extrusion (Todd and Ono 2016).
With this add-back assay, we found that addition of 1 μg/μL of yeast total tRNA is able to completely inhibit Gag binding down to levels observed prior to treatment with RNase A (Fig. 1). At 100 ng/μL yeast total tRNA, Gag binding is inhibited ∼50% relative to the RNase-treated condition (Fig. 1). HeLa total tRNA was also able to inhibit Gag binding to a similar degree as yeast total tRNA (data not shown). Subsequent add-back experiments were performed at 100 ng/μL of each RNA species to compare the inhibitory effect of RNAs with roughly the same total negative charge as yeast total tRNA. Using this approach, we examined the effects of various RNA species on Gag–membrane binding including specific tRNA species and RNAs derived from regions of HIV-1 viral RNA.
FIGURE 1.
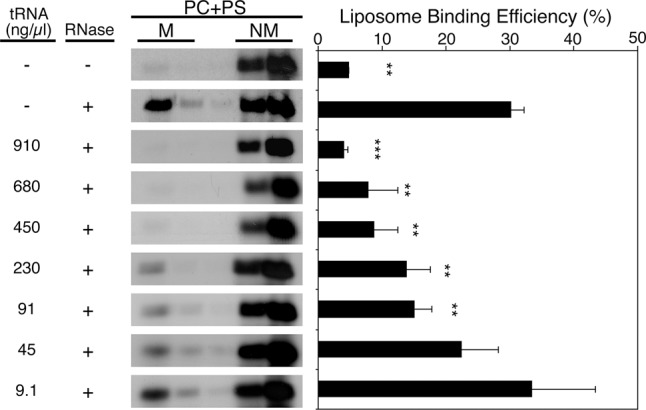
Yeast total tRNA inhibits Gag binding to PC + PS liposomes. [35S]-labeled Gag synthesized using rabbit reticulocyte lysates was treated (or left untreated) with RNase A at 37°C for 20 min. After inactivation of RNase A with RNasin, the mixtures were further incubated with varying concentrations of yeast tRNA at 37°C for 30 min. Subsequently, PC + PS liposomes were added and incubated further for 15 min before performing equilibrium flotation centrifugation. Fractions were analyzed using SDS-PAGE followed by phosphorimager analysis. (M) Membrane-bound, (NM) non-membrane-bound. The liposome binding efficiency was calculated as the amount of membrane-bound Gag as a fraction of total Gag. Data from three different experiments are shown as mean ± standard deviation. P-values were determined in comparison to the no RNA add-back condition using Student's t-test. (**) P < 0.01; (***) P < 0.001. Reduction of liposome binding efficiency to 50% was observed when RNase-treated Gag was incubated with 91 ng/µL yeast tRNA prior to mixing with liposomes.
tRNAPro, but not tRNALys3, can suppress Gag–membrane interactions
Because yeast total tRNA was able to inhibit Gag–membrane interactions, we sought to test whether specific tRNA species are able to function similarly. In particular, we were interested in human tRNALys3, which is selectively packaged into virions and anneals to the primer binding site in the 5′ UTR of the HIV-1 genome where it serves as a primer for reverse transcription (Kleiman et al. 2010). Furthermore, tRNALys3 was one of the tRNAs found to interact with MA in the cell (Kutluay et al. 2014). tRNALys3 acting as a MA-bound regulator of Gag–membrane binding could represent an additional role played by the reverse transcription primer and help to ensure its incorporation into virions. However, relative to yeast total tRNA, unmodified human tRNALys3 is unable to suppress Gag–membrane interactions efficiently (Figs. 2A, 3).
FIGURE 2.

Sequences and secondary structures of all RNA constructs used in this study. (A) Human tRNALys3 and human tRNAPro. (B) MA-binding RNA aptamer (“SELEX”) and a random sequence control (“Random”). (C) HIV-1 5′ UTR with TAR/polyA, PBS, and Psi domains indicated. Psi-ΔDIS is the same as Psi RNA with the exception of the loop nucleotide replacement indicated to eliminate dimerization. To facilitate in vitro transcription, the PBS RNA contains three additional G and C nucleotides at its 5′ and 3′ termini, respectively. Psi and Psi-ΔDIS RNAs contain two additional G residues at their 5′ ends. Note that several 5′ UTR secondary structures for NL4-3 in addition to the one shown have recently been reported (Abd El-Wahab et al. 2014; Keane et al. 2015; Smyth et al. 2015). (D) Psi-SL1 and Psi-SL1,2 RNAs are truncated Psi RNA constructs also containing three extra G residues at their 5′ ends.
FIGURE 3.
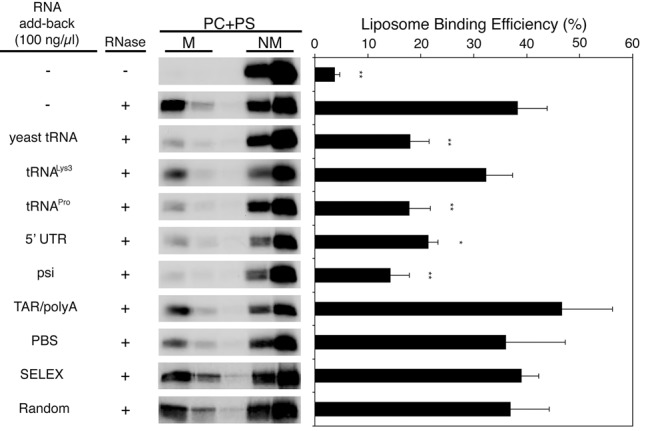
Specific RNA species inhibit Gag binding to PC + PS liposomes. RNA constructs shown in Figure 2 were examined for their effects on Gag binding to PC + PS liposomes using the add-back assay as described in Figure 1. Data from three different experiments are shown as mean ± standard deviation. P values were determined in comparison to the no RNA add-back condition using Student's t-test. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
Other retroviruses use alternative tRNAs as primers for reverse transcription (e.g., Moloney murine leukemia virus uses tRNAPro) (Harada et al. 1979). We tested add-back with unmodified human tRNAPro, and, in contrast to human tRNALys3, tRNAPro was able to inhibit Gag binding to PC + PS liposomes to a similar extent as yeast total tRNA (Figs. 2A, 3). These results indicate that a specific subset of tRNAs can serve as regulators of MA-membrane interactions. These data are consistent with our earlier results (Chukkapalli et al. 2013), as well as with the RNA CLIP study showing that MA predominantly interacts with tRNAs in HEK293T cells (Kutluay et al. 2014).
An RNA sequence selected in vitro for its affinity to MA is unable to suppress Gag interactions with negatively charged membranes
In addition to the MA-binding tRNA species identified in cells (Kutluay et al. 2014), alternative RNA sequences that bind to MA in vitro have been identified using systematic evolution of ligands by exponential enrichment (SELEX) (Lochrie et al. 1997; Purohit et al. 2001; Ramalingam et al. 2011). One of these selected RNAs shares a 13-nt sequence with the pol open reading frame (ORF) in the HIV-1 genome. This RNA species interacts with the MA domain via amino acid residues that overlap with those at the PI(4,5)P2 binding site (Alfadhli et al. 2011) and binds purified MA with an affinity of ∼500 nM in a buffer containing 50 mM NaCl (Purohit et al. 2001; Alfadhli et al. 2011). Importantly, this oligonucleotide is displaced from MA by a water-soluble PI(4,5)P2 analog but not by a water-soluble analog of PS, a more prevalent acidic phospholipid in cellular membranes (Alfadhli et al. 2011). These results suggest that this SELEX-derived RNA, and possibly the corresponding portion of the pol ORF, might inhibit Gag interactions with nonspecific membranes.
To examine the effect of this RNA sequence on Gag–membrane binding, we compared a 25-mer SELEX-derived RNA oligonucleotide containing either the 13-nt consensus sequence or a random sequence (Purohit et al. 2001; Alfadhli et al. 2011) in the add-back assay at 100 ng/µL (Fig. 2B). As anticipated, the random RNA had no impact on Gag–membrane binding. Surprisingly, we found that the SELEX RNA was also unable to inhibit Gag binding to PC + PS liposomes (Fig. 3). These results suggest that this MA-specific RNA is either unable to completely shield the HBR residues from the charged liposomes or that Gag–membrane interactions are stronger than MA-SELEX RNA binding.
The 5′ UTR of HIV-1 RNA is able to suppress Gag interactions with negatively charged membranes
In addition to the SELEX RNA sequence overlapping with the pol region of the HIV-1 genome, we also tested RNAs derived from the 5′ UTR of the gRNA (the first 356 nt, pNL4-3 numbering), which is highly structured and known to regulate many steps of the HIV-1 life cycle (Fig. 2C). HIV-1 gRNA is recognized by the NC domain of Gag and packaged into viral particles. The psi region of the 5′ UTR initiates dimerization and is also critical for NC recognition and genome packaging (Wilkinson et al. 2008; Watts et al. 2009; Lu et al. 2011; Abd El-Wahab et al. 2014; Keane et al. 2015; Smyth et al. 2015). The 5′ UTR is not known to interact with MA, but, surprisingly, we found that the 5′ UTR inhibited Gag–membrane interactions to a similar extent as yeast total tRNA (Fig. 3). This observation suggests a possibility that the 5′ UTR may contain a sequence or structure with high affinity to MA and/or an ability to bind in a specific conformation that blocks MA interactions with negatively charged lipid bilayers.
SL1 of the psi region can regulate Gag–membrane interactions
To further delineate the functional region that suppresses Gag–membrane interactions, we tested three fragments of the 5′ UTR: (i) the transactivation response and polyA hairpins [TAR/polyA; nucleotides 1–104], (ii) the primer binding site (PBS; nucleotides 125–223), and (iii) the psi sequence (nucleotides 234–329), which contains the dimerization initiation (DIS) stem–loop (SL1), the 5′ major splice donor hairpin (SL2), and the psi hairpin (SL3) (Fig. 2C). Of the three 5′ UTR fragments, only the psi region inhibited Gag interactions with PC + PS liposomes (Fig. 3).
We further tested shorter fragments of psi that contain SL1 (Psi-SL1) or SL1 and SL2 (Psi-SL1,2) (Fig. 2D). We also tested a full-length psi mutant whose palindromic dimerization loop sequence has been mutated to GAGA (Psi-ΔDIS), a mutation that completely eliminates RNA dimerization (Fig. 2C; Jones et al. 2014). All of these fragments inhibited Gag interactions with liposomes to a similar extent as the full-length, wild-type psi sequence (Fig. 4). Thus, we conclude that suppression of Gag–membrane interactions only requires SL1 and that dimerization is unlikely to play a role in this suppression.
FIGURE 4.
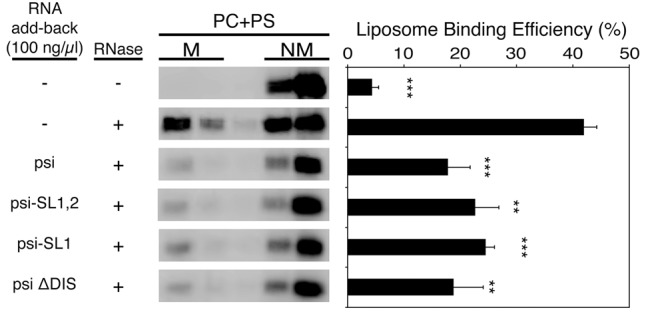
Psi SL1 is capable of inhibiting Gag binding to PC + PS liposomes. Psi and its derivatives shown in Figure 2 were examined for their effects on Gag binding to PC + PS liposomes using the add-back assay as described in Figure 1. Data from three different experiments are shown as mean ± standard deviation. P values were determined in comparison to the no RNA add-back condition using Student's t-test. (**) P < 0.01; (***) P < 0.001.
Psi RNA-mediated suppression of interactions between Gag and negatively charged membranes does not depend on the exact MA HBR sequence, but does require the NC domain
To elucidate the molecular determinants within Gag that are required for RNA-mediated suppression of membrane binding, we used the add-back assay to compare the impact of psi RNA addition on the membrane binding abilities of Gag with mutations in the MA or NC domains (Fig. 5A). To determine whether MA HBR recognizes specific RNAs in a manner dependent on its amino acid sequence rather than just electrostatic charge, we examined the “HBR switch mutant” in which the basic residues Arg and Lys between MA residues 14 and 31 have been swapped with one another in this region (Llewellyn et al. 2013). We found that the pattern of add-back inhibition by yeast tRNA, tRNAPro and psi RNA was identical for both WT Gag and the HBR switch mutant (Fig. 5B,C). We observed that tRNALys3 has a minor but statistically significant inhibitory effect on the HBR switch mutant, suggesting that the basic residue arrangement in the HBR may influence the tRNA specificity. Together with our previous observations (Llewellyn et al. 2013; Inlora et al. 2014), these results suggest that while the charge in the HBR is critical, a specific RNA binding motif or structure in the WT HBR is not required for RNA-mediated suppression of Gag membrane binding.
FIGURE 5.

The NC domain is required for inhibition of Gag binding to PC + PS liposomes mediated by psi, yeast tRNA, and tRNAPro. Gag constructs containing MA or NC changes were examined for susceptibility of their liposome binding ability to various RNA species. (A) Schematic representation of Gag derivatives used in these experiments. (B) [35S]-labeled WT Gag, HBR switch Gag, or delNC Gag, synthesized using rabbit reticulocyte lysates, were treated (or left untreated) with RNase A and examined for susceptibility of their liposome binding ability to yeast tRNA or psi in the add-back experiments performed as described in Figure 1. In these experiments, liposome binding efficiency normalized to the binding efficiency observed upon RNase treatment is shown as 100% for each Gag construct. The actual mean liposome binding efficiencies under this condition for WT Gag, HBR switch Gag, and delNC Gag are 47%, 42%, and 46%, respectively. Data from five different experiments are shown as mean ± standard deviation. P values were determined in comparison to the no RNA add-back condition using Student's t-test. (***) P < 0.001. (C,D) Dependence of the inhibition of Gag–liposome binding on the MA HBR sequence (C) and NC (D) was examined for yeast tRNA, tRNALys3, and tRNAPro as in panel B. The actual mean liposome binding efficiencies upon RNase treatment are 33% (C) and 31% (D) for WT Gag, 35% for HBR switch Gag, and 72% for delNC Gag. Data from three different experiments are shown as mean ± standard deviation. P-values were determined in comparison to the no RNA add-back condition using Student's t-test. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
Since psi RNA is known to interact specifically with NC, we next examined the effect of psi RNA on liposome binding of delNC Gag, a mutant in which most residues of the NC domain (NC residues 5–52) have been deleted. Interestingly, although delNC Gag binding to PC + PS liposomes also increased upon RNase treatment, relative to WT Gag this derivative exhibited higher liposome binding prior to RNase treatment, suggesting that RNA-dependent suppression of Gag–liposome binding is somewhat attenuated in the absence of NC. In addition, we found that upon add-back neither yeast total tRNA nor psi RNA suppressed delNC Gag interactions with negatively charged liposomes lacking PI(4,5)P2 (Fig. 5B). Likewise, tRNAPro, which blocked WT Gag binding to these liposomes (Fig. 3), is no longer inhibitory to membrane binding of Gag upon NC deletion (Fig. 5D). These results strongly support the possibility that the NC domain is required for recognizing specific RNAs and imparting inhibition of Gag interactions with nonspecific acidic membranes.
NC likely facilitates psi- and tRNA-mediated suppression of Gag binding to negatively charged membranes via different mechanisms
The above results suggest that NC plays an important role in recruiting specific RNAs that suppress Gag binding to acidic lipid bilayers lacking PI(4,5)P2. We hypothesized that selective binding of RNA to NC promotes binding of RNAs to MA, which in turn shields the HBR from nonspecific interactions with negatively charged membranes.
In order to examine which domains of Gag were involved in specific binding of each RNA species, we monitored binding between purified GagΔp6, MA, and NC domains and RNAs by fluorescence anisotropy (FA). We compared binding of TAR/polyA and psi, as well as two human tRNAs, tRNALys3 and tRNAPro. Measurements were performed at varying salt concentrations, which allowed determination of the number of electrostatic interactions (Zeff) and the strength of the nonelectrostatic/specific component of binding, Kd(1M) (Table 1; Rye-McCurdy et al. 2015).
TABLE 1.
Parameters for HIV-1 GagΔp6, NC, and MA proteins binding to tRNAs and HIV-1 genome-derived RNAs
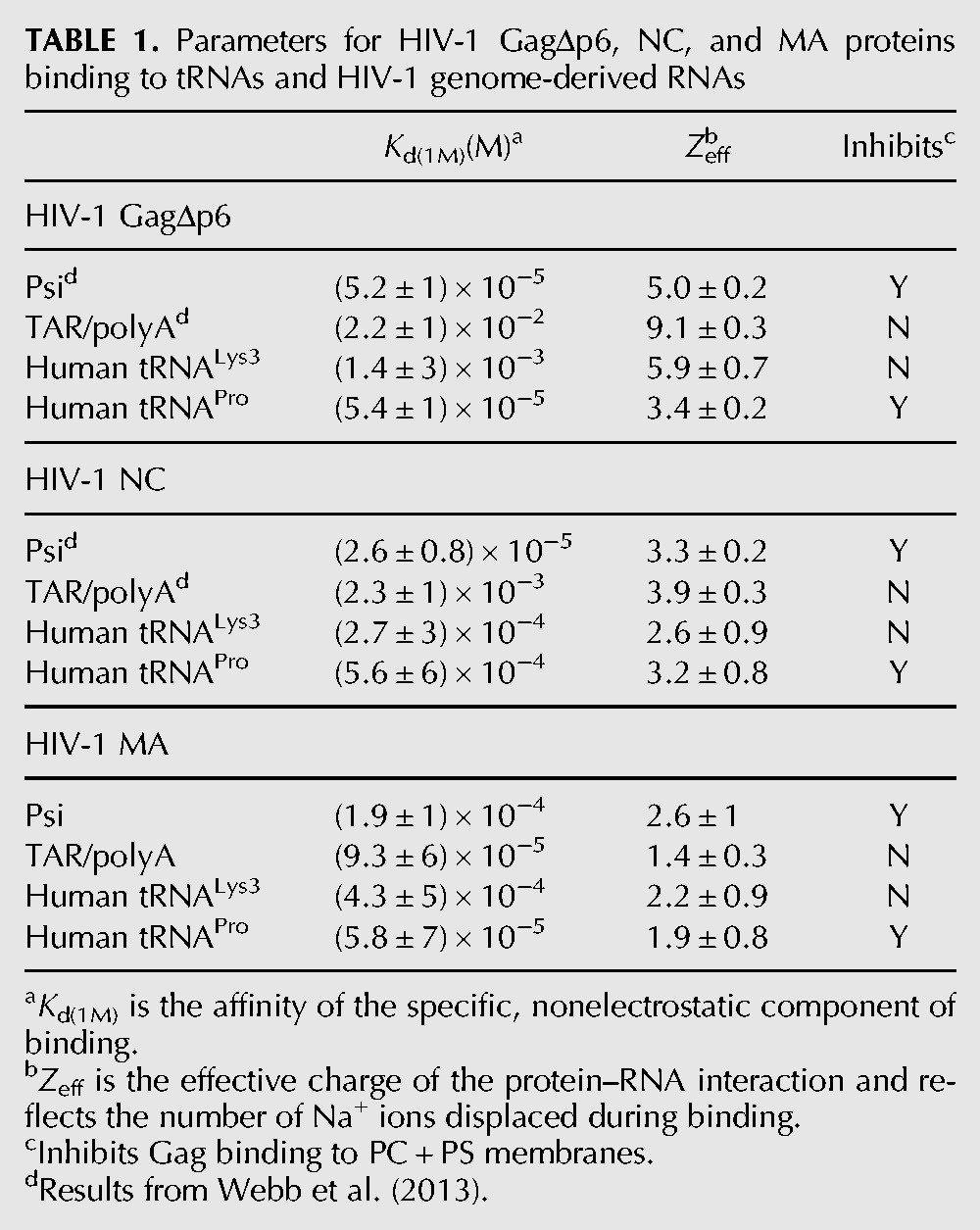
As previously reported (Webb et al. 2013), both GagΔp6 and NC display specific binding to psi that is characterized by a greater nonelectrostatic binding component (Kd(1M) values ∼10−5) relative to TAR/polyA (Kd(1M) values ∼10−2–10−3) and lower effective charge in the case of GagΔp6. The latter is consistent with GagΔp6 binding primarily via the NC domain to psi and with both the NC and MA domains to TAR/polyA. In contrast, MA interactions with TAR/polyA and psi RNAs were found to have similar values of Kd(1M), and the interactions were mediated by a similar number of electrostatic contacts in each case (∼2), suggesting that MA alone is not able to distinguish between these RNAs (Table 1). Considering that psi RNA, but not TAR/polyA, is able to suppress Gag–membrane interactions, these results suggest that NC recognition of psi RNA is a critical step for proper Gag–membrane regulation by this RNA.
MA demonstrated only a modest preference for tRNAPro over tRNALys3, with an approximately sevenfold lower Kd(1M) value. Both tRNAPro and tRNALys3 have similar electrostatic and specific binding interactions with NC, yet only tRNAPro is able to block Gag binding to PC + PS liposomes. Interestingly, the Kd(1M) value determined for binding of tRNAPro to GagΔp6 was ∼25-fold lower than that for tRNALys3, suggesting that full-length Gag interacts more specifically with tRNAPro relative to tRNALys3. Taken together, these results suggest that while both psi and tRNAPro suppress MA-acidic lipid interactions in an NC-dependent manner, NC may facilitate this process via different mechanisms; the NC domain alone is sufficient for the specificity of psi RNA, whereas for tRNAPro, NC determines the specificity in the context of Gag, potentially in combination with MA.
DISCUSSION
In this study, we demonstrated that specific RNA species are necessary to suppress MA-mediated Gag binding to negatively charged lipid membranes lacking PI(4,5)P2. We find that yeast total tRNA and HeLa total tRNA (results not shown), as well as in vitro synthesized human tRNAPro and RNAs containing SL1 of the psi region of the 5′ UTR, are able to inhibit Gag binding to PC + PS liposomes. In contrast, in vitro synthesized human tRNALys3, an RNA aptamer selected for MA binding and the TAR/polyA domain of the 5′ UTR have no impact on membrane binding (Figs. 2, 3). While MA mutagenesis in this and previous studies (Fig. 5; Llewellyn et al. 2013; Inlora et al. 2014) suggest that specific MA sequences are unlikely to be a key determinant for the susceptibility to RNA-mediated suppression, the sequence and/or structure of the RNA is likely to play an important role in inhibition of MA-acidic lipid interactions.
Importantly, we found that the ability of psi RNA to inhibit Gag–membrane interactions is dependent on the NC domain (Fig. 5B). Considering that MA and NC domains are in a close proximity within a single Gag molecule in solution (Datta et al. 2007; Munro et al. 2014), these results suggest a model for NC-dependent “loading” of RNA onto the MA domain in which recognition of RNA by the NC domain precedes or facilitates binding of the same RNA to MA. The results of salt-titration experiments (Table 1) are consistent with the possibility that specific recognition of the psi sequence by NC, rather than MA, plays the major role in the inhibitory effect of the psi-containing RNAs on Gag membrane binding. This effect is not dependent on psi dimerization, which is consistent with our previous observation that dimerization of psi RNA does not play a role in NC or Gag binding specificity (Webb et al. 2013). However, MA may also play a role in this effect, since we have previously shown that an HIV-1 CANC construct that lacks the MA domain binds to psi RNA with reduced specificity (Webb et al. 2013). Irrespective of whether the mechanism involves one or both Gag domains, it is tempting to speculate that this psi-mediated suppression of Gag interactions with acidic lipids reduces the association of Gag–psi complexes to non-PM membranes, thus ensuring productive assembly and release of viral particles containing the RNA genome from cells.
In apparent contrast to our study, a recent study by Carlson et al. (2016) reported that the 5′ UTR RNA promotes binding of recombinant Gag protein to giant unilamellar vesicle membranes. Among many differences in the experimental conditions between that study and ours, the presence or absence of PI(4,5)P2 in lipid membranes is likely a key difference. As described in the Introduction, in the presence of PI(4,5)P2 RNA can no longer mediate inhibition of Gag membrane binding (Chukkapalli et al. 2010, 2013). Carlson et al. (2016) used lipid vesicles containing PI(4,5)P2, which we speculate attenuated the inhibitory effect of 5′ UTR RNA that we observe for membranes lacking PI(4,5)P2. Instead, they observed an enhancement of membrane binding due to Gag multimerization induced by the 5′ UTR. As the focus of the present study was to compare the ability of different RNAs to inhibit nonspecific Gag binding to membranes, we used liposomes lacking PI(4,5)P2. Under these conditions, the inhibitory effect of the 5′ UTR is readily observed. It is also possible that the presence of 5′ UTR RNA in excess of Gag in our system may have prevented Gag multimerization and hence enhancement of membrane binding.
We also observed that human tRNAPro efficiently suppresses Gag binding to acidic membranes in an NC-dependent manner (Figs. 3, 5C). However, in contrast to psi RNA, NC alone did not exhibit strong nonelectrostatic/specific binding to tRNAPro, especially when compared to tRNALys3, which does not suppress Gag interactions with acidic lipids. Binding between MA and tRNAPro is slightly more specific than the interaction between MA and tRNALys3 (approximately sevenfold lower Kd(1M), Table 1). Therefore, it is conceivable that tRNAPro, but not tRNALys3, may interact with a feature in MA shared by WT and HBR switch mutant, both of which are sensitive to tRNAPro. However, the specificity for tRNAPro relative to tRNALys3 was much more pronounced in the context of GagΔp6.
Although deletion of the NC domain did not significantly affect the identity of tRNAs preferentially bound to MA in cells (Kutluay et al. 2014), deletion of NC abolished suppression of Gag–liposome binding by all RNAs tested in the add-back assay, including total yeast tRNA and tRNAPro. Therefore, it is likely that NC modulates RNA–MA interactions in the context of the in vitro liposome binding assay, either directly by binding the same RNA molecule as MA or indirectly via alteration of the MA/Gag conformation. Conversely, in cells, it is conceivable that the presence of PI(4,5)P2 or other cellular components may facilitate dissociation of tRNAs that are differentially bound to WT and NC-deleted Gag.
Interestingly, RNAs that have been shown to bind MA in vitro or in cells (Purohit et al. 2001; Alfadhli et al. 2011; Kutluay et al. 2014), i.e., the SELEX-derived RNA and tRNALys3, did not inhibit Gag binding to PC + PS liposomes in the add-back experiments. This difference is likely due at least in part to the difference in assays. The outcomes of the add-back assay represent a combination of MA-RNA interactions and their competition with MA-liposome binding. Therefore, it is possible that even when an RNA species can bind MA under a given condition, the binding may be insufficient for suppressing MA-mediated Gag binding to negatively charged liposomes. The SELEX oligonucleotide has been shown to compete well against a water-soluble PS analog (Alfadhli et al. 2011). However, this RNA may no longer be able to compete when Gag binding to liposomes occurs in the presence of hydrophobic interaction via MA's myristoyl moiety as is the case in this study. It is also possible that the smaller size of the SELEX RNA relative to the other RNAs investigated in this study precludes simultaneous interactions with both MA and NC domains of Gag. The ability of tRNALys3 to outcompete interactions between Gag and PS has not been examined previously. While we do not find tRNALys3 capable of regulating Gag–membrane interactions, it is possible that the chemical modifications missing from in vitro synthesized tRNA used in this study may play an important role in MA binding and/or competition with negatively charged liposomes (Graham et al. 2011). Studies aimed at elucidating the factors involved in tRNA loading to MA in cells are underway.
In summary, the current study demonstrates that specific RNA motifs or structures, rather than mere electrostatic interactions between MA and RNA, are necessary for blocking Gag binding to nonspecific membranes. Furthermore, NC-mediated RNA interactions contribute to inhibition of Gag–membrane binding. These findings serve as the initial investigation into the molecular determinants of RNA-mediated suppression of Gag–membrane interactions and will inform efforts to develop therapeutic MA-binding aptamers that robustly inhibit Gag membrane binding even in the presence of PI(4,5)P2.
MATERIALS AND METHODS
Plasmids
WT Gag and all mutants thereof used in the RNA add-back experiments were expressed from a pGEM vector containing a fragment from the pNL4-3 strain of HIV-1 from nucleotides 639–5748 (Gag is 790–2292) oriented downstream from an SP6 promoter (Chukkapalli et al. 2008). The mutants HBR switch (Llewellyn et al. 2013) and delNC (a gift from David Ott [AIDS and Cancer Virus Program, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research] [Ott et al. 2003]) were all prepared by standard molecular biology techniques.
RNA
Yeast total tRNA was obtained from Ambion. For HeLa tRNA, RNA of ∼80 nt was isolated by gel excision from low molecular weight RNA (<200 nt) prepared using the QIAGEN miRNeasy kit according to the manufacturer's instructions. The SELEX and Random 25-mers were synthesized and HPLC purified by IDT. All other RNA constructs were in vitro transcribed from linearized plasmids using T7 RNA polymerase and purified by previously established methods (Milligan et al. 1987). Human tRNALys3 and human tRNAPro were in vitro transcribed from pLysf119 (Shiba et al. 1997) and pPUC119htRNAPro (Stehlin et al. 1998), respectively. HIV-1 TAR/polyA (104 nt), PBS (105 nt), Psi (109 nt), Psi-ΔDIS (105 nt), Psi-SL1 (51 nt), and Psi-SL1,2 (68 nt) RNAs were in vitro transcribed from linearized plasmids cloned from the pMSMΔEnv containing HIV NL4-3 cDNA as previously described (Webb et al. 2013). RNA concentrations were determined by measuring the absorbance at 260 nm and the following molar extinction coefficients: 60.4 × 104 M−1 cm−1 (htRNALys3); 66.5 × 104 M−1 cm−1 (htRNAPro); 92.6 × 104 M−1 cm−1 [HIV-1 TAR/polyA]; 93.6 × 104 M−1 cm−1 (HIV-1 PBS); 97.3 × 104 M−1 cm−1 (HIV-1 Psi); 93.6 × 104 M−1 cm−1 (HIV-1 Psi-ΔDIS); 43.1 × 104 M−1 cm−1 (HIV-1 Psi-SL1); 59.0 × 104 M−1 cm−1 (HIV-1 Psi-SL1,2).
Proteins
All recombinant proteins were expressed and purified from Escherichia coli. Purified HIV NC was a gift from Robert J. Gorelick (AIDS and Cancer Virus Program, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research) and was prepared as previously described (Wu et al. 1996; Carteau et al. 1999). The plasmid encoding His-tagged HIV-1 MA (HIV-1 H6.MA) in a pET16B vector was a gift from Louis M. Mansky (University of Minnesota). HIV-1 H6.MA was purified using an established protocol (Massiah et al. 1994), with additional steps of polyethylenimine (5% v/v) precipitation followed by ammonium sulfate (60% saturation) precipitation to remove nucleic acids. The plasmid encoding HIV-1 GagΔp6 was a gift from Dr. Alan Rein (HIV Drug Resistance Program, Center for Cancer Research, National Cancer Institute). HIV-1 GagΔp6 was expressed and purified essentially as previously described, with the addition of a polyethylenimine (0.15% v/v) precipitation step immediately after cell lysis (Datta and Rein 2009). Protein concentrations were determined by measuring the absorbance at 280 nm and using the following molar extinction coefficients: 1.702 × 104 M−1 cm−1 (HIV-1 MA), 5.690 × 103 M−1 cm−1 (HIV-1 NC), and 6.309 × 104 M−1 cm−1 (HIV-1 GagΔp6).
Preparation of liposomes
Lipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (sodium salt) (POPS) were purchased from Avanti Polar Lipids, Inc. dissolved in chloroform. Using a glass Hamilton syringe, desired amounts of lipids were transferred to a glass vial and dried down with a stream of N2(g). Films were further dried overnight under vacuum. Lipid films were resuspended in 150 μL of 20 mM HEPES, pH 7.0, (HB) by vortexing for 1 min and then allowed to hydrate at room temperature for ∼1 h. Sizing of liposomes was achieved via ten freeze/thaw cycles and subsequent extrusion through a 100 nm polycarbonate membrane using a mini extruder (Avanti). Fresh liposomes were prepared for each experiment.
RNA add-back liposome flotation assay
Gag was prepared by in vitro transcription/translation (TNT) in a rabbit reticulocyte lysate (Promega, L4600) as per manufacturer's instructions except that a homemade stock of amino acid (-Met) was used (Sigma, LAA21-1KT, 1 mM each amino acid in 10 mM Tris pH 7.0) in some experiments. TNT reactions were carried out as a master mix at 30°C for 90 min at which point a small aliquot (25 μL) of the TNT reaction was removed as a negative control. HB (1.5 μL) was added to the negative control while RNase A (Thermo Scientific, diluted to 1 μg/μL in HB) was added to the pool, 1.5 μL per reaction. Following incubation at 37°C for 20 min, RNase A was inactivated by addition of 8 μL of RNasin (Promega) per reaction and incubated at 37°C for 10 min. RNAs of interest were refolded in reticulocyte lysis buffer (1× RRL: 20 mM HEPES-KOH, pH 7.0, 100 mM KCl, 0.5 mM MgCl2) (Pelham and Jackson 1976). Briefly, RNA dissolved in water was heated to 95°C for 1 min, snap cooled on ice, adjusted to 1× RRL buffer by addition of an appropriate amount of 10× RRL, and refolded at 37°C for 15 min. Refolded RNA was subsequently added to RNase-treated Gag and incubated at 37°C for 30 min; 1× RRL buffer was added to negative control and RNase-treated control reactions. Finally 7.5 μL of liposomes were added to each reaction and incubated at 37°C for 15 min. Total reaction volume was 50 μL.
Reactions were diluted to 200 μL with HB and, in an ultracentrifuge tube, vortexed with 1 mL of HB containing 85% (w/v) sucrose. 2.8 mL of HB containing 65% (w/v) sucrose and subsequently 1 mL of HB containing 10% (w/v) sucrose were layered on top of the reaction. Samples were subjected to ultracentrifugation at 35,000 rpm at 4°C for 16 h. The gradient was then divided into five 1 mL fractions. One hundred microliters of well-vortexed fractions were added to 50 μL of 3× SDS loading buffer (188 mM Tris–HCl, pH 6.8, 30% [v/v] glycerol, 15.2% [v/v] β-mercaptoethanol, 9.4% [w/v] SDS, 0.02% bromophenol blue), boiled for 5 min and then subjected to SDS-PAGE. Gels were fixed with 40% methanol and 10% acetic acid for 30 min then soaked in 1 M salicylic acid with 2% (v/v) glycerol for 30 min followed by drying at 80°C for 2 h. Dried gels were exposed overnight to a phosphorimaging screen and then quantified using a GE Typhoon scanner and ImageQuant software. The percentage of Gag bound to membranes is taken as the amount of Gag in the first two fractions (top of the gradient, membrane bound) over the total amount of Gag in all fractions.
Fluorescence anisotropy binding assays
FA was used to determine the binding affinity of HIV-1 MA and NC for various RNAs as previously described (Stewart-Maynard et al. 2008). All RNAs were labeled with fluorescein-5-thiosemicarbazide (FTSC) at the 3′ end using established protocols (Rye-McCurdy et al. 2015). The concentration and labeling efficiency was determined by measuring the absorbance at 260 nm and 495 nm and using the molar extinction coefficient of 8.5 × 104 M−1 cm−1 (FTSC, ε495) and the molar extinction coefficients at 260 nm for the nucleic acids as listed above. HIV-1 H6.MA FA assays were performed using 5 nM FTSC-labeled RNA, 50 mM monovalent ions, 1 mM MgCl2 and 20 mM Tris–HCl, pH 8. HIV-1 NC FA assays were performed using 5 nM FTSC-labeled RNA, 50 mM monovalent ions, 1 mM MgCl2, 20 mM HEPES, pH 7.4, 20 µM tris(2-carboxyethyl)phosphine (TCEP), and 5 mM β-mercaptoethanol. HIV-1 GagΔp6 FA assays were performed using 5 nM FTSC-labeled RNA, 50 mM monovalent ions, 1 mM MgCl2, and 20 mM HEPES, pH 7.4. Binding affinities were determined by fitting FA data to a 1:1 binding model as previously described (Rye-McCurdy et al. 2015).
Fluorescence anisotropy salt-titration binding assays
All RNAs were fluorescently labeled as described in the previous section. Salt-titration assays were performed as previously described (Rye-McCurdy et al. 2015). Salt titrations with HIV-1 H6.MA were carried out using 5 nM FTSC-labeled RNA in the same buffer as for the FA binding assays, varying the monovalent ions from 16 mM to 1.3 M and using 680 nM (Psi and tRNALys3) or 5 µM [TAR/polyA and tRNAPro] HIV-1 H6.MA to prevent aggregation. Salt titrations with HIV-1 NC were performed with 5 nM FTSC-labeled RNA in the same buffer as for the FA binding assays, varying the monovalent ions from 16 mM to 1.3 M and using 500 nM (tRNAPro) or 750 nM (tRNALys3) HIV-1 NC. Salt titrations with HIV-1 GagΔp6 were performed with 5 nM FTSC-labeled RNA in the same buffer as for the FA binding assays, varying the monovalent ions from 30 mM to 1.3 M and using 1.7 µM HIV-1 GagΔp6. The extrapolated dissociation constant at 1 M salt (Kd(1M)) and effective charge (Zeff) were determined by fitting data to a 1:1 binding model as previously described (Rye-McCurdy et al. 2015). All fluorescence measurements were performed on a SpectraMax M5 plate reader (Molecular Devices).
ACKNOWLEDGMENTS
We thank the members of our laboratories for helpful discussions and critical review of the manuscript. We thank Drs. D. Ott and R. Gorelick for reagents. This work was supported by National Institutes of Health (NIH) training grant T32 AI007527 (to G.C.T.); the Ohio State University Center for RNA Biology Fellowship (to A.A.D.); American Heart Association Midwest Predoctoral Fellowship 13PRE17060006 and University of Michigan Rackham Predoctoral Fellowship (to J.I.); NIH training grant T32 GM008512 and NIH Predoctoral Fellowship F31 AI120868 (to E.D.O.); NIH grant R01 GM065056 (to K.M.-F.); and NIH grant R01 AI071727 (to A.O.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.058453.116.
REFERENCES
- Abd El-Wahab EW, Smyth RP, Mailler E, Bernacchi S, Vivet-Boudou V, Hijnen M, Jossinet F, Mak J, Paillart JC, Marquet R. 2014. Specific recognition of the HIV-1 genomic RNA by the Gag precursor. Nat Commun 5: 4304. [DOI] [PubMed] [Google Scholar]
- Alfadhli A, Still A, Barklis E. 2009. Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J Virol 83: 12196–12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfadhli A, McNett H, Tsagli S, Bachinger HP, Peyton DH, Barklis E. 2011. HIV-1 matrix protein binding to RNA. J Mol Biol 410: 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anraku K, Fukuda R, Takamune N, Misumi S, Okamoto Y, Otsuka M, Fujita M. 2010. Highly sensitive analysis of the interaction between HIV-1 Gag and phosphoinositide derivatives based on surface plasmon resonance. Biochemistry 49: 5109–5116. [DOI] [PubMed] [Google Scholar]
- Balasubramaniam M, Freed EO. 2011. New insights into HIV assembly and trafficking. Physiology (Bethesda) 26: 236–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros M, Heinrich F, Datta SA, Rein A, Karageorgos I, Nanda H, Losche M. 2016. Membrane binding of HIV-1 matrix protein: dependence on bilayer composition and protein lipidation. J Virol 90: 4544–4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson LA, Bai Y, Keane SC, Doudna JA, Hurley JH. 2016. Reconstitution of selective HIV-1 RNA packaging in vitro by membrane-bound Gag assemblies. Elife 5: e14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carteau S, Gorelick RJ, Bushman FD. 1999. Coupled integration of human immunodeficiency virus type 1 cDNA ends by purified integrase in vitro: stimulation by the viral nucleocapsid protein. J Virol 73: 6670–6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan R, Uchil PD, Jin J, Shui G, Ott DE, Mothes W, Wenk MR. 2008. Retroviruses human immunodeficiency virus and murine leukemia virus are enriched in phosphoinositides. J Virol 82: 11228–11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chukkapalli V, Hogue IB, Boyko V, Hu WS, Ono A. 2008. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. J Virol 82: 2405–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chukkapalli V, Oh SJ, Ono A. 2010. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc Natl Acad Sci 107: 1600–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chukkapalli V, Inlora J, Todd GC, Ono A. 2013. Evidence in support of RNA-mediated inhibition of phosphatidylserine-dependent HIV-1 Gag membrane binding in cells. J Virol 87: 7155–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimarelli A, Luban J. 1999. Translation elongation factor 1-α interacts specifically with the human immunodeficiency virus type 1 Gag polyprotein. J Virol 73: 5388–5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SA, Rein A. 2009. Preparation of recombinant HIV-1 gag protein and assembly of virus-like particles in vitro. Methods Mol Biol 485: 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SA, Curtis JE, Ratcliff W, Clark PK, Crist RM, Lebowitz J, Krueger S, Rein A. 2007. Conformation of the HIV-1 Gag protein in solution. J Mol Biol 365: 812–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick RA, Kamynina E, Vogt VM. 2013. Effect of multimerization on membrane association of Rous sarcoma virus and HIV-1 matrix domain proteins. J Virol 87: 13598–13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber PP, Cabrini M, Jancic C, Paoletti L, Banchio C, von Bilderling C, Sigaut L, Pietrasanta LI, Duette G, Freed EO, et al. 2015. Rab27a controls HIV-1 assembly by regulating plasma membrane levels of phosphatidylinositol 4,5-bisphosphate. J Cell Biol 209: 435–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham WD, Barley-Maloney L, Stark CJ, Kaur A, Stolarchuk C, Sproat B, Leszczynska G, Malkiewicz A, Safwat N, Mucha P, et al. 2011. Functional recognition of the modified human tRNALys3UUU anticodon domain by HIV's nucleocapsid protein and a peptide mimic. J Mol Biol 410: 698–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada F, Peters GG, Dahlberg JE. 1979. The primer tRNA for Moloney murine leukemia virus DNA synthesis. Nucleotide sequence and aminoacylation of tRNAPro. J Biol Chem 254: 10979–10985. [PubMed] [Google Scholar]
- Hearps AC, Wagstaff KM, Piller SC, Jans DA. 2008. The N-terminal basic domain of the HIV-1 matrix protein does not contain a conventional nuclear localization sequence but is required for DNA binding and protein self-association. Biochemistry 47: 2199–2210. [DOI] [PubMed] [Google Scholar]
- Inlora J, Chukkapalli V, Derse D, Ono A. 2011. Gag localization and virus-like particle release mediated by the matrix domain of human T-lymphotropic virus type 1 Gag are less dependent on phosphatidylinositol-(4,5)-bisphosphate than those mediated by the matrix domain of HIV-1 Gag. J Virol 85: 3802–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inlora J, Collins DR, Trubin ME, Chung JY, Ono A. 2014. Membrane binding and subcellular localization of retroviral Gag proteins are differentially regulated by MA interactions with phosphatidylinositol-(4,5)-bisphosphate and RNA. MBio 5: e02202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CP, Datta SA, Rein A, Rouzina I, Musier-Forsyth K. 2011. Matrix domain modulates HIV-1 Gag's nucleic acid chaperone activity via inositol phosphate binding. J Virol 85: 1594–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CP, Cantara WA, Olson ED, Musier-Forsyth K. 2014. Small-angle X-ray scattering-derived structure of the HIV-1 5′ UTR reveals 3D tRNA mimicry. Proc Natl Acad Sci 111: 3395–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane SC, Heng X, Lu K, Kharytonchyk S, Ramakrishnan V, Carter G, Barton S, Hosic A, Florwick A, Santos J, et al. 2015. RNA structure. Structure of the HIV-1 RNA packaging signal. Science 348: 917–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman L, Jones CP, Musier-Forsyth K. 2010. Formation of the tRNALys packaging complex in HIV-1. FEBS Lett 584: 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutluay SB, Zang T, Blanco-Melo D, Powell C, Jannain D, Errando M, Bieniasz PD. 2014. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell 159: 1096–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewellyn GN, Grover JR, Olety B, Ono A. 2013. HIV-1 Gag associates with specific uropod-directed microdomains in a manner dependent on its MA highly basic region. J Virol 87: 6441–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochrie MA, Waugh S, Pratt DG Jr, Clever J, Parslow TG, Polisky B. 1997. In vitro selection of RNAs that bind to the human immunodeficiency virus type-1 gag polyprotein. Nucleic Acids Res 25: 2902–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Heng X, Summers MF. 2011. Structural determinants and mechanism of HIV-1 genome packaging. J Mol Biol 410: 609–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massiah MA, Starich MR, Paschall C, Summers MF, Christensen AM, Sundquist WI. 1994. Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J Mol Biol 244: 198–223. [DOI] [PubMed] [Google Scholar]
- Mercredi PY, Bucca N, Loeliger B, Gaines CR, Mehta M, Bhargava P, Tedbury PR, Charlier L, Floquet N, Muriaux D, et al. 2016. Structural and molecular determinants of membrane binding by the HIV-1 matrix protein. J Mol Biol 428: 1637–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. 1987. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res 15: 8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monde K, Chukkapalli V, Ono A. 2011. Assembly and replication of HIV-1 in T cells with low levels of phosphatidylinositol-(4,5)-bisphosphate. J Virol 85: 3584–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro JB, Nath A, Farber M, Datta SA, Rein A, Rhoades E, Mothes W. 2014. A conformational transition observed in single HIV-1 Gag molecules during in vitro assembly of virus-like particles. J Virol 88: 3577–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olety B, Veatch SL, Ono A. 2015. Phosphatidylinositol-(4,5)-bisphosphate acyl chains differentiate membrane binding of HIV-1 Gag from that of the phospholipase Cδ1 Pleckstrin homology domain. J Virol 89: 7861–7873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. 2004. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci 101: 14889–14894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott DE, Coren LV, Chertova EN, Gagliardi TD, Nagashima K, Sowder RC II, Poon DT, Gorelick RJ. 2003. Elimination of protease activity restores efficient virion production to a human immunodeficiency virus type 1 nucleocapsid deletion mutant. J Virol 77: 5547–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham HR, Jackson RJ. 1976. An efficient mRNA-dependent translation system from reticulocyte lysates. Eur J Biochem 67: 247–256. [DOI] [PubMed] [Google Scholar]
- Purohit P, Dupont S, Stevenson M, Green MR. 2001. Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA 7: 576–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam D, Duclair S, Datta SA, Ellington A, Rein A, Prasad VR. 2011. RNA aptamers directed to human immunodeficiency virus type 1 Gag polyprotein bind to the matrix and nucleocapsid domains and inhibit virus production. J Virol 85: 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rye-McCurdy T, Rouzina I, Musier-Forsyth K. 2015. Fluorescence anisotropy-based salt-titration approach to characterize protein-nucleic acid interactions. Methods Mol Biol 1259: 385–402. [DOI] [PubMed] [Google Scholar]
- Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. 2006. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci 103: 11364–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba K, Stello T, Motegi H, Noda T, Musier-Forsyth K, Schimmel P. 1997. Human lysyl-tRNA synthetase accepts nucleotide 73 variants and rescues Escherichia coli double-defective mutant. J Biol Chem 272: 22809–22816. [DOI] [PubMed] [Google Scholar]
- Shkriabai N, Datta SA, Zhao Z, Hess S, Rein A, Kvaratskhelia M. 2006. Interactions of HIV-1 Gag with assembly cofactors. Biochemistry 45: 4077–4083. [DOI] [PubMed] [Google Scholar]
- Smyth RP, Despons L, Huili G, Bernacchi S, Hijnen M, Mak J, Jossinet F, Weixi L, Paillart JC, von Kleist M, et al. 2015. Mutational interference mapping experiment (MIME) for studying RNA structure and function. Nat Methods 12: 866–872. [DOI] [PubMed] [Google Scholar]
- Stehlin C, Burke B, Yang F, Liu H, Shiba K, Musier-Forsyth K. 1998. Species-specific differences in the operational RNA code for aminoacylation of tRNAPro. Biochemistry 37: 8605–8613. [DOI] [PubMed] [Google Scholar]
- Stewart-Maynard KM, Cruceanu M, Wang F, Vo MN, Gorelick RJ, Williams MC, Rouzina I, Musier-Forsyth K. 2008. Retroviral nucleocapsid proteins display nonequivalent levels of nucleic acid chaperone activity. J Virol 82: 10129–10142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundquist WI, Krausslich HG. 2012. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med 2: a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd GC, Ono A. 2016. Methods to study determinants for membrane targeting of HIV-1 Gag in vitro. Methods Mol Biol 1354: 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr, Swanstrom R, Burch CL, Weeks KM. 2009. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460: 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JA, Jones CP, Parent LJ, Rouzina I, Musier-Forsyth K. 2013. Distinct binding interactions of HIV-1 Gag to Psi and non-Psi RNAs: implications for viral genomic RNA packaging. RNA 19: 1078–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM. 2008. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol 6: e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Henderson LE, Copeland TD, Gorelick RJ, Bosche WJ, Rein A, Levin JG. 1996. Human immunodeficiency virus type 1 nucleocapsid protein reduces reverse transcriptase pausing at a secondary structure near the murine leukemia virus polypurine tract. J Virol 70: 7132–7142. [DOI] [PMC free article] [PubMed] [Google Scholar]