

Abstract
Aim: Antrodia Camphorate (AC) is a mushroom that is widely used in Asian countries to prevent and treat various diseases, including liver diseases. However, the active ingredients that contribute to the biological functions remain elusive. The purpose of the present study is to test the hepatoprotective effect of Antcin H, a major triterpenoid chemical isolated from AC, in murine models of acute liver injury.
Results: We found that Antcin H pretreatment protected against liver injury in both acetaminophen (APAP) and galactosamine/tumor necrosis factor (TNF)α models. More importantly, Antcin H also offered a significant protection against acetaminophen-induced liver injury when it was given 1 h after acetaminophen. The protection was verified in primary mouse hepatocytes. Antcin H prevented sustained c-Jun-N-terminal kinase (JNK) activation in both models. We excluded an effect of Antcin H on acetaminophen metabolism and TNF receptor signaling and excluded a direct effect as a free radical scavenger or JNK inhibitor. Since the sustained JNK activation through its interaction with mitochondrial Sab, leading to increased mitochondrial reactive oxygen species (ROS), is pivotal in both models, we examined the effect of Antcin H on p-JNK binding to mitochondria and impairment of mitochondrial respiration. Antcin H inhibited the direct effect of p-JNK on isolated mitochondrial function and binding to isolated mitochondria.
Innovation and Conclusion: Our study has identified Antcin H as a novel active ingredient that contributes to the hepatoprotective effect of AC, and Antcin H protects against liver injury through disruption of the binding of p-JNK to Sab, which interferes with the ROS-dependent self-sustaining activation of MAPK cascade. Antioxid. Redox Signal. 26, 207–220.
Keywords: : Antrodia Camphorate, Antcin H, liver injury, c-Jun-N-terminal kinase, mitochondria, ROS
Introduction
Acetaminophen (APAP) is one of the most commonly used analgesic drugs. APAP hepatotoxicity in humans was first reported in 1960s (7). Since then, APAP has become the leading cause of acute liver failure in many developed countries, including the United States and most of the European countries (11, 22, 23). N-acetyl-p-benzoquinone (NAPQI), an electrophilic reactive metabolite of APAP produced mainly by CYP2E1 metabolism, plays a pivotal role in activation of downstream toxic signaling pathways, including glutathione (GSH) depletion, NAPQI-protein adduct formation (6, 13), reactive oxygen species (ROS) accumulation, sustained c-Jun-N-terminal kinase (JNK) activation and its mitochondria translocation (12, 15), and increased mitochondrial membrane permeability transition, which eventually leads to necrotic cell death (43–45).
Tumor necrosis factor (TNF)α, a proinflammatory cytokine, is suggested to play a critical role in hepatocellular death in many conditions, such as immune-mediated and alcoholic and nonalcoholic fatty liver diseases (3, 36, 38). Galactosamine (GalN), as a hepatocyte-specific sensitizer, enhances TNFα-mediated hepatotoxicity. GalN/TNFα-induced liver injury is a useful model for investigating innate immune-mediated apoptotic hepatotoxicity. In the GalN/TNFα model, sustained JNK activation has also been identified as a key event to trigger mitochondrial pathway-dependent apoptotic death of hepatocytes (14, 41, 43, 45). Targeting the key signaling pathways, such as JNK activation or its effects on mitochondria, may be an attractive approach to manage drug-induced and immune-mediated hepatotoxicity.
Innovation.
Antrodia Camphorate (AC) is a mushroom which is widely used in Asian countries to prevent and treat various diseases, including liver diseases. However, the active ingredients that contribute to the biological functions of AC remain elusive. We found for the first time that Antcin H is highly effective against either acetaminophen (APAP) or galactosamine (GalN)/tumor necrosis factor (TNF)α-mediated hepatotoxicity. Our study has identified Antcin H as a novel active ingredient that contributes to the hepatoprotective effect of AC, and Antcin H protects against liver injury through the disruption of the binding of p-JNK to Sab, which consequently interferes with the mitochondrial reactive oxygen species-dependent self-sustaining activation of MAPK cascade.
Folk medicines have long been used to manage various diseases, including liver disease. However, the scientific validation of their efficacy and the mechanistic interpretations are needed. Folk medicine is suggested to be a rich source for developing evidence-based chemopreventive or therapeutic agents. Antrodia Camphorate (AC) is a rare and precious medicinal mushroom found in Taiwan. As a folk medicine, AC is used widely in Asian countries for the treatment of various diseases, including liver disease (24). The chemistry of AC has been extensively investigated and it has been found that AC is rich in ergostane and lanostane triterpenoids (25, 31).
Antcin H is one of the main terpenoids isolated from AC. It has been shown that Antcin H possesses anti-inflammatory and insecticidal activities (5, 26, 35). We hypothesized that Antcin H could be a major active component of AC that may contribute to its hepatoprotective activity. In present study, the protective effect of Antcin H against drug-induced and immune-mediated hepatotoxicity has been evaluated using both APAP- and GalN/TNFα-induced acute liver injury in mouse models. The results demonstrate for the first time that Antcin H is highly effective against either APAP- or GalN/TNFα-mediated liver toxicity. Mechanistically, the hepatoprotective activity of Antcin H was attributed to its ability to directly disrupt the interaction between JNK and mitochondria and prevent JNK-mediated mitochondrial dysfunction.
Results
Antcin H protects against APAP- or GalN/TNFα-induced hepatotoxicity in vivo and in vitro
Our pilot study demonstrated that the ethanol extract of AC offered a significant protective effect on APAP-induced liver injury (data not shown). To identify the active compound that contributed to the hepatoprotective activity of AC, Antcin H, a major component of AC, was evaluated using both APAP- and GalN/TNFα-induced liver injury models.
For the APAP-induced liver injury model, we first investigated the protective effect of pretreatment with Antcin H. Antcin H was given by i.p. injection 1 h before APAP treatment. As shown in Figure 1A, 24 h after APAP treatment, the very high serum alanine aminotransferase (ALT) in vehicle controls was nearly abolished by pretreatment with 25 mg/kg or 50 mg/kg Antcin H. In line with the changes of ALT, the histological analysis showed that APAP-induced severe zone III necrosis of hepatic lobules was dramatically attenuated by the pretreatment with 25 or 50 mg/kg Antcin H (Fig. 1B).
FIG. 1.

Antcin H protects against liver injury induced by APAP. (A, B) Mice received 25 and 50 mg/kg Antcin H i.p. in corn oil 1 h before APAP (300 mg/kg), and serum ALT and representative histology (H&E) determined at 24 h. ***p < 0.001, n = 3–4 mice per group. (C, D) Antcin H (50 mg/kg) was administered 1 h after APAP (300 mg/kg), and serum ALT and histology determined at 24 h. **p < 0.01, n = 6 mice per group. (E) Cultured PMH were pretreated with 50 and 75 μM Antcin H for 2 h and then switched to APAP (10 mM) plus Antcin H. Necrotic cells (SYTOX Green positive) were counted in 10 different fields per dish at 24 h. *p < 0.05 (n = 3 experiments). ALT, alanine aminotransferase; APAP, acetaminophen; H&E, hematoxylin and eosin. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Having established the preventive efficacy of Antcin H, we next explored the therapeutic potential of Antcin H against APAP-induced hepatotoxicity by administering APAP 1 h before Antcin H. Since maximal covalent binding and GSH depletion are seen by 1 h after APAP (27), a protective effect is unlikely to be due to inhibition of NAPQI production. As shown in Figure 1C and D, a significant protection was also observed in this therapeutic setting. To confirm that the effect of Antcin H was on hepatocytes, we assessed the effect of Antcin H 2 h pretreatment of cultured mouse hepatocytes followed by 2 h of exposure to APAP and then changed to Antcin H containing media without APAP. Cell death at 24 h was decreased by Antcin H in a dose-dependent manner (Fig. 1E).
We next determined if Antcin H is also capable of counteracting the hepatotoxicity triggered by GalN/TNFα, which occurs by a different mode of cell death (apoptosis) and is independent of drug metabolism. Mice received Antcin H i.p. twice, 6 and 1 h, before GalN/TNFα administration. The liver damage was examined 6 h after GalN/TNFα exposure. Increased ALT level (Fig. 2A) and histological injury (Fig. 2B) in controls were significantly ameliorated or negligible in Antcin H pretreated mice. In the primary mouse hepatocyte culture model, cotreatment with Antcin H significantly inhibited apoptosis induced by actinomycin D/TNFα (Fig. 2C). Thus, these results demonstrate that Antcin H has protective effects against either APAP necrosis- or TNFα-mediated apoptotic hepatotoxicity in vivo and in vitro.
FIG. 2.

Antcin H protects against the liver injury induced by GalN/TNFα. (A, B) Mice were pretreated with Antcin H (25, 50 mg/kg in DMSO) twice, 6 and 1 h, before GalN (800 mg/kg)/TNFα (12 μg/kg) i.p. After 6 h, serum ALT level and liver histology (H&E) were determined ***p < 0.001(n = 3 mice per group). (C) Cultured PMH were treated with different concentrations of Antcin H (50, 75, and 100 μM) and Act D (0.5 μg/ml)/TNFα (5 ng/ml) simultaneously. After 6 h, percent apoptosis was determined (Hoechst 33248). *p < 0.05, **p < 0.01 (n = 3 experiments). GalN, galactosamine; TNF, tumor necrosis factor. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Antcin H inhibits sustained JNK activation induced by APAP or GalN/TNFα
To decipher the mechanisms of the hepatoprotective activity of Antcin H in the APAP model, we assessed the effect of Antcin H on APAP-induced GSH depletion and covalent binding. As shown in Figure 3A, vehicle-treated and Antcin H-treated mice were found to have a comparable level of GSH, whereas APAP treatment led to a significant reduction of GSH at 1 h as expected, but GSH depletion by APAP was not affected by Antcin H exposure. Furthermore, the formation of NAPQI-protein adducts by APAP was also not significantly affected by Antcin H (Fig. 3B). These results suggested that protection of Antcin H was not due to blocking the toxic metabolism of APAP.
FIG. 3.

The effects of Antcin H on the metabolism of APAP and JNK activation. (A) Liver homogenate GSH 1 h after APAP injection was determined in control mice with or without Antcin H (50 mg/kg) pretreatment 1 h before APAP (300 mg/kg) i.p. **p < 0.01, n = 3. (B) Immunoblot of time course of NAPQI-protein adducts in liver extracts obtained 1 h after APAP (300 mg/kg) with or without pretreatment with Antcin H (50 mg/kg). Bar graph shows densitometry of total adducts/GAPDH of n = 3. (C) Initial p-JNK in samples with or without pretreatment with Antcin H. (D, E) Time course of p-JNK in samples from B, using whole liver homogenate (D) and cytoplasm and mitochondria (E). Bar graphs show densitometry of p-JNK/JNK or p-JNK/PHB1 of n = 3. (F) PMH were pretreated with Antcin H or DMSO vehicle for 2 h. Media then changed to APAP (10 mM) without Antcin H, and whole cell lysates were collected at 1, 2, and 4 h for Western blot. Bar graph shows densitometry of p-JNK/JNK of n = 3. **p < 0.01, ***p < 0.001. JNK, c-Jun-N-terminal kinase; GSH, glutathione; NAPQI, N-acetyl-p-benzoquinone; PMH, primary mouse hepatocytes. *p < 0.5, **p < 0.01.
Activation of JNK and its mitochondrial translocation are important in exacerbating APAP-induced hepatotoxicity (12, 15, 41, 43, 45). It is well established that APAP induces rapid mitochondrial impairment and increased O2•− production; released H2O2 then activates MAP3K leading to JNK activation. p-JNK then interacts with the mitochondrial outer membrane protein, Sab, which induces further mitochondrial impairment and ROS production (29, 42, 43).
We therefore investigated whether APAP-induced JNK activation was attenuated by Antcin H. JNK activation was not seen in phosphate-buffered saline (PBS)-treated and Antcin H-treated control mice. As expected, APAP exposure caused a rapid JNK activation (as early as 15 min). Treatment with Antcin H did not significantly inhibit the early activation of JNK after APAP injection at 15 and 30 min (Fig. 3C), but nearly abolished activation of JNK after APAP injection at 1, 2, and 4 h (Fig. 3D), suggesting Antcin H might mainly target the sustained JNK activation rather than the initial or transient event of JNK activation induced by APAP. The latter is known to be a consequence of direct toxicity of APAP on mitochondria leading to ROS release and activation of MAPK (15).
In addition, the inhibitory effect of Antcin H on JNK activation was also found in the primary mouse hepatocyte culture model (Fig. 3F). We further evaluated mitochondrial JNK translocation using cytoplasmic and mitochondrial fractions from mice treated with APAP and/or Antcin H. Consistent with the above results, a dramatic reduction in p-JNK by Antcin H was observed in both the mitochondria and cytoplasm (Fig. 3E).
For GalN/TNFα-induced liver injury, we first assessed if Antcin H affected TNF receptor signaling. As shown in Figure 4A, TNFα alone induced a transient increase of p-IκBα and decrease of IκBα, which were not influenced by Antcin H treatment. In addition, the transient JNK phosphorylation after TNFα treatment was not suppressed by Antcin H. It is also well established that sustained JNK activation also plays a key role in GalN/TNFα-mediated toxicity (14, 41). In this case, the p-JNK interaction with Sab leads to mitochondrial ROS production and sustained JNK activation, which then modulates Bcl-2 family to induce mitochondria outer membrane permeabilization and apoptosis (20).
FIG. 4.

Effects of Antcin H on initial TNFα receptor signaling in PMH and expression of p-JNK in GalN/TNFα model. (A) Hepatocytes from wild-type mice were cultured with 20 ng/ml TNFα and DMSO or Antcin H (100 μM) for 5, 10, and 30 min. Immunoblotting of PMH extracts was performed using antisera against p-IκBα, IκBα, p-JNK, JNK, and GAPDH. (B, C) Mice were pretreated with Antcin H (50 mg/kg i.p. or DMSO vehicle) and whole liver homogenate (B) or cytoplasm and mitochondria (C) at various times after GalN/TNFα were immunoblotted for p-JNK and total JNK with GAPDH or PHB1 as a loading control. Bar graphs at right are as in Figure 3 (n = 3). *p < 0.5, **p < 0.01.
We therefore measured the effect of Antcin H on the sustained JNK activation. Liver samples were collected 30 min and 1 and 2 h after GalN/TNFα treatment, and the phosphorylation level of JNK was measured by Western blot. Phosphorylation of JNK in whole liver tissue homogenate and cytoplasmic and mitochondrial fractions (Fig. 4B, C) was substantially inhibited by Antcin H exposure.
Thus, inhibition of JNK by Antcin H was found in both APAP- and GalN/TNFα-induced acute liver injury. Given the similar role of activated JNK on mitochondria in both models, the APAP model was used to further decipher the mechanisms of JNK inhibition by Antcin H in the subsequent experiments.
JNK is not the direct target of Antcin H
The above data showed that Antcin H markedly inhibited the sustained activation of JNK. We therefore assessed if Antcin H is a direct inhibitor of p-JNK activity. To test this hypothesis, we carried out an in vitro kinase assay using c-Jun fusion protein as a substrate of JNK and SP600125, a known p-JNK inhibitor, as a positive control. Cytoplasmic fraction containing p-JNK was isolated from APAP-treated (2 h) mice and then incubated with different concentrations of SP600125 or Antcin H or vehicle control. The activity of JNK was detected by immunoblot analysis of p-c-Jun. As shown in Figure 5, the JNK inhibitor (SP600125) completely inactivated JNK, while Antcin H did not inhibit JNK activity.
FIG. 5.

Effect of Antcin H on JNK activity. Cytoplasm containing endogenous p-JNK was prepared 2 h after APAP (300 mg/kg, i.p.). Kinase activity was measured as production of p-c-Jun in the absence of c-Jun substrate (lane 1) or in the absence (lane 2) or presence of JNK inhibitor (SP600124), DMSO vehicle, or Antcin H as indicated. All assays contained ATP (50 μM). The lower panel confirms equal amount of p-JNK and GAPDH in the various assay conditions (representative of n = 3 experiments).
Antcin H blocks mitochondrial ROS generation induced by APAP, but is not a direct scavenger or antioxidant
JNK is intricately regulated by a network of kinases and phosphatases. We and others have previously shown that the interaction of p-JNK with Sab increases mitochondrial ROS (4, 42). We next measured ROS levels by using flow cytometry in primary mouse hepatocytes (PMH) after 1 h APAP treatment. Increased levels of cellular ROS were observed in APAP-treated PMH, while Antcin H significantly alleviated ROS accumulation to the basal level, although not directly addressing mitochondrial ROS (Fig. 6A).
FIG. 6.

Effect of Antcin H on ROS generation. (A) Antcin H decreases ROS level of hepatocytes in vitro. Three hours after plating hepatocytes and pretreatment with Antcin H for 2 h, PMH were exposed to APAP (10 mM) for 1 h. Hepatocytes were then incubated for 30 min with DHE probe at 37°C and ROS detected by flow cytometry (left panel). “FL2-H” represents the intensity of fluorescence of DHE, and “Counts” is the cell numbers. Right panel shows summary of data from three experiments normalized to control. (*p < 0.05, n = 3 experiments). (B) Mitochondrial superoxide measured by MitoSOX increased after incubation of liver cytoplasm harvested 2 h after APAP (p-JNKc) containing p-JNK with normal mitochondria. Vehicle (DMSO) had no effect on ROS level, while Antcin H in DMSO significantly inhibited mitochondrial ROS production. “no MitoSOX” and “no p-JNK (normal cytoplasm)” are controls. (*p < 0.05 “no p-JNK” vs. “p-JNKc”; #p < 0.05 “p-JNK” vs. “p-JNKc+Antcin H,” n = 3 experiments). (C) Fifty micromolar DPPH with different Antcin H concentrations (0, 2.5, 5, 10, 24, 50, 75, 100, 150, 50, and 300 μM) in triplicate were wrapped in aluminum foil and kept at 30°C for 30 min in the dark. All measurements were done under dim light. Spectrophotometric measurements were done at 517 nm. BHA (50, 100, 50, and 300 μM) was used as a positive control. Radical scavenging activity (%) = {OD (Blank) − OD (Sample)}/OD (Control) × 100%. (D) Antimycin A inhibition of Complex III generated superoxide from mitochondria. FeTCCP (iron protoporphyrin) effectively inhibited superoxide production, whereas Antcin H did not affect superoxide level. ROS, reactive oxygen species. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
However, when liver cytoplasm harvested 2 h after APAP (containing p-JNK) was incubated with MitoSOX-loaded normal liver mitochondria, the expected (42) increase in O2•− was observed and was blocked by Antcin H (Fig. 6B). This effect could be due to Antcin H be a radical or ROS scavenger causing removal of ROS or it could be due to Antcin H interfering with the production of ROS by interfering with p-JNK interaction with Sab.
Therefore, we addressed whether Antcin H is a radical scavenger or directly scavengers ROS. Scavenging of DPPH radical, which is a stable free radical, has been used for assessing if a compound is a radical scavenger (46). We compared Antcin H with BHA, a well-known antioxidant, and we found no evidence that Antcin H is a radical scavenger compared to BHA, which was a potent inhibitor (Fig. 6C). To further verify that Antcin H does not directly inhibit intramitochondrial ROS production, we examined its effect on MitoSOX-loaded mitochondria treated with antimycin A. The expected increased ROS readout was not altered by preincubation of the mitochondria with Antcin H (Fig. 6D).
Many investigators have shown that APAP induces mitochondrial superoxide (O2•−) and NO, which then react to form peroxynitrite (ONOO−), which reacts with protein tyrosine (1, 16–18, 21). In addition, others have shown that the increased peroxynitrite formation exclusively in mitochondria in response to p-JNK depends on APAP increasing superoxide production (34). Therefore, as further support for Antcin H suppressing mitochondrial ROS production, we assessed mitochondrial 3-NT levels by Western blot (Fig. 7A) and immunohistochemistry (Fig. 7B). Antcin H inhibited the formation of 3-NT, which is further evidence confirming inhibition of mitochondrial superoxide anion production.
FIG. 7.
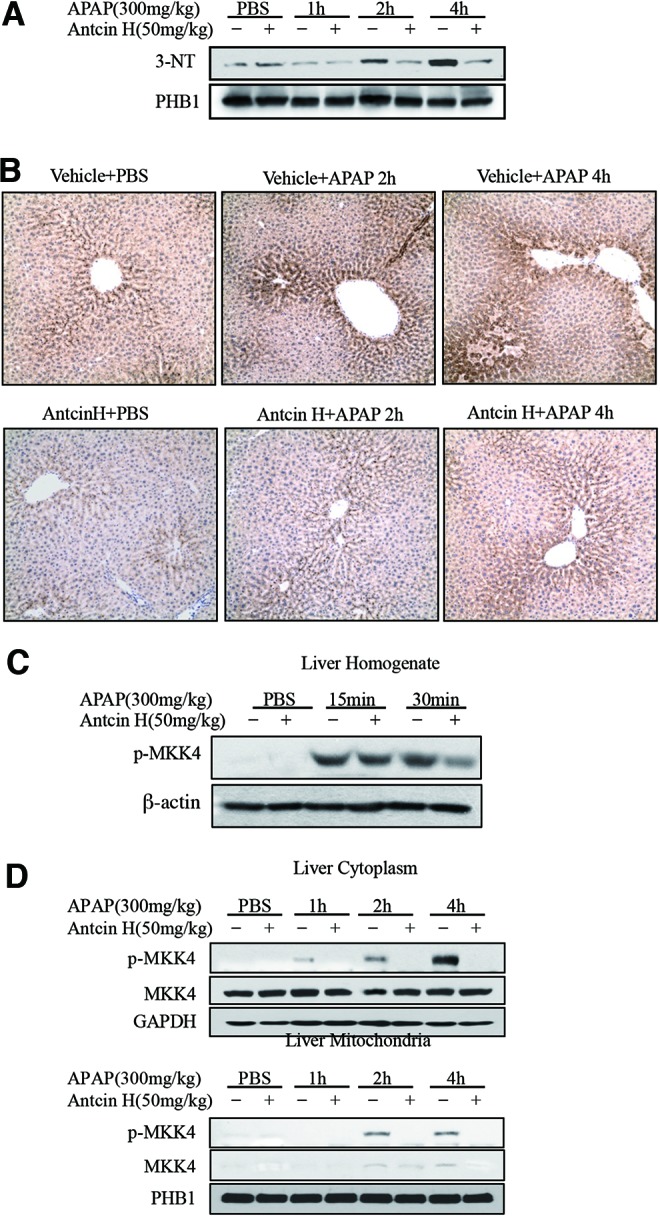
Effect of Antcin H on 3-NT generation and MKK4 activation. (A) Western blotting of mitochondrial 3-NT. (B) Immunohistochemical staining for 3-NT after APAP treatment in vivo with or without Antcin H. Representative of n = 3. (C) Immunoblotting of p-MKK4 in liver homogenate from Figure 3C. (D) Cytoplasmic and mitochondrial p-MKK4 were detected by Western blot in samples from Figure 3E. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The increased ROS generation is known to sustain the activation of MAPK upstream of JNK. We reported previously (43, 45) that sustained MKK4 (MAP2K) activation was seen in the cytoplasm after APAP treatment, and p-MKK4 translocated to the mitochondria. In presence of Antcin H, early activation of p-MKK4 was not significantly inhibited by Antcin H after APAP injection at 15 and 30 min (Fig. 7C), and no p-MKK4 activation and translocation at 1, 2, and 4 h after APAP injection was observed (Fig. 7D). Thus, the data in Figures 6 and 7 suggest that Antcin H is not a direct antioxidant or ROS scavenger and its effect likely is to decrease the production of mitochondrial ROS, which sustains JNK activation, and is not due to the removal or detoxification of ROS.
Antcin H directly disrupts the binding of JNK to mitochondria
The blockade of any step in the sustained JNK activation loop (MAPK cascade→p-JNK→Sab→mitochondrial ROS→MAPK cascade) (45) would limit p-JNK and provide protection against APAP hepatotoxicity. However, since we observed that Antcin H inhibited p-JNK-induced ROS production in isolated mitochondria, we hypothesized that Antcin H interferes with the binding or action of p-JNK on mitochondrial Sab. This interaction activates an intramitochondrial signaling pathway, which inhibits respiration and increases ROS production.
Binding of p-JNK to Sab is the first step in initiating this pathway (42). We examined the mitochondrial Sab protein level by Western blot (Fig. 8A) and found no effect of Antcin H. We treated normal liver mitochondria with cytoplasm containing p-JNK (isolated from 2 h APAP) in the presence of different concentrations of Antcin H (12.5, 50, 100, 300 μM). The washed mitochondria were then analyzed for bound p-JNK and total JNK protein level by immunoblotting. In the presence of Antcin H, both p-JNK and total JNK binding to mitochondria were decreased (Fig. 8B). PHB1 was used as mitochondria protein loading level, while GAPDH was used to assess any cytoplasmic protein contamination.
FIG. 8.

Effects of Antcin H on the interaction between JNK and mitochondria. (A) Western blotting of Sab in mitochondria isolated from control and Antcin H treated alone at different times after APAP treatment, PHB1 is loading control. (B) Isolated liver mitochondria were incubated at 30°C for 1 h with cytoplasm obtained 2 h after APAP (300 mg/kg), with or without Antcin H (preincubated for 30 min). Extracts of washed mitochondria were then immunoblotted for p-JNK, total JNK, and loading control for mitochondria (PHB1) and cytoplasmic contamination (GAPDH). (C) Comparison of the effect of Antcin H and Dexa on p-JNK binding performed as B. Bar graphs in (B, C) represent densitometry of p-JNK/PHB1 (n = 3). *p < 0.05, **p < 0.01. (D) Mitochondria were incubated with Antcin H (25 μM) or Dexa (25 μM) in vehicle for 30 min and OCR measured as described in the Materials and Methods section in Seahorse apparatus in the presence of JNK1/2 plus ATP or p-JNK1/2 plus ATP (upper panel). The data are presented as mean ± SD of five wells for one representative experiment. Lower panel, bar graphs of oxidative phosphorylation and maximal respiration with or without Dexa and Antcin H derived from Seahorse data. Neither Dexa nor Antcin H affected OCR in the absence of p-JNK (Supplementary Fig. S2). Bar graphs summarized the results of three Seahorse experiments (oxidative phosphorylation: OCR with ADP minus oligomycin; Maximal respiration: CCCP minus AA) *p < 0.05. AA, Antimycin A; OCR, oxygen consumption rate. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Since Dexa has a similar structure as Antcin H (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars), we compared their effects. Antcin H inhibited p-JNK binding, but Dexa had no effect (Fig. 8C). To confirm the link between Antcin H and p-JNK interaction with mitochondria, we examined the changes of mitochondrial OCR induced by recombinant p-JNK1/2 by using Seahorse XF24 analyzer.
Normal mitochondria were incubated with Antcin H or Dexa for 30 min and then exposed to p-JNK1/2 plus ATP in the presence of glutamate and malate. Neither Antcin H nor Dexa inhibited respiration in the presence of unactivated JNK1/2 plus ATP. However, Antcin H inhibited the impairment of oxidative phosphorylation and maximum respiratory capacity in the presence of p-JNK1/2 plus ATP, whereas Dexa did not (Fig. 8D).
Thus, our findings suggest that Antcin H directly inhibits the binding and effects of p-JNK on mitochondria. This leads to decreased mitochondrial ROS production, which abrogates the effect of mitochondrial ROS to induce MAPK cascade activation of JNK.
Discussion
In preclinical and clinical studies, AC has been shown to have multiple biological activities, including hepatoprotective effects (19, 24, 30). Although the chemistry of AC has been extensively studied (10), the active components that contribute to its biological functions are far from clear. Identification of active ingredients from AC is needed for a better utilization of this medicinal mushroom. Antcin H is a major triterpenoid compound isolated from AC (31), which possesses a favorable pharmacokinetic feature (32).
In the current study, we have demonstrated the hepatoprotective effect of Antcin H using mouse models of both APAP necrotic and GalN/TNFα-induced apoptotic liver injury. Antcin H was still highly effective against APAP-induced liver injury when given after APAP in vivo, suggesting it has therapeutic potential. Because these two liver injury models exhibit JNK dependence while differing in mode of cell death, we explored the effect of Antcin H on the JNK signaling pathway.
JNK can be initially activated by extracellular death receptor signaling (TNF) or by intracellular mitochondrial stress (e.g., APAP). However, sustained JNK activation in cell death scenarios depends upon the binding of p-JNK to and its phosphorylation of Sab, an outermembrane p-JNK “receptor” and substrate. This leads to an intramitochondrial signaling pathway that disrupts electron transport leading to the release of ROS, which continues to activate the MAPK cascade through a self-sustained MLK3/ASK1→p-MKK4→p-JNK→Sab→ROS pathway (43–45). It is currently believed that continued H2O2 release from mitochondria in response to p-JNK interaction with Sab leads to mitochondrial ROS release, which activates MAP3K and inhibits MAPK phosphatases, so JNK activation is sustained (29, 42).
Antcin H inhibited sustained JNK activation and ROS production. We excluded effects on APAP metabolic activation and the possibility that Antcin H is a radical scavenger. In addition, Antcin H did not inhibit antimycin A-induced O2•−, or act as a direct JNK inhibitor.
However, in normal mitochondria, Antcin H inhibited the Sab-dependent p-JNK-mediated inhibition of mitochondrial oxidative phosphorylation and maximum respiratory capacity, as well as the p-JNK-induced mitochondrial O2•−. Since the p-JNK-mediated mitochondrial ROS production is self-sustaining (13), we therefore addressed the possibility that Antcin H might interfere with the binding of p-JNK to mitochondrial Sab, which would block downstream ROS production. Using normal liver mitochondria incubated with cytoplasmic extracts from APAP-treated mice, we found that Antcin H decreased the binding of p-JNK to mitochondria without affecting p-JNK levels during the incubation. This leads us to the conclusion that the protective effect of Antcin H is mediated by inhibition of the binding of p-JNK to Sab (Fig. 9).
FIG. 9.

Mechanism of protection by Antcin H: Inhibition of sustained JNK activation and blocking the binding of p-JNK to mitochondria. The interplay of p-JNK and mitochondrial Sab leads to increased ROS release, which activates the MAPKinase cascade and may inactivate JNK phosphatase (MKP1), leading to a self-perpetuating pathway, which activates mitochondrial ROS production and cytoplasmic JNK activation. Thus, the ROS may then amplify the effects of APAP on mitochondria leading to MPT induction and necrosis. In the APAP model, GSH and ATP depletion and a high level of ROS incapacitate caspase activity. Sustained JNK activation also plays an important role in apoptosis by its known actions on c-FLIP degradation, allowing caspase-8 to cleave Bid to t-Bid, as well as activation of Bax and inactivation of Bcl-XL and Mcl-1, thereby promoting MOMP and cytochrome c release. Antcin H disrupts the ability of p-JNK to interact with mitochondrial Sab, thereby abrogating the mechanism for sustained JNK activation. MOMP, mitochondrial outer membrane permeabilization; MPT, mitochondrial permeability transition. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
There are a few caveats about our work. We cannot at this point definitively conclude that Antcin H binds to Sab (as a competitive or allosteric inhibitor) or if it indirectly causes decreased binding by affecting another mitochondrial protein, which changes the binding of p-JNK to Sab, or by affecting the physicochemical properties of the outer membrane lipids. More work will be required to elucidate this mechanism for inhibition of p-JNK binding to Sab. In addition, more work will be needed on pharmacokinetic properties of Antcin H and on the accumulation of Antcin H on the outer membrane of mitochondria in vivo. In addition, the effect of other Antcin compounds from AC will need to be assessed in future studies.
However, we believe that this is a very novel finding as Antcin H is the first identified compound that can interfere with the binding of p-JNK to mitochondria. This is particularly important since it is not a direct JNK inhibitor and therefore would not be expected to inhibit the protective/beneficial aspects of JNK signaling (2, 40), such as AP-1 transcription factor activation (39, 47). This has been a theoretical limitation in the therapeutic application of JNK inhibitors, particularly in liver injury, because of the potential for inhibition of liver regeneration and other beneficial effects of transient JNK activation. Therefore, Antcin H may offer a novel therapeutic to prevent or treat liver injury and perhaps other diseases associated with the adverse effects of sustained JNK activation, which require the interaction of JNK and mitochondrial Sab.
Materials and Methods
Chemicals and reagents
Antcin H (purity 95% or higher) was isolated from AC by Ye's laboratory as reported previously (31). Acetaminophen (APAP), dexamethasone (Dexa), galactosamine (GalN), actinomycin D, ADP, ATP, dihydroethidium, oligomycin, CCCP, FeTCCP, antimycin A, and GAPDH antibody were purchased from Sigma-Aldrich (St. Louis, MO). TNFα was purchased from Calbiochem (Billerica, MA). Antibodies specific for p-JNK, JNK, p-MKK4, MKK4, and β-actin were purchased from Cell Signaling Technology (Denvers, MA). NAPQI adduct antiserum was provided by Laura James at the University of Arkansas. 3-Nitrotyrosine antibody for immunohistochemistry and immunoblotting was obtained from Abcam (Cambridge, MA). Sab antibody was purchased from Santa Cruz Biotechnology (Dallas, TX).
Animal experiments
Male C57BL/6NHsd mice (6 to 8 weeks of age) were purchased from Harlan Bioproducts for Science Inc. (Indianapolis, IN). All mice were housed in a temperature-, light-, and humidity-controlled environment that is accredited by the Associate for Assessment and Accreditation of Laboratory Animal Care. Mice were maintained on standard laboratory chow, had free access to water, and acclimatized for at least 1 week. APAP was dissolved in 55°C warm PBS. Overnight-fasted mice received APAP 300 mg/kg body weight by intraperitoneal (i.p.) injection as described before (43). Antcin H dissolved in corn oil (to avoid DMSO) was injected i.p. 1 h before or after APAP treatment.
Plasma and liver tissue were collected 1, 2, 4, and 24 h after APAP. Overnight-fasted mice received GalN (800 mg/kg)/TNFα (12 μg/kg) dissolved in sterile PBS by i.p. injection as described before (43). Mice were pretreated with Antcin H, 6 and 1 h in DMSO, before GalN/TNFα injection. Plasma and liver tissue were collected 30 min, 1, 2, and 6 h after GalN/TNFα. A portion of the liver was fixed in 10% neutral formalin for histology, and the rest of the tissue was used for subcellular fractionation and immunoblotting. Alanine aminotransferase (ALT) was measured using reagent kits from Teco Diagnostics (Anaheim, CA).
Immunohistochemical and chemical staining
After deparaffinization and rehydration of neutral buffered formalin-fixed paraffin-embedded tissue sections (5 μm thickness), primary antibody to 3-Nitrotyrosine immunostaining was performed using the Leica BOND Stainer apparatus as described before (43). Liver sections were also stained with hematoxylin and eosin.
Cell culture and treatments
PMHs were isolated from C57BL/6 N mice as previously described (8). The viability of isolated hepatocytes was assessed by trypan blue dye exclusion. The cells used for all experiments had a viability exceeding 88%. Three hours after plating hepatocytes, the medium containing serum and phenol red was replaced with serum-free DMEM/F12 medium containing 100 U/mL penicillin and 0.1 mg/mL streptomycin. After pretreatment with DMSO or Antcin H for 2 h, the medium was replaced with 10 mM APAP dissolved in prewarmed DMEM/F12 medium. Two hours after APAP exposure, the medium was removed and replaced with DMEM/F12 medium supplemented with Antcin H. Cell lysates were collected at 1, 2, or 4 h, and cell death assessed at 24 h by double staining of SYTOX Green (1 μM) and Hoechst 33258 (8 μg/mL) (28).
In other experiments, cultured hepatocytes were incubated with actinomycin D (Act D; 0.5 μg/mL)/TNFα (20 ng/mL) with Antcin H or TNFα with Antcin H. After the indicated time points, cell lysates were stained with Hoechst 33258 for apoptotic counts (9).
GSH measurement
GSH was measured by spectrophotometric/microplate reader using the enzymatic recycling method as previously described (33).
JNK kinase assay measured by c-Jun phosphorylation assay
JNK activity was determined according to the modified protocol described below using the nonradioactive SAPK/JNK Kinase Assay Kit (No. 8794; Cell Signaling Technology). Two hours after APAP 300 mg/kg i.p., liver was homogenized in the isolation buffer supplemented with protease and phosphatase inhibitor cocktail dissolved in DMSO or water. Cytoplasm was obtained after two times centrifugation at 15,000 × g, 4°C. Thirty micrograms of cytoplasm was incubated with different concentrations of DMSO, SP600125, or Antcin H at 30°C for 10 min followed by addition of 50 μM ATP and kinase substrate, c-Jun fusion protein (1 μg), and incubated at 30°C for 30 min. Reaction was terminated with SDS sample buffer and boiled for 3 min. Western blot for p-c-Jun, c-Jun, p-JNK, and GAPDH was then performed.
p-JNK and mitochondria binding assay
Liver mitochondria from PBS-treated mouse were isolated as described before (45). Liver cytoplasm from APAP 300 mg/kg i.p.-treated mouse was isolated 2 h after treatment as described before (45). Mitochondria (1 mg/100 μl, 5% BSA) was preincubated at room temperature for 15 min. Cytoplasm (5 mg/600 μl) with Antcin H or structurally similar dexamethasone was preincubated at room temperature for 30 min and then added to mitochondria and incubated for 1 h at room temperature. Mitochondria were then washed in 10 mL of isolation buffer with protease and phosphatase inhibitor cocktail. Mitochondria pellets were dissolved in RIPA buffer for Western blot.
Mitochondrial respiration measurement
Livers were homogenized in mitochondrial assay solution (MAS) (70 mM sucrose, 25 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, pH 7.2 at 37°C) supplemented with pyruvate and malate, without protease and phosphatase inhibitors. Mitochondria were isolated by differential centrifugation and resuspended in MAS.
Fifty nanograms (160 U/mg) activated or unactivated JNK was diluted in 25 μl of MAS and incubated with Antcin H or dexamethasone (Dexa) at room temperature for 30 min. Five micrograms of mitochondria was diluted in 24 μl MAS supplemented with ATP 6 μM and then JNK was added to with Antcin H or Dexa. Fifty microliters premixed mitochondria 5 μg and 50 ng JNK with 24–50 μM Antcin H or Dexa was then loaded into each well of Seahorse XF24 analyzer cell culture microplate on ice and was centrifuged at 3000 × g for 5 min at 8°C, and then incubated at 37°C in CO2-free incubator for 15 min.
After incubation, each well was fed with 450 μl of 37°C prewarmed MAS supplemented with substrate, and oxygen consumption rate (OCR) was measured. The program was set to equilibrate at 37°C for 5 min before sequential measurement of basal OCR (State 2 respiration), followed by injection of ADP (4 mM; final), oligomycin (2.5 μg/ml; final), CCCP (4 μM; final), and antimycin A (4 μM; final), and OCR was measured. After ADP injection, mitochondrial oxidative phosphorylation (State 3 respiration) was determined. OCR after oligomycin injection was defined as State 4 respiration, and CCCP induced OCR as maximal respiratory capacity (37, 42, 45).
Subcellular fractionation for cytoplasm and mitochondria from liver
Livers were homogenized in ice cold homogenizing buffer (250 mM sucrose, 10 mM Tris, 2 mM EGTA, pH 7.4) supplemented with protease and phosphatases inhibitor cocktail. The homogenate was centrifuged at 1000 × g for 5 min; the centrifugation was repeated again for 10 min. The supernatant was centrifuged at 9000 × g for 10 min. The resulting supernatant is referred to as cytoplasm. The mitochondria pellet was washed with isolation buffer and recentrifuged. The mitochondria were resuspended in RIPA buffer for Western blot analysis (45).
Western blotting
Thirty micrograms of protein samples was denatured, and SDS-PAGE electrophoresis was performed in Bio-Rad 7.5%, 10%, or 4%–20% Mini-PROTEAN® TGX™ Precast Gel. After transfer to 0.2 μm nitrocellulose membrane using iBlot transfer device (Invitrogen), immunoblot was performed using specific antibodies as described.
Measurement of antimycin A and p-JNK-induced mitochondrial ROS
Mitochondria from C57BL/6 N mice liver were isolated in respiration buffer (MAS buffer supplemented with pyruvate and malate without protease and phosphatase inhibitors). Twenty micrograms of mitochondria in 50 μl of respiration buffer was loaded into precooled 96-well solid bottom Corning Costar® plate (Cat. No. 3917) on ice. One hundred microliters of ice cold respiration buffer was supplemented with MitoSOX (2 μM; final), antimycin A (5 μM; final), and ADP (4 μM; final). Antcin H (25 μM; final) or FeTCCP (1 μM; final) was added into indicated wells on ice just before loading into 37°C prewarmed FLUOstar Omega Microplate Reader. Six minutes after equilibration at 37°C in a dark chamber, basal level of fluorescence intensity was measured at Ex355nm/Em590nm and fluorescence intensity was measured at 6-min intervals.
Mitochondria and cytoplasm from APAP 2 h-treated mice or untreated mice were isolated as described above. Twenty micrograms of control mitochondria in 50 μl of respiration buffer was loaded into precooled 96-well plates on ice. MitoSOX (2 μM; final), ADP (4 μM; final), and 100 μg APAP-treated cytoplasm, which provided activated JNK or normal cytoplasm, were added into indicated wells on ice as above and then fluorescence intensity was measured.
Statistical analysis
Data are presented as mean ± SD. T-test was used for comparison of two groups. p < 0.05 was considered statistically significant.
Supplementary Material
Abbreviations Used
- 3-NT
3-Nitrotyrosine
- AC
Antrodia Camphorate
- Act D
actinomycin D
- ALT
alanine aminotransferase
- APAP
acetaminophen
- BHA
butyl hydroxy anisd
- CCCP
carbonyl cyanide 3-chlorophenylhydrazone
- Dexa
dexamethasone
- DMEM
Dulbecco's modified Eagle's medium
- DPPH
2,2-diphenyl-1picrylhydrazyl
- GalN
galactosamine
- GSH
glutathione
- JNK
c-Jun-N-terminal kinase
- MAS
mitochondrial assay solution
- MKK4
mitogen-activated protein kinase kinase 4
- NAPQI
N-acetyl-p-benzoquinone
- OCR
oxygen consumption rate
- PBS
phosphate-buffered saline
- PHB1
prohibitin
- PMH
primary mouse hepatocytes
- RIPA
radioimmunoprecipitation
- ROS
reactive oxygen species
- Sab
Src homology 3 domain-binding protein 5
- TNF
tumor necrosis factor
Acknowledgments
This work was supported by the NIH grant R01DK067215 (NK) and Microscopy, Histology, and Cell Separation and Culture Core of the USC Research Center (P30DK48522) and by a grant from the Ministry of Science and Technology of China (2012BAD33B09). The authors thank the China Scholarship Council for financial support of Y.H.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Abdelmegeed MA, Jang S, Banerjee A, Hardwick JP, and Song BJ. Robust protein nitration contributes to acetaminophen-induced mitochondrial dysfunction and acute liver injury. Free Radical Bio Med 60: 211–222, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amir M, Liu K, Zhao E, and Czaja MJ. Distinct functions of JNK and c‐Jun in oxidant‐induced hepatocyte death. J Cell Biochem 113: 3254–3265, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.An L, Wang X, and Cederbaum AI. Cytokines in alcoholic liver disease. Arch Toxicol 86: 1337–1348, 2012 [DOI] [PubMed] [Google Scholar]
- 4.Chambers JW. and LoGrasso PV. Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J Biol Chem 286: 16052–16062, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen CH, Yang SW, and Shen YC. New steroid acids from Antrodia cinnamomea, a fungal parasite of Cinnamomum micranthum. J Nat Prod 1655–1661, 1995 [DOI] [PubMed] [Google Scholar]
- 6.Coen M. Metabolic phenotyping applied to pre-clinical and clinical studies of acetaminophen metabolism and hepatotoxicity. Drug Metab Rev 47: 29–44, 2015 [DOI] [PubMed] [Google Scholar]
- 7.Davidson DG. and Eastham WN. Acute liver necrosis following overdose of paracetamol. Br Med J 2: 497–499, 1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng G. and Kaplowitz N. Colchicine protects mice from the lethal effect of an agonistic anti-Fas antibody. J Clin Invest 105: 329–339, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng G. and Kaplowitz N. Mechanism of staurosporine-induced apoptosis in murine hepatocytes. Am J Physiol-Gastrointest Liver Physiol 282: G825–G834, 2002 [DOI] [PubMed] [Google Scholar]
- 10.Geethangili M. and Tzeng YM. Review of pharmacological effects of Antrodia camphorata and its bioactive compounds. Evid Based Complement Alternat Med 212641, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gulmez SE, Larrey D, Pageaux GP, Bernuau J, Bissoli F, Horsmans Y, Thorburn D, McCormick PA, Stricker B, Toussi M, Lignot-Maleyran S, Micon S, Hamoud F, Lassalle R, Jové J, Blin P, and Moore N. Liver transplant associated with paracetamol overdose: results from the seven‐country SALT study. Br J Clin Pharmacol 80: 599–606, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, and Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 131: 165–178, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Han D, Shinohara M, Ybanez MD, Saberi B, and Kaplowitz N. Signal transduction pathways involved in drug-induced liver injury. Adverse Drug React 267–310, 2010 [DOI] [PubMed] [Google Scholar]
- 14.Han D, Ybanez MD, Ahmadi S, Yeh K, and Kaplowitz N. Redox regulation of tumor necrosis factor signaling. Antioxid Redox Signal 11: 2245–2263, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, and Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem 283: 13565–13577, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinson JA, Michael SL, Ault SG, and Pumford NR. Western blot analysis for Nitrotyrosine protein adducts in livers of saline-treated and acetaminophen-treated mice. Toxicol Sci 53: 467–473, 2000 [DOI] [PubMed] [Google Scholar]
- 17.Hinson JA, Pike SL, Pumford NR, and Mayeux PR. Nitrotyrosine-protein adducts in hepatic centrilobular areas following toxic doses of acetaminophen in mice. Chem Res Toxicol 11: 604–607, 1998 [DOI] [PubMed] [Google Scholar]
- 18.James LP, Mayeux PR, and Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos 31: 1499–1506, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Ker YB, Peng CC, Chang WL, Chyau CC, and Peng RY. Hepatoprotective bioactivity of the glycoprotein, antrodan, isolated from Antrodia cinnamomea mycelia. PLoS One 9: e93191, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, Cheng K, Chen YN, Campbell A, Sudha T, Yuan ZM, Narula J, Weichselbaum R, Nalin C, and Kufe D. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-xL in response to DNA damage. J Biol Chem 275: 322–327, 2000 [DOI] [PubMed] [Google Scholar]
- 21.Knight TR, Ho YS, Farhood A, and Jaeschke H. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J Pharmacol Exp Ther 303: 468–475, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiødt FV, Ostapowicz G, Shakil AO, and Lee WM. Acetaminophen‐induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 42: 1364–1372, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Lee WM, Squires RH, Nyberg SL, Doo E, and Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 47: 1401–1415, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu YW, Lu KH, Ho CT, and Sheen LY. Protective effects of Antrodia cinnamomea against liver injury. J Tradit Complement Med 2: 284, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu MC, El-Shazly M, Wu TY, Du YC, Chang TT, Chen CF, Hsu YM, Lai KH, Chiu CP, Chang FR, and Wu YC. Recent research and development of Antrodia cinnamomea. Pharmacol Ther 139: 124–156, 2013 [DOI] [PubMed] [Google Scholar]
- 26.Male KB, Rao YK, Tzeng YM, Montes J, Kamen A, and Luong JH. Probing inhibitory effects of Antrodia camphorata isolates using insect cell-based impedance spectroscopy: inhibition vs chemical structure. Chem Res Toxicol 21: 2127–2133, 2008 [DOI] [PubMed] [Google Scholar]
- 27.McGill MR, Lebofsky M, Norris HRK, Slawson MH, Bajt ML, Xie Y, Williams CD, Wilkins DG, Rollins DE, and Jaeschke H. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: dose–response, mechanisms, and clinical implications. Toxicol Appl Pharmacol 269: 240–249, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagai H, Matsumaru K, Feng G, and Kaplowitz N. Reduced glutathione depletion causes necrosis and sensitization to tumor necrosis factor‐α-induced apoptosis in cultured mouse hepatocytes. Hepatology 36: 55–64, 2002 [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa H, Maeda S, Hikiba Y, Ohmae T, Shibata W, Yanai A, Sakamoto K, Ogura K, Noguchi T, Karin M, Ichijo H, and Omata M. Deletion of apoptosis signal-regulating kinase 1 attenuates acetaminophen-induced liver injury by inhibiting c-Jun N-terminal kinase activation. Gastroenterology 135: 1311–1321, 2008 [DOI] [PubMed] [Google Scholar]
- 30.Phuong DT, Ma CM, Hattori M, and Jin JS. Inhibitory effects of antrodins A-E from Antrodia cinnamomea and their metabolites on hepatitis C virus protease. Phytother Res 23: 582–584, 2009 [DOI] [PubMed] [Google Scholar]
- 31.Qiao X, An R, Huang Y, Ji S, Li L, Tzeng YM, Guo DA, and Ye M. Separation of 25R/S-ergostane triterpenoids in the medicinal mushroom Antrodia camphorata using analytical supercritical-fluid chromatography. J Chromatogr A 1358: 252–260, 2014 [DOI] [PubMed] [Google Scholar]
- 32.Qiao X, Wang Q, Ji S, Huang Y, Liu KD, Zhang ZX, Bo T, Tzeng YM, Guo DA, and Ye M. Metabolites identification and multi-component pharmacokinetics of ergostane and lanostanetriterpenoids in the anticancer mushroom Antrodiacinnamomea. J Pharm Biomed Anal 111: 266–276, 2015 [DOI] [PubMed] [Google Scholar]
- 33.Rahman I, Kode A, and Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc 1: 3159–3165, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Saito C, Lemasters JJ, and Jaeschke H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol 246: 8–17, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen YC, Wang YH, Chou YC, Chen CF, Lin LC, Chang TT, Tien JH, and Chou CJ. Evaluation of the anti-inflammatory activity of zhankuic acids isolated from the fruiting bodies of Antrodia camphorata. Planta Med 70: 310–314, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Tahir M, Rehman MU, Lateef A, Khan R, Khan AQ, Qamar W, Ali F, O'Hamiza O, and Sultana S. Diosmin protects against ethanol-induced hepatic injury via alleviation of inflammation and regulation of TNF-α and NF-κB activation. Alcohol 47: 131–139, 2013 [DOI] [PubMed] [Google Scholar]
- 37.Than TA, Win S, and Kaplowitz N. In vitro assays of mitochondrial function/dysfunction. Clin Pharmacol Ther 96: 665–668, 2014 [DOI] [PubMed] [Google Scholar]
- 38.Tilg H. and Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. New Engl J Med 343: 1467–1476, 2000 [DOI] [PubMed] [Google Scholar]
- 39.Tsou TC, Tsai FY, Wu MC, and Chang LW. The protective role of NF-κB and AP-1 in arsenite-induced apoptosis in aortic endothelial cells. Toxicol Appl Pharmacol 191: 177–187, 2003 [DOI] [PubMed] [Google Scholar]
- 40.Twumasi‐Boateng K, Wang TW, Tsai L, Lee KH, Salehpour A, Bhat S, Tan MW, and Shapira M. An age-dependent reversal in the protective capacities of JNK signaling shortens Caenorhabditis elegans lifespan. Aging cell 11: 659–667, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weston CR. and Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol 19: 142–149, 2007 [DOI] [PubMed] [Google Scholar]
- 42.Win S, Than TA, Fernandez-Checa JC, and Kaplowitz N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis 5: e989, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Win S, Than TA, Han D, Petrovic LM, and Kaplowitz N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J Biol Chem 286: 35071–35078, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Win S, Than TA, Le BH, García-Ruiz C, Fernandez-Checa JC, and Kaplowitz N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J Hepatol 62: 1367–1374, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Win S, Than TA, Min RWM, Aghajan M, and Kaplowitz N. JNK mediates mouse liver injury through a novel Sab (SH3BP5) dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 63: 1987–2003, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang L, Sun G, Guo Y, Hou Z, and Chen S. Holistic evaluation of quality consistency of Ixeris sonchifolia (Bunge) Hance injectables by quantitative fingerprinting in combination with antioxidant activity and chemometric methods. PLoS One 11: e0148878, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ye X, Liu SF, and Liu G. NF-κB to Ap-1 switch: a mechanism regulating the transition from endothelial barrier injury to barrier repair following endotoxemic lung injury. Lung 1: 2, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.