

SUMMARY
Neural circuits involving midbrain dopaminergic (DA) neurons regulate reward and goal-directed behaviors. Although local GABAergic input is known to modulate DA circuits, the mechanism that controls excitatory/inhibitory synaptic balance in DA neurons remains unclear. Here, we show that DA neurons use autocrine transforming growth factor β (TGF-β) signaling to promote the growth of axons and dendrites. Surprisingly, removing TGF-β type II receptor in DA neurons also disrupts the balance in TGF-β1 expression in DA neurons and neighboring GABAergic neurons, which increases inhibitory input, reduces excitatory synaptic input, and alters phasic firing patterns in DA neurons. Mice lacking TGF-β signaling in DA neurons are hyperactive and exhibit inflexibility in relinquishing learned behaviors and re-establishing new stimulus-reward associations. These results support a role for TGF-β in regulating the delicate balance of excitatory/inhibitory synaptic input in local microcircuits involving DA and GABAergic neurons and its potential contributions to neuropsychiatric disorders.
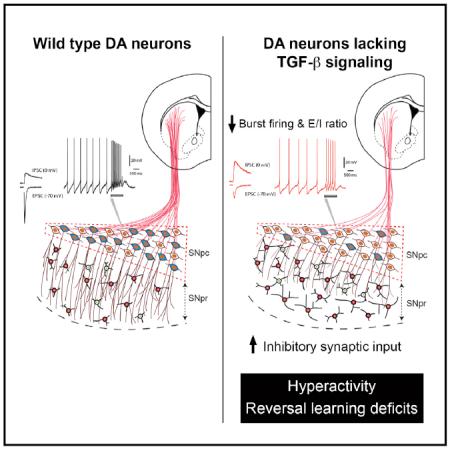
Graphical Abstract
INTRODUCTION
Dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) and ventral tegmental area (VTA) originate from the neurogenic niche in the embryonic ventral mesencephalon and require extrinsic factors to achieve proper growth and differentiation. During development, DA neurons send their axons to the striatum and prefrontal cortex and their dendrites to substantia nigra pars reticulata (SNpr) to establish neural circuits that impact motor-learning, reward, motivation, and goal-directed behaviors (Luo and Huang, 2016). In addition, DA neurons acquire characteristic rhythmic phasic (or burst) firing patterns, which serve as a critical driver of DA neuron performance (Blythe et al., 2009; Hyland et al., 2002; Overton and Clark, 1997). Perturbations to phasic firing of DA neurons and their circuitry undermine our ability to learn and unlearn and contribute to the pathogenesis of impulsive behaviors and other neuropsychiatric diseases (Dagher and Robbins, 2009; Everitt and Robbins, 2005).
Three closely related members of the transforming growth factor β (TGF-β) family promote the survival of DA neurons in culture (Krieglstein et al., 1995; Poulsen et al., 1994; Roussa et al., 2006; Zhang et al., 2007). Interestingly, however, mice with single null mutations of TGF-β isoforms do not display severe phenotypes in midbrain DA neurons during embryonic development, suggesting that TGF-β isoforms may functionally compensate for each other (Kaartinen et al., 1995; Sanford et al., 1997; Shull et al., 1992). In contrast, double knockout of TGF-β2 and TGF-β3 leads to a loss of DA neurons at E14.5 in mouse embryos (Roussa et al., 2006), supporting that different TGF-β isoforms are functionally redundant in regulating the survival of DA neurons. Despite these robust phenotypes, the embryonic and perinatal lethality of TGF-β null mice precludes investigation into the functions of TGF-β in the maturation and postnatal development of the nigrostriatal and mesocortical system. Thus, it remains unclear whether TGF-β ligands can promote synaptic plasticity and maintenance of neural circuits in mature DA neurons similar to that described in the Drosophila neuromuscular junction and Aplysia sensory neurons (Chin et al., 2002; Fuentes-Medel et al., 2012; Goold and Davis, 2007; Zhang et al., 1997). In support of this idea, a recent study identifies TGF-β3 as a potent astrocyte-derived factor that promotes the transcription of complement genes C1qa, C1qb, and C1qc in retinal ganglion cells (RGCs). Expression of TGF-β ligands in the developing retina coincides with the period of synapse refinement in the retinogeniculate system. Indeed, deletion of TGF-β type II receptor (TβRII) in RGCs results in reduced C1q expression and microglia-mediated synaptic pruning in the dorsal lateral geniculate nucleus (Bialas and Stevens, 2013). In addition, mice with brain-specific deletion of TGF-β1 show reduced dendritic spines and long-term potentiation (LTP) in hippocampal neurons (Koeglsperger et al., 2013). However, the mechanism leading to the synaptic phenotypes in these mutants remains unclear.
To circumvent the pro-survival role of TGF-β signaling, we generated a conditional mutant in which DAT-iCre selectively removed TβRII in mature DA neurons. Our results showed that removing TβRII in mature DA neurons does not affect survival but causes significant reductions in axonal and dendritic growth and a concomitant upregulation of TGF-β1 expression in neighboring h (PV)+ GABAergic neurons. These phenotypes result in a significant increase in inhibitory inputs, a marked reduction in excitatory-inhibitory (E-I) ratio, and reduced phasic firing frequency in DA neurons. Behaviorally, adult mice lacking TGF-β signaling in DA neurons are persistently hyperactive and exhibit profound defects in behavioral flexibility and fail to re-establish new stimulus-reward associations in a reversal-learning paradigm. These results support the role for TGF-β in controlling the development of dopaminergic neural circuits in early postnatal life and its implications in neuropsychiatric disorders.
RESULTS
Loss of TβRII in DA Neurons Reduces Axonal Growth
To examine the in vivo function of TGF-β signaling in mature DA neurons, we generated conditional DAT-iCre;TbRIIfl/fl mutants in which the floxed exon 2 of the TβRII gene was deleted using the BAC transgenic DAT-iCre (Chytil et al., 2002; Turiault et al., 2007). Using the Ai14 reporter line, we showed that the Cre-mediated recombination could be detected in DA neurons as early as embryonic day (E) 15.5 and was complete in every DA neurons at postnatal day (P) 0. Consistent with the timing of DAT-iCre recombination, confocal microscopy revealed that, compared to control TbRIIfl/fl DA neurons, DA neurons in DAT-iCre;TbRIIfl/fl mutants showed a modest reduction of TβRII at E15.5 (Figures 1A–1D, arrowheads). By P0, no TβRII proteins could be detected in the DA neurons of DAT-iCre;TbRIIfl/fl mice (Figures 1E–1H′and S1). Despite the loss of TβRII, there was no reduction in DA neuron number in DAT-iCre;TbRIIfl/fl mutants from E15.5 to P28 (Figure 1I).
Figure 1. Loss of TβRII in DA Neurons Does Not Affect Survival, but Reduces Axonal Growth.

(A–H) Immunostaining of TβRII (green), TH (red), and DAPI (blue) in ventral midbrain of E15.5 and P0 TbRIIfl/fl and DAT-iCre;TbRIIfl/fl. (F′ and H′) Higher-magnification images show TβRII proteins in the DA neurons of control P0 TbRIIfl/fl but not in DAT-iCre;TbRIIfl/fl.
(I) Stereology quantification of TH+ neurons in the SNpc of TbRIIfl/fl and DAT-iCre;TbRIIfl/fl mice at E15.5, P0, P14, and P28 shows no difference in DA neuron numbers (n = 4 per genotype per age). Student's t test, n.s., not significant.
(J and K) Immunofluorescent images show reduced TH+ axons in the coronal sections of DAT-iCre;TbRIIfl/fl mice at E15.5. Scale bar, 200 μm.
(L–O) Sagittal sections of GFP and TH immunostaining in E17.5 and P14 DAT-iCre;R26Ryfp/+ and DAT-iCre;TbRIIfl/fl;R26Ryfp/+ brains. Arrowheads highlight the TH+ and GFP+ axons innervating the striatum and frontal cortex. Scale bar, 500 μm.
(P) Quantification of TH+ axonal density in the striatum of TbRIIfl/fl and DAT-iCre;TbRIIfl/fl mice at E15.5, E17.5, and P14. n = 3 per genotype per age, Student's t test, ***p < 0.0005.
(Q and R) Immunogold EM of TH-labeled terminals in the striatum of P14 DAT-iCre and DAT-iCre;TbRIIfl/fl mice. Scale bar, 1 μm.
(S) Quantification of TH+ terminals in control DAT-iCre and DAT-iCre;TbRIIfl/fl mice (n = 3 per genotype), *p = 0.059, Student's t test. Data are presented as mean± SD.
Given the reported role of TGF-β signaling in specification of axon in cortical neurons (Yi et al., 2010), we asked whether loss of TβRII in DA neurons likewise affected axon specification. To this end, we analyzed TH+ axons in the striatum of control TbRIIfl/fl and DAT-iCre;TbRIIfl/fl mice from E15.5 to P14. In addition, we analyzed DAT-iCre;TbRIIfl/fl mice that also carried the R26Ryfp (DAT-iCre;TbRIIfl/fl;R26Ryfp/+) and found no axon specification defects in DA neurons (Figures 1J–1M). Instead, TH+ axon density in the striatum of DAT-iCre;TbRIIfl/fl mutants showed ~60%, ~34%, and ~25% reduction at E15.5, E17.5, and P14, respectively (Figures 1N–1P). Consistent with these results, TH immunogold electron microscopy also showed ~40% reduction in TH+ terminals in the striatum of DAT-iCre;TbRIIfl/fl mice at P14 (Figures 1Q–1S).
TGF-β Signaling Promotes Dendritic Growth and Spine Formation in DA Neurons
In addition to the reduced axonal growth phenotype, we noticed that DA neurons in DAT-iCre;TbRIIfl/fl mutants showed reduced density and length of TH+ dendrites in SNpr at P0 and P14 (Figures 2A and 2B). To quantify this phenotype, we prepared brain slices from P14 control (DAT-iCre) and DAT-iCre;TbRIIfl/fl;Ai14 mice and injected biocytin into individual DA neurons to highlight their dendritic arbors (Figures 2C and 2D). Neurolucida 3D reconstruction of DA neurons in SNpc showed a consistent reduction in dendritic complexity and cumulative surface areas in DAT-iCre;TbRIIfl/fl;Ai14 mutant (Figures 2E and 2F). In addition, the density of dendritic spines in DA neurons of DAT-iCre; TbRIIfl/fl;Ai14 mutants were markedly reduced, affecting all four spine categories (Figures 2G–2N).
Figure 2. Loss of TβRII in DA Neurons Reduces Dendritic Length and Complexity.

(A and B) TH immunofluorescent stain highlights DA neuron dendrites extending into SNpr in P14 TbRIIfl/fl and DAT-iCre;TbRIIfl/fl mice. Scale bars, 200 μm in (A) and 20 μm in insets.
(C and D) Neurolucida 3D reconstructions of biocytin-labeled individual DA neuron in P14 DAT-iCre and DAT-iCre;TbRIIfl/fl brains.
(E and F) Quantification of dendritic complexity by Sholl analysis (genotype: F1,2554 = 86.97, p < 0.0001, two-way ANOVA) and cumulative surface area (genotype: F1,2554 = 161.90, p < 0.0001, two-way ANOVA) of biocytin-labeled neurons (n = 9 from three DAT-iCre;Ai14 and n = 11 for four DAT-iCre;TbRIIfl/fl;Ai14 mice).
(G–J) Spine density and morphology in proximal and distal dendrites of biocytin-labeled DA neurons in P14 DAT-iCre and DAT-iCre;TbRIIfl/fl brains.
(K–M) Number of spines plotted against distance from the cell body (n = 9 neurons per group, genotype: F1,2496 = 773.54, p < 0.0001, two-way ANOVA) (K), spine density (p < 0.005, Student's t test) (M) and total number of spines (p < 0.005, Student's t test) (N). Data are presented as mean ± SEM.
(N) Quantification of thin, stubby, mushroom and branched spines in biocytin-labeled DA neurons in P14 TbRIIfl/fl and DAT-iCre;TbRIIfl/fl mice (n = 9 neurons per group). Data are presented as mean ± SEM, Student's t test. p < 0.005.
To determine whether TGF-β1 is sufficient to promote neurite outgrowth in DA neurons, we cultured primary DA neurons from E13.5 control TbRIIfl/fl and DAT-iCre;TbRIIfl/fl embryos and showed that control DA neurons elaborated long, branching dendrites that could be further stimulated by recombinant TGF-β1 (10 ng/mL) or completely blocked by TβRI inhibitor SB-431542 (Figures 3A–3C). In contrast, DAT-iCre;TbRIIfl/fl DA neurons had shorter and less complex dendrites that did not respond to TGF-β1 treatment (Figures 3D and 3E). Sholl analyses showed that TGF-β1 (10 ng/mL) treatment increased dendritic branches in control DA neurons, especially within a 50- to 200-μm radius of the cell body, whereas the same treatment did not increase dendritic branches in DAT-iCre;TbRIIfl/fl DA neurons (Figure 3F). Consistent with these findings, control DA neurons showed much higher cumulative surface areas at basal condition and after TGF-β1 treatment than DAT-iCre;, TbRIIfl/fl DA neurons (Figure 3G). Finally, the dendritic growth in control DA neurons responded to TGF-β in a dose-dependent manner, and this effect could be completely blocked by SB-431542 to the level similar to DAT-iCre;TbRIIfl/fl DA neurons (Figure 3H).
Figure 3. TGF-β Signaling in Midbrain Dopaminergic Neurons Promotes Neurite Outgrowth In Vitro.

(A) Experimental design for recombinant TGFβ1 ligand and SB-431542 treatment of DA primary neuron cultures.
(B–E) Neurolucida tracings of TH+ neurons from TbRIIfl/fl and DAT-iCre;TbRIIfl/fl cultures that were untreated or treated with TGF-β1.
(F–H) Quantification of neurite phenotype using Sholl analysis, cumulative surface area, and total dendrite length. For each condition, 30 neurons from three separate cultures using E13.5 embryos were analyzed. Data are presented as mean ± SEM, two-way ANOVA (F and G), Student's t test (H). *p < 0.05.
Autoregulation of TGF-β1 in DA Neurons Support Dendritic Growth
Given the robust effects of TGF-β signaling in the development of axons and dendrites in DA neurons, we asked whether TGF-β ligands could function as target-derived trophic factors. To this end, we developed in situ hybridization to characterize the expression of each of the three TGF-β ligands, and showed that Tgfb1, Tgfb2, and Tgfb3 mRNAs were either in very low abundance or undetectable in the caudate/putamen of P28 control mouse brains (Figures 4A–4C). In contrast, Tgfb1 mRNA showed abundant expression in DA neurons in SNpc and in GABAergic neurons in SNpr, whereas Tgfb2 and Tgfb3 mRNA expression was not detectable in DA or GABAergic neurons (Figures 4A′–4C′). Compared to DA neurons, Tgfb1, Tgfb2, and Tgfb3 mRNAs were below detectable levels in astrocytes and microglia in SNpc (Figure S2).
Figure 4. Autoregulation of Tgfb1 Expression in DA Neurons by Smad2 and c-Jun.

(A–C and A′–C′) In situ hybridization for Tgfb1, Tgfb2, and Tgfb3 mRNA using isoform-specific antisense RNA probes showed high abundance of Tgfb1 mRNA in SNpc, but no detectable Tgfb2 and Tgfb3 mRNA. A very low level of Tgfb1 mRNA can also be detected in the caudate/putamen. Scale bar, 500 μm.
(D–O) Tgfb1 mRNA expression in P28 control and DAT-iCre;TbRIIfl/fl mutant DA and GABAergic neurons. Confocal images of TH and GAD67 immunostaining (green) combined with fluorescent in situ hybridization for Tgfb1 mRNA expression (red) in TbRIIfl/fl and DAT-iCre;TbRIIfl/fl DA neurons. Scale bars, 100 μm. Higher magnification of single cells shown as insets in (F), (I), (L), and (O).
(P) Percentage of Tgfb1+;TH+ neurons across five bregma levels (n = 4 for TbRIIfl/fl and DAT-iCre-TbRIIfl/fl mice, genotype: F1,28 = 54.45, p < 0.005, two-way ANOVA).
(Q) Tgfb1 mRNA fluorescence intensity in individual GAD67+ neurons (n = 3 for TbRIIfl/fl and n = 4 for DAT-iCre-TbRIIfl/fl mice), p = 0.05, Student's t test). Data are presented as mean ± SD.
(R–U) Confocal images show reduced p-Smad2 and increased p-c-jun in TH+ DA neurons (red) in P14 DAT-iCre;TbRIIfl/fl mice. Insets show higher-magnification images. Scale bar, 60 μm.
(V and W) Fluorescent intensity of p-Smad2 and p-c-Jun in DA neurons in TbRIIfl/fl and DAT-iCre;TbRIIfl/fl at P14 (n = 3 per genotype). Data are presented as mean ± SD p < 0.001, Student's t test.
(X) Schematic diagram of the mouse Tgfb1 promoter region and summary of luciferase assays using reporter constructs generated from 5 kb upstream of the transcriptional start site in mouse Tgfb1 locus reveal positive and negative regulations of Tgfb1 transcription by Smad and c-Jun, respectively.
(Y) Relative luciferase activity of reporter constructs, (−4 kb~−3 kb)-Luc, (−3 kb~−2 kb)-Luc, (−2 kb~+754 bp)-Luc and (−417 bp~+754 bp)-Luc (normalized to pGL4.10-Luc activity).
(Z) Neuro-2A cells were co-transfected with (−417 bp~+754 bp)-Luc and plasmids encoding Smad2/3/4 or c-Jun. *p < 0.05 and **p < 0.01, when compared to (−417 bp~+745 bp)-Luc, and #p < 0.05 and ##p < 0.01, when compared to (−417 bp~+745 bp)-Luc. n = 3 for each reaction, Student's t test.
The co-expression of TGF-β1 and TβRII in DA neurons suggested that TGF-β might use an autoregulatory mechanism to sustain its own expression. To test this, we developed fluorescent in situ hybridization and showed that, compared to control (TbRIIfl/fl) DA neurons, Tgfb1 mRNA was drastically reduced in DAT-iCre;TbRIIfl/fl DA neurons across all bregma levels in the ventral midbrain (Figures 3P and 4D–4I). Despite the reduced number of Tgfb1+;TH+ DA neurons in DAT-iCre;TbRIIfl/fl mutants, the signal intensity of Tgfb1 mRNA in the few Tgfb1+;TH+ neurons remained unchanged compared to control. Similar to DA neurons, ~60% GABAergic neurons in SNpr of control mice also expressed Tgfb1 mRNA (Figures 4J–4L, arrowheads). Contrary to the marked reduction of Tgfb1 mRNA in DA neurons of DAT-iCre;TbRIIfl/fl mutants, a significantly higher percentage of GABAergic neurons were positive for Tgfb1 mRNA (Figures 4M–4O). On average, the Tgfb1 mRNA signal intensity in DAT-iCre;TbRIIfl/fl GAD67+ neurons increased by ~77% compared to control neurons (Figure 4Q). Given the opposing roles of Smad2/3 and AP-1 transcription factors in TGF-β receptor downstream signaling, we examined the level of p-Smad2 and p-c-Jun in DA neurons of control and DAT-iCre;TbRIIfl/fl mutants by confocal microscopy. Our results showed that the level of p-Smad2 in DA neurons was drastically reduced in DAT-iCre;TbRIIfl/fl mutants, whereas total Smad2 levels remained unchanged (Figures 4R, 4S, 4V, and S3). In contrast, p-c-Jun was undetectable in control DA neurons but showed a significant increase in DAT-iCre;TbRIIfl/fl mutant DA neurons (Figures 4T, 4U, and 4W).
To further characterize the opposing effects of Smad2 and c-Jun in the transcription of Tgfb1, we used bioinformatics to identify four Smad-binding elements (SBEs) and four AP-1 binding sites in the 5-kb sequence upstream of the transcriptional start site in mouse Tgfb1 locus (Figure 4X). We then generated four luciferase reporters that contained non-overlapping regions of this 5-kb Tgfb1 promoter/enhancer sequences. Luciferase reporter assays showed that construct (−417~+754 bp)-Luc contained the core SBE and AP-1 binding sites required for basal transcriptional activity of Tgfb1 promoter/enhancer (Figure 4Y). Furthermore, Smad2 dose-dependently activated, whereas c-Jun suppressed, transcription of (−417~+754 bp)-Luc activity (Figure 4Z). These results support the idea that the co-expression of TGF-β1 and TβRII in DA neurons to maintain a delicate balance of TGF-β1 expression in DA neurons and GABAergic neurons.
Loss of TβRII in DA Neurons Alters Excitation-Inhibition Synaptic Inputs
The changes of Tgfb1 mRNA levels in DA neurons and GABAergic neurons in substantia nigra lead us to hypothesize that these changes might alter the delicate balance of excitatory and inhibitory input to DA neurons. This hypothesis is further supported by a recent study indicating that the extension of DA neuron dendrites into SNpr may dictate GABAergic innervation and inhibitory response to DA neurons (Henny et al., 2012). These results motivated us to investigate how the dendrite phenotype in DA neurons of DAT-iCre;TbRIIfl/fl mutants might influence the excitatory and inhibitory synaptic inputs to DA neurons (Watabe-Uchida et al., 2012). To characterize changes in the local GABAergic microcircuits within ventral midbrain, we examined several markers and found that the majority of GABAergic neurons in SNpr expressed parvalbumin (PV), but not calbindin, calretinin, or neuropeptide Y (NPY) (Figure S4). Similar to the development of PV+ neurons in the cerebral cortex, no PV+ neurons were detected at P0 in the substantia nigra, but the number of PV+ neurons showed a significant increase in control mice at P14 and remained stable in adult life. Compared to control, the number of PV+ neurons in SNpr was significantly increased in DAT-iCre;TbRIIfl/fl mutants at P14 and P28, with distinct increases in PV+ dendrites (Figures 5A–5E).
Figure 5. Loss of TGF-β Signaling in DA Neurons Reduces Excitatory Synaptic Inputs but Increases Inhibitory Synaptic Inputs.

(A–D) Immunohistochemistry of PV+ neurons in SNpr of P14TbRIIfl/fl and DAT-iCre;TbRIIfl/fl mice. Scale bars, 200 μm (A) and 50 μm (B).
(E) Stereological quantification of PV+ neuron number at P14 (n = 3) and P28 (n = 4) *p < 0.05, Student's t test.
(F–I) Confocal images of DA neurons immunostained for VGlut2 (green) and TH (red) in TbRIIfl/fl and DAT-iCre;TbRIIfl/fl mice at P0 and P14. Scale bars, 100 μm and 20 μm (inset).
(J) Density of VGlut2+ synapses at P0 (n = 3, 9 fields per genotype, ***p < 0.01, Student's t test).
(K) Percentage of VGlut2+;TH+ puncta per unit TH+ dendrite area at P14 (n = 3 for TbRIIfl/fl and DAT-iCre;TbRIIfl/fl mice, 35–38 fields analyzed per mouse, p = 0.005, Student's t test).
(L–O) Immunogold electron micrographs of TH-labeled synapses in P14 TbRIIfl/fl and DAT-iCre;TbRIIfl/fl. Examples of asymmetric (L and M) and symmetric (N and O) synapses onto TH+ dendrites. Scale bar, 500 nm.
(P–S) Number of TH+ terminals with synapses in SNpc and SNpr (P), synapse number normalized to TH+ terminal circumference in SNpc and SNpr (Q), asymmetric and symmetric synapse density in SNpc (R), and the ratio of symmetric versus asymmetric synapses in TH+ and non-TH+ terminals (S) (n = 3 per genotype). *p < 0.05, **p < 0.01, ***p < 0.005, n.s., not significant. Data are presented as mean ± SD, Student's t test.
To determine how changes in PV+ neurons might altered excitatory and inhibitory synaptic input to DA neurons in DAT-iCre;TbRIIfl/fl mutants, we used vesicular glutamate transporter type 2 (VGlut2) as a presynaptic excitatory marker and showed an abundant VGlut2+ puncta in SNpc and SNpr in control TbRIIfl/fl mice at P0 (Figure 5F). These VGlut2+ puncta persisted at P14 with many identified along TH+ dendrites (Figure 5G). In contrast, SNpc and SNpr of DAT-iCre;TbRIIfl/fl mice showed an overall reduction of VGlut2 density at P0 and a marked reduction of VGlut2+;TH+ synapses along TH+ dendrites at P14 (Figures 5H–5K). Immunogold electron microscopy using tyrosine hydroxylase (TH) antibody showed that in control mice, ~86% of TH+ dendrites in SNpc and ~93% in SNpr contained definitive synapses (Figures 5L–5P). In contrast, fewer TH+ dendrites (~64%) in SNpc and SNpr of DAT-iCre;TbRIIfl/fl mutants contained definitive synapses. Even after normalizing the number of synapses by the circumference of TH+ dendrites, the synaptic density in DAT-iCre;TbRIIfl/fl mutants was significantly reduced in SNpc and SNpr (Figure 5Q). We then analyzed ultrastructural features that distinguish inhibitory (symmetric) from excitatory (asymmetric) synapses and found that the excitatory synapse density was reduced and the inhibitory-to-excitatory synapse ratio was higher in TH+ and non-TH+ dendrites in DAT-iCre;TbRIIfl/fl (Figures 5R and 5S).
Loss of TGF-β Signaling Reduces E-I Ratio and Burst Firing in DA Neurons
To analyze the functional consequence of the altered ratio of excitatory and inhibitory synapses in DAT-iCre;TbRIIfl/fl mutants, we performed electrophysiological recordings and found a significant, albeit modest, decrease in the frequency of mEPSC in DA neurons of DAT-iCre;Ai14;TbRIIfl/fl mutants (Figures 6A and 6C), while the amplitude was comparable between the genotypes (Figures S5A and S5C). In contrast, mIPSC frequency in DA neurons of DAT-iCre;Ai14;TbRIIfl/fl mutants showed a dramatic increase compared with control littermates (Figures 6B and 6D), without affecting the amplitude (Figures S5B and S5D). We then examined both input-output relationships for evoked EPSC and IPSC from the same neuron by locally stimulating synaptic inputs to the dendrites of DA neurons to measure the excitation/inhibition ratio (EPSC/IPSC ratio). Consistent with the mEPSC/mIPSC results, we found that the excitatory input-output curve was decreased in DAT-iCre;Ai14;TbRIIfl/fl DA neurons, whereas the inhibitory input-output curve was enhanced, resulting in a marked reduction in the E/I ratio (Figures 6E–6G).
Figure 6. DA Neurons Lacking TβRII Showed Excitation-Inhibition Imbalance and Reduced Synaptic Stimulation-Induced Burst Firing.

(A and B) Traces of mEPSC (A) and mIPSC (B) recorded from DA neurons from littermate control (top) and DAT-iCre;Ai14;TbRIIfl/fl mutants (bottom).
(C) Cumulative probability plots of mEPSC inter-event intervals (left, *p < 0.05, Kolmogorov-Smirnov [KS] test) and summary of mean mEPSC frequency (right, p > 0.21, Mann-Whitney). Number of neuron recorded, n = 16 from three DAT-iCre;Ai14;TbRII+/+ mice and n = 17 from four DAT-iCre;Ai14;TbRIIfl/fl mice.
(D) Cumulative probability plots of mIPSC inter-event intervals (left, p < 0.05, K.S test) and mean mIPSC frequency (right, p < 0.05, Mann-Whitney). Number of neuron recorded, DAT-iCre;Ai14;TbRII+/+: n = 16; DAT-iCre;Ai14;TbRIIfl/fl: n = 17.
(E–G) Evoked EPSCs (left) and input/output curve for evoked EPSC (right) (E), IPSCs (left) and input/output curve for evoked IPSC (right) (F), and excitation/inhibition ratios (EPSC/IPSC ratios) (G). Number of neuron recorded, DAT-iCre;Ai14;TbRII+/+: n = 16 and DAT-iCre;Ai14;TbRIIfl/fl: n = 17. *p < 0.05, Mann-Whitney.
(H) Spontaneous and burst firing of DA neurons recorded from littermate control (left) and DAT-iCre;Ai14;TbRIIfl/fl conditional knockout mice (right) in response to high-frequency stimulation. Rectangular bar indicates stimulation (100 Hz, 1 s).
(I) Number of AP of burst firing, maximal frequency of burst firing, and mean frequency of burst firing evoked by high-frequency stimulation (*p < 0.001, Mann-Whitney). Number of neuron recorded, n = 12 from three DAT-iCre;Ai14;TbRII+/+ mice, and n = 13 from four DAT-iCre;Ai14;TbRIIfl/fl mice. Mann-Whitney test, ***p < 0.001. All data are presented as mean ± SEM.
DA neurons in SNpc can exhibit both low-frequency tonic firing activity (<4 Hz) and high-frequency phasic burst-firing activity (>10 Hz), determined by their intrinsic properties and synaptic activities (Blythe et al., 2009; Hyland et al., 2002; Overton and Clark, 1997). This burst-firing property is governed by the balance of excitation and inhibition through regulation of membrane potential and input resistance (Isaacson and Scanziani, 2011). To assess whether changes of excitation/inhibition ratio in DAT-iCre;Ai14;TbRIIfl/fl mutants lead to defects in the generation of burst-firing, we recorded spontaneous and burst-firing of DA neurons in SNpc in response to a brief, local electrical stimulation (100 Hz, 1 s). While high-frequency stimulation delivered to control (DAT-iCre;Ai14) DA neurons induced an increase in the firing frequency, the same stimulation induced a significant reduction in burst firing in DAT-iCre;Ai14;TbRIIfl/fl DA neurons, reflected by reduced number of action potentials (APs), reduced maximal frequency, and reduced mean frequency during burst firing evoked by high-frequency stimulation (Figures 6H and 6I). There was no difference in spontaneous firing frequency or other physiological characteristics of action potentials between the genotypes (Figures S5E–S5G), suggesting the defects in burst firing are not caused by changes in DA neuron intrinsic properties but due to changes in synaptic inputs.
DAT-iCre;TbRIIfl/fl Mice Are Hyperactive and Exhibit Reversal-Learning Defects
We next examined whether the perturbations of excitation-inhibition balance in DAT-iCre;TbRIIfl/fl DA neurons impacted psychomotor behaviors and reward learning. In open-field tests, 2-month-old DAT-iCre;TbRIIfl/fl mice exhibited features of hyperactivity with increases in total distance traveled, average velocity, and rearing, but no difference in proportion of time spent in the periphery versus center (Figures 7A, 7B, and S6A–S6C). The hyperactivity phenotype in DAT-iCre;TbRIIfl/fl mice persisted at 5 and 7 months old, even though overall activity for both groups declined with age (Figure 7C). Similarly, DAT-iCre;TbRIIfl/fl mice were also hyperactive on an elevated plus maze with a marked increase in total distance traveled, but no differences in proportion of time spent in either arm (Figure S6D). The lack of difference in spatial preference in the open field and elevated plus maze suggest that DAT-iCre;TbRIIfl/fl mice do not have anxiety-like behaviors. Gross motor function and motor learning on Rotarod were also not impaired in DAT-iCre; TbRIIfl/fl mice (Figure S6E).
Figure 7. DAT-iCre;TbRIIfl/fl Mice Are Hyperactive and Display Deficits in Reversal Learning.

(A) Representative tracing of open-field test for control and DAT-iCre;TbRIIfl/fl mice.
(B) Distance traveled by 2-month-old male control and DAT-iCre;TbRIIfl/fl mice in the first 60 min of the open-field test. Data are presented at a 10-min interval. n = 12 mice per genotype, genotype: F2,44 = 10.44, *p < 0.05, two-way ANOVA.
(C) Distance traveled by 2-, 5- and 7-month-old male control and DAT-iCre;TbRIIfl/fl mice in the open-field test. n = 12 mice per genotype for each age, genotype: F2,44 = 10.44, ***p < 0.005, two-way ANOVA. The same cohort is tested at 2, 5, and 7 months old.
(D) Four-choice discrimination reversal learning. The star icon represents food reward. In the shaping phase, the shavings are unscented. In the discrimination and reversal phases, shavings are scented with four different odors (O1–O4).
(E) Number of trials required to reach criterion in 3-month-old male control (n = 20) and DAT-iCre;TbRIIfl/fl (n = 16) mice during discrimination (Disc.) and reversal (Rev.) phases (n.s., not significant, Student's t test between genotypes and paired t test within genotypes).
(F) Total errors, reversal errors (returns to anise) and regressive errors in 3-month-old male control and DAT-iCre;TbRIIfl/fl mice during reversal phase. ***p < 0.005, Student's t test.
(G and H) Cumulative latency (time taken to make a digging choice per trial) over the first ten trials in the discrimination (genotype: F1,160 = 14.91, ***p = 0.0002, two-way ANOVA) (G) and reversal phases (genotype: F1,160 = 9.88, ***p = 0.002, two-way ANOVA) (H). Data are presented as mean ± SEM.
Given the important role of DA neuron burst firing in reward learning (Adamantidis et al., 2011; Tsai et al., 2009), we investigated whether DAT-iCre;TbRIIfl/fl mice showed impairments in a four-choice discrimination reversal-learning paradigm (Izquierdo and Jentsch, 2012; Johnson and Wilbrecht, 2011). Both control and DAT-iCre;TbRIIfl/fl mice were trained to dig for food reward (the habituation and shaping phases) and then learned to associate a specific odor with the food reward (the discrimination phase) (Figure 7D). In the reversal phase, the food reward was paired with a different odor to determine whether there were genotype-specific differences in behavioral flexibility. In addition, a new odor never paired with food was introduced to assess their response to novelty (Figure 7D). Mice were considered to have reached criterion in both the discrimination and reversal phases, once they successfully identified food reward in eight out of ten consecutive trials. DAT-iCre;TbRIIfl/fl mice showed no difference from controls in the number of trials to criterion (TTC) during the discrimination phase (Figure 7E), suggesting no deficit in the acquisition of reward association. However, in the reversal phase, DAT-iCre;TbRIIfl/fl mice required approximately twice the number of trials to establish the new odor-reward association (Figure 7E). DAT-iCre;TbRIIfl/fl mice committed more total errors, the majority of which were due to digging in the previously rewarded odor (“reversal errors”) (Figure 7F). DAT-iCre;TbRIIfl/fl mice showed a high propensity to return to the previously rewarded odor even after a correct trial in the new odor (“regressive errors”). Consistent with their hyperactive phenotype, DAT-iCre;TbRIIfl/fl mice tended to show a shorter latency to dig for food in both the discrimination and reversal phases (Figures 7G and 7H). There was no significant difference in “perseverative errors” (digging in the first odor before trying a new strategy), “irrelevant errors” (digging in an odor that was never rewarded), or “novel errors” (digging in the novel odor). These data suggest that changes in excitation/inhibition balance in DA neurons do not affect the ability of DAT-iCre;TbRIIfl/fl mice to learn a reward association but have dramatic effects on the ability of these mice to update the reward association as new information becomes available.
DISCUSSION
Previous studies have shown that TGF-β signaling plays critical roles in astrocytes and microglia under physiological and injury conditions (Bialas and Stevens, 2013; Butovsky et al., 2014). However, neuronal expression and function of TGF-β in postnatal brains remains poorly characterized. Using Tgfb isoform-specific in situ hybridization probes, we show that Tgfb1 mRNA, but not Tgfb2 or Tgfb3, is abundantly expressed in DA neurons in SNpc and GABAergic neurons in SNpr in postnatal brains. In contrast, Tgfb1 mRNA is below detectable level in astrocytes and microglia in substantia nigra. The robust co-expression of Tgfb1 mRNA and TβRII in postnatal DA neurons supports the idea that DA neurons likely use an autocrine mechanism to maintain a relatively high level of TGF-β1 expression during the maturation of DA neurons. In support of this idea, bioinformatics and luciferase reporter analyses identify several Smad and AP-1 transcription factor binding elements in the promoter sequence of Tgfb1 locus, which positively and negatively regulate Tgfb1 mRNA expression, respectively. Consistent with these results, control DA neurons contain high p-Smad2 and low p-c-Jun levels in the nucleus, whereas DA neurons in DAT-iCre;TbRIIfl/fl mutants show a marked reduction in p-Smad2 level and an increase in p-c-Jun level. These results support the idea that DA neurons use a positive feedback mechanism to maintain a high level of TGF-β1 expression.
Both TGF-β and TGF-β downstream transcriptional cofactor HIPK2 regulate DA neuron survival during embryonic development (Krieglstein et al., 1995; Poulsen et al., 1994; Roussa et al., 2006; Zhang et al., 2007). In addition, TGF-β signaling has been shown to regulate axonal specification in the embryonic cortical neurons and dendritic arborization in newborn neurons in the dentate gyrus (He et al., 2014; Yi et al., 2010). Using electroporation of Cre recombinase plasmids in the cerebral cortex of E14.5 TbRIIfl/fl mice, it has been reported that developing cortical neurons lacking TβRII exhibit retardation in neuronal migration and no axonal outgrowth. Conversely, when exposed to TGF-β-coated substrates in vitro, embryonic cortical neurons show robust growth of supernumerary axons (Yi et al., 2010). Our results indicate that removal of TβRII from DA neurons between E15.5-P0 does not affect axonal specification but causes a modest delay in axonal growth. Several factors may contribute to the different phenotypes caused by TβRII removal in cortical neurons and DA neurons. These include the timing of Cre expression and the functional redundancy of non-Par6-dependent pathways may result in a very modest axonal phenotype in the DA neurons of DAT-iCre;TbRIIfl/fl mutants.
Compared to the axonal phenotype, DA neurons in DAT-iCre;TbRIIfl/fl mice exhibit a more robust reduction in dendritic length and complexity. Using dissociated cultures, we show that TGF-β1 dose-dependently promotes neurite outgrowth in control DA neurons and that this effect can be completely blocked by TβRI inhibitor SB-431542. In contrast, DA neurons from DAT-iCre;TbRIIfl/fl mice do not respond to TGF-β1 treatment. In fact, the dendritic length and complexity of DAT-iCre;TbRIIfl/fl DA neurons are identical to control neurons treated with TβRI inhibitor SB-431542. The dendritic phenotypes in the DA neurons of DAT-iCre;TbRIIfl/fl mice are similar to those reported in the newborn neurons of the dentate gyrus in adult ALK5fl/fl mutants using lentivirus expressing Cre recombinase or wild-type mice receiving retroviruses expressing shRNA to downregulate ALK5 expression in progenitors (He et al., 2014). These results support the important role of TGF-β signaling in neurite outgrowth and maintenance and further suggest that TGF-β signaling may have similar effects in different neuron subtypes.
Morphological and electrophysiological data in DA neurons of DAT-iCre;TbRIIfl/fl mutants show that loss of TGF-β signaling causes a significant reduction in excitatory synaptic inputs and a robust increase in inhibitory synaptic inputs, leading to a drastic reduction in excitation-inhibition ratio and reduced burst firing. At least two mechanisms may contribute to the increase in inhibitory synaptic inputs to DA neurons in DAT-iCre;TbRIIfl/fl mutants. First, electrophysiological recording of individual DA neurons followed by Neurolucida 3D reconstruction of their dendritic structure indicate that the somato-dendritic architecture of DA neurons correlates with their afferent synaptic organization (Henny et al., 2012). It is estimated that DA neurons receive approximately 8,000 synaptic inputs, of which ~30% are glutamatergic and 40%–70% are GABAergic. Interestingly, the proportion of inhibitory synaptic inputs to DA neurons appears to be dictated by the relative extension of DA dendrites into the SNpr. It is possible that the reduced DA dendritic extension into SNpr in DAT-iCre;TbRIIfl/fl mutants may trigger compensatory GABAergic synaptogenesis. Another mechanism causing the increase in inhibitory synapses could be due to the increase in Tgfb1 mRNA expression in GABAergic neurons in SNpr of DAT-iCre;TbRIIfl/fl mutants. TGF-β is known to promote synaptogenesis in invertebrate and vertebrate nervous system (Chin et al., 2002; Fuentes-Medel et al., 2012; Goold and Davis, 2007). Furthermore, two recent studies indicate that TGF-β may preferentially promote GABAergic synapse formation. In C. elegans, an RNAi screen identifies genes encoding TGF-β, receptor-regulated R-Smad, and Co-Smad as key factors in promoting GABAergic synapse inputs to body muscles (Vashlishan et al., 2008). In addition, intraventricular infusion of TGF-β1 in adult mice enhances inhibitory synapse density in cerebral cortex (Diniz et al., 2014). Our results show a significant increase of Tgfb1 mRNA in GABAergic neurons in the SNpr of DAT-iCre;TbRIIfl/fl mutants, which coincides with the prominent growth of PV+ dendrites in SNpr in postnatal life. It is likely that the increase of TGF-β1 expression can promote neurogenesis, dendritic growth, and synapse formation in late-born PV+ neurons in SNpr, which form “high-PV configurations” that can interfere with reward and goal-directed learning in DA neurons similar its ability to interfere with the acquisition of new memories and structural synaptic plasticity in hippocampus-based learning (Donato et al., 2013, 2015).
Previous studies show that removing NMDA receptor NR1 in DA neurons causes reduced burst firing and habit-learning deficits. In many aspects, the electrophysiological and behavioral phenotypes in DAT-iCre;TbRIIfl/fl are similar to mice lacking NMDA receptor in DA neurons (Engblom et al., 2008; Wang et al., 2011; Zweifel et al., 2009), thus providing compelling evidence that loss of TGF-β signaling in DA neurons can cause a marked reduction in the excitation/inhibition synaptic ratio in the local neural circuits involving DA neurons. In the reversal-learning paradigm, DAT-iCre;TbRIIfl/fl mutants have no problem acquiring a new task but do show profound deficits in relinquishing learned behaviors and re-establishing new reward associations. Unlike mice lacking NR1 in DA neurons, however, DAT-iCre;TbRIIfl/fl mutants show persistent hyperactivity in open-field and elevated plus maze tests. This hyperactivity phenotype may be caused by the imbalance of excitation-inhibition synaptic input to DA neurons in DAT-iCre;TbRIIfl/fl mutants. Alternatively, the upregulation of TGF-β1 in PV+ GABAergic neurons in DAT-iCre;TbRIIfl/fl mutants may upregulate potential TGF-β downstream targets, which may influence hyperactivity phenotype. Regardless the cause, it is interesting to note that the hyperactivity and behavioral inflexibility phenotypes in DAT-iCre;TbRIIfl/fl mutants are similar to symptoms observed in neuropsychiatric disorders, including attention deficit hyperactivity disorder (ADHD), post-traumatic stress disorder (PTSD), and schizophrenia. Indeed, misexpression of members of the TGF-β signaling pathway, including TGF-β2 and TβRI, have been linked to the GABAergic synaptic defects in the hippocampus of schizophrenia patients (Benes et al., 2007). These results suggest that TGF-β signaling mechanism may serve as a therapeutic target for GABAergic synaptic dysfunction in neuropsychiatric diseases.
EXPERIMENTAL PROCEDURES
Mice
DAT-iCre mice were obtained from Dr. Ken Nakamura (GIND/UCSF) and mice harboring the conditional (“floxed”) allele for TGF-β type II receptor (TbRIIfl mice) were obtained from Dr. Sam Pleasure (UCSF). The Ai14 reporter line (Jackson Laboratory, stock 007914) was obtained from Dr. Allison Xu (UCSF). Animal care followed the Institutional of Animal Care and Use Committee (IACUC) and the NIH guidelines.
Primary DA Neuron Cultures
Primary DA neuron cultures were prepared from embryonic ventral midbrain using microisland methods according to published procedures (Takeshima et al., 1996; Tang et al., 2010). Briefly, E13.5 mouse embryos were collected from timed-pregnant females. The ventral midbrain was dissected, dissociated after treatment with trypsin and cultured on coverslips coated with poly-D-ornithine (Sigma) and laminin (Sigma) at the density of 1.0 × 106/mL. The dissociated cells were maintained in DMEM/F-12 (1:1) medium containing 10% FBS overnight. Then, the media was changed to DMEM/F-12 (1:1) medium containing N2 supplements (Invitrogen), 20 ng/mL FGF2 (Millipore Corporation), 100 ng/mL FGF8 (PeproTech), and designated factors, including 1–10 ng/mL TGF-b1 (R&D), TGF-β3 (R&D), and 1 μM SB-431542 (Sigma) for additional 2 days in vitro (DIV) before they were fixed and processed for immunofluorescence. For each treatment condition, 30 neurons from three separate embryonic brain cultures were analyzed.
Immunohistochemistry and Image Quantification
Immunohistochemistry was performed as described previously with minor modifications (Tang et al., 2010). Specifically, mouse embryos and tissues were collected at embryonic day 15.5 (E15.5), E17.5, postnatal day 0 (P0), P14, and P28 were fixed in 4% paraformaldehyde (PFA) overnight at 4°C. To eliminate observers' bias, the tissue samples were de-identified before being processed for histology and the genotypes were not revealed until after the analyses were completed. Samples sectioned using a Leica cryostat were cryoprotected in 15% and 30% sucrose solutions and embedded in optimal cutting temperature (OCT). Samples sectioned using Leica vibratome were embedded in 6% Agarose. Sections were prepared at 50 μm. Primary antibodies in this study included anti-tyrosine hydroxylase (1:1,000, AB152, Millipore, or 1:1000, AB113, Abcam), anti-VGAT (1:500, #13011, Synaptic Systems), anti-VGLUT2 (1:500, #135402, Synaptic Systems), anti-GFAP (1:1,000, #3670, Cell Signaling Technology), anti-Iba1 (1:2,000, #019-19741, Wako), and anti-Gad67 (1:500, mAB5406, Millipore), anti-Parvalbumin (1:2,000, ab11427, Abcam), anti-Neuropeptide Y (1:1,000, No. 22940, Immunostar), anti-Calbindin (1:1,000, C2724, Sigma), anti-Calretinin (1:1,000, AB5054, Millipore), anti-GFP (1:500, GFP-1020, Aves), anti-Synaptophysin (1:500, S5768, Sigma), anti-Gephyrn (1:500, #147011, Synaptic Systems), and anti-TGFβ receptor type II (1:200, ab61213, Abcam). For immunofluorescent staining, sections were incubated with primary antibody overnight, followed by secondary antibodies conjugated with Alexa fluorophores 488 and 568 (1:300, Invitrogen) for 2 hr to detect signals. For chromogen staining, sections were incubated with primary antibody overnight, followed by incubation for 1 hr with biotinylated immunoglobulin G (IgG) and avidin-biotin complex (Vector Laboratories). Tissue sections were incubated with diaminobenzidine (DAB) solution to visualize the staining results.
Confocal images for the immunofluorescent stains of TβRII, Tgfb1 mRNA, p-Smad2, p-c-Jun, and total Smad2/3 were captured using a 60× objective on Nikon C2 microscope, and imported into NIH ImageJ to calculate corrected total cell fluorescence (CTCF, expressed as arbitrary units [a.u.]) for the relative abundance of each antigen (Lee et al., 2016). Similar approaches were used to quantify TH+ dopaminergic axon density in the striatum of control and DAT-iCre;TbRIIfl/fl mutants at E15.5, E17.5, P0, and P14.
In Situ Hybridization for Tgfb mRNA Expression
To detect the expression of Tgfb1, Tgfb2, and Tgfb3 mRNA in embryonic and postnatal mouse brains, we prepared anti-sense RNA probes for in situ hybridization using plasmids that contained cDNA for Tgfb1 (Open Biosystems), Tgfb2, or Tgfb3. The plasmids were linearized with appropriate restriction enzymes and transcribed with SP6 or T7 polymerase using digoxigenin (DIG)-labeling reagents and a DIG RNA labeling kit (Roche). Tissue and embryos were fixed overnight at 4°C in 4% PFA in diethylpyrocarbonate (DEPC)-treated PBS, cryoprotected in 15% and 30% sucrose, embedded in OCT, and sectioned at 40 μm thickness. During hybridization, free-floating sections were first washed with acetylation solution and 1% Triton X-100. Then sections were incubated with hybridization buffer (AMRESCO) for 2–4 hr before applying hybridization buffer containing DIG-labeled riboprobes (200–300 ng/mL) at 65°C overnight. For in situ hybridization using alkaline phosphatase (AP), sections were washed twice on day 2 with 0.2 × saline sodium citrate (SSC) solution in 0.1% Tween 20 (pH 4.5) at 65°C for 30 min (high-stringency wash), and with 100 mM maleic acid, 150 mM NaCl, 2 mM levamisole, and 0.1% Tween 20 (pH 7.5) for 10 min. Then, the sections were incubated with anti-DIG antibody overnight at 4°C, and the in situ hybridization results were developed using BM purple (Boehringer Mannheim). For fluorescence in situ hybridization (FISH), we used reagents from TSA Plus DNP (horseradish peroxidase [HRP]) system (NEL747A001KT, PerkinElmer) to detect mRNA. On day 2, sections were subjected to high-stringency wash at 65°C for 30 min twice, blocked with Tris-HCl, pH 7.5 (0.1M), NaCl (0.15M), blocking reagent (PerkinElmer FP1020, 0.5% w/v) (TNB) buffer for 30 min, and incubated with anti-DIG-HRP (1:100, #11207733910, Roche), followed by incubation with DNP Amplification Reagent (1:50) for 10 min, and by secondary antibody anti-dinitrophenyl-KLH conjugated with Alexa fluorophore 488 (A11097, Invitrogen) or anti-DNP-HRP conjugate (1:100) and Cyanine 3 Tyramide reagent (1:500, Perkin Elmer). Finally, the sections were mounted and dried under room temperature and mounted with either Clear Mount (Electron Microscopy Sciences) or Fluoromount-G (0100-01, Southern-Biotech). Images were captured using a Nikon C2 confocal microscope system or a BX53 Olympus microscope equipped with Olympus DP72 camera.
Stereology and Quantification for Dendrite Phenotype
Confocal images of cultured neurons stained with TH antibody were used for Neurolucida (MBF Bioscience) tracing and reconstruction. Sholl analysis was performed using Neurolucida Explorer software and biocytin injection used published methods (Marx et al., 2012). Briefly, 300-μM slices were prepared from P14 DAT-iCre;Ai14 or DAT-iCre;Ai14;TβRIIfl/fl mouse brains. After single tdTomato-positive DA neurons were filled with biocytin, sections were fixed with 4% PFA and washed in PBS. After quenching with 3% H2O2 solution, slices were incubated in 1% avidin-biotinylated HRP complex (ABC solution, Vector Labs) and developed using nickel- and cobalt-intensified DAB solution. Slices were mounted and dried on gelatinized microscope slides then dehydrated through ten ethanol concentrations and embedded in Eukitt embedding medium (Fluka). Manual 3D reconstruction of labeled neurons was done using an Olympus BX51 microscope and Neurolucida software.
Whole-Cell Patch-Clamp Recording of DA Neurons
Acute brain slices from both hemispheres containing SNpc were obtained from P14~P16 DAT-iCre;Ai14;TbRII+/+ (control) and DAT-iCre;Ai14;TbR2fl/fl mice of both genders. Animals were anesthetized with isoflurane, decapitated, and briefly exposed to chilled artificial cerebrospinal fluid (ACSF) containing 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 15 mM glucose, 2 mM CaCl2, and 1 mM MgCl2 oxygenated with 95% O2 and 5% CO2 (300~305 mOsm [pH 7.4]). Coronal or sagittal slices containing SNc were prepared (275 μm) using tissue vibratome (VT1200S, Leica), and slices were first maintained in ACSF for 30 min at 34°C and then for an additional 30 min at room temperature before recording. After recovery, slices were transferred to submerge recording chamber perfused with ACSF at a rate of 2~4 mL/min at 30~31°C (Warner Instrument).
Statistical Analyses
Statistical analyses for Figures 1P, 4V, and 4W used a mixed effects model (Stata) with mouse as the random effect and conditional knockout (DAT-iCre;TbRIIfl/fl) versus control (TbRIIfl/fl) as the fixed effect. The p values given are testing the hypothesis that the coefficient on the conditional knockout indicator variable is zero. Statistical analyses for other panels used the unpaired t test to compare means (GraphPad Prism 5, GraphPad Software).
Supplementary Material
Highlights.
Loss of TGF-β1 regulates excitatory-inhibitory inputs in DA neurons via autocrine mechanism
TGF-β signaling reduces axonal and dendritic growth in DA neurons
Mice lacking TGF-β signaling in DA neurons are persistently hyperactive
Mice lacking TGF-β signaling in DA neurons exhibit reversal-learning deficits
In Brief.
Luo et al. show that loss of TGF-β signaling in mature DA neurons leads to defects in axonal and dendritic growth and in the maintenance of balanced excitation-inhibition synaptic input in early postnatal life. Mice lacking TGF-β signaling in DA neurons exhibit hyperactivity and behavioral inflexibility in reversal learning.
ACKNOWLEDGMENTS
We thank Ivy Hsieh for help with the electron microscope (EM), Dr. Linda Noble and UCSF Neurobehavioral Core Facility for help with motor behavioral analyses, and Dr. Michael Kohn and Dr. Isabel Elaine Allen for statistical analyses. This work has been supported by grants from NIH NS081485 and NS098516 (E.J.H.), NS091144 (J.B.D.), DA037963 (J.L.W.), and from the Department of Veterans Affairs BX001108 and RX002133 (E.J.H.), the Klingenstein Foundation (J.B.D.), and funds provided by the state of California for medical research on alcohol and substance abuse through UCSF (J.L.W.). S.X.L. was supported by the A*STAR Scholar Program.
Footnotes
SUPPLEMENTAL INFORMATION Supplemental Information includes Supplemental Experimental Procedures and six figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.11.068.
AUTHOR CONTRIBUTIONS S.X.L. performed histology, cell biology, ultrastructural, and behavioral analyses on DAT-iCre;TbRIIfl/fl mice with assistance from L.T., K.S., and A.A.T. J.L.W. provided training and facility for the reversal learning and helped with data interpretations. J.-I.K. performed electrophysiological recordings in DA neurons, with input and supervision from J.B.D. Y.S. generated luciferase constructs to characterize the transcriptional control of Tgfb1 expression by R-Smad and c-Jun. E.J.H. provided project oversight. S.X.L. and E.J.H. wrote the manuscript.
REFERENCES
- Adamantidis AR, Tsai HC, Boutrel B, Zhang F, Stuber GD, Budygin EA, Touriño C, Bonci A, Deisseroth K, de Lecea L. Optogenetic interrogation of dopaminergic modulation of the multiple phases of reward-seeking behavior. J. Neurosci. 2011;31:10829–10835. doi: 10.1523/JNEUROSCI.2246-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM, Lim B, Matzilevich D, Walsh JP, Subburaju S, Minns M. Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc. Natl. Acad. Sci. USA. 2007;104:10164–10169. doi: 10.1073/pnas.0703806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialas AR, Stevens B. TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat. Neurosci. 2013;16:1773–1782. doi: 10.1038/nn.3560. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Blythe SN, Wokosin D, Atherton JF, Bevan MD. Cellular mechanisms underlying burst firing in substantia nigra dopamine neurons. J. Neurosci. 2009;29:15531–15541. doi: 10.1523/JNEUROSCI.2961-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Angers A, Cleary LJ, Eskin A, Byrne JH. Transforming growth factor beta1 alters synapsin distribution and modulates synaptic depression in Aplysia. J. Neurosci. 2002;22:RC220. doi: 10.1523/JNEUROSCI.22-09-j0004.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chytil A, Magnuson MA, Wright CV, Moses HL. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis. 2002;32:73–75. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
- Dagher A, Robbins TW. Personality, addiction, dopamine: Insights from Parkinson's disease. Neuron. 2009;61:502–510. doi: 10.1016/j.neuron.2009.01.031. [DOI] [PubMed] [Google Scholar]
- Diniz LP, Tortelli V, Garcia MN, Araújo AP, Melo HM, Silva GS, Felice FG, Alves-Leon SV, Souza JM, Romão LF, et al. Astrocyte transforming growth factor beta 1 promotes inhibitory synapse formation via CaM kinase II signaling. Glia. 2014;62:1917–1931. doi: 10.1002/glia.22713. [DOI] [PubMed] [Google Scholar]
- Donato F, Rompani SB, Caroni P. Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature. 2013;504:272–276. doi: 10.1038/nature12866. [DOI] [PubMed] [Google Scholar]
- Donato F, Chowdhury A, Lahr M, Caroni P. Early- and late-born parvalbumin basket cell subpopulations exhibiting distinct regulation and roles in learning. Neuron. 2015;85:770–786. doi: 10.1016/j.neuron.2015.01.011. [DOI] [PubMed] [Google Scholar]
- Engblom D, Bilbao A, Sanchis-Segura C, Dahan L, Perreau-Lenz S, Balland B, Parkitna JR, Luján R, Halbout B, Mameli M, et al. Glutamate receptors on dopamine neurons control the persistence of cocaine seeking. Neuron. 2008;59:497–508. doi: 10.1016/j.neuron.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: From actions to habits to compulsion. Nat. Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Fuentes-Medel Y, Ashley J, Barria R, Maloney R, Freeman M, Budnik V. Integration of a retrograde signal during synapse formation by glia-secreted TGF-β ligand. Curr. Biol. 2012;22:1831–1838. doi: 10.1016/j.cub.2012.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, Davis GW. The BMP ligand Gbb gates the expression of synaptic homeostasis independent of synaptic growth control. Neuron. 2007;56:109–123. doi: 10.1016/j.neuron.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zhang H, Yung A, Villeda SA, Jaeger PA, Olayiwola O, Fainberg N, Wyss-Coray T. ALK5-dependent TGF-β signaling is a major determinant of late-stage adult neurogenesis. Nat. Neurosci. 2014;17:943–952. doi: 10.1038/nn.3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henny P, Brown MT, Northrop A, Faunes M, Ungless MA, Magill PJ, Bolam JP. Structural correlates of heterogeneous in vivo activity of midbrain dopaminergic neurons. Nat. Neurosci. 2012;15:613–619. doi: 10.1038/nn.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyland BI, Reynolds JN, Hay J, Perk CG, Miller R. Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience. 2002;114:475–492. doi: 10.1016/s0306-4522(02)00267-1. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Scanziani M. How inhibition shapes cortical activity. Neuron. 2011;72:231–243. doi: 10.1016/j.neuron.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo A, Jentsch JD. Reversal learning as a measure of impulsive and compulsive behavior in addictions. Psychopharmacology (Berl.) 2012;219:607–620. doi: 10.1007/s00213-011-2579-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C, Wilbrecht L. Juvenile mice show greater flexibility in multiple choice reversal learning than adults. Dev. Cogn. Neurosci. 2011;1:540–551. doi: 10.1016/j.dcn.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- Koeglsperger T, Li S, Brenneis C, Saulnier JL, Mayo L, Carrier Y, Selkoe DJ, Weiner HL. Impaired glutamate recycling and GluN2B-mediated neuronal calcium overload in mice lacking TGF-β1 in the CNS. Glia. 2013;61:985–1002. doi: 10.1002/glia.22490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieglstein K, Suter-Crazzolara C, Fischer WH, Unsicker K. TGF-beta superfamily members promote survival of midbrain dopaminergic neurons and protect them against MPP+ toxicity. EMBO J. 1995;14:736–742. doi: 10.1002/j.1460-2075.1995.tb07052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Shang Y, Redmond SA, Urisman A, Tang AA, Li KH, Burlingame AL, Pak RA, Jovičić A, Gitler AD, et al. Activation of HIPK2 Promotes ER Stress-Mediated Neurodegeneration in Amyotrophic Lateral Sclerosis. Neuron. 2016;91:41–55. doi: 10.1016/j.neuron.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo SX, Huang EJ. Dopaminergic neurons and brain reward pathways: From neurogenesis to circuit assembly. Am. J. Pathol. 2016;186:478–488. doi: 10.1016/j.ajpath.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx M, Günter RH, Hucko W, Radnikow G, Feldmeyer D. Improved biocytin labeling and neuronal 3D reconstruction. Nat. Protoc. 2012;7:394–407. doi: 10.1038/nprot.2011.449. [DOI] [PubMed] [Google Scholar]
- Overton PG, Clark D. Burst firing in midbrain dopaminergic neurons. Brain Res. Brain Res. Rev. 1997;25:312–334. doi: 10.1016/s0165-0173(97)00039-8. [DOI] [PubMed] [Google Scholar]
- Poulsen KT, Armanini MP, Klein RD, Hynes MA, Phillips HS, Rosenthal A. TGF beta 2 and TGF beta 3 are potent survival factors for midbrain dopaminergic neurons. Neuron. 1994;13:1245–1252. doi: 10.1016/0896-6273(94)90062-0. [DOI] [PubMed] [Google Scholar]
- Roussa E, Wiehle M, Dünker N, Becker-Katins S, Oehlke O, Krieglstein K. Transforming growth factor beta is required for differentiation of mouse mesencephalic progenitors into dopaminergic neurons in vitro and in vivo: Ectopic induction in dorsal mesencephalon. Stem Cells. 2006;24:2120–2129. doi: 10.1634/stemcells.2005-0514. [DOI] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima T, Shimoda K, Johnston JM, Commissiong JW. Standardized methods to bioassay neurotrophic factors for dopaminergic neurons. J. Neurosci. Methods. 1996;67:27–41. [PubMed] [Google Scholar]
- Tang M, Villaescusa JC, Luo SX, Guitarte C, Lei S, Miyamoto Y, Taketo MM, Arenas E, Huang EJ. Interactions of Wnt/beta-catenin signaling and sonic hedgehog regulate the neurogenesis of ventral midbrain dopamine neurons. J. Neurosci. 2010;30:9280–9291. doi: 10.1523/JNEUROSCI.0860-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HC, Zhang F, Adamantidis A, Stuber GD, Bonci A, de Lecea L, Deisseroth K. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science. 2009;324:1080–1084. doi: 10.1126/science.1168878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turiault M, Parnaudeau S, Milet A, Parlato R, Rouzeau JD, Lazar M, Tronche F. Analysis of dopamine transporter gene expression pattern – generation of DAT-iCre transgenic mice. FEBS J. 2007;274:3568–3577. doi: 10.1111/j.1742-4658.2007.05886.x. [DOI] [PubMed] [Google Scholar]
- Vashlishan AB, Madison JM, Dybbs M, Bai J, Sieburth D, Ch'ng Q, Tavazoie M, Kaplan JM. An RNAi screen identifies genes that regulate GABA synapses. Neuron. 2008;58:346–361. doi: 10.1016/j.neuron.2008.02.019. [DOI] [PubMed] [Google Scholar]
- Wang LP, Li F, Wang D, Xie K, Wang D, Shen X, Tsien JZ. NMDA receptors in dopaminergic neurons are crucial for habit learning. Neuron. 2011;72:1055–1066. doi: 10.1016/j.neuron.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron. 2012;74:858–873. doi: 10.1016/j.neuron.2012.03.017. [DOI] [PubMed] [Google Scholar]
- Yi JJ, Barnes AP, Hand R, Polleux F, Ehlers MD. TGF-beta signaling specifies axons during brain development. Cell. 2010;142:144–157. doi: 10.1016/j.cell.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Endo S, Cleary LJ, Eskin A, Byrne JH. Role of transforming growth factor-beta in long-term synaptic facilitation in Aplysia. Science. 1997;275:1318–1320. doi: 10.1126/science.275.5304.1318. [DOI] [PubMed] [Google Scholar]
- Zhang J, Pho V, Bonasera SJ, Holtzman J, Tang AT, Hellmuth J, Tang S, Janak PH, Tecott LH, Huang EJ. Essential function of HIPK2 in TGFbeta-dependent survival of midbrain dopamine neurons. Nat. Neurosci. 2007;10:77–86. doi: 10.1038/nn1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, Darvas M, Kim MJ, Mizumori SJ, Paladini CA, et al. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc. Natl. Acad. Sci. USA. 2009;106:7281–7288. doi: 10.1073/pnas.0813415106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.