

Abstract
The 3-methylcytidine (m3C) modification is ubiquitous in eukaryotic tRNA, widely found at C32 in the anticodon loop of tRNAThr, tRNASer, and some tRNAArg species, as well as in the variable loop (V-loop) of certain tRNASer species. In the yeast Saccharomyces cerevisiae, formation of m3C32 requires Trm140 for six tRNA substrates, including three tRNAThr species and three tRNASer species, whereas in Schizosaccharomyces pombe, two Trm140 homologs are used, one for tRNAThr and one for tRNASer. The occurrence of a single Trm140 homolog is conserved broadly among Ascomycota, whereas multiple Trm140-related homologs are found in metazoans and other fungi. We investigate here how S. cerevisiae Trm140 protein recognizes its six tRNA substrates. We show that Trm140 has two modes of tRNA substrate recognition. Trm140 recognizes G35-U36-t6A37 of the anticodon loop of tRNAThr substrates, and this sequence is an identity element because it can be used to direct m3C modification of tRNAPhe. However, Trm140 recognition of tRNASer substrates is different, since their anticodons do not share G35–U36 and do not have any nucleotides in common. Rather, specificity of Trm140 for tRNASer is achieved by seryl-tRNA synthetase and the distinctive tRNASer V-loop, as well as by t6A37 and i6A37. We provide evidence that all of these components are important in vivo and that seryl-tRNA synthetase greatly stimulates m3C modification of tRNASer(CGA) and tRNASer(UGA) in vitro. In addition, our results show that Trm140 binding is a significant driving force for tRNA modification and suggest separate contributions from each recognition element for the modification.
Keywords: 3-methylcytidine, methyltransferase, modification, tRNA, specificity, anticodon loop
INTRODUCTION
tRNA undergoes extensive post-transcriptional modifications in all domains of life to ensure the efficiency and accuracy of translation. In the yeast Saccharomyces cerevisiae, each cytoplasmic tRNA has an average of 12.6 modifications, with ∼10 modifications in the main body of the tRNA and ∼2.6 modifications in the anticodon loop region, comprising residues N32–N38 of the anticodon loop and the neighboring N31–N39 base pair of the anticodon stem (Juhling et al. 2009). Modifications within the tRNA body often contribute to folding or stability (Helm et al. 1999; Kadaba et al. 2004; Alexandrov et al. 2006; Whipple et al. 2011). In contrast, modifications in the anticodon (primarily at the wobble nucleotide N34) or at N37 often contribute to accurate decoding and reading frame maintenance (Gerber and Keller 1999; Bjork et al. 2001, 2007; Urbonavicius et al. 2001; Murphy and Ramakrishnan 2004; Esberg et al. 2006; Agris et al. 2007; Waas et al. 2007; Weixlbaumer et al. 2007; Johansson et al. 2008; Chen et al. 2011; El Yacoubi et al. 2011).
Modifications occurring at other residues within the anticodon loop region also have important roles in tRNA function. For example, a yeast pus3Δ mutant, which lacks pseudouridine at U38 or U39, is temperature sensitive, primarily due to reduced tRNAGln(UUG) function (Han et al. 2015); and a yeast trm7Δ mutant, which lacks 2′-O-methylation at C32 (Cm) as well as Gm34, grows poorly due to reduced translation and reduced tRNAPhe function (Pintard et al. 2002; Guy et al. 2012). N32 and N38 are at the borders of the anticodon loop, often form a noncanonical base pair (Auffinger and Westhof 1999), and have been shown to be critical for ribosome binding and decoding (Lustig et al. 1993; Olejniczak et al. 2005; Olejniczak and Uhlenbeck 2006; Ledoux et al. 2009).
The 3-methylcytidine (m3C) modification is also found at C32 in the anticodon loop of tRNAs and likely has an important role. m3C is found at C32 of all four characterized eukaryotic cytoplasmic tRNAThr species, 18 of the 20 characterized cytoplasmic tRNASer species, and two of five characterized eukaryotic tRNAArg species that have an encoded C32, as well as at residue e2 of the tRNASer variable loop (V-loop) in animals (Weissenbach et al. 1977; Juhling et al. 2009; Machnicka et al. 2013; Arimbasseri et al. 2016). The m3C modification is formed by members of the Trm140 m3C methyltransferase family. In S. cerevisiae, TRM140 is required for m3C32 modification of six tRNA species, including all three tRNAThr species, with anticodons IGU, CGU, and UGU, and the three tRNASer species with anticodons CGA, UGA, and GCU (D'Silva et al. 2011; Noma et al. 2011), while tRNASer(IGA) does not have C32. In contrast, in Schizosaccharomyces pombe there are two TRM140 homologs; trm140+ is required for modification of all three tRNAThr species, whereas trm141+ is required for modification of all four tRNASer species (Arimbasseri et al. 2016). Although S. cerevisiae trm140Δ mutants and S. pombe mutants lacking TRM140 and/or TRM141 have no growth defect in a variety of media (D'Silva et al. 2011; Arimbasseri et al. 2016), the importance of m3C32 is underscored by the growth defect of S. cerevisiae trm140Δ trm1Δ mutants in the presence of cycloheximide (D'Silva et al. 2011), and by the broad conservation of the TRM140 family and the m3C modification in eukaryotes.
One important question about S. cerevisiae Trm140 is how it recognizes its specific tRNA substrates. For enzymes such as Pus3 that modify the same residues in every tRNA, recognition typically exploits a common structural feature of tRNAs (Hur and Stroud 2007). For enzymes such as the tRNAHis guanylyltransferase Thg1, which only modifies tRNAHis, recognition of the unique GUG anticodon drives modification (Jackman and Phizicky 2006; Nakamura et al. 2013). However, several modifying enzymes recognize a specific subset of tRNAs without obvious recognition elements; examples include the m1G9 modification catalyzed by Trm10 for 13 of 24 species with G9 (Swinehart et al. 2013; Swinehart and Jackman 2015) and the Cm32 and Gm34 modification catalyzed by Trm7 on three tRNA species (Pintard et al. 2002), as well as the m3C32 modification catalyzed by S. cerevisiae Trm140 on its six substrates (D'Silva et al. 2011; Noma et al. 2011). Indeed, simple sequence inspection suggests no common theme that would direct Trm140 modification of its three tRNAThr substrates and its three tRNASer substrates that distinguishes these tRNAs from the 17 other S. cerevisiae tRNAs with C32.
Here we define the specificity of S. cerevisiae Trm140 by both in vivo and in vitro approaches. We show that there are two distinct modes of Trm140 recognition of tRNA substrates for m3C32 modification. For tRNAThr species, Trm140 reads the anticodon nucleotides and the t6A modification, whereas for tRNASer species, recognition is achieved through seryl-tRNA synthetase (SerRS, encoded by SES1) and the V-loop region, as well as i6A37 or t6A37.
RESULTS
An XGU anticodon and t6A37 are necessary and sufficient for m3C modification of tRNAThr species
To begin to elucidate the specificity of Trm140, we designed a scaffold based on tRNAThr(CGU) [gene name: tT(CGU)] that would enable analysis of m3C in variants with this target scaffold independent of other tRNAs present in the cell. We altered the identity of base pairs 28:42 and 50:64 to allow us to analyze this tRNA species (tRNAThr(CGU)28,50) with oligonucleotides spanning these residues (Fig. 1A). This variant was fully functional because an otherwise lethal tT(CGU)Δ strain was completely rescued upon integration of tT(CGU)28,50, with no discernable growth defect in the range of 30°C to 37°C, compared to the corresponding strain bearing wild-type tT(CGU) at the same locus (Fig. 1B). The tRNAThr(CGU)28,50 variant was easily analyzed by primer extension independent of the wild-type tRNAThr(CGU) (Fig. 1C), and was efficiently modified to m3C; the vast majority of this tRNA has m3C32, based on the prominent primer extension block at N33, compared to the relatively minor read-through signal (Fig. 1C), similar to the amount of m3C modification observed for wild-type tRNAThr(CGU) (Fig. 2).
FIGURE 1.

The tRNAThr(CGU)28,50 scaffold used for analysis of tRNAThr variants is fully functional and efficiently modified to m3C. (A) Schematic of tRNAThr(CGU)28,50. The secondary structure of tRNAThr(CGU) is shown. The base pairs of residues 28:42 and 50:64 are switched as indicated in the boxes. The primer complementary to residues 55–36 is indicated with a 3′ arrow. (B) tRNAThr(CGU)28,50 is functional in vivo. tT(CGU)Δ cells containing the integrated WT tT(CGU) or tT(CGU)28,50 as indicated were grown overnight in YPD medium, serially diluted and spotted onto YPD medium, and plates were incubated at indicated temperatures for 3 d. (C) tRNAThr(CGU)28,50 is efficiently modified to m3C32. Bulk RNA from cells containing the integrated WT tT(CGU) or tT(CGU)28,50 as indicated were analyzed by primer extension assay, as described in Materials and Methods, using the primer shown in A. The vast majority of tRNAThr(CGU)28,50 has m3C32 based on the primer extension stop at U33, compared to the amount of read-through (RT).
FIGURE 2.

An XGU anticodon and t6A37 are important for m3C modification of tRNAThr(CGU). (A) G35 and U36 of tRNAThr(CGU) are required for m3C formation. Bulk RNA from strains containing integrated tT(CGU)28,50 variants with mutations as indicated were analyzed for m3C by primer extension, using a primer complementary to residues 51–37 of tRNAThr(CGU)28,50. The major read-through band on the top of the gel corresponds to the full-length tRNA. Note that for variants with G35 and U36, the primer extension is slightly displaced, presumably due to a different primer extension sequence. (B) Summary of primer extension results of m3C modification in A. +++, WT levels of m3C; +, low but detectable m3C; ND, m3C not detected. (C) t6A37 is important for m3C formation of tRNAThr species. Bulk RNA from SUA5+ and sua5Δ cells was analyzed by primer extension with primers as indicated, to evaluate the importance of t6A for m3C modification of tRNAThr(CGU), tRNAThr(UGU), and tRNAThr(IGU).
Since the tRNAThr species bearing m3C32 all have similar anticodons (CGU, UGU, and IGU), we made and analyzed a set of yeast strains, each with an integrated tRNAThr(CGU)28,50 variant bearing a single mutation in the anticodon loop (Fig. 2A,B). Primer extension analysis showed that alteration of C34 to other nucleotides resulted in retention of the major primer extension block at N33, indicative of m3C32. In contrast, substitution of residue G35 or U36 with each of the other nucleotides resulted in almost complete loss of m3C32. These results suggest that the conserved G35 and U36 residues in the anticodons of tRNAThr species are required for m3C formation. As expected, substitution of C32 to U32 resulted in no primer extension stop.
Since U36 was important for m3C modification activity, and U36 is always followed by t6A37 (Machnicka et al. 2013) or, as frequently occurs in yeast and some other organisms, a cyclized derivative of t6A called ct6A (Miyauchi et al. 2013), it was possible that t6A37 or ct6A37 (collectively referred to as t6A for simplicity) also had a role in m3C formation. To examine the contribution of t6A to m3C modification, we analyzed m3C levels in wild-type tRNAThr species from an sua5Δ strain, which lacks t6A37 (El Yacoubi et al. 2009). The m3C levels in each of the three tRNAThr species from the sua5Δ strain were substantially reduced compared to those from the corresponding wild-type strain (Fig. 2C). This result suggested that t6A37 was important, but not absolutely required, for m3C modification of the three tRNAThr species in vivo, along with the required G35–U36 anticodon sequence.
To probe the connection between Trm140 tRNA modification specificity and Trm140 binding, we developed a pull-down assay. In a strain in which Trm140 was overproduced with a C-terminal tandem affinity purification tag (PT), we observed highly specific copurification of tRNA substrates. Trm140 was purified using IgG Sepharose in buffer conditions predicted to maintain native protein–RNA interactions, and then bound protein was washed once and eluted with protease to release Trm140 and retained RNAs. Northern analysis of the eluted tRNAs (Fig. 3A) showed highly efficient retention of Trm140 substrate tRNAs, including tRNAThr(IGU), tRNAThr(UGU), tRNAThr(CGU), relative to that in a vector control, whereas several nonsubstrate tRNAs examined did not copurify with Trm140, including tRNALeu(UAA), tRNAPhe(GAA), and tRNATrp(CCA). Similar copurification results were obtained upon overproduction of ORF240 (Fig. 3B), the C-terminal domain of Trm140 (comprising residues 277–628), which is necessary and sufficient for the methyltransferase activity in vivo (D'Silva et al. 2011); with either Trm140 or ORF240, we observed efficient retention of tRNAThr(IGU), tRNASer(GCU), and tRNASer(UGA)/(CGA) (which were not distinguished by the hybridization probe we used), compared to the vector control. Because ORF240 was expressed at higher levels, had slightly stronger tRNA binding signals than Trm140, and also lacked the unnecessary N-terminal domain of Trm140, we continued binding experiments with ORF240. To quantify the specificity of ORF240 tRNA binding, we analyzed biological triplicate samples for ORF240 binding of a number of different tRNAs, using two washes before the release of bound protein by proteolytic cleavage. Under these conditions, tRNAThr(CGU) bound very efficiently and reproducibly, whereas four nonsubstrate tRNAs did not, including tRNAPhe(GAA), tRNALys(CUU), tRNAiMet (CAU), and tRNAAla(UGC) (Fig. 3C). For these and subsequent experiments we calculated binding as the percentage of tRNA in the combined second wash and the elution step, relative to that in the crude extract, because it was apparent that tRNAThr(CGU) was prominent in the second wash and the elution step, relative to the first wash, whereas the negative controls were much reduced in these fractions. These results establish that copurification of tRNA with Trm140 or ORF240 is a highly specific assay for substrate tRNA binding.
FIGURE 3.

ORF240 binding of tRNAThr(CGU) requires G35 and U36. (A) Trm140 specifically binds tRNAThr substrates. Trm140 was purified from a WT strain expressing TRM140-PT (or a vector control) by pull-down using IgG Sepharose, followed by a wash step and by elution of bound Trm140 by 3C protease treatment. Then, copurifying RNAs were resolved by PAGE, transferred to nitrocellulose, and analyzed by hybridization with probes as indicated. V, vector control; T, Trm140-PT. (B) ORF240 binds tRNASer substrates as efficiently as Trm140. Trm140 and ORF240 were purified from WT strains expressing TRM140-PT or ORF240-PT (or a vector control) by pull-down, and copurifying tRNAs were analyzed as in A. O, ORF240-PT. (C) ORF240 binds specifically and reproducibly to tRNAThr(CGU) and not to four nonsubstrate tRNAs. ORF240-PT was expressed in WT cells in biological triplicate and purified by pull-down, two wash steps, and 3C protease elution of bound ORF240, and copurifying tRNAs were analyzed as in A. The percentage of tRNA retained in the combined second wash and the elution step, relative to that in the crude extract was calculated for each tRNA, as indicated. CE, crude extract (0.4% loaded); W1, wash 1 (10% loaded); W2, wash 2, (10% loaded); E, elution (10% loaded). (D) G35 and U36 of tRNAThr(CGU) are important for binding of tRNA to ORF240. Strains containing integrated tRNAThr(CGU)28,50 variants as indicated were grown and analyzed for ORF240 tRNA binding by the pull-down assay as described in C. (E) Schematic of pull-down results in D.
Consistent with our results from analysis of in vivo methyltransferase activity, we found that anticodon residues G35 and U36 were important for ORF240 binding as measured by this pull-down assay (Fig. 3D,E). Under these conditions, we observed reproducible copurification of wild-type tRNAThr(CGU) (23 ± 5%) and reproducible lack of copurification of tRNAPhe(GAA) (1 ± 0.3%). For the tRNAThr(CGU)28,50 variants, substitution of G35 or U36 with each of the other three nucleotides almost completely abolished the copurification of tRNA, whereas substitution of C34 with other nucleotides led to high levels of variant copurification. Based on these results, we conclude that Trm140 binds tRNAs with a G35–U36 anticodon. Since these are the same residues that are important for m3C modification, we infer that m3C modification is driven in large part by Trm140 tRNA binding. Because we found that copurification of the C34 variants was distinctly more efficient than the other three variants (35% versus 11%–14%), we infer that C34 is a modestly preferred wobble base nucleotide for ORF240 recognition. Residue 38 is likewise modestly influential for Trm140 m3C modification, since the A38U or A38C mutations reduced binding modestly, from 35% to 13%–15%, similar to the effects of C34 mutations.
To examine the sufficiency of G35–U36–t6A37 for m3C formation, we replaced the anticodon of the nonsubstrate tRNAPhe(GAA) with a CGU anticodon and a G37A mutation to allow for t6A37 modification, and assayed the variant for m3C modification and tRNA binding by ORF240. As we did for tRNAThr(CGU) variants, we altered 2 bp in the stem to allow for unique detection of the tRNAPhe(GAA) variants; in this case, we flipped the 29:41 and 50:64 pairs to make tRNAPhe(GAA)29,50 (Fig. 4A). ORF240 pull-down experiments resulted in efficient copurification (29%) of the tRNAPhe(GAA)29,50 [CGUA]34-37 variant, but only background copurification (1%) of tRNAPhe(GAA)29,50 [GAAG]34-37 (Fig. 4B,C). Consistent with the binding results, primer extension of RNA from cells containing the tRNAPhe(GAA)29,50 [CGUA]34-37 variant revealed a strong primer extension block at N33, indicative of m3C modification (Fig. 4D). These binding and primer extension results of tRNAPhe(GAA)29,50 indicate that a CGU anticodon and A37 (presumably modified to t6A) are sufficient for m3C modification of tRNAPhe(GAA) and establish that this sequence is an identity element for the modification.
FIGURE 4.
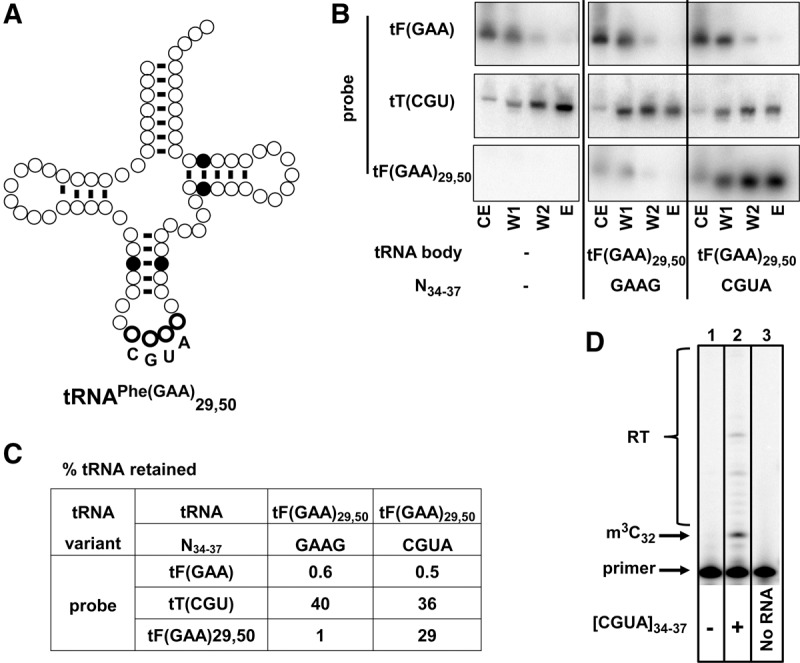
An XGU anticodon and t6A37 are sufficient for ORF240 binding and for m3C modification of tRNAPhe. (A) Schematic of tRNAPhe(GAA)29,50 variant. The secondary structure of tRNAPhe is shown, with highlighting of base pairs 29:41 and 50:64 that were flipped, and bold outline of residues N34–37 that were changed from GAAG to CGUA. (B) A CGU anticodon and t6A37 are sufficient for binding of tRNAPhe to ORF240. Strains with integrated tF(GAA)29,50 variants as indicated were analyzed by the ORF240 tRNA pull-down assay as described in Figure 3C. (C) Summary of pull-down results in B. (D) A CGU anticodon and t6A37 are sufficient for m3C modification of tRNAPhe. Bulk RNA from strains without (lane 1) or with a tRNAPhe(GAA)29,50 [CGUA]34-37 variant (lane 2) was analyzed by primer extension for m3C, using a primer complementary to residues 52–36 of the variant. Lane 3, primer alone.
t6A37 and i6A37 are important for m3C modification of tRNASer species
Unlike the three tRNAThr Trm140 substrates, which all share the anticodon residues G35 and U36, the three tRNASer substrates tRNASer(CGA), tRNASer(UGA), and tRNASer(GCU) do not share any anticodon residues with one another, and therefore the major Trm140 specificity determinant for tRNASer must be elsewhere. Furthermore, although t6A37 is important for Trm140 modification of tRNAThr, only tRNASer(GCU) has t6A, whereas tRNASer(CGA) and tRNASer(UGA) have i6A37.
Consistent with the importance of t6A for m3C modification of tRNAThr species, we found that the m3C modification level of tRNASer(GCU) was substantially lower in the sua5Δ strain compared to the WT strain (Fig. 5A). We also found that m3C was substantially reduced in tRNASer(CGA)/(UGA) in a mod5Δ strain, which lacks the i6A37 modification (Fig. 5B; Dihanich et al. 1987), as also found for the m3C modification of tRNASer substrates in S. pombe (Arimbasseri et al. 2016). Thus, it appears that both t6A and i6A contribute to efficient m3C modification.
FIGURE 5.

t6A37 and i6A37 are important for m3C modification of tRNASer species. (A) t6A37 is important for m3C modification of tRNASer(GCU). Bulk RNA from SUA5+ and sua5Δ cells was analyzed by primer extension with a primer annealing to residue e7–35 of tRNASer(GCU). (B) i6A37 is important for m3C modification of tRNASer(UGA) and tRNASer(CGA). Bulk RNA from MOD5+ and mod5Δ cells was analyzed by primer extension with a primer annealing to residue e8–36 of tRNASer(CGA). (C) i6A37 is important for tRNASer(CGA) binding to ORF240. ORF240-PT was expressed in MOD5+ and mod5Δ strains, and ORF240 tRNA binding was analyzed by the IgG Sepharose pull-down assay as described in Figure 3C. (D) Schematic of tRNASer with highlighting of the V-loop containing residues Ne1–e9 (black) and five other nucleotides (gray).
We further showed that i6A37 was important for tRNASer(CGA) binding to ORF240 (Fig. 5C). While ORF240 bound 50% of the tRNASer(CGA) in a MOD5+ strain, it only bound 1% of this tRNA in a mod5Δ strain. Similarly, although tRNASer(UGA) bound more weakly to ORF240 in a MOD5+ strain (6%), this binding was undetectable in a mod5Δ strain. As expected, binding of tRNASer(GCU) was not affected in the mod5Δ strain, because this tRNA is not a Mod5 substrate. Because i6A37 is crucial for ORF240 binding and for m3C modification of tRNASer(CGA) and tRNASer(UGA), we infer that ORF240 binding is important for m3C formation.
Although i6A and t6A are important for recognition of tRNASer substrates by Trm140, they could not be the sole determinants for m3C modification, since there are nine other S. cerevisiae tRNAs with both C32 and t6A37 that lack the m3C modification, and one other tRNA with C32 and i6A that lacks m3C.
Ses1 copurifies with Trm140
One unique feature of tRNASer species that could in principle be important for m3C modification is the distinctive long V-loop. In yeast, the tRNASer V-loops are all 14 nt long (Fig. 5D), whereas the only other tRNAs with a long V-loop are members of the tRNALeu family, with V-loops of 13 or 15 nt. Trm140 could recognize the distinctive tRNASer V-loop, or Trm140 might cooperate with other proteins such as seryl-tRNA synthetase, Ses1, to recognize the V-loop. Ses1 is known to recognize a V-loop of the appropriate length for its charging activity (Himeno et al. 1997).
To define proteins that interact with Trm140, we purified ORF240 under identical conditions to those we used for copurification of tRNAs, and compared the copurifying polypeptides with those of the negative control glutaminyl-tRNA synthetase, encoded by GLN4. Coomassie staining revealed three polypeptide bands that copurified with ORF240, one of which, labeled A, appeared to be shared by Gln4 (Fig. 6A). Mass spectrometry analysis showed that the major proteins in band A from Ses1 and Gln4 were the same (heat shock protein Ssa1, Ssa2, and Ssc1), and that band C corresponded to ORF240 breakdown polypeptides. However, the major polypeptides in band B were Ses1 and YNL040W, with convincing Mascot scores of 1737 and 660, respectively, and these proteins were not found at all in the corresponding Gln4 purification. Examination of a ynl040wΔ strain showed no alteration of m3C levels of tRNAThr(IGU) or tRNASer(UGA)/(CGA). We therefore focused on Ses1.
FIGURE 6.
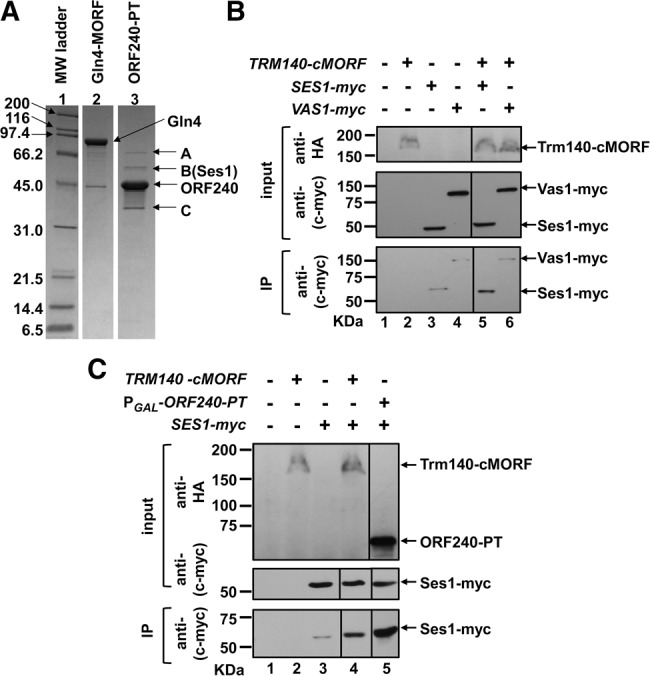
Trm140 interacts with Ses1. (A) SDS-PAGE analysis reveals three polypeptide bands that copurify with Trm140. Strains containing either PGAL-ORF240-PT or PGAL-GLN4-MORF were grown as described in Materials and Methods, ORF240 and Gln4 were affinity purified under conditions identical to those used for pull-down assays, and proteins were analyzed by SDS-PAGE and Coomassie staining. Three distinct copurifying bands that were found in the ORF240 preparation (lane 3) are labeled A–C. (B) Chromosomally expressed Ses1-Myc and Trm140-cMORF interact. Trm140 was affinity purified from crude extracts of the indicated strains, using IgG Sepharose and 3C protease cleavage. Crude extract (input) was subjected to immunoblot analysis with anti-HA and anti-(c-myc) antibody, and IgG-purified protein (immunoprecipitate, IP) was analyzed by immunoblot with anti-(c-myc) antibody. (C) Overproduction of ORF240-PT results in increased copurification of Ses1. Trm140 was purified from a chromosomally tagged Trm140-cMORF strain, ORF240 was purified from a strain overexpressing ORF240-PT, and the amount of copurifying Ses1 was compared by immunoblot, as described in B.
We found that Trm140 interacted with Ses1 in wild-type cells, because affinity purification of Trm140 with a chromosomal C-terminal MORF tag (Trm140-cMORF) resulted in copurification of chromosomally tagged Ses1-myc (Fig. 6B, cf. lanes 3 and 5), but no obvious copurification of the control Vas1-Myc (valyl-tRNA synthetase) (Fig. 6B, lanes 4,6). Moreover, overproduction of ORF240-PT under PGAL control resulted in copurification of substantially increased amounts of Ses1-myc (Fig. 6C, lanes 4,5). This interaction of Trm140 with Ses1 suggested the possibility that Ses1 might have a role in the m3C modification reaction.
Ses1 stimulates m3C formation of tRNASer species
To determine if Ses1 had a role in m3C modification, we examined the methyltransferase activity of Trm140 in vitro. We expressed and purified His6-ORF240 from Escherichia coli and assayed its activity with tRNA purified from a trm140Δ strain. After incubation of ORF240 with S-adenosylmethionine (SAM) and tRNA, we analyzed m3C modification by primer extension in the presence of ddTTP, and calculated the efficiency of m3C modification based on the intensities of the primer extension blocks at U33, due to m3C32, and at A31, due to read-through and termination by ddTTP incorporation. Using this assay, we found that the three tRNAThr species were equally efficiently modified by ORF240 (Table 1; Fig. 7A,B), in each case requiring ∼0.02 µM ORF240 for one-half-maximal modification (ORF2401/2). In contrast, ORF240 was very inefficient at m3C modification of tRNASer(UGA) and tRNASer(CGA), with ORF2401/2 values of ∼15 µM and ∼0.3 µM, respectively, while the ORF2401/2 value was ∼0.05 µM for tRNASer(GCU). As anticipated, no m3C was detected upon ORF240 assay of tRNATyr(GUA), which normally bears an unmodified C32 (Table 1).
TABLE 1.
Summary of the efficiency of ORF240 m3C methyltransferase activity on tRNAThr and tRNASer substrates
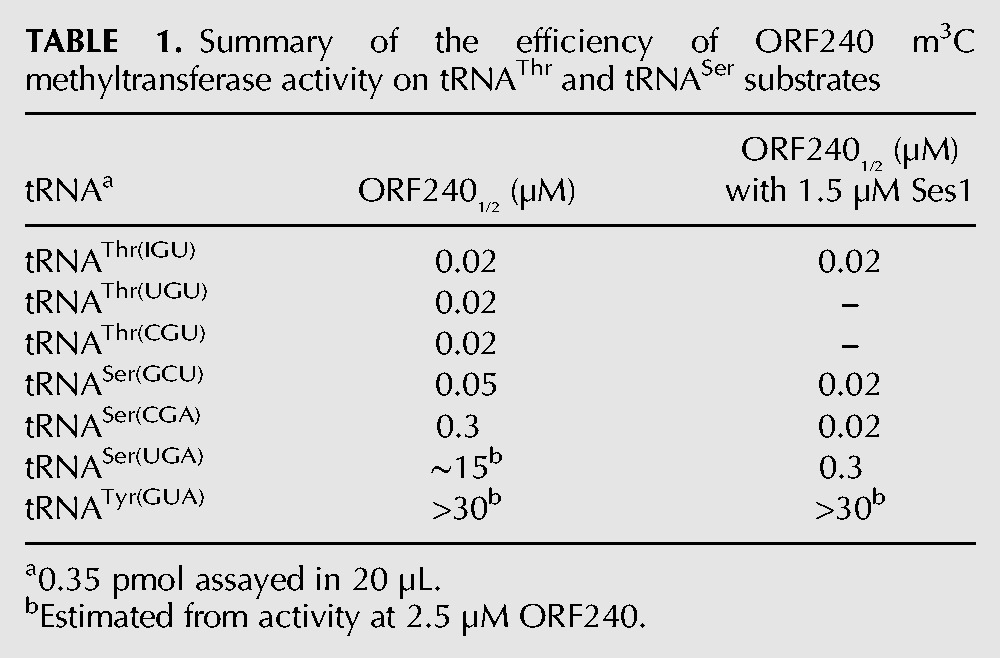
FIGURE 7.

ORF240 catalyzes efficient m3C modification of tRNAThr(IGU) but inefficient modification of tRNASer(CGA). (A) Titration of ORF240 m3C methyltransferase activity on tRNAThr(IGU) and tRNASer(CGA). Of note, 0.35 pmol of tRNAThr(IGU) or tRNASer(CGA) purified from a trm140Δ strain was incubated with serial fivefold dilutions of ORF240 (2.5, 0.5, 0.1, 0.02, 0.004 µM) purified from E. coli or with buffer (−) in the presence of SAM at 30°C for 1 h, and tRNA was analyzed for m3C by primer extension in the presence of ddTTP, as described in Materials and Methods. (B) Plot of m3C modification as a function of the concentration of ORF240 in A.
Consistent with an important biological role for the interaction of Trm140 with Ses1, we found that purified Ses1 stimulated the Trm140 m3C modification activity on tRNASer species in vitro. Using Ses1 purified from a trm140Δ strain, to avoid Trm140 contamination, we found that 1.5 µM Ses1 reduced the ORF2401/2 of tRNASer(CGA) from ∼0.3 µM to ∼0.02 µM, of tRNASer(UGA) from ∼15 µM to ∼0.3 µM, and of tRNASer(GCU) from 0.05 µM to 0.02 µM, but had little or no effect on tRNAThr(IGU) or tRNATyr(GUA) (Table 1; Fig. 8A,B).
FIGURE 8.
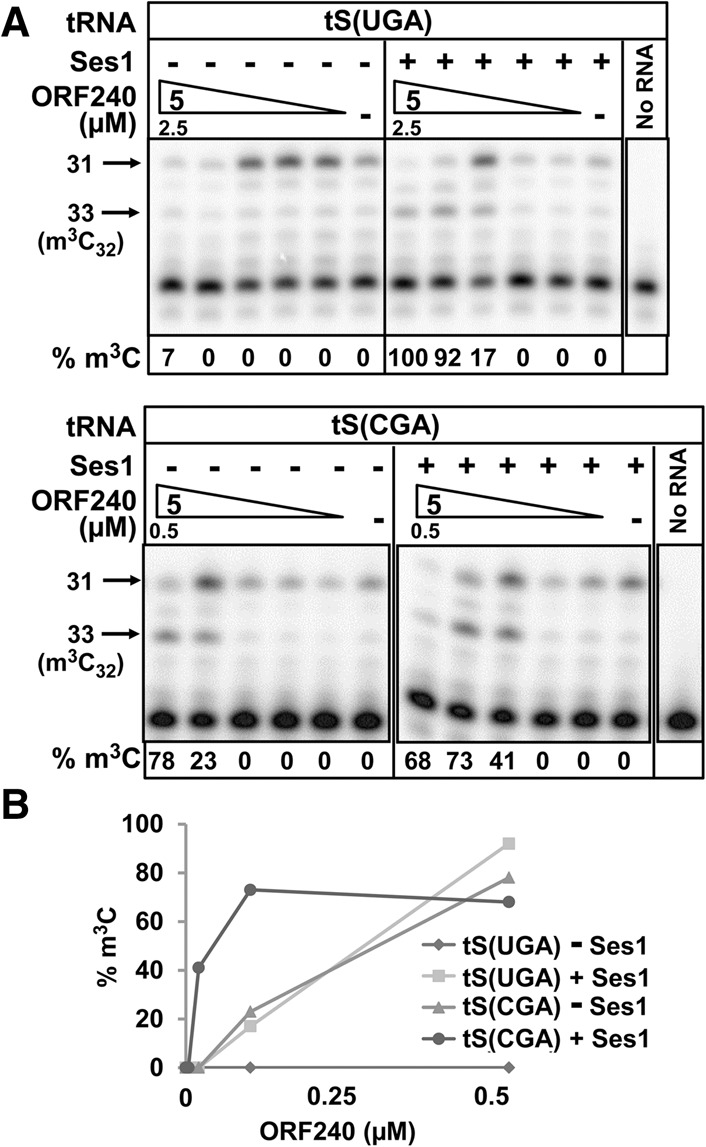
Ses1 stimulates ORF240 m3C modification of tRNASer(UGA) and tRNASer(CGA). (A) Effect of Ses1 on the titration of ORF240 m3C methyltransferase activity on tRNASer(CGA) and tRNASer(UGA). Of note, 0.35 pmol of tRNASer(UGA) and tRNASer(CGA) purified from a trm140Δ strain was assayed for m3C formation by ORF240 in the presence or absence of 1.5 µM Ses1 purified from a trm140Δ strain, as described in Figure 7A. (B) Plot of m3C modification as a function of the concentration of ORF240 in A.
Consistent with these in vitro results, we observed Ses1 stimulation of m3C modification in vivo. To address this question, we assayed m3C modification after overproduction of Ses1-MORF in a mod5Δ strain, in which m3C32 modification of tRNASer(CGA)/(UGA) was reduced due to lack of i6A (Fig. 5B). We found that overproduction of Ses1 resulted in a substantial increase in m3C levels of tRNASer(CGA)/(UGA) relative to that in the vector control, from 2% to 24% (Fig. 9A). Overproduction of ORF240 also improved the m3C modification, consistent with the fact that i6A was important for ORF240 binding. As expected, the m3C levels of tRNAThr(IGU), which has t6A and not i6A, were not affected in the mod5Δ strain or by overproduction of either Ses1 or ORF240.
FIGURE 9.
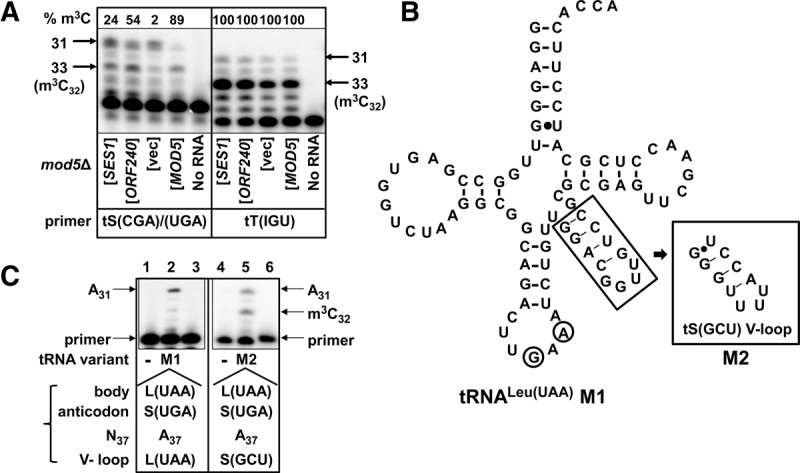
Ses1 stimulates m3C modification of tRNASer(CGA) and tRNASer(UGA) in vivo and the unique tRNASer V-loop that Ses1 recognizes is important for m3C modification. (A) Ses1 stimulates m3C formation in vivo. Bulk RNA from mod5Δ cells expressing SES1, ORF240, vector, or MOD5 as indicated was analyzed for m3C of tRNASer(CGA)/(UGA) or tRNAThr(IGU) by primer extension in the presence of ddTTP, as described in Materials and Methods. (B) Schematic of tRNALeu(UAA) M1 and M2 variants. The secondary structure of tRNALeu(UAA) M1 is shown, with anticodon loop mutations as indicated (circled). Nucleotides in the V-loop that were changed to the V-loop of tRNASer(GCU) to make the tRNALeu(UAA) M2 variant are boxed. (C) The variable loop of tRNASer(GCU) is sufficient to confer m3C modification in vivo of tRNALeu(UAA) with G35 and A37 mutations. Variants described in B were integrated into a trm732Δ strain, to eliminate the possibility of 2′-O-methylation at C32 (Guy et al. 2012), and bulk RNA from strains containing either variants M1 or M2 was analyzed for m3C by primer extension, using ddTTP and primers extending from residue e8 to 36 of M1 and from residue 47 to 36 of M2.
To determine whether the important region for m3C modification of tRNASer species was the V-loop, we tested the modification of a chimeric tRNALeu species with a V-loop based on tRNASer. We constructed a variant with the body sequence of tRNALeu(UAA), the anticodon UGA, and A37, called variant M1 (Fig. 9B). This M1 variant was not a substrate for m3C modification (Fig. 9C, lane 2). However, the variant M2, which differs from M1 due to replacement of the V-loop with that of tRNASer(GCU), was significantly modified to m3C32 (Fig. 9C, lane 5). This result shows that the tRNASer V-loop is sufficient to confer m3C32 modification of the M1 variant in the context of this tRNA species.
DISCUSSION
We have provided evidence here that S. cerevisiae Trm140 has two strikingly different recognition modes for tRNA m3C modification. Trm140 explicitly recognizes the XGU anticodon and t6A37 of tRNAThr species as an identity element for m3C modification in vivo. Thus, substitution of either G35 or U36 of tRNAThr(CGU) with any other residue resulted in the complete absence of m3C modification, and removal of t6A substantially reduced m3C modification, whereas introduction of a CGU anticodon and A37 to tRNAPhe resulted in efficient m3C modification in vivo. Consistent with these results, ORF240 expressed and purified from E. coli efficiently catalyzed modification of all three tRNAThr species, but not tRNATyr.
In contrast, the CGA, UGA, and GCU anticodons of the three tRNASer species that are modified with m3C32 do not have a common feature, and Trm140 recognition of these species is instead propelled by interaction with Ses1 and the tRNASer V-loop, as well as by i6A37 of tRNASer(CGA) and tRNASer(UGA), and t6A37 of tRNASer(GCU). Thus, purified Ses1 stimulated the efficiency of ORF240 m3C modification of tRNASer(CGA), decreasing ORF2401/2 by 20-fold, to a value comparable to that of tRNAThr substrates. Similarly, Ses1 stimulated the efficiency of ORF240 m3C modification of tRNASer(UGA) by ∼50-fold. Furthermore, this Ses1 stimulation of m3C modification was also observed in vivo, since Ses1 overproduction significantly increased m3C modification of tRNASer(CGA)/(UGA) in a strain lacking i6A.
Our results suggest that m3C modification activity is driven in part by binding. We showed that all six known Trm140 substrate tRNAs copurified with Trm140 or ORF240, whereas each of six tested nonsubstrate tRNAs did not copurify. We also showed that ORF240 binding of tRNAThr anticodon loop variants tracked perfectly with m3C modification, since all six possible variants with mutations in G35 or U36 lacked modification and also did not bind, whereas each of the three variants with mutations in C34 were modified and bound efficiently, although not as efficiently as with C34. Furthermore, ORF240 bound tRNAPhe efficiently only when its anticodon was altered to CGU-A37, correlated with m3C modification, and i6A37 was important for binding of tRNASer(CGA) to ORF240 as well as for m3C modification. These results suggest a modular binding mechanism to explain substrate specificity, in which contributions are made by G35, U36, i6A37, or t6A37, and the V-loop, as well as C34. C34 is favored over other N34 residues based on preferential binding of tRNAThr C34 variants and preferential binding of tRNASer(CGA) compared to tRNASer(UGA) since these two tRNAs are identical other than U34 and the N28–N42 pair. This preferential binding of tRNASer(CGA) relative to that of tRNASer(UGA) is also consistent with the preferential ORF240 modification activity of tRNASer(CGA) compared to tRNASer(UGA).
Proteins like Trm140 that bind nucleic acids by two very different modes are rare. Notable examples include TFIIIA, which recognizes 5S promoter DNA and 5S RNA using distinctive zinc fingers (Nolte et al. 1998; Lu et al. 2003; Hall 2005), and phage λ integrase protein, which recognizes two different DNA sequences with different domains (Moitoso de Vargas et al. 1988). It is intriguing that bovine mitochondrial Ses1 recognizes tRNASer substrates with and without a D-stem–loop by different mechanisms, but involving the same region of the T-loop (Shimada et al. 2001); the different modes of Trm140 recognition of tRNAThr compared to tRNASer(CGA) and tRNASer(UGA) also appear to involve common elements including C34 and i6A37 or t6A37, as well as the specific involvement of Ses1 for tRNASer and G35–U36 for tRNAThr.
The observation that i6A and t6A are important for m3C modification is a clear demonstration of ordered modification circuitry in the anticodon loops of tRNAThr and tRNASer, in which either A37 modification drives m3C formation. Maraia and coworkers recently observed that i6A modification was important for m3C32 modification in tRNASer species of S. pombe and speculated that t6A modification would be important for m3C32 modification of tRNAThr and tRNASer substrates with U36 (Arimbasseri et al. 2016). Because we observed that i6A is important for both tRNA binding and m3C modification, this suggests that tRNA binding efficiency is at least partially responsible for the observed ordering of these two modifications. It remains to be determined how both t6A and i6A drive the same m3C modification reaction, and in particular if t6A37 stimulates m3C modification due to increased Trm140 tRNA binding. Our preliminary attempts to express ORF240 in an sua5 mutant strain have been unsuccessful.
The ordered modification circuitry described above for m3C modification of tRNASer and tRNAThr is reminiscent of the conserved ordered modification circuitry found in the anticodon loop of tRNAPhe. We previously showed that complete modification of the anticodon loop of S. cerevisiae tRNAPhe required 2′-O-methylation of C32 and G34 to drive efficient formation of wybutosine at m1G37 (Guy et al. 2012), and that this circuitry for tRNAPhe anticodon loop modification was conserved in S. pombe and human lymphoblastoid cell lines (Guy and Phizicky 2015; Guy et al. 2015). The similar conservation of modification order for i6A/t6A and m3C in the anticodon loops of tRNAThr and tRNASer species in S. cerevisiae and S. pombe suggests that this ordered modification circuitry will be preserved in other eukaryotes, perhaps by conserved mechanisms, and suggests the existence of other circuits for anticodon loop modifications.
The stimulation by Ses1 of Trm140 m3C modification activity that we observe in vitro and in vivo could occur in two ways. One possibility is that Trm140, Ses1, and tRNA form three binary interactions, which might stabilize weak enzyme–substrate binding. We have shown that Ses1-myc copurified with Trm140-cMORF, although it remains to be determined whether the interaction is tRNA mediated. Alternatively, it is possible that binding of tRNASer to Ses1 rearranges the local conformation of the anticodon loop to facilitate Trm140 recognition of C32, although the cocrystal structures of both Thermus thermophilus SerRS-tRNASer and human SerRS-tRNASec lack any direct interaction between the synthetases and the anticodon loop (Biou et al. 1994; Wang et al. 2015). It is known that S. cerevisiae Ses1 recognizes the V-loop of tRNASer for its synthetase activity (Himeno et al. 1997), providing a probable explanation for the V-loop dependence of m3C modification, but not casting light on its mechanism of m3C stimulation.
The two modes of tRNA substrate recognition for S. cerevisiae Trm140 seems likely to be found widely in the Saccharomycotina and Pezizomycotina subdivisions of the phylum Ascomycota, and to a more limited extent in Basidiomycota, based on the occurrence of a single highly similar TRM140 homolog in a large fraction of these organisms that we examined (Candida albicans, Candida tropicalis, Yarrowia lipolytica, Saccharomyces castellii, Debaryomyces hansenii, Candida glabrata, Saccharomyces mikatae, Saccharomyces paradoxus, Saccharomyces bayanus, Kluyveromyces lactis, Ashbya gossypii, Saccharomyces kluyveri, Kluyveromyces waltii, Aspergillus nidulans, Aspergillus oryzae, Neurospora crassa, Coccidioides posadasii, Coccidioides immitis, Coprinopsis cinerea, Phanerochaete chrysosporium, Uncinocarpus reesii. Chaetomium globosum, Fusarium verticillioides, Botrytis cinerea). However, split substrate recognition by Trm140 and related proteins is also likely widely found since S. pombe was recently shown to have separate trm140+ and trm141+ (METTL6) homologs dedicated to tRNAThr and to tRNASer substrates, respectively, and multiple homologs appear to be the rule rather than the exception in metazoans, plants, and other groups of fungi (Arimbasseri et al. 2016). Thus, a large number of eukaryotes, including vertebrates, have two or three TRM140 family members, drawn from the phylogenetic clades of TRM140, METTL6, METTL2, and METTL8 homologs, which might be used for modification of specific tRNA species with m3C32 or with m3C in the V-loop (Arimbasseri et al. 2016). In view of our findings, we speculate that tRNASer m3C modification activity in other organisms might also be stimulated by SerRS. In support of this, we note that the human SerRS has the same V-loop recognition element as yeast Ses1 (Achsel and Gross 1993). Alternatively, it is possible that the different homologs have evolved separate tRNA recognition elements in S. pombe and other organisms. The recent finding that the METTL6 homolog TbMTase37 of T. brucei is important for ribosome stability and cytokinesis emphasizes the importance of this family of proteins, although the proximate cause is not yet known (Fleming et al. 2016).
The Ses1 requirement for efficient Trm140 m3C modification of tRNASer species adds to a small subset of the two-subunit modification enzymes (Guy and Phizicky 2014) in which one interacting partner seemingly directs the enzyme to different residues or substrates, including Trm7/Trm732 for Nm32 formation, Trm7/Trm734 for Nm34 formation (Guy et al. 2012; Guy and Phizicky 2015), and Kre33/Tan1 for ac4C12 formation (Johansson and Bystrom 2004; Sharma et al. 2015). The finding of Ses1 as an interacting partner for Trm140 m3C modification is also another example of the remarkable range of different noncanonical functions of tRNA synthetases (Wakasugi and Schimmel 1999; Guo et al. 2010; Smirnova et al. 2012; Yao and Fox 2013), at least some of which exert these roles through RNA interactions (Herbert et al. 1988; Sampath et al. 2004; Sarkar et al. 2012). It remains to be determined precisely how Ses1 recognizes Trm140 to stimulate m3C modification activity of tRNASer, and the connection between tRNASer charging and modification.
MATERIALS AND METHODS
Yeast strains
Strains (listed in Table 2) used for genetic tests and/or analysis of tRNA were derivatives of strain BY4741 or BY4742, strains used for immunoblotting analysis were derivatives of BCY123, and strains used for pull-down experiments and protein purification were derivatives of YLH126.
TABLE 2.
Strains used in this study

TRM140 was deleted by PCR amplification of the trm140Δ::bleR cassette from ySD179 using primers containing sequences 5′ and 3′ of TRM140 (TRM140 − 409 and TRM140 + 307), followed by transformation, selection on YPD media containing 8 mg/L Bleocin and verification by PCR using appropriate primers. MOD5 was deleted in a similar fashion.
The tT(CGU)Δ[CEN URA3 tT(CGU)] strain was constructed by transformation of BY4741 with the [CEN URA3 tT(CGU)] plasmid containing the tT(CGU) gene with its own flanking sequence, followed by PCR amplification of the bleR marker and linear transformation to delete the tT(CGU) gene.
Strains with the chromosomal cMORF tag (His6-HA-3C site-ZZ domain of protein A) were generated by PCR amplification of a gene-specific product from a cMORF∷URA3 cassette of pAVA0258, followed by linear transformation and selection (Gelperin et al. 2005; Guy et al. 2012). C-terminal myc-tagged strains were generated in a similar fashion from the pYM46 1myc-7His∷KanR cassette (Janke et al. 2004).
Plasmids
Plasmids used in this study are listed in Table 3. Plasmids expressing tRNAs were constructed by ligation-independent cloning (LIC) of a tRNA with its own flanking sequence into the [2μ LEU2] LIC vector pAVA577. LIC was also used to build the [CEN URA3 MOD5] plasmid. Integration vectors for tRNA variants were made by insertion of a BglII, XhoI fragment encoding the tRNA variant into plasmid pAB230-1, as previously described (Guy et al. 2014). ORF240 was cloned by LIC into a [2µ URA3 PGAL1,10] expression vector, in which ORF240 is expressed under PGAL1 control with a C-terminal PT tag (as ORF240-3C site-HA epitope-His6-ZZ domain of protein A) essentially as previously described (Quartley et al. 2009).
TABLE 3.
Plasmids used in this study
Expression and affinity purification of Ses1-MORF from yeast
To purify Ses1 from yeast without interacting Trm140, strain YLH974-1 (trm140Δ) was transformed with a [2µ URA3 PGAL-SES1-MORF] plasmid encoding a SES1-MORF fusion protein (Ses1-His6-HA-3C site-ZZ domain of protein A), and the resulting strain was grown in selective media containing raffinose and induced for 6 h by addition of one-half volume of 3× YP media containing 6% galactose and 6% raffinose. Then Ses1-MORF was affinity purified using IgG Sepharose chromatography, followed by Ses1 elution with GST-3C protease, removal of the protease with glutathione Sepharose resin, and dialysis into buffer containing 50% glycerol, 20 mM Tris–Cl pH 7.5, 150 mM NaCl, 1 mM MgCl2, and 1 mM DTT, as previously described (Quartley et al. 2009).
Growth and affinity purification of His6-ORF240 from E. coli
The E. coli expression plasmid pSD248, which expresses the entire C-terminal domain of TRM140 (residues 277–628, called ORF240) as a His6-3C-ORF240 fusion, was transformed into pLys(S)BL21(DE3) cells. Transformants were grown, induced, and harvested, and then tagged protein was purified using immobilized metal ion affinity chromatography (IMAC), followed by imidazole elution, cleavage of the affinity tag with protease 3C, removal of contaminants by passage through the same IMAC resin, and dialysis into buffer containing 50% glycerol, 20 mM Tris–Cl, pH 7.5, 200 mM NaCl, and 1 mM DTT, essentially as previously described (D'Silva et al. 2011).
Extraction of bulk RNA from yeast and purification of tRNA
Strains were grown to an OD600 1–2, and bulk RNA was extracted from ∼300 OD pellets (for tRNA purification) or from ∼3 OD pellets (for primer extension analysis) using hot phenol. tRNA was purified from ∼1.25 mg bulk RNA using 5′-biotinylated oligonucleotides (Integrated DNA Technologies), as previously described (Jackman et al. 2003).
Primer extension assays
Primers were 5′ end labeled and purified as previously described (D'Silva et al. 2011). In a 5 μL annealing reaction, 0.25–1 pmol of labeled primers were annealed to 0.4–3 μg of bulk RNA or ∼3 ng of purified tRNA by incubation for 3 min at 95°C followed by slow cooling and incubation for 30 min at 50°C–55°C. The annealing product was then extended using 64 U Superscript III (Invitrogen) in a 10 μL reaction containing 1× First Strand buffer, 1 mM of each dNTP, and 10 mM MgCl2 at 50°C–55°C for 1 h. For reactions containing ddTTP, dTTP was replaced by 2 mM ddTTP, and other dNTPs were reduced to 0.5 mM. Reactions were stopped by addition of 2× RNA loading dye containing 98% formamide, 10 mM EDTA, 1 mg/mL bromophenol blue, and 1 mg/mL xylene cyanol, resolved on a 7M urea–15% polyacrylamide gel, and the dried gel was imaged on a Typhoon phosphorimager and quantified as previously described (Jackman et al. 2003).
ORF240 and Trm140 pull-down assays of tRNA binding
Strains expressing PGAL-ORF240-PT or PGAL-TRM140-PT plasmid were grown in selective media containing raffinose to OD600 ∼0.75 and induced for 6 h with one-half volume 3× YP media containing 6% galactose and 6% raffinose. Then tagged proteins were affinity purified from 320–360 OD pellets using IgG Sepharose, followed by one or two washes with 1 mL buffer and 3 min of mixing, and then overnight incubation with GST-3C protease to release bound protein and copurifying tRNAs. Then RNA was purified from each fraction, resolved by PAGE, and tRNAs were analyzed by hybridization, as previously described (Alexandrov et al. 2006).
Mass spectrometry
For mass spectrometry analysis of ORF240 binding proteins, ORF240-PT purification was done as for a pull-down assay, proteins were analyzed by SDS-PAGE, and copurifying polypeptides were analyzed by the Mass Spectrometry Resource Center of the University of Rochester Medical Center.
Immunoblotting analysis
Yeast strains with C-terminal chromosomal tags (cMORF or myc) were grown in YPD to OD600 1–2.5 and crude extracts were made from 500–600 OD-mL pellets, followed by IgG Sepharose affinity purification and elution of bound protein with GST-3C protease. Then samples were subject to SDS-PAGE, transferred to nitrocellulose membrane (Bio-Rad), and probed with appropriate antibodies: mouse monoclonal anti-[c-myc] (Roche), followed by goat anti-mouse IgG-HRP (Bio-Rad); or rat anti-HA (Roche), followed by goat anti-rat IgG-HRP (Bio-Rad). Strains containing a PGAL-ORF240-PT plasmid were grown as described in pull-down experiment.
Assay of m3C methyltransferase activity in vitro
Reaction mixtures (20 μL) contained 60–67.5 mM NaCl, 50 mM Hepes, pH 7.5, 3 mM MgCl2, 55–60 µg/mL BSA, 50 µM EDTA, 2.5–3.5 mM Tris–Cl pH 7.5, 2.5–5% glycerol, 0.5 mM SAM, 50 μg/mL Poly(A), 10 ng of purified and refolded tRNA from a trm140Δ strain, ORF240 purified from E. coli, and where indicated, Ses1 purified from a trm140Δ strain or compensating buffer. Reactions were incubated at 30°C for 1 h, stopped by phenol extraction, and then tRNA was precipitated with ethanol, resuspended in water, and subjected to primer extension analysis to analyze m3C32.
ACKNOWLEDGMENTS
We thank E. O'Shea and M. Hampsey for strains, S. Ghaemmaghami and K. Welle of the Mass Spectrometry Resource Center of the University of Rochester Medical Center, and E. Grayhack for valuable discussions and comments during the course of this work. This research was supported by National Institutes of Health grant GM052347 to E.M.P.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.059667.116.
REFERENCES
- Achsel T, Gross HJ. 1993. Identity determinants of human tRNASer: sequence elements necessary for serylation and maturation of a tRNA with a long extra arm. EMBO J 12: 3333–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agris PF, Vendeix FA, Graham WD. 2007. tRNA's wobble decoding of the genome: 40 years of modification. J Mol Biol 366: 1–13. [DOI] [PubMed] [Google Scholar]
- Alexandrov A, Chernyakov I, Gu W, Hiley SL, Hughes TR, Grayhack EJ, Phizicky EM. 2006. Rapid tRNA decay can result from lack of nonessential modifications. Mol Cell 21: 87–96. [DOI] [PubMed] [Google Scholar]
- Arimbasseri AG, Iben J, Wei FY, Rijal K, Tomizawa K, Hafner M, Maraia RJ. 2016. Evolving specificity of tRNA 3-methyl-cytidine-32 (m3C32) modification: a subset of tRNAsSer requires N6-isopentenylation of A37. RNA 22: 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffinger P, Westhof E. 1999. Singly and bifurcated hydrogen-bonded base-pairs in tRNA anticodon hairpins and ribozymes. J Mol Biol 292: 467–483. [DOI] [PubMed] [Google Scholar]
- Biou V, Yaremchuk A, Tukalo M, Cusack S. 1994. The 2.9 A crystal structure of T. thermophilus seryl-tRNA synthetase complexed with tRNASer. Science 263: 1404–1410. [DOI] [PubMed] [Google Scholar]
- Bjork GR, Jacobsson K, Nilsson K, Johansson MJ, Bystrom AS, Persson OP. 2001. A primordial tRNA modification required for the evolution of life? EMBO J 20: 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork GR, Huang B, Persson OP, Bystrom AS. 2007. A conserved modified wobble nucleoside (mcm5s2U) in lysyl-tRNA is required for viability in yeast. RNA 13: 1245–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Huang B, Eliasson M, Ryden P, Bystrom AS. 2011. Elongator complex influences telomeric gene silencing and DNA damage response by its role in wobble uridine tRNA modification. PLoS Genet 7: e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dihanich ME, Najarian D, Clark R, Gillman EC, Martin NC, Hopper AK. 1987. Isolation and characterization of MOD5, a gene required for isopentenylation of cytoplasmic and mitochondrial tRNAs of Saccharomyces cerevisiae. Mol Cell Biol 7: 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Silva S, Haider SJ, Phizicky EM. 2011. A domain of the actin binding protein Abp140 is the yeast methyltransferase responsible for 3-methylcytidine modification in the tRNA anti-codon loop. RNA 17: 1100–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Yacoubi B, Lyons B, Cruz Y, Reddy R, Nordin B, Agnelli F, Williamson JR, Schimmel P, Swairjo MA, de Crecy-Lagard V. 2009. The universal YrdC/Sua5 family is required for the formation of threonylcarbamoyladenosine in tRNA. Nucleic Acids Res 37: 2894–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Yacoubi B, Hatin I, Deutsch C, Kahveci T, Rousset JP, Iwata-Reuyl D, Murzin AG, de Crécy-Lagard V. 2011. A role for the universal Kae1/Qri7/YgjD (COG0533) family in tRNA modification. EMBO J 30: 882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esberg A, Huang B, Johansson MJ, Bystrom AS. 2006. Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol Cell 24: 139–148. [DOI] [PubMed] [Google Scholar]
- Fleming IM, Paris Z, Gaston KW, Balakrishnan R, Fredrick K, Rubio MA, Alfonzo JD. 2016. A tRNA methyltransferase paralog is important for ribosome stability and cell division in Trypanosoma brucei. Sci Rep 6: 21438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelperin DM, White MA, Wilkinson ML, Kon Y, Kung LA, Wise KJ, Lopez-Hoyo N, Jiang L, Piccirillo S, Yu H, et al. 2005. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev 19: 2816–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AP, Keller W. 1999. An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science 286: 1146–1149. [DOI] [PubMed] [Google Scholar]
- Grant TD, Snell EH, Luft JR, Quartley E, Corretore S, Wolfley JR, Snell ME, Hadd A, Perona JJ, Phizicky EM, et al. 2012. Structural conservation of an ancient tRNA sensor in eukaryotic glutaminyl-tRNA synthetase. Nucleic Acids Res 40: 3723–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Yang XL, Schimmel P. 2010. New functions of aminoacyl-tRNA synthetases beyond translation. Nat Rev Mol Cell Biol 11: 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy MP, Phizicky EM. 2014. Two-subunit enzymes involved in eukaryotic post-transcriptional tRNA modification. RNA Biol 11: 1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy MP, Phizicky EM. 2015. Conservation of an intricate circuit for crucial modifications of the tRNAPhe anticodon loop in eukaryotes. RNA 21: 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy MP, Podyma BM, Preston MA, Shaheen HH, Krivos KL, Limbach PA, Hopper AK, Phizicky EM. 2012. Yeast Trm7 interacts with distinct proteins for critical modifications of the tRNAPhe anticodon loop. RNA 18: 1921–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy MP, Young DL, Payea MJ, Zhang X, Kon Y, Dean KM, Grayhack EJ, Mathews DH, Fields S, Phizicky EM. 2014. Identification of the determinants of tRNA function and susceptibility to rapid tRNA decay by high-throughput in vivo analysis. Genes Dev 28: 1721–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy MP, Shaw M, Weiner CL, Hobson L, Stark Z, Rose K, Kalscheuer VM, Gecz J, Phizicky EM. 2015. Defects in tRNA anticodon loop 2′-O-methylation are implicated in nonsyndromic X-linked intellectual disability due to mutations in FTSJ1. Hum Mutat 36: 1176–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TM. 2005. Multiple modes of RNA recognition by zinc finger proteins. Curr Opin Struct Biol 15: 367–373. [DOI] [PubMed] [Google Scholar]
- Han L, Kon Y, Phizicky EM. 2015. Functional importance of Ψ38 and Ψ39 in distinct tRNAs, amplified for tRNAGln(UUG) by unexpected temperature sensitivity of the s2U modification in yeast. RNA 21: 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M, Giege R, Florentz C. 1999. A Watson-Crick base-pair-disrupting methyl group (m1A9) is sufficient for cloverleaf folding of human mitochondrial tRNALys. Biochemistry 38: 13338–13346. [DOI] [PubMed] [Google Scholar]
- Herbert CJ, Labouesse M, Dujardin G, Slonimski PP. 1988. The NAM2 proteins from S. cerevisiae and S. douglasii are mitochondrial leucyl-tRNA synthetases, and are involved in mRNA splicing. EMBO J 7: 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himeno H, Yoshida S, Soma A, Nishikawa K. 1997. Only one nucleotide insertion to the long variable arm confers an efficient serine acceptor activity upon Saccharomyces cerevisiae tRNALeu in vitro. J Mol Biol 268: 704–711. [DOI] [PubMed] [Google Scholar]
- Hur S, Stroud RM. 2007. How U38, 39, and 40 of many tRNAs become the targets for pseudouridylation by TruA. Mol Cell 26: 189–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman JE, Phizicky EM. 2006. tRNAHis guanylyltransferase adds G-1 to the 5′ end of tRNAHis by recognition of the anticodon, one of several features unexpectedly shared with tRNA synthetases. RNA 12: 1007–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman JE, Montange RK, Malik HS, Phizicky EM. 2003. Identification of the yeast gene encoding the tRNA m1G methyltransferase responsible for modification at position 9. RNA 9: 574–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, et al. 2004. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21: 947–962. [DOI] [PubMed] [Google Scholar]
- Johansson MJ, Bystrom AS. 2004. The Saccharomyces cerevisiae TAN1 gene is required for N4-acetylcytidine formation in tRNA. RNA 10: 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson MJ, Esberg A, Huang B, Bjork GR, Bystrom AS. 2008. Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol Cell Biol 28: 3301–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhling F, Morl M, Hartmann RK, Sprinzl M, Stadler PF, Putz J. 2009. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res 37: D159–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadaba S, Krueger A, Trice T, Krecic AM, Hinnebusch AG, Anderson J. 2004. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev 18: 1227–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux S, Olejniczak M, Uhlenbeck OC. 2009. A sequence element that tunes Escherichia coli tRNAAlaGGC to ensure accurate decoding. Nat Struct Mol Biol 16: 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Searles MA, Klug A. 2003. Crystal structure of a zinc-finger-RNA complex reveals two modes of molecular recognition. Nature 426: 96–100. [DOI] [PubMed] [Google Scholar]
- Lustig F, Boren T, Claesson C, Simonsson C, Barciszewska M, Lagerkvist U. 1993. The nucleotide in position 32 of the tRNA anticodon loop determines ability of anticodon UCC to discriminate among glycine codons. Proc Natl Acad Sci 90: 3343–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL. 2005. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science 309: 1534–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, et al. 2013. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res 41: D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi K, Kimura S, Suzuki T. 2013. A cyclic form of N6-threonylcarbamoyladenosine as a widely distributed tRNA hypermodification. Nat Chem Biol 9: 105–111. [DOI] [PubMed] [Google Scholar]
- Moitoso de Vargas L, Pargellis CA, Hasan NM, Bushman EW, Landy A. 1988. Autonomous DNA binding domains of λ integrase recognize two different sequence families. Cell 54: 923–929. [DOI] [PubMed] [Google Scholar]
- Murphy FVt, Ramakrishnan V. 2004. Structure of a purine–purine wobble base pair in the decoding center of the ribosome. Nat Struct Mol Biol 11: 1251–1252. [DOI] [PubMed] [Google Scholar]
- Na JG, Pinto I, Hampsey M. 1992. Isolation and characterization of SUA5, a novel gene required for normal growth in Saccharomyces cerevisiae. Genetics 131: 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Nemoto T, Heinemann IU, Yamashita K, Sonoda T, Komoda K, Tanaka I, Soll D, Yao M. 2013. Structural basis of reverse nucleotide polymerization. Proc Natl Acad Sci 110: 20970–20975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte RT, Conlin RM, Harrison SC, Brown RS. 1998. Differing roles for zinc fingers in DNA recognition: structure of a six-finger transcription factor IIIA complex. Proc Natl Acad Sci 95: 2938–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A, Yi S, Katoh T, Takai Y, Suzuki T, Suzuki T. 2011. Actin-binding protein ABP140 is a methyltransferase for 3-methylcytidine at position 32 of tRNAs in Saccharomyces cerevisiae. RNA 17: 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olejniczak M, Uhlenbeck OC. 2006. tRNA residues that have coevolved with their anticodon to ensure uniform and accurate codon recognition. Biochimie 88: 943–950. [DOI] [PubMed] [Google Scholar]
- Olejniczak M, Dale T, Fahlman RP, Uhlenbeck OC. 2005. Idiosyncratic tuning of tRNAs to achieve uniform ribosome binding. Nat Struct Mol Biol 12: 788–793. [DOI] [PubMed] [Google Scholar]
- Pintard L, Lecointe F, Bujnicki JM, Bonnerot C, Grosjean H, Lapeyre B. 2002. Trm7p catalyses the formation of two 2′-O-methylriboses in yeast tRNA anticodon loop. EMBO J 21: 1811–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartley E, Alexandrov A, Mikucki M, Buckner FS, Hol WG, DeTitta GT, Phizicky EM, Grayhack EJ. 2009. Heterologous expression of L. major proteins in S. cerevisiae: a test of solubility, purity, and gene recoding. J Struct Funct Genomics 10: 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath P, Mazumder B, Seshadri V, Gerber CA, Chavatte L, Kinter M, Ting SM, Dignam JD, Kim S, Driscoll DM, et al. 2004. Noncanonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell 119: 195–208. [DOI] [PubMed] [Google Scholar]
- Sarkar J, Poruri K, Boniecki MT, McTavish KK, Martinis SA. 2012. Yeast mitochondrial leucyl-tRNA synthetase CP1 domain has functionally diverged to accommodate RNA splicing at expense of hydrolytic editing. J Biol Chem 287: 14772–14781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Langhendries JL, Watzinger P, Kotter P, Entian KD, Lafontaine DL. 2015. Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res 43: 2242–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada N, Suzuki T, Watanabe K. 2001. Dual mode recognition of two isoacceptor tRNAs by mammalian mitochondrial seryl-tRNA synthetase. J Biol Chem 276: 46770–46778. [DOI] [PubMed] [Google Scholar]
- Smirnova EV, Lakunina VA, Tarassov I, Krasheninnikov IA, Kamenski PA. 2012. Noncanonical functions of aminoacyl-tRNA synthetases. Biochemistry (Mosc) 77: 15–25. [DOI] [PubMed] [Google Scholar]
- Swinehart WE, Jackman JE. 2015. Diversity in mechanism and function of tRNA methyltransferases. RNA Biol 12: 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinehart WE, Henderson JC, Jackman JE. 2013. Unexpected expansion of tRNA substrate recognition by the yeast m1G9 methyltransferase Trm10. RNA 19: 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbonavicius J, Qian Q, Durand JM, Hagervall TG, Bjork GR. 2001. Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J 20: 4863–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waas WF, Druzina Z, Hanan M, Schimmel P. 2007. Role of a tRNA base modification and its precursors in frameshifting in eukaryotes. J Biol Chem 282: 26026–26034. [DOI] [PubMed] [Google Scholar]
- Wakasugi K, Schimmel P. 1999. Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science 284: 147–151. [DOI] [PubMed] [Google Scholar]
- Wang C, Guo Y, Tian Q, Jia Q, Gao Y, Zhang Q, Zhou C, Xie W. 2015. SerRS-tRNASec complex structures reveal mechanism of the first step in selenocysteine biosynthesis. Nucleic Acids Res 43: 10534–10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenbach J, Kiraly I, Dirheimer G. 1977. Primary structure of tRNA Thr 1a and b from brewer's yeast. Biochimie 59: 381–391. [DOI] [PubMed] [Google Scholar]
- Weixlbaumer A, Murphy FVt, Dziergowska A, Malkiewicz A, Vendeix FA, Agris PF, Ramakrishnan V. 2007. Mechanism for expanding the decoding capacity of transfer RNAs by modification of uridines. Nat Struct Mol Biol 14: 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whipple JM, Lane EA, Chernyakov I, D'Silva S, Phizicky EM. 2011. The yeast rapid tRNA decay pathway primarily monitors the structural integrity of the acceptor and T-stems of mature tRNA. Genes Dev 25: 1173–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao P, Fox PL. 2013. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol Med 5: 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]