

Abstract
Background
Familial breast cancer (BC) represents 5 to 10% of all BC cases. Mutations in two high susceptibility BRCA1 and BRCA2 genes explain 16–40% of familial BC, while other high, moderate and low susceptibility genes explain up to 20% more of BC families. The Lebanese reported prevalence of BRCA1 and BRCA2 deleterious mutations (5.6% and 12.5%) were lower than those reported in the literature.
Methods
In the presented study, 45 Lebanese patients with a reported family history of BC were tested using Whole Exome Sequencing (WES) technique followed by Sanger sequencing validation.
Results
Nineteen pathogenic mutations were identified in this study. These 19 mutations were found in 13 different genes such as: ABCC12, APC, ATM, BRCA1, BRCA2, CDH1, ERCC6, MSH2, POLH, PRF1, SLX4, STK11 and TP53.
Conclusions
In this first application of WES on BC in Lebanon, we detected six BRCA1 and BRCA2 deleterious mutations in seven patients, with a total prevalence of 15.5%, a figure that is lower than those reported in the Western literature. The p.C44F mutation in the BRCA1 gene appeared twice in this study, suggesting a founder effect. Importantly, the overall mutation prevalence was equal to 40%, justifying the urgent need to deploy WES for the identification of genetic variants responsible for familial BC in the Lebanese population.
Electronic supplementary material
The online version of this article (doi:10.1186/s12920-017-0244-7) contains supplementary material, which is available to authorized users.
Keywords: Breast cancer, BRCA, Next-generation sequencing, Exome, Familial, Lebanon, Germline, Mutation
Background
Breast cancer (BC) is the most common cancer in women, accounting for around 25% of all new cases of cancer [1]. Most BC cases are sporadic, while 5 to 10% of all BC cases are inherited and cluster in families [1]. While mutations in BRCA1 and BRCA2 genes explain 16–40% of all familial BC cases [1–3], other genes have also been found to increase BC susceptibility, which highlights the polygenic nature of many BC cases [4]. Some of these genes including CDH1, TP53, PTEN and STK11, although less frequently altered compared to the BRCA1/2 genes, they have been linked to high-penetrance autosomal dominant BC [5–7]. Moderate penetrance genes are implicated in around 5% of familial BC. These genes include the Fanconi anemia pathway genes: FANCA, PALB2, BRIP1, RAD51C and XRCC2 [8–10] and non-Fanconi anemia genes: ATM, CHEK2, NBN, RAD50, RAD51B, and RAD51D [11–15].
In Lebanon, BC is the most common cancer type in females and it constitutes one-third of all reported cancer cases. BC incidence rates are expected to reach 137 per 100,000 by 2018 [16]. Yet, to date, only two studies have investigated the role of BRCA1 and BRCA2 mutations in the Lebanese population. These studies reported varied prevalence of pathogenic BRCA mutations ranging between 5.6 to 12.5% in BC cases [17, 18]. The reported prevalences of both BRCA1 and BRCA2 deleterious mutations were lower than those reported for the Western populations, which suggest the involvement of other genes in the pathogenesis of BC cases [19]. The reported low prevalence does not support the hypothesis that BRCA1 and BRCA2 mutations alone are responsible for the majority of the observed Lebanese women with early-onset BC. This finding could well explain the fact that BC is a disease with a high level of genetic heterogeneity and that monogenic and polygenic models of inheritance may exist.
Since the completion of the human genome project, massive leaps have reshaped the field of clinical genomics. The development of Next-generation sequencing (NGS) platforms allowed a more robust, fast and accurate analysis of diseases and syndromes with polygenic nature. NGS platforms including WES are believed to enhance and improve diagnosis and therapy development of many diseases including BC [20–23].
In the presented study, we utilized WES to investigate germline genetic variations in 45 Lebanese cases diagnosed with familial BC and unknown BRCA1 or BRCA2 status. We found several rare variants that can potentially explain BC susceptibility in the analyzed cases.
Methods
Inclusion criteria
From 2012 to 2015, 45 unrelated patients with inherited BC were selected to undergo DNA testing. They were referred from a wide variety of settings from all over the country, ranging from private physicians’ clinics to major academic medical centers because of hereditary BC. The patients fulfilled a personal history of invasive BC and at least one of the following criteria: A) diagnosis at age ≤ 40 years, B) BC at any age at onset with at least 2 first- and/or second-degree relatives, C) BC < 50 years in a first- or second-degree relative, D) ovarian cancer in at least 2 first- and/or second-degree relatives, E) breast and ovarian cancer in at least 2 first- and/or second-degree relatives, F) both breast and ovarian cancer in a single first- or second-degree relative.
Approval to conduct the study was obtained from the Ethics Committee of Saint-Joseph University-Lebanon. After an informed consent was signed and all ethical requirements were fulfilled, a 10 ml of peripheral blood was isolated from each individual enrolled and the DNA was extracted using the salting out methods [24]. All patients signed the informed consent and agreed to share their variant data.
Whole exome sequencing
Exon capture and sequencing: Samples were prepared for whole Exome sequencing and enriched according to the manufacturer’s standard protocol. The concentration of each library was determined using Agilent’s QPCR NGS Library Quantification Kit (G4880A). Samples were pooled prior to sequencing with each sample at a final concentration of 10nM. Sequencing was performed on the Illumina HiSeq2000 platform using TruSeq v3 chemistry.
Mapping and alignment: Reads files (FASTQ) were generated from the sequencing platform via the manufacturer’s proprietary software. Reads were aligned to the hg19/b37 reference genome using the Burrows-Wheeler Aligner (BWA) package v0.6.1 [25]. Local realignment of the mapped reads around potential insertion/deletion (Indel) sites was carried out with the Genome Analysis Tool Kit (GATK) v1.6 [26]. Duplicate reads were marked using Picard v1.62. Additional BAM file manipulations were performed with Samtools 0.1.18 [27]. Base quality (Phred scale) scores were recalibrated using GATK’s covariance recalibration. SNP and Indel variants called using the GATK Unified Genotyper for each sample [28]. SNP novelty is determined against dbSNP. A list of 134 genes known to be associated with hereditary BC and other cancers were studied (Additional file 1).
Variants evaluation
Variants obtained were reported using five categories according to the Human Genome Mutation Database (HGMD Professional) [29]. These categories are listed in Table 1.
Table 1.
Variants reported in five categories according to the HGMD Professional
| Category | Category | Variation reported as |
|---|---|---|
| DM | Disease-causing mutations | Pathological mutation |
| DM? | Disease-causing mutations | Likely pathological mutation |
| DP | Disease-associated polymorphism | Polymorphism in significant association with a disease/phenotype (p < 0.05) that is assumed to be functional |
| DFP | Disease-associated polymorphism | Polymorphism in significant association with disease (p < 0.05) that has evidence of being of direct functional importance |
| FP | In vitro/laboratory or in vivo functional polymorphism | Polymorphism that affects the structure, function or expression of the gene (or gene product), but with no disease association reported as yet |
The first variant category consists of alleles labeled as disease causing mutations (DM) in HGMD Professional. These alleles must be rare: <1% allele frequency in 6,500 exomes from the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (“Exome Variant Server” 2015) and the 1,000 Genomes Project Genomes [30].
The BRCA gene variants identified were checked for pathogenicity in 4 databases: Breast Cancer Information Core (BIC) [31], Leiden Open Variation Database (LOVD) [32], the Catalogue of Somatic Mutations in Cancer database (COSMIC) [33] and BRCA Exchange website (http://brcaexchange.org) providing data from the ENIGMA consortium [34].
Variants confirmation
Sanger’s sequencing was utilized to confirm the relevant variants identified by WES and to study the segregation of these variants with the disease phenotype in members of families included in the study. PCR reactions were run in final volumes of 50 μl containing 100 ng DNA, 0.25 mM dNTPs, 100 ng of each primer and 0.02 unit of Taq polymerase (Invitrogen Life Technologies, Carlsbad, CA, USA). PCR was performed in an ABI9700 thermocycler (Applied Biosystems, Foster City, CA) with initial denaturation at 94 °C for 5 min, followed by 35 cycles of 95 °C for 30s, specific annealing temperature for 30s, 72 °C for 30s. Primer sequences are available on request as well as annealing temperatures of each exon. PCR products were purified using the illustraTM GFX PCR DNA and Gel Band Purification Kit (GE Healthcare, Buckinghamshire, UK). Both strands of the products were sequenced using the BigDyeW Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) under standard conditions. The labeled products were subjected to electrophoresis on an ABI3130 and ABI3500 Genetic Analyzer sequencing system (Applied Biosystems, Foster City, CA, USA). Electropherograms were analyzed using Sequence Analysis Software v5.2 (Applied Biosystems, Foster City, CA, USA) and compared to reference sequences using ChromasPro v1.7.6.1 (Technelysium, Queensland, Australia).
Results
Patient characteristics and sequencing statistics
The mean age at diagnosis of BC for the 45 patients was 44 years (range 29–79). Sixteen patients provided us with their histopathological results. Seven BC were estrogen-receptor (ER) and progesterone-receptor (PR) positive, 5 patients had negative ER and PR disease and 2 patients had negative ER and positive PR disease. Two patients had triple negative disease from which one patient (Family 30) carried p.C44F mutation in BRCA1 (Fig. 1).
Fig. 1.
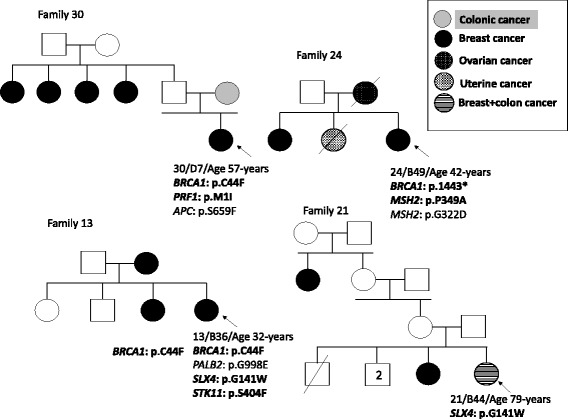
Pedigrees of the families presenting DM mutations. Filled squares (males) and circles (females) indicate the affected individuals. Probands are marked with arrows. DM mutations are bolded
We obtained an average of 44 million reads per sample, with a mean coverage of 94% at a mean X coverage of 20X.
WES analysis
Within this cohort, a total of 126 variants were detected by WES and these are listed in Table 2. In 7 of the 45 patients, not listed in Table 2, no variants in cancer predisposing genes (Additional file 1) were identified.
Table 2.
Mutations in BC associated genes detected by NGS in a series of 45 Lebanese patients
| Family number/Patient code | Genes | Results | HGMD Professional | ExAC allele frequency |
|---|---|---|---|---|
| 1/B19 | XRCC3 | c.C722T p.T241M | DFP Association with melanoma | 0.3075 |
| XRCC1 | c.G839A p.R280H | DFP Association with increased lung cancer | 0.08811 | |
| CASP8 | c.*429A > G | Not found | Not found | |
| BRCA2 | c.C65T p.A22V | Not found | Not found | |
| MUTYH | c.C1258G p.L420V | Not found | Not found | |
| SLX4 | c.C1837T p.Q613X | Not found | Not found | |
| 2/B21 | BRCA1 | c.A536G p.Y179C | DM Breast and/or ovarian cancer | 0.0002718 |
| ATM | c.T2572C p.F858L | DP Association with breast cancer | 0.009149 | |
| ATM | c.C3161G p.P1054R | DFP Association with breast cancer | 0.01692 | |
| TP53 | c.C215G p.P72R | DFP Association with Lung cancer | 0.6600 | |
| 3/B22 | BRCA2 | c.C5744T p.T1915M | DP Association with breast cancer risk | 0.01790 |
| ARL11 | c.G446A p.W149X | DP Association with cancer | 0.009898 | |
| TP53 | c.C215G p.P72R | DFP Association with Lung cancer | 0.6600 | |
| ERCC3 | c.C508T p.R170X | Not found | Not found | |
| VHL | c.A631C p.M211L | Not found | 0.00004623 | |
| MRE11A | c.C1491T p.I497I | Not found | 0.0006514 | |
| PTCH1 | c.G4054A p.V1352I | Not found | Not found | |
| 4/B23 | TP53 | c.C215G p.P72R | DFP Association with Lung cancer | 0.6600 |
| TP53 | c.C245T p.P82L | DM Breast cancer | 0.00001657 | |
| BRCA2 | c.A1114C p.N372H | DFP Association with Breast cancer | 0.2779 | |
| FANCA | c.G1038C p.W346C | Not found | 0.00006621 | |
| POLE | c.C3890T p.S1297L | Not found | 0.00002580 | |
| POLD1 | c.T2257C p.Y753H | Not found | Not found | |
| GATA2 | c.C1040T p.T347I | Not found | Not found | |
| 5/B24 | BRCA2 | c.G8775C p.Q2925H | Not found | 0.000008322 |
| APC | c.C6821T p.A2274V | DM Adenomatous polyposis coli | 0.0009917 | |
| EZH2 | c.C349T p.Q117X | Not found | Not found | |
| 6/B25 | XRCC3 | c.C722T p.T241M | DFP Association with melanoma | 0.3075 |
| MLH1 | c.A655G p.I219V | DP Colorectal cancer, non-polyposis | 0.2325 | |
| RAD51D | c.G494A p.R165Q | Not found | Not found | |
| ATM | c.496 + 4 T > C | Not found | 0.00009891 | |
| PPM1D | c.G275C p.C92S | Not found | Not found | |
| STK11 | c.375-1C > T | DM Colorectal cancer | Not found | |
| 7/B26 | XRCC3 | c.C722T p.T241M | DFP Association with melanoma | 0.3075 |
| MSH2 | c.C1045G p.P349A | DM Renal cell carcinoma | 0.00009062 | |
| MUTYH | c.C1174A p.L392M | Not found | Not found | |
| MUTYH | c.C1258A p.L420M | DM? Colorectal cancer | Not found | |
| ATM | c.496 + 4 T > C | Not found | 0.00009891 | |
| RB1 | c.C1505T p.T502I | Not found | 0.00001098 | |
| PPM1D | c.G275C p.C92S | Not found | Not found | |
| 8/B27 | XRCC3 | c.C722T p.T241M | DFP Association with melanoma | 0.3075 |
| BRCA1 | c.A1067G p.Q356R | DP Association with breast and/or ovarian cancer | 0.04407 | |
| XRCC1 | c.C580T p.R194W | DFP Benign breast disease | 0.09276 | |
| CDH1 | c.G1774A p.A592T | DM? Breast cancer | 0.003212 | |
| 35/B28 | BARD1 | c.1071_1091del p.357_364del | Not found | Not found |
| ABCC12 | c.G490T p.G164X | DM Bladder cancer | 0.003185 | |
| MCC | c.G152T p.G51V | Not found | 0.0001346 | |
| 9/B31 | ATM | c.T2119C p.S707P | DFP Association with Breast cancer | 0.007927 |
| FANCA | c.C4232T p.P1411L | Not found | 0.0001318 | |
| 36/B32 | ATM | c.C2770T p.R924W | Not found | 0.00004942 |
| ALK | c.T4211C p.L1404P | Not found | 0.00008370 | |
| 10/B33 | BRIP1 | c.A3571G p.I1191V | Not found | 0.00004967 |
| NSD1 | c.2224_2243del p.P742fs | Not found | Not found | |
| FANCG | c.G1298C p.R433P | Not found | 0.00004118 | |
| FLCN | c.T1387C p.Y463H | Not found | 0.00003298 | |
| PTCH1 | c.A3749G p.Y1250C | Not found | Not found | |
| 11/B34 | XRCC4 | c.T401C p.I134T | DP Association with Lung cancer | 0.02505 |
| RAD51C | c.G376A p.A126T | FP Reduced activity | 0.003529 | |
| 12/B35 | ARL11 | c.G571A p.G191R | Not found | 0.00002188 |
| Rad50 | c.A280C p.I94L | DM? Breast and/or ovarian cancer | 0.003473 | |
| POLE | c.G2276A p.R759H | Not found | 0.00001647 | |
| 13/B36 | BRCA1 | c.G131T p.C44F | DM Breast and/or ovarian cancer | Not found |
| SLX4 | c.G421T p.G141W | DM Breast and/or ovarian cancer | 0.0008237 | |
| STK11 | c.C1211T p.S404F | DM Peutz-Jeghers syndrome | 0.0009281 | |
| PALB2 | c.G2993A p.G998E | DP Breast cancer, increased risk- | 0.01579 | |
| BRCA2 | c.C1151T p.S384F | DM? Breast cancer | 0.0006789 | |
| DICER1 | c.A5276G p.K1759R | Not found | 0.00004942 | |
| CEBPA | c.T122C p.I41T | Not found | Not found | |
| RECQL4 | c.G3314A p.G1105D | Not found | 0.005430 | |
| 14/B37 | RAD50 | c.G379A p.V127I | Not found | 0.001653 |
| CASP8 | c.A1117G p.I373V | Not found | Not found | |
| RECQL4 | c.C3184T p.R1062W | Not found | 0.0003129 | |
| WRN | c.G4129A p.G1377S | Not found | 0.00002483 | |
| 15/B38 | BARD1 | c.C119T p.A40V | Not found | 0.00004775 |
| PTCH1 | c.169_170delCT p.57_57del | Not found | 0.000008913 | |
| PTCH1 | c.A3749G p.Y1250C | Not found | Not found | |
| PTCH1 | c.C4126T p.R1376W | Not found | Not found | |
| ERCC5 | c.A1904G p.H635R | Not found | Not found | |
| DICER1 | c.C3811T p.L1271F | Not found | Not found | |
| 16/B39 | CDKN2A | c.G442A p.A148T | DP Association with melanoma | 0.02278 |
| RAD51D | c.A758G p.E253G | Not found | 0.01144 | |
| ERCC6 | c.C2800A p.P934T | DM Cockayne syndrome | Not found | |
| 17/B40 | MSH2 | c.A1787G p.N596S | DM Colorectal cancer, non-polyposis | 0.0002558 |
| ATM | c.A1982C p.D661A | Not found | Not found | |
| PMS2 | c.G1688T p.R563L | DM? Colorectal cancer, non-polyposis | 0.005813 | |
| GPC3 | c.78_79insCCG p.P27delinsPP | Not found | Not found | |
| 18/B41 | BRCA2 | c.658_659delGT p.V220I* | DM Breast and/or ovarian cancer | 0.00006119 |
| SLX4 | c.G3337C p.G1113R | Not found | 0.000008237 | |
| SMARCA4 | c.C1098G p.I366M | Not found | 0.00002715 | |
| EPHX1 | c.G1040C p.R347T | Not found | 0.00003296 | |
| 19/B42 | wwoxtv2 | c.A544G p.K182E | DM? cancer | Not found |
| ATM | c.A5558T p.D1853V | DP Association with breast cancer, contralateral | 0.005186 | |
| RET | c.C2508T p.S836S | DP Association with thyroid cancer | 0.04666 | |
| BRCA1 | c.5090_5093delGTTA p.L1697fs | Not found | Not found | |
| 20/B43 | PALB2 | c.G2014C p.E672Q | DM? Breast cancer? (common variant) | 0.02239 |
| PALB2 | c.G2993A p.G998E | DP Breast cancer (common variant) | 0.01579 | |
| RAD51C | c.G376A p.A126T | FP Reduced activity | 0.003529 | |
| Tp53 | c.673-36G > C | DFP Breast cancer | Not found | |
| 21/B44 | SLX4 | c.G421T p.G141W | DM Breast and/or ovarian cancer | 0.0008237 |
| SLX4 | c.C1919A p.T640N | Not found | Not found | |
| FANCM | c.A5224G p.I1742V | Not found | 0.008398 | |
| POLD1 | c.G2793C p.K931N | Not found | Not found | |
| 22/B45 | ATM | c.A5071C p.S1691R | DM Ataxia telangiectasia | 0.002019 |
| BRIP1 | c.G2220T p.Q740H | DM? Breast and/or ovarian cancer | 0.0004614 | |
| RET | c.C2508T p.S836S | DP Association with thyroid cancer | 0.04666 | |
| FANCA | c.A796G p.T266A | DP Associated with breast cancer | 0.5166 | |
| 23/B46 | BARD1 | c.1071_1091del p.357_364del | Not found | Not found |
| FANCA | c.C3412G p.L1138V | Not found | 0.001533 | |
| MRE11A | c.A1728T p.R576R | Not found | 0.000008238 | |
| SLX4 | c.C1186G p.L396V | Not found | Not found | |
| 37/B47 | SLX4 | c.A5501G p.N1834S | Not found | 0.005542 |
| ERCC4 | c.G1633C p.G545R | Not found | 0.000008243 | |
| 38/B48 | SDHC | c.C31T p.R11C | Not found | 0.000008252 |
| FANCD2 | c.A1348G p.I450V | Not found | 0.0003871 | |
| FANCF | c.C959T p.P320L | Not found | 0.01264 | |
| TSC2 | c.A2834G p.K945R | Not found | Not found | |
| DIS3L2 | c.1651_1652insGGG p.A551delinsGA | Not found | Not found | |
| GNAS | c.C1046T p.P349L | Not found | Not found | |
| 24/B49 | BRCA1 | c.C4327T p.R1443* | DM Breast cancer | Not found |
| MSH2 | c.C1045G p.P349A | DM Renal cell carcinoma | 0.00009062 | |
| MSH2 | c.G965A p.G322D | DM? Colorectal cancer, non-polyposis | 0.01411 | |
| BARD1 | c.G253T p.V85L | Not found | 0.001068 | |
| NBN | c.G340T p.V114F | Not found | Not found | |
| RET | c.C2249G p.A750G | Not found | 0.000008238 | |
| XRCC3 | c.C260T p.P87L | Not found | 0.00006286 | |
| 25/B50 | POLH | c.A2074G p.T692A | DM Xeroderma pigmentosum | 0.0001824 |
| Tp53 | c.673-36G > C | DFP Breast cancer | Not found | |
| CTNNB1 | c.A2315G p.N772S | Not found | 0.00003355 | |
| POLD1 | c.C519G p.S173R | Not found | 0.009212 | |
| 26/D1 | ARL11 | c.G446A p.W149X | DP Association with cancer | 0.009898 |
| MSH2 | c.T1182G p.F394L | Not found | 0.00001648 | |
| 27/D4 | CHEK2 | c.T470C p.I157T | DFP Li-Fraumeni syndrome | Not found |
| 28/D5 | CDH1 | c.G2387A p.R796Q | Not found | 0.00003300 |
| 29/D6 | BUB1B | c.A1535G p.E512G | Not found | 0.000008239 |
| 30/D7 | APC | c.C2876T p.S959F | Not found | Not found |
| BRCA1 | c.G131T p.C44F | DM Breast and/or ovarian cancer | Not found | |
| PRF1 | c.G3A p.M1I | DM Haemophagocytic lymphohistiocytosis, familial | Not found | |
| 31/D8 | TP53 | c.G469A p.V157I | DM Sarcoma, adult-onset | 0.00005776 |
| 32/III_4 | CDH1 | c.G3A p.M1I | DM Gastric cancer | Not found |
| BRCA2 | c.C4061T p.T1354M | DM Breast cancer | 0.000008328 | |
| BRCA2 | c.G4258T p.D1420Y | DM? Breast and/or ovarian cancer | 0.006796 | |
| 33/D12 | CDH1 | c.A160G p.R54G | Not found | 0.00005916 |
| 34/D13 | BRCA2 | c.G223C p.A75P | DM? Breast cancer | 0.0001650 |
DM disease-causing mutation, DM? likely disease-causing mutation, DP disease-associated polymorphism, FP in vitro or in vivo functional polymorphism, DFP disease-associated polymorphism with additional functional evidence
We were able to detect 19 HGMD DM variations of which 9 are specifically associated with breast cancer (Table 2). The distribution of the remaining variants in the HGMD categories was: 11 DM?, 11 DP, 1 FP, and 9 DFP. In addition, 75 novel variations were detected in this study (Table 2).
Six BRCA1 and BRCA2 DM mutations were detected in 5 and 2 patients, respectively in a total prevalence of 15.5% (Table 2).
Nine truncating mutations were detected in 9 different patients (Table 2). Three of these mutations were DM in HGMD: The first woman carried p.R1443* in BRCA1, the second one carried p.V220I* in BRCA2 and the third one carried p.G164X in ABCC12 (Table 2). The six remaining truncating mutations were not found in HGMD: p.Q613X in SLX4, p.R170X in ERCC3, p.Q117X in EZH2, p.P742fs in NSD1, p.357_364del in BARD1 and p.L1697fs in BRCA1 (Table 2).
Three DM mutations were found, each one, in 2 different patients: p.C44F in BRCA1 (Families 13 and 30), p.P349A in MSH2 (Families 7 and 24) and p.G141W in SLX4 (Fig. 1 and Table 2).
In some families where different variants were found, in order to consider, which variant is pathogenic, we analyzed the co-segregation of the variations found with the cancer phenotype within 3 families 12, 13, and 32 (Figs. 1 and 2).
Fig. 2.
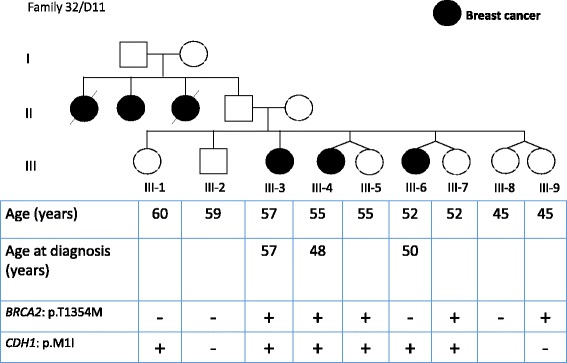
Pedigree of one family segregated for BRCA2 p.T1354M and CDH1 p.M1I variants. Filled squares (males) and circles (females) indicate the affected individuals. + sign indicates the presence of the variant and – sign the absence of the variant in tested individuals
Two members of family 12 were diagnosed with BC, their mother and maternal uncle were diagnosed with primary lung cancer and bone cancer, respectively. The nonsmoking mother was affected at the age of 63 but the age of the maternal uncle at diagnostic was not accessible. WES, in proband 12/B35 diagnosed with BC at the age of 42, identified 2 variants including one DM? p.I94L in RAD50, according to HGMD Professional database, and one novel variation p.G191R in ARL11 (data not shown). Prediction tool Polyphen2 indicated that both changes are benign and SIFT prediction tool indicated that p.I94L in RAD50 is tolerated and that p.G191R in ARL11 is damaging. Only p.I94L in RAD50 segregated in the affected sisters, diagnosed with BC at the age of 48, but it was also found in their third youngest 51 years old unaffected sister.
Three members of Family 13 were diagnosed with BC. WES identified 3 DM according to HGMD Professional database, including p.C44F in BRCA1, p.G141W in SLX4 and p.S404F in STK11 (Table 2). Leiden Open Variation Database indicated that p.C44F in BRCA1 affects protein function and it segregated with the disease (Table 3) (Fig. 1).
Table 3.
BRCA variations found and their evaluations in BRCA databases
| Gene | Variation | BIC database Clinically Importance/ Clinical Classification | COSMIC | Leiden Open Variation Database (LOVD) | BRCA Exchange |
|---|---|---|---|---|---|
| BRCA1 | c.G131T p.C44F | unknown/ pending | Not found | Affects function | Not found |
| c.A536G p.Y179C | unknown/ pending | Not found | Does not affect function | Benign | |
| c.C4327T p.R1443* | yes/ class 5 | Neutral | Affects function | Not found | |
| c.A1067G p.Q356R | unknown/ pending | Pathogenic | Does not affect function | Benign | |
| c.5090_5093delGTTA p.L1697fs | Not found | Not found | Not found | Not found | |
| BRCA2 | c.C65T p.A22V | unknown/pending | Not found | Effect unknown | Not found |
| c.G223C p.A75P | unknown/ pending | Not found | Does not affect function | Benign | |
| c.658_659delGT p.V220I* | yes/ class 5 | Not found | Affects function | Not found | |
| c.C4061T p.T1354M | unknown/ pending | Neutral | Does not affect function | Benign | |
| c.G4258T p.D1420Y | no/ pending | Neutral | Does not affect function | Benign | |
| c.C5744T p.T1915M | no/ class 1 | Neutral | Does not affect function | Not found | |
| c.G8775C p.Q2925H | unknown/ pending | Not found | Effect unknown | Not found | |
| c.A1114C p.N372H | no/ class 1 | Neutral | Not found | Benign | |
| c.C1151T p.S384F | no/ pending | Not found | Not found | Benign |
Descriptions of the classes in the BIC database:
Class 1: Not pathogenic/low clinical significance: There is significant evidence against this variant being a dominant high-risk pathogenic mutation
Class 5: Pathogenic: There is significant evidence to suggest that this variant is a dominant high-risk pathogenic mutation
Six members of family 32 were diagnosed with BC (Fig. 2). Members III-3, III-4 and III-6 were diagnosed with BC at the age of 56, 48 and 50, respectively. WES in proband III-4 identified 2 relevant variants including p.M1I in CDH1 and p.T1354M in BRCA2. Prediction tool SIFT indicated that both changes are damaging and are DM according to HGMD Professional database (Table 2 and Fig. 2). The analysis of this family showed that these variations were carried by affected and siblings that are not affected to date (Fig. 2). However, they were advised to join our screening program.
We have noted that the most frequently altered genes involved in our familial cases are DNA repair genes (Fig. 3a) and that some variants were recurrent in our cohort: p.W149X in ARL11, p.S836S in RET, p.A126T in RAD51C, p.T241M in XRCC3, p.G998E in PALB2 and c.673-36G > C in TP53 (Table 2 and Fig. 3b). In four cases, like the 4 families shown in Fig. 1, individuals appear to co-inherit multiple cancer causing or predisposing gene mutations. Unlike, the old strategy where one stops the investigation once a pathogenic mutation was identified, NGS gives us the capability of collating all known mutations/variants in a sample, which may permit a more comprehensive understanding of the polygenic landscape model of cancer. An important question to be answered is: Does an individual in Family 13 harboring all three DM mutation have different penetrance, genotype to phenotype correlation, type or age of onset of cancer than a sibling with only one DM variant? This critical question can only be answered when we start to combine all germline variant data of cancer patients and their comprehensive phenotypes from around the world in well-curated databases.
Fig. 3.
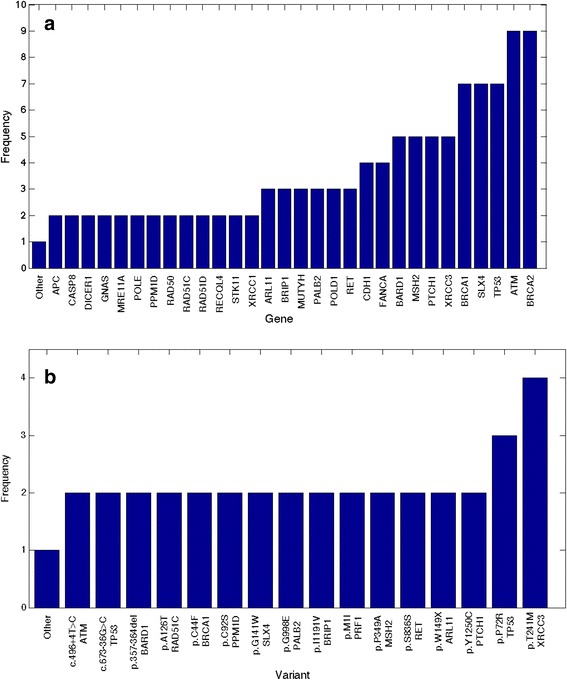
Frequencies of all variants in breast cancer predisposing genes from our 45 patients’ cohort (a) and the details of the most frequent variants shown in (b)
Discussion
We identified, in 45 patients with familial BC, 19 pathogenic mutations that are DM mutations according to the HGMD Professional database (Table 2). These 19 mutations were found in 13 different genes including ABCC12, APC, ATM, BRCA1, BRCA2, CDH1, ERCC6, MSH2, POLH, PRF1, SLX4, STK11, and TP53. Six mutations were found in BRCA1 and BRCA2 presenting a lower prevalence (15.5%) of deleterious BRCA mutations compared to the published literature [21–23].
In the Lebanese population, p.C44F mutation in the BRCA1 gene was found twice in this study and 5 times in previous studies [17, 18] in a total of 7 from 367 cases studied (1.9%). In fact, 2 of 9 patients carried a deleterious BRCA mutation in a cohort of 72 patients and 3 of 14 patients carried a deleterious BRCA mutation in a cohort of 250 patients. Our findings suggest it is the most recurrent mutation in the Lebanese population.
In families 23 and 35, we identified the truncating mutation p.357_364del in BARD1 (Table 2). A previous study, on this variation, showed the absence of co-segregation with the disease and it was considered as neutral polymorphisms [35]. We have observed this variant in our population and breast cancer patients and it is recommended that a more thorough and functional examination of this variant be conducted in the future.
In families 12, 13 and 32, we identified 7 variants in ARL11, BRCA1, BRCA2, CDH1, RAD50, SLX4, and STK11. The association of which variation towards increasing predisposition to BC remains unknown. Therefore, we analyzed the segregation of these variations and BC within the families. In family 13, only p.C44F in BRCA1 segregated with BC in the family. In family 12, p.I94L in RAD50 (a DM? mutation) was found in affected and healthy sisters and could therefore not lead to a conclusion regarding predisposition to BC. In family 32, p.M1I in CDH1 and p.T1354M in BRCA2 are implicated in gastric cancer and BC respectively and knowing that the family presented with only BC, two hypothesis can be formulated. First, III-6 can be considered as phenocopy and second healthy, till now, sisters III-5, III-7 and III-9 are at high risk (Fig. 2). In fact, in high-risk families, women testing negative for the familial BRCA mutation have an increased risk of BC and should be considered for continued surveillance [36]. Interestingly, two members of this family, III-4 and III-6 presented with invasive lobular breast cancer (Fig. 2). The association between CDH1 gene mutation and lobular cancer has been well established previously [37], and it is not unrealistic to suggest that this CDH1 variant may be the cause of lobular breast cancer in this family.
The pathogenic status of the majority of novel substitutions found and the 6 variations considered as DM? according to HGMD professional, remains problematic (Table 2). In fact, HGMD professional reports DM? as likely pathological mutation reported to be disease causing in the corresponding report, but the author has indicated that there may be some degree of doubt, or subsequent evidence has come to light in the literature, calling the deleterious nature of the variant into question [29]. Further studies are needed to define the pathogenic status of the novel substitutions and the DM? variations that have been found in our cohort of patients with BC. These future studies have to be analyzed in a larger number of affected families and control population samples.
NGS and traditional sequencing methods are not proficient in detecting BRCA genomic rearrangements including large deletions or duplications. Deletion and duplication genomic rearrangements vary significantly among countries and within ethnic groups [38]. We admit, therefore, that our reported BRCA mutation prevalence is underestimated.
Among the DM mutations found, several were associated with syndromes (Peutz-Jeghers), different cancer types (renal cell carcinoma, gastric cancer) and with diseases (Xeroderma pigmentosa, ataxia telangiectasia) (Table 2). Clinically, none of the symptoms found in these diseases were manifested in the different studied families except for family 24. In this family, proband 24/B49 carried the mutation p.R1443* in BRCA1 and two MSH2 variants (Fig. 1). Her mother had ovarian cancer and her sister uterine cancer, both are deceased and could not consequently be tested for these variants. MSH2 mutation is reported in families with endometrial cancer (Lynch syndrome) and breast cancer from Kuwait [39].
This is the first application of NGS on BC in Lebanon. In this study, we showed that the prevalence of deleterious BRCA mutations (15.5%) is lower than expected [17, 18] and that the overall mutation prevalence is equal to 40%, justifying the urgent need for the adoption of high-throughput NGS technologies to identify genes responsible for familial BC in the Lebanese population. Indeed, additional to BRCA mutations, highly penetrant mutations in genes associated with various hereditary cancer syndromes, such as CDH1, TP53, MSH2, ATM and POLH were found in the Lebanese population. Finally, we cannot rule out that some of these families shift a putative explanation towards a polygenic model where moderate and low penetrance alleles, acting together, may play a predominant role [20, 40, 41]. Our findings support the eligibility of performing genetic testing by massively parallel sequencing on Lebanese familial BC cases. Moreover, we would like to use this technology for tumor genome sequencing, in order to identify somatic alterations, which would be a valuable guidance towards individualized cancer therapy of Lebanese patients with BC. However, it is worthy of note that our study reports a small number of variants that are clinically actionable. Given the high rate of novel variants identified in BRCA1/2 and other breast cancer-associated genes, the clinical usefulness of the data is currently limited. Unless larger and rigorous studies are committed in this area of the world to correctly classify variants identified here or in other studies, the diagnosis and treatment of breast cancer will remain suboptimal.
Conclusion
This is the first study that utilized NGS technology to study genetic variants in 45 patients with familial breast cancer from Lebanon. Our deleterious mutation prevalence was 40% with only 15.5% accounted for by the BRCA1 and BRCA2 genes. This data should encourage a different strategy for familial breast cancer genetic screening in Lebanon, one that is based on WES rather than the initial screening of BRCA1/2 genes. We report here novel and rare variants in breast cancer predisposing genes, which will be valuable to researchers and clinicians around the world for variants’ classification and patients’ care in general.
Acknowledgements
We would like to thank all the patients and their family members who contributed their samples and information for this study.
Funding
This work was supported by a grant from the Kuwait Foundation for the Advancement of Sciences (KFAS No. 2011-1302-06) given to Prof. Fahd Al-Mulla and by grants from the Saint Joseph University and the Mohamad Cheaib Foundation.
Availability of data and materials
Data generated or analyzed during this study are included in this published article and its Additional file. The link to the compiled data will be shared on our website http://www.al-mulla.org, which will be made available once it is connected to international databases in the near future. Meanwhile, data supporting the manuscript can be requested from the any of the two corresponding authors.
Authors' contributions
NJ, FM, AM: drafted the manuscript. NJ, EC, ZH, HA, FM, MRM: carried out the molecular genetic studies and participated in the sequence alignment. CK, DA, MJM: acquisition of data. FM, HA, MRM, NJ: performed statistical analysis and interpretation of data. FM, AM conceived of the study, participated in the design of the study, and in its design and coordination. All authors read and approved the final manuscript.
Competing interests
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Consent for publication
All patients signed the informed consent during the counseling sessions and agreed to share their variant data in a reputable scientific journal.
Ethics approval and consent to participate
Approval to conduct the study was obtained from the Ethics Committee of Saint-Joseph University-Lebanon. Written informed consent was obtained at the time of enrollment for genetic sample collection.
Abbreviations
- BC
Breast cancer
- BIC
Breast cancer information core
- BWA
Burrows-wheeler aligner
- COSMIC
Catalog of somatic mutations in cancer database
- DFP
Disease-associated polymorphism
- DM
Disease-causing mutations
- DM?
Disease-causing mutations
- DP
Disease-associated polymorphism
- FP
In vitro/laboratory or in vivo functional polymorphism
- GATK
Genome analysis tool kit
- HGMD Professional
Human genome mutation database
- LOVD
Leiden open variation database
- NGS
Next generation sequencing
- NHLBI
National heart, lung, and blood institute
- WES
Whole exome sequencing.
Additional file
Cancer genes explored in this study. (DOCX 11 kb)
Contributor Information
Nadine Jalkh, Email: nadine.jalkh@usj.edu.lb.
Eliane Chouery, Email: eliane.chouery@usj.edu.lb.
Zahraa Haidar, Email: zahraa.haidar@net.usj.edu.lb.
Christina Khater, Email: ckhatermd@gmail.com.
David Atallah, Email: david.atallah@gmail.com.
Hamad Ali, Email: hamad.ali@hsc.edu.kw.
Makia J. Marafie, Email: mj_marafie@yahoo.com
Mohamed R. Al-Mulla, Email: mrhalm@sci.kuniv.edu.kw
Fahd Al-Mulla, Phone: +96567771040, Email: fahd@al-mulla.org.
Andre Megarbane, Email: andre.megarbane@yahoo.fr.
References
- 1.Melchor L, Benitez J. The complex genetic landscape of familial breast cancer. Hum Genet. 2013;132(8):845–863. doi: 10.1007/s00439-013-1299-y. [DOI] [PubMed] [Google Scholar]
- 2.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 3.Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559):789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 4.Scalia-Wilbur J, Colins BL, Penson RT, Dizon DS. Breast Cancer Risk Assessment: Moving Beyond BRCA 1 and 2. Semin Radiat Oncol. 2016;26(1):3–8. doi: 10.1016/j.semradonc.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 5.Borresen AL, Andersen TI, Garber J, Barbier-Piraux N, Thorlacius S, Eyfjord J, et al. Screening for germ line TP53 mutations in breast cancer patients. Cancer Res. 1992;52(11):3234–3236. [PubMed] [Google Scholar]
- 6.Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119(6):1447–1453. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- 7.Lynch ED, Ostermeyer EA, Lee MK, Arena JF, Ji H, Dann J, et al. Inherited mutations in PTEN that are associated with breast cancer, cowden disease, and juvenile polyposis. Am J Hum Genet. 1997;61(6):1254–1260. doi: 10.1086/301639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39(2):165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38(11):1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 10.Shamseldin HE, Elfaki M, Alkuraya FS. Exome sequencing reveals a novel Fanconi group defined by XRCC2 mutation. J Med Genet. 2012;49(3):184–186. doi: 10.1136/jmedgenet-2011-100585. [DOI] [PubMed] [Google Scholar]
- 11.Loveday C, Turnbull C, Ramsay E, Hughes D, Ruark E, Frankum JR, et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 2011;43(9):879–882. doi: 10.1038/ng.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002;31(1):55–59. doi: 10.1038/ng879. [DOI] [PubMed] [Google Scholar]
- 13.Orr N, Lemnrau A, Cooke R, Fletcher O, Tomczyk K, Jones M, et al. Genome-wide association study identifies a common variant in RAD51B associated with male breast cancer risk. Nat Genet. 2012;44(11):1182–1184. doi: 10.1038/ng.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porhanova NV, Sokolenko AP, Sherina NY, Ponomariova DN, Tkachenko NN, Matsko DE, et al. Ovarian cancer patient with germline mutations in both BRCA1 and NBN genes. Cancer Genet Cytogenet. 2008;186(2):122–124. doi: 10.1016/j.cancergencyto.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 15.Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38(8):873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 16.Shamseddine A, Saleh A, Charafeddine M, Seoud M, Mukherji D, Temraz S, et al. Cancer trends in Lebanon: a review of incidence rates for the period of 2003–2008 and projections until 2018. Popul Health Metr. 2014;12(1):4. doi: 10.1186/1478-7954-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El Saghir NS, Zgheib NK, Assi HA, Khoury KE, Bidet Y, Jaber SM, et al. BRCA1 and BRCA2 mutations in ethnic Lebanese Arab women with high hereditary risk breast cancer. Oncologist. 2015;20(4):357–364. doi: 10.1634/theoncologist.2014-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jalkh N, Nassar-Slaba J, Chouery E, Salem N, Uhrchammer N, Golmard L, et al. Prevalance of BRCA1 and BRCA2 mutations in familial breast cancer patients in Lebanon. Hered Cancer Clin Pract. 2012;10(1):7. doi: 10.1186/1897-4287-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sundquist M, Thorstenson S, Brudin L, Wingren S, Nordenskjold B. Incidence and prognosis in early onset breast cancer. Breast. 2002;11(1):30–35. doi: 10.1054/brst.2001.0358. [DOI] [PubMed] [Google Scholar]
- 20.Gracia-Aznarez FJ, Fernandez V, Pita G, Peterlongo P, Dominguez O, de la Hoya M, et al. Whole exome sequencing suggests much of non-BRCA1/BRCA2 familial breast cancer is due to moderate and low penetrance susceptibility alleles. PLoS One. 2013;8(2):e55681. doi: 10.1371/journal.pone.0055681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall MJ, Reid JE, Burbidge LA, Pruss D, Deffenbaugh AM, Frye C, et al. BRCA1 and BRCA2 mutations in women of different ethnicities undergoing testing for hereditary breast-ovarian cancer. Cancer. 2009;115(10):2222–2233. doi: 10.1002/cncr.24200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malone KE, Daling JR, Neal C, Suter NM, O'Brien C, et al. Frequency of BRCA1/BRCA2 mutations in a population-based sample of young breast carcinoma cases. Cancer. 2000;88(6):1393–1402. doi: 10.1002/(SICI)1097-0142(20000315)88:6<1393::AID-CNCR17>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 23.Uhrhammer N, Abdelouahab A, Lafarge L, Feillel V, Ben Dib A, Bignon YJ. BRCA1 mutations in Algerian breast cancer patients: high frequency in young, sporadic cases. Int J Med Sci. 2008;5(4):197–202. doi: 10.7150/ijms.5.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25(21):2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133(1):1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clarke L, Zheng-Bradley X, Smith R, Kulesha E, Xiao C, Toneva I, et al. The 1000 Genomes Project: data management and community access. Nat Methods. 2012;9(5):459–462. doi: 10.1038/nmeth.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szabo C, Masiello A, Ryan JF, Brody LC. The breast cancer information core: database design, structure, and scope. Hum Mutat. 2000;16(2):123–131. doi: 10.1002/1098-1004(200008)16:2<123::AID-HUMU4>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 32.Fokkema IF, den Dunnen JT, Taschner PE. LOVD: easy creation of a locus-specific sequence variation database using an "LSDB-in-a-box" approach. Hum Mutat. 2005;26(2):63–68. doi: 10.1002/humu.20201. [DOI] [PubMed] [Google Scholar]
- 33.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805–811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spurdle AB, Healey S, Devereau A, Hogervorst FB, Monteiro AN, Nathanson KL, et al. ENIGMA-evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat. 2012;33(1):2–7. doi: 10.1002/humu.21628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Brakeleer S, De Greve J, Loris R, Janin N, Lissens W, Sermijn E, et al. Cancer predisposing missense and protein truncating BARD1 mutations in non-BRCA1 or BRCA2 breast cancer families. Hum Mutat. 2010;31(3):E1175–1185. doi: 10.1002/humu.21200. [DOI] [PubMed] [Google Scholar]
- 36.Smith A, Moran A, Boyd MC, Bulman M, Shenton A, Smith L, et al. Phenocopies in BRCA1 and BRCA2 families: evidence for modifier genes and implications for screening. J Med Genet. 2007;44(1):10–15. doi: 10.1136/jmg.2006.043091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masciari S, Larsson N, Senz J, Boyd N, Kaurah P, Kandel MJ, et al. Germline E-cadherin mutations in familial lobular breast cancer. J Med Genet. 2007;44(11):726–731. doi: 10.1136/jmg.2007.051268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ewald IP, Ribeiro PL, Palmero EI, Cossio SL, Giugliani R, Ashton-Prolla P. Genomic rearrangements in BRCA1 and BRCA2: A literature review. Genet Mol Biol. 2009;32(3):437–446. doi: 10.1590/S1415-47572009005000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marafie MJ, Al-Awadi S, Al-Mosawi F, Elshafey A, Al-Ali W, Al-Mulla F. Impact of 226C > T MSH2 gene mutation on cancer phenotypes in two HNPCC-associated highly-consanguineous families from Kuwait: emphasis on premarital genetic testing. Fam Cancer. 2009;8(4):289–298. doi: 10.1007/s10689-009-9275-3. [DOI] [PubMed] [Google Scholar]
- 40.Johnson N, Fletcher O, Palles C, Rudd M, Webb E, Sellick G, et al. Counting potentially functional variants in BRCA1, BRCA2 and ATM predicts breast cancer susceptibility. Hum Mol Genet. 2007;16(9):1051–1057. doi: 10.1093/hmg/ddm050. [DOI] [PubMed] [Google Scholar]
- 41.Sehl ME, Langer LR, Papp JC, Kwan L, Seldon JL, Arellano G, et al. Associations between single nucleotide polymorphisms in double-stranded DNA repair pathway genes and familial breast cancer. Clin Cancer Res. 2009;15(6):2192–2203. doi: 10.1158/1078-0432.CCR-08-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data generated or analyzed during this study are included in this published article and its Additional file. The link to the compiled data will be shared on our website http://www.al-mulla.org, which will be made available once it is connected to international databases in the near future. Meanwhile, data supporting the manuscript can be requested from the any of the two corresponding authors.