

Abstract
The emergence of hydrogen sulfide (H2S) as a new signalling molecule able to control vasodilation, neurotransmission and immune response, prompted questions about its possible cross-talk with the other gasontransmitter, nitric oxide (NO). It has been shown that H2S reacts with NO and its metabolites and several potentially biologically active species have been identified. Thionitrous acid (HSNO) was proposed to be an intermediate product of the reaction of S-nitrosothiols with H2S capable of crossing the membranes and causing further trans-nitrosation of proteins. Alternatively, formation of nitrosopersulfide (SSNO−) has been proposed in this reaction. SSNO− was claimed to be particularly stable and inert to H2S, thiols and cyanides. It is suggested that this putative SSNO− slowly decomposes to give NO, HNO and polysulfides. However, the chemical studies with pure SSNO− salts showed some conflicting observations. In this study, we work with pure PNP+SSNO− to show that contrary to everything that is claimed for the yellow reaction product of GSNO with H2S, pure SSNO− decomposes readily in the presence of cyanide, H2S and glutathione to form SNO−. Based on literature overview and chemical data about the structures of HSNO/SNO− and SSNO− we discuss the biological role these two species could have.
Keywords: nitric oxide, hydrogen sulfide, nitrosopersulfide, thionitrous acid
1. Introduction
Hydrogen sulfide (H2S) emerged as the third gaseous signalling molecule (gasotransmitter), alongside nitric oxide (NO) and carbon monoxide (CO) [1–3]. The similarity of its physiological effects (vasodilation [4,5], cardioprotection [6,7], neurotransmission [8,9], immunomodulation [10], etc.) to those previously reported for NO, prompted questions about possible cross-talk with NO [11].
It is now well established that H2S strongly interferes with NO signalling, either by reacting directly with NO [12] and its metabolites (S-nitrosothiols (RSNO) [13], peroxynitrite [14,15] and nitrite [16–18]) or by modulating NO production [19] and cGMP production [20] and degradation [21,22]. Namely, even the first reports on H2S-induced vasodilatory effects showed that endogenous H2S functions as a smooth muscle tone regulating factor in synergy with NO [23]. Inhibition of NO synthase by non-specific inhibitor L-NAME led to inhibition of H2S-induced vasodilation [24], while the deletion of cystathionine gamma lyase (CSE; an H2S producing enzyme) prevented vasodilatory effects of acetylcholine and NO [21]. Finally, cardioprotective effects of H2S are abolished in eNOS−/− mice [25], although mitochondria targeted H2S donors remain active even in the absence of NO [26,27].
In their initial studies, Whiteman and co-workers [28,29] observed that different donors of NO or nitrosonium species (NO+) react with H2S to give a new S-nitrosothiol, which they proposed to be HSNO. Before that, HSNO was reported as a product of cis-HNSO photolysis in argon matrices [30,31]. Its isomers (cis and trans) were studied under these conditions by the means to IR spectroscopy. Seel and co-workers [32] managed to crystalize SNO− salts with PNP+ showing charge delocalization over all three atoms. Computational studies suggested that although slightly less stable than RSNOs, HSNO could exist under physiologically relevant conditions [33].
By the means of pulse radiolysis (to generate NO• and HS•), or by mixing H2S with either acidified nitrite or GSNO, we also observed that HSNO could be formed under physiologically relevant conditions [13], confirming previous assumptions by Whiteman and co-workers [28]. Furthermore, we showed that HSNO could serve as a shuttle of ‘NO+’ from one thiol to another, being thus involved in trans-nitrosation processes [13].
Recently, McCarthy and co-workers gathered highly accurate molecular parameters (rotational, centrifugal distortion and hyperfine constants) of HSNO using Fourier-transform microwave (FTMW) spectroscopy [34]. They confirmed the existence of both cis and trans isomers and estimated S–N bond length to 1.834 ± 0.002 Å for cis-HSNO and 1.852 ± 0.002 Å for trans-HSNO, the latter being the longest experimentally measured S–N bond in an S-nitrosothiol.
Contrary to that, Feelisch and co-workers failed to identify HSNO when mixing GSNO and H2S and proposed the formation of another stable S-nitroso species, nitrosopersulfide (SSNO−) [35]. In their paper, Cortesse-Krott et al. claim that (i) HSNO is too short lived to be identified, (ii) that ‘enriched’ solutions of SSNO− could be prepared by having an excess of H2S over GSNO, (iii) that SSNO− is stable in the presence of thiols (such as glutathione which is one of the products formed in the reaction of GSNO with H2S) and (iv) resistant to cyanides [35]. Parallel with the publication of this study, we reported the (bio)chemical characterization of pure crystalline SSNO− (prepared as PNP+ SSNO− salt) [36], following the synthetic protocol reported by Seel et al. [32], to show that SSNO− could not really exist under physiological conditions.
Here, we extend our previous work with pure PNP+SSNO− to show that contrary to everything that is claimed for the yellow reaction product of GSNO with H2S, pure SSNO− decomposes readily in the presence of cyanide, H2S and glutathione to form SNO−, as confirmed by 15N NMR and cryo-spray mass spectrometry. Furthermore, based on, often mis-cited, old literature and actual chemical data about the structures and chemical reactivity of both HSNO/SNO− and SSNO−, we discuss the biological role these two species could have.
2. Material and methods
2.1. Chemicals
All chemicals were obtained from Sigma Aldrich if not stated otherwise. All aqueous solutions were prepared using nanopure water. All buffers were supplemented with Chelex-100 resin and kept over the resin to remove traces of transition metal ions. Organic solvents were purchased dry, additionally dried with MgSO4 and stored in argon box.
2.2. Preparation of PNP+SSNO–
PNP+SSNO− was prepared as previously described [32,36]. Briefly, 0.525 g of 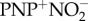 (900 µmol) and 57 mg of elemental sulfur (1.8 mmol as referred to ‘S’) were weighed in a vial in the argon box, 15 ml of dry acetone was added. The reaction mixture was stirred overnight, in the argon box, yielding a dark orange-to-red solution. This solution was PTFE-filtered to remove remnants of unreacted sulfur. To the reaction mixture, 10 ml of dried diethylether was added to initiate crystallization. The majority of the crystals were red, with some trace amounts of yellow ones. Crystals were dissolved in acetone and recrystallized by addition of diethylether at least twice.
(900 µmol) and 57 mg of elemental sulfur (1.8 mmol as referred to ‘S’) were weighed in a vial in the argon box, 15 ml of dry acetone was added. The reaction mixture was stirred overnight, in the argon box, yielding a dark orange-to-red solution. This solution was PTFE-filtered to remove remnants of unreacted sulfur. To the reaction mixture, 10 ml of dried diethylether was added to initiate crystallization. The majority of the crystals were red, with some trace amounts of yellow ones. Crystals were dissolved in acetone and recrystallized by addition of diethylether at least twice.
2.3. Absorption spectroscopy
UV/Vis measurements were performed in anaerobic cuvettes (i.e. cuvettes sealable with a screw cap equipped with a silicon/PTFE septum) using an HP 8452A diode array spectrophotometer connected to a computer equipped with Olis Spectralworks software. Additional measurements were performed on a Specord 200 spectrophotometer by Jena Analytics, connected to a computer equipped with Winaspect software.
2.4. NMR spectroscopy
14N- and 15N-NMR spectra were recorded on a Bruker AVANCE DRX 400WB spectrometer equipped with a Spectrospin superconducting wide-bore magnet operating at a resonance frequency of 28.90 MHz and 40.54 MHz, respectively, at a magnetic induction of 9.4 T. The measurements were performed with a commercial 5 mm Bruker broadband probe thermostated with a Bruker B-VT 3000 variable temperature unit. Chemical shifts given were referenced to nitromethane.
2.5. Electron spray ionization mass spectrometry (ESI-MS)
MS measurements were performed on a UHR-ToF Bruker Daltonik (Bremen, Germany) maXis, which was coupled to a Bruker cryo-spray unit. Detection was in positive and negative ion mode. The flow rates were 200 µl h−1. The dry-gas (N2) temperature was held at −20°C and the spray-gas temperature was held at −15°C. The instrument was calibrated prior to every experiment via direct infusion of the Agilent ESI-TOF low concentration tuning mixture, which provided an m/z range of singly charged peaks up to 2700 Da in both ion modes.
3. Results and discussion
3.1. UV/Vis spectral properties of SSNO− in water
A history of the identification of S–S bond-containing compounds in solutions was rich in contradicting conclusions [37], and ‘some of those who dared to tackle this challenging task were victims of delusions because such species that were optically (or even by other methods) observed in non-aqueous solutions could not easily be established as defined substances’ [38]. This is a translated quotation from Seel & Wagner [38], who were the first to synthesize SSNO− salt under exclusion of water and dioxygen [32]. Seel and co-workers tackled this challenging chemistry before either NO or H2S were known as signalling molecules. Besides crystalizing PNP+SSNO−, they observed that when NO is bubbled into alkaline solutions of sulfide (pH > 10) a yellow species is formed with an absorbance maximum at 412 nm [39]. Based on their observation that solutions of crystalline SSNO− absorbs at 448 nm in acetone, they assumed that this was a solvent-induced shift and that the 412 nm product, observed in the reaction of NO with alkaline sulfide solutions, might also be SSNO−. They estimated that the yield of this putative SSNO− is not more than 10%, but the authors clearly stated that the proposed chemical nature of this product was questionable, because SSNO− could not be confirmed by 15N NMR [39].
When studying the reaction of S-nitrosoglutathione with H2S, at pH 7.4 we observed the formation of yellow product with absorbance at 412 nm but because the time-resolved 15N NMR studies and time-resolved IR studies confirmed that the 15N NMR signal and N = O vibrational band, respectively, were only present for short amount of time, while the ‘yellow’ product remained stable for couple of hours (slowly decaying with sulfur precipitation), we proposed that this is a mixture of polysulfides/sulfur sols [13,36]. Working with pure SSNO− we could not test whether there was a water-induced hypsochromic shift, as PNP+SSNO−, as in the Seel's report [32], decomposed when 10% of water was added into the acetone solution of the salt [36]. The matured mixture of GSNO and H2S, called ‘SSNO− mix’, has been referred to as enriched SSNO− in several papers [35,40–42], solely based on 412 nm absorbance, although no real chemical evidence was provided to prove that this ‘yellow’ mixture consists of SSNO−. Recently, Olabe and co-workers performed a computational study to predict the UV/Vis spectrum of SSNO− in water and they also suggested that SSNO− should indeed absorb at 412 nm when dissolved in water [43].
We wanted to address this experimentally, by recording absorbance maximum shift in different solvents and then plotting the data over the dielectric constant (ɛ) of the particular solvent. First obvious observation is that it is impossible to establish any correlation for most of the tested solvents (figure 1). For example, the absorbance maximum of SSNO− in acetonitrile and dichloromethane is quite similar (442 nm) while the difference in their ɛ values is quite large (ɛ = 8.93 for dichloromethane and ɛ = 37.5 for acetonitrile) (figure 1). Therefore, the absorbance at 448 nm that others and we reported for the acetone solution can hardly be used as a reference point for predicting the UV/Vis features of SSNO− in other solvents, particularly in water. Several of the tested alcohols however showed nice linear dependence (figure 1b). Assuming that water will behave in a similar way, we used the obtained correlation between absorbance maximum and dielectric constant of alcohols to estimate the former value for water, being 402 ± 2 nm. It should be stressed that if we consider a linear correlation observed only for small water-soluble alcohols, the widely used 412 ± 2 nm value is quite off the observed linear correlation (figure 1b). By way of comparison, the estimated absorbance maximum is even lower than what we calculated and experimentally measured for HSNO2 (408 nm; [14,15]). It is worth mentioning that SSNO− solutions in methanol and ethanol were not as stable as they were in acetone with 250 µM SSNO− disappearing from the ethanol solution in 30 min (not shown). This contradicts the statement that SSNO− is stable for hours in aqueous solutions at pH 7.4, especially in high concentrations in ‘SSNO−-enriched mixtures’ (‘SSNO− mix’) [35].
Figure 1.

UV/Vis spectral changes of SSNO− in different solvents. (a) Overlaid UV/Vis spectra of PNP+SSNO− in acetone (200 µM, black line) and methanol (saturated solution, maximally 380 µM, red line). The absorbance maxima are located at 448 nm and 422 nm, respectively. (b) The wavelengths of the absorbance maxima of PNP+SSNO− in different organic solvents plotted against the relative permittivity (dielectric constant) of the respective solvent. The putative value of SSNO− absorbance in water is given as 412 nm according to the proposal of Cortese-Krott et al. Contrary to the other tested solvents, the alcohols displayed a pronounced linearity and we used an extrapolated linear fit derived from those three solvents (red line) to estimate the real value for H2O. The values for relative permittivity of solvents are taken from: Maryott & Smith, 1951 [44]. (c) UV/Vis spectra of PNP+SSNO− (maximal starting concentration of 200 µM, black) in methanol, recorded 15 s later (red line) and 30 s later (green line). The relatively fast decay of the characteristic SSNO− peak at 422 nm is clearly visible.
3.2. Biologically relevant reactivity of SSNO−
In their paper Cortese-Krott and co-workers claim (i) that ‘enriched’ solutions of SSNO− could be prepared by having an excess of H2S over GSNO, (ii) that SSNO− is stable in the presence of thiols (such as glutathione which is one of the products formed in the reaction of GSNO with H2S) and (iii) resistant to cyanides [35]. This is surprising, considering that X-ray structural analysis of SSNO− suggests a structure that could best be described as cage structure of [S2----NO−] [36], therefore one of the two sulfur atoms should be available for nucleophilic attack by either HS−, GS− or CN−.
We previously reported that SSNO− reacts rapidly with sulfide [36]. Now, we combined 15N NMR with ESI-TOF-MS data to observe the reaction product of SSNO− with sulfide. When acetone solutions of PNP+SS15NO− were mixed with Na2S and the reaction monitored by 15N NMR, a clear shift in 15N NMR from 354 ppm to 314 ppm is noticeable. The NMR (314 ppm) signal was also observed previously in the reaction of GS15NO and H2S and we assigned it to the 15N NMR of HSNO/SNO− (figure 2a). To confirm this we performed cryo-electron spray ionization mass spectrometry, spraying the acetonitrile mixture of PNP+SSNO− (figure 2b) or PNP+SS15NO− (figure 2c) and H2S at −20°C. Recording spectra in positive mode we could clearly demonstrate the formation of HSNO, either as [HSNO + H]+ or [HSNO + CH3CN + H]+. The observed and predicted masses matched nicely (figure 2b). When we sprayed the reaction mixture in the negative ion mode, we could see the formation of SNO− with both PNP+SSNO− (not shown) and PNP+SS15NO− (figure 2c). These data clearly confirm that SNO− is the product of the reaction (equation (3.1); figure 2b). The use of cryo-ESI-TOF-MS once again proved to be a useful tool for facile detection of HSNO/SNO−.
| 3.1 |
Figure 2.

SNO− is the reaction product of HS− with nitrosopersulfide. (a) Overlayed 15N NMR spectra of PNP+SS15NO− (100 mM in acetone, black line) as well as of a mixture of PNP+SS15NO− (30 mM) and H2S (50 mM) in a solution of acetone : water 9 : 1, recorded 1 h after mixing (red line). While we ascribe the signal at 354 ppm to SS15NO−, the newly appearing signal at 314 ppm in the nitrosopersulfide/hydrogen sulfide mixture is assigned to S15NO− (figure 4b). (b) Identification of HSNO as a reaction product of PNP+SSNO− with H2S. 250 µM PNP+SSNO− was mixed with an equimolar amount of H2S (solution in THF) and reaction monitored in positive mode. Simulated mass spectra for some of the species identified during the measurement are shown in black. (c) Negative ion mode ionization of a reaction mixture containing 250 µM PNP+SS15NO− and H2S. Recorded spectrum is shown in red, and simulated mass spectra of S15NO− and 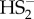 are shown in blue and green, respectively.
are shown in blue and green, respectively.
Sulfur precipitated from this reaction, most probably from 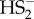 . We previously reported that synthetic H2S2 explodes when exposed to the air humidity giving H2S and elemental sulfur [36]. But after filtration of this mixture of decomposition products, the yellow species remained and it absorbs exactly at 412 nm, just like putative ‘SSNO− mix’. This yellow species was resistant to reducing agents such as DTT (reacting with it rather slowly) [36], just like what Cortese-Krott et al. reported for their ‘SSNO− mix’ [35]. Based on 15N NMR and cryo-ESI-TOF-MS data that show only SNO− as the nitrogen containing species, and knowing that SNO− absorbs at 340 nm, this chemical evidence suggests that 412 nm absorbance may only be attributed to the presence of the mixture of polysulfides/sulfur sols. Importantly, contrary to previous claims [35,40], these results demonstrate that real SSNO− cannot be stabilized in solutions containing excess of H2S because it reacts with it.
. We previously reported that synthetic H2S2 explodes when exposed to the air humidity giving H2S and elemental sulfur [36]. But after filtration of this mixture of decomposition products, the yellow species remained and it absorbs exactly at 412 nm, just like putative ‘SSNO− mix’. This yellow species was resistant to reducing agents such as DTT (reacting with it rather slowly) [36], just like what Cortese-Krott et al. reported for their ‘SSNO− mix’ [35]. Based on 15N NMR and cryo-ESI-TOF-MS data that show only SNO− as the nitrogen containing species, and knowing that SNO− absorbs at 340 nm, this chemical evidence suggests that 412 nm absorbance may only be attributed to the presence of the mixture of polysulfides/sulfur sols. Importantly, contrary to previous claims [35,40], these results demonstrate that real SSNO− cannot be stabilized in solutions containing excess of H2S because it reacts with it.
Comparably, thiols should also be capable of nucleophilic attack. In fact, in our previous work we estimated that the redox potential of SSNO− is −0.2 V (versus NHE) [36], a value that is easily reached in the biological environment by simple thiol buffers such as the glutathione buffer. We showed previously that SSNO− decomposes readily in the presence of DTT and we wanted now to address its ability to react with GSH. Owing to the limited solubility of GSH in other solvents, the reaction had to be carried out in DMSO. Addition of 4 mM GSH into 400 µM SSNO− resulted in a steady decay of SSNO− with a half-life of 10 min at 21°C (figure 3). Taking into account that GSH is not deprotonated under these conditions, its nucleophilicity is lower than what it would be under physiological conditions, where the reaction would probably proceed even faster. But even with such a reaction rate and by assuming that the physiological concentration of GSH is 5 mM, and that there is only a fivefold increase in the reaction rate at 37°C when compared with 21°C, the half-life of SSNO− becomes less than 100 s. The isosbestic point and the shoulder at approximately 340 nm is indicative of formation of another species, most likely HSNO/SNO− (equation (3.2)) [13].
| 3.2 |
and
| 3.3 |
Figure 3.
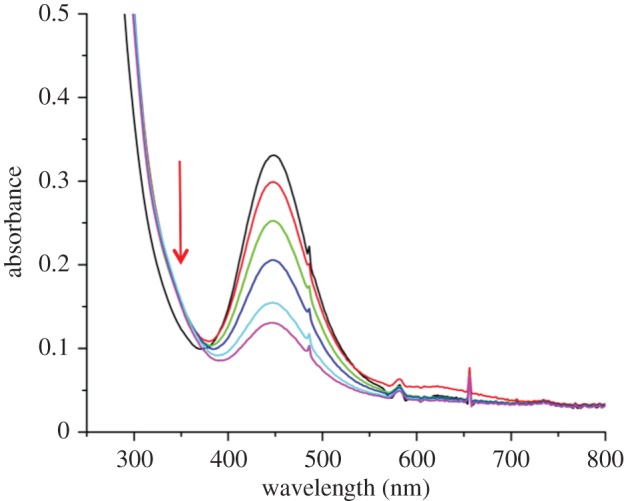
Glutathione causes decomposition of SSNO−. Time-resolved UV/Vis spectra of the reaction mixture containing SSNO− (400 µM PNP+SSNO−) with glutathione (4 mM) in DMSO over 30 min. The colours of the lines correspond to the respective observation times as follows: immediately after mixing (black line), 1 min later (red line), 5 min later (green line), 10 min later (deep blue line), 20 min later (light blue line), 30 min later (purple line).
A trans-nitrosation reaction to give GSNO and 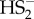 (equation (3.3) is a possibility but considering that there were no characteristic UV/Vis features of polysulfides/sulfur sols formed as decomposition products of HS2− (as in the case of the reaction with HS−) we concluded that reaction described by equation (3.2) is the most likely one. In addition, when we studied the reaction of H2S2 with NO+BF4, we observed that although the product is SSNO− it is not nitrosopersulfide but its isomer, dithionitrate, with distinct UV/Vis, 15N NMR and IR characteristics and different chemical reactivity [36].
(equation (3.3) is a possibility but considering that there were no characteristic UV/Vis features of polysulfides/sulfur sols formed as decomposition products of HS2− (as in the case of the reaction with HS−) we concluded that reaction described by equation (3.2) is the most likely one. In addition, when we studied the reaction of H2S2 with NO+BF4, we observed that although the product is SSNO− it is not nitrosopersulfide but its isomer, dithionitrate, with distinct UV/Vis, 15N NMR and IR characteristics and different chemical reactivity [36].
Finally, cyanides are known to react with sulfane sulfur [45]; however, the putative ‘SSNO− mix’ was resistant to the cyanide treatment [35]. In order to experimentally address this contradiction, we firstly followed the decay of PNP+SSNO− upon addition of potassium cyanide into the acetone solution. SSNO− started decaying rather fast with the reaction being over in approximately 200 s (figure 4a,b). If CN− indeed reacted with sulfane sulfur then the reaction products should be SCN− and SNO− (equation (3.4)). To resolve the product identity, we mixed SS15NO− with C14N− and recorded both 14N and 15N NMR (figure 4; equation (3.4)).
| 3.4 |
Figure 4.

SSNO− reacts with cyanide to give SNO− and SCN−. (a) Time-resolved spectra of SSNO− decomposition upon addition of KCN. (b) Kinetics of SSNO− decomposition in acetone upon addition of KCN. (c) 14N NMR spectrum of a mixture of 32 mM PNP+SS15NO− and 200 mM NaCN in 5 : 2 acetone : methanol (black line) solution, recorded 4 h after mixing. The two broad signals at about −173 ppm and at about −107 ppm are ascribed to SCN− and unreacted CN−, respectively, by measuring 14N NMR spectra of solutions of NaSCN (about 1 M, red line) and NaCN (about 200 mM, blue line) in the same solvent mixture. (d) 15N NMR spectrum of a mixture of 32 mM PNP+SS15NO− and 200 mM NaCN in 5 : 2 acetone : methanol solution (the same sample as shown in the figure 4c). The signal at 314 ppm is ascribed to S15NO− (figure 2).
In the 15N NMR, we again observed the 314 ppm signal which we also saw when sulfide reacted with SS15NO− and which corresponds to S15NO− (figure 4c). In 14N NMR, we could see two signals, one corresponding to the unreacted CN− and the other to newly formed SCN− as confirmed by comparing the 14N NMR chemical shifts of separately recorded cyanide and thiocyanate salts (figure 4d). These results unambiguously showed that SSNO− does react readily with CN− forming more stable SNO−, contrary to the chemical behaviour of putative ‘SSNO− mix’ described in literature [35].
3.3. HSNO versus SSNO−: relevance for cell signalling
In our previous attempt to prove that HSNO could be formed under physiologically relevant conditions, (water, pH 7.4) we used three approaches to generate HSNO: pulse radiolysis (to monitor the reaction of HS• with NO•), reaction of H2S with acidified nitrite, and trans-nitrosation reaction between S-nitrosoglutathione and H2S [13]. Using pulse radiolysis to generate HS• and NO•, we observed transient formation of a new species with spectral characteristics of an S-nitrosothiol, i.e. absorbance maximum at approximately 340 nm.
Nitrite and H2S do not react at neutral pH [16], but acidified nitrite is used to induce S-nitrosothiol formation through the formation of [H2NO2]+ species.
| 3.5 |
By adding H2S to acidified nitrite, we could detect transient red-brown coloration and identify HSNO by ESI-TOF-MS. Finally, using ESI-TOF-MS, 14N/15N IR and 15N NMR we also studied the trans-nitrosation reaction between S-nitrosoglutathione and H2S. HSNO, more precisely cis-HSNO has been shown to be formed. HSNO was not stable and decayed in time, particularly depending on the GSNO/H2S ratio [13]. In addition, coordinated HSNO has been observed in nitroprusside [46], water-soluble haem centres [17] and Ru(III)EDTA [47].
The most surprising was the ability of HSNO to act as a carrier of ‘NO+’ from one protein to another, even through the cell membrane. Namely, when S-nitrosated bovine serum albumin (BSA-SNO) was placed in a dialysis bag, which was immersed into the solution of BSA, addition of H2S led to nitrosation of the outer solution containing BSA. A similar observation was made with haemoglobin and red blood cells (RBC): RBC incubated with BSA-SNO in the presence of H2S showed increased haemoglobin S-nitrosation [13].
Protein S-nitrosation is considered to be an important post-translational modification. The role of S-nitrosation in controlling the protein function has been extensively covered in the past decade [48–54]. S-nitrosation has been implicated in the regulation of proteins involved in muscle contractility, neuronal transmission, host defence, cell trafficking, apoptosis, cardioprotection, etc. [48–54]. One of the unsolved questions, however, is the means by which trans-nitrosation (formal transfer of ‘NO+’ moiety) proceeds from one S-nitrosothiol to another [54–56]. Most S-nitrosothiols do not freely diffuse into the cells. A protein transporter for S-nitrosocysteine has been reported but it cannot explain [54], for example, almost immediate S-nitrosation of complex I in mitochondria achieved by treating the cells with S-nitrosothiols [57,58]. By reacting with H2S, low-molecular weight (LMW) and protein S-nitrosothiols could generate HSNO and we showed previously that HSNO could fulfil the criteria of acting as a shuttle of ‘NO+’, which classifies it as a new signalling species [13]. Indeed, inhibition of H2S production led to lower intracellular S-nitrosation, while the combination of H2S with RSNO increased the total S-nitrosothiol content when compared with a treatment with RSNO alone [59].
Recently, HSNO has been made at room temperature by diluting NO and H2S gases with Ne [34]. The reaction took place due to the fact that under this experimental condition there is a surface-catalysed NO disproportionation and formation of N2O3 which then reacted with H2S to give HSNO. McCarthy and co-workers used FTMW spectroscopy to prove facile HSNO formation from N2O3 and H2S [34]. Maximal HSNO concentration was reached when all N2O3 was consumed. Further addition of H2S led to disappearance of HSNO and formation of N2O. Using 18O labelled N2O3 (equation (3.6); * represents the 18O) the authors detected HSN18O and then N218O, which suggests that HNO was generated in between (equation (3.7)), confirming our previous findings from the buffered solutions [13].
| 3.6 |
| 3.7 |
| 3.8 |
Although reaction between HSNO and H2S to give HNO (equation (3.7)) is thermodynamically unfavourable (ΔrxnG0′ = +32 kJ mol−1) [60], if the products get removed/decomposed (e.g. considering the dimerization of HNO to N2O (equation (3.8)), the reaction of HNO with RSH or H2S2 decomposition) reaction could be physiologically relevant. Interesting computational studies by Timerghazin and co-workers proposed that HSNO could isomerize to form Y-isomer SN(H)O, a process that is additionally favoured by water [61]. This Y-isomer could then react with H2S to give HNO. The lowering of the barrier to isomerization by water and formation of the Y-isomer, as well as other isomers such as HONS, could explain the peculiar observation that (i) HSNO ionizes as [HSNO + H]+ in the MS experiments even at pH 7.4 ([13], this study), (ii) that it remains protonated when coordinated [17,47] and (iii) that it can cross the membrane freely [13,59]. Namely, chemical logic implies that the pKa of HSNO should be lower than that of HNO2 (4.1), therefore the predominant species in the solution should be SNO−. However, other isomers would have a higher pKa.
Facile formation of HSNO from N2O3 and H2S could be of importance for intracellular RSNO generation. Although intracellular N2O3 formation is kinetically/statistically improbable, N2O3 can easily be formed in lipid bilayers where both NO and O2 accumulate [62]. The H2S concentration should also be higher in lipid bilayers than in the cytosol [63] creating ideal conditions for HSNO formation that can act as further trans-nitrosating agent.
Contrary to all these claims where the existence of HSNO was proved by all available analytical methods [13,17,29,31,32,34], Cortese-Krott et al. proposed that SSNO− has to be one of three key bioactive reaction products of the reaction of H2S with S-nitrosothiols [35,40]. Based on these authors' claims, SSNO− can be obtained in high yields in aqueous solutions at pH 7.4 (even in the presence of oxygen) and it is stable for hours. HSNO/SNO−, however, was marked as unstable to be detected even at low temperatures as it reacts fast to eventually form a stable SSNO− [35]. This is in contradiction with the previous study by the same authors where HSNO formation was observed at room temperature from a 1 : 1 mixture of RSNO and sulfide in water (pH 7.4) at even higher yield than the putative ‘SSNO− mix’ [40].
It is claimed that HSSNO is more stable than HSNO, because HSSNO supposedly has increased electron density on the proximal sulfur (although no experimental support for such claim was provided) and therefore does not easily react with HS− and positive metal centres [35]. This putative SSNO− was reported to be relatively stable in general, and particularly stable in H2S, so the synthetic procedure for preparation of its enriched solutions from RSNO and H2S was proposed [35,40]. Furthermore, this putative SSNO− showed no reactivity with biological thiols or with cyanide. The facts are quite different: (i) the proximal-S has a +0.24 charge, (ii) the S–N bonds in HSSNO and SSNO− (calculated BDE 16.0 and 22.1 kcal mol−1, respectively; B3LYP/aug-cc-pv5z, in the presence of solvent/water) are weaker than those in HSNO and SNO− (BDE 27.74 and 36.21 kcal mol−1, respectively), which makes (H)SSNO more prone to homolysis than (H)SNO [36]. We also demonstrate here quite the opposite reactivity, when using the crystalline SSNO− (characterized previously by 15N NMR, IR, EPR, MS, X-ray analysis and electrochemical methods). Namely, real SSNO− reacts with all three, HS−, CN− and GSH to actually produce SNO−, which appears to be more stable species than SSNO−.
SSNO− formation from RSNO and H2S is proposed to happen through following reaction steps:
| 3.9 |
| 3.10 |
| 3.11 |
The first two reactions (equation (3.9) and (3.10)) have been characterized in our previous studies and confirmed recently by McCarthy and co-workers [34]. The reaction shown in equation (3.11) is in equilibrium and under physiological conditions or the experimental conditions where thiols/H2S are in excess it will proceed in an opposite direction, as demonstrated in the figures 2 and 3 and equations (3.1)–(3.2). Both thiols and H2S react with SSNO− to give HSNO/SNO− and HS2− as confirmed by 15N NMR and ESI-TOF-MS (figures 2 and 3).
The ‘SSNO− mix’ has been used to treat animals and cells, and is proposed to represent storage of NO and to be an activator of Keap-1/Nrf-2 signalling [35,40,41,64]. Reanalysing the original data published by Cortese-Krott et al. [35], Koppenol & Bounds discussed recently the probability for SSNO− formation under physiological conditions following the reaction steps shown in equations (3.9)–(3.11) [60]. The kinetics of SSNO− formation, based on the reported experimental data [35], was estimated to be 10−14 M−1 s−1 [60]. In other words, even if we assume that SSNO− remains stable in the cell, it would take more than 1 day to generate 1 nM concentration from 1 µM H2S, which is, for most of the tissues, an unreachable concentration under physiological conditions. Instability aside, given the starting concentrations of NO and H2S inside a cell and the likelihood of them interacting at a chemical level in the presence of outcompeting substrates, it is more likely that it would take even more than 1 day to form 1 nM SSNO−. Therefore, it becomes unclear what would be the physiological (and even pharmacological) relevance of treating the cells and animals with 2 mM combination of NO and H2S [35] or 40 µM of ‘SSNO− mix’ [41], for example. If 1 nM SSNO− cannot be formed in the cells what would be the physiological relevance of 40 µM?
In conclusion, it is of utmost importance that eventual studies with SSNO− are performed using clean and defined substance, which is simple to prepare, than to use reaction mixtures of unidentified composition, as the latter might lead to erroneous conclusions.
Furthermore, it is very important to separate the physiological relevance of all these proposed reactions from those that are just possible in vitro. Considering the very low HS− concentration [65,66] and very high thiol concentrations, the reaction shown in equation (3.10) is less likely to occur (although the rate constant for that reaction is approximately 107 M−1 s−1 [60]), let alone form 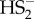 , which is intrinsically unstable [67,68] and could easily be reduced by the intracellular thiol pool, and wait to meet another S-nitrosothiol (or, even less probable, another HSNO) to form SSNO−. HSNO/SNO− remains therefore the most probable signalling molecule able to react with the abundant thiol pool to either cause further trans-nitrosation or HNO and RSS− formation, in specific protein environments [69].
, which is intrinsically unstable [67,68] and could easily be reduced by the intracellular thiol pool, and wait to meet another S-nitrosothiol (or, even less probable, another HSNO) to form SSNO−. HSNO/SNO− remains therefore the most probable signalling molecule able to react with the abundant thiol pool to either cause further trans-nitrosation or HNO and RSS− formation, in specific protein environments [69].
Competing interests
We declare we have no competing interests.
Funding
The authors would like to acknowledge the support from Friedrich-Alexander University Erlangen-Nuremberg (Emerging Field Initiative, MRIC) and CNRS/INSERM Atip-Avenir and the ‘Investments for the future’ Programme IdEx Bordeaux (ANR-10-IDEX-03-02).
References
- 1.Wang R. 2002. Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 16, 1792–1798. ( 10.1096/fj.02-0211hyp) [DOI] [PubMed] [Google Scholar]
- 2.Li L, Hsu A, Moore PK. 2009. Actions and interactions of nitric oxide, carbon monoxide and hydrogen sulphide in the cardiovascular system and in inflammation—a tale of three gases! Pharmacol. Ther. 123, 386–400. ( 10.1016/j.pharmthera.2009.05.005) [DOI] [PubMed] [Google Scholar]
- 3.Mustafa AK, Gadalla MM, Snyder SH. 2009. Signaling by gasotransmitters. Sci. Signal. 2, re2. ( 10.1126/scisignal.268re2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang G, et al. 2008. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322, 587–590. ( 10.1126/science.1162667) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mustafa AK, et al. 2011. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 109, 1259–1268. ( 10.1161/CIRCRESAHA.111.240242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, Kevil CG, Lefer DJ. 2009. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 105, 365–374. ( 10.1161/CIRCRESAHA.109.199919) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calvert JW, Elston M, Nicholson CK, Gundewar S, Jha S, Elrod JW, Ramachandran A, Lefer DJ. 2010. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation 122, 11–19. ( 10.1161/CIRCULATIONAHA.109.920991) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abe K, Kimura H. 1996. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 16, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kimura H, Nagai Y, Umemura K, Kimura Y. 2005. Physiological roles of hydrogen sulfide: synaptic modulation, neuroprotection, and smooth muscle relaxation. Antioxidants Redox Signal. 7, 795–803. ( 10.1089/ars.2005.7.795) [DOI] [PubMed] [Google Scholar]
- 10.Li L, et al. 2005. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 19, 1196–1198. ( 10.1096/fj.04-3583fje) [DOI] [PubMed] [Google Scholar]
- 11.Kolluru GK, Yuan S, Shen X, Kevil CG. 2015. H2S regulation of nitric oxide metabolism. Methods Enzymol. 554, 271–297. ( 10.1016/bs.mie.2014.11.040) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eberhard M, et al. 2014. H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO-TRPA1-CGRP signalling pathway. Nat. Commun. 5, 4381 ( 10.1038/ncomms5381) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filipovic MR, Miljkovic JLJ, Nauser T, Royzen M, Klos K, Shubina T, Koppenol WH, Lippard SJ, Ivanovic-Burmazovic I. 2012. Chemical characterization of the smallest S-nitrosothiol, HSNO; cellular cross-talk of H2S and S-nitrosothiols. J. Am. Chem. Soc. 134, 12 016–12 027. ( 10.1021/ja3009693) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filipovic MR, Milijkovic J, Allgaeuer A, Chaurio R, Shubina T, Herrmann M, Ivanovic-Burmazovic I. 2012. Biochemical insight into physiological effects of H2S: reaction with peroxynitrite and formation of a new nitric oxide donor, sulfinyl nitrite. Biochem. J. 441, 609–621. ( 10.1042/BJ20111389) [DOI] [PubMed] [Google Scholar]
- 15.Cuevasanta E et al. . 2015. Insights into the mechanism of the reaction between hydrogen sulfide and peroxynitrite. Free Radic. Biol. Med. 80, 93–100. ( 10.1016/j.freeradbiomed.2014.12.017) [DOI] [PubMed] [Google Scholar]
- 16.Wedmann R, Bertlein S, Macinkovic I, Boeltz S, Miljkovic J, Munoz L, Herrmann M, Filipovic MR. 2014. Working with ‘H2S’: facts and apparent artifacts. Nitric Oxide 41, 85–96. ( 10.1016/j.niox.2014.06.003) [DOI] [PubMed] [Google Scholar]
- 17.Miljkovic JLJ, Ivanovic-Burmazovic I, Filipovic MR. 2013. Generation of HNO and HSNO from nitrite by heme-iron-catalyzed metabolism with H2S. Angew. Chem. Int. Ed. 52, 12 061–12 064. ( 10.1002/anie.201305669) [DOI] [PubMed] [Google Scholar]
- 18.Bir SC, Kolluru GK, McCarthy P, Shen X, Pardue S, Pattillo CB, Kevil CG. 2012. Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1α and vascular endothelial growth factor-dependent angiogenesis. J. Am. Heart Assoc. 1, e004093 ( 10.1161/JAHA.112.004093) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Altaany Z, Ju Y, Yang G, Wang R. 2014. The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci. Signal. 7, ra87 ( 10.1126/scisignal.2005478) [DOI] [PubMed] [Google Scholar]
- 20.Zhou Z, Martin E, Sharina I, Esposito I, Szabo C, Bucci M, Cirino G, Papapetropoulos A. 2016. Regulation of soluble guanylyl cyclase redox state by hydrogen sulfide. Pharmacol. Res. 111, 556–562. ( 10.1016/j.phrs.2016.06.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coletta C et al. . 2012. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl Acad. Sci. USA 109, 9161–9166. ( 10.1073/pnas.1202916109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang R, Szabo C, Ichinose F, Ahmed A, Whiteman M, Papapetropoulos A. 2015. The role of H2S bioavailability in endothelial dysfunction. Trends Pharmacol. Sci. 36, 568–578. ( 10.1016/j.tips.2015.05.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hosoki R, Matsuki N, Kimura H. 1997. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 237, 527–531. ( 10.1006/bbrc.1997.6878) [DOI] [PubMed] [Google Scholar]
- 24.Zhao W, Zhang J, Lu Y, Wang R. 2001. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 20, 6008–6016. ( 10.1093/emboj/20.21.6008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King AL, et al. 2014. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase–nitric oxide dependent. Proc. Natl Acad. Sci. USA 111, 3182–3187. ( 10.1073/pnas.1321871111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chatzianastasiou A, et al. 2016. Cardioprotection by 2S donors: nitric oxide-dependent and -independent mechanisms. J. Pharmacol. Exp. Ther. 358, 431–440. ( 10.1124/jpet.116.235119) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karwi GQ, Bornbaum J, Boengler K, Torregrossa R, Whiteman M, Wood ME, Schulz R, Baxter GF. In press. Ap39, a mitochondria-targeting hydrogen sulfide (H2S) donor, protects against myocardial reperfusion injury independently of salvage kinase signalling. Br. J. Pharmacol. ( 10.1111/bph.13688) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whiteman M, Li L, Kostetski I, Chu SH, Siau JL, Bhatia M, Moore PK. 2006. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem. Biophys. Res. Commun. 343, 303–310. ( 10.1016/j.bbrc.2006.02.154) [DOI] [PubMed] [Google Scholar]
- 29.Ali MY, Ping CY, Mok YY, Ling L, Whiteman M, Bhatia M, Moore PK. 2006. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br. J. Pharmacol. 149, 625–634. ( 10.1038/sj.bjp.0706906) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nonella M, Huber JR, Ha T-K. 1987. Photolytic preparation and isomeriation of HNSO, HOSN, HSNO, and HONS in an argon matrix. An experimental and theoretical study. J. Phys. Chem. 91, 5203–5209. ( 10.1021/j100304a014) [DOI] [Google Scholar]
- 31.Mueller RP, Nonella M, Russegger P, Huber JR. 1984. UV-, VIS- and IR-light-induced isomerization of HSNO in a low-temperature matrix. Chem. Phys. 87, 351–361. ( 10.1016/0301-0104(84)85116-2) [DOI] [Google Scholar]
- 32.Seel F, Kuhn R, Simon G, Wagner M. 1985. PNP-Perthionitrit und PNP-Monothionitrit. Z. Naturforsch. 40b, 1607–1617. [Google Scholar]
- 33.Timerghazin QK, Peslherbe GH, English AM. 2008. Structure and stability of HSNO, the simplest S-nitrosothiol. Phys. Chem. Chem. Phys. 10, 1532–1539. ( 10.1039/b715025c) [DOI] [PubMed] [Google Scholar]
- 34.Nava M, et al. 2016. Spontaneous and selective formation of HSNO, a crucial intermediate linking H2S and nitroso chemistries. J. Am. Chem. Soc. 138, 11 441–11 444. ( 10.1021/jacs.6b05886) [DOI] [PubMed] [Google Scholar]
- 35.Cortese-Krott MM, et al. 2015. Key bioactive reaction products of the NO/H2S interaction are S/N-hybrid species, polysulfides, and nitroxyl. Proc. Natl Acad. Sci. USA 112, E4651–E4660. ( 10.1073/pnas.1509277112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wedmann R, Zahl A, Shubina TE, Dürr M, Heinemann FW, Bugenhagen BE, Burger P, Ivanovic-Burmazovic I, Filipovic MR. 2015. Does perthionitrite (SSNO(-)) account for sustained bioactivity of NO? a (bio)chemical characterization. Inorg. Chem. 54, 9367–9380. ( 10.1021/acs.inorgchem.5b00831) [DOI] [PubMed] [Google Scholar]
- 37.Seel F, Guttler H-J, Simon G, Wieckowski A. 1977. Colored sulfur species in EPD-solvents. Pure Appl. Chem. 49, 45–54. ( 10.1351/pac197749010045) [DOI] [Google Scholar]
- 38.Seel F, Wagner M. 1985. The reaction of polysulfides with nitrogen monoxide in non-acqueous solvents -nitrosodisulfides. Z. Naturforsch. 40b, 762–764. [Google Scholar]
- 39.Seel F, Wagner M. 1988. Reaction of sulfides with nitrogen monoxide in aqueous solution. Z. Anorg. Allg. Chem. 558, 189–192. ( 10.1002/zaac.19885580118) [DOI] [Google Scholar]
- 40.Cortese-Krott MM, Fernandez BO, Santos JLT, Mergia E, Grman M, Nagy P, Kelm M, Butler A, Feelisch M. 2014. Nitrosopersulfide (SSNO(−)) accounts for sustained NO bioactivity of S-nitrosothiols following reaction with sulfide. Redox Biol. 2, 234–244. ( 10.1016/j.redox.2013.12.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cortese-Krott MM, Pullmann D, Feelisch M. 2016. Nitrosopersulfide (SSNO-) targets the Keap-1/Nrf2 redox system. Pharmacol. Res. 113, 490–499. ( 10.1016/j.phrs.2016.09.022) [DOI] [PubMed] [Google Scholar]
- 42.Bolden C, King SB, Kim-Shapiro DB. 2016. Reactions between nitrosopersulfide and heme proteins. Free Radic. Biol. Med. 99, 418–425. ( 10.1016/j.freeradbiomed.2016.09.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marcolongo JP, Morzan UN, Zeida A, Scherlis DA, Olabe JA. 2016. Nitrosodisulfide [S2NO]- (perthionitrite) is a true intermediate during the ‘cross-talk’ of nitrosyl and sulfide. Phys. Chem. Chem. Phys. 18, 30 047–30 052. ( 10.1039/C6CP06314D) [DOI] [PubMed] [Google Scholar]
- 44.Maryott AA, Smith ER. 1951. Table of dielectric constants of pure liquids. NBS Circular 514, 1–7. [Google Scholar]
- 45.Wood JL. 1987. Sulfane sulfur. Methods Enzymol. 143, 25–29. ( 10.1016/0076-6879(87)43009-7) [DOI] [PubMed] [Google Scholar]
- 46.Filipovic MR, Eberhardt M, Prokopovic V, Mijuskovic A, Orescanin-Dusic Z, Reeh P, Ivanovic-Burmazovic I. 2013. Beyond H2S and NO interplay: hydrogen sulfide and nitroprusside react directly to give nitroxyl (HNO). A new pharmacological source of HNO. J. Med. Chem. 56, 1499–1508. ( 10.1021/jm3012036) [DOI] [PubMed] [Google Scholar]
- 47.Chatterjee D, Sarkar P, Oszajca M, van Eldik R. 2016. Formation of [Ru(III)(edta)(SNO)](2-) in Ru(III)(edta)-Mediated S-Nitrosylation of Bisulfide Ion. Inorg. Chem. 55, 5037–5040. ( 10.1021/acs.inorgchem.6b00615) [DOI] [PubMed] [Google Scholar]
- 48.Foster MW, Hess DT, Stamler JS. 2009. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol. Med. 15, 391–404. ( 10.1016/j.molmed.2009.06.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hess DT, Stamler JS. 2012. Regulation by S-nitrosylation of protein post-translational modification. J. Biol. Chem. 287, 4411–4418. ( 10.1074/jbc.R111.285742) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seth D, Stamler JS. 2011. The SNO-proteome: causation and classifications. Curr. Opin. Chem. Biol. 15, 129–136. ( 10.1016/j.cbpa.2010.10.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lima B, Forrester MT, Hess DT, Stamler JS. 2010. S-nitrosylation in cardiovascular signaling. Circ. Res. 106, 633–646. ( 10.1161/CIRCRESAHA.109.207381) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Broniowska KA, Hogg N. 2012. The chemical biology of S-nitrosothiols. Antioxidants Redox Signal. 17, 969–980. ( 10.1089/ars.2012.4590) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chouchani ET, et al. 2013. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 19, 753–759. ( 10.1038/nm.3212) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Hogg N. 2004. The mechanism of transmembrane S-nitrosothiol transport. Proc. Natl Acad. Sci. USA 101, 7891–7896. ( 10.1073/pnas.0401167101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anand P, Stamler JS. 2012. Enzymatic mechanisms regulating protein S-nitrosylation: implications in health and disease. J. Mol. Med. 90, 233–244. ( 10.1007/s00109-012-0878-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hogg N. 2002. The biochemistry and physiology of S-nitrosothiols. Annu. Rev. Pharmacol. Toxicol. 42, 585–600. ( 10.1146/annurev.pharmtox.42.092501.104328) [DOI] [PubMed] [Google Scholar]
- 57.Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. 2006. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem. J. 394, 627–634. ( 10.1042/BJ20051435) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dahm CC, Moore K, Murphy MP. 2006. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: implications for the interaction of nitric oxide with mitochondria. J. Biol. Chem. 281, 10 056–10 065. ( 10.1074/jbc.M512203200) [DOI] [PubMed] [Google Scholar]
- 59.Sun J, Aponte AM, Menazza S, Gucek M, Steenbergen C, Murphy E. 2016. Additive cardioprotection by pharmacological postconditioning with hydrogen sulfide and nitric oxide donors in mouse heart: S-sulfhydration vs. S-nitrosylation. Cardiovasc. Res. 110, 96–106. ( 10.1093/cvr/cvw037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koppenol WH, Bounds PL. In press. Signaling by sulfur-containing molecules. Quantitative aspects. Arch. Biochem. Biophys. ( 10.1016/j.abb.2016.09.012) [DOI] [PubMed] [Google Scholar]
- 61.Ivanova LV, Anton BJ, Timerghazin QK. 2014. On the possible biological relevance of HSNO isomers: a computational investigation. Phys. Chem. Chem. Phys. 16, 8476–8486. ( 10.1039/C4CP00469H) [DOI] [PubMed] [Google Scholar]
- 62.Koppenol W. 2012. Nitrosation, thiols, and hemoglobin: energetics and kinetics. Inorg. Chem. 51, 5637–5641. ( 10.1021/ic202561f) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cuevasanta E, Denicola A, Alvarez B, Moller MN. 2012. Solubility and permeation of hydrogen sulfide in lipid membranes. PLoS ONE 7, e34562 ( 10.1371/journal.pone.0034562) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berenyiova A, et al. 2015. The reaction products of sulfide and S-nitrosoglutathione are potent vasorelaxants. Nitric Oxide 46, 123–130. ( 10.1016/j.niox.2014.12.008) [DOI] [PubMed] [Google Scholar]
- 65.Kabil O, Banerjee R. 2014. Enzymology of H2S biogenesis, decay and signaling. Antioxidants Redox Signal. 20, 770–782. ( 10.1089/ars.2013.5339) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Olson KR. 2012. A practical look at the chemistry and biology of hydrogen sulfide. Antioxidants Redox Signal. 17, 32–44. ( 10.1089/ars.2011.4401) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giggenbach W. 1972. Optical spectra and equilibrium distribution of polysulfide ions in aqueous solution at 20°. Inorg. Chem. 11, 1201–1207. ( 10.1021/ic50112a009) [DOI] [Google Scholar]
- 68.Schwarzenbach G, Fischer A. 1960. Die aciditaet der sulfane und die zusammensetzung waesseriger polysulfidloesungen. Helvetica Chim. Acta. 169, 1365–1390. ( 10.1002/hlca.19600430521) [DOI] [Google Scholar]
- 69.Talipov MR, Timerghazin QK. 2013. Protein control of S-nitrosothiol reactivity: interplay of antagonistic resonance structures. J. Phys. Chem. B 117, 1827–1837. ( 10.1021/jp310664z) [DOI] [PubMed] [Google Scholar]