

SUMMARY
Regulated proteolysis is essential for the normal physiology of all organisms. While all eukaryotes and archaea use proteasomes for protein degradation, only certain orders of bacteria have proteasomes, whose functions are likely as diverse as the species that use them. In this review, we discuss the most recent developments in the understanding of how proteins are targeted to proteasomes for degradation, including ATP-dependent and -independent mechanisms, and the roles of proteasome-dependent degradation in protein quality control and the regulation of cellular physiology. Furthermore, we explore newly established functions of proteasome system accessory factors that function independently of proteolysis.
KEYWORD: proteasome
INTRODUCTION
Proteolysis is a fundamental process in all cells, playing essential roles in posttranslational regulation, responses to environmental changes and intracellular stress, and removal of unfolded proteins. The proteasome, a proteolytic machine present in all three domains of life, is a barrel-shaped protease complex that performs both ATP-dependent and -independent proteolysis (reviewed in reference 1). In many cases, proteins are targeted to the proteasome through specific posttranslational modifications; in eukaryotes, a small protein called ubiquitin (Ub) tags proteins for ATP-dependent proteasomal degradation. Three classes of enzymes carry out ubiquitylation: a Ub-activating enzyme (E1) uses ATP to adenylate the C-terminal glycine (Gly) of Ub, priming it for attack by a cysteine (Cys) in the E1 enzyme to form a Ub-E1 thioester bond. Ub is then transferred to a Ub-conjugating enzyme (E2), which allows a Ub ligase (E3) to catalyze the formation of an isopeptide bond between the Ub C terminus and a substrate lysine (Lys) residue. Substrates are selected through recognition by dedicated E3 ligases, of which there are up to several hundred representatives in a given eukaryotic genome (reviewed in reference 2). As the major pathway of ATP-dependent proteolysis in the eukaryotic cytosol and nucleus, the Ub-proteasome system is essential for eukaryotes and is involved in numerous cellular processes (reviewed in reference 3).
The protease component of all proteasomes, called the 20S core particle (20S CP) based on its sedimentation coefficient, is gated to prevent nonspecific entry of proteins; substrates must be fed into the proteolytic core by a proteasome activator (4–6). In eukaryotes, the 19S regulatory particle (RP) caps the 20S CP, where it functions to both recognize and unfold ubiquitylated proteins prior to degradation (7–10). Additionally, the 19S RP removes Ub from substrates as a recycling mechanism (11, 12). While the 19S RP uses ATP hydrolysis to provide energy for protein unfolding, several ATP-independent proteasome activators have also been described for eukaryotes (13, 14), although their substrate specificities and biological functions are poorly understood.
Proteasomal genes are represented in nearly every lineage of the bacterial orders Actinomycetales and Nitrospirales, whose members include the human pathogen Mycobacterium tuberculosis and a major source of natural antibiotics, Streptomyces coelicolor (reviewed in reference 15). Although proteasomes perform critical physiological functions in all proteasome-bearing bacteria characterized to date, they are not absolutely essential as in eukaryotes. To target proteins to a proteasome, bacteria employ a mechanism similar to ubiquitylation, called pupylation. Several roles for pupylation in bacterial physiology have been identified, and most notably, the degradation of pupylated substrates is required for the full virulence of M. tuberculosis. Beyond pupylation, the study of bacterial proteasomes has also revealed the importance of ATP-independent proteasomal degradation for the normal physiology of M. tuberculosis, and possibly for other species.
20S PROTEASOME CORE PARTICLES
Structure and Assembly
In all domains of life, 20S CPs are 28-subunit complexes consisting of four stacked rings (16–19) (Fig. 1A). Two identical outer rings, each constituting a heptamer of α-subunits, provide the entry point at which substrates are delivered. Likewise, two identical inner rings, each a heptamer of β-subunits, form the active site of the protease (19–22). The β-subunits belong to the amino (N)-terminal nucleophilic hydrolase family of proteins (23). Notably, eukaryotic 20S CPs are formed from 14 unique gene products, while archaea and proteasome-bearing bacteria encode only a single type each of the α- and β-subunits (16, 17, 20, 24) (Fig. 1B). The only known exception to this is a single sequenced strain of Rhodococcus erythropolis (strain NI86/21) that encodes two functionally interchangeable variants of each subunit (20).
FIG 1.
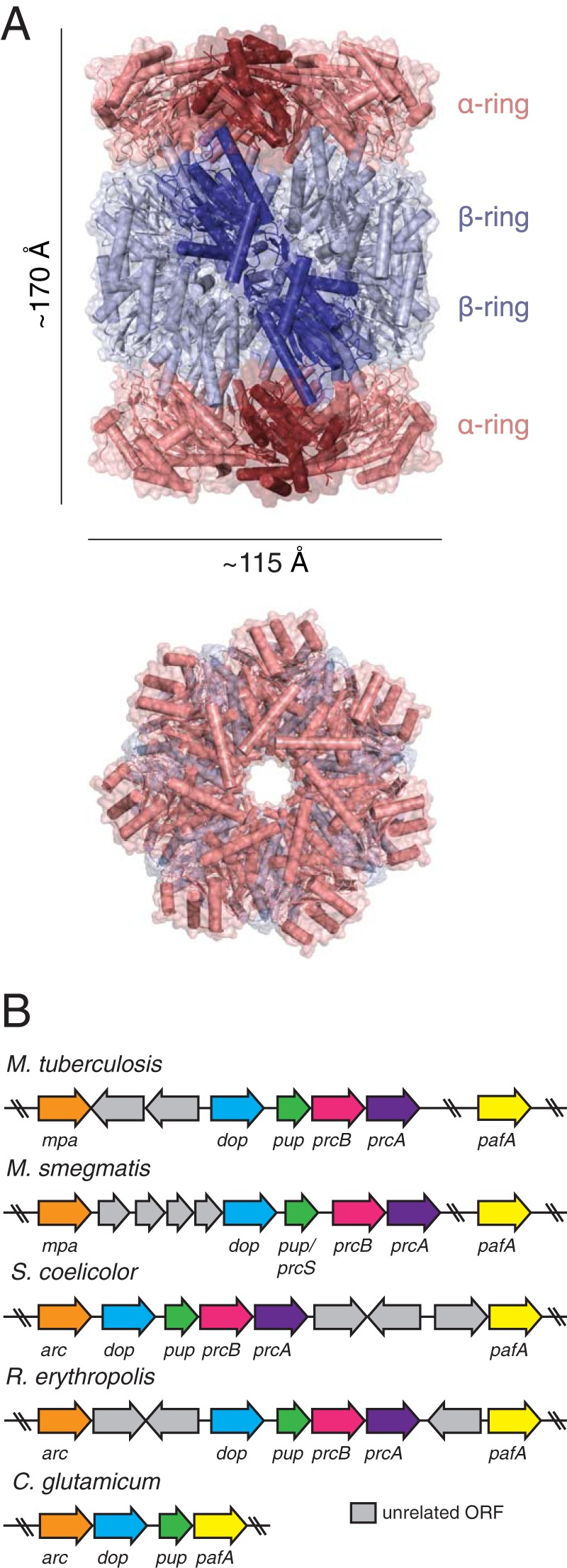
Structure of a bacterial proteasome and proteasome gene loci in select actinobacteria. (A) (Top) Crystal structure of the 20S CP from M. tuberculosis (PDB entry 2FHG). An individual subunit from each ring is shown in a darker tint. (Bottom) Top-down view of the 20S CP. (B) Proteasome gene loci in five representative actinobacterial species. C. glutamicum does not carry prcBA.
In bacteria, assembly of a 20S CP is preceded by the formation of a half-proteasome consisting of an α- and a β-ring (25). All β-subunits are produced with an N-terminal propeptide; autocatalytic processing of β-subunits proceeds upon β-ring-to-β-ring joining of two half-proteasomes (25, 26). Removal of the propeptides is catalyzed by β-subunit threonines (Thr), which become the N-terminal catalytic residues of the assembled holoproteasome. Initial insight into the function of propeptides was elucidated upon studies of 20S CP assembly in R. erythropolis, which encodes α- and β-subunits called PrcA and PrcB (proteasome components A and B), respectively. Production of a propeptideless PrcB subunit significantly lowers the rate of 20S CP assembly; furthermore, supplementation of PrcA and propeptideless PrcB with propeptides in trans results in functional proteasomes that assemble more rapidly than their wild-type counterparts (25). These observations suggest that in R. erythropolis, the propeptide promotes structural maturation of the proteasome. In contrast, the propeptide of M. tuberculosis PrcB is dispensable for the assembly of functional 20S CPs; in fact, production of PrcB lacking catalytic residues for processing halts assembly at the half-proteasome stage (24, 26). Because propeptide processing appears to be a thermodynamic barrier to M. tuberculosis 20S CP assembly (24), we speculate that the propeptide is conserved in M. tuberculosis PrcB in order to reduce unregulated peptidase activity that may have detrimental effects on bacterial physiology.
Catalytic Activity
The mechanism of proteolysis by 20S CPs appears to be universal among proteasomes (27). The β-subunit active site facilitates hydrolysis of both its own propeptide and protein substrates entering the proteasome. In the preprocessed state, Thr1 (numbered according to its position in the processed β-subunit) hydrolyzes the amide bond linking it to the adjacent −1 residue, thereby releasing the propeptide; Thr1 subsequently hydrolyzes substrates in an identical manner. Several conserved residues participate in this reaction: an aspartate (Asp) or glutamate (Glu) orients Thr1 toward the substrate, and a lysine (Lys) is responsible for deprotonating the terminal amine of Thr1 to carry out hydrolysis (19, 27, 28). Together, Thr, Lys, and Asp/Glu form a set of catalytic residues that is conserved in proteasomes from all domains of life. In mature bacterial proteasomes, all 14 β-subunits are processed and catalytically active (24); this is in contrast to the hetero-oligomeric proteasome of eukaryotes, in which only three of seven β-subunits harbor a functional catalytic Thr (16).
While the proteasome active site residues are universally conserved, the peptide sequences targeted for proteolysis are variable. Proteolytic specificity is determined by the chemical nature of the pocket surrounding the active site. In the 20S CPs of eukaryotes, the three active β-subunits each have a unique specificity for hydrophobic, basic, or acidic sequences (29). Because prokaryotes, for the most part, carry a single allele each for the α- and β-subunits, specificity is predictably more limited; indeed, 20S CPs in both R. erythropolis and Thermoplasma acidophilum have an overall hydrophobic central chamber and thus appear to selectively process hydrophobic substrates (19, 20). Remarkably, however, the 20S CP of M. tuberculosis has a highly unique active site pocket with both hydrophobic and hydrophilic surfaces (19). This affords the M. tuberculosis proteasome the ability to efficiently degrade all three peptide sequence types (24).
Gating
The α-subunits of 20S CPs harbor an N-terminal extension that prevents substrates, including most peptides, from freely entering the central protease chamber (4, 24, 26). In M. tuberculosis PrcA, this extension consists of several hydrophobic residues, and the N termini of PrcA monomers stack atop one another to occlude the CP entrance (24). This structure is capable of a wide range of movement, which is a requisite for the large positional shifts it must undergo upon binding to proteasome activators that allow the entry of proteins into a 20S CP (26). Many studies using N-terminally truncated α-subunits, which result in the assembly of so-called “open gate” proteasomes, have demonstrated that the absence of a gate allows for the degradation of peptides and unfolded proteins without the need for a proteasome activator (4, 24, 30). Moreover, activators appear to associate more strongly with 20S CPs in the absence of a gate (31, 32), altogether indicating that the gate acts as a barrier to substrate translocation.
ATP-DEPENDENT PROTEASOME ACTIVATION
Proteasomal ATPases
ATPase proteasome activators are present in all domains of life. In bacteria, they perform three essential functions: specific recognition of substrates, ATPase-driven unfolding, and engagement with 20S CPs to induce gate opening. The two best-characterized bacterial ATPase activators are M. tuberculosis Mpa (mycobacterial proteasome ATPase) and its orthologue, R. erythropolis ARC (AAA ATPase forming ring-shaped complexes). Mpa and ARC form homohexameric rings, highly similar to the previously characterized archaeal proteasome activator PAN (proteasome-activating nucleotidase) (32–34) (Fig. 2A). The eukaryotic 19S RP, which consists of at least 19 different gene products, includes a base subcomplex that is structurally and functionally analogous to the prokaryotic ATPase activators (10, 35). It therefore appears that the general features of ATP-dependent delivery of proteins into a 20S CP are evolutionarily conserved from bacteria to eukaryotes.
FIG 2.
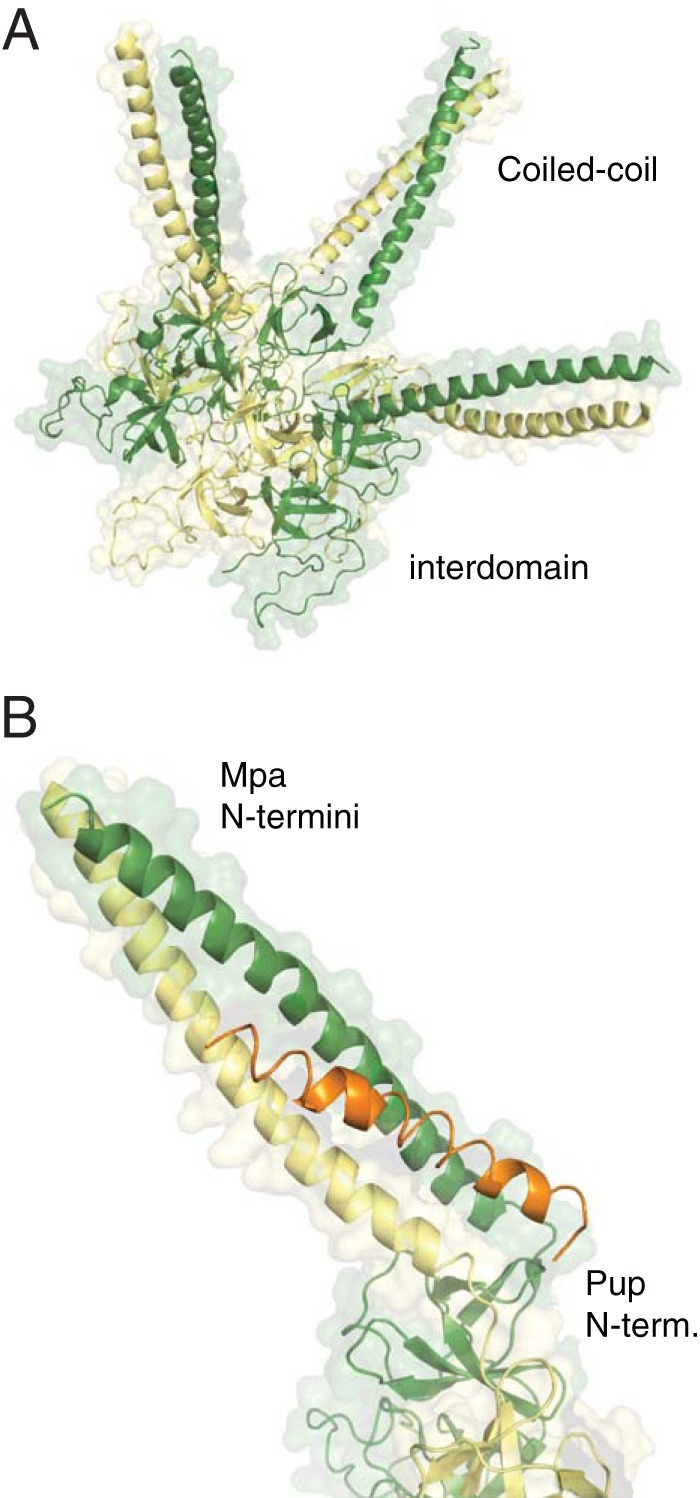
ATP-dependent degradation requires a hexameric chaperone. (A) Crystal structure of a hexamer composed of Mpa1–234 monomers from M. tuberculosis (PDB entry 3M9D). Shading is used to mark individual subunits; two domains of Mpa are indicated. (B) Crystal structure of an Mpa coiled-coil domain (green shades) bound to Pup21–64 (orange), demonstrating binding-induced folding of Pup by Mpa.
Mpa and ARC consist of at least the following three domains that, in an assembled hexamer, form a channel: an N-terminal coiled-coil domain, an interdomain, and an ATPase domain (32, 34). Atop the entrance to the channel, the N-terminal helices of each monomer pair up to form three coiled coils, which extend from the hexamer (32, 34) (Fig. 2A). In Mpa, the coiled-coil domain is responsible for substrate recognition (32). In the absence of comprehensive studies, little is known about the substrate selection of ARC or other bacterial proteasome activators, but considering that the coiled-coil domain is a conserved feature of proteasomal ATPases and is absent from nonproteasomal protease activators (reviewed in reference 36), it is likely that this domain is required for substrate recognition in most or all other bacterial proteasomal ATPases.
The interdomain of Mpa and ARC harbors two oligosaccharide/oligonucleotide-binding-fold (OB) subdomains. Despite this connotation, the only known function of the interdomain is to promote hexamer assembly and stability. In accordance with the high degree of structural rigidity and intersubunit contacts found within the OB subdomains, the interdomain can uniquely self-assemble (32, 37).
Mpa and ARC are representative of a class of enzymes called AAA ATPases (ATPases associated with various cellular activities) (38, 39). The AAA domain powers substrate delivery into the 20S CP. The initial characterization of the role of AAA ATPases in protein degradation was performed on the ATPases of nonproteasomal bacterial proteases, such as ClpP and HslV (ClpQ) (reviewed in references 40 and 41). The study of both nonproteasomal and proteasomal activators has proven to be mutually beneficial, as these are thought to carry out highly similar mechanisms of protein unfolding and translocation. The defining feature of AAA ATPase unfoldases is a conserved aromatic-hydrophobic-glycine (Ar-Φ) motif, commonly referred to as the pore loop, which extends into the channel of the ATPase and makes direct contact with substrates (31, 32, 38, 42). Through mechanistic studies of ClpX, an AAA ATPase activator of ClpP, a model has been proposed whereby individual pore loops “grab” an exposed peptide and pull the substrate in the direction of the protease opening, an action driven by ATP hydrolysis (42). Furthermore, a recent investigation into PAN and the 19S RP put forth a mechanism by which ATP hydrolysis (and thus pore loop mobilization) cycles in a counterclockwise fashion to facilitate the sequential grabbing and pulling of polypeptides (43). This is thought to be a mechanism utilized by all degradation-associated ATPases; indeed, Mpa mutants lacking conserved pore loop residues are unable either to unfold proteins or to facilitate protein degradation in M. tuberculosis (31, 32).
As discussed previously, 20S CPs have gated entries, formed by N-terminal helices extending from each subunit of the α-subunit ring, which must be opened to allow for proteolysis. To accomplish this, PAN and several eukaryotic activators have C-terminal extensions that are required to open proteasome gates in order to facilitate proteolysis (5, 6, 33). The C-terminal three amino acids in these activators are a hydrophobic amino acid, tyrosine, and any amino acid (HbYX motif) (6). Structural and biochemical studies have demonstrated that these C-terminal residues insert into grooves between α-subunits, inducing rigid-body movements that widen the 20S CP entrance (5, 6). Although such scrutinizing experiments have not been performed specifically with bacterial proteasome activators, it is nonetheless reasonable to hypothesize that gate opening in mycobacteria occurs through a similar mechanism. In support of this hypothesis, Mpa/ARC hexamers also have C-terminal extensions, ending with the sequence glycine-glutamine-tyrosine-leucine (GQYL), which are required for engaging 20S CPs (44–46). By sequence and location, the GQYL motif is similar to the HbYX motif; indeed, truncation or amino acid substitutions within the GQYL sequence render Mpa nonfunctional in M. tuberculosis (45, 47–49). Furthermore, M. tuberculosis PrcA has a Lys residue that is highly conserved among proteasomes, and this residue in other proteasomes allows the HbYX motif of a proteasome activator to open the 20S CP (44). Collectively, these data suggest that the GQYL motif inserts between α-subunits in order to open the M. tuberculosis 20S CP gate.
Despite harboring a C-terminal GQYL motif, Mpa is unable to activate the proteolysis of even small peptides by wild-type 20S CPs in vitro. Furthermore, several reports have demonstrated that the degradation of substrates by use of Mpa and 20S CPs in vitro is much slower than the degradation observed in bacteria, a phenomenon that has long confounded the field (31, 50, 51). The failure of Mpa to facilitate robust in vitro degradation is likely explained by the fact that Mpa has very poor in vitro affinity for the 20S CP (32); however, the structural explanation for this phenomenon has yet to be defined.
Pupylation Targets Proteins for ATP-Dependent Degradation
To begin to understand how doomed proteins are targeted to a bacterial proteasome, Pearce et al. performed a genetic screen to identify proteins that interact with Mpa; this led to the discovery of prokaryotic ubiquitin-like protein (Pup). Pup covalently links to several known proteasome substrates in mycobacteria, and this “pupylation” is essential for their degradation (51, 52). Around the same time, the authors of another study noted that a gene, orf7, found directly upstream of the 20S CP genes of mycobacteria and other members of the Actinomycetes (21), was predicted to encode a small protein with a diglycine motif near its C terminus, a feature found in Ub. Hypothesizing that this gene might encode a degradation tag, an epitope-tagged Orf7 protein (later named Pup) was produced in Mycobacterium smegmatis, resulting in the visualization of pupylation of numerous proteins (53). The “pupylomes” of M. smegmatis and M. tuberculosis include at least 50 proteins in which a pupylated site has been identified (52, 54, 55).
Pup is ligated to substrates via a covalent linkage between its C-terminal residue and a substrate Lys residue (51, 53, 56). This is comparable to the linkage of Ub, whose C terminus also attaches to Lys. Remarkably, however, Pup itself has no structural similarity to Ub: rather than harboring the β-grasp fold characteristic of Ub and its related modifiers, free Pup is mostly unstructured (57, 58).
In mycobacteria, Pup is produced with a C-terminal glutamine (Gln), yet mass spectrometry and nuclear magnetic resonance (NMR) studies revealed that Pup attaches to a substrate via a C-terminal glutamate (Glu), which is suggestive of a deamidation event on the Gln side chain (51, 53, 56). It was discovered that two enzymes, Dop (deamidase of Pup) and PafA (proteasome accessory factor A), are required to carry out the pupylation reaction. Dop catalyzes the ATP-dependent deamidation of the C-terminal Gln of Pup, generating PupGlu (59, 60); PafA subsequently phosphorylates the C-terminal Glu, rendering it available for nucleophilic attack by the ε-amino group of a specific Lys residue in a substrate protein (59, 61) (Fig. 3A and B). Reconstitution of the mycobacterial pupylation system in Escherichia coli, which lacks pupylation, demonstrated that Dop and PafA are necessary and sufficient for pupylation of known mycobacterial substrates as well as E. coli proteins (62). Notably, the requirement of Dop for pupylation can be bypassed upon production of PupGlu, a variant found in several actinobacteria, including S. coelicolor (60–63).
FIG 3.

ATP-dependent degradation requires pupylation. (A) From left to right, Pup is deamidated at its C terminus by Dop, producing PupGlu; PafA ligates PupGlu to a substrate; and a pupylated substrate can either be depupylated by Dop or be degraded by the proteasome. (B) Proposed mechanisms of deamidation and pupylation. Starting from top left, the Pup C-terminal Gln is attacked by a nucleophilic Asp in Dop, forming a Pup-Dop intermediate; Pup undergoes a nucleophilic attack by water, activated by Dop, to yield PupGlu; PafA uses ATP to phosphorylate the γ-carboxylate of PupGlu; and nucleophilic attack by a substrate Lys residue results in the covalent linkage of Pup to a substrate. Another mechanism of Pup deamidation has been proposed, in which the Dop Asp residue coordinates a water molecule and nucleophilic attack by this activated water results in deamidation (66). (C) Depupylation of a pupylated substrate in M. smegmatis requires an intact Pup N terminus as well as Mpa. The top labels indicate the M. smegmatis strain background, as follows: WT, wild-type strain; ΔprcBA, deletion mutant lacking the prcBA genes; and mpa, a strain in which the gene has been disrupted with an integrated plasmid. Lanes: vec., empty vector; Pup, overproduced full-length Pup; Pup91, overproduced truncated Pup missing 30 N-terminal amino acids. Immunoblotting (IB) was performed on cell lysates from the indicated strains by using an antibody to M. tuberculosis Ino1 (inositol-1-phosphate synthetase), a known pupylated substrate. Bands corresponding to Ino1 and its Pup or Pup91 conjugate are indicated at the right. (Reprinted from reference 50 with permission from Elsevier.)
While bacterial 20S CPs and ATP-dependent proteasome activators are homologous to their eukaryotic counterparts, Dop and PafA are completely unrelated to the E1, E2, and E3 enzymes required for the attachment of Ub to proteins in eukaryotes. Dop and PafA are in the glutamine synthetase (GS) family of proteins, whose members carry out condensation reactions between Glu and amino groups (61, 64). GS proteins primarily use ATP to phosphorylate the γ-carboxylate of Glu, preparing it for attack by an amino group, as is the case with PafA (65). In contrast, Dop performs deamidation of Pup without ATP hydrolysis, rather using ATP as a cofactor (59). Because Dop and PafA share high sequence and structural identities (64, 66), one might hypothesize that one of the two proteins arose from a genetic duplication event in order to provide a second activity in the pupylation pathway.
Efforts to define the regions of Pup that are recognized by Dop and PafA have established that the C-terminal half of Pup is sufficient for deamidation and substrate ligation (67, 68). The crystal structure of PafA bound to the C-terminal fragment of Pup revealed that PafA binding causes Pup to adopt two short, helical folds, allowing it to be positioned correctly within the ligase active site (69). As Dop harbors the same Pup-stabilizing pocket, it is predicted to associate with Pup in a similar manner. In line with this observation, the Pup C terminus is highly conserved, while the N-terminal half of Pup is variable among bacterial genera and species (58).
Another major difference between the Ub and Pup systems is that while polyubiquitylation is generally required for substrate recognition and degradation in eukaryotes (70), polypupylation has been observed only in vitro or in bacteria overproducing Pup (71–73). While we cannot rule out the occurrence of polypupylation in a natural setting, the fusion of a single Pup to a protein is sufficient to target it for efficient proteasomal degradation (68). Thus, it is generally accepted that a single Pup is necessary and sufficient to doom a protein to a bacterial proteasome.
While a consensus pupylation motif has not been identified in substrates, a comparison of the pupylation of free Lys versus that of a known mycobacterial substrate, PanB (ketopantenoate hydroxymethyltransferase) (49), demonstrated that while both occur through the same reaction, PanB pupylation proceeds more quickly (65). Moreover, PafA has an affinity for PanB that is approximately 3 orders of magnitude higher than that for free lysine, suggesting that intrinsic features of substrate proteins significantly influence their pupylation by PafA (65). Additionally, once a protein is pupylated, it appears to have a weaker affinity for PafA, suggesting that it will not be subjected to additional rounds of pupylation (71).
Pup Interacts with Mpa/ARC To Target Substrates for Proteolysis
In mycobacteria, substrates are targeted to the proteasome through the interaction of Pup with the N-terminal coiled-coil domains of Mpa (31, 37). The crystal structure of the Pup-Mpa complex revealed that, similar to that of PafA and Dop, Mpa binding causes Pup to adopt a partially helical structure, which in this case orients the N terminus toward the ATPase channel (Fig. 2B). Mutations that disrupt Pup-Mpa helical interactions abolish degradation of a model substrate in M. tuberculosis (37).
Numerous studies collectively suggest that the flexible nature of Pup allows it to be manipulated by Mpa in order to initiate the process of substrate unfolding. Using in-cell NMR analysis, the disordered N-terminal region of Pup can be observed within the central channel of Mpa when in complex with a 20S CP (74). Because the N terminus of Pup is required both for unfolding by Mpa and for substrate degradation (31, 60), it is hypothesized that headfirst translocation of Pup through the ATPase channel of Mpa is required for degradation of pupylated substrates. It is not known how Mpa interacts with substrates themselves once Pup is pulled through the hexameric channel. In the case of E. coli ClpXP and other proteases, association of substrates with the protease complex is enhanced by adaptors that bind to both the protease and the degradation tag (reviewed in reference 41); it is possible that similar adaptors function for Mpa.
Dop Is a Depupylase
There have been several observations suggesting that Dop might have a function beyond deamidation of Pup. First, several bacterial species containing Dop encode PupGlu rather than PupGln, thus obviating the need for a deamidase. Second, evidence for the existence of depupylation emerged during the phenotypic characterization of an M. tuberculosis dop mutant. An attempt was made to rescue pupylation in a dop mutant by ectopically expressing pupGlu; however, the amount of pupylated proteins was only partially restored, and free Pup was undetectable. It was proposed that pupylation had been rescued but that the vast majority of Pup was degraded along with substrates, suggesting a possible requirement of Dop for the removal and recycling of Pup prior to proteasomal degradation. To test this hypothesis, the strain was treated with a proteasome inhibitor, which restored the pupylome (61). It was subsequently determined that Dop indeed catalyzes the removal of Pup from pupylated substrates (50, 75).
The mechanisms of deamidation and depupylation by Dop are proposed to be similar; both processes involve the hydrolysis of an amide bond at the C terminus of Pup (66, 76). The use of a molecular trap to capture a Dop-Pup intermediate revealed that an aspartate (Asp) in the Dop active site could be a direct nucleophile. Based on these studies, it is proposed that the Asp residue in Dop attacks the side chain amide of the Pup C-terminal Gln residue, causing the exit of ammonia (in the case of deamidation) (Fig. 3B) or the substrate Lys residue (in the case of depupylation) (76).
Additional observations led to the conclusion that Mpa participates in depupylation. In wild-type M. smegmatis, overproduction of Pup results in the disappearance of the proteasome substrate Ino1 (inositol-1-phosphate synthetase), likely driven by increased pupylation; in contrast, overproduction of Pup in an mpa mutant results in the robust accumulation of pupylated Ino1 (Pup∼Ino1) (Fig. 3C). This was surprising considering that in a strain lacking 20S CP genes, only Ino1, not Pup∼Ino1, is detectable. These data suggest that, in bacteria, depupylation occurs robustly only in the presence of Mpa. Supporting this hypothesis, production of an N-terminally truncated Pup variant (′Pup) which lacks several key Mpa-interacting residues results in the accumulation of ′Pup∼Ino1 in both wild-type and mpa mutant M. smegmatis strains (50) (Fig. 3C). Taken together, these observations strongly suggest that in the absence of proteasome activity, substrates are depupylated rather than maintained in a pupylated state. Furthermore, depupylation by Dop appears to require the N-terminal half of Pup as well as Mpa in vitro and in vivo (Fig. 3C) (75).
The apparent necessity of Mpa for efficient depupylation might be explained by the ability of Mpa to partially unfold Pup or its bound substrate, which may allow easier access of Dop to the isopeptide bond formed between Pup and its substrate. Notably, another study demonstrated that Mpa enhances the in vitro pupylation activity of PafA, with Mpa, Pup, and PafA forming a ternary complex (77); Dop may likewise form such a complex, as it is structurally similar to PafA. Further studies are needed to define the mechanism by which Mpa facilitates depupylation.
In contrast to the proven necessity for Ub recycling in eukaryotes (11), the importance of Pup recycling in bacteria is unclear. Unlike in M. tuberculosis, ectopic expression of pupGlu in an M. smegmatis dop mutant appears to fully restore the pupylome (60, 61). Additionally, an M. tuberculosis dop mutant expressing pupGlu grows normally (our unpublished data). However, there may be certain conditions in which depupylation confers an overall advantage to the bacterium. It remains to be determined whether Pup recycling occurs or is important in other bacterial species.
The current model of substrate degradation indicates that the N terminus of Pup is threaded through the Mpa pore (31). What is unclear is whether or not Pup is then threaded into the 20S CP and how Dop fits into this picture. A possibility is that Dop, with assistance from Mpa, depupylates substrates independently of proteasome engagement; this is supported by the observation that Pup∼Ino1 is undetectable in an M. smegmatis 20S CP mutant (50) (Fig. 3C). However, because Mpa is itself a proteasome substrate (49), it is possible that this result is artificial: if Mpa facilitates depupylation, then the accumulation of Mpa in a 20S CP mutant may result in hyperdepupylation regardless of proteasome association. Further experiments are needed to determine if depupylation is coupled to proteasomal degradation.
Degradation-Independent Functions of Pupylation
In several bacterial species, pupylation components are not always encoded by 20S CP subunit genes. One such example is Corynebacterium glutamicum, a relative of mycobacteria, which expresses pupGlu, pafA, arc, and dop but lacks prcBA (Fig. 1B) (78). This bacterium has generated interest in the hope of establishing a degradation-independent role for Pup. A recent study of C. glutamicum found that ferritin, a protein that assembles into multimeric complexes to sequester ferrous iron (Fe2+), is a pupylation substrate; engagement of pupylated ferritin by ARC causes complex disassembly, presumably to release Fe2+. It is hypothesized that ferritin disassembly by pupylation and ARC unfolding is a requisite for survival during iron starvation, as a Pup-deficient mutant of C. glutamicum grows poorly under Fe-depleted conditions (79).
Interestingly, the ferritin homologue BfrB in M. smegmatis is also pupylated, and it is potentially pupylated in M. tuberculosis (52, 55). While it is unknown if prcBA, mpa, and pafA mutants are more sensitive to Fe limitation, M. tuberculosis mutants are more resistant to peroxide, for reasons that have never been determined (47, 80). It is possible that M. tuberculosis mutants that cannot effectively degrade or disassemble ferritin complexes have lower levels of exchangeable Fe2+, which could otherwise catalyze Fenton chemistry with peroxide to generate highly toxic reactive oxygen species (81). Supporting the notion that BfrB levels contribute to peroxide sensitivity, an M. tuberculosis bfrB null mutant is more sensitive to Fe-generated oxidative stress (82).
S. coelicolor is a relative of Mycobacterium spp. that has a Pup-proteasome system and a sizeable pupylome (21, 63). Although proteasomal degradation appears to be functional in this species, several phenotypes have been identified for S. coelicolor that depend exclusively on pupylation rather than degradation. S. coelicolor pup and pafA mutants, but not 20S CP mutants, cannot properly sporulate, and they undergo changes in the production of several secondary metabolites (63, 83). It is unknown how pupylation regulates these processes.
It is notable that several proteins that were identified as confirmed or putative pupylated substrates in M. tuberculosis do not accumulate in mpa or pafA mutants under routine culture conditions (52). While it is plausible that these proteins are not targeted to the 20S CP for degradation, it is also possible that these proteins are degraded only under specific conditions that remain to be determined. Thus, further work is needed to establish whether pupylation has functions independent of proteolysis in mycobacteria and other proteasome-bearing bacteria.
ATP-INDEPENDENT PROTEASOMAL ACTIVATION
In M. tuberculosis, disruption of mpa results in only a slight growth defect in culture; however, chemical inhibition of proteasome activity or deletion of prcBA results in marked growth defects under all conditions tested (47, 80). This observation suggests that the proteasome provides functions that are independent of pupylation. To identify new proteins that interact with the M. tuberculosis proteasome, a catalytically inactive 20S CP was used to “trap” potential proteasome cofactors. This approach resulted in the discovery of a novel, non-ATPase proteasome activator, proteasome accessory factor E (PafE) (45), also known as Bpa (bacterial proteasome activator) (46), that is not genetically linked to the Pup proteasomal genes (pafE is annotated Rv3780 for M. tuberculosis). PafE assembles into an unprecedented dodecameric ring with 12-fold symmetry (Fig. 4), though monomers within the ring adopt helical structures similar to those of the eukaryotic ATP-independent 11S activators PA26 and PA28, which form heptamers (44). Notably, PafE, like Mpa, contains a C-terminal GQYL motif required for 20S CP gate opening. This feature also allowed for the independent identification of PafE by a homology search (46). Both studies found that PafE could stimulate the degradation of short peptides and a model unfolded protein (45, 46).
FIG 4.

PafE/Bpa is an ATP-independent proteasome activator. (Top) Crystal structure of dodecameric PafE/Bpa from M. tuberculosis (PDB entry 5IET). Dashed lines approximate the extended C termini of PafE in contact with the 20S CP α-ring. (Bottom) Top-down view of PafE.
Rather than acting as an unfoldase to deliver proteins into a 20S CP, PafE seems to function simply as a gate opener. In line with this, the PafE-proteasome efficiently degrades unfolded proteins in vitro without ATP (45, 46). The PafE ring is lined with hydrophobic residues that may allow unfolded proteins, which tend to expose normally buried hydrophobic amino acids, to slip into the 20S CP (44). Furthermore, proteomic analysis of a pafE mutant identified an inherently unstable transcriptional repressor, HspR (heat shock protein repressor), as a PafE-proteasome substrate. Remarkably, native HspR can be degraded robustly by PafE and 20S CPs in the absence of ATP; this experiment presented the first example of rapid and robust degradation of an intrinsic substrate by an intact prokaryotic proteasome in vitro. Because recombinant HspR produced in a heterologous system (E. coli) can be degraded efficiently in vitro, it does not appear that a mycobacterium-specific posttranslational modification is required for degradation by the PafE-proteasome; this suggests that PafE-proteasome substrates have inherent features that target them for degradation (45). The identification of additional PafE-proteasome substrates should ultimately reveal features in common among these proteins, which may indicate the presence of a PafE-dependent degron.
CONTRIBUTIONS OF PROTEASOMES TO BACTERIAL PHYSIOLOGY
Proteasomes degrade potentially dozens, if not hundreds, of different bacterial proteins and are thus essential for the normal physiology of all proteasome-bearing bacteria studied to date. Here we focus on the pathways that have so far been determined to be affected either directly or indirectly by proteasomal degradation in both nonpathogenic and pathogenic bacterial species.
Nitric Oxide Resistance
The study that reinvigorated the bacterial proteasome field described a screen for M. tuberculosis mutants hypersensitive to nitric oxide (NO). Nathan and colleagues discovered that Mpa and PafA, which were uncharacterized at the time, are essential for NO resistance in M. tuberculosis (47). This and other studies further demonstrated that NO resistance is substantially dependent on proteasomal degradation (47, 80, 84). M. tuberculosis is the causative agent of the human disease tuberculosis, which is currently the global leading cause of death by a single infectious agent (85). M. tuberculosis is contracted by the inhalation of aerosolized droplets released from an infected individual by coughing or sneezing. In the lungs, M. tuberculosis is phagocytosed by macrophages and dendritic cells, which are sentinels of the immune system (reviewed in reference 86). An important mechanism by which phagocytes control pathogen growth is through the production of NO by inducible nitric oxide synthase (iNOS or NOS2) (87). The importance of NO for tuberculosis has been demonstrated most clearly in iNOS-deficient mice, which succumb rapidly to M. tuberculosis infection (88). However, despite its obligate residence within macrophages, M. tuberculosis persists, avoiding clearance, often for the lifetime of the host. The identification of proteasome activity in M. tuberculosis as a protective factor against NO toxicity suggested that proteolysis of one or more proteins serves to defend this bacterium against host immunity.
NO can damage proteins, lipids, and nucleic acids to inhibit microbial growth (reviewed in reference 89). While it was often presumed that the Pup-proteasome system was needed to degrade NO-damaged proteins, this was never demonstrated. Furthermore, as previously mentioned, mpa and pafA mutants are more resistant to peroxide stress and have wild-type resistance to heat shock, suggesting that the Pup-proteasome system is not required for a general proteotoxic stress response (47, 80). It would be over a decade before the mechanism of Pup-proteasome-mediated NO protection was elucidated. A screen for suppressor mutants of the NO hypersensitivity of an mpa mutant identified a new Pup-proteasome substrate, Log (lonely guy), whose accumulation renders bacteria hypersensitive to NO (90). Log is an orthologue of a plant enzyme, LONELY GUY, which catalyzes the production of adenine-based hormones called cytokinins (91). Intriguingly, Samanovic et al. determined that M. tuberculosis secretes cytokinins and that cytokinin production is significantly increased in an mpa strain (90). Cytokinins can be broken down into toxic aldehydes (92); indeed, at least one aldehyde, para-hydroxybenzaldehyde, accumulates in an mpa mutant. Addition of this or another aldehyde, 2-methyl-3-butanal, along with NO effectively kills wild-type M. tuberculosis. Because aldehydes and NO are unlikely to react with each other, it is proposed that aldehyde-induced cellular damage sensitizes M. tuberculosis to nitrosative stress through a mechanism that has yet to be elucidated (Fig. 5A). Notably, disruption of Log leads to a partial restoration of virulence in an mpa mutant, indicating that NO sensitivity resulting from cytokinin accumulation is a significant but not exclusive cause of the attenuated in vivo phenotype of this strain (90).
FIG 5.

Contributions of proteasomes to the physiology of M. tuberculosis. (A) Proteasomal degradation of the cytokinin synthase Log keeps levels of cytokinins and aldehydes low. Accumulation of Log in a Pup-proteasome mutant causes increased cytokinin production. Cytokinins break down into aldehydes, which cause cytotoxicity in the presence of NO. (B) In wild-type bacteria, the transcriptional repressor RicR binds Cu+, causing its dissociation from promoters controlling the expression of Cu homeostasis genes at five loci on the M. tuberculosis chromosome. In the absence of proteasomal degradation, RicR appears to sense low intracellular Cu levels and binds to DNA, repressing expression of these genes. In the presence of elevated Cu levels, the RicR regulon may not be induced fully in a Pup-proteasome mutant. It is unclear if it is the weak induction of this regulon or another unidentified pathway that results in the hypersensitivity of Pup-proteasome system mutants to Cu. (C) The heat shock-responsive transcriptional repressor HspR, which controls expression of the protein chaperone-encoding genes dnaK, clpB, and hsp, is degraded by the PafE-proteasome. The abundance of misfolded proteins is kept low by DnaK, ClpB, and Hsp, and possibly by direct degradation by the PafE-proteasome. When degradation is disrupted, it is presumed that the failure to degrade HspR causes incomplete derepression of chaperone genes, leading to an accumulation of toxic unfolded proteins. See the text for proteasome-regulated pathways in other bacterial species.
It is notable that treatment of an mpa mutant with a proteasome protease inhibitor can exacerbate the NO-sensitive phenotype of the strain (47). Barring off-target effects of the proteasome inhibitor, this suggests that there may be additional, proteasome-dependent, Pup-independent mechanisms of NO resistance in M. tuberculosis.
Copper Resistance
A transcriptional analysis of genes misregulated in pafA and mpa mutants led to the discovery of a group of copper-inducible genes whose expression is controlled by a single transcriptional repressor, RicR (regulated in copper repressor), thus linking proteasome-dependent degradation to copper (Cu) homeostasis (93). Cu is both an essential nutrient and a potent source of toxicity; it is a cofactor for several enzymes involved in aerobic respiration and the oxidative stress response, but it can also be detrimental to cells by nonspecifically binding to and destabilizing proteins and may promote the generation of reactive oxygen species by Fenton-like chemistry (94). In wild-type bacteria, the RicR regulon is activated under conditions of elevated Cu, as Cu+ induces the dissociation of RicR from its respective promoters, thereby alleviating repression of gene expression (Fig. 5B). Several proteins encoded within the RicR regulon have been characterized functionally, including MmcO (mycobacterial multicopper oxidase), which oxidizes Cu+ to the less reactive species Cu2+ (95), and MymT (mycobacterial metallothionein), which sequesters Cu+ ions from other cellular components to mitigate toxicity (96). There are four additional genes of unknown function that are also regulated by RicR (Fig. 5B). While disruption or deletion of any single RicR-regulated gene does not affect the virulence of M. tuberculosis, simultaneous repression of the entire regulon leads to Cu hypersensitivity and attenuation in mice (97). Notably, only Mycobacterium species that infect mammals are known to have the RicR regulon in its entirety (93).
Where does M. tuberculosis, which is naturally found only in humans, encounter Cu? In macrophages, Cu is imported through a plasma membrane transporter, Ctr1 (reviewed in reference 98), while an intracellular Cu transporter, ATP7A, traffics Cu into intracellular vesicles (99). Ctr1 and ATP7A expression is induced during hypoxia (100, 101), a defining feature of M. tuberculosis lung infection; additionally, proinflammatory stimulation of macrophages induces localization of ATP7A to phagosomal vesicles, a mechanism required for antimicrobial activity in vitro (99). It has been proposed that the host Cu transport machinery is induced and mobilized during M. tuberculosis infections (reviewed in reference 102). Perhaps supporting this hypothesis, Niederweis and colleagues showed in a guinea pig infection model that Cu concentrations are significantly increased at sites of M. tuberculosis infection compared to healthy lung tissue and that supplementation of food with Cu promotes increased control of bacterial growth (103). Interestingly, M. tuberculosis-infected macrophages accumulate intraphagosomal copper to a degree that is not observed in cells infected with nonpathogenic mycobacteria (104).
The RicR regulon is repressed in pafA and mpa null mutants, and the induction of this regulon in response to Cu is impaired in these strains (93; our unpublished observations). Consistent with this observation, both pafA and mpa mutants are markedly more sensitive to Cu than wild-type M. tuberculosis (97), presumably due to a failure to fully induce expression of the RicR regulon. While the simplest model suggests that RicR itself is a Pup-proteasome substrate, RicR levels are not conspicuously different between wild-type and mpa strains (93). Importantly, we cannot rule out that there are RicR-independent reasons for the Cu hypersensitivity of Pup-proteasome mutants. Thus, a molecular link remains to be established between proteasomal degradation and Cu homeostasis.
Cu is somewhat unique among physiologically relevant metals in that free Cu is essentially nonexistent in cells; rather, it is passed between proteins or other small-molecule ligands (105–107). Because both the RicR regulon and other independently regulated Cu-inducible genes are repressed in Pup-proteasome strains (93, 108), it is possible that the accumulation of an unknown Cu-binding proteasome substrate results in a decrease in bioavailable Cu in the cytoplasm, which could explain the failure of RicR and other Cu-sensing repressors to disassociate from DNA. Furthermore, we cannot yet rule out that pupylation affects Cu homeostasis independently of proteasomal degradation, as described for iron homeostasis in C. glutamicum (79).
Metabolism
In eukaryotes, proteolysis is an essential source of amino acids for the synthesis of new proteins, and proteasome activity increases when amino acids are withheld from growth media (109–111). Several lines of evidence suggest that the same may be true for bacteria. In M. smegmatis, mutants in the Pup-proteasome system have survival defects in carbon- or nitrogen-limited media, and a model Pup-proteasome substrate in M. smegmatis is degraded more rapidly when bacteria are grown under nitrogen starvation than when they are grown in standard media (73). To explain how proteolysis for amino acid recycling might be regulated, Gur and colleagues suggested that in M. smegmatis, PafA preferentially pupylates high-molecular-weight proteins, while Dop preferentially depupylates low-molecular-weight proteins. This may constitute a mechanism whereby proteolysis for amino acid recycling is optimized: to obtain the same amount of recycled material, degradation of a few large proteins would be preferential to the degradation of many small proteins in order to avoid disruption of essential cellular systems (112). While this is a reasonable hypothesis, it remains unclear if the defective growth of M. smegmatis Pup-proteasome mutants during nutrient starvation is directly and only caused by a failure to recycle proteins or if other metabolic processes required for growth under starvation are regulated by proteasomal degradation. Along these lines, the Gur group found that complementation of a ΔprcSBA mutation (prcS is another name for pup) with prcS alone could restore part of the growth defect observed in this strain (73). In light of the observation that C. glutamicum uses pupylation to disassemble ferritin complexes under low-iron conditions (79), it is thus possible that pupylation, independently of degradation, regulates one or more pathways needed for growth of mycobacteria under nutrient-limiting conditions.
Deletion of pup in S. coelicolor results in drastic defects in cell morphology and division. Additionally, the production of specific antibiotics is abolished in this strain, demonstrating roles for pupylation in distinct cellular processes in this species (83). Notably, in contrast to the increased peroxide resistance of M. tuberculosis Pup-proteasome mutants, degradation-defective S. coelicolor is hypersensitive to peroxide (63, 83). Thus, defects in Pup-proteasome system components can result in markedly different phenotypes among bacterial genera.
Proteotoxic Stress Response
A role for ATP-independent proteasomal degradation was established with the identification of the M. tuberculosis PafE-proteasome substrate HspR (45). HspR is a repressor of several genes encoding proteins involved in the proteotoxic stress response, including the protein chaperones DnaK (Hsp70), ClpB, and Hsp (Acr2) (113). Based on the observation that this regulon is required for heat shock resistance, it was presumed that the failure to degrade HspR in a pafE mutant would contribute to increased heat sensitivity in this strain. Indeed, a pafE null mutant is hypersensitive to heat shock and is also slow to grow under normal conditions (45). HspR itself is a somewhat unstable protein, a feature that may allow it to act as a sensor of proteotoxic stress: Das Gupta and colleagues proposed that unfolding of HspR contributes to derepression of the clpB, dnaK, and hsp gene promoters (114). It was thus proposed that PafE-proteasome-dependent degradation of HspR enhances the proteotoxic stress response by preventing HspR from prematurely rebinding to these promoters before all proteotoxic stress has been alleviated (Fig. 5C). In support of this hypothesis, we found that under certain conditions, a pafE mutant will acquire spontaneous suppressor mutations to restore wild-type-like growth. Most of the suppressor mutants have loss-of-function mutations in hspR, resulting in complete derepression of the HspR regulon (J. Jastrab, M. Samanovic, and K. H. Darwin, unpublished data). These suppressor mutants are also as resistant to heat shock as the parental pafE+ strain. Thus, these results suggest that the inability of a pafE mutant to fully derepress the hspR regulon both slows the growth of a pafE mutant under routine culture conditions and hinders recovery after heat shock.
An M. tuberculosis pafE mutant was significantly attenuated in mice, suggesting that bacteria encounter proteotoxic stress in vivo (45). This stress may include not only damage induced by phagocyte-derived reactive oxygen and nitrogen species but also dramatic changes in temperature when an infected host releases bacteria in aerosolized droplets into the environment; transitions from body temperature (∼37°C) to room temperature (∼25°C) and back into a new host may potentially induce protein misfolding.
It is important that we cannot rule out other pathways affected by PafE-dependent protein degradation that are unrelated to the proteotoxic stress response but are nonetheless important for the pathogenesis of M. tuberculosis. Several proteins accumulate in a pafE mutant, and it remains to be determined if some or all of them are proteasome substrates and if any contribute to the pathogenesis of tuberculosis.
REMAINING QUESTIONS
While much has been learned in the past decade about proteasomal degradation in bacteria, there remain significant gaps in our current knowledge. Below are several of the most pertinent questions in the field.
What Are the Requirements for Mpa/ARC-Mediated Degradation in Bacteria?
An issue that has frequently complicated the study of bacterial proteasome function is that ATP-dependent proteasomal degradation is very inefficient in vitro; recombinant Mpa associates poorly (if at all) with wild-type 20S CPs and therefore fails to induce gate opening (32). How, then, does this proteasome maintain a stable association with Mpa in bacteria? While it is possible that optimal in vitro reaction conditions have simply not yet been established, another possibility is that there are other accessory factors that bind to or otherwise promote a closer association between Mpa and 20S CPs. It may also be that additional proteins are required to maintain substrates in an unfolded state. The lack of these elements in vitro may result in the instability of the complex during proteolysis.
How Are Substrates Selected for Pupylation, Depupylation, and Degradation?
To date, it is unknown how proteins are selected for pupylation in any species. Production of an M. tuberculosis substrate in E. coli results in the pupylation of several different Lys residues rather than one specific Lys residue as observed in mycobacteria. Furthermore, the production of M. tuberculosis PafA and PupGlu in E. coli results in the pupylation of numerous E. coli proteins, most of which do not overlap the mycobacterial pupylomes (62). While it might seem that the mere presence of an exposed lysine would be sufficient to destine proteins for pupylation, this is not the case, as not all proteins with lysines are necessarily PafA targets. Taken together, these observations suggest the existence of other factors as well as intrinsic features of substrates that target them for pupylation. The structures of intermediate complexes of PafA, Pup, and substrates will hopefully reveal the physical interactions among the proteins that facilitate pupylation.
A possible mechanism of selection or negative regulation is Lys acetylation, an emerging phenomenon in M. tuberculosis that is known to directly affect the activities of several enzymes (115–117). It is possible that Lys acetylation protects exposed lysines on nonsubstrates, as well as nonideal lysines on proteins that are pupylated at another, preferred site. It is also possible that phosphorylation or other posttranslational modifications affect interactions between substrates and PafA.
Relatedly, how are pupylated substrates selected for degradation? We know of several pupylated proteins, for example, several enzymes of the M. tuberculosis fatty acid synthase complex II pathway, that do not seem to be robustly degraded under routine culture conditions (52). This particular example may offer a potential clue: if Pup modifications are sheltered within a complex, Mpa might be unable to access them or to otherwise unfold them in the presence of multiple interacting proteins.
How Do Bacteria Balance Degradation and Depupylation?
It is intriguing to imagine that the processes of degradation and depupylation are dynamically regulated in response to signals such as environmental stress or nutrient starvation, as suggested previously (73). While we have not observed any changes in the relative abundances of pupylation system components in M. tuberculosis under different culture conditions (our unpublished observations), studies of M. smegmatis have shown that the levels of Mpa, Dop, PafA, and PrcA oscillate during stationary growth phase and upon nitrogen starvation (73, 112). It is possible that the activities of PafA, Dop, and Mpa change in the presence of cofactors or posttranslational modifications that have yet to be identified. It has additionally been proposed that phosphorylation of the 20S CP regulates degradation of pupylated substrates. In M. smegmatis, overproduction of M. tuberculosis protein kinase B (PknB) leads to phosphorylation of PrcA and is associated with the increased degradation of a pupylated substrate (118). However, these data must be taken with caution, as the overproduction of PknB was required to observe this phenomenon.
Do PafE and Mpa/ARC Compete for Binding to the 20S CP?
PafE inhibits the degradation of pupylated substrates by the Mpa-proteasome in vitro (46). This observation, along with the apparently stronger in vitro binding of 20S CPs to PafE than to Mpa (44), suggests that PafE may compete with Mpa-mediated degradation in bacteria. While this is a reasonable hypothesis, overproduction of a 20S CP trap in wild-type and mpa mutant M. tuberculosis strains showed that PafE binding to CPs was not enriched in the mpa mutant, as would be expected if PafE and Mpa competed for access to 20S CPs (45). However, we cannot rule out the possibility that there are conditions in which the activity or interaction of either activator with 20S CPs is altered based on need.
What Are the Roles of Proteasomal Degradation in Broader Bacterial Physiology?
While several pathways affected by proteasomal degradation have been identified, there are dozens, if not hundreds, of other proteins reliant on ATP-dependent and -independent proteasomal degradation. M. tuberculosis strains with a deletion or knockdown of prcBA are viable but unable to grow normally in vitro (80, 84); notably, because a high-density transposon screen predicted that prcBA is essential in M. tuberculosis (80, 119), we propose that the prcBA deletion mutant harbors one or more suppressor mutations that allow it to survive in culture. Curiously, while deletion of 20S CP activity impairs M. tuberculosis growth under almost all conditions tested, it appears to have a minimal effect on M. smegmatis growth under most conditions, with the exception of nitrogen and carbon limitation (53, 73, 120). S. coelicolor is also viable upon deletion and disruption of its proteasome genes (83). Thus, it may be that the proteasome has more specialized functions in environmental bacterial strains while being essential for the normal physiology of M. tuberculosis, which is found only in humans.
CONCLUDING REMARKS
In addition to revealing several new contributions of regulated proteolysis to bacterial physiology, the study of bacterial proteasomes may potentially translate broadly to the understanding of proteasomal degradation in general. For one, despite extensive structural and mechanistic studies of eukaryotic ATP-independent proteasomal activators, the physiological role of ATP-independent protein degradation in eukaryotes has remained elusive. Ongoing studies of PafE in bacteria may finally reveal functions of ATP-independent degradation that are relevant to eukaryotes as well as other prokaryotes. The study of proteasome-dependent degradation has also revealed insights into the challenges faced by bacterial pathogens, such as M. tuberculosis, during infections. From proteotoxic stress responses to copper resistance, there is little doubt that the study of bacterial proteasome-regulated pathways will reveal new avenues of study for years to come.
ACKNOWLEDGMENTS
We are grateful to Huilin Li for providing the images presented in Fig. 4. We thank Jordan Jastrab and Marie Samanovic for reviewing a draft version of this review. We are grateful to the anonymous reviewers, who made several critical corrections and helpful suggestions.
Proteasome research in the Darwin lab is supported by NIH grants HL92774 and AI088075. K.H.D. holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. S.H.B. received support from the Jan T. Vilcek Endowed Fellowship Fund and is supported by NIH grant T32AI007180-34.
REFERENCES
- 1.Kish-Trier E, Hill CP. 2013. Structural biology of the proteasome. Annu Rev Biophys 42:29–49. doi: 10.1146/annurev-biophys-083012-130417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Streich FC Jr, Lima CD. 2014. Structural and functional insights to ubiquitin-like protein conjugation. Annu Rev Biophys 43:357–379. doi: 10.1146/annurev-biophys-051013-022958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forster F, Unverdorben P, Sledz P, Baumeister W. 2013. Unveiling the long-held secrets of the 26S proteasome. Structure 21:1551–1562. doi: 10.1016/j.str.2013.08.010. [DOI] [PubMed] [Google Scholar]
- 4.Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D. 2000. A gated channel into the proteasome core particle. Nat Struct Biol 7:1062–1067. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- 5.Rabl J, Smith DM, Yu Y, Chang SC, Goldberg AL, Cheng Y. 2008. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol Cell 30:360–368. doi: 10.1016/j.molcel.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith DM, Chang SC, Park S, Finley D, Cheng Y, Goldberg AL. 2007. Docking of the proteasomal ATPases' carboxyl termini in the 20S proteasome's alpha ring opens the gate for substrate entry. Mol Cell 27:731–744. doi: 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schreiner P, Chen X, Husnjak K, Randles L, Zhang N, Elsasser S, Finley D, Dikic I, Walters KJ, Groll M. 2008. Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature 453:548–552. doi: 10.1038/nature06924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, Dikic I. 2008. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453:481–488. doi: 10.1038/nature06926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu CW, Millen L, Roman TB, Xiong H, Gilbert HF, Noiva R, DeMartino GN, Thomas PJ. 2002. Conformational remodeling of proteasomal substrates by PA700, the 19 S regulatory complex of the 26 S proteasome. J Biol Chem 277:26815–26820. doi: 10.1074/jbc.M201782200. [DOI] [PubMed] [Google Scholar]
- 10.Braun BC, Glickman M, Kraft R, Dahlmann B, Kloetzel PM, Finley D, Schmidt M. 1999. The base of the proteasome regulatory particle exhibits chaperone-like activity. Nat Cell Biol 1:221–226. doi: 10.1038/12043. [DOI] [PubMed] [Google Scholar]
- 11.Verma R, Aravind L, Oania R, McDonald WH, Yates JR III, Koonin EV, Deshaies RJ. 2002. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 298:611–615. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- 12.Yao T, Cohen RE. 2002. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 419:403–407. doi: 10.1038/nature01071. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt M, Haas W, Crosas B, Santamaria PG, Gygi SP, Walz T, Finley D. 2005. The HEAT repeat protein Blm10 regulates the yeast proteasome by capping the core particle. Nat Struct Mol Biol 12:294–303. doi: 10.1038/nsmb914. [DOI] [PubMed] [Google Scholar]
- 14.Yao Y, Huang L, Krutchinsky A, Wong ML, Standing KG, Burlingame AL, Wang CC. 1999. Structural and functional characterizations of the proteasome-activating protein PA26 from Trypanosoma brucei. J Biol Chem 274:33921–33930. doi: 10.1074/jbc.274.48.33921. [DOI] [PubMed] [Google Scholar]
- 15.Darwin KH. 2009. Prokaryotic ubiquitin-like protein (Pup), proteasomes and pathogenesis. Nat Rev Microbiol 7:485–491. doi: 10.1038/nrmicro2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. 1997. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 17.Lowe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. 1995. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 18.Kwon YD, Nagy I, Adams PD, Baumeister W, Jap BK. 2004. Crystal structures of the Rhodococcus proteasome with and without its pro-peptides: implications for the role of the pro-peptide in proteasome assembly. J Mol Biol 335:233–245. doi: 10.1016/j.jmb.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 19.Hu G, Lin G, Wang M, Dick L, Xu RM, Nathan C, Li H. 2006. Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol Microbiol 59:1417–1428. doi: 10.1111/j.1365-2958.2005.05036.x. [DOI] [PubMed] [Google Scholar]
- 20.Tamura T, Nagy I, Lupas A, Lottspeich F, Cejka Z, Schoofs G, Tanaka K, De Mot R, Baumeister W. 1995. The first characterization of a eubacterial proteasome: the 20S complex of Rhodococcus. Curr Biol 5:766–774. doi: 10.1016/S0960-9822(95)00153-9. [DOI] [PubMed] [Google Scholar]
- 21.Nagy I, Tamura T, Vanderleyden J, Baumeister W, De Mot R. 1998. The 20S proteasome of Streptomyces coelicolor. J Bacteriol 180:5448–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pouch MN, Cournoyer B, Baumeister W. 2000. Characterization of the 20S proteasome from the actinomycete Frankia. Mol Microbiol 35:368–377. doi: 10.1046/j.1365-2958.2000.01703.x. [DOI] [PubMed] [Google Scholar]
- 23.Brannigan JA, Dodson G, Duggleby HJ, Moody PC, Smith JL, Tomchick DR, Murzin AG. 1995. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature 378:416–419. doi: 10.1038/378416a0. [DOI] [PubMed] [Google Scholar]
- 24.Lin G, Hu G, Tsu C, Kunes YZ, Li H, Dick L, Parsons T, Li P, Chen Z, Zwickl P, Weich N, Nathan C. 2006. Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity. Mol Microbiol 59:1405–1416. doi: 10.1111/j.1365-2958.2005.05035.x. [DOI] [PubMed] [Google Scholar]
- 25.Zuhl F, Seemuller E, Golbik R, Baumeister W. 1997. Dissecting the assembly pathway of the 20S proteasome. FEBS Lett 418:189–194. doi: 10.1016/S0014-5793(97)01370-7. [DOI] [PubMed] [Google Scholar]
- 26.Li D, Li H, Wang T, Pan H, Lin G, Li H. 2010. Structural basis for the assembly and gate closure mechanisms of the Mycobacterium tuberculosis 20S proteasome. EMBO J 29:2037–2047. doi: 10.1038/emboj.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huber EM, Heinemeyer W, Li X, Arendt CS, Hochstrasser M, Groll M. 2016. A unified mechanism for proteolysis and autocatalytic activation in the 20S proteasome. Nat Commun 7:10900. doi: 10.1038/ncomms10900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCormack T, Baumeister W, Grenier L, Moomaw C, Plamondon L, Pramanik B, Slaughter C, Soucy F, Stein R, Zuhl F, Dick L. 1997. Active site-directed inhibitors of Rhodococcus 20 S proteasome. Kinetics and mechanism. J Biol Chem 272:26103–26109. [DOI] [PubMed] [Google Scholar]
- 29.Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf DH. 1997. The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J Biol Chem 272:25200–25209. doi: 10.1074/jbc.272.40.25200. [DOI] [PubMed] [Google Scholar]
- 30.Choi WH, de Poot SA, Lee JH, Kim JH, Han DH, Kim YK, Finley D, Lee MJ. 2016. Open-gate mutants of the mammalian proteasome show enhanced ubiquitin-conjugate degradation. Nat Commun 7:10963. doi: 10.1038/ncomms10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Striebel F, Hunkeler M, Summer H, Weber-Ban E. 2010. The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup's N-terminus. EMBO J 29:1262–1271. doi: 10.1038/emboj.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang T, Li H, Lin G, Tang C, Li D, Nathan C, Darwin KH, Li H. 2009. Structural insights on the Mycobacterium tuberculosis proteasomal ATPase Mpa. Structure 17:1377–1385. doi: 10.1016/j.str.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Y, Smith DM, Kim HM, Rodriguez V, Goldberg AL, Cheng Y. 2010. Interactions of PAN′s C-termini with archaeal 20S proteasome and implications for the eukaryotic proteasome-ATPase interactions. EMBO J 29:692–702. doi: 10.1038/emboj.2009.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Djuranovic S, Hartmann MD, Habeck M, Ursinus A, Zwickl P, Martin J, Lupas AN, Zeth K. 2009. Structure and activity of the N-terminal substrate recognition domains in proteasomal ATPases. Mol Cell 34:580–590. doi: 10.1016/j.molcel.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 35.Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, Martin A. 2012. Complete subunit architecture of the proteasome regulatory particle. Nature 482:186–191. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Striebel F, Kress W, Weber-Ban E. 2009. Controlled destruction: AAA+ ATPases in protein degradation from bacteria to eukaryotes. Curr Opin Struct Biol 19:209–217. doi: 10.1016/j.sbi.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 37.Wang T, Darwin KH, Li H. 2010. Binding-induced folding of prokaryotic ubiquitin-like protein on the Mycobacterium proteasomal ATPase targets substrates for degradation. Nat Struct Mol Biol 17:1352–1357. doi: 10.1038/nsmb.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erzberger JP, Berger JM. 2006. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct 35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 39.Ogura T, Wilkinson AJ. 2001. AAA+ superfamily ATPases: common structure-diverse function. Genes Cells 6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 40.Barkow SR, Levchenko I, Baker TA, Sauer RT. 2009. Polypeptide translocation by the AAA+ ClpXP protease machine. Chem Biol 16:605–612. doi: 10.1016/j.chembiol.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sauer RT, Baker TA. 2011. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem 80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 42.Martin A, Baker TA, Sauer RT. 2008. Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat Struct Mol Biol 15:1147–1151. doi: 10.1038/nsmb.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim YC, Snoberger A, Schupp J, Smith DM. 2015. ATP binding to neighbouring subunits and intersubunit allosteric coupling underlie proteasomal ATPase function. Nat Commun 6:8520. doi: 10.1038/ncomms9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bai L, Hu K, Wang T, Jastrab JM, Darwin KH, Li H. 2016. Structural analysis of the dodecameric proteasome activator PafE in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 113:E1983–E1992. doi: 10.1073/pnas.1512094113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jastrab JB, Wang T, Murphy JP, Bai L, Hu K, Merkx R, Huang J, Chatterjee C, Ovaa H, Gygi SP, Li H, Darwin KH. 2015. An adenosine triphosphate-independent proteasome activator contributes to the virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 112:E1763–E1772. doi: 10.1073/pnas.1423319112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delley CL, Laederach J, Ziemski M, Bolten M, Boehringer D, Weber-Ban E. 2014. Bacterial proteasome activator bpa (rv3780) is a novel ring-shaped interactor of the mycobacterial proteasome. PLoS One 9:e114348. doi: 10.1371/journal.pone.0114348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. 2003. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 48.Darwin KH, Lin G, Chen Z, Li H, Nathan CF. 2005. Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol Microbiol 55:561–571. [DOI] [PubMed] [Google Scholar]
- 49.Pearce MJ, Arora P, Festa RA, Butler-Wu SM, Gokhale RS, Darwin KH. 2006. Identification of substrates of the Mycobacterium tuberculosis proteasome. EMBO J 25:5423–5432. doi: 10.1038/sj.emboj.7601405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burns KE, Cerda-Maira FA, Wang T, Li H, Bishai WR, Darwin KH. 2010. “Depupylation” of prokaryotic ubiquitin-like protein from mycobacterial proteasome substrates. Mol Cell 39:821–827. doi: 10.1016/j.molcel.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, Darwin KH. 2008. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 322:1104–1107. doi: 10.1126/science.1163885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Festa RA, McAllister F, Pearce MJ, Mintseris J, Burns KE, Gygi SP, Darwin KH. 2010. Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis [corrected]. PLoS One 5:e8589. doi: 10.1371/journal.pone.0008589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burns KE, Liu WT, Boshoff HI, Dorrestein PC, Barry CE III. 2009. Proteasomal protein degradation in Mycobacteria is dependent upon a prokaryotic ubiquitin-like protein. J Biol Chem 284:3069–3075. doi: 10.1074/jbc.M808032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poulsen C, Akhter Y, Jeon AH, Schmitt-Ulms G, Meyer HE, Stefanski A, Stuhler K, Wilmanns M, Song YH. 2010. Proteome-wide identification of mycobacterial pupylation targets. Mol Syst Biol 6:386. doi: 10.1038/msb.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watrous J, Burns K, Liu WT, Patel A, Hook V, Bafna V, Barry CE III, Bark S, Dorrestein PC. 2010. Expansion of the mycobacterial “PUPylome.” Mol Biosyst 6:376–385. doi: 10.1039/b916104j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sutter M, Damberger FF, Imkamp F, Allain FH, Weber-Ban E. 2010. Prokaryotic ubiquitin-like protein (Pup) is coupled to substrates via the side chain of its C-terminal glutamate. J Am Chem Soc 132:5610–5612. doi: 10.1021/ja910546x. [DOI] [PubMed] [Google Scholar]
- 57.Chen X, Solomon WC, Kang Y, Cerda-Maira F, Darwin KH, Walters KJ. 2009. Prokaryotic ubiquitin-like protein pup is intrinsically disordered. J Mol Biol 392:208–217. doi: 10.1016/j.jmb.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sutter M, Striebel F, Damberger FF, Allain FH, Weber-Ban E. 2009. A distinct structural region of the prokaryotic ubiquitin-like protein (Pup) is recognized by the N-terminal domain of the proteasomal ATPase Mpa. FEBS Lett 583:3151–3157. doi: 10.1016/j.febslet.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 59.Striebel F, Imkamp F, Sutter M, Steiner M, Mamedov A, Weber-Ban E. 2009. Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat Struct Mol Biol 16:647–651. doi: 10.1038/nsmb.1597. [DOI] [PubMed] [Google Scholar]
- 60.Imkamp F, Rosenberger T, Striebel F, Keller PM, Amstutz B, Sander P, Weber-Ban E. 2010. Deletion of dop in Mycobacterium smegmatis abolishes pupylation of protein substrates in vivo. Mol Microbiol 75:744–754. doi: 10.1111/j.1365-2958.2009.07013.x. [DOI] [PubMed] [Google Scholar]
- 61.Cerda-Maira FA, Pearce MJ, Fuortes M, Bishai WR, Hubbard SR, Darwin KH. 2010. Molecular analysis of the prokaryotic ubiquitin-like protein (Pup) conjugation pathway in Mycobacterium tuberculosis. Mol Microbiol 77:1123–1135. doi: 10.1111/j.1365-2958.2010.07276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cerda-Maira FA, McAllister F, Bode NJ, Burns KE, Gygi SP, Darwin KH. 2011. Reconstitution of the Mycobacterium tuberculosis pupylation pathway in Escherichia coli. EMBO Rep 12:863–870. doi: 10.1038/embor.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Compton CL, Fernandopulle MS, Nagari RT, Sello JK. 2015. Genetic and proteomic analyses of pupylation in Streptomyces coelicolor. J Bacteriol 197:2747–2753. doi: 10.1128/JB.00302-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iyer LM, Burroughs AM, Aravind L. 2008. Unraveling the biochemistry and provenance of pupylation: a prokaryotic analog of ubiquitination. Biol Direct 3:45. doi: 10.1186/1745-6150-3-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guth E, Thommen M, Weber-Ban E. 2011. Mycobacterial ubiquitin-like protein ligase PafA follows a two-step reaction pathway with a phosphorylated pup intermediate. J Biol Chem 286:4412–4419. doi: 10.1074/jbc.M110.189282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ozcelik D, Barandun J, Schmitz N, Sutter M, Guth E, Damberger FF, Allain FH, Ban N, Weber-Ban E. 2012. Structures of Pup ligase PafA and depupylase Dop from the prokaryotic ubiquitin-like modification pathway. Nat Commun 3:1014. doi: 10.1038/ncomms2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smirnov D, Dhall A, Sivanesam K, Sharar RJ, Chatterjee C. 2013. Fluorescent probes reveal a minimal ligase recognition motif in the prokaryotic ubiquitin-like protein from Mycobacterium tuberculosis. J Am Chem Soc 135:2887–2890. doi: 10.1021/ja311376h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burns KE, Pearce MJ, Darwin KH. 2010. Prokaryotic ubiquitin-like protein provides a two-part degron to Mycobacterium proteasome substrates. J Bacteriol 192:2933–2935. doi: 10.1128/JB.01639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barandun J, Delley CL, Ban N, Weber-Ban E. 2013. Crystal structure of the complex between prokaryotic ubiquitin-like protein and its ligase PafA. J Am Chem Soc 135:6794–6797. doi: 10.1021/ja4024012. [DOI] [PubMed] [Google Scholar]
- 70.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. 2000. Recognition of the polyubiquitin proteolytic signal. EMBO J 19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Regev O, Roth Z, Korman M, Khalaila I, Gur E. 2015. A kinetic model for the prevalence of mono- over poly-pupylation. FEBS J 282:4176–4186. doi: 10.1111/febs.13413. [DOI] [PubMed] [Google Scholar]
- 72.Chen X, Li C, Wang L, Liu Y, Li C, Zhang J. 2016. The mechanism of Mycobacterium smegmatis PafA self-pupylation. PLoS One 11:e0151021. doi: 10.1371/journal.pone.0151021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Elharar Y, Roth Z, Hermelin I, Moon A, Peretz G, Shenkerman Y, Vishkautzan M, Khalaila I, Gur E. 2014. Survival of mycobacteria depends on proteasome-mediated amino acid recycling under nutrient limitation. EMBO J 33:1802–1814. doi: 10.15252/embj.201387076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maldonado AY, Burz DS, Reverdatto S, Shekhtman A. 2013. Fate of pup inside the Mycobacterium proteasome studied by in-cell NMR. PLoS One 8:e74576. doi: 10.1371/journal.pone.0074576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Imkamp F, Striebel F, Sutter M, Ozcelik D, Zimmermann N, Sander P, Weber-Ban E. 2010. Dop functions as a depupylase in the prokaryotic ubiquitin-like modification pathway. EMBO Rep 11:791–797. doi: 10.1038/embor.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burns KE, McAllister FE, Schwerdtfeger C, Mintseris J, Cerda-Maira F, Noens EE, Wilmanns M, Hubbard SR, Melandri F, Ovaa H, Gygi SP, Darwin KH. 2012. Mycobacterium tuberculosis prokaryotic ubiquitin-like protein-deconjugating enzyme is an unusual aspartate amidase. J Biol Chem 287:37522–37529. doi: 10.1074/jbc.M112.384784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Forer N, Korman M, Elharar Y, Vishkautzan M, Gur E. 2013. Bacterial proteasome and PafA, the pup ligase, interact to form a modular protein tagging and degradation machine. Biochemistry 52:9029–9035. doi: 10.1021/bi401017b. [DOI] [PubMed] [Google Scholar]
- 78.Kuberl A, Franzel B, Eggeling L, Polen T, Wolters DA, Bott M. 2014. Pupylated proteins in Corynebacterium glutamicum revealed by MudPIT analysis. Proteomics 14:1531–1542. doi: 10.1002/pmic.201300531. [DOI] [PubMed] [Google Scholar]
- 79.Kuberl A, Polen T, Bott M. 2016. The pupylation machinery is involved in iron homeostasis by targeting the iron storage protein ferritin. Proc Natl Acad Sci U S A 113:4806–4811. doi: 10.1073/pnas.1514529113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gandotra S, Schnappinger D, Monteleone M, Hillen W, Ehrt S. 2007. In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat Med 13:1515–1520. doi: 10.1038/nm1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Halliwell B, Gutteridge JM. 1985. The importance of free radicals and catalytic metal ions in human diseases. Mol Aspects Med 8:89–193. doi: 10.1016/0098-2997(85)90001-9. [DOI] [PubMed] [Google Scholar]
- 82.Pandey R, Rodriguez GM. 2012. A ferritin mutant of Mycobacterium tuberculosis is highly susceptible to killing by antibiotics and is unable to establish a chronic infection in mice. Infect Immun 80:3650–3659. doi: 10.1128/IAI.00229-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boubakri H, Seghezzi N, Duchateau M, Gominet M, Kofronova O, Benada O, Mazodier P, Pernodet JL. 2015. The absence of pupylation (prokaryotic ubiquitin-like protein modification) affects morphological and physiological differentiation in Streptomyces coelicolor. J Bacteriol 197:3388–3399. doi: 10.1128/JB.00591-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gandotra S, Lebron MB, Ehrt S. 2010. The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLoS Pathog 6:e1001040. doi: 10.1371/journal.ppat.1001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.World Health Organization. 2015. Global tuberculosis report 2015, 20th ed World Health Organization, Geneva, Switzerland. [Google Scholar]
- 86.Philips JA, Ernst JD. 2012. Tuberculosis pathogenesis and immunity. Annu Rev Pathol 7:353–384. doi: 10.1146/annurev-pathol-011811-132458. [DOI] [PubMed] [Google Scholar]
- 87.MacMicking J, Xie QW, Nathan C. 1997. Nitric oxide and macrophage function. Annu Rev Immunol 15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 88.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. 1997. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci U S A 94:5243–5248. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bowman LA, McLean S, Poole RK, Fukuto JM. 2011. The diversity of microbial responses to nitric oxide and agents of nitrosative stress close cousins but not identical twins. Adv Microb Physiol 59:135–219. doi: 10.1016/B978-0-12-387661-4.00006-9. [DOI] [PubMed] [Google Scholar]
- 90.Samanovic MI, Tu S, Novak O, Iyer LM, McAllister FE, Aravind L, Gygi SP, Hubbard SR, Strnad M, Darwin KH. 2015. Proteasomal control of cytokinin synthesis protects Mycobacterium tuberculosis against nitric oxide. Mol Cell 57:984–994. doi: 10.1016/j.molcel.2015.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kurakawa T, Ueda N, Maekawa M, Kobayashi K, Kojima M, Nagato Y, Sakakibara H, Kyozuka J. 2007. Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature 445:652–655. doi: 10.1038/nature05504. [DOI] [PubMed] [Google Scholar]
- 92.Frebort I, Kowalska M, Hluska T, Frebortova J, Galuszka P. 2011. Evolution of cytokinin biosynthesis and degradation. J Exp Bot 62:2431–2452. doi: 10.1093/jxb/err004. [DOI] [PubMed] [Google Scholar]
- 93.Festa RA, Jones MB, Butler-Wu S, Sinsimer D, Gerads R, Bishai WR, Peterson SN, Darwin KH. 2011. A novel copper-responsive regulon in Mycobacterium tuberculosis. Mol Microbiol 79:133–148. doi: 10.1111/j.1365-2958.2010.07431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ladomersky E, Petris MJ. 2015. Copper tolerance and virulence in bacteria. Metallomics 7:957–964. doi: 10.1039/C4MT00327F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rowland JL, Niederweis M. 2013. A multicopper oxidase is required for copper resistance in Mycobacterium tuberculosis. J Bacteriol 195:3724–3733. doi: 10.1128/JB.00546-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gold B, Deng H, Bryk R, Vargas D, Eliezer D, Roberts J, Jiang X, Nathan C. 2008. Identification of a copper-binding metallothionein in pathogenic mycobacteria. Nat Chem Biol 4:609–616. doi: 10.1038/nchembio.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shi X, Festa RA, Ioerger TR, Butler-Wu S, Sacchettini JC, Darwin KH, Samanovic MI. 2014. The copper-responsive RicR regulon contributes to Mycobacterium tuberculosis virulence. mBio 5:e00876–13. doi: 10.1128/mBio.00876-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang Y, Hodgkinson V, Zhu S, Weisman GA, Petris MJ. 2011. Advances in the understanding of mammalian copper transporters. Adv Nutr 2:129–137. doi: 10.3945/an.110.000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.White C, Lee J, Kambe T, Fritsche K, Petris MJ. 2009. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J Biol Chem 284:33949–33956. doi: 10.1074/jbc.M109.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.White C, Kambe T, Fulcher YG, Sachdev SW, Bush AI, Fritsche K, Lee J, Quinn TP, Petris MJ. 2009. Copper transport into the secretory pathway is regulated by oxygen in macrophages. J Cell Sci 122:1315–1321. doi: 10.1242/jcs.043216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zimnicka AM, Tang H, Guo Q, Kuhr FK, Oh MJ, Wan J, Chen J, Smith KA, Fraidenburg DR, Choudhury MS, Levitan I, Machado RF, Kaplan JH, Yuan JX. 2014. Upregulated copper transporters in hypoxia-induced pulmonary hypertension. PLoS One 9:e90544. doi: 10.1371/journal.pone.0090544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Neyrolles O, Wolschendorf F, Mitra A, Niederweis M. 2015. Mycobacteria, metals, and the macrophage. Immunol Rev 264:249–263. doi: 10.1111/imr.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, Nolan S, Lamichhane G, Wang Y, Bossmann SH, Basaraba RJ, Niederweis M. 2011. Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 108:1621–1626. doi: 10.1073/pnas.1009261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wagner D, Maser J, Lai B, Cai Z, Barry CE III, Honer Zu Bentrup K, Russell DG, Bermudez LE. 2005. Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell's endosomal system. J Immunol 174:1491–1500. doi: 10.4049/jimmunol.174.3.1491. [DOI] [PubMed] [Google Scholar]
- 105.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O'Halloran TV. 1999. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 106.Cobine PA, George GN, Jones CE, Wickramasinghe WA, Solioz M, Dameron CT. 2002. Copper transfer from the Cu(I) chaperone, CopZ, to the repressor, Zn(II)CopY: metal coordination environments and protein interactions. Biochemistry 41:5822–5829. doi: 10.1021/bi025515c. [DOI] [PubMed] [Google Scholar]
- 107.Maryon EB, Molloy SA, Kaplan JH. 2013. Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am J Physiol Cell Physiol 304:C768–C779. doi: 10.1152/ajpcell.00417.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ward SK, Hoye EA, Talaat AM. 2008. The global responses of Mycobacterium tuberculosis to physiological levels of copper. J Bacteriol 190:2939–2946. doi: 10.1128/JB.01847-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vabulas RM, Hartl FU. 2005. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 310:1960–1963. doi: 10.1126/science.1121925. [DOI] [PubMed] [Google Scholar]
- 110.Fuertes G, Martin De Llano JJ, Villarroya A, Rivett AJ, Knecht E. 2003. Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem J 375:75–86. doi: 10.1042/bj20030282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Suraweera A, Munch C, Hanssum A, Bertolotti A. 2012. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol Cell 48:242–253. doi: 10.1016/j.molcel.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Elharar Y, Roth Z, Hecht N, Rotkopf R, Khalaila I, Gur E. 2016. Posttranslational regulation of coordinated enzyme activities in the Pup-proteasome system. Proc Natl Acad Sci U S A 113:E1605–E1614. doi: 10.1073/pnas.1525185113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stewart GR, Snewin VA, Walzl G, Hussell T, Tormay P, O'Gaora P, Goyal M, Betts J, Brown IN, Young DB. 2001. Overexpression of heat-shock proteins reduces survival of Mycobacterium tuberculosis in the chronic phase of infection. Nat Med 7:732–737. doi: 10.1038/89113. [DOI] [PubMed] [Google Scholar]
- 114.Bandyopadhyay B, Das Gupta T, Roy D, Das Gupta SK. 2012. DnaK dependence of the mycobacterial stress-responsive regulator HspR is mediated through its hydrophobic C-terminal tail. J Bacteriol 194:4688–4697. doi: 10.1128/JB.00415-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ghosh S, Padmanabhan B, Anand C, Nagaraja V. 2016. Lysine acetylation of the Mycobacterium tuberculosis HU protein modulates its DNA binding and genome organization. Mol Microbiol 100:577–588. doi: 10.1111/mmi.13339. [DOI] [PubMed] [Google Scholar]
- 116.Singhal A, Arora G, Virmani R, Kundu P, Khanna T, Sajid A, Misra R, Joshi J, Yadav V, Samanta S, Saini N, Pandey AK, Visweswariah SS, Hentschker C, Becher D, Gerth U, Singh Y. 2015. Systematic analysis of mycobacterial acylation reveals first example of acylation-mediated regulation of enzyme activity of a bacterial phosphatase. J Biol Chem 290:26218–26234. doi: 10.1074/jbc.M115.687269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xie L, Wang X, Zeng J, Zhou M, Duan X, Li Q, Zhang Z, Luo H, Pang L, Li W, Liao G, Yu X, Li Y, Huang H, Xie J. 2015. Proteome-wide lysine acetylation profiling of the human pathogen Mycobacterium tuberculosis. Int J Biochem Cell Biol 59:193–202. doi: 10.1016/j.biocel.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 118.Anandan T, Han J, Baun H, Nyayapathy S, Brown JT, Dial RL, Moltalvo JA, Kim MS, Yang SH, Ronning DR, Husson RN, Suh J, Kang CM. 2014. Phosphorylation regulates mycobacterial proteasome. J Microbiol 52:743–754. doi: 10.1007/s12275-014-4416-2. [DOI] [PubMed] [Google Scholar]
- 119.Sassetti CM, Boyd DH, Rubin EJ. 2001. Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci U S A 98:12712–12717. doi: 10.1073/pnas.231275498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Knipfer N, Shrader TE. 1997. Inactivation of the 20S proteasome in Mycobacterium smegmatis. Mol Microbiol 25:375–383. doi: 10.1046/j.1365-2958.1997.4721837.x. [DOI] [PubMed] [Google Scholar]