

Abstract
Background
Disturbed amyloid precursor protein (APP) processing is considered to be central to the pathogenesis of Alzheimer’s disease (AD). The autosomal dominant form of the disease, familial AD (FAD), may serve as a model for the sporadic form of AD. In FAD the diagnosis of AD is reliable and presymptomatic individuals carrying FAD mutations can give valuable insights into the earliest stages of the disease where therapeutic interventions are thought to be the most effective.
Methods
In the current cross-sectional study, products of APP processing (e.g., sAPPα, sAPPβ, Aβ38, Aβ40 and Aβ42) were measured in the cerebrospinal fluid (CSF) of individuals carrying one of three FAD mutations, APPswe (p.KM670/671NL), APParc (p.E693G) and PSEN1 (p.H163Y), as well as in non-mutation carriers from the same families.
Results
We observed pathological APP processing in presymptomatic carriers of FAD mutations, with different profiles of APP and Aβ isoforms in the three mutation carrier groups, APPswe (p.KM670/671NL), APParc (p.E693G) and PSEN1 (p.H163Y), except for the well-established decrease in CSF Aβ42 that was found with all mutations.
Conclusions
These findings add to the current evidence that AD pathophysiology differs between disease-causing mutations and can be monitored in the presymptomatic disease stage by CSF analyses. This may also be important from a therapeutic standpoint, by opening a window to monitor effects of disease-modifying drugs on AD pathophysiology.
Keywords: Alzheimer’s disease, Amyloid precursor protein, Biomarkers, Cerebrospinal fluid, Genetics
Background
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder with pathological hallmarks in the brain, including extracellular amyloid-beta (Aβ) plaques and intracellular tangles of hyperphosphorylated tau protein. According to the amyloid cascade hypothesis, accumulation of Aβ in the brain is the driver of the disease process [1]. Aβ isoforms ending at amino acid 42 (Aβ42) are the most prone to aggregate, both into synaptotoxic oligomers and into fibrils, which are the main components of neuritic plaques [2]. Aβ is a product of the cleavage of the amyloid precursor protein (APP) by the proteases β-secretase (BACE1) and γ-secretase. Following APP proteolysis at the cell surface, Aβ and sAPPβ, are shed into the brain interstitial fluid, which communicates freely with the cerebrospinal fluid (CSF). APP can also be cleaved within its Aβ sequence by α-secretase in a nonamyloidogenic pathway, releasing a different N-terminal APP fragment, sAPPα [3].
The processing of APP is interesting from a therapeutic standpoint because modulation/inhibition at different stages can be feasible to direct APP processing towards the nonamyloidogenic pathway. Measuring the products of APP processing in CSF can clarify how the processing differs in vivo in individuals with AD pathology, compared with healthy controls. This is of particular interest in patients who are still in the preclinical stage of AD, where disease-modifying drug candidates are most likely to be successful.
To learn more about APP and Aβ metabolism in familial AD (FAD), we measured sAPPα, sAPPβ, Aβ38, Aβ40 and Aβ42 in the CSF of individuals carrying autosomal dominant FAD-causing mutations, comparing them with noncarriers from the same families. Most of the subjects were still in the preclinical stage of the disease. We included carriers of three different mutations, APPswe [4, 5], APParc [6] and PSEN1 H163Y [7, 8], each of which causes distinct changes in APP processing in vitro. The effects of the different FAD mutations are illustrated in Fig. 1.
Fig. 1.
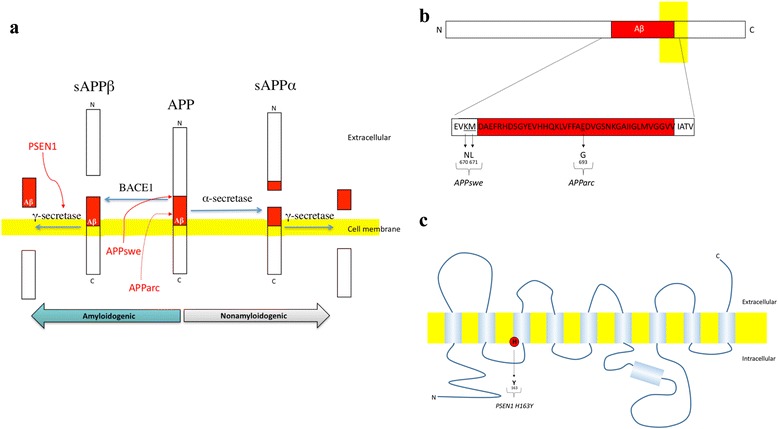
Effects of the APPswe, APParc and PSEN1 H163Y mutations on APP processing in vitro. a APPswe mutation is located in the extracellular domain of APP, at the BACE1 cleavage site, causing a 5–10-fold increase in the production of Aβ40 and Aβ42 [34]. Presenilin 1 is a subunit of the γ-secretase and most PSEN1 mutations modulate the γ-secretase cleavage site-preference in a disease-promoting manner by inhibiting cleavages at Gly37, Gly38 and Val39 in the Aβ sequence, without affecting the production of Aβ42 and Aβ40 significantly [35]. Finally, the arctic APP mutation, located within the Aβ sequence, leads to the production of Aβ with higher propensity for protofibril formation than wild-type Aβ [6]. b More detailed overview of the position of the APPswe and APParc mutations within APP. Letters, amino acids; numbers, position of the amino acids within the APP sequence. c Schematic illustration of the transmembrane protein presenilin 1, a subunit of γ-secretase. The PSEN1 H163Y mutation is located in the third transmembrane domain of the protein. APP amyloid precursor protein
Aβ42 is a well-established biomarker, which is decreased in the CSF of individuals with AD, most probably due to the accumulation of Aβ42 in the brain [9]. The same does not apply to CSF Aβ40, which has been shown to be unchanged in AD [10–12]. Previous studies have shown conflicting results regarding the levels of other APP processing products in AD. CSF Aβ38 has been reported unchanged or increased in sporadic AD (SAD) [13, 14] but decreased in FAD [15, 16]. Aβ16 has been shown to be increased both in SAD and FAD [15, 16]. The levels of sAPPα and sAPPβ have been reported to be similar in FAD, SAD and controls [17, 18], but an increase in sAPPβ has been observed when comparing SAD with frontotemporal dementia [19]. When patients with SAD were compared with patients assessed as having other dementias than AD, the SAD patients had increased levels of both sAPPα and sAPPβ [20]. In contrast, one study reported a decrease in sAPPα in carriers of the APPswe mutation [21] and another a decrease both in sAPPα and sAPPβ in patients with advanced AD compared with healthy controls [10].
Here we speculated that the levels of the products of APP processing would already be abnormal in preclinical FAD and differ depending on the underlying mutation: that the carriers of the APPswe mutation would have increased levels of sAPPβ, Aβ38 and Aβ40 as well as decreased levels of sAPPα due to the increased affinity of BACE1 to the mutated APP sequence; that the APParc mutation carriers would have normal sAPPα and sAPPβ levels but decreased levels of Aβ42, and potentially Aβ40 due to fibril formation; and that subjects with the PSEN1 H163Y mutation would also have normal levels of sAPPα and sAPPβ, but decreased levels of Aβ38 as the mutated γ-secretase preferentially produces Aβ40 and Aβ42.
In the therapeutic sense it was interesting to see whether these hypotheses would be correct or whether we would find evidence of a common pathological aspect of APP processing in carriers of the different FAD mutations.
Finally, we calculated the sAPPβ/sAPPα ratio and the Aβ42/Aβ40 ratio, both of which are of interest as potential biomarkers.
Methods
Study population
Members of three Swedish families segregating three known mutations leading to autosomal dominant AD, APPswe (p.KM670/671NL), APParc (p.E693G) and PSEN1 (p.H163Y), were included in the study. The average age at onset of the first clinically relevant symptoms for each family is: 54 ± 4 years for APPswe (based on 19 affected cases), 56 ± 4 years for APParc (based on 12 affected cases) and 51 ± 7 years for PSEN1 H163Y (based on 11 affected cases). The average age at onset in each family is normally distributed and is therefore presented as a mean with standard deviation. Participants were recruited to a longitudinal clinical and experimental FAD study through the Genetics Unit, which provides genetic counseling at the Memory Clinic at the Karolinska University Hospital, and were examined between 1993 and 2011. The FAD study is a prospective study including a thorough clinical evaluation, a comprehensive neuropsychological test battery, neuroimaging (3Tesla MRI), electroencephalography and biochemical assessments, including collection of CSF. For AD diagnosis, the criteria for dementia according to the National Institute of Neurological Disorders and Stroke, Washington DC, and Alzheimer Disease and Related Disorders Association, Washington DC (NINCDS-ADRDA) were used [22]; and for the diagnosis of mild cognitive impairment (MCI) we used the criteria of the International Working Group on Mild Cognitive Impairment [23]. In a person with MCI the ability to perform activities of daily living is preserved, while at the same time there is evidence of cognitive decline either objectively measured over time or subjectively reported by the individual/an informant in conjunction with objective deficits. The study subjects are a subsample of individuals at 50% risk of carrying one of the mutations leading to FAD who gave informed written consent to and underwent lumbar puncture (n = 36). The CSF was collected at baseline/the entrance of each participant into the study, during the time period of 1993–2011, and the age of the participants presented in the following sections is the age at CSF collection. Of the 36 subjects who underwent the procedure, 18 came from a family segregating the APPswe mutation, 10 from a family segregating the APParc mutation and eight from a family segregating the PSEN1 H163Y mutation. All of the subjects received genetic counseling in conjunction with their participation in the study. The participants, clinicians and researchers involved in the study were blind to the mutation status of the asymptomatic participants unless the participant opted for presymptomatic genetic testing (in a clinical genetics setting). All study procedures were approved by the Regional Ethical Review Board in Stockholm, Sweden.
Cerebrospinal fluid sampling
CSF samples were collected by lumbar puncture in the L3/L4 or L4/L5 interspace at variable time points during the day. According to current recommendations on CSF sampling and handling, diurnal variation of AD biomarkers should not be a concern [24]. Some of the participants received premedication with 5 mg diazepam and 1 g paracetamol prior to the procedure. Immediately after collection the CSF was centrifuged at 2200 × g at room temperature for 10 minutes. The supernatant was collected and aliquoted into polypropylene cryotubes. The aliquots used in the current study were stored at −80 °C and had been thawed and refrozen once before being thawed for analysis in this study.
Assay of sAPPα, sAPPβ, Aβ38, Aβ40 and Aβ42
The CSF samples were all analyzed at the same time by electrochemiluminescence technology (Meso Scale Discovery, Gaithersburg, MD, USA). An analysis of the stability of the frozen CSF samples had been performed previously and did not raise concerns. Concentrations of Aβ38, Aβ40 and Aβ42 were analyzed using the MS6000 Human Abeta 3-Plex Ultra-Sensitive Kit (the 6E10 version), while β-secretase cleaved soluble APP (sAPP-β) and α-secretase cleaved soluble APP (sAPP-α) in CSF were analyzed using the MS6000 Human sAPPalpha/sAPPbeta Kit, following the recommendations by the manufacturer, and as described previously [25]. Because the APPswe mutation changes the neo-epitope recognized by the capturing antibody in the sAPPβ assay, CSF sAPPβ levels in carriers of the APPswe mutation were not included in the study.
Genetic analysis
Apolipoprotein E
The APOE genotyping was performed for SNPs rs7412 and rs429358 using TaqMan® SNP Genotyping Assays (ABI, Foster City, CA, USA) according to the manufacturer’s protocol. The amplified products were run on 7500 fast Real-Time PCR Systems (ABI).
Mutation analyses in APP and PSEN1
Exons 16 and 17 in APP were sequenced to screen for the KM670/671NL and the E693G mutations [12, 14]. To confirm the H163Y mutation in PSEN1, exon 6 was sequenced [26]. DNA was amplified using AmpliTaq Gold® 360 PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Primer sequences and PCR conditions are available upon request. The Big Dye® terminator v3.1 Cycle sequencing Kit (Applied Biosystems, Austin, TX, USA) was used for Sanger sequencing. The exons in APP and PSEN1 were sequenced in both directions and analyzed on an ABI3500 Genetic Analyzer (Applied Biosystems, Foster City).
Statistical analysis
Because of the fact that the age at symptom onset in FAD varies between families, each subject’s age relative to the mean age at clinical onset in their respective family was calculated. Unpaired t tests and Pearson correlations were applied to normally distributed data, while the Mann–Whitney U test and Spearman correlations were performed on data that was not normally distributed. The distribution of data was assessed with the D’Agostino–Pearson normality test. Thus age, years to predicted family specific clinical onset and CSF biomarker levels were compared between noncarriers of the FAD mutations and the mutation carrier group as a whole as well as the noncarriers and carriers of each of the three mutations separately. Fisher’s exact test was used to compare gender and the frequency of the APOE ε4 allele between the noncarriers and each of the mutation carrier groups.
Exploratory Pearson/Spearman correlations were performed between biomarker levels, as well as between biomarker levels and predicted age at symptom onset. Two-tailed p < 0.05 was considered significant.
Results
Demographics of the study population
Of the 36 subjects who underwent a lumbar puncture, 19 were mutation carriers (MC) and 17 were noncarriers (NC). Of the MC, six carried a PSEN1 H163Y mutation, four carried an APParc mutation and nine carried an APPswe mutation (see Table 1 for a summary of the demographic data of the study population). There was no significant difference between the MC group as a whole, or any of the MC subgroups, and the NC regarding gender distribution, mean age or mean number of years to predicted family specific onset. For the sake of anonymity, the gender distribution in each mutation group is not revealed in Table 1, because this could cause certain participants to be able to deduct their mutation status from the data. The PSEN1 carriers were somewhat younger than the NC, but this difference did not reach statistical significance. Furthermore, there was no significant difference in the prevalence of APOE ε4 carriers between the NC group and the different MC groups and there were only two individuals, a noncarrier and a carrier of the APPswe mutation, who were APOE ε4 homozygotes. At the time of CSF sampling, three of the APPswe carriers fulfilled the diagnostic criteria for AD with dementia, two APPswe carriers and one PSEN1 carrier fulfilled the criteria for MCI and the other 30 subjects (13 MC and 17 NC) were free of symptoms.
Table 1.
Demographics of the study population
| All MC (n = 19) | APPswe (n = 9) | APParc (n = 4) | PSEN1 (n = 6) | NC (n = 17) | |
|---|---|---|---|---|---|
| Age (SD) | 46.7 (11.9) | 51.2 (12.1) | 50 (6.6) | 33.7 (7.0) | 47.8 (10.2) |
| Years to onset (SD) | −7.7 (12.3) | −1.8 (12.1) | −7 (6.6) | −21.3 (7.0) | −6.9 (9.9) |
| ApoE ε4 carrier | 9/19 | 5/9 | 1/4 | 3/6 | 7/17 |
| Normal cognition | 13/19 | 4/9 | 4/4 | 5/6 | 17/17 |
| MCI | 3 | 2 | - | 1 | - |
| AD | 3 | 3 | - | - | - |
AD Alzheimer’s disease, MC mutation carriers, MCI mild cognitive impairment, NC noncarriers
Levels of sAPPα, sAPPβ, Aβ38, Aβ40 and Aβ42
There was no significant difference in the levels of sAPPα when comparing the MC group as a whole with the NC. The same applied to sAPPβ when the PSEN1 and APParc carriers were grouped together and compared with the NC. In contrast, all of the measured Aβ isoforms, Aβ38, Aβ40 and Aβ42, were significantly lower in the MC group. When the MC were grouped by their specific mutation, a somewhat different pattern emerged. The carriers of the APPswe mutation had significantly lower levels of sAPPα than NC (note that APPswe MC were not included in the analysis of CSF sAPPβ because the end-specific capture antibody in the assay will not react with sAPPβ modified at positions 670/671 by the mutation), normal levels of Aβ38 and Aβ40 and significantly decreased levels of Aβ42. The APParc carriers shared the same pattern of the Aβ isoforms as the APPswe carriers, but had normal levels of sAPPα and high sAPPβ levels. Finally, the PSEN1 carriers had low levels of both Aβ38 and Aβ42, and normal levels of Aβ40, sAPPα and sAPPβ. See Fig. 2 for a summary of the levels of the APP processing products.
Fig. 2.
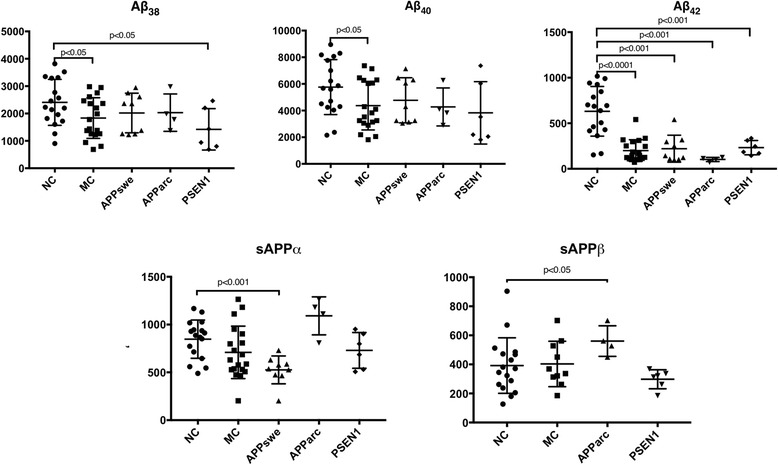
CSF levels of sAPPα, sAPPβ, Aβ38, Aβ40 and Aβ42 in carriers of three FAD mutations. Scatter plots showing levels of sAPPα, sAPPβ, Aβ38, Aβ40 and Aβ42 in carriers of three different mutations leading to FAD compared with healthy family members (NC). The mutation carriers were compared with the NC as one group (MC) and by type of mutation (APPswe, APParc and PSEN1). Only carriers of the APParc and PSEN1 mutations are included in the calculation of the mean level of sAPPβ. The biomarker levels in the NC group were normally distributed, except for sAPPβ. Thus, the Mann–Whitney U test was used when comparing sAPPβ between groups. Also, the Mann–Whitney U test was used when comparing the APParc and PSEN1 carriers with the NC due to the small sample sizes of these two MC groups. The unpaired t test was used for the rest of the data, except when comparing Aβ42 between the NC and MC and comparing sAPPα between the NC and APPswe (here we used the Mann–Whitney U test because these data were not normally distributed). Aβ amyloid beta, MC mutation carriers, NC noncarriers
These calculations were repeated after excluding all subjects with MCI or AD diagnosis. This did not change the results for the levels of sAPPα, sAPPβ and Aβ42, with all of the significant differences in Fig. 2 remaining significant. The levels of Aβ38 were no longer significantly lower in the MC group as a whole when subjects with MCI and AD had been excluded, but remained significantly low in the PSEN1 carrier group. The results for Aβ40 remained unchanged after removing MCI and AD subjects, except the Aβ40 levels of the PSEN1 carriers, which were significantly lower than in the NC when the PSEN1 carrier with MCI diagnosis was excluded.
sAPPβ/sAPPα ratio and Aβ42/Aβ40 ratio
There was no significant difference in the sAPPβ/sAPPα ratio when comparing the PSEN1 and APParc carriers with the NC, irrespective of whether the NC were compared with the MC as a whole or with a subgroup with a specific mutation. The same applied when the NC were compared with the presymptomatic PSEN1 carriers only (data not shown).
The Aβ42/Aβ40 ratio was significantly lower in the MC group as a whole than in the NC group (0.05 vs 0.11, p < 0.0001). The same applied to all the subgroups, the APPswe carriers vs the NC (0.04 vs 0.11 p < 0.0001), the presymptomatic APPswe carriers vs the NC (0.05 vs 0.11, p = 0.002), the APParc carriers vs the NC (0.03 vs 0.11, p < 0.001), the PSEN1 carriers vs the NC (0.07 vs 0.11, p < 0.01) and the presymptomatic PSEN1 carriers vs the NC (0.07 vs 0.11, p < 0.01).
Correlations between the different products of APP processing
Because of the small size of the APParc mutation carrier group (n = 4) no correlation calculations were performed involving that group alone. As has been reported previously [27], there was a positive correlation between sAPPα and sAPPβ in the MC group as a whole (excluding the APPswe carriers as explained earlier), as well as in the NC group. When looking only at the PSEN1 carriers, this correlation failed to reach significance, probably due to the small number of individuals in the group.
A strong positive correlation was observed between Aβ38 and Aβ40 in all groups. There was a significant correlation between Aβ40 and Aβ42 as well, in all groups except the PSEN1 carriers. Interestingly, there was a positive correlation between sAPPα and all the measured Aβ isoforms in the NC group, but not in any of the mutation carrier groups. Also, the NC group was the only group showing a significant positive correlation between Aβ38 and Aβ42.
The correlations between sAPPα, sAPPβ, Aβ38, Aβ40 and Aβ42 are summarized in Table 2.
Table 2.
Correlations between the products of APP processing in FAD mutation carriers
| CSF biomarker | sAPPα | sAPPβ | Aβ38 | Aβ40 | Aβ42 | |
|---|---|---|---|---|---|---|
| MC | sAPPα | 0.87 (<0.01) | 0.2 (0.4) | 0.09 (0.7) | -0.2 (0.4) | |
| sAPPβ | 0.87 (<0.01) | 0.2 (0.3) | 0.1 (0.6) | -0.2 (0.5) | ||
| Aβ38 | 0.2 (0.4) | 0.2 (0.3) | 0.93 (<0.0001) | 0.44 (0.06) | ||
| Aβ40 | 0.09 (0.7) | 0.1 (0.6) | 0.93 (<0.0001) | 0.6 (0.01) | ||
| Aβ42 | -0.2 (0.4) | -0.2 (0.5) | 0.44 (0.06) | 0.6 (0.01) | ||
| APPswe | sAPPα | 0.2 (0.6) | 0.2 (0.7) | -0.03 (0.9) | ||
| sAPPβ | n.a. | n.a. | n.a. | n.a. | n.a. | |
| Aβ38 | 0.2 (0.6) | 0.9 (<0.001) | 0.7 (0.06) | |||
| Aβ40 | 0.2 (0.7) | 0.9 (<0.001) | 0.8 (0.01) | |||
| Aβ42 | -0.03 (0.9) | 0.7 (0.06) | 0.8 (0.01) | |||
| PSEN1 | sAPPα | 0.66 (0.18) | 0.20 (0.7) | 0.20 (0.7) | 0.086 (0.9) | |
| sAPPβ | 0.66 (0.18) | 0.60 (0.2) | 0.60 (0.2) | 0.54 (0.3) | ||
| Aβ38 | 0.20 (0.7) | 0.60 (0.2) | 1 (<0.01) | 0.83 (0.06) | ||
| Aβ40 | 0.20 (0.7) | 0.60 (0.2) | 1 (<0.01) | 0.83 (0.06) | ||
| Aβ42 | 0.086 (0.9) | 0.54 (0.3) | 0.83 (0.06) | 0.83 (0.06) | ||
| NC | sAPPα | 0.64 (<0.01) | 0.71 (<0.01) | 0.72 (<0.01) | 0.65 (<0.01) | |
| sAPPβ | 0.64 (<0.01) | 0.28 (0.3) | 0.33 (0.2) | 0.29 (0.3) | ||
| Aβ38 | 0.71 (<0.01) | 0.28 (0.3) | 0.98 (<0.0001) | 0.95 (<0.0001) | ||
| Aβ40 | 0.72 (<0.01) | 0.33 (0.2) | 0.98 (<0.0001) | 0.98 (<0.0001) | ||
| Aβ42 | 0.65 (<0.01) | 0.29 (0.3) | 0.95 (<0.0001) | 0.98 (<0.0001) |
Correlations between the products of APP processing in the mutation carrier group as a whole (MC), carriers of the APPswe and PSEN1 mutations separately and in the nonmutation carriers (NC)
Numbers represent Pearson/Spearman correlation coefficients (p values) (significant correlations in italics)
APP amyloid precursor protein, FAD familial Alzheimer’s disease, Aβ amyloid beta, MC mutation carriers, NC noncarriers
Correlations between sAPPα, sAPPβ, Aβ38, Aβ40 and Aβ42 and years to expected symptom onset
Correlations between sAPPα, sAPPβ, Aβ38, Aβ40, Aβ42 and years to expected symptom onset were calculated to see whether the levels of these markers changed as the onset of symptoms approached. No such changes were observed in any of the five markers in the whole MC group, the PSEN1 group or in the NC group. In the APPswe mutation carriers, the concentration of Aβ38 (r = −0.69, p = 0.04), Aβ40 (r = −0.67, p = 0.05) and Aβ42 (r = −0.86, p < 0.01) decreased significantly when the expected symptom onset approached and beyond (see Fig. 3). When subjects with a dementia diagnosis were excluded from the APPswe mutation carrier group these correlations failed to reach significance, apart from Aβ42, which still showed a decrease as age at onset approached (r = −0.85, p = 0.03). It was not possible to repeat this calculation with only presymptomatic APPswe mutation carriers because their number is too small.
Fig. 3.
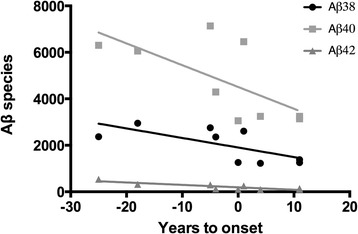
APPswe: correlations between years to onset and Aβ species. Pearson correlations between the Aβ isoforms Aβ38, Aβ40 and Aβ42 and years to expected onset of the first clinically relevant symptoms of FAD in carriers of the APPswe mutation. X axis, −10 represents an individual with 10 years left to the expected onset (which is set to 0) while 10 represents an individual 10 years past the expected onset. All three correlations were significant, Aβ38 (r = −0.69, p = 0.04), Aβ40 (r = −0.67, p = 0.05) and Aβ42 (r = −0.86, p < 0.01), decreasing in subjects sampled closer to the expected onset. Aβ amyloid beta
Correction for multiple comparisons
All of the significant correlations presented in Table 2 remained significant after correcting for multiple comparisons using the false discovery rate (FDR) correction. The same applied to the biomarker levels presented in Fig. 2, except for the low Aβ38 levels in the MC and the PSEN1 groups and the low Aβ40 levels in the MC group which did not survive this correction.
Discussion
Compared with noncarriers, carriers of the APPswe mutation have significantly decreased CSF levels of sAPPα and Aβ42 whereas the levels of Aβ38 and Aβ40 were not significantly different. The decreased levels of Aβ42 are expected, because Aβ42 is a well-established AD biomarker and has been shown to be decreased in AD patients in numerous studies [9]. The sAPPα levels have also been shown to be reduced in a previous study on some of the same individuals [21]. This could be due to the increased activity of the amyloidogenic pathway of APP processing in APPswe carriers at the expense of the nonamyloidogenic pathway in which sAPPα is produced.
In the PSEN1 carriers the CSF levels of sAPPα and sAPPβ were unchanged, whereas sAPPβ was increased in the APParc carriers. The APParc carriers also showed the expected decrease in Aβ42 levels, while both Aβ38 and Aβ42 were decreased in the PSEN1 carriers. The decrease in Aβ38 has been observed in previous studies on PSEN1 carriers [15, 16] and the same applies to the normal levels of sAPPα and sAPPβ in the PSEN1 carriers [17, 18]. Amyloid plaques including Aβ38 deposits, have been shown to be limited to FAD cases and this accumulation of Aβ38 in the brain could explain the low levels Aβ38 in CSF [28]. Another, possibly coexisting, explanation could be that the mutated γ-secretase preferentially produces Aβ40 and Aβ42, at the expense of Aβ38. The decrease in Aβ38 levels seems quite robust, remaining significant when only presymptomatic PSEN1 carriers were included. In contrast, the observed reduced levels of Aβ38 for the whole MC group were no longer significantly low when looking only at presymptomatic individuals. Taken together our results emphasize the differences in production and metabolism of Aβ isoforms in different FAD mutations. The results for the PSEN1 and APParc carriers fit our hypothesis quite nicely, apart from the observed increased levels of sAPPβ in the APParc carriers. A possible explanation for this observation might be that increased fibril formation and lack of monomeric Aβ isoforms somehow stimulates amyloidogenic APP processing.
When looking at correlations between years to symptom onset and the different APP processing products it is interesting to see that the products remain stable in subjects at different time points from expected onset in the MC group as a whole and in the PSEN1 carriers alone. The Aβ isoforms remained low in the MC compared with the NC, while there was no difference in the levels of sAPPα and sAPPβ in the MC and the NC. Very few studies have looked at APP processing in preclinical FAD [15–17] and to our knowledge none have correlated the APP processing products to years to symptom onset.
According to one study on SAD, there were no differences in BACE1 activity, sAPPα, sAPPβ and Aβ40 concentrations between AD patients and controls until the levels of all of these biomarkers dropped in advanced stages of AD [10]. The only mutation carrier group in the current study that showed a change in these biomarkers in relation to expected symptom onset was the APPswe group, in which the levels of all of the Aβ isoforms decreased with progression of the disease. This resonates quite well with the study by Rosen et al. [10] on SAD, because the APPswe mutation carriers were the only group including individuals that already had a dementia diagnosis. The correlation ceased to be significant when the subjects with dementia were excluded, apart from Aβ42, which still showed a decrease over time. A longitudinal study on the subjects included here is ongoing and may shed light on the changes in these markers over time through repeated sampling from the same individuals.
We replicated the previously observed positive correlation between sAPPα and sAPPβ in both the MC group (excluding the APPswe carriers) and the NC group. This observation correlates well with other studies including healthy individuals as well as subjects with MCI, FAD and SAD, suggesting that the α and β pathways are noncompetitive [17, 20, 26, 27, 29, 30]. This positive correlation also applies to the MC in this study, possibly excluding the APPswe carriers because they had low levels of sAPPα. Because of limitations in the sAPPβ assay we do not have information on the levels of sAPPβ in the APPswe carriers and therefore can only speculate on the correlation between sAPPα and sAPPβ in this group. It is interesting that all three Aβ isoforms are positively correlated to sAPPα but not to sAPPβ in the NC group, as sAPPα is not a product of the same APP cleaving pathway as Aβ. A possible explanation would be that all of the measured Aβ isoforms are more abundant in the CSF of healthy individuals with high levels of sAPPα, as the threshold for Aβ aggregation in the brain has not yet been reached. Aβ38 and Aβ40 have a positive correlation to each other and the same is true for Aβ40 and Aβ42. This suggests that the Aβ42 decrease in CSF in FAD may be accompanied by a decrease in Aβ40 and possibly in Aβ38 as well, although the current study failed to show a significant correlation between Aβ42 and Aβ38.
Finally, we did not observe a difference in the sAPPβ/sAPPα ratio between the MC and the NC. The sAPPβ/sAPPα ratio has been suggested as a biomarker for brain amyloidosis [30], but our findings do not support it as a marker of early pathology. This should be interpreted with caution, however, because the possibility remains that the results were negative due to the small sample size in this instance (only 10 MC from the PSEN1 and APParc families because data on sAPPβ were not included from the APPswe MC). As expected, the Aβ42/Aβ40 ratio was low in the MC group as a whole and in the MC subgroups compared with the NC. This difference remained statistically significant when symptomatic individuals had been excluded, supporting its role as a robust early AD biomarker.
The method used in the current study to measure the potential biomarkers sAPPβ and sAPPα has recently been analytically validated using novel standard operating procedures developed within the BIOMARKAPD project [31]. This validation increases the credibility of the results obtained with this method and decreases the likelihood of analysis bias.
The results discussed were not corrected for multiple comparisons and the differences observed in Aβ38 and Aβ40 levels between groups did not survive a FDR correction. When interpreting the results of the FDR correction one should keep in mind that the study was hypothesis driven and the biomarkers carefully selected before any statistical analysis was performed. Therefore, the study design itself reduces the likelihood of false positives. Also, the small sample sizes, which often require nonparametric testing, already reduce the power to detect true differences between groups. The possibility still remains that the differences observed in Aβ38 and Aβ40 levels between groups were significant by chance, as suggested by the FDR correction. However, we feel that this possibility is not great enough to disregard the results altogether and that they warrant further study in larger cohorts.
A limitation to this study is the number of included subjects and this should be taken into account when interpreting the results. The current study, however, is larger than previous studies on other aspects of APP processing which have included FAD subjects and replicates some of their results, underlining the robustness of the data acquired from this population. Even though the small sample size might have had some effect on the outcomes presented here, they are at the very least strongly hypothesis generating. Another limitation is the cross-sectional design of the study, which makes it impossible to assess changes in APP processing products over time. This will be addressed in a longitudinal study on the same subjects, which is ongoing. The usefulness of FAD subjects as models for SAD has been supported by several studies [32], but the possibility still exists that these results might not be generalizable to SAD.
Conclusion
We observed pathological APP processing in presymptomatic carriers of FAD mutations, which adds to the current evidence that AD pathogenesis is present before the development of clinical symptoms [33]. We found no apparent common aspect of APP processing in the three mutation carrier groups, apart from the well-known decrease in CSF Aβ42, which is an early event in AD pathogenesis. Perhaps this decrease is secondary to the aggregation of brain amyloid, rather than a result of the APP processing steps, and thus a poor marker for the primary etiology. If the same type of treatment was to be used in presymptomatic FAD mutation carriers regardless of the type of mutation, these results suggest that a treatment interfering with the final product of APP processing, Aβ, would be a feasible option.
Acknowledgements
The authors thank all of the participants in the FAD study. They would also like to thank the staff employed at the Center for Alzheimer Disease Research at Karolinska Institutet and at the Karolinska University Hospital Memory Clinic in the Department of Geriatric Medicine, including Lena Lilius, MS, for technical assistance, Charlotte Forsell, MS, for statistical assistance, Mette Bergman, social worker, for counseling the participants and Dr Niels Andreasen for assisting in the acquisition of CSF samples.
Funding
The study was financially supported by Swedish Brain Power, The Swedish Alzheimer Foundation, The Torsten Söderberg foundation, The Swedish Dementia Association, Gun and Bertil Stohne’s Foundation, Gamla Tjänarinnors Foundation, The Regional Agreement on Medical Training and Clinical Research (ALF) between Stockholm County Council and Karolinska Institutet, The Swedish Research Council, Karolinska Institutet PhD-student funding, The Swedish Brain Foundation and King Gustaf V and Queen Victoria’s Free Mason Foundation. The funding bodies had no role in the design of the study, the collection, analysis and interpretation of data or in writing the manuscript.
Availability of data and materials
The datasets generated and analyzed during the current study are not publicly available due to individual privacy of the study participants. However, they are available from the corresponding author upon reasonable request.
Authors’ contributions
ST contributed to the conception and design of the study, acquired the cerebrospinal fluid and clinical data and analyzed and interpreted them. ST drafted the manuscript. AKS was involved in the conception and design of the study, the analysis and interpretation of data and in revising the manuscript critically for intellectual content. OA contributed to the conception and design of the study, acquired neuropsychological data and revised the manuscript critically for intellectual content. HT was responsible for analysis and interpretation of data and for critical revision of the manuscript for intellectual content. ME was involved in the conception and design of the study, the analysis and interpretation of data and in revising the manuscript critically for intellectual content. HZ was involved in the conception and design of the study, the analysis and interpretation of data and in revising the manuscript critically for intellectual content. KB was involved in the conception and design of the study, the analysis and interpretation of data and in revising the manuscript critically for intellectual content. CG was principal investigator in the conception and design of the study, the acquisition of data, the analysis and interpretation of data and in revising the manuscript critically for intellectual content. All authors have given final approval of the version to be published. All authors have agreed to be accountable for all aspects of the work ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All study procedures were approved by the Regional Ethical Review Board in Stockholm, Sweden. Informed, written consent was obtained from all participants prior to their participation in the study.
Abbreviations
- AD
Alzheimer’s disease
- APP
Amyloid precursor protein
- CSF
Cerebrospinal fluid
- FAD
Familial Alzheimer’s disease
- FDR
False discovery rate
- MC
Mutation carrier
- MCI
Mild cognitive impairment
- MRI
Magnetic resonance imaging
- NC
Noncarrier
- PCR
Polymerase chain reaction
- SAD
Sporadic Alzheimer’s disease
- SD
Standard deviation
Contributor Information
Steinunn Thordardottir, Phone: 354 696 7699, Email: steinunn.thordardottir@ki.se.
Anne Kinhult Ståhlbom, Email: anne.kinhult-stahlbom@karolinska.se.
Ove Almkvist, Email: ove.almkvist@ki.se.
Håkan Thonberg, Email: hakan.thonberg@karolinska.se.
Maria Eriksdotter, Email: maria.eriksdotter@karolinska.se.
Henrik Zetterberg, Email: henrik.zetterberg@clinchem.gu.se.
Kaj Blennow, Email: kaj.blennow@neuro.gu.se.
Caroline Graff, Email: caroline.graff@ki.se.
References
- 1.Hardy J, Selkoe DJ. Medicine—The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Zetterberg H, Blennow K, Hanse E. Amyloid beta and APP as biomarkers for Alzheimer's disease. Exp Gerontol. 2010;45(1):23–9. doi: 10.1016/j.exger.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 3.O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. In: Hyman SE, Jessell TM, Shatz CJ, Stevens CF, Zoghbi HY, editors. Annual Review of Neuroscience. Palo Alto: Annual Reviews; 2011. pp. 185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, et al. A pathogenic mutation for probable Alzheimers-disease in the APP gene at the N-terminus of beta-amyloid. Nature Genet. 1992;1(5):345–7. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 5.Axelman K, Basun H, Winblad B, Lannfelt L. A large Swedish family with Alzheimers-disease with a codon-670/671 amyloid precursor protein mutation—a clinical and genealogical investigation. Arch Neurol. 1994;51(12):1193–7. doi: 10.1001/archneur.1994.00540240037013. [DOI] [PubMed] [Google Scholar]
- 6.Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, et al. The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced A beta protofibril formation. Nat Neurosci. 2001;4(9):887–93. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 7.Clark RF, Hutton M, Fuldner RA, Froelich S, Karran E, Talbot C, et al. The structure of the presenilin-1 (S182) gene and identification of 6 novel mutations in early-onsET AD families. Nature Genet. 1995;11(2):219–22. doi: 10.1038/ng1095-219. [DOI] [PubMed] [Google Scholar]
- 8.Axelman K, Basun H, Lannfelt L. Wide range of disease onset in a family with Alzheimer disease and a His163Tyr mutation in the presenilin-1 gene. Arch Neurol. 1998;55(5):698–702. doi: 10.1001/archneur.55.5.698. [DOI] [PubMed] [Google Scholar]
- 9.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6(3):131–44. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- 10.Rosen C, Andreasson U, Mattsson N, Marcusson J, Minthon L, Andreasen N, et al. Cerebrospinal fluid profiles of amyloid beta-related biomarkers in Alzheimer's disease. Neuromolecular Med. 2012;14(1):65–73. doi: 10.1007/s12017-012-8171-4. [DOI] [PubMed] [Google Scholar]
- 11.Mehta PD, Pirttila T, Mehta SP, Sersen EA, Aisen PS, Wisniewski HM. Plasma and cerebrospinal fluid levels of amyloid beta proteins 1–40 and 1–42 in Alzheimer disease. Arch Neurol. 2000;57(1):100–5. doi: 10.1001/archneur.57.1.100. [DOI] [PubMed] [Google Scholar]
- 12.Kanai M, Matsubara E, Isoe K, Urakami K, Nakashima K, Arai H, et al. Longitudinal study of cerebrospinal fluid levels of tau, A beta 1–40, and A beta 1-42(43) in Alzheimer's disease: a study in Japan. Ann Neurol. 1998;44(1):17–26. doi: 10.1002/ana.410440108. [DOI] [PubMed] [Google Scholar]
- 13.Schoonenboom NS, Mulder C, Van Kamp GJ, Mehta SP, Scheltens P, Blankenstein MA, et al. Amyloid beta 38, 40, and 42 species in cerebrospinal fluid: more of the same? Ann Neurol. 2005;58(1):139–42. doi: 10.1002/ana.20508. [DOI] [PubMed] [Google Scholar]
- 14.Lewczuk P, Esselmann H, Meyer M, Wollscheid V, Neumann M, Otto M, et al. The amyloid-beta (A beta) peptide pattern in cerebrospinal fluid in Alzheimer's disease: evidence of a novel carboxyterminally elongated A beta peptide. Rapid Commun Mass Spectrom. 2003;17(12):1291–6. doi: 10.1002/rcm.1048. [DOI] [PubMed] [Google Scholar]
- 15.Portelius E, Andreasson U, Ringman JM, Buerger K, Daborg J, Buchhave P, et al. Distinct cerebrospinal fluid amyloid beta peptide signatures in sporadic and PSEN1 A431E-associated familial Alzheimer's disease. Mol Neurodegener. 2010;5:10. doi: 10.1186/1750-1326-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Portelius E, Fortea J, Molinuevo JL, Gustavsson MK, Andreasson U, Sanchez-Valle R. The amyloid-beta isoform pattern in cerebrospinal fluid in familial PSEN1 M139T-and L286P-associated Alzheimer's disease. Mol Med Rep. 2012;5(4):1111–5. doi: 10.3892/mmr.2012.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pera M, Alcolea D, Sanchez-Valle R, Guardia-Laguarta C, Colom-Cadena M, Badiola N, et al. Distinct patterns of APP processing in the CNS in autosomal-dominant and sporadic Alzheimer disease. Acta Neuropathol. 2013;125(2):201–13. doi: 10.1007/s00401-012-1062-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olsson A, Hoglund K, Sjogren M, Andreasen N, Minthon L, Lannfelt L, et al. Measurement of alpha- and beta-secretase cleaved amyloid precursor protein in cerebrospinal fluid from Alzheimer patients. Exp Neurol. 2003;183(1):74–80. doi: 10.1016/S0014-4886(03)00027-X. [DOI] [PubMed] [Google Scholar]
- 19.Perneczky R, Tsolakidou A, Arnold A, Diehl-Schmid J, Grimmer T, Forstl H, et al. CSF soluble amyloid precursor proteins in the diagnosis of incipient Alzheimer disease. Neurology. 2011;77(1):35–8. doi: 10.1212/WNL.0b013e318221ad47. [DOI] [PubMed] [Google Scholar]
- 20.Lewczuk P, Kamrowski-Kruck H, Peters O, Heuser I, Jessen F, Popp J, et al. Soluble amyloid precursor proteins in the cerebrospinal fluid as novel potential biomarkers of Alzheimer's disease: a multicenter study. Mol Psychiatry. 2010;15(2):138–45. doi: 10.1038/mp.2008.84. [DOI] [PubMed] [Google Scholar]
- 21.Lannfelt L, Basun H, Wahlund LO, Rowe BA, Wagner SL. Decreased alpha-secretase-cleaved amyloid precursor protein as a diagnostic marker for Alzheimers-disease. Nat Med. 1995;1(8):829–32. doi: 10.1038/nm0895-829. [DOI] [PubMed] [Google Scholar]
- 22.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical-diagnosis of Alzheimers-disease—report of the NINCDS-ADRDA work group under the auspices of Department-of-Health-and-Human-Services Task-Force on Alzheimers-Disease. Neurology. 1984;34(7):939–44. doi: 10.1212/WNL.34.7.939. [DOI] [PubMed] [Google Scholar]
- 23.Winblad B, Palmer K, Kivipelto M, Jelic V, Fratiglioni L, Wahlund LO, et al. Mild cognitive impairment—beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med. 2004;256(3):240–6. doi: 10.1111/j.1365-2796.2004.01380.x. [DOI] [PubMed] [Google Scholar]
- 24.Vanderstichele H, Bibl M, Engelborghs S, Le Bastard N, Lewczuk P, Molinuevo JL, et al. Standardization of preanalytical aspects of cerebrospinal fluid biomarker testing for Alzheimer's disease diagnosis: a consensus paper from the Alzheimer's Biomarkers Standardization Initiative. Alzheimers Dement. 2012;8(1):65–73. doi: 10.1016/j.jalz.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 25.Zetterberg H, Andreasson U, Hansson O, Wu G, Sankaranarayanan S, Andersson ME, et al. Elevated cerebrospinal fluid BACE1 activity in incipient Alzheimer disease. Arch Neurol. 2008;65(8):1102–7. doi: 10.1001/archneur.65.8.1102. [DOI] [PubMed] [Google Scholar]
- 26.Hartl D, Gu W, Mayhaus M, Pichler S, Schope J, Wagenpfeild S, et al. Amyloid-beta protein precursor cleavage products in postmortem ventricular cerebrospinal fluid of Alzheimer's disease patients. J Alzheimers Dis. 2015;47(2):365–72. doi: 10.3233/JAD-150191. [DOI] [PubMed] [Google Scholar]
- 27.Gabelle A, Roche S, Geny C, Bennys K, Labauge P, Tholance Y, et al. Correlations between soluble alpha/beta forms of amyloid precursor protein and Abeta38, 40, and 42 in human cerebrospinal fluid. Brain Res. 2010;1357:175–83. doi: 10.1016/j.brainres.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 28.Reinert J, Martens H, Huettenrauch M, Kolbow T, Lannfelt L, Ingelsson M, et al. A beta(38) in the brains of patients with sporadic and familial Alzheimer's disease and transgenic mouse models. J Alzheimers Dis. 2014;39(4):871–81. doi: 10.3233/JAD-131373. [DOI] [PubMed] [Google Scholar]
- 29.Alexopoulos P, Tsolakidou A, Roselli F, Arnold A, Grimmer T, Westerteicher C, et al. Clinical and neurobiological correlates of soluble amyloid precursor proteins in the cerebrospinal fluid. Alzheimers Dement. 2012;8(4):304–11. doi: 10.1016/j.jalz.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 30.Dobrowolska JA, Kasten T, Huang YF, Benzinger TLS, Sigurdson W, Ovod V, et al. Diurnal patterns of soluble amyloid precursor protein metabolites in the human central nervous system. Plos One. 2014;9(3):12. doi: 10.1371/journal.pone.0089998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Doorn L, Koel-Simmelink MJ, Haussmann U, Klafki H, Struyfs H, Linning P, et al. Validation of soluble amyloid- precursor protein assays as diagnostic CSF biomarkers for neurodegenerative diseases. J Neurochem. 2016;137(1):112–21. doi: 10.1111/jnc.13527. [DOI] [PubMed] [Google Scholar]
- 32.Ringman JM. What the study of persons at risk for familial Alzheimer's disease can tell us about the earliest stages of the disorder: a review. J Geriatr Psychiatry Neurol. 2005;18(4):228–33. doi: 10.1177/0891988705281878. [DOI] [PubMed] [Google Scholar]
- 33.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goate AM. Monogenetic determinants of Alzheimer's disease: APP mutations. Cell Mol Life Sci. 1998;54(9):897–901. doi: 10.1007/s000180050218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, et al. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. Embo J. 2012;31(10):2261–74. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and analyzed during the current study are not publicly available due to individual privacy of the study participants. However, they are available from the corresponding author upon reasonable request.