

Abstract
Background
Genome wide association studies (GWAS) have identified multiple loci associated with epithelial ovarian cancer (EOC) susceptibility, but further progress requires integration of epidemiology and biology to illuminate true risk loci below genome-wide significance levels (P<5×10−8). Most risk SNPs lie within non-protein-encoding regions, and we hypothesize that long non-coding RNA (lncRNA) genes are enriched at EOC risk regions and represent biologically relevant functional targets.
Methods
Using imputed GWAS data from ~18,000 invasive EOC cases and 34,000 controls of European ancestry, the GENCODE (v19) lncRNA database was used to annotate SNPs from 13,442 lncRNAs for permutation-based enrichment analysis. Tumor expression quantitative trait locus (eQTL) analysis was performed for sub-genome-wide regions (1×10−5>P>5×10−8) overlapping lncRNAs.
Results
Of 5,294 EOC-associated SNPs (P<1.0×10−5), 1,464 (28%) mapped within 53 unique lncRNAs and an additional 3,484 (66%) SNPs were correlated (r2>0.2) with SNPs within 115 lncRNAs. EOC-associated SNPs comprised 130 independent regions, of which 72 (55%) overlapped with lncRNAs, representing a significant enrichment (P = 5.0×10−4) that was more pronounced among a subset of 5,401 lncRNAs with active epigenetic regulation in normal ovarian tissue. EOC-associated lncRNAs and their putative promoters and transcription factors were enriched for biologically relevant pathways and eQTL analysis identified five novel putative risk regions with allele-specific effects on lncRNA gene expression.
Conclusion
lncRNAs are significantly enriched at EOC risk regions suggesting a mechanistic role for lncRNAs in driving predisposition to EOC.
Impact
lncRNAs represent key candidates for integrative epidemiological and functional studies. Further research on their biological role in ovarian cancer is indicated.
Keywords: lncRNA, ovarian cancer risk, germline polymorphisms, epigenomics, eQTL
INTRODUCTION
Epithelial ovarian cancer (EOC) risk has a significant genetic component that is not fully characterized. Risk is greatly increased by rare inherited mutations in highly penetrant genes like BRCA1 and BRCA2 that segregate in an autosomal dominant manner and confer lifetime risks as high as 39 and 17 percent, respectively(1, 2). Risk is modestly increased by uncommon mutations in genes with moderate penetrance, such as RAD51C/D and BRIP1(3, 4). That the known genetic risk factors account for <50 percent of the heritable risk of EOC suggests that additional risk alleles await discovery(5). The advent of genome wide association studies (GWAS) has enabled the international Ovarian Cancer Association Consortium (OCAC) to discover approximately 22 single nucleotide polymorphisms (SNPs) with mild effects(6–14). Since OCAC includes virtually every large case-control study of EOC in the world, which precludes a substantial increase in sample size, innovative approaches are needed to evaluate the thousands of risk SNPs at sub genome-wide levels of statistical significance (1×10−5>P>5×10−8).
Most risk SNPs identified by GWAS are located in non-coding regions of the genome (3) and the functional biofeatures and target genes remain unknown for many loci. Data suggest that a significant proportion coincide with long non-coding RNAs (lncRNAs)(15), a class of transcripts emerging as significant contributors to ovarian carcinogenesis(16–21). We hypothesize that lncRNAs represent functional targets of some EOC risk SNPs and that the integration of genotyping and lncRNA expression datasets will enable identification of additional susceptibility alleles and help unravel the etiology.
Non-coding RNAs (ncRNAs) resemble protein-coding transcripts but without functional open reading frames(22) and are typically classified according to size; thus small ncRNAs are less than 200 nucleotides in length whereas lncRNAs contain at least 200 nucleotides. While small ncRNAs, including microRNAs (miRNAs), small interfering RNAs (siRNAs), and PIWI-interacting RNAs (piRNAs) have recognized functional importance in carcinogenesis(23), lncRNAs remain under studied even though they are the most common type of transcribed RNAs(24). Recent studies have shown that lncRNAs can act in cis- or trans- to regulate gene expression and promote tumorigenesis through transcriptional regulation, initiation of chromatin remodeling, modulating alternative splicing, altering protein activity or localization, and genomic imprinting(16, 22, 25–27). Similar to other regulatory elements, lncRNAs exhibit cell-type specificity with varying expression and activity across different tissue types(28).
Given the likely role of lncRNAs in EOC pathogenesis(16–21, 29, 30) and growing evidence implicating inherited variants in lncRNAs with cancer susceptibility(31–34), we sought to systematically test the hypothesis that genetic variants associated with EOC risk are enriched at lncRNA gene regions, particularly those with active epigenomic profiles in ovarian tissue. We further investigated whether lncRNAs represent potential functional target genes of sub-genome-wide EOC risk regions by integrating lncRNA expression data and performing expression quantitative trait loci (eQTL) analyses. Our results suggest that lncRNAs are significantly enriched at EOC risk regions and that variants within these regions have functional effects on lncRNA expression. More comprehensive testing of this hypothesis and candidate lncRNA associated with EOC risk is warranted.
MATERIALS AND METHODS
Genetic association studies and lncRNA Annotation
Analyses were based on four pooled GWAS totaling 46,213 subjects of European ancestry (15,397 invasive EOC cases, 30,816 controls) from 43 independent studies in the international Ovarian Cancer Association Consortium (OCAC)(14, 35). A meta-analysis was performed to combine results across studies. Details of the study participants, genotyping, quality control, imputation, and meta-analysis have been previously described(6, 12). Briefly, cases were women with histologic-confirmed primary invasive EOC, fallopian tube cancer, or peritoneal cancer, and for most studies were frequency-matched to controls on age group and self-reported race. Specimens and data were collected according to protocols approved by local institutional review and ethics boards. Genotype data from the contributing GWAS were imputed separately using IMPUTE2 software(36) and 1000 Genomes Project phase 3 as the reference panel with pre-phasing of the genotypes performed using SHAPEIT(37). For each study, log-additive models were fit to estimate SNP associations with EOC risk. The meta-analysis used a fixed-effect model weighted by the inverse variance and only study results for SNPs imputed with r2>0.25 were included. Association analyses were performed for all invasive EOC cases versus controls and by histologic subtype. A SNP with a significance level of p < 1.0×10−5 was defined as EOC-associated.
Coordinates for 13,870 human lncRNAs with biological functions in eukaryotes were downloaded from the publicly available GENCODE (v19) database based on Genome Build 37(38). Of the 13,870 lncRNA genes, we excluded 70 genes on the Y chromosome and 4 genes annotated for multiple locations (n=21 observations), but retained 17 genes that were less than 200 bp in length leaving 13,779 unique gene name and positions for analysis. There were 337 lncRNA genes without variants for a total of 1,757,495 variants mapping to intron and exon coordinates of 13,442 unique lncRNA genes.
Identification of Ovarian-Active lncRNAs
Since activity of lncRNAs can be tissue specific (39, 40), we annotated lncRNA genes based on their epigenomic profiles in ovarian tissue from the NIH Epigenome Roadmap(41) to select those with active enhancer, promoter, or transcription profiles (Supplemental Figure 1). We quantified histone H3 lysine 4 monomethylation (H3K4me1) and trimethylation (H3K4me3) marks in the transcription start site (TSS; +/− 3kb) of each gene to identify active enhancers and promoters, respectively(28). To identify transcriptionally active lncRNA we quantified histone 3 lysine 36 trimethlyation (H3K36me3) marks in the gene body(26). For each gene, we computed the average signal density (rpm/bp) for H3K4me1 and H3K4me3 histone marks within the TSS and for H3K36me3 within the gene coordinates using the Genboree Workbench Epigenomic Slicer tool(41). We considered lncRNAs as ‘active’ in ovarian tissue when either the H3K4me1, H3K4me3, or H3K36me3 average density was higher than a threshold (7 rpm/bp, 4 rpm/bp, and 4 rpm/bp respectively), which was determined by a P value (< 0.05) taken from the background model of Poisson distribution for each histone mark, parameterized by the signal density of all lncRNAs(Supplementary Figure 2). Finally, we similarly computed average H3K4me1, H3K4me3, and H3K36me3 signal density for 11 highly uniform and strongly expressed housekeeping genes as controls(42). All 11 housekeeping genes were defined as active using the above criteria.
Enrichment of lncRNA at EOC Risk Regions
To determine if EOC risk regions were more likely to be found near encoded lncRNAs than expected by chance, we compared the observed proportion of EOC-associated SNPs (P < 1.0×10−5) within lncRNAs to an expected proportion based on the whole genome. A permutation-based approach was used to obtain the expected proportion and the level of significance for enrichment. An empirical p-value for the observed proportion of lncRNA risk SNPs was calculated from a background (null) distribution obtained by randomly shuffling lncRNA-sized regions on the chromosome and computing the proportion of risk SNPs for the shuffled regions (10,000 times). We also compared ovarian-active lncRNAs to all lncRNAs by permuting ovarian active/inactive classification 10,000 times to generate the background distribution and calculated empirical p-values.
If an EOC risk region harbors multiple SNPs due to linkage disequilibrium (LD), this can inflate the potential significance of lncRNAs in that region. To correct for that, analyses were performed to identify independent EOC-associated regions and to test for enrichment of lncRNAs within these regions. An independent region was defined as a genomic interval containing at least one EOC-associated SNP at P < 1.0×10−5 (index) and all surrounding, nominally significant LD SNPs (P < 0.05 and r2 > 0.2) within a +/−250kb window. We then performed permutation-based testing to determine if lncRNAs were enriched for overlap with these independent risk regions. Analysis was performed using the gene-set enrichment analysis tool INRICH (INterval enRICHment analysis)(43). Briefly, the number of lncRNA genes that overlap at least one risk region was compared to a null distribution generated by permuting the risk regions to random genomic locations with the constraint that each randomized region matches the original region’s number of SNPs tested and SNP density (+/−10%). Additionally, the permuted regions were constrained to lie within the meta-analysis SNP positions. We also compared ovarian-active lncRNA to all lncRNA genes to determine if the subset of lncRNA active in ovarian tissue were more enriched for overlap with independent regions of association than all lncRNA. For this comparison, we permuted only the regions that overlapped with lncRNA (e.g. 72 intervals for all invasive analysis) and constrained the permuted regions to overlap the same number of lncRNA genes.
Biological Processes of lncRNAs associated with EOC
For the subset of lncRNAs that contained EOC-associated SNPs, we tested for enrichment of specific biological processes, pathways, and promoter motifs using the GREAT tool (Genomic Regions Enrichment of Annotations)(44). GREAT uses gene set collections from the Molecular Signatures Database (MSigDB)(45) and calculates a binomial test for enrichment over genomic regions and a hypergeometric test for enrichment over genes within 500kb of the region. We also tested for enrichment of ENCODE transcription factor (TF) binding sites using HOMER (Hypergeometric Optimization of Motif EnRichment)(46). For GREAT analyses, we required a False Discovery Rate (FDR) of 15% for both binomial and hypergeometric tests to determine significance. For HOMER analysis of ENCODE TF binding sites, we required the more restrictive Bonferroni corrected p-value of 1.0×10−4. We also examined the set of significant promoters and TFs, identified by GREAT and HOMER, using the PANTHER classification system and tool set(47) to determine if they were enriched for specific biological processes.
We additionally annotated EOC-associated lncRNAs for gene expression in 35 normal ovarian tissue samples from the Genotype-Tissue Expression Project (GTEx)(48) web portal(49) and for tumor tissue expression from 412 HGS tumor samples from The Cancer Genome Atlas (TCGA)(50).
eQTL Analysis of Novel EOC-associated SNPs in Primary Ovarian Tumors
For the EOC-associated SNPs within novel sub-genome-wide risk regions, we sought to identify potential lncRNA targets. We performed eQTL analysis of primary ovarian tumor tissues from TCGA (50). Germline genotypes for 402 high grade serous (HGS) cases of European ancestry with non-missing stage and grade data were downloaded and imputed to 1000 genome project phase 3 reference panel (March, 2012) using MACH and Minimac software(51–53). Analyses were limited to SNPs with imputation quality r2 > 0.3 and with at least 5 minor allele carriers (MAF>0.0075). Analysis of lncRNA gene expression was performed with lncRNA RPKM (reads per kilobase per million reads) data for 12,727 intergenic lncRNAs, which was generated from RNA-sequencing reads using GENCODE v19 annotations and was downloaded through the TANRIC platform v1.0(54). A total of 334 HGS cases with germline genotype and gene expression data were available for analyses. Unadjusted linear regression was used to estimate minor allele dose effect on gene expression (log2 transformed RPKM) for genes with ≥0.1 RPKM in at least 2 individuals. We performed cis-eQTL analysis for genes within 1MB of a SNP with a significant association defined by an FDR of less than 5%.
RESULTS
The genome-wide association meta-analysis participants are detailed in Supplementary Table S1. As expected, most cases (62%) had tumors with serous histology, followed by endometrioid (14%) and mucinous (7%) and clear cell (7%). Of the ~15 million genotyped and imputed SNPs, 5,294 (0.035%) were associated with invasive EOC risk (P < 10−5) (Table 1). These SNPs mapped to 130 independent regions, 78 of which are below genome-wide significance and more than 500kb from previously reported risk SNPs. Fourteen of the 22 reported EOC risk loci associate with invasive EOC and 13 of these were replicated here, the lone exception being a locus identified in high risk BRCA1/2 mutation carriers (55). Additionally, subtype analyses replicated four out of four serous risk loci, two out of three mucinous risk loci, and the sole clear cell risk loci, for a total of 21 out of 22 previously reported ovarian cancer risk loci represented in our data.
Table 1.
SNPs associated with epithelial ovarian cancer risk are enriched in lncRNA genes
| Tumor Histology | Whole Genome (WG) (15,159,372 SNPs) |
13,442 lncRNA Genes (1,757,495 SNPs) |
5,287 Ovarian-Active lncRNA Genes (457,227 SNPs) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| SNPs p<10−5 |
Independent Regionsb |
SNPs p<10−5 |
P-valuea | Independent Regionsb | P-valuec | SNPs p<10−5 |
P-valued | Independent Regionsb | P-valuee | |
| All Invasive | 5294 | 130 | 1464 | 0.047 | 72 | 0.0005 | 873 | 0.009 | 46 | 0.043 |
| Serous | 5922 | 147 | 1572 | 0.044 | 81 | 0.009 | 960 | 0.002 | 51 | 0.16 |
| High Grade | 5367 | 178 | 1467 | 0.045 | 89 | 0.002 | 899 | 0.004 | 50 | 0.25 |
| Low Grade | 1916 | 1104 | 219 | 0.48 | 385 | 0.64 | 50 | 0.70 | 169 | 0.15 |
Proportion of risk SNPs within lncRNA genes compared to whole genome distrbution. Empirical p-values based on 10,000 permutations of lncRNA genes across the whole genome.
SNPs with r2>0.2 and within a 250kb distance were grouped into independent regions.
Proportion of regions overlapping lncRNA genes compared to whole genome distribution. Empirical p-values based on 10,000 permutations of independent regions across the whole genome.
Proportion of risk SNPs in ovarian-active lncRNA genes compared to all lncRNAs. Empirical p-values based on 10,000 permutations of active/inactive classification of lncRNAs.
Proportion of regions overlapping ovarian-active lncRNA genes compared to all lncRNAs. Empirical p-values based on 10,000 permutations of independent regions that overlapped lncRNAs.
Globally, 1.76 million SNPs (12%) mapped to 13,442 lncRNA genes and nearly all (97.55%) lncRNA genes contained genotyped or imputed SNPs. Most (75%) of the 13,442 lncRNAs annotated were greater than 1700 bp in length and classified as long-intergenic ncRNAs (lincRNAs; n=7,048), followed by antisense (n=5,257), sense intronic (n=741), sense overlapping (n=202), processed transcript (n=511), and 3′ overlapping (n=21). We additionally identified a subset of 5,401 lncRNAs (~40%) with active histone modification profiles in ovarian tissue and annotated them as ‘ovarian-active’ (Supplementary Fig. S1 and S2). Just over 457,000 SNPs (26% of lncRNA SNPs and 3% of all genotyped or imputed SNPs) mapped to 5,287 of the ovarian-active lncRNAs and most were antisense (n=2,926) or lincRNA (n=1,651).
Enrichment of lncRNA at EOC risk regions
Of the 130 independent regions associated with EOC risk, 72 (55%) overlapped encoded lncRNAs, of which 39 regions are novel (>500kb from previously reported loci). These 72 regions consisted of 28% percent (n=1,464) of the SNP hits which directly mapped to 53 unique lncRNA genes and an additional 3,484 (66%) SNP hits in linkage disequilibrium (LD; r2>0.2) with SNPs located in 115 lncRNAs. The proportion of risk SNPs that mapped to lncRNA gene coordinates was approximately 2 times higher than the proportion observed across the whole genome (0.083% versus 0.035%; Figure 1A) and was significantly higher than the expected proportion based on a random distribution across the genome (PPERM = 0.047; Supplementary Fig. S3). We also compared the number of independent regions that overlapped with lncRNA genes to a random distribution of regions across the genome and the observed overlap (55%) was significantly higher than expected (PINRICH = 0.0005), providing further evidence for enrichment even when accounting for LD structure. To determine if the r2 threshold used to define LD regions had any undue influence on our results, we repeated analyses with a more stringent r2=0.8 criterion and while the observed overlap with lncRNA was lower (28%), it remained significantly higher than expected (P INRICH = 0.01).
Figure 1. Proportion of EOC-associated SNPs in lncRNA genes compared to whole genome.
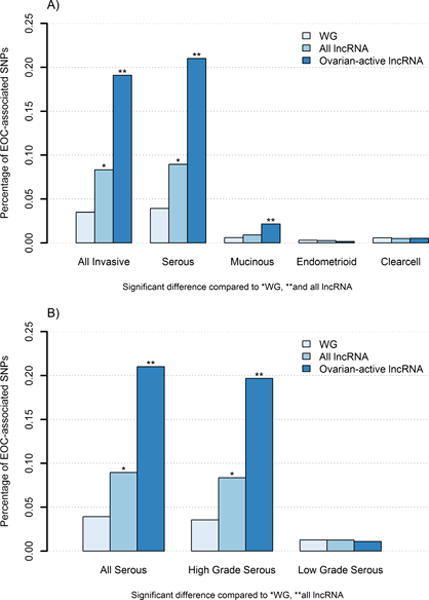
Histograms of the proportion of EOC-associated SNPs (P < 1.0×10−5) from analysis of A) main histologic subgroups and B) serous histology by grade for whole genome (WG), all lncRNA, and ovarian-active lncRNA regions.
The subset of 5,287 ovarian-active lncRNAs encompassed 60% (n=873) of the lncRNA SNP hits and overlapped 64% (n=46) of lncRNA independent regions of association. When limiting to ovarian-active lncRNAs, the enrichment for EOC-associated SNPs was significantly increased from 0.083% to 0.20% (PPERM=0.009; Figure 1A); a 5-fold higher proportion of SNP hits than observed across the whole genome. Similarly, enrichment for independent risk regions was higher for ovarian-active lncRNAs versus all lncRNAs (PINRICH=0.04; Table 1). Analyses stratified by tumor histology revealed that SNPs associated with high grade serous tumors, the most predominant subtype, are enriched in lncRNA (Table 1), but not other, less common histologic subtypes (Figure 1A; Supplementary Table S2) or low grade serous histology (Figure 1B). Although SNPs associated with mucinous EOC were not enriched within all lncRNA, when subset to ovarian-active lncRNAs the difference became more pronounced (PPERM=0.0001; Supplementary Table S2).
Having determined that the proportion of EOC-associated SNPs were over-represented at genomic regions harboring lncRNA, we sought to assess whether this enrichment was influenced by the length (KB) or SNP coverage (# tested SNPs) of the lncRNA regions. The lncRNAs containing SNP hits were comparable in length and SNP coverage to the overall catalogue of lncRNA genes, suggesting an absence of bias due to gene coverage (Supplementary Fig. S4). Moreover, we compared the density of SNP hits between lncRNA regions and whole genome to assess enrichment while accounting for coverage. This analysis demonstrated a significant enrichment of EOC-associated SNPs in lncRNA regions compared to whole genome and protein-coding genes (223 KB/hit versus 536 KB/hit and 339.1 KB/hit, respectively) and further supported our findings overall and by histologic subtype (Supplemental Methods; Supplementary Table S3 and S4).
Biological pathways of EOC-associated lncRNAs
A total of 53 lncRNA genes contained EOC-associated SNP(s) within their coordinates. These genes were located within 36 of the 72 independent risk regions and contained 1,464 EOC-associated SNPs. Most of the 53 genes (83%) were expressed in normal (GTeX; n=35) or tumor tissues (TCGA; n=412) with the majority showing expression in both (57%) or in tumor tissues only (30%) (Figure 2A). Roughly half (n=25) had active epigenomic profiles in normal ovarian tissue.
Figure 2. Ovarian tissue expression and biological processes of EOC-associated lncRNA genes.
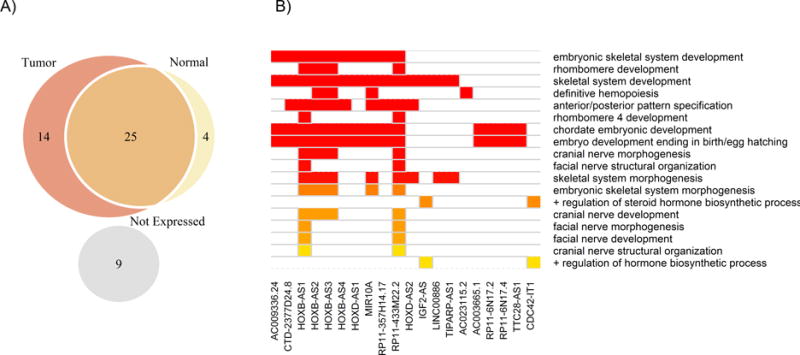
Figure A shows the expression of 53 EOC-associated lncRNA for 412 ovarian tumor tissues and 35 normal ovarian tissues. Figure B shows a heatmap of significant p-values (FDR<15%) for 18 biological processes associated with 20 out of 53 EOC-associated lncRNAs.
Pathway analysis of the 53 lncRNA genes revealed significant enrichment for multiple embryonic development and morphogenesis pathways as well as positive regulation of hormone/steroid biosynthesis (FDR < 15%) (Figure 2B). The lncRNA regions were enriched for five predicted promoter motifs, including androgen receptor (AR; P = 3.6×10−6), signal transducer and activator of transcription 3 (STAT3; P = 5.8×10−5) and paired box 8 (PAX8; P = 1.3×10−3) and five transcription factor binding sites were over-represented within their sequences, including n-MYC and c-MYC (Supplementary Table S5). Taken together, these promoter and transcription factors were enriched for regulatory pathways of transcription, cell differentiation, and epithelial development (Supplementary Table S6).
eQTL analysis of novel lncRNA risk SNPs in primary ovarian tumors
To potentially inform biological significance, we conducted eQTL analyses of primary tumor tissue for EOC-associated SNPs within the 39 novel sub-GW regions that overlapped lncRNA genes. TCGA gene expression data were available for 334 HGS EOC cases with genotype data imputed to 1KGP density. A total of 8,763 lncRNAs were at least minimally expressed (≥0.1 RPKM in ≥2 individuals) in the tumor tissues and were retained for analysis. The 39 novel regions contained 158 EOC-associated SNPs located within or in LD to 78 lncRNA genes; of these we analyzed 143 SNPs that met the inclusion criteria (imputation r2 > 0.3 and ≥ 5 minor allele carriers). Cis-eQTL analysis revealed that 5 novel regions (24 SNPs) were associated with expression of 6 lncRNAs in tumor tissue (FDR<5%; Table 2). Expression of the lncRNA at four of these loci was associated with reduced EOC risk (11p15, 11p13, 16q21, 16q22.1) and one (19q13.12–13) was associated with an increase in EOC risk. The locus 19q13.12–13 where risk alleles were associated with increased expression contained 9 SNPs with the top signal observed for chr19:38451511 TA>T (OR (95% CI) = 1.12 (1.09–1.15), P=5.74×10−6; Supplementary Fig. S5A). Seven of the 9 SNPs were associated with differential expression of AC012309.5 (P = 0.0003; Figure 3A) that is located 695 KB from the top regional SNP. The reduced risk locus 16q21 exhibited the strongest SNP association (P = 8.57×10−8) located within the coordinates of RP11-410D17.2 (Supplementary Fig. S5B) and eQTL analysis revealed four SNPs with minimal allele-specific effects on two distal lncRNAs (Figure 3B–C). The 11p15.5 locus contained the only other significant SNP associations located within an encoded lncRNA. The strongest signal was seen for rs3741205 A>C (OR (95% CI) = 0.93 (0.91–0.94), P = 3.94×10−6) located within an exon of IGF2-AS and introns of IGF2 and INS-IGF2 (Supplementary Fig. S5C). Two other loci were also associated with reduced EOC risk, 11p13 and 16q22.1, and contained eQTL SNPs associated with the expression of proximal lncRNA genes (<50kb) (Supplementary Fig. S5D–E).
Table 2.
Novel EOC risk regions (P < 1.0×10−5) with allele-specific effects on lncRNA gene expression
| Locus | Independent Regions (Length KB) | Overlap lncRNA a (Ovarian-active?) | Top SNP | MAF | R2 | OR (95% CI) |
P-value | SNP Hits/eQTL SNPs | lncRNA eQTL Targets (Distance KB) |
|---|---|---|---|---|---|---|---|---|---|
| 11p15.5 | chr11:2116492-2190591 (74) |
IGF2-AS (Y) AC132217.4 (N) |
rs3741205 (A>C) | 0.28 | 0.99 | 0.93 (0.91–0.94) |
3.94E-06 | 4/2 | FAM99A (429) |
| 11p13 | chr11:36325764-36396678 (71) | RP11-514F3.4 (N) | rs10501153 (C>T) | 0.34 | 0.82 | 0.92 (0.90-0.93) |
3.76E-07 | 12/8 | RP11-219O3.2 (50) |
| 16q21 | chr16:58944508-59028237 (84) | RP11-410D17.2 (Y) | rs6499994 (A>G) | 0.09 | 0.71 | 0.91 (0.89-0.93) |
8.57E-08 | 28/4 | CTB-134F13.1 (−791) RP11-430C1.1 (+861) |
| 16q22.1 | chr16:67625872-68029739 (404) | RP11-167P11.2 (Y) CTC-479C5.10 (Y) CTC-479C5.17 (N) AC009095.4 (Y) |
rs12325430 (T>C) | 0.44 | 0.75 | 0.92 (0.90-0.93) |
2.91E-07 | 2/2 | RP11-167P11.2 (0) |
| chr16:67950613-68429047 (478) | RP11-96D1.5 (Y) RP11-96D1.9 (N) RP11-96D1.6 (Y) RP11-96D1.7 (Y) RP11-96D1.10 (Y) RP11-96D1.11 (Y) RP11-96D1.3 (Y) RP11-67A1.2 (Y) CTC-479C5.6 (N) CTC-479C5.17 (Y) |
chr16:68187782 (CT>C) | 0.40 | 0.73 | 0.92 (0.90-0.93) |
2.10E-07 | 11/1 | RP11-167P11.2 (177) |
|
| 19q13.12-13 | chr19:38201712-38474127 (272) | CTD-2554C21.3 (N) CTD-2554C21.2 (Y) CTD-2528L19.6 (N) CTC-244M17.1 (N) AC016582.2 (N) |
chr19:38451511 (TA>T) | 0.14 | 0.81 | 1.12 (1.09-1.15) |
5.74E-06 | 9/7 | AC012309.5 (459) |
lncRNAs in bold contain SNP hits within their coordinates and non-bolded lncRNAs are in LD with SNP hits
MAF=Minor-alllele frequency; R2=Imputation quality r2
Figure 3.
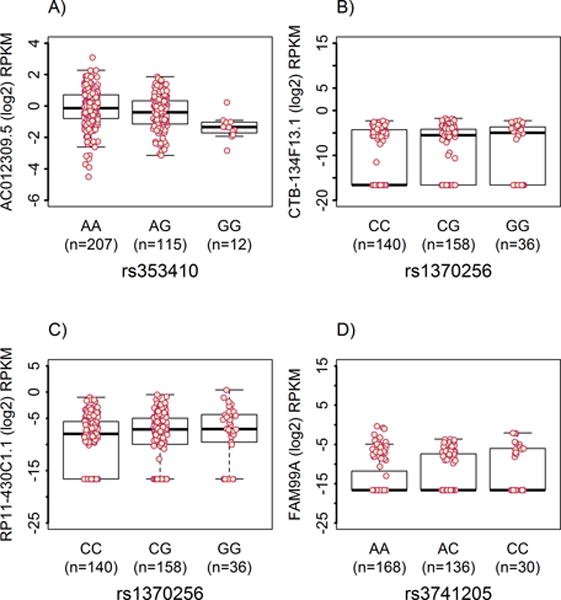
Selected boxplots for significant eQTL within novel regions associated with EOC risk
DISCUSSION
Evidence for a prominent role of lncRNA in carcinogenesis is rapidly accumulating (56). This study represents the first genome-wide evaluation of germline lncRNA variants in epithelial ovarian cancer susceptibility. We performed an enrichment analysis of genome-wide association data from 46,213 subjects and show that lncRNA regions are significantly enriched for EOC risk loci (P<10−5) with a two-fold higher proportion of risk variants than across the whole genome. Moreover, among the 40% of lncRNA genes with active epigenetic regulation in ovarian tissue, the risk variant enrichment was five-fold higher than whole genome. This high concentration of risk loci at ovarian-active lncRNAs aligns with previous studies that have shown an overrepresentation of disease-associated variants at enhancers(57) and within tissue-specific long-intergenic ncRNAs (lincRNAs)(28). Similar to these studies(28, 57), we focused on identifying lncRNA activity based on epigenomic profiles and did not focus on analyzing lncRNAs expressed in ovarian tissue that did not have active histone modification marks. Although this approach may have missed areas of lncRNA risk SNP enrichment, the subset of ovarian-active lncRNAs we analyzed were regulatory specific and potentially include overlapping enhancer and lncRNA sequences that could be functionally interrelated given that lncRNA expression associates with tissue-specific enhancers(27) and lncRNAs can mediate enhancer function(26, 58, 59). Taken together, our findings provide further support for a predominant regulatory role of EOC risk variants(57, 60) and reveal that lncRNAs may account for a significant proportion of such variation, particularly where tissue-specific regulatory elements are present. Further mechanistic studies are needed to confirm these findings.
Our pathway analysis demonstrates that lncRNAs containing EOC risk SNPs within their coordinates are enriched for developmental and regulatory pathways relevant to ovarian cancer pathogenesis. While the majority of pathways were developmental, most EOC-associated lncRNAs were expressed in adult tumor and/or normal ovarian tissues suggesting their role in EOC development likely extends beyond developmental pathways. Our ‘upstream’ enrichment analyses revealed that EOC-associated lncRNAs were enriched for putative targets of AR, STAT3, and PAX8 transcription factors (TF), all of which have been implicated in EOC pathogenesis. Androgen receptor is expressed in most ovarian tumors, and androgens promote ovarian tumor growth(61, 62) although prospective studies have not identified a clear association between androgens and EOC risk(63). STAT3 is over-activated in ovarian cancer cells and inhibition is subsequently accompanied by tumor growth suppression(64). Additionally, the Jak/STAT3 pathway has been linked with cancer cell survival and chemoresistance(65, 66) and recent work suggests germline polymorphisms within STAT3 predict poor response to platinum-based therapy(67). Lastly, PAX8 is a member of the paired box family of transcription factors (PAX1-9) that are primarily expressed in the embryo with persistent expression observed in ovarian tumors(68). PAX8 specifically is expressed in fallopian tube secretory epithelial and ovarian surface epithelial cells(69) and in vitro knockdown of PAX8 expression reduces ovarian cancer cell proliferation, migration, and invasion(70). Importantly, a recent gene set enrichment analysis (GSEA) of genome-wide association data revealed enrichment of putative PAX8 targets near serous EOC risk loci(71). Our analysis correlates well with the GSEA finding and shows that this enrichment of putative PAX8 targets can also be observed near the subset of invasive EOC risk loci that overlap lncRNAs. Given that several lncRNAs can alter the binding and/or activity of transcription factors as well as interact with them directly(72), it is possible that EOC-associated lncRNA variants may influence these transcription factors. Further studies evaluating the role of lncRNA risk variants could help elucidate the underlying etiology of EOC susceptibility and possibly identify opportunities for therapeutic intervention.
Our integration of GWAS, GENCODE, and TCGA gene expression data identified five novel sub-genome-wide regions with suggested functional effects on lncRNA targets. These novel regions contained common SNPs (MAF > 0.10) with small effect sizes that may represent true associations previously undetected due to limited power. Studies well-powered (≥ 80% power) to detect an odds ratio ≥ 1.1 among common SNPs (assuming a rare disease prevalence of 1.4%) would require a sample size of 110,176 matched cases and controls, almost double the sample size of previous EOC GWAS(6, 12). Thus for such small effects, sample size is a rate-limiting step and we demonstrate an integrative approach to select and provide biological support for candidate loci in the absence of increased sample sizes. Our in silico biological investigations consisted of eQTL analyses of lncRNA expression data to identify candidate loci although this was also limited by sample size. As expected, the 12,727 lincRNAs analyzed were expressed at lower levels (average = 0.29 RPKM, median = 0.03 RPKM) compared to mRNAs (average = 21.09 RPKM, median = 3.88 RPKM) which makes detection of cis-eQTL more difficult(73). We also observed relatively small yet potentially relevant fold changes in lncRNA expression that further hampered eQTL detection. A more comprehensive eQTL analysis of lncRNA expression with the larger samples sizes needed to overcome the difficulty of low expression and small fold changes is warranted; including adjustment for copy number variation, methylation, and/or batch effects. Confirmation of candidate regions will require functional validation through analysis of allele-specific effects on the lncRNA and in vivo and in vitro studies to determine the lncRNAs role in the initiation and development of EOC.
In summary, the current study implicates SNPs in lncRNAs as plausible candidates for risk regions that show evidence of EOC association but fail to reach genome-wide statistical significance. Integrative molecular studies provide biological support for the hypothesis and reveal connections between germline and tissue-level expression.
Supplementary Material
Acknowledgments
We thank all of the women who participated in this research along with all of the researchers, clinicians, and staff who have contributed to the participating studies.
Financial Support
The Ovarian Cancer Association Consortium is supported by a grant from the Ovarian Cancer Research Fund thanks to donations by the family and friends of Kathryn Sladek Smith. Funding for this project was supported by the National Institute of Health and the Genetic Associations and Mechanisms in Oncology (GAME-ON), a NCI Cancer Post-GWAS Initiative (U19-CA148112 to T. A. Sellers). This study made use of data generated by the Wellcome Trust Case Control consortium that was funded by the Wellcome Trust under award 076113. In addition, we acknowledge the following agencies for funding of constituent studies: AOC/ACS: U.S. Army Medical Research and Materiel Command (DAMD17-01-1-0729, to D. Bowtell and A. Green), National Health & Medical Research Council of Australia, Cancer Councils of New South Wales, Victoria, Queensland, South Australia and Tasmania, Cancer Foundation of Western Australia; National Health and Medical Research Council of Australia (199600 and 400281)BAV: ELAN funds of the University of Erlangen-Nuremberg. BEL: Nationaal Kankerplan; DOV: U.S. National Cancer Institute (NCI) (R01-CA112523 and R01-CA87538, to M.A. Rossing); GER: German Federal Ministry of Education and Research, Programme of Clinical Biomedical Research (01 GB 9401) and the German Cancer Research Center; GRR: Roswell Park Cancer Institute Alliance Foundation (P30 CA016056, to K. Odunsi). HAW: U.S. National Institutes of Health (R01-CA58598, N01-CN-55424 and N01-PC-67001 to M. T. Goodman); HJO and HMO: Intramural funding; Rudolf-Bartling Foundation; HOC: Helsinki University Research Fund; HOP: US Army Medical Research and Material Command (DAMD17-02-1-0669 to R.B. Ness); US NCI (K07-CA080668 to F. Modugno; R01-CA095023 to R.B. Ness; P50-CA159981, to K. B. Moysich); NIH/National Center for Research Resources/General Clinical Research Center (M01-RR000056 to F. Modugno, R. B. Ness); LAX: American Cancer Society Early Detection Professorship (SIOP-06-258-01-COUN to B.Y. Karlan); National Center for Advancing Translational Sciences (NCATS) (UL1TR000124 to S.M. Dubinett); MAL: US NCI (R01- CA61107 to S. K. Kjaer); Danish Cancer Society (94-222-52); Mermaid I project; MAY: US NCI (R01-CA122443, P30-CA15083, P50-CA136393 to E.L. Goode); Mayo Foundation; Minnesota Ovarian Cancer Alliance; Fred C. and Katherine B. Andersen Foundation; MCC: Cancer Council Victoria; NHMRC (209057, 251533, 396414, and 504715, to G. Giles); MDA: US DOD Ovarian Cancer Research Program (W81XWH-07-0449 to M. Hildebrandt); NEC: US NCI (R01-CA54419 and P50-CA105009 to D. Cramer); US DOD (W81XWH-10-1-02802 to K.L. Terry); NHS: US NCI (UM1-CA176726 and R01-CA67262 to W. C. Willett); NJO: US NCI (K07 CA095666 and K22-CA138563 to E.V. Bandera, R01-CA83918 to S. Olson,, and Rutgers Cancer Institute of New Jersey Cancer Center Support Grant P30-CA072720)NOR: Helse Vest; The Norwegian Cancer Society; The Research Council of Norway; NTH: Radboud University Medical Centre; ORE: OHSU Foundation; OVA: Canadian Institutes of Health Research (MOP-86727 to N. Le); US NCI (R01CA160669 to L. S. Cook); POC: Pomeranian Medical University; POL: Intramural Research Program of the NCI; PVD: Herlev Hospitals Forskningsrad; Herlev Hospitals Forskningsrad; Danish Cancer Society; RMH: Cancer Research UK; SEA: Cancer Research UK (C490/A10119 and C490/A10124 to P. Pharoah); UK National Institute for Health Research Biomedical Research Centres at the University of Cambridge; SRO: Cancer Research UK (C536/A13086 and C536/A6689 to S. Banerjee); Imperial Experimental Cancer Research Centre (C1312/A15589 to S. Banerjee)STA: US NCI (U01-CA71966 and U01-CA69417 to A. S. Whittemore, R01-CA16056 to K. B. Moysich, K07-CA143047 to W. Sieh,); TOR: US NCI (R01-CA063678 to S. A. Narod, R01-CA063682 to H. A. Risch); UCI: US NCI (R01-CA058860 to H. A. Anton-Culver); Lon V Smith Foundation (LVS-39420 to H. A. Anton-Culver); UKO: The Eve Appeal (The Oak Foundation); National Institute for Health Research University College London Hospitals Biomedical Research Centre; UKR: Cancer Research UK (C490/A6187 to P. Pharoah); UK National Institute for Health Research Biomedical Research Centres at the University of Cambridge; USC: US NIH (P01-CA17054 to A. H. Wu, P30-CA14089 (to C. L. Pearce and S.J. Ramus), R01-CA61132 to M. C. Pike, N01-PC67010 and N01-CN025403 to R. K. Ross, R03-CA113148 and R03-CA115195 to C. L. Pearce,); California Cancer Research Program (00-01389V-20170, 2II0200); WOC: National Science Centren (N N301 5645 40); The Maria Sklodowska-Curie Memorial Cancer Center; Institute of Oncology (Warsaw, Poland).
Footnotes
Conflicts of Interest
None
References
- 1.Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117–30. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25:1329–33. doi: 10.1200/JCO.2006.09.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song H, Dicks E, Ramus SJ, Tyrer JP, Intermaggio MP, Hayward J, et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J Clin Oncol. 2015;33:2901–7. doi: 10.1200/JCO.2015.61.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramus SJ, Song H, Dicks E, Tyrer JP, Rosenthal AN, Intermaggio MP, et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J Natl Cancer Inst. 2015;107 doi: 10.1093/jnci/djv214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bahcall O. Common variation and heritability estimates for breast, ovarian and prostate cancers. Nat Genet. 2013 [Google Scholar]
- 6.Kuchenbaecker KB, Ramus SJ, Tyrer J, Lee A, Shen HC, Beesley J, et al. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat Genet. 2015;47:164–71. doi: 10.1038/ng.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371–84. 84e1–2. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolton KL, Tyrer J, Song H, Ramus SJ, Notaridou M, Jones C, et al. Common variants at 19p13 are associated with susceptibility to ovarian cancer. Nat Genet. 2010;42:880–4. doi: 10.1038/ng.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen K, Ma H, Li L, Zang R, Wang C, Song F, et al. Genome-wide association study identifies new susceptibility loci for epithelial ovarian cancer in Han Chinese women. Nature communications. 2014;5:4682. doi: 10.1038/ncomms5682. [DOI] [PubMed] [Google Scholar]
- 10.Goode EL, Chenevix-Trench G, Song H, Ramus SJ, Notaridou M, Lawrenson K, et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet. 2010;42:874–9. doi: 10.1038/ng.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Permuth-Wey J, Lawrenson K, Shen HC, Velkova A, Tyrer JP, Chen Z, et al. Identification and molecular characterization of a new ovarian cancer susceptibility locus at 17q21.31. Nature communications. 2013;4:1627. doi: 10.1038/ncomms2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pharoah PD, Tsai YY, Ramus SJ, Phelan CM, Goode EL, Lawrenson K, et al. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet. 2013;45:362–70. 70e1–2. doi: 10.1038/ng.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song H, Ramus SJ, Tyrer J, Bolton KL, Gentry-Maharaj A, Wozniak E, et al. A genome-wide association study identifies a new ovarian cancer susceptibility locus on 9p22.2. Nat Genet. 2009;41:996–1000. doi: 10.1038/ng.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelemen LE, Lawrenson K, Tyrer J, Li Q, Lee JM, Seo JH, et al. Genome-wide significant risk associations for mucinous ovarian carcinoma. Nat Genet. 2015;47:888–97. doi: 10.1038/ng.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheetham SW, Gruhl F, Mattick JS, Dinger ME. Long noncoding RNAs and the genetics of cancer. Br J Cancer. 2013;108:2419–25. doi: 10.1038/bjc.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer. 2011;10:38. doi: 10.1186/1476-4598-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy SK, Huang Z, Wen Y, Spillman MA, Whitaker RS, Simel LR, et al. Frequent IGF2/H19 domain epigenetic alterations and elevated IGF2 expression in epithelial ovarian cancer. Mol Cancer Res. 2006;4:283–92. doi: 10.1158/1541-7786.MCR-05-0138. [DOI] [PubMed] [Google Scholar]
- 18.Perez DS, Hoage TR, Pritchett JR, Ducharme-Smith AL, Halling ML, Ganapathiraju SC, et al. Long, abundantly expressed non-coding transcripts are altered in cancer. Hum Mol Genet. 2008;17:642–55. doi: 10.1093/hmg/ddm336. [DOI] [PubMed] [Google Scholar]
- 19.Richards EJ, Permuth-Wey J, Li Y, Chen YA, Coppola D, Reid BM, et al. A functional variant in HOXA11-AS, a novel long non-coding RNA, inhibits the oncogenic phenotype of epithelial ovarian cancer. Oncotarget. 2015;6:34745–57. doi: 10.18632/oncotarget.5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silva JM, Boczek NJ, Berres MW, Ma X, Smith DI. LSINCT5 is over expressed in breast and ovarian cancer and affects cellular proliferation. RNA Biol. 2011;8:496–505. doi: 10.4161/rna.8.3.14800. [DOI] [PubMed] [Google Scholar]
- 21.Tanos V, Prus D, Ayesh S, Weinstein D, Tykocinski ML, De-Groot N, et al. Expression of the imprinted H19 oncofetal RNA in epithelial ovarian cancer. Eur J Obstet Gynecol Reprod Biol. 1999;85:7–11. doi: 10.1016/s0301-2115(98)00275-9. [DOI] [PubMed] [Google Scholar]
- 22.Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer Lett. 2013;339:159–66. doi: 10.1016/j.canlet.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 23.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 24.Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–8. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- 25.Guil S, Esteller M. Cis-acting noncoding RNAs: friends and foes. Nat Struct Mol Biol. 2012;19:1068–75. doi: 10.1038/nsmb.2428. [DOI] [PubMed] [Google Scholar]
- 26.Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vucicevic D, Corradin O, Ntini E, Scacheri PC, Orom UA. Long ncRNA expression associates with tissue-specific enhancers. Cell Cycle. 2015;14:253–60. doi: 10.4161/15384101.2014.977641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amin V, Harris RA, Onuchic V, Jackson AR, Charnecki T, Paithankar S, et al. Epigenomic footprints across 111 reference epigenomes reveal tissue-specific epigenetic regulation of lincRNAs. Nature communications. 2015;6:6370. doi: 10.1038/ncomms7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akrami R, Jacobsen A, Hoell J, Schultz N, Sander C, Larsson E. Comprehensive analysis of long non-coding RNAs in ovarian cancer reveals global patterns and targeted DNA amplification. PLoS One. 2013;8:e80306. doi: 10.1371/journal.pone.0080306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Permuth-Wey J, Kim D, Tsai YY, Lin HY, Chen YA, Barnholtz-Sloan J, et al. LIN28B polymorphisms influence susceptibility to epithelial ovarian cancer. Cancer Res. 2011;71:3896–903. doi: 10.1158/0008-5472.CAN-10-4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung S, Nakagawa H, Uemura M, Piao L, Ashikawa K, Hosono N, et al. Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci. 2011;102:245–52. doi: 10.1111/j.1349-7006.2010.01737.x. [DOI] [PubMed] [Google Scholar]
- 32.Jendrzejewski J, He H, Radomska HS, Li W, Tomsic J, Liyanarachchi S, et al. The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc Natl Acad Sci U S A. 2012;109:8646–51. doi: 10.1073/pnas.1205654109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer KB, Maia AT, O’Reilly M, Ghoussaini M, Prathalingam R, Porter-Gill P, et al. A functional variant at a prostate cancer predisposition locus at 8q24 is associated with PVT1 expression. PLoS genetics. 2011;7:e1002165. doi: 10.1371/journal.pgen.1002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasmant E, Sabbagh A, Vidaud M, Bieche I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 2011;25:444–8. doi: 10.1096/fj.10-172452. [DOI] [PubMed] [Google Scholar]
- 35.Fasching PA, Gayther S, Pearce L, Schildkraut JM, Goode E, Thiel F, et al. Role of genetic polymorphisms and ovarian cancer susceptibility. Mol Oncol. 2009;3:171–81. doi: 10.1016/j.molonc.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–9. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2012;9:179–81. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 38.Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–74. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ravasi T, Suzuki H, Pang KC, Katayama S, Furuno M, Okunishi R, et al. Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res. 2006;16:11–9. doi: 10.1101/gr.4200206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibb EA, Vucic EA, Enfield KS, Stewart GL, Lonergan KM, Kennett JY, et al. Human cancer long non-coding RNA transcriptomes. PLoS One. 2011;6:e25915. doi: 10.1371/journal.pone.0025915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet. 2013;29:569–74. doi: 10.1016/j.tig.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 43.Lee PH, O’Dushlaine C, Thomas B, Purcell SM. INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics. 2012;28:1797–9. doi: 10.1093/bioinformatics/bts191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mi H, Lazareva-Ulitsky B, Loo R, Kejariwal A, Vandergriff J, Rabkin S, et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005;33:D284–8. doi: 10.1093/nar/gki078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, et al. Human genomics. The human transcriptome across tissues and individuals. Science. 2015;348:660–5. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.September 4, 2015; Available from www.gtexportal.org
- 50.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fuchsberger C, Abecasis GR, Hinds DA. minimac2: faster genotype imputation. Bioinformatics. 2015;31:782–4. doi: 10.1093/bioinformatics/btu704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu Rev Genomics Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li J, Han L, Roebuck P, Diao L, Liu L, Yuan Y, et al. TANRIC: An Interactive Open Platform to Explore the Function of lncRNAs in Cancer. Cancer Res. 2015;75:3728–37. doi: 10.1158/0008-5472.CAN-15-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Couch FJ, Wang X, McGuffog L, Lee A, Olswold C, Kuchenbaecker KB, et al. Genome-wide association study in BRCA1 mutation carriers identifies novel loci associated with breast and ovarian cancer risk. PLoS genetics. 2013;9:e1003212. doi: 10.1371/journal.pgen.1003212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yan X, Hu Z, Feng Y, Hu X, Yuan J, Zhao SD, et al. Comprehensive Genomic Characterization of Long Non-coding RNAs across Human Cancers. Cancer Cell. 2015;28:529–40. doi: 10.1016/j.ccell.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, et al. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature. 2013;494:497–501. doi: 10.1038/nature11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–4. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coetzee SG, Shen HC, Hazelett DJ, Lawrenson K, Kuchenbaecker K, Tyrer J, et al. Cell-type-specific enrichment of risk-associated regulatory elements at ovarian cancer susceptibility loci. Hum Mol Genet. 2015;24:3595–607. doi: 10.1093/hmg/ddv101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Modugno F. Ovarian cancer and polymorphisms in the androgen and progesterone receptor genes: a HuGE review. Am J Epidemiol. 2004;159:319–35. doi: 10.1093/aje/kwh046. [DOI] [PubMed] [Google Scholar]
- 62.Risch HA. Hormonal etiology of epithelial ovarian cancer, with a hypothesis concerning the role of androgens and progesterone. J Natl Cancer Inst. 1998;90:1774–86. doi: 10.1093/jnci/90.23.1774. [DOI] [PubMed] [Google Scholar]
- 63.Ose J, Fortner RT, Rinaldi S, Schock H, Overvad K, Tjonneland A, et al. Endogenous androgens and risk of epithelial invasive ovarian cancer by tumor characteristics in the European Prospective Investigation into Cancer and Nutrition. Int J Cancer. 2015;136:399–410. doi: 10.1002/ijc.29000. [DOI] [PubMed] [Google Scholar]
- 64.Cai L, Zhang G, Tong X, You Q, An Y, Wang Y, et al. Growth inhibition of human ovarian cancer cells by blocking STAT3 activation with small interfering RNA. Eur J Obstet Gynecol Reprod Biol. 2010;148:73–80. doi: 10.1016/j.ejogrb.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 65.Abubaker K, Luwor RB, Zhu H, McNally O, Quinn MA, Burns CJ, et al. Inhibition of the JAK2/STAT3 pathway in ovarian cancer results in the loss of cancer stem cell-like characteristics and a reduced tumor burden. BMC Cancer. 2014;14:317. doi: 10.1186/1471-2407-14-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quintas-Cardama A, Verstovsek S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res. 2013;19:1933–40. doi: 10.1158/1078-0432.CCR-12-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Permuth-Wey J, Fulp WJ, Reid BM, Chen Z, Georgeades C, Cheng JQ, et al. STAT3 polymorphisms may predict an unfavorable response to first-line platinum-based therapy for women with advanced serous epithelial ovarian cancer. Int J Cancer. 2016;138:612–9. doi: 10.1002/ijc.29799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muratovska A, Zhou C, He S, Goodyer P, Eccles MR. Paired-Box genes are frequently expressed in cancer and often required for cancer cell survival. Oncogene. 2003;22:7989–97. doi: 10.1038/sj.onc.1206766. [DOI] [PubMed] [Google Scholar]
- 69.Adler E, Mhawech-Fauceglia P, Gayther SA, Lawrenson K. PAX8 expression in ovarian surface epithelial cells. Hum Pathol. 2015;46:948–56. doi: 10.1016/j.humpath.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Di Palma T, Lucci V, de Cristofaro T, Filippone MG, Zannini M. A role for PAX8 in the tumorigenic phenotype of ovarian cancer cells. BMC Cancer. 2014;14:292. doi: 10.1186/1471-2407-14-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kar SP, Adler E, Tyrer J, Anton-Culver H, Bandera EV, Beckmann MW, et al. Enrichment of putative PAX8 target genes at serous epithelial ovarian cancer susceptibility loci. Br J Cancer. 2016 doi: 10.1038/bjc.2016.426. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013;14:699–712. doi: 10.1038/nrm3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kumar V, Westra HJ, Karjalainen J, Zhernakova DV, Esko T, Hrdlickova B, et al. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS genetics. 2013;9:e1003201. doi: 10.1371/journal.pgen.1003201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.