

Abstract
Neural stem cells (NSCs) have the capacity to differentiate into neurons, astrocytes, and oligodendrocytes, and therefore represent a promising donor tissue source for treating neurodegenerative diseases and repairing injuries of the nervous system. However, it remains unclear how canonical microRNAs (miRNAs), the subset of miRNAs requiring the Drosha-Dgcr8 microprocessor and the type III RNase Dicer for biogenesis, regulate NSCs. In this study, we established and characterized Dgcr8−/− NSCs from conditionally Dgcr8-disrupted mouse embryonic brain. RNA-seq analysis demonstrated that disruption of Dgcr8 in NSCs causes a complete loss of canonical miRNAs and an accumulation of pri-miRNAs. Dgcr8−/− NSCs can be stably propagated in vitro, but progress through the cell cycle at reduced rates. When induced for differentiation, Dgcr8−/− NSCs failed to differentiate into neurons, astrocytes, or oligodendrocytes under permissive conditions. Compared to Dgcr8+/− NSCs, Dgcr8−/− NSCs exhibit significantly increased DNA damage. Comparative RNA-seq analysis and gene set enrichment analysis (GSEA) revealed that Dgcr8−/− NSCs significantly downregulate genes associated with neuronal differentiation, cell cycle progression, DNA replication, protein translation, and DNA damage repair. Furthermore, we discovered that Dgcr8−/− NSCs significantly downregulate genes responsible for cholesterol biosynthesis and demonstrated that Dgcr8−/− NSCs contain lower levels of cholesterol. Together, our data demonstrate that canonical miRNAs play essential roles in enabling lineage specification, protecting DNA against damage, and promoting cholesterol biosynthesis in NSCs.
Keywords: : neural stem cells, miRNA, Dgcr8, cholesterol
Introduction
Neural stem cells (NSCs) have the capacity to differentiate into neurons, astrocytes, and oligodendrocytes, and therefore represent a promising donor tissue source for treating neurodegenerative diseases and repairing injuries of the nervous system [1,2]. During development, NSCs first appear as the highly proliferative neuroepithelial cells lining the lateral ventricular wall. They subsequently transform into radial glial cells in the ventricular zone. Then they become the proliferation-inert adult NSCs in the subventricular zone of the lateral ventricles and the subgranular zone of the dentate gyrus of the hippocampus [3]. Loss of NSCs could lead to inadequate neural regeneration [2], while unrestrained NSC proliferation may cause brain tumors [4]. Therefore, the precise regulation on proliferation, differentiation, and genomic integrity of NSCs is essential for the formation, regeneration, and function of the nervous system.
Cholesterol is an essential structural component of cellular membranes and a precursor for biosynthesis of steroid hormones, oxysterols, and bile acids [5]. In the nervous system, cholesterol is also essential for the formation of myelin, the oligodendrocyte-derived insulating layer that enwraps axons and enables saltatory conduction [5]. Dysregulation of cholesterol leads to various nervous system disorders such as Alzheimer's disease, Smith–Lemli–Optiz syndrome, and Niemann–Pick Type C disease [6]. Because of the blood-brain barrier, cholesterol in the nervous system is primarily de novo synthesized, which is markedly different from other periphery organs in which cholesterol can be acquired from dietary intake [7]. Biosynthesis of cholesterol requires activities of more than twenty enzymes, all of which are transcriptionally regulated by the sterol regulatory element binding factors (SREBFs) encoded by Srebf1 and Srebf2 [8]. Furthermore, cholesterol also plays a pivotal role in the maturation of the Hedgehog ligands [9], which initiate the Hedgehog pathways and regulate normal brain development and brain tumor formation [10].
MicroRNAs (miRNAs) are small noncoding RNAs that play critical roles in embryogenesis, tissue homeostasis, and human diseases [11–14]. miRNAs may be categorized as canonical or noncanonical based on the biogenesis pathways. In the biogenesis of canonical miRNAs, primary miRNA transcripts (pri-miRNAs) are processed by the Drosha-Dgcr8 microprocessor into ∼70 nt stem-loop structured pre-miRNAs, which are further processed into ∼22 nt mature miRNAs by the type III RNase Dicer [15]. In contrast, biogenesis of noncanonical miRNAs bypasses the requirement of the Drosha-Dgcr8 complex and only requires Dicer to produce mature miRNAs by cleaving endogenous shRNAs and mirtrons [16]. In addition to miRNA biogenesis, Dicer is also required for the biogenesis of endogenous small interfering RNAs (endo-siRNAs) by cleaving double-stranded RNAs (dsRNAs) [16].
miRNAs play critical roles in neural development and the regulation of NSCs. Conditional disruption of Dicer in the developing cortex or other developing brain tissues by tissue-specific Cre results in a decreased number of neural progenitor cells (NPCs), increased apoptosis, and abnormal neuronal differentiation [17–21]. Although ablation of the microprocessor activity by Dgcr8 inactivation in developing brain leads to similar phenotypes, the phenotypic defects are significantly milder than those caused by Dicer inactivation [22]. This suggests that noncanonical miRNAs or endo-siRNAs, which are Dicer dependent but Drosha-Dgcr8-independent, are functionally important to neural development.
Dicer−/− NSCs can be established from the cortex of conditional Dicer−/− embryos, but are deficient in differentiation [23,24]. Because disruption of Dicer leads to a complete loss of several small RNA species, it remains unclear how disruption of canonical miRNAs alone affects NSCs. Furthermore, it has been implicated that Drosha−/− NPCs would quickly undergo neuronal differentiation because of accumulation of Neurog2 transcripts, which encode a differentiation-promoting transcription factor and are cleaved by the Drosha-Dgcr8 microprocessor [25]. Therefore, it is not entirely clear whether NSCs lacking microprocessor activities can self-renew and be stably maintained in culture. In this study, we established and characterized Dgcr8−/− NSCs from developing brains of conditional Dgcr8−/− embryos. Our data demonstrated that Dgcr8−/− NSCs can be stably propagated in vitro, but fail to undergo lineage specification. We demonstrated that genes regulating neuronal differentiation, cell cycle progression, DNA replication, protein translation, and DNA damage repair are significantly downregulated in Dgcr8−/− NSCs. Furthermore, we discovered that disruption of Dgcr8 in NSCs leads to an increase of DNA damage and a decrease of cholesterol biosynthesis. Together, our data demonstrated that canonical miRNAs play essential roles in enabling differentiation, protecting DNA against damage, and promoting cholesterol biosynthesis in NSCs.
Materials and Methods
Mice breeding and genotyping
All animal experiments were performed in accordance with guidelines from the University of Alabama at Birmingham and National Institutes of Health. Mice embryos with neural specific disruption of Dgcr8 were generated by crossing Dgcr8flox/flox mice [26] with Nestin-Cre mice [27]. NSCs were genotyped by PCR analysis as described [28].
Derivation and differentiation of NSCs
NSCs were isolated from the lateral ventricles of E13.5 mouse embryonic brain as described [29] and were maintained in Mouse NSC Expansion medium (EMD Millipore) on tissue culture plate coated with polyornithine (Sigma-Aldrich) and laminin (BD Biosciences). Dgcr8+/− NSCs isolated from littermates were used as controls. For neuronal differentiation, NSCs were cultured for 7 days in Neurobasal Medium with 2% B-27 Serum-Free Supplement and 2 mM GlutaMAX-I (Thermo Scientific). For astrocyte differentiation, NSCs were cultured for 7 days in DMEM with 1% FBS (Gemini Bio), 1% N2 Supplement (Thermo Scientific), and 2 mM GlutaMAX-I. For oligodendrocyte differentiation, NSCs were cultured for 7 days in Neurobasal medium supplemented with 2% B-27, 2 mM GlutaMAX-1, and 30 ng/mL T3 (Sigma-Aldrich).
Immunostaining and immunoblotting
Immunostaining and immunoblotting were performed as described [30]. For immunostaining, NSCs, neurons, or astrocytes were fixed in 4% paraformaldehyde, blocked in Protein Block (Dako), and incubated with the appropriate primary antibodies overnight at 4°C and secondary antibodies for 1 h at room temperature. Nuclei were stained by 0.5 μg/mL DAPI (Thermo Scientific) at room temperature for 10 min. For oligodendrocyte staining, live cells were incubated with O1 antibody (MAB344; Millipore) at 37°C for 30 min, washed with PBS twice, and incubated with secondary antibody (A21042; Thermo Scientific) at 37°C for 30 min. Images were acquired by a Nikon Ti-S microscope and processed by Photoshop CS6. For immunoblotting, whole cell extracts were prepared in RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS), separated on a 4%–20% SDS-polyacrylamide gel (Bio-Rad), and transferred to PVDF membrane (Thermo Scientific). Primary antibodies used were Dgcr8 (10996-1-AP; Proteintech), SOX2 (130-095-636; Miltenyi Biotec), Nestin (MAB353; EMD Millipore), Tuj1 (MMS-435P, BioLegend), γH2AX (9718S; Cell Signaling), GAPDH, MAP2, and GFAP (sc-25778, sc-20172, and sc-6170, Santa Cruz Biotech). Secondary antibodies used for immunostaining were Alexa Fluor 488- or 568-conjugated anti-mouse or -rabbit IgG (Thermo Scientific) and for immunoblotting were HRP-conjugated anti-mouse or -rabbit IgG (BioLegend).
Cell cycle and apoptosis analyses
Cell cycle and apoptosis analyses were performed as described [31]. For cell cycle analysis, cells at 30%–50% confluency were pulse-labeled with 10 μM BrdU (Sigma-Aldrich) for 30 min before being trypsinized and fixed in cold 70% ethanol at −20°C overnight. Cells were denatured in 2 N HCl/0.5% Triton X-100 solution for 30 min at room temperature, neutralized by 0.1 M sodium borate (pH 8.5), washed twice by PBS with 1% BSA and 0.5% Tween-20, and incubated with APC-conjugated anti-BrdU antibody (BioLegend) for 30 min at room temperature. Before being analyzed on a BD Fortessa flow cytometer, cells were stained with 1 μg/mL of propidium iodide (Sigma-Aldrich) at room temperature for 5 min. For Annexin V staining, NSCs were trypsinized, stained with APC-conjugated Annexin V (BD Biosciences) on ice for 15 min, and then stained by 1 μg/mL of propidium iodide at room temperature for 5 min. The cells were analyzed on a BD Fortessa flow cytometer. All data were analyzed by the FlowJo VX software (FlowJo, LLC).
Comet assay
Comet Assay was performed using the OxiSelect Comet Assay Kit according to the manufacturer (Cell Biolabs). Cells at 30%–50% confluency were trypsinized and embedded in low melting agarose gel at density of 1 × 104 cells/mL on glass slides. The slides were incubated in prechilled Lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Trizma base, 1% Triton X-100, pH 10.0) for 60 min and alkaline buffer (300 mM sodium hydroxide and 1 mM EDTA) for 30 min at 4°C in the dark before electrophoresis in alkaline buffer for 30 min at 1 volt/cm and 300 mA. The slides were then washed by prechilled distilled water, fixed by 70% ethanol, air-dried, and stained with Vista Green DNA dye. Images were acquired by a Nikon Ti-S microscope using a FITC filter, and the percentage of tail DNA of individual cells was determined by the OpenComet plugin of ImageJ [32]. The graph and two-tailed Student's t-test were performed using the GraphPad Prism 7 software.
Lentiviral production and NSC transduction
Lentivirus expressing a human DGCR8 cDNA (pSIN-EF2-DGCR8-Pur) was prepared as described [30]. Dgcr8−/− NSCs were transduced with DGCR8 lentivirus (MOI = 3) in the presence of 5 μg/mL protamine sulfate (Sigma-Aldrich) 24 h after seeding onto polyornithine (Sigma-Aldrich) and laminin (EMD Millipore)-coated tissue culture plates.
RNA extraction and qRT-PCR analysis
Total RNA was isolated using the Direct-zol RNA Kit (Zymo Research), and cDNA was synthesized by the Verso cDNA Synthesis Kit (Thermo Scientific). qRT-PCR was performed using 2× Absolute Blue Q-PCR Master Mix (Thermo Scientific) on a ViiA 7 Real-Time PCR system (Thermo Scientific). Primers used are listed in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/scd).
RNA-seq and bioinformatics
For mRNA-seq and small RNA-seq analyses, total RNA samples prepared from Dgcr8+/− and Dgcr8−/− NSCs were submitted to the Genomic Services Laboratory at HudsonAlpha Institute (Huntsville, AL) for library construction and sequencing. For mRNA-seq analysis, transcripts with poly(A) tails were enriched by poly(A) selection and at least 25 million, 50-bp paired-end (PE) reads were acquired from each sample. For small RNA-seq analysis, miRNAs were enriched and at least 15 million, 50-bp single-end (SE) reads were acquired from each sample. For mRNA-seq, sequence alignment was performed using TopHat v2.0.14 against the UCSC mm10 Assembly. Expression values were calculated with featureCounts v1.4.6-p2, and differential expression analysis was determined by DESeq2. GSEA was performed according to Subramanian et al. [33]. For small RNA-seq, adapters were removed from the reads using cutadapt (v1.8.1) [34]. All the reads were mapped to the mouse reference genome (GRCm38.74/mm10) using STAR aligner guided by a Gene Transfer File (Ensembl GTF version GRCm38.74) [35]. Read count tables for miRNA genes were made using HT-seq (v0.6.0). Deferential Expression (DE) analysis was performed using DESeq2, and the downstream statistical analyses and plots were made in R (v3.1.1; www.r-project.org/). RNA-seq data were deposited at Gene Expression Omnibus (GSE88709).
Cholesterol measurement
Cholesterol measurement was performed using the Total Cholesterol Assay Kit according to the manufacturer (Cell Biolabs). 1 to 5 × 105 cells were homogenized with 200 μL of extraction mixture (chloroform: isopropanol: NP-40 = 7: 11: 0.1), and the extracts were centrifuged at 15,000 g for 10 min. The liquid phase was collected, dried at 50°C, and dissolved in 200 μL of 1× Diluent. For free cholesterol measurement, 15–40 μL of each sample was incubated with the Cholesterol Reaction Reagent for 45 min at 37°C before measuring the absorbance at 540 nm on a Synergy H1 Multi-Mode Plate Reader (BioTek). For total cholesterol measurement, cholesterol esterase was included in the Cholesterol Reaction Reagent. Esterified cholesterol was determined by subtraction of free cholesterol from total cholesterol. Cholesterol level was normalized to protein concentration, which was prepared by a previously reported method [31] and determined by measuring absorbance at 562 nm using BCA assays (Thermo Scientific) on a Synergy H1 Multi-Mode Plate Reader (BioTek).
Results
Dgcr8−/− NSCs can be stably propagated in vitro
To investigate how canonical miRNAs regulate NSCs, we attempted to isolate NSCs from the developing cortex of E13.5 embryos crossed from Nes-Cre; Dgcr8+/− mice, and Dgcr8flox/flox mice. The established NSC lines, each from a single embryo of a same litter, exhibited two distinct cell morphologies (Fig. 1A, B). Genotyping demonstrated that NSCs with smaller cell bodies, but thinner and longer protrusions, retain functional Dgcr8 (Dgcr8+/−), while NSCs forming aggregates and with thicker and shorter protrusions are knockout mutants (Dgcr8−/−) (Fig. 1C). qRT-PCR confirmed that Dgcr8 transcripts are absent in Dgcr8−/− NSCs (Fig. 1D). Immunostaining demonstrated that Dgcr8−/− NSCs, like the control Dgcr8+/− NSCs, express the NSC-specific markers SOX2 and Nestin (Fig. 1E, F).
FIG. 1.
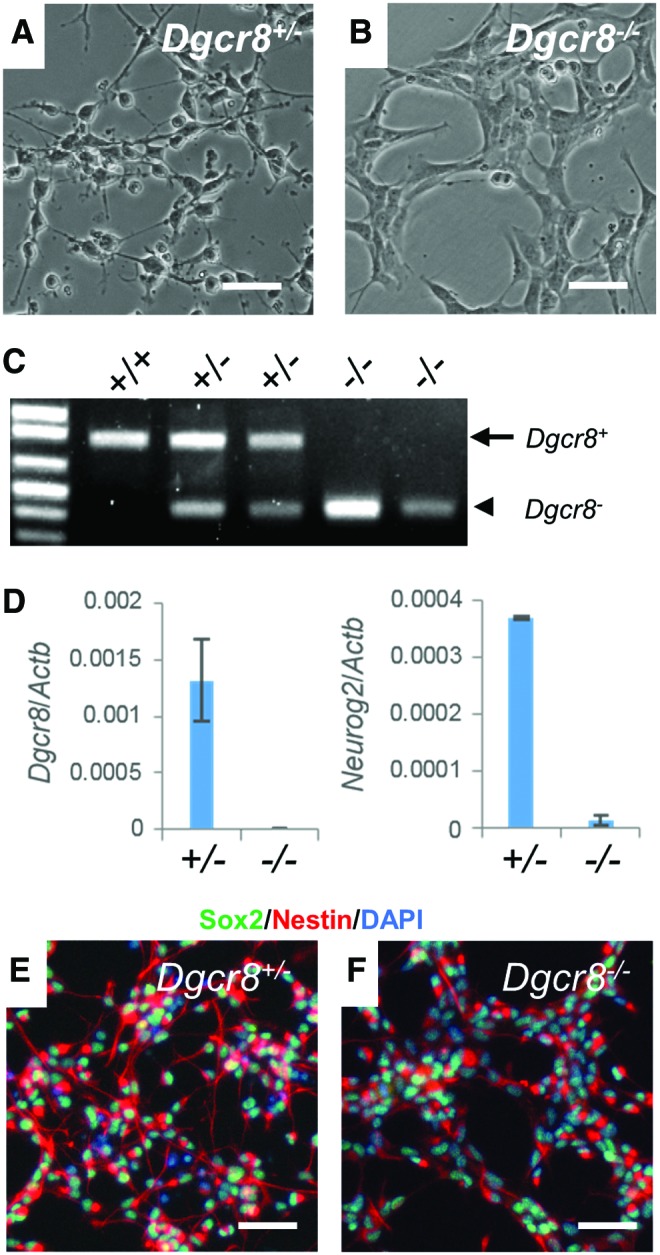
Isolation of Dgcr8−/− NSCs from conditionally Dgcr8-disrupted mouse embryonic brain. (A, B) Bright field images of NSCs isolated from mouse E13.5 embryonic brain. (A) Dgcr8+/− and (B) Dgcr8−/− NSCs. Scale bars, 50 μm. (C) PCR genotyping of NSCs. Shown are genotyping results of a wild-type control (mouse tail tip fibroblasts), two independent clones of Dgcr8+/− NSCs, and two independent clones of Dgcr8−/− NSCs. Arrow, wild-type Dgcr8 allele (Dgcr8+); arrowhead, Dgcr8 mutant allele (Dgcr8−). (D) qRT-PCR analyses of Dgcr8 (left) and Neurog2 (right) in Dgcr8+/− and Dgcr8−/− NSCs. Data were normalized to the mRNA levels of β-actin gene Actb. n = 3 independent biological repeats. Error bar, SD. (E, F) Immunostaining of NSC markers SOX2 (green) and NESTIN (red) in (E) Dgcr8+/− and (F) Dgcr8−/− NSCs. Cell nuclei were counterstained with DAPI (blue). Scale bars, 50 μm. NSC, neural stem cell. Color images available online at www.liebertpub.com/scd
Because it has been implicated that Drosha−/− NPCs would quickly differentiate into neurons in embryonic brain due to accumulation of Neurog2 transcripts [25], we examined whether Dgcr8−/− NSCs are similarly defective in self-renewal by long term in vitro passaging. We found that Dgcr8−/− NSCs can be stably cultured for at least 20 passages without noticeable changes in cell morphology or stem cell marker expression (Supplementary Fig. S1). qRT-PCR analysis revealed that, instead of an increase of Neurog2 transcripts as reported in Drosha−/− NPCs [25], Neurog2 expression is significantly downregulated in Dgcr8−/− NSCs (Fig. 1D). Our data therefore demonstrated that Dgcr8−/− NSCs can be established from the embryonic cortex and stably propagated in vitro.
Dgcr8−/− NSCs do not express canonical miRNAs
To investigate how Dgcr8 inactivation affects miRNA expression in NSCs, we performed small RNA-seq analysis of Dgcr8+/− and Dgcr8−/− NSCs. We obtained 58.9 million single-end reads from Dgcr8+/− NSCs and 26.8 million reads from Dgcr8−/− NSCs. Among these, 5.5 million reads from Dgcr8+/− NSCs and 0.08 million reads of Dgcr8−/− NSCs were mapped to miRNA genes, which account for 9.4% and 0.3% of total small RNA reads, respectively (Fig. 2A). As expected, the great majority of miRNAs expressed in Dgcr8+/− NSCs are significantly downregulated in Dgcr8−/− NSCs (Fig. 2B and Supplementary Table S2). Noncanonical miRNAs, which are microprocessor-independent for biogenesis, have been previously defined as those miRNAs that decrease less than twofold when Dgcr8 is disrupted [36]. By this criteria, noncanonical miRNAs only account for 0.66% of all miRNA reads obtained from Dgcr8+/− NSCs, but account for 92.6% of all miRNA reads from Dgcr8−/− NSCs (Fig. 2C and Supplementary Table S3).
FIG. 2.

Small RNA-seq of Dgcr8+/− and Dgcr8−/− NSCs. (A) Percentage miRNA reads in total small RNA-seq reads of Dgcr8+/− and Dgcr8−/− NSCs. (B) Heatmap of miRNA expression in Dgcr8+/− and Dgcr8−/− NSCs. (C) Percentage noncanonical miRNA reads (Dgcr8-independent) in total miRNA reads of Dgcr8+/− and Dgcr8−/− NSCs. (D) Heatmaps of mature miRNAs (left) and corresponding pri-miRNAs (right) in Dgcr8+/− and Dgcr8−/− NSCs. Shown are those miRNAs with differential expression of corresponding pri-miRNA (>2-fold, P < 0.01) in mRNA-seq analysis of Dgcr8+/− and Dgcr8−/− NSCs. (E) Pie graph of the most abundantly expressed miRNAs in Dgcr8+/− NSCs. (F) Pie graph of the most abundantly expressed miRNAs in Dgcr8−/− NSCs. Color images available online at www.liebertpub.com/scd
Intriguingly, while mature miRNAs are significantly downregulated in Dgcr8−/− NSCs, we detected that many corresponding pri-miRNAs are significantly upregulated in RNA-seq analysis of mRNA transcripts, demonstrating that a lack of Dgcr8 leads to accumulation of pri-miRNAs (Fig. 2D). The only exception is miR-3093, both mature and pri-miRNAs of which are downregulated in Dgcr8−/− NSCs (Fig. 2D). These data suggest that additional microprocessor-related mechanism regulates the transcription of pri-miR-3093.
In Dgcr8+/− NSCs, the most abundantly expressed miRNAs are miR-21 and let-7 family members (let-7i, g, d, and b), which account for 31% and 19% of total miRNA reads, respectively (Fig. 2E and Supplementary Table S2). Other abundantly expressed miRNAs include members from miR-30, miR-17/92, and miR-99 families, which together with miR-21 and let-7s account for nearly 80% of all miRNA reads in Dgcr8+/− NSCs (Fig. 2E and Supplementary Table S2). In Dgcr8−/− NSCs, the most abundantly expressed miRNAs are miR-344 family members (miR-344f, c, b, and g), miR-1981, miR-484, miR-320, and miR-5099, which account for 80% of all miRNA reads (Fig. 2F and Supplementary Table S3). Among these miRNAs, miR-344f, miR-1981, miR-484, miR-320, and miR-5099 have previously been identified as noncanonical miRNAs [22,36,37]. Our data demonstrate that other members of the miR-344 family (miR-344c, b, and g) also belong to noncanonical miRNAs that do not require Dgcr8 for biogenesis (Fig. 2F and Supplementary Table S3). Together, our data demonstrate that the great majority of NSC-expressed miRNAs are canonical miRNAs that require Dgcr8 for biogenesis.
Canonical miRNAs are required for lineage specification of NSCs
To investigate how canonical miRNAs regulate differentiation of NSCs, we attempted to differentiate Dgcr8−/− NSCs. We demonstrated that Dgcr8+/− NSCs can be readily differentiated into neurons, astrocytes, or oligodendrocytes, as indicated by the expression of neuronal specific markers Tuj1 and MAP2, astrocyte marker GFAP, and oligodendrocyte marker O1, respectively (Fig. 3A–A″). However, Dgcr8−/− NSCs fail to undergo lineage specification under permissive conditions (Fig. 3B–B″). When differentiated into neurons or oligodendrocytes, Dgcr8−/− NSCs rapidly undergo cell death (Fig. 3B, B″). When induced to astrocytes, Dgcr8−/− NSCs differentiated into cells weakly stained by the astrocyte-specific marker GFAP in nucleus and cytoplasm (Fig. 3B′), which is markedly different from the cytoplasmic staining of Dgcr8+/− NSC-derived astrocytes (Fig. 3A′).
FIG. 3.

Dgcr8−/− NSCs cannot differentiate into neurons, astrocytes, or oligodendrocytes. (A–A″) Induction of Dgcr8+/− NSCs into neurons, astrocytes, and oligodendrocytes. Immunostaining of (A) neuronal markers Tuj1 (green) and MAP2 (red), (A′) astrocyte-specific marker GFAP (red) and DAPI (blue), and (A″) oligodendrocyte-specific marker O1 (green). Scale bars, 50 μm. (B–B″) Induction of Dgcr8−/− NSCs into neurons, astrocytes, and oligodendrocytes. (B) Bright field image showing cell death 48 h after inducing Dgcr8−/− NSCs into neurons. (B′) Immunostaining of astrocyte-specific marker GFAP (red) and DAPI (blue). Note that GFAP is weakly stained in both nuclei and cytoplasm, which is different from the cytoplasmic staining of Dgcr8+/− NSC-derived astrocytes. (B″) Bright field image showing cell death 48 h after inducing Dgcr8−/− NSCs into oligodendrocytes. Scale bars, 50 μm. (C–C″) Induction of Dgcr8−/− NSCs rescued by a DGCR8 cDNA into neurons, astrocytes, and oligodendrocytes. Immunostaining of (C) neuronal markers Tuj1 (green) and MAP2 (red), (C′) astrocyte-specific marker GFAP (red) and DAPI (blue), and (C″) oligodendrocyte-specific marker O1 (green). Scale bars, 50 μm. Color images available online at www.liebertpub.com/scd
The lack of differentiation potential of Dgcr8−/− NSCs can be interpreted either as canonical miRNAs are required to execute lineage specification, or as the isolated Dgcr8−/− NSCs are not true multipotent stem cells. To distinguish these two possibilities, we reintroduced a functional DGCR8 cDNA into the Dgcr8−/− NSCs using lentivirus. The rescued NSCs can efficiently differentiate into neurons, astrocytes, or oligodendrocytes like control Dgcr8+/− NSCs (Fig. 3C–C″), demonstrating that Dgcr8−/− NSCs retain differentiation potential and the failure in lineage specification is due to a lack of canonical miRNAs.
Canonical miRNAs promote proliferation of NSCs
Next, we investigated whether proliferation and survival of NSCs are affected by the lack of canonical miRNAs. Dgcr8−/− NSCs progress through the cell cycle at reduced rates compared to Dgcr8flox/+ controls, as demonstrated by a significant increase of cells in the G1 phase but a decrease in the S phase in BrdU-pulse labeling experiments (Fig. 4A, B). These data therefore demonstrate that canonical miRNAs promote proliferation of NSCs. However, apoptosis rate, as measured by Annexin V staining, does not significantly differ between Dgcr8flox/+ and Dgcr8−/− NSCs (Fig. 4C, D). These data agree with the previous reports that disruption of Dicer in NSCs leads to reduced proliferation, but not increased apoptosis [23,24].
FIG. 4.

Dgcr8−/− NSCs exhibit decreased proliferation. (A) Representative flow cytometry plots of cell cycle analysis of Dgcr8+/− (left) and Dgcr8−/− (right) NSCs by BrdU pulse-labeling and PI staining. (B) Quantification of cell cycle analysis of Dgcr8+/− and Dgcr8−/− NSCs. n = 3 independent biological repeats. N.S., not significant; **P < 0.01, two-tailed Student's t-test. (C) Representative flow cytometry plots of apoptosis analysis of Dgcr8+/− (left) and Dgcr8−/− (right) NSCs by Annexin V and propidium iodide (PI) staining. (D) Quantification of apoptosis analysis of Dgcr8+/− and Dgcr8−/− NSCs. n = 3 independent biological repeats. N.S., not significant, two-tailed Student's t-test. PI, propidium iodide. Color images available online at www.liebertpub.com/scd
Loss of canonical miRNAs causes DNA damage in NSCs
Conditional disruption of Dicer or Dgcr8 can lead to increased DNA damage and apoptosis in cerebellum neural progenitors [38]. Because an increase of apoptosis in Dgcr8−/− (Fig. 4C, D) or Dicer−/− NSCs was not observed [24], we examined whether NSCs lacking Dgcr8 exhibit increased DNA damage. Immunostaining of γH2AX, the phosphorylated H2A.X histones that label DNA strand breakage [39], revealed that a higher percentage of Dgcr8−/− NSCs are positive for γH2AX (Fig. 5A, C). Consistently, immunoblotting demonstrated that Dgcr8−/− NSCs express higher levels of γH2AX than the control Dgcr8+/− NSCs (Fig. 5D). We further evaluated the degree of DNA damage using the comet assay, which allows quantitatively measuring DNA strand breaks of individual cells [40]. In agreement with the γH2AX staining result, Dgcr8−/− NSCs contain significantly more damaged DNA than the control Dgcr8+/− NSCs (Fig. 5E–G). These data demonstrated that, just like the cerebellum neural progenitors [38], loss of canonical miRNAs also causes DNA damage in embryonic cortex-derived NSCs. Because Dgcr8−/− NSCs do not exhibit increased apoptosis (Fig. 4C, D), but undergo cell death when differentiated into neurons or oligodendrocytes (Fig. 3B, B″), our data suggest that NSCs are more tolerant to DNA damage than differentiated cells, a feature that has been reported in several other adult stem cells and cancer stem cells [41–43].
FIG. 5.

Dgcr8−/− NSCs exhibit increased DNA damage. (A–A″) Representative images of γH2AX immunostaining in Dgcr8+/− NSCs. (A) Immunostaining of γH2AX (red), (A′) DAPI staining, and (A″) merged. Scale bars, 50 μm. (B–B″) Representative images of γH2AX immunostaining in Dgcr8−/− NSCs. (B) Immunostaining of γH2AX (red), (B′) DAPI staining, and (B″) merged. Scale bars, 50 μm. (C) Quantification of γH2AX positive cells of Dgcr8+/− and Dgcr8−/− NSCs. n = 3 independent biological repeats. P < 0.05, two-tailed Student's t-test. (D) Immunoblotting of Dgcr8 and γH2AX in Dgcr8+/− and Dgcr8−/− NSCs. Immunoblotting of GAPDH and Ponceau S staining are used as loading controls. (E, F) Representative Comet assay images of (E) Dgcr8+/− and (F) Dgcr8−/− NSCs. (G) A Box-and-Whisker plot quantifies comet assays of Dgcr8+/− (n = 126) and Dgcr8−/− (n = 121) NSCs. P < 0.001, two-tailed Student's t-test. Color images available online at www.liebertpub.com/scd
Loss of canonical miRNAs substantially alters expression profiles of NSCs
To gain understanding on how canonical miRNAs regulate NSCs on a molecular level, we performed mRNA profiling between Dgcr8+/− and Dgcr8−/− NSCs by RNA-seq analysis. We identified that 1287 genes are downregulated and 845 genes are upregulated in Dgcr8−/− NSCs (>fourfold; P < 0.01; q < 0.01) (Fig. 6A and Supplementary Tables S4 and S5). Among the upregulated genes, we observed an enrichment of predicated targets of miR-21 and let-7 (Fig. 6B), which are the most abundantly expressed miRNAs in NSCs (Fig. 2E). Gene set enrichment analysis (GSEA) further revealed that genes associated to neuronal system, cell cycle regulation, DNA replication, and translation are among the most significantly downregulated biological processes in Dgcr8−/− NSCs (Fig. 6C). This is consistent with the observation that Dgcr8−/− NSCs are unable to undergo lineage specification (Fig. 3) and proliferate slower than Dgcr8+/− NSCs (Fig. 4A, B). Furthermore, genes associated with DNA repair are also significantly downregulated in Dgcr8−/− NSCs (Fig. 6C), suggesting that the downregulation of DNA repair pathway is at least partially responsible for the increased DNA damage in Dgcr8−/− NSCs (Fig. 5).
FIG. 6.

RNA-seq analysis of Dgcr8+/− and Dgcr8−/− NSCs. (A) Unsupervised clustering analysis of Dgcr8+/− and Dgcr8−/− NSCs. (B) GSEA demonstrated de-repression of transcription targets of miR-21 (left) and let-7 (right) in Dgcr8−/− NSCs. (C) GSEA demonstrated downregulation of genes associated with neuronal system, cell cycle, DNA replication, translation, and DNA repair in Dgcr8−/− NSCs. GSEA, gene set enrichment analysis. Color images available online at www.liebertpub.com/scd
Dgcr8−/− NSCs significantly downregulate cholesterol biosynthesis
Unexpectedly, GSEA revealed that genes associated with cholesterol biosynthesis are significantly downregulated in Dgcr8−/− NSCs (Fig. 7A, B), suggesting that canonical miRNAs promote cholesterol biosynthesis in NSCs while a complete miRNA loss leads to a reduction in cholesterol levels. Intriguingly, while the genes responsible for cholesterol biosynthesis are downregulated, Abca1 and Cyp46a1, which encode important factors reducing intracellular cholesterol by exporting and metabolizing cholesterol [6], are significantly upregulated in Dgcr8−/− NSCs (Fig. 7B). These data suggest that Dgcr8−/− NSCs express a transcriptional program that coordinately reduces intracellular cholesterol.
FIG. 7.

Dgcr8−/− NSCs decrease cholesterol biosynthesis. (A) GSEA revealed that Dgcr8−/− NSCs downregulate genes associated with cholesterol biosynthesis, target genes of SREBFs, Hedgehog signaling, and Alzheimer's disease. (B) Heatmap of selected genes involved in cholesterol biosynthesis and metabolism. (C) Total, free, and esterified cholesterol levels in Dgcr8+/− and Dgcr8−/− NSCs. Levels of cholesterol (μg) were normalized to total amount of protein (μg). n = 4 independent biological repeats. *P < 0.05; N.S., not significant; two-tailed Student's t-test. SREBF, sterol regulatory element binding factor. Color images available online at www.liebertpub.com/scd
In addition to being a basic structural component of animal cell membranes and myelin, cholesterol also plays a pivotal role in the maturation of Hedgehog ligands [9]. Furthermore, dysregulation of cholesterol is closely associated with Alzheimer's disease [44]. Consistent with these functions of cholesterol, GSEA revealed that genes associated with the Hedgehog pathway and Alzheimer's disease are significantly downregulated in Dgcr8−/− NSCs (Fig. 7A). Interestingly, Apoe, which mediates cholesterol metabolism and is one of the strongest genetic risk factors of Alzheimer's disease [45], is strongly downregulated in Dgcr8−/− NSCs (Fig. 7B). These data suggest that the canonical miRNA-mediated cholesterol biosynthesis plays a critical role in the normal function of NSCs and likely in the molecular etiology of Alzheimer's disease.
To further investigate how canonical miRNAs regulate cholesterol biosynthesis, we measured cholesterol levels in NSCs. Our data demonstrated that the great majority of cholesterol in NSCs is present as free cholesterol (Fig. 7C), which is consistent with the notion that free cholesterol is the major form of cholesterol in brain tissue [46]. We found that Dgcr8−/− NSCs contain significantly lower levels of total and free cholesterol than Dgcr8+/− NSCs (Fig. 7C), which is consistent with the mRNA-seq data (Fig. 7A, B).
To gain insights into how Dgcr8−/− NSCs downregulate cholesterol biosynthesis, we examined expression of Srebf1 and Srebf2, which encode the master transcriptional regulators of enzymes involved in cholesterol biosynthesis [8]. We discovered that Dgcr8−/− NSCs downregulate both genes (Fig. 7B). Consistently, GSEA revealed that transcriptional targets of SREBFs are significantly downregulated in Dgcr8−/− NSCs (Fig. 7A). These data demonstrated that canonical miRNAs modulate cholesterol biogenesis at least partially through regulation of SREBF activities. Together, our data revealed a role of canonical miRNAs in the regulation of cholesterol biogenesis and suggested that modulation of canonical miRNA activities could serve as potential strategies to regulate cholesterol homeostasis and treat diseases related to cholesterol dysregulation such as Alzheimer's disease.
Discussion
Precise regulation of proliferation and differentiation of NSCs is essential for the generation and regeneration of the nervous system. The defects in proliferation and differentiation of Dicer−/− NSCs have been reported previously [23,24]. Because Dicer also participates in the biogenesis of noncanonical miRNAs and endo-siRNAs, it is not entirely clear whether the observed phenotypic defects of Dicer−/− NSCs are due to canonical miRNAs or other Dicer-dependent small RNAs. In this study, we demonstrated that canonical miRNAs, which require the activities of both Drosha-Dgcr8 microprocessor and Dicer for biogenesis [22,36], are mainly responsible for the reported proliferation and differentiation defects (Figs. 3 and 4). Consistently, Dgcr8−/− NSCs significantly downregulated genes associated with cell cycle progression, DNA replication, protein translation, and neuronal differentiation (Fig. 6B).
Drosha−/− NPCs have been implicated to rapidly undergo neuronal differentiation due to the accumulation of Neurog2 transcripts, which are degraded after cleavage by the Drosha-Dgcr8 microprocessor [25]. However, our data demonstrated that Dgcr8−/− NSCs downregulate Neurog2 and can be stably propagated in vitro (Fig. 1 and Supplementary Fig. S1). The discrepancy between our observation and the previous finding may be explained by two mechanisms. Although always forming the microprocessor complex with Drosha, Drosha-independent Dgcr8 function has been reported in the neuronal morphogenesis in Drosophila [47]. It is therefore possible that a similar Drosha-independent Dgcr8 pathway maintains expression of Neurog2 and regulates self-renewal of mouse NSCs. Alternatively, the difference between Drosha−/− NPCs and Dgcr8−/− NSCs may lie in the cell populations that were analyzed. We analyzed Dgcr8−/− NSCs that were expanded and passaged in vitro, in which no residual canonical miRNAs were detected (Fig. 2). However, the previous study analyzed in vivo NPCs immediately after Drosha disruption, in which a significant amount of miRNAs likely still persisted due to the long half-lives of miRNAs [25]. Therefore, the regulation of Neurog2 by the Drosha-Dgcr8 microprocessor and canonical miRNAs may contain two aspects. On the one hand, transcription of Neurog2 is positively regulated by canonical miRNAs while a complete canonical miRNA loss leads to downregulation of Neurog2 (Fig. 1D). On the other hand, the existing Neurog2 transcripts may be subjected to negative regulation by the Drosha-Dgcr8 microprocessor [25].
DNA damage and apoptosis caused by depletion of Dicer or Dgcr8 have been reported in cerebellum neuronal precursors, which makes the miRNA biogenesis pathway a potential target to inhibit cerebellar tumors [38]. In this study, we demonstrated that disruption of Dgcr8 in cortex-derived NSCs also exhibit increased DNA damage (Fig. 5), but not increased apoptosis (Fig. 4C, D), which agrees with the previous report that Dicer−/− NSCs do not show increased apoptosis [24]. However, differentiation of Dgcr8−/− NSCs into neurons or oligodendrocytes leads to cell death (Fig. 3B, B″), suggesting that NSCs, like several other adult stem cells and cancer stem cells [41–43], are more tolerant to DNA damage than differentiated cells. Because brain tumor stem cells share many features with normal NSCs [4], it would be of great interest and importance to determine whether brain tumor stem cells show similar tolerance to miRNA depletion-induced DNA damage.
As a basic structural component of animal cell membrane and myelin, cholesterol is highly enriched in the brain, which contains approximately 25% of total cholesterol of the body [6]. Because of the blood-brain barrier, brain cholesterol is primarily synthesized de novo and is generally considered as distinct from cholesterol of periphery organs [5,7]. Biosynthesis of cholesterol is through the isoprenoid pathway, which involves more than twenty enzymes [48]. 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase, which is encoded by the Hmgcr gene, is the major rate-limiting enzyme of cholesterol biosynthesis [6]. Defects in several other genes dedicated to cholesterol biosynthesis, such as MVK, DHCR7, DHCR24, EBP, and CYP51, are associated with a number of human inherited diseases such as Smith–Lemli–Opitz syndrome (MIM 270400), Desmosterolosis (MIM 602398), and Conradi–Hünermann–Happle syndrome (MIM 302960) [49]. Intriguingly, all these genes, as well as most other genes involved in cholesterol biosynthesis, are significantly downregulated in Dgcr8−/− NSCs (Fig. 7A, B). Downregulation of Srebf1 and Srebf2, which encode the master regulators for cholesterol biosynthesis [8], is likely at least partially responsible for the reduced cholesterol levels in Dgcr8−/− NSCs. Furthermore, Cyp46a1 and Abca1, which encode factors that metabolize and/or export brain cholesterol out of cells or across the blood-brain barrier [6], are significantly upregulated in Dgcr8−/− NSCs (Fig. 7B). These data suggest that canonical miRNAs maintain a transcriptional program that promotes expression of enzymes producing cholesterol, but repress factors that eliminate cellular cholesterol.
Several miRNAs, including miR-122, miR-33, miR-758, miR-106b, and miR-218, have been demonstrated to negatively regulate cholesterol metabolism [50]. Recently, the miR-183/96/182 cluster has been demonstrated to positively regulate cholesterol biosynthesis in liver cells by inhibiting expression of ISIG2 and FBXW7 [51–53], which encode factors that negatively regulate SREBF2. Because mature miR-183, miR-96, and miR-182 are either not detected or at very low levels in Dgcr8+/− NSCs (Supplementary Table S2), it is unlikely that the absence of the miR-183/96/182 cluster is responsible for the reduced cholesterol biosynthesis in Dgcr8−/− NSCs. Therefore, our data suggest that NSCs likely use a different set of canonical miRNAs to promote cholesterol biosynthesis (Fig. 7).
Loss of cholesterol homeostasis is closely correlated with Alzheimer's disease [44]. Apoe, which encodes an apoprotein, regulates metabolism of cholesterol and has been identified as the strongest genetic risk factor for Alzheimer's disease [44], is significantly downregulated in Dgcr8−/− NSCs (Fig. 7B). Furthermore, genes associated with Alzheimer's disease have been significantly downregulated in Dgcr8−/− NSCs (Fig. 7A). These data suggest that the canonical biogenesis pathway could serve as potential targets to modulate brain cholesterol levels and genes associated with Alzheimer's diseases.
Supplementary Material
Acknowledgments
The authors thank Drs. Hairi Su and Xinyang Zhao for their help in cholesterol measurement. RZ is supported by the University of Alabama at Birmingham Faculty Development Fund. HL is supported by NIH (CA196631-01A1) and Mayo Clinic Center for Individualized Medicine. KK is supported by NIH (R00HL093212), NIH (R01AG043531), TriStem-Star Foundation (2013-049), Louis V. Gerstner, Jr. Young Investigators awards, Geoffrey Beene Junior Chair Award, Sidney Kimmel Scholar Award, Alfred W. Bressler Scholars Endowment Fund, and MSKCC Society Fund. Xiaosi Han is supported by NIH (R01NS095626).
Author Disclosure Statement
The authors indicate no potential conflicts of interest.
References
- 1.Martino G. and Pluchino S. (2006). The therapeutic potential of neural stem cells. Nat Rev Neurosci 7:395–406 [DOI] [PubMed] [Google Scholar]
- 2.Gage FH. and Temple S. (2013). Neural stem cells: generating and regenerating the brain. Neuron 80:588–601 [DOI] [PubMed] [Google Scholar]
- 3.Merkle FT. and Alvarez-Buylla A. (2006). Neural stem cells in mammalian development. Curr Opin Cell Biol 18:704–709 [DOI] [PubMed] [Google Scholar]
- 4.Vescovi AL, Galli R. and Reynolds BA. (2006). Brain tumour stem cells. Nat Rev Cancer 6:425–436 [DOI] [PubMed] [Google Scholar]
- 5.Orth M. and Bellosta S. (2012). Cholesterol: its regulation and role in central nervous system disorders. Cholesterol 2012:292598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin M, Dotti CG. and Ledesma MD. (2010). Brain cholesterol in normal and pathological aging. Biochim Biophys Acta 1801:934–944 [DOI] [PubMed] [Google Scholar]
- 7.Pitas RE, Boyles JK, Lee SH, Hui D. and Weisgraber KH. (1987). Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem 262:14352–14360 [PubMed] [Google Scholar]
- 8.Horton JD, Goldstein JL. and Brown MS. (2002). SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109:1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wendler F, Franch-Marro X. and Vincent JP. (2006). How does cholesterol affect the way Hedgehog works? Development 133:3055–3061 [DOI] [PubMed] [Google Scholar]
- 10.Ruiz i Altaba A, Sanchez P. and Dahmane N. (2002). Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer 2:361–372 [DOI] [PubMed] [Google Scholar]
- 11.Ivey KN. and Srivastava D. (2010). MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cell 7:36–41 [DOI] [PubMed] [Google Scholar]
- 12.Lee YS. and Dutta A. (2009). MicroRNAs in cancer. Annu Rev Pathol 4:199–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pauli A, Rinn JL. and Schier AF. (2011). Non-coding RNAs as regulators of embryogenesis. Nat Rev Genet 12:136–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun K. and Lai EC. (2013). Adult-specific functions of animal microRNAs. Nat Rev Genet 14:535–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ha M. and Kim VN. (2014). Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15:509–524 [DOI] [PubMed] [Google Scholar]
- 16.Cook MS. and Blelloch R. (2013). Small RNAs in germline development. Curr Top Dev Biol 102:159–205 [DOI] [PubMed] [Google Scholar]
- 17.Kawase-Koga Y, Otaegi G. and Sun T. (2009). Different timings of Dicer deletion affect neurogenesis and gliogenesis in the developing mouse central nervous system. Dev Dyn 238:2800–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Pietri Tonelli D, Pulvers JN, Haffner C, Murchison EP, Hannon GJ. and Huttner WB. (2008). miRNAs are essential for survival and differentiation of newborn neurons but not for expansion of neural progenitors during early neurogenesis in the mouse embryonic neocortex. Development 135:3911–3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makeyev EV, Zhang J, Carrasco MA. and Maniatis T. (2007). The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell 27:435–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi PS, Zakhary L, Choi WY, Caron S, Alvarez-Saavedra E, Miska EA, McManus M, Harfe B, Giraldez AJ, et al. (2008). Members of the miRNA-200 family regulate olfactory neurogenesis. Neuron 57:41–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis TH, Cuellar TL, Koch SM, Barker AJ, Harfe BD, McManus MT. and Ullian EM. (2008). Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J Neurosci 28:4322–4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Babiarz JE, Hsu R, Melton C, Thomas M, Ullian EM. and Blelloch R. (2011). A role for noncanonical microRNAs in the mammalian brain revealed by phenotypic differences in Dgcr8 versus Dicer1 knockouts and small RNA sequencing. RNA 17:1489–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andersson T, Rahman S, Sansom SN, Alsio JM, Kaneda M, Smith J, O'Carroll D, Tarakhovsky A. and Livesey FJ. (2010). Reversible block of mouse neural stem cell differentiation in the absence of dicer and microRNAs. PLoS One 5:e13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawase-Koga Y, Low R, Otaegi G, Pollock A, Deng H, Eisenhaber F, Maurer-Stroh S. and Sun T. (2010). RNAase-III enzyme Dicer maintains signaling pathways for differentiation and survival in mouse cortical neural stem cells. J Cell Sci 123:586–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knuckles P, Vogt MA, Lugert S, Milo M, Chong MM, Hautbergue GM, Wilson SA, Littman DR. and Taylor V. (2012). Drosha regulates neurogenesis by controlling neurogenin 2 expression independent of microRNAs. Nat Neurosci 15:962–969 [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Medvid R, Melton C, Jaenisch R. and Blelloch R. (2007). DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet 39:380–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R. and Schutz G. (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23:99–103 [DOI] [PubMed] [Google Scholar]
- 28.Suh N, Baehner L, Moltzahn F, Melton C, Shenoy A, Chen J. and Blelloch R. (2010). MicroRNA function is globally suppressed in mouse oocytes and early embryos. Curr Biol 20:271–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Currle DS, Hu JS, Kolski-Andreaco A. and Monuki ES. (2007). Culture of mouse neural stem cell precursors. J Vis Exp 2:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z, Skamagki M, Kim K. and Zhao R. (2015). Canonical microRNA activity facilitates but may be dispensable for transcription factor-mediated reprogramming. Stem Cell Rep 5:1119–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao R, Deibler RW, Lerou PH, Ballabeni A, Heffner GC, Cahan P, Unternaehrer JJ, Kirschner MW. and Daley GQ. (2014). A nontranscriptional role for Oct4 in the regulation of mitotic entry. Proc Natl Acad Sci U S A 111:15768–15773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D. and Clement MV. (2014). OpenComet: an automated tool for comet assay image analysis. Redox Biol 2:457–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES. and Mesirov JP. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anders S, Pyl PT. and Huber W. (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M. and Gingeras TR. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Babiarz JE, Ruby JG, Wang Y, Bartel DP. and Blelloch R. (2008). Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev 22:2773–2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, et al. (2010). Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev 24:992–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swahari V, Nakamura A, Baran-Gale J, Garcia I, Crowther AJ, Sons R, Gershon TR, Hammond S, Sethupathy P. and Deshmukh M. (2016). Essential function of dicer in resolving DNA damage in the rapidly dividing cells of the developing and malignant cerebellum. Cell Rep 14:216–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan J, Adamski R. and Chen J. (2010). Focus on histone variant H2AX: to be or not to be. FEBS Lett 584:3717–3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olive PL. and Banath JP. (2006). The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 1:23–29 [DOI] [PubMed] [Google Scholar]
- 41.Liu JC, Lerou PH. and Lahav G. (2014). Stem cells: balancing resistance and sensitivity to DNA damage. Trends Cell Biol 24:268–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sperka T, Wang J. and Rudolph KL. (2012). DNA damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol Cell Biol 13:579–590 [DOI] [PubMed] [Google Scholar]
- 43.Blanpain C, Mohrin M, Sotiropoulou PA. and Passegue E. (2011). DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell 8:16–29 [DOI] [PubMed] [Google Scholar]
- 44.Di Paolo G. and Kim TW. (2011). Linking lipids to Alzheimer's disease: cholesterol and beyond. Nat Rev Neurosci 12:284–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J, Basak JM. and Holtzman DM. (2009). The role of apolipoprotein E in Alzheimer's disease. Neuron 63:287–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang J. and Liu Q. (2015). Cholesterol metabolism and homeostasis in the brain. Protein Cell 6:254–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luhur A, Chawla G, Wu YC, Li J. and Sokol NS. (2014). Drosha-independent DGCR8/Pasha pathway regulates neuronal morphogenesis. Proc Natl Acad Sci U S A 111:1421–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waterham HR. (2006). Defects of cholesterol biosynthesis. FEBS Lett 580:5442–5449 [DOI] [PubMed] [Google Scholar]
- 49.Waterham HR. (2002). Inherited disorders of cholesterol biosynthesis. Clin Genet 61:393–403 [DOI] [PubMed] [Google Scholar]
- 50.Rotllan N. and Fernandez-Hernando C. (2012). MicroRNA Regulation of Cholesterol Metabolism. Cholesterol 2012:847849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miao J, Ling AV, Manthena PV, Gearing ME, Graham MJ, Crooke RM, Croce KJ, Esquejo RM, Clish CB, et al. (2015). Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat Commun 6:6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jeon TI, Esquejo RM, Roqueta-Rivera M, Phelan PE, Moon YA, Govindarajan SS, Esau CC. and Osborne TF. (2013). An SREBP-responsive microRNA operon contributes to a regulatory loop for intracellular lipid homeostasis. Cell Metab 18:51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jeon TI. and Osborne TF. (2016). miRNA and cholesterol homeostasis. Biochim Biophys Acta 1861:2041–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.