

Abstract
To elucidate the function of a gene in bacteria it is vital that targeted gene inactivation (allelic replacement) can be achieved. Allelic replacement is often carried out by disruption of the gene of interest by insertion of an antibiotic-resistance marker followed by subsequent transfer of the mutant allele to the genome of the host organism in place of the wild-type gene. However, due to their intrinsic resistance to many antibiotics only selected antibiotic-resistance markers can be used in members of the genus Burkholderia, including the Burkholderia cepacia complex (Bcc). Here we describe the construction of improved antibiotic-resistance cassettes that specify resistance to kanamycin, chloramphenicol or trimethoprim effectively in the Bcc and related species. These were then used in combination with and/or to construct a series enhanced suicide vectors, pSHAFT2, pSHAFT3 and pSHAFT-GFP to facilitate effective allelic replacement in the Bcc. Validation of these improved suicide vectors was demonstrated by the genetic inactivation of selected genes in the Bcc species Burkholderia cenocepacia and B. lata, and in the non-Bcc species, B. thailandensis.
Abbreviations: BHR, broad host-range; MCS, multiple cloning site; TIR, translation initiation region; CmR, chloramphenicol resistance/resistant; CmS, chloramphenicol sensitive; KmR, kanamycin resistance; TcR, tetracycline resistance; TpR, trimethoprim resistance
Keywords: Marked mutation, Gene inactivation, Burkholderia cepacia complex, Suicide vector, Antibiotic-resistance
Highlights
-
•
We have constructed antibiotic-resistance cassettes and suicide vectors for use in Burkholderia and related species.
-
•
These vectors facilitate construction of mutants by gene disruption with antibiotic-resistance markers.
-
•
We have validated the utility of the vectors for marked genetic inactivation in members of the genus Burkholderia.
1. Introduction
Burkholderia cepacia complex (Bcc) are a group of at least twenty closely related Gram-negative bacterial species of the Burkholderia genus, which are phenotypically similar but genetically different (Vandamme et al., 1997, De Smet et al., 2015). These species are found ubiquitously in the environment, including soil, water and the rhizosphere of some plants (Coenye and Vandamme, 2003). Many Bcc species have also been isolated from the sputum of cystic-fibrosis (CF) patients, identifying them as opportunistic pathogens of those with lung disease and weakened immune systems (Henry et al., 1997, Speert et al., 2002). Colonisation of the lungs by Bcc can result in several clinical outcomes, including chronic-infection with gradual lung deterioration and a rare adverse reaction known as cepacia syndrome, which leads to rapid lung decline, septicaemia, necrotizing pneumonia and is often fatal (Isles et al., 1984). The most prevalent causative agents of Bcc infections in CF-patients in the UK, Canada and USA are B. cenocepacia and B. multivorans (Drevinek and Mahenthiralingam, 2010, De Soyza et al., 2010, Zlosnik et al., 2015). Unfortunately, Bcc infections can be difficult to treat due to the high intrinsic resistance of these bacteria to many antibiotics. Over recent years restrictions have been placed on individuals colonized with Bcc to prevent transmission of Bcc infections, by limiting their contact with at risk members of the population (Ledson et al., 1998).
Efforts have been made to understand the virulence mechanisms used by Bcc bacteria that allow them to be successful pathogens. These include mechanisms of bacterial cell invasion, intracellular survival, quorum sensing, iron acquisition, and protein secretion (Martin and Mohr, 2000, Sokol et al., 2003, Visser et al., 2004, Mullen et al., 2007, Aubert et al., 2008, Uehlinger et al., 2009, Somvanshi et al., 2010). A key factor to aid in the characterization of these traits is the ability to manipulate the genome of Bcc species. Methods for targeted gene inactivation in Burkholderia have largely relied on the disruption of the target chromosomal gene with an antibiotic-resistance marker, either through employing an integrative vector (Flannagan et al., 2007, Chapalain et al., 2013) or by allelic replacement whereby the wild type gene is replaced by a copy of the target gene that is inactivated with an antibiotic-resistance cassette (Huber et al., 2001, Malott et al., 2005, Agnoli et al., 2006). In the latter, two recombination crossover events are selected for (one occurring either side of the lesion in the target gene) and no vector sequences remain on the chromosome in the mutant. Vectors for generation of such marked mutants include plasmids in the pEX18 and pEX19 series (Hoang et al., 1998), and a vector constructed in our lab, pSHAFT (Agnoli et al., 2006).
Due to their high level of intrinsic resistance to many antibiotics, the number of commonly employed resistance markers that can be used for genetic manipulation of Bcc species is generally restricted to those specifying increased resistance to trimethoprim, chloramphenicol and tetracycline. All three markers can be selected for in single copy in B. cenocepacia (Farmer and Thomas, 2004, Asghar et al., 2011, Ryan et al., 2013). For aminoglycoside-sensitive strains, whether naturally occurring (such as B. cenocepacia strain H111) or engineered, the aac and aph markers, which specify resistance to the aminoglycosides gentamycin and kanamycin, respectively, are selectable in single copy and can therefore also be used in allelic replacement experiments (Huber et al., 2001, Hamad et al., 2010, Inhülsen et al., 2012, Carlier et al., 2014).
Allelic replacement in bacteria involving disruption of a target gene with a selectable resistance marker can be problematic if the host bacterium already exhibits a high degree of intrinsic resistance to the antibiotic, thereby precluding selection of the marker. This can be ameliorated if the antibiotic-resistance gene is efficiently expressed. However, the use of strong promoters to drive high-level transcription of the marker gene can exert detrimental polar effects that include destabilisation of plasmids housing such markers or unwanted or toxic expression of downstream genes following insertion of the marker in the host genome (Stueber and Bujard, 1982, Stassi and Lacks, 1982, Schrecke et al., 2013; our published observations). In these situations the negative effects of very active promoters can be circumvented by placement of a strong transcription terminator downstream of the selectable marker gene (Gentz et al., 1981, Fellay et al., 1987). Here we describe the construction of novel antibiotic-resistance cassettes where we have employed these principles either to increase expression of the antibiotic-resistance marker to facilitate its selection in a single copy or to prevent detrimental polar effects by blocking transcriptional read through into downstream genes. These cassettes were used to build improved suicide vectors derived from the vector pSHAFT and were also used in conjunction with these vectors for allelic replacement in the Bcc and other members of the genus Burkholderia.
2. Materials and methods
2.1. Strains, plasmids, and growth conditions
The bacterial strains and plasmids used in this study are given in Supplementary Table S1. For cultivation of bacteria, strains were routinely grown in/on LB medium at 37 °C. For selection of trimethoprim resistance in E. coli isosensitest agar (Oxoid) was employed, whereas for B. cenocepacia M9-minimal salts agar containing 0.5% glucose was used (except where indicated). M9-minimal salts used here contained 42 mM Na2HPO4, 22 mM KH2PO4, 19 mM NH4Cl, 9 mM NaCl, 1 mM MgSO4 and 100 μM CaCl2. For selection of kanamycin resistance in B. cenocepacia Lennox agar was utilised. Antibiotics were used, when appropriate, at the following concentrations for selection of plasmids and antibiotic-resistance cassettes in E. coli and Burkholderia species, as indicated: ampicillin, 100 μg/ml (E. coli); kanamycin, 50 μg/ml (E. coli and B. cenocepacia); chloramphenicol, 25 μg/ml (E. coli) and 50 μg/ml (B. cenocepacia); trimethoprim, 25 μg/ml (E. coli, B. cenocepacia and B. lata) and 50 μg/ml (B. thailandensis). For B. cenocepacia strains exhibiting a high intrinsic resistance to trimethoprim, such as K56-2, this antibiotic was included in the medium at 100 μg/ml.
2.2. DNA preparation and manipulation
Recombinant DNA techniques were performed essentially as described in Sambrook et al. (1989). DNA amplification by PCR was performed as standard according to the manufacturer's instructions using KOD DNA polymerase enzyme (Millipore) and a G-storm thermocycler. Primers used in this study are indicated in Supplementary table S2, and were purchased from Eurogentec, Belgium. To extract DNA for PCR, bacterial colonies were resuspended in 200 μl TE buffer (10 mM Tris, 1.0 mM EDTA (pH 8.0)), boiled for 7 min, and then centrifuged to pellet cell debris. The supernatant was retained for use in PCR (referred to as ‘boiled lysate’). PCR products were purified from solution or by agarose gel-extraction using a QIAquick PCR purification kit (Qiagen). DNA restriction enzymes were purchased from Promega or New England Biolabs. DNA was ligated using T4 DNA ligase (Promega). 5′ DNA overhangs were filled-in using DNA I polymerase Klenow fragment (Promega). Sequencing was performed by the Core Genomic Facility at The University of Sheffield, UK.
2.3. Plasmid constructions
Plasmids were extracted from saturated bacterial cultures using a spin-column-based plasmid mini-prep extraction kit (Thermo Scientific). Plasmids were transferred into E. coli by heat‐shock assisted transformation according to Hanahan (1983).
To construct p34E-Km, the Tn5-derived aphA2 gene, including its promoter (Rothstein and Reznikoff, 1981), was excised from pKNOCK-Km as a MluI fragment, end-filled with DNA polymerase I Klenow fragment and ligated between the filled EcoRI sites of p34E-Tp.
To generate p34E-Cm2, the catA2 gene of the plasmid pSHAFT was amplified with primers p34E-Cmfor2 and p34E-Cmrev, which incorporated a synthetic promoter (− 10 and − 35 elements) upstream of the catA2 TIR in the amplified product. The 823 bp amplicon was restricted with EcoRI and used to replace the EcoRI-excised KmR cassette of p34E-Km.
To construct p34E-TpTer, first a 306 bp DNA fragment containing the rrnB T1 T2 terminators was amplified from the plasmid pEA302T with rrnBterfor and rrnBterrev and restricted with BamHI and HindIII, following which it was ligated between the corresponding restriction sites of pUC18 to give pUC18Ter. Second, the megaprimer variation of SOE PCR (Perrin and Gilliland, 1990) was used to fuse the rrnB T1 T2 terminators to the 3′ end of dfrB2. To do this, the dfrB2 gene (and native promoter) was amplified from p34E-Tp with primer Tp(forward), and the fusion primer Tp(reverse) that was complementary to the 3′ end of the dfrB2 gene, including the stop codon, and contained a 5′ tail with 19 bp of homology to the upstream region of the rrnB T1 T2 DNA fragment. The resultant amplicon served as a forward megaprimer in combination with reverse primer rrn(R), to amplify a 903 bp dfrB2-rrnB T1BT2 fusion fragment from pUC18Ter. This product was then ligated between the EcoRI and HindIII sites of pUC19, giving rise to pUC19TpTer. Last, the TpTer cassette of pUC19TpTer was excised as a StuI fragment and ligated between the filled-in EcoRI sites of p34E-Km, thereby replacing the KmR cassette.
To construct pSHAFT2, the catA2 region of pSHAFT was amplified with a pair of primers, pSHOOTERfor2 and pSHOOTERrev. The 871 bp product was digested with BamHI and SalI and ligated between the BglII and SalI sites of pSHAFT, thereby replacing the Ω-Cm interposon.
To construct pSHAFT3, first, a double-stranded oligonucleotide was generated by annealing oligonucleotides pSHAFT3MCSfor and pSHAFT3MCSrev. This was achieved by combining 45 μM of each oligonucleotide in 1 × KOD DNA polymerase reaction buffer (Millipore) and 1 mM MgCl2, incubating at 90 °C for 10 min followed by incubation at room temperature for 1 h and subsequent purification of the double-stranded oligonucleotide. pSHAFT2 was restricted with NotI and EcoRI and ligated to the double-stranded oligonucleotide, to generate pSHAFT3.
To construct pSHAFT-GFP, an 866 bp DNA fragment containing the gfp coding sequence and Shine-Dalgarno was amplified from pBHR1-GFP with primers GFPfor and GFPrev, purified and digested with BamHI and SalI. This was then ligated between BglII-SalI sites of pSHAFT to replace the 3.7 kb Ω-Cm fragment of this vector.
The sequences of p34E-Km, p34E-Cm2, p34E-TpTer, pSHAFT, pSHAFT2, pSHAFT3 and pSHAFT-GFP were deposited in GenBank database, and given the accession numbers KX485327, KX485328, KX485329, KX485330, KX485332, KX485333 and KX485331, respectively. As we observed that the nucleotide sequence of the dfrB2 cassette in p34E-Tp as deposited in the database is incorrect, we have also submitted an amended sequence of this plasmid that has been assigned accession number KX485326. pUC18Ter and pUC19TpTer were assigned accession numbers KX527623 and KX527624, respectively.
2.4. Gene replacement in B. cenocepacia
To inactivate chromosomal genes in Burkholderia using pSHAFT2 or pSHAFT3, the antibiotic counter-selection-based strategy used for allelic replacement by pSHAFT was employed (Agnoli et al., 2006). For isolation of a B. cenocepacia BCAM0195::Tp mutant, a 3.64 kb DNA fragment containing the BCAM0195 gene orthologue in strain H111 was amplified from a boiled lysate using primers BCAM0195for and BCAM0195rev, restricted with HindIII and BamHI and ligated between the corresponding sites of pBBR1MCS-1, generating pBBR1-BCAM0195’. The cloned 3.11 kb gene fragment was then disrupted by ligation of the p34E-Tp-derived TpR cassette as a SmaI-fragment between two PmlI sites located 1.58 kb apart within BCAM0195, generating pBBR1-ΔBCAM0195'::Tp. The 2.04 kb ΔBCAM0195'::Tp allele was transferred to pSHAFT2 as a XhoI-XbaI fragment to give pSHAFT2-ΔBCAM0195'::Tp. This plasmid was conjugated into B. cenocepacia H111 using E. coli donor strain S17-1(λpir) according to Herrero et al. (1990) and de Lorenzo and Timmis (1994), and B. cenocepacia were selected on M9-glucose agar containing trimethoprim. Exconjugants were patched onto the same medium and also LB agar containing chloramphenicol. Exconjugants that were chloramphenicol-sensitive were identified as candidate ΔBCAM0195::Tp mutants. Due to the loss of short regions of DNA at the 5′ and 3′ ends of BCAM0195 during the construction of pSHAFT2-ΔBCAM0195'::Tp, potential ΔBCAM0195::Tp mutants could be verified by PCR using the original BCAM0195for and BCAM0195rev primers, as they annealed to genomic sequences located a short distance either side of the region of DNA that was also present in pSHAFT2-ΔBCAM0195'::Tp.
For isolation of a BCAL1709::TpTer mutant in B. cenocepacia AHA27, a 1.354 kb DNA fragment containing the 3′ region of the BCAL1709 orthologue was amplified from B. cenocepacia Pc715j genomic DNA using primers BCAL1709for and BCAL1709rev, restricted with XbaI and XhoI, and the resulting 1.297 kb amplicon was ligated between the corresponding sites of pSHAFT-GFP to give rise to pSHAFT-GFP-BCAL1709. The BCAL1709 gene was then disrupted by ligation of the p34E-TpTer-derived TpR cassette as a SmaI fragment into a unique ZraI site within BCAL1709, resulting in pSHAFT-GFP-BCAL1709::TpTer. This plasmid was then conjugated into AHA27 and exconjugants containing the BCAL1709::TpTer allele within the genome were selected for as described for construction of the BCAM0195::Tp mutant. The presence of gfp on pSHAFT-GFP causes recipient colonies to fluoresce under UV light, and was used to distinguish between recombinants that arose through a single crossover (i.e. plasmid integration - fluorescent) and a double crossover (i.e. allelic replacement - non-fluorescent). 50 trimethoprim-resistant exconjugants were patched on duplicate IST agar plates containing trimethoprim. One of each pair of plates was exposed to UV light on a transilluminator in the dark to identify non-fluorescent candidate AHA27 BCA1709::TpTer mutants. Candidate mutants from the non-irradiated duplicate plate(s) were verified by PCR using primers BCAL1709forOut and BCAL1709revOut, which annealed to genomic sequences a short distance either side of the region of DNA that was also present in pSHAFT-GFP-BCAL1709::TpTer.
3. Results and discussion
3.1. Construction of antibiotic resistance cassettes for genetic manipulation of Burkholderia
To facilitate the generation of mutants in Burkholderia through insertional inactivation of chromosomal target genes by antibiotic resistance markers and the construction of useful plasmid vectors that would allow an investigation into gene function and control in members of this genus, we made derivatives of the cassette cloning vector, p34E (Tsang et al., 1991), harbouring the kanamycin- (aphA2), chloramphenicol- (catA2) and trimethoprim- (dfrB2), resistance genes.
First, p34E-Km was constructed by replacing the trimethoprim-resistance (TpR) cassette of p34E-Tp with the Tn5-derived aphA2 (aph(3′)-II) gene from pKNOCK-Km (Fig. 1). Although a similar kanamycin-resistance cassette (KmR) plasmid, p34S-Km (which contains the Tn903-derived aph(3′)-Ia gene), has been previously described (Dennis and Zylstra, 1998a), the Tn5-derived antibiotic resistance marker in p34E-Km offers the advantage that it makes available the HindIII and SmaI sites flanking the cassette (as they do not cut within the aphA2 gene) and also contains flanking EcoRI sites. Both types of cassette are of a similar size (~ 1.3 kb). A shorter variant of the Tn903-based KmR cassette lacking the internal HindIII and SmaI sites was subsequently constructed, but it lacks the flanking EcoRI sites and the possibility for directional cloning associated with p34E-Km (Dennis and Zylstra, 1998b).
Fig. 1.

Maps of novel antibiotic-cassette vectors. The location of antibiotic resistance-conferring genes, bla (ampicillin-resistance), aphA2 (kanamycin-resistance), catA2 (chloramphenicol-resistance) and dfrB2 (trimethoprim-resistance), and the plasmid ColE1 origin of replication (oriR) are indicated for plasmids p34E-Km (top), p34E-Cm2 (left) and p34E-TpTer. Dual-cutting restriction sites that can be utilised to excise the antibiotic resistance cassette are also shown, as are internal sites that can be used for determination of the orientation of the cassette following insertion into a target gene. Restriction sites that occur only once in each plasmid are shown in bold. Note that additional sites for AseI, BsrGI, NheI and XmnI occur in the backbone of all three vectors that for clarity are not shown. Promoters for the cassette antibiotic-resistance genes are shown (Paph, Pcat, PcS). Maps created with SnapGene® software (from GSL Biotech; available at snapgene.com).
Most plasmids used for genetic manipulation that specify chloramphenicol resistance (CmR) harbour the Tn9-derived catA1 gene (or one that is closely related) that specifies an enzyme belonging to group 1 of the type A chloramphenicol acetyltransferases (CATs) (Shaw, 1983, Schwarz et al., 2004). A notable exception is mini-Tn5Cm, which contains the catA2 gene (de Lorenzo et al., 1990). Pertinently, chloramphenicol resistance conferred by this mini-transposon is selectable in single copy in B. cenocepacia (Farmer and Thomas, 2004). However, the catA2 gene specifying this resistance is located on a large DNA fragment (the ~ 3.7 kb Ω-Cm interposon) that was used to assemble the transposon and is not available as a small cassette (Fellay et al., 1987, de Lorenzo et al., 1990). The Ω-Cm interposon includes a 2.8 kb DNA fragment derived from the cloning vector pKT210 which contains the 214 codon catA2 ORF and several other predicted ORFs found on the naturally occurring BHR plasmid pSa (Bagdasarian et al., 1981, Tait et al., 1982, Shaw, 1983, Fellay et al., 1987). In order to generate a compact CmR cassette containing an efficiently expressed cat gene, the catA2 gene contained on pSHAFT (a suicide plasmid derived from pUTmini-Tn5Cm (see below)), was modified to incorporate a synthetic σ70-dependent promoter upstream of the catA2 TIR. The modified catA2 gene was used to replace the KmR cassette of p34E-Km, to generate p34E-Cm2 (Fig. 1). The CmR cassette in p34E-Cm2, can be transferred to other vectors using any of the flanking restriction sites except SmaI (Fig. 1), although even here, selection for chloramphenicol resistance would permit selection for the products of a tripartite ligation between the target vector and the two SmaI-generated cassette fragments. As an alternative, either of the blunt end-generating SacI isoschizomers, Eco53kI or Ecl136II, may be used.
The most similar vectors available as sources of a CmR cassette are p34S-Cm and -Cm2 (Dennis and Zylstra, 1998a, Dennis and Zylstra, 1998b). Rather than the catA2 gene present in p34E-Cm2, these other plasmids contain the Tn9-derived catA1 gene and the cassettes are slightly longer (918 bp rather than 823 bp). p34S-Cm contains an internal EcoRI site whereas in p34S-Cm2 this site has been removed. However, unlike p34E-Cm2, p34S-Cm2 lacks flanking EcoRI sites.
The trimethoprim-resistance gene (dfrB2) present on the cassette vectors p34E-Tp and p34S-Tp is under control of the very strong PcS integron promoter (also known as P1) (Lévesque et al., 1994, DeShazer and Woods, 1996, Dennis and Zylstra, 1998a, Jové et al., 2010). We have observed that insertion of the TpR cassette in certain orientations in some plasmid vectors is not possible (see below) and when used in allelic replacement experiments it can result in impaired growth of the resultant mutants, presumably due to overexpression of chromosomal genes located downstream of the integrated cassette (S.S. and M.S.T., unpublished observations). To circumvent this problem, we modified the TpR cassette by placing the efficient T1 T2 transcription terminators from the E. coli rrnB ribosomal RNA operon, downstream of the dfrB2 coding sequence (Brosius, 1984, Orosz et al., 1991). To do this, we started with pEA302T, a plasmid into which a 500 bp EcoRI fragment containing the 5S rRNA gene and the T1 T2 terminators was cloned in order to prevent read through transcription of the replication region (Amann et al., 1983). A DNA fragment containing only the rrnB T1 T2 terminators was amplified using primers that also incorporated additional restriction sites at the flanking regions of the fragment, and was subsequently transferred to pUC18 to give pUC18Ter.
The rrnB terminators (from pUC18Ter) were then fused to the dfrB2 gene (from p34E-Tp) by the megaprimer variation of the SOE PCR technique (Perrin and Gilliland, 1990), and the amplicon was ligated into pUC19, giving rise to pUC19TpTer. Although the TpTer DNA fragment in pUC19TpTer can be transferred as a StuI or NdeI cassette, to increase its versatility, it was transferred into p34E-Km, substituting for the KmR cassette. The resultant plasmid, p34E-TpTer, is analogous to p34E-Tp but has strong transcription termination signals located downstream of the dfrB2 gene and includes the addition of NdeI sites in the flanking MCSs (Fig. 1).
3.2. Construction of allelic replacement vectors, pSHAFT2 and pSHAFT3, for generation of marked mutants in Burkholderia
We sought to improve upon existing vectors used for allelic replacement in the Bcc. A useful vector for insertional inactivation of chromosomal genes with selective markers (usually antibiotic resistance cassettes) is pSHAFT, an R6K-based suicide plasmid (Fig. 2) (Agnoli et al., 2006). This vector was derived from the transposon delivery plasmid, pUTmini-Tn5Cm (de Lorenzo et al., 1990), by deletion of the tnp (transposase) gene and one of the 19 bp repeat sequences (the ‘I end’) that flank mini-Tn5Cm, and therefore it cannot be mobilised in the presence of transposase provided in trans. pSHAFT is also devoid of the pir gene, which specifies the plasmid replication initiator protein, π, and can therefore only replicate in bacteria containing the pir gene, such as E. coli CC118(λpir) (Herrero et al., 1990, Rakowski and Filutowicz, 2013). Along with the chloramphenicol-resistance marker carried by mini-Tn5Cm, pSHAFT also contains the origin of transfer (oriT) from RP4 (RK2) that allows for efficient conjugal transfer of the plasmid to a variety of Gram-negative bacterial species. Following transfer of a Bcc gene (or gene fragment) to pSHAFT, the gene is disrupted by insertion an antibiotic-resistance cassette (other than CmR) that is selectable in the Bcc. The plasmid is then introduced into a Bcc strain and selection for the presence of the antibiotic-resistance cassette (AbR) is applied. As the plasmid cannot replicate in Bcc, only recombinants are obtained in which the plasmid has recombined with the host genome at the locus that is homologous to the Bcc DNA present on the plasmid. This may result in integration of the plasmid into the genome (single crossover, CmR AbR) or allelic replacement (double crossover, CmS AbR).
Fig. 2.

Maps of the gene replacement vectors of the pSHAFT-series utilised for marked mutagenesis in Burkholderia. The location of antibiotic-resistance conferring genes, bla (ampicillin-resistance) and catA2 (chloramphenicol-resistance), RP4 origin of transfer (oriT), R6K origin of replication (oriR6K) and GFP-encoding gene (gfp) are indicated for pSHAFT, (left), pSHAFT2 (top right), pSHAFT3 (centre right) and pSHAFT-GFP (bottom right). The transcriptional orientation for each gene and restriction sites within the multiple cloning site of each vector are shown. Additional restriction sites in pSHAFT that flank the Ω-Cm interposon (dashed line) are also indicated. Restriction sites that occur only once in each plasmid are shown in bold. Maps created with SnapGene® software (from GSL Biotech; available at snapgene.com).
However, the major drawback with pSHAFT is the limited number of unique restriction sites that can be used for inserting mutant alleles for subsequent chromosomal targeting. One reason for this is the duplication of restriction sites either side of the CmR element during the assembly of mini-Tn5Cm in the progenitor plasmid pUT (de Lorenzo and Timmis, 1994). Moreover, as discussed above, the cassette specifying resistance to chloramphenicol is unnecessarily large, as mini-Tn5Cm was originally constructed by inserting the ~ 3.7 kb Ω-Cm interposon between the 19 bp I and O ends of Tn5 carried by pUT (de Lorenzo et al., 1990).
To improve the utility of pSHAFT, the vector was modified by replacing the interposon with a much shorter DNA fragment containing the catA2 gene under control of a synthetic σ70-dependent promoter as also incorporated in p34E-Cm2, thereby decreasing the plasmid size from 7.5 to 4.6 kb. The new plasmid, pSHAFT2, contains 11 unique restriction sites located downstream of the catA2 gene, one of which (StuI) allows the cloning of blunt-ended fragments, while the BglII site can also accommodate BamHI-generated fragments. At the catA2-distal end of the MCS are three closely spaced EcoRI sites that can be considered as an additional single unique site for cloning purposes (Fig. 2).
The versatility of pSHAFT2 was then improved by replacing the region extending from the NotI site in the MCS to the most distant of the three EcoRI sites by a double-stranded oligonucleotide that contained internal SpeI and ApaI sites. This also resulted in a net loss of two of the three EcoRI sites present in pSHAFT2 and removal of the mini-Tn5 O end that originated from the progenitor plasmid of pSHAFT. The new plasmid, pSHAFT3 (4.5 kb), is shown in Fig. 2.
3.3. Construction of an allelic replacement vector, pSHAFT-GFP, that allows fluorogenic detection of integration events in Burkholderia species during generation of marked mutants
To allow for inactivation of chromosomal genes where the Burkholderia strain already harbours a chloramphenicol resistance marker or to disrupt chromosomal genes with a CmR cassette we constructed pSHAFT-GFP (Fig. 2). This plasmid is analogous to pSHAFT2 but with the catA2 gene replaced by the gfp gene, which therefore allows for fluorogenic detection of recombinants containing the genomically integrated vector. The forward primer used to amplify the gfp gene specified recognition sites for the restriction enzymes SalI, SmaI, BglII, XbaI, KpnI, XhoI and StuI and an artificial promoter containing the consensus − 35 and − 10 elements for σ70-dependent promoters, whereas the reverse primer specified only a BamHI site. This vector is used in the same way as pSHAFT2 except for the fact that single and double crossover recombinants harbouring the selectable marker used to inactivate the target chromosomal gene are distinguished from each other by screening colonies for the absence of yellow-green fluorescence. Although this usually requires exposure to UV in the dark (a UV transilluminator works well in this regard), in some mutagenesis experiments the presence of the GFP marker can be discerned without recourse to UV exposure by the yellow-green colour of the colonies.
3.4. Isolation of marked mutants in B. cenocepacia and related species using the pSHAFT-vector series
To inactivate chromosomal genes in Burkholderia using the pSHAFT-vector series, DNA fragments of ≥ 1.0 kb containing the target gene (or gene fragment) are inserted into the MCS and then subsequently disrupted by insertion of an antibiotic resistance cassette (usually TpR, KmR or TcR) such that at least 0.5 kb of homology occurs between the cloned DNA target region and the chromosome either side of the lesion (Fig. 3A). Depending on the availability of restriction sites, in some cases we have found it more convenient to first clone the target DNA sequence into a general-purpose plasmid vector and then introduce the antibiotic resistance cassette before transferring the disrupted DNA fragment to the suicide vector. In using these plasmids to inactivate chromosomal genes in Burkholderia, we have observed that the TpR cassette can often only be inserted into target genes in one orientation, such that transcription is directed towards the catA2 gene, and away from the origin of replication. We assume this is due to transcriptional destabilisation of plasmid replication functions (Gentz et al., 1981, Stueber and Bujard, 1982, Stassi and Lacks, 1982) as this problem does not occur with the TpTer cassette (our unpublished observations).
Fig. 3.
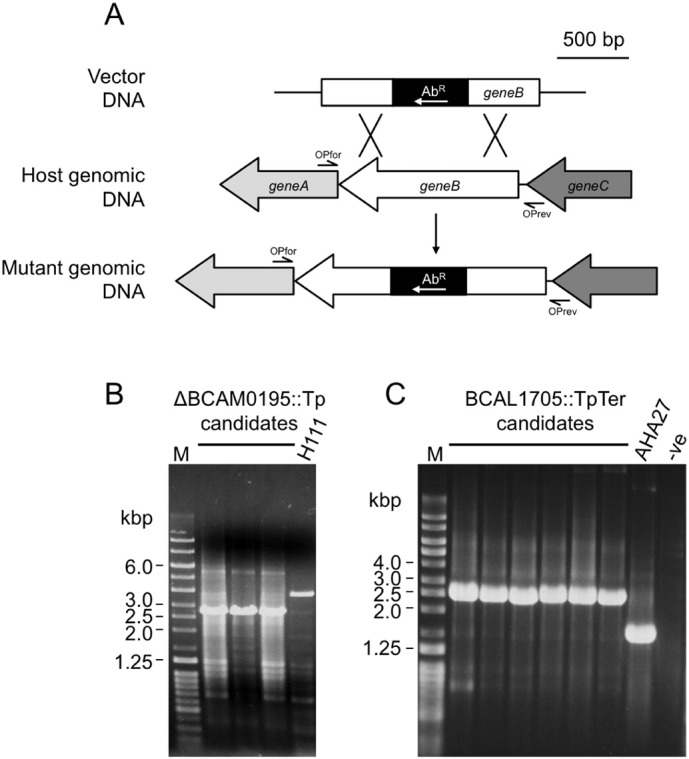
Generation of marked mutants in B. cenocepacia using pSHAFT2 and pSHAFT-GFP derivatives. (A) ≥ 1.0 kb of DNA containing the target gene (or gene fragment) is cloned into a pSHAFT vector and then disrupted by insertion of an antibiotic resistance cassette, ensuring there is at least 0.5 kb of homology between the cloned DNA target region and the chromosome on either side of the cassette. Following transfer of the pSHAFT-derived construct into Burkholderia, double crossover recombinants are selected for based on their resistance to the antibiotic specified by the antibiotic resistance cassette, and either sensitivity to chloramphenicol (pSHAFT2 and pSHAFT3) or the absence of fluorescence (pSHAFT-GFP). Candidate mutants are then verified by PCR using primers that anneal to genomic sequences located either side of the region cloned into the allelic replacement vector (‘outside’ primers), indicated as OPfor and OPrev. Drawn to scale. (B) PCR screening of candidate H111-ΔBCAM0195::Tp mutants following allelic replacement with pSHAFT2-ΔBCAM0195’::Tp. (C) PCR screening of candidate AHA27-BCAL1709::TpTer mutants following allelic replacement with pSHAFT-GFP-BCAL1709::TpTer.
Once the disrupted gene or gene fragment has been introduced into pSHAFT2, it is transferred to Burkholderia and selection is applied for the antibiotic resistance marker that was used to disrupt the gene or gene fragment present in the suicide vector, thus identifying strains that have integrated the disrupted allele into the genome by homologous recombination. Recombinants are of two types: those that are the result of a single crossover, in which the entire plasmid has integrated into the genome at the target gene locus, and those that are the result of a double crossover in which only the mutant allele has been transferred to the genome in place of the original wild type gene (Fig. 3A). In the former, wild type and mutated copies of the gene (or gene fragment) are present in the genome of the recipient bacterium, which will also specify increased resistance to chloramphenicol due to the integrated vector. These are identified by ‘patching’ out recombinants on LB agar containing 50 μg/ml chloramphenicol. Chloramphenicol-sensitive recombinants are screened for the presence of the mutated allele (and absence of the wild type allele) by PCR using primers that anneal to genomic sequences flanking the region that was originally cloned in the suicide vector.
We have successfully used pSHAFT2 to generate several marked mutants within Bcc species. This included the insertional inactivation of the I35_0520 gene in B. cenocepacia strain H111 (the orthologue of BCAM0195 in strain J2315 and will be referred to as such hereon), which was disrupted by the TpR cassette derived from p34E-Tp. BCAM0195 is predicted to encode a non-ribosomal peptide synthetase (NRPS) of unknown function. During the isolation of the ΔBCAM0195::Tp mutant, we observed that out of 50 trimethoprim resistant exconjugants, 16 were chloramphenicol sensitive. Three of these were verified as ΔBCAM0195::Tp mutants by PCR, where the DNA fragment amplified from BCAM0195 in the mutants was ~ 900 bp smaller than the wild-type, due to replacement of a large segment of BCAM0195 by the TpR cassette in the ΔBCAM0195::Tp allele (Fig. 3B). We have also used this plasmid to introduce an orbI::Tp allele into the genome of B. lata using the same selection conditions employed for allelic replacement in B. cenocepacia (results not shown). The applicability of the pSHAFT vectors to other non-Bcc species within the genus was established by generating a type VI secretion system mutant of B. thailandensis (a member of the ‘pseudomallei’ group) in an analogous fashion using pSHAFT3 as the vector and the TpTer cassette to disrupt the tssK gene (results not shown).
To demonstrate the utility of pSHAFT-GFP it was used to inactivate the orthologue of the B. cenocepacia J2315 BCAL1709 gene that is present in the siderophore-negative B. cenocepacia mutant AHA27. AHA27 is a derivative of B. cenocepacia strain Pc715j that contains a mini-Tn5CmlacZYA insertion in the pobA gene encoding a phosphopantetheinyl transferase (PPTase) required for activating NRPS enzymes, and thereby specifies higher levels of resistance to chloramphenicol than the parent strain (Asghar et al., 2011). Based on amino acid sequence alignment, BCAL1709 is very likely to encode a putative TonB-dependent receptor thought to be involved in the uptake of an unknown xenosiderophore complexed with ferric iron (M.S.T., unpublished results). Following cloning of a BCAL1709 gene fragment in pSHAFT-GFP and its disruption with the TpR cassette derived from p34E-TpTer, the resultant plasmid (pSHAFT-GFP-BCAL1709::TpTer) was introduced into AHA27 and candidate BCAL1709 mutants were selected by screening trimethoprim-resistant exconjugants for the absence of GFP-mediated fluorescence. Among 50 ex-conjugants that were screened in this way, 6 non-fluorescent recombinants were identified which were subsequently verified as BCAL1709 mutants by PCR, as indicated by a ~ 900 bp increase in the size of the DNA fragment amplified from the BCAL1709 locus due to the presence of the TpR cassette (Fig. 3C).
It should also be noted that use of the TpR cassette in allelic replacement experiments is also applicable to Burkholderia strains that exhibit higher levels of intrinsic resistance to trimethoprim, such as members of the B. cenocepacia ET-12 lineage, i.e. strains J2315 and K56-2. Due to the highly active promoter located upstream of the dfrB2 ORF, expression of the gene is sufficiently strong to allow its selection in such strains by increasing the concentration of trimethoprim in the medium, and we have used pSHAFT2 to transfer TpR cassette-disrupted iron acquisition and type VI secretion system genes into K56-2 (unpublished results).
4. Conclusion
To conclude, we have constructed a series of versatile suicide vectors and antibiotic resistance cassettes that allow for the efficient and simple generation of marked mutants in Burkholderia species. We have improved upon our original vector pSHAFT by reducing the size of the CmR marker and increasing the availability of unique restriction site for cloning in vectors pSHAFT2 and pSHAFT3. The versatility of this plasmid series has been further enhanced by incorporating a fluorescent marker in pSHAFT-GFP to provide an alternative means of distinguishing recombinants that have undergone allelic replacement events from those in which the suicide vector remains integrated in the genome. All vectors described here are a useful addition to the molecular toolkit required for the manipulation and subsequent characterization of important genes in not only Burkholderia species, but potentially other Proteobacteria, such as enterobacteria and pseudomonads, where they may be used in combination with the antibiotic-resistance cassettes described herein or other available antibiotic-resistance markers.
Acknowledgements
This work was supported by a BBSRC research grant (BB/M003531/1) awarded to M.S.T. and a scholarship from the University of Umm Al-qura awarded to A.H.A. that was administered through the Saudi Arabian Ministry of Higher Education.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.plasmid.2016.11.002.
Appendix A. Supplementary data
Bacterial strains, plasmids and oligonucleotides.
References
- Agnoli K., Lowe C.A., Farmer K.L., Husnain S.I., Thomas M.S. The ornibactin biosynthesis and transport genes of Burkholderia cenocepacia are regulated by an extracytoplasmic function sigma factor which is a part of the Fur regulon. J. Bacteriol. 2006;188(10):3631–3644. doi: 10.1128/JB.188.10.3631-3644.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann E., Brosius J., Ptashne M. Vectors bearing a hybrid trp-lac promoter useful for regulated expression of cloned genes in Escherichia coli. Gene. 1983;25(2–3):167–178. doi: 10.1016/0378-1119(83)90222-6. [DOI] [PubMed] [Google Scholar]
- Asghar A.H. The pobA gene of Burkholderia cenocepacia encodes a group I Sfp-type phosphopantetheinyl transferase required for biosynthesis of the siderophores ornibactin and pyochelin. Microbiology. 2011;157(2):349–361. doi: 10.1099/mic.0.045559-0. [DOI] [PubMed] [Google Scholar]
- Aubert D.F., Flannagan R.S., Valvano M.A. A novel sensor kinase-response regulator hybrid controls biofilm formation and type VI secretion system activity in Burkholderia cenocepacia. Infect. Immun. 2008;76(5):1979–1991. doi: 10.1128/IAI.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagdasarian M. Specific-purpose plasmid cloning vectors. II. Broad host range, high copy number, RSF1010-derived vectors, and a host-vector system for gene cloning in pseudomonas. Gene. 1981;16(1–3):237–247. doi: 10.1016/0378-1119(81)90080-9. [DOI] [PubMed] [Google Scholar]
- Brosius J. Toxicity of an overproduced foreign gene product in Escherichia coli and its use in plasmid vectors for the selection of transcription terminators. Gene. 1984;27(2):161–172. doi: 10.1016/0378-1119(84)90137-9. [DOI] [PubMed] [Google Scholar]
- Carlier A. Genome sequence of Burkholderia cenocepacia H111, a cystic fibrosis airway isolate. Genome Announc. 2014;2(2) doi: 10.1128/genomeA.00298-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapalain A. Identification of quorum sensing-controlled genes in Burkholderia ambifaria. Microbiologyopen. 2013;2(2):226–242. doi: 10.1002/mbo3.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coenye T., Vandamme P. Diversity and significance of Burkholderia species occupying diverse ecological niches. Environ. Microbiol. 2003;5(9):719–729. doi: 10.1046/j.1462-2920.2003.00471.x. [DOI] [PubMed] [Google Scholar]
- de Lorenzo V., Herrero M., Jakubzik U., Timmis K.N. Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J. Bacteriol. 1990;172(11):6568–6572. doi: 10.1128/jb.172.11.6568-6572.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lorenzo V., Timmis K.N. Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol. 1994;235:386–405. doi: 10.1016/0076-6879(94)35157-0. [DOI] [PubMed] [Google Scholar]
- Dennis J.J., Zylstra G.J. Plasposons: modular self-cloning minitransposon derivatives for rapid genetic analysis of gram-negative bacterial genomes. Appl. Environ. Microbiol. 1998;64(7):2710–2715. doi: 10.1128/aem.64.7.2710-2715.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis J.J., Zylstra G.J. Improved antibiotic-resistance cassettes through restriction site elimination using Pfu DNA polymerase PCR. Biotechniques. 1998;25(5):772–774. doi: 10.2144/98255bm04. (776) [DOI] [PubMed] [Google Scholar]
- DeShazer D., Woods D.E. Broad-host-range cloning and cassette vectors based on the R388 trimethoprim resistance gene. Biotechniques. 1996;20(5):762–764. doi: 10.2144/96205bm05. [DOI] [PubMed] [Google Scholar]
- De Smet B. Burkholderia stagnalis sp. nov. and Burkholderia territorii sp. nov., two novel Burkholderia cepacia complex species from environmental and human sources. Int. J. Syst. Evol. Microbiol. 2015;65(7):2265–2271. doi: 10.1099/ijs.0.000251. [DOI] [PubMed] [Google Scholar]
- De Soyza A. Lung transplantation for patients with cystic fibrosis and Burkholderia cepacia complex infection: a single-center experience. J. Heart Lung Transplant. 2010;29(12):1395–1404. doi: 10.1016/j.healun.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Drevinek P., Mahenthiralingam E. Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clin. Microbiol. Infect. 2010;16(7):821–830. doi: 10.1111/j.1469-0691.2010.03237.x. [DOI] [PubMed] [Google Scholar]
- Farmer K.L., Thomas M.S. Isolation and characterization of Burkholderia cenocepacia mutants deficient in pyochelin production: pyochelin biosynthesis is sensitive to sulfur availability. J. Bacteriol. 2004;186(2):270–277. doi: 10.1128/JB.186.2.270-277.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellay R., Frey J., Krisch H. Interposon mutagenesis of soil and water bacteria: a family of DNA fragments designed for in vitro insertional mutagenesis of gram-negative bacteria. Gene. 1987;52(2–3):147–154. doi: 10.1016/0378-1119(87)90041-2. [DOI] [PubMed] [Google Scholar]
- Flannagan R.S., Aubert D., Kooi C., Sokol P.A., Valvano M.A. Burkholderia cenocepacia requires a periplasmic HtrA protease for growth under thermal and osmotic stress and for survival in vivo. Infect. Immun. 2007;75(4):1679–1689. doi: 10.1128/IAI.01581-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentz R., Langner A., Chang A.C., Cohen S.N., Bujard H. Cloning and analysis of strong promoters is made possible by the downstream placement of a RNA termination signal. Proc. Natl. Acad. Sci. U. S. A. 1981;78(8):4936–4940. doi: 10.1073/pnas.78.8.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamad M.A., Skeldon A.M., Valvano M.A. Construction of aminoglycoside-sensitive Burkholderia cenocepacia strains for use in studies of intracellular bacteria with the gentamicin protection assay. Appl. Environ. Microbiol. 2010;76(10):3170–3176. doi: 10.1128/AEM.03024-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 1983;166(4):557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- Henry D.A., Campbell M.E., LiPuma J.J., Speert D.P. Identification of Burkholderia cepacia isolates from patients with cystic fibrosis and use of a simple new selective medium. J. Clin. Microbiol. 1997;35(3):614–619. doi: 10.1128/jcm.35.3.614-619.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero M., de Lorenzo V., Timmis K.N. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J. Bacteriol. 1990;172(11):6557–6567. doi: 10.1128/jb.172.11.6557-6567.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang T.T., Karkhoff-Schweizer R.R., Kutchma A.J., Schweizer H.P. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998;212(1):77–86. doi: 10.1016/s0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- Huber B. The cep quorum-sensing system of Burkholderia cepacia H111 controls biofilm formation and swarming motility. Microbiology. 2001;147(Pt 9):2517–2528. doi: 10.1099/00221287-147-9-2517. [DOI] [PubMed] [Google Scholar]
- Inhülsen S. Identification of functions linking quorum sensing with biofilm formation in Burkholderia cenocepacia H111. Microbiologyopen. 2012;1(2):225–242. doi: 10.1002/mbo3.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isles A. Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J. Pediatr. 1984;104(2):206–210. doi: 10.1016/s0022-3476(84)80993-2. [DOI] [PubMed] [Google Scholar]
- Jové T., Da Re S., Denis F., Mazel D., Ploy M.-C. Inverse correlation between promoter strength and excision activity in class 1 integrons. In: Casadesús J., editor. PLoS Genet. Vol. 6. 2010. p. e1000793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledson M.J., Gallagher M.J., Corkill J.E., Hart C.A., Walshaw M.J. Cross infection between cystic fibrosis patients colonised with Burkholderia cepacia. Thorax. 1998;53(5):432–436. doi: 10.1136/thx.53.5.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévesque C., Brassard S., Lapointe J., Roy P.H. Diversity and relative strength of tandem promoters for the antibiotic-resistance genes of several integrons. Gene. 1994;142(1):49–54. doi: 10.1016/0378-1119(94)90353-0. [DOI] [PubMed] [Google Scholar]
- Malott R.J., Baldwin A., Mahenthiralingam E., Sokol P.A. Characterization of the cciIR quorum-sensing system in Burkholderia cenocepacia. Infect. Immun. 2005;73(8):4982–4992. doi: 10.1128/IAI.73.8.4982-4992.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.W., Mohr C.D. Invasion and intracellular survival of Burkholderia cepacia. Infect. Immun. 2000;68(1):24–29. doi: 10.1128/iai.68.1.24-29.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen T., Markey K., Murphy P., McClean S., Callaghan M. Role of lipase in Burkholderia cepacia complex (Bcc) invasion of lung epithelial cells. Eur. J. Clin. Microbiol. Infect. Dis. 2007;26(12):869–877. doi: 10.1007/s10096-007-0385-2. [DOI] [PubMed] [Google Scholar]
- Orosz A., Boros I., Venetianer P. Analysis of the complex transcription termination region of the Escherichia coli rrnB gene. Eur. J. Biochem. 1991;201(3):653–659. doi: 10.1111/j.1432-1033.1991.tb16326.x. [DOI] [PubMed] [Google Scholar]
- Perrin S., Gilliland G. Site-specific mutagenesis using asymmetric polymerase chain reaction and a single mutant primer. Nucleic Acids Res. 1990;18(24):7433–7438. doi: 10.1093/nar/18.24.7433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakowski S.A., Filutowicz M. Plasmid R6K replication control. Plasmid. 2013;69(3):231–242. doi: 10.1016/j.plasmid.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein S.J., Reznikoff W.S. The functional differences in the inverted repeats of Tn5 are caused by a single base pair nonhomology. Cell. 1981;23:191–199. doi: 10.1016/0092-8674(81)90284-1. [DOI] [PubMed] [Google Scholar]
- Ryan G.T., Wei Y., Winans S.C. A LuxR-type repressor of Burkholderia cenocepacia inhibits transcription via antiactivation and is inactivated by its cognate acylhomoserine lactone. Mol. Microbiol. 2013;87(1):94–111. doi: 10.1111/mmi.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Cold Spring Harbor Laboratory Press. Cold Spring Harbor; New York: 1989. Molecular cloning: a laboratory manual. [Google Scholar]
- Schrecke K., Jordan S., Mascher T. Stoichiometry and perturbation studies of the LiaFSR system of Bacillus subtilis. Mol. Microbiol. 2013;87(4):769–788. doi: 10.1111/mmi.12130. [DOI] [PubMed] [Google Scholar]
- Schwarz S., Kehrenberg C., Doublet B., Cloeckaert A. Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol. Rev. 2004;28(5):519–542. doi: 10.1016/j.femsre.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Shaw W.V. Chloramphenicol acetyltransferase: enzymology and molecular biology. CRC Crit. Rev. Biochem. 1983;14(1):1–46. doi: 10.3109/10409238309102789. [DOI] [PubMed] [Google Scholar]
- Sokol P.A. The CepIR quorum-sensing system contributes to the virulence of Burkholderia cenocepacia respiratory infections. Microbiology. 2003;149(Pt 12):3649–3658. doi: 10.1099/mic.0.26540-0. [DOI] [PubMed] [Google Scholar]
- Somvanshi V.S. The type 2 secretion Pseudopilin, gspJ, is required for multihost pathogenicity of Burkholderia cenocepacia AU1054. Infect. Immun. 2010;78(10):4110–4121. doi: 10.1128/IAI.00558-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speert D.P., Henry D., Vandamme P., Corey M., Mahenthiralingam E. Epidemiology of Burkholderia cepacia complex in patients with cystic fibrosis, Canada. Emerg. Infect. Dis. 2002;8(2):181–187. doi: 10.3201/eid0802.010163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stassi D.L., Lacks S.A. Effect of strong promoters on the cloning in Escherichia coli of DNA fragments from Streptococcus pneumoniae. Gene. 1982;18(3):319–328. doi: 10.1016/0378-1119(82)90170-6. [DOI] [PubMed] [Google Scholar]
- Stueber D., Bujard H. Transcription from efficient promoters can interfere with plasmid replication and diminish expression of plasmid specified genes. EMBO J. 1982;1(11):1399–1404. doi: 10.1002/j.1460-2075.1982.tb01329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait R.C., Close T.J., Rodriguez R.L., Kado C.I. Isolation of the origin of replication of the IncW-group plasmid pSa. Gene. 1982;20(1):39–49. doi: 10.1016/0378-1119(82)90085-3. [DOI] [PubMed] [Google Scholar]
- Tsang T., Copeland V., Bowden G.T. A set of cassette cloning vectors for rapid and versatile adaptation of restriction fragments. Biotechniques. 1991;10(3):330. [PubMed] [Google Scholar]
- Uehlinger S. Identification of specific and universal virulence factors in Burkholderia cenocepacia strains by using multiple infection hosts. Infect. Immun. 2009;77(9):4102–4110. doi: 10.1128/IAI.00398-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandamme P. Occurrence of multiple genomovars of Burkholderia cepacia in cystic fibrosis patients and proposal of Burkholderia multivorans sp. nov. Int. J. Syst. Bacteriol. 1997;47(4):1188–1200. doi: 10.1099/00207713-47-4-1188. [DOI] [PubMed] [Google Scholar]
- Visser M.B., Majumdar S., Hani E., Sokol P.A. Importance of the ornibactin and pyochelin siderophore transport systems in Burkholderia cenocepacia lung infections. Infect. Immun. 2004;72(5):2850–2857. doi: 10.1128/IAI.72.5.2850-2857.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlosnik J.E.A. Burkholderia species infections in patients with cystic fibrosis in British Columbia, Canada. 30 years' experience. Ann. Am. Thorac. Soc. 2015;12(1):70–78. doi: 10.1513/AnnalsATS.201408-395OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacterial strains, plasmids and oligonucleotides.