

Abstract
Although neuronal activity can be modulated using a variety of techniques, there are currently few methods for controlling neuronal connectivity. We introduce a tool (GFE3) that mediates the fast, specific and reversible elimination of inhibitory synaptic inputs onto genetically determined neurons. GFE3 is a fusion between an E3 ligase, which mediates the ubiquitination and rapid degradation of proteins, and a recombinant, antibody-like protein (FingR) that binds to Gephyrin. Expression of GFE3 leads to a strong and specific reduction of Gephyrin in culture or in vivo and to a substantial decrease in phasic inhibition onto cells that express GFE3. By temporarily expressing GFE3 we showed that inhibitory synapses regrow following ablation. Thus, we have created a simple, reversible method for modulating inhibitory synaptic input onto genetically determined cells.
Editorial summary
When studying neural circuitry, the ablation of synapses may be an alternative to optogenetic manipulation of neurons. Arnold et al. report a genetically encoded tool called GFE3 that eliminates inhibitory inputs into neurons expressing GFE3.
We sought to create a method for eliminating inhibitory inputs onto neurons by ablating Gephyrin, a postsynaptic protein that clusters GABAA and Glycine receptors1–3. Traditional methods for eliminating protein expression, such as gene deletion4 or siRNA5, work by means of cutting off protein supply, and thus become effective only as the protein degrades, which can take a week or longer6,7. Furthermore, they are not easily reversible. Therefore, our approach uses an E3 Ligase enzyme, which marks proteins for degradation by mediating the covalent attachment of ubiquitin polypeptides to lysine residues, causing the marked proteins to be transported to the proteasome and degraded8. E3 ligases can be engineered to have arbitrary specificity by fusing their ubiquitin transferase domains with proteins that bind to specific targets. For instance, a fusion of an E3 ligase with a GFP nanobody degrades exogenously expressed GFP in vivo9. However, endogenous proteins have been targeted with genetically encoded molecules in few examples only10.
We have generated a protein, GFE3, that mediates the fast and specific degradation of Gephyrin, eliminating the inhibitory synaptic inputs to the neurons in which it is expressed. Gephyrin degradation is complete within 5 hours of inducing GFE3 expression, and cessation of GFE3 expression results in inhibitory synapses reappearing within 48 hours. Thus, GFE3 can specifically and reversibly alter synaptic connectivity.
Results
GFE3 mediates degradation of Gephyrin
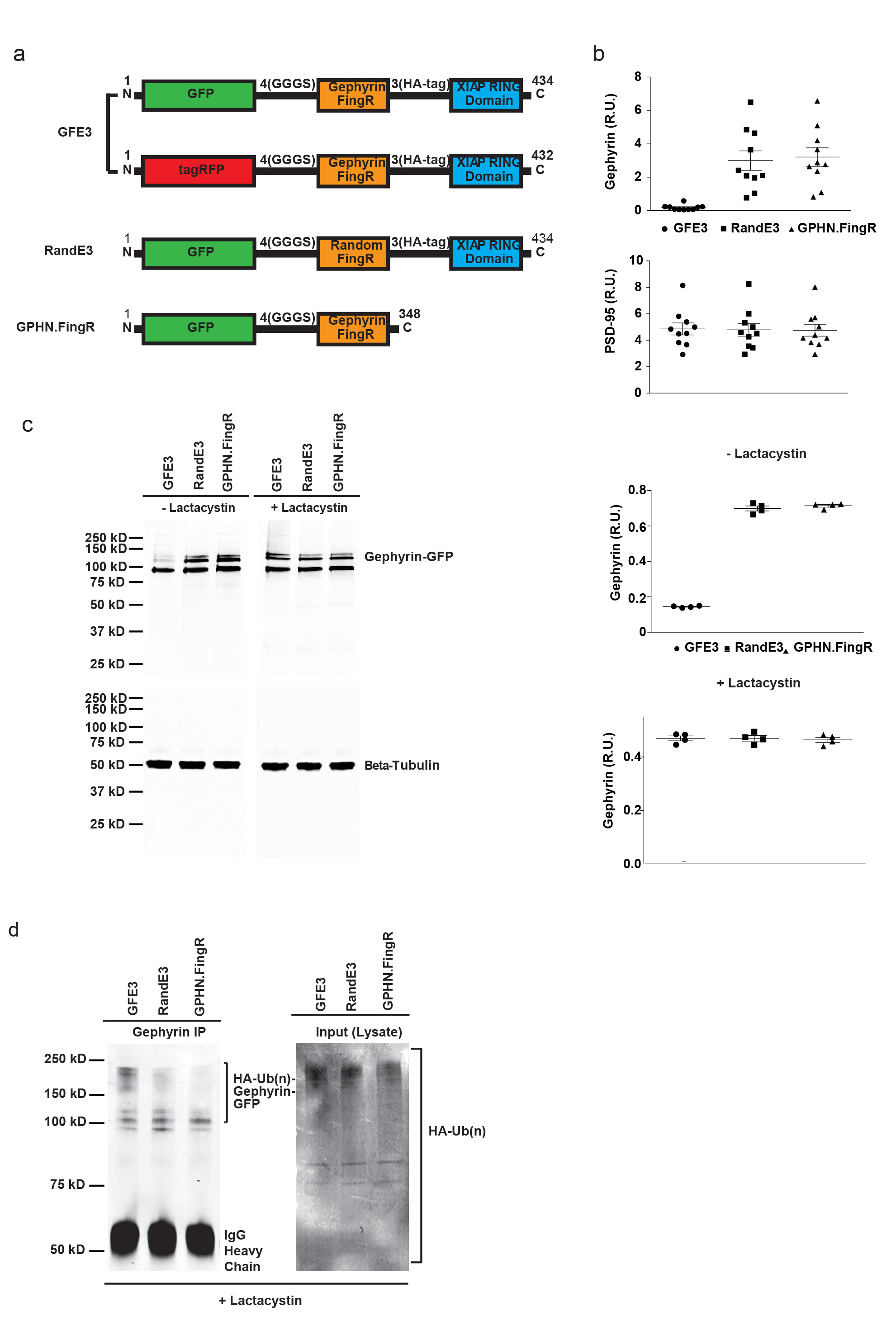
We generated GFE3 by fusing GFP-GPHN.FingR, an antibody-like protein based on the 10FNIII domain of fibronectin that binds Gephyrin with high affinity11, with the RING domain of XIAP, an E3 ubiquitin ligase12 (Supplementary Fig. 1). To test whether GFE3 can mediate the elimination of endogenous Gephyrin, we compared the expression of synaptic proteins from cultures of cortical neurons expressing GFE3 and cultures expressing control constructs. Neurons that expressed either GPHN.FingR only or a fusion of the RING domain with a FingR containing random residues within its target binding domains (RandE3) displayed both Gephyrin and PSD-95 puncta, whereas neurons expressing GFE3 displayed PSD-95 puncta, but no Gephyrin puncta (Fig. 1a–c, Supplementary Fig. 1).
Figure 1. GFE3 specifically ablates Gephyrin.

(a–c) Immunostaining showing Gephyrin (green) and PSD-95 (magenta) localization at inhibitory and excitatory synapses, respectively, after expression of GPHN.FingR (a), RandE3 (b) or GFE3 (c) in cultured rat cortical neurons. (d) Western blot showing the expression of Gephyrin, the alpha1 subunit of the GABAA receptor, PSD-95, the GluA1 subunit of the AMPA receptor, the GluN2B subunit of the NMDA receptor, or the Gephyrin interacting proteins Collybistin II and GABARAP in neurons not transduced (Control) or transduced with GFE3, RandE3 or GPHN.FingR. Gephyrin expression is reduced in neurons expressing GFE3 (80 ± 3%, P = 0.01, Kruskal-Wallis). No significant change is observed in the steady-state levels of the other proteins (P > 0.8, Kruskal-Wallis; n = 4 replicate blots). (e, f) Staining for the alpha1 subunit of the GABAA receptor (magenta) and Gephyrin (green) in cortical neuron expressing RandE3 (e) or GFE3 (f). Scale bar represents 10 µm.
To investigate GFE3’s mode of action, we co-expressed GFE3, GPHN.FingR or RandE3 in COS-7 cells with Gephyrin-GFP. In the course of 24 hours, GFE3 reduced exogenous Gephyrin-GFP by ~ 80% compared to the controls. This reduction was dependent on proteasome function as shown by treatment with Lactacystin (clasto-Lactacystin-[beta]-lactone), a highly specific and irreversible proteasome inhibitor (Supplementary Fig. 1). We also co-expressed Gephyrin-GFP in COS-7 cells with HA-Ubiquitin and either GFE3, GPHN.FingR or RandE3 in the presence of Lactacystin and immunoprecipitated with an anti-Gephyrin antibody. Anti-HA staining of the precipitates revealed a prominent high molecular weight band in the GFE3 lane that likely corresponds to poly-ubiquitinated Gephyrin (Supplementary Fig. 1). Thus, our results are consistent with GFE3 causing poly-ubiquitination of Gephyrin followed by degradation in the proteasome.
To quantify the effect of expressing GFE3 in neurons, we compared uninfected (control) neuronal cultures to cultures infected with a lentivirus encoding either GFE3, GPHN.FingR or RandE3. Lentiviral infection led to efficient expression of GFE3 and a sharp reduction of endogenous Gephyrin, whereas infection with RandE3 or GPHN.FingR, while equally efficient, did not affect Gephyrin expression (Supplementary Fig. 2). GFE3 expressing cultures expressed 80% less Gephyrin compared with neurons expressing GPHN.FingR (Fig. 1d, Supplementary Fig. 3). Despite this effect on Gephyrin expression, GFE3 had a negligible effect on expression levels of the GABAA receptor, PSD-95, GluA1, GluN2B, or the Gephyrin interactors Collybistin II13 and GABARAP14 (Fig. 1d). Indeed, expression levels of these proteins are not significantly different in cells expressing GFE3 versus the three types of control cells (Supplementary Fig. 4). Thus, expression of GFE3 leads to the efficient, specific reduction of endogenous Gephyrin levels.
Because Gephyrin clusters GABAA receptors, we examined the effect of GFE3 on the receptors’ distribution. In control cells GABAA receptors were clustered and colocalized with Gephyrin, whereas in neurons transfected with GFE3 GABAA receptors were diffusely distributed and Gephyrin staining was not visible (Fig. 1e, f; Supplementary Fig. 4). Thus, reducing endogenous Gephyrin by expressing GFE3 appears to release GABAA receptors from postsynaptic clusters, allowing them to distribute through the dendrites, but does not substantially reduce their expression, consistent with results obtained with siRNA against Gephyrin15. Furthermore, although expression of GFE3 causes a large change in Gephyrin expression levels, its expression is not associated with changes in either cell viability or overall proteasome activity (Supplementary Fig. 5).
GFE3 expression eliminates phasic inhibition
To examine the functional consequences of GFE3 expression, we transfected cultured hippocampal neurons with GFE3, GFP (control) or RandE3. We recorded from neurons with access resistance below 30 MΩ that varied by 20% or less using whole cell patch clamp. GFE3-expressing cells exhibited miniature inhibitory postsynaptic currents (mIPSCs) with a roughly 65% decrease in the frequency and 30% reduction in amplitude when compared to control cells or to cells expressing RandE3 (Fig. 2a–d). In contrast, miniature EPSCs recorded under similar conditions in cells expressing GFE3 showed no significant difference in either frequency or amplitude when compared with control cells (P > 0.75, Mann Whitney; Supplementary Fig. 6). To test whether functional GABAA receptors are still present on the neuronal membrane in cells expressing GFE3, we applied a high concentration of GABA (1 mM) to the bath. Under these conditions the amplitude of evoked currents was not significantly different between cells expressing GFE3 and controls (P > 0.5, Mann-Whitney; Fig. 2e, f). This suggests that unclustered GABAA receptors are on the cell surface and remain functional. Furthermore, noise analysis of these data is consistent with the kinetics of GABAA receptors being similar in the two conditions (Supplementary Fig. 7).
Figure 2. Gephyrin ablation reduces the amplitude and frequency of mIPSCs without affecting GABA-evoked currents.

(a) Representative traces of mIPSCs obtained from dissociated rat hippocampal neurons expressing GFE3 or RandE3, or control neurons. Cells expressing GFE3 exhibit a significant reduction in frequency (b; P < 0.002, Kruskal-Wallis) and amplitude (c; P < 0.02, Kruskal-Wallis) of miniature inhibitory postsynaptic currents (mIPSCs) vs. cells expressing RandE3 or control cells. (d) Histogram of the distribution of mIPSC amplitude in a control cell and in a cell expressing GFE3. n = 5 cells for control, n = 6 cells for GFE3 and RandE3. (e). Quantification of the peak amplitude of evoked GABA currents recorded in GFE3 transfected and control cells (f; P > 0. 5, Mann-Whitney). n = 7 independent experiments. Bars indicate means ± SEM.
To test the efficiency of GFE3 in abolishing GABAergic synaptic transmission in the intact brain, we virally infected indirect spiny projection neurons (iSPNs) to induce GFE3 or GPHN.FingR expression in a cre-dependent manner in the dorsal striatum of Adora2A-cre mice, while simultaneously expressing Channelrhodopsin2 (ChR2) in non-cre-expressing striatal cells (Fig. 3a). This approach resulted in light-evoked synaptic GABAergic transmission onto iSPNs, with GABA being released presumably mostly from direct SPNs, with some contribution from GABAergic interneurons16. A single light pulse reliably evoked GABAA receptor mediated synaptic currents under control conditions where we expressed GPHN.FingR, but currents were abolished in iSPNs expressing GFE3 (Fig. 3b). We also examined spontaneous IPSCs, and found a significant reduction in both frequency (P = 0.0008, Mann-Whitney) and amplitude (P < 0.03, Mann-Whitney), compared to control conditions (Fig. 3c). This reduction in spontaneous synaptic activity was selective for GABAergic transmission, as spontaneous excitatory inputs were unchanged (Fig. 3c). Thus, expression of GFE3 specifically eliminates functional GABAergic synapses, without affecting glutamatergic synapses. In contrast, the amplitude of tonic GABA currents17–19 showed a tendency towards reduction, but this change did not rise to the level of significance (Fig. 3d, e). These results indicate that while there might possibly be a partial reduction in total (synaptic and extra-synaptic) GABAA receptor expression at the cell surface, elimination of GABAA receptors from the synapse by GFE3 is not due to a general elimination of GABAA receptors from the cell, consistent with our results in dissociated cultures (Figs. 1e, f; 2e, f). Thus, our results are consistent with GFE3 specifically eliminating the phasic inhibitory input onto neurons in the intact brain.
Figure 3. Loss of synaptic GABAergic currents in the intact mouse brain from expression of GFE3.

(a) Schematic of experimental layout. (b) Evoked synaptic currents in control and GFE3-expressing indirect pathway spiny projection neurons (iSPNs) in control conditions (black) and after slices were exposed to SR95331 (10 µM), a GABAA receptor antagonist (red). (c) Spontaneous inhibitory currents (sIPSCs, top) were reduced by GFE3 compared with control in both frequency (P < 0.0008, Mann-Whitney) and amplitude (P < 0.03, Mann-Whitney), but no effect was observed on the frequency (P > 0.9, Mann-Whitney) or amplitude (P > 0.17, Mann-Whitney) of excitatory currents (sEPSCs, bottom). Representative example traces are shown on the left, quantification summaries on the right. (d) Tonic GABAA receptor current was quantified as the change in baseline holding current following application of SR95331 (10 µM). (e) Expression of GFE3 resulted in a tendency to reduction of tonic GABAA receptor current, (P = 0.12, Mann-Whitney). For all experiments, grey circles indicate individual cells from a total of 4 animals (inhibitory inputs) or 2 animals (excitatory inputs), bars indicate means ± SEM.
GFE3 in zebrafish spinal neurons causes motor defects
To further test the efficacy of GFE3 in vivo, we injected zebrafish zygotes with a plasmid containing GFE3-GFP driven by the mnx1 enhancer, which restricts expression to spinal cord neurons20. Expression of GFE3-GFP was both efficient and specific as indicated by numerous labeled cells along the midline in the spinal cord 2 days post-injection (Fig. 4a). In fish expressing RandE3-GFP, GFP is visible within the cell bodies of spinal cord neurons, while at the outer edge of these cell bodies there is a ring of Gephyrin staining (Fig. 4b). In contrast, Gephyrin staining is sharply reduced in the cell bodies of spinal cord neurons expressing GFE3-GFP (Fig. 4c). Discontinuous staining on the edge of the GFP staining likely arises from unlabeled neighboring cells. Furthermore, the average level of Gephyrin staining surrounding the cell bodies of cells expressing GFE3-GFP was significantly lower than that of cells expressing RandE3-GFP alone (P < 0.008, Mann-Whitney; Fig. 4d).
Figure 4. GFE3 expression in zebrafish spinal cord results in Gephyrin ablation and behavioral deficits.

(a) A confocal image of a live zebrafish age 2 days post fertilization (dpf) shows mosaic expression of GFE3-GFP driven by an enhancer element of the mnx1 gene that restricts expression to spinal motoneurons. (b,c) Immunostaining of Gephyrin (magenta) and GFP (green) in motoneurons expressing RandE3-GFP (b) or GFE3 (c). Scale bar represents 5 µm. (d) Gephyrin levels in cells expressing RandE3-GFP (64 ± 5 a.u.; n = 5 fish, 6 cells per fish, 3 independent experiments) and in cells expressing GFE3-GFP (23 ± 2 a.u.; P < 0.008, Mann-Whitney; n = 5 fish, 6 cells per fish, 3 independent experiments). (e) Zebrafish expressing RandE3-GFP, age 24 hours post fertilization, exhibit normal spontaneous tail flicking, including touching tip of the tail to the base. (f) Zebrafish expressing GFE3-GFP in motoneurons are unable to curl their tails sufficiently to touch the tip to the base. (g) Quantitation of the minimum distance between the base of the tail and the tip, measured during 5 spontaneous flicks, reveals a > 100% increase in zebrafish that express GFE3-GFP vs. those expressing RandE3-GFP (P < 0.003, Mann-Whitney; n = 5 fish, 5 flicks per fish). Bars indicate means ± SEM.
A major role of inhibitory neurons in the zebrafish spinal cord is to prevent co-contractions of opposing muscles by inhibiting contralateral motoneurons21. Accordingly, zebrafish expressing mutant Glycine receptors in the spinal cord that fail to cluster at inhibitory synapses exhibit co-contraction of opposing muscles within the tail22,23. Zebrafish expressing GFE3-GFP (Supplementary Videos 1, 2) exhibited a lack of coordination when compared to zebrafish expressing RandE3-GFP (Supplementary Videos 3, 4), which would be expected if the lack of Gephyrin caused co-contraction of opposing muscles. To quantify these effects we took advantage of a peculiarity of the stereotyped tail flick behavior24,25. Fish expressing RandE3-GFP bend their tails in a tight circle so that the tip of the tail touches the base of the tail (Fig. 4e, Supplementary Fig. 8), where it connects with the abdomen of the fish. In contrast, fish that express GFE3-GFP do not touch the bases of their tails with the tips (Fig. 4f, Supplementary Fig. 8). Indeed, the minimum distance between the base and the tip of the tail during 5 tail flicks in zebrafish expressing GFE3-GFP was more than twice that of the same distance measured in zebrafish expressing RandE3-GFP (Fig. 4g). These results are consistent with expression of GFE3 causing the efficient ablation of Gephyrin in vivo, resulting in the loss of phasic inhibition within spinal cord neurons in zebrafish.
GFE3-mediated loss of Gephyrin is reversible
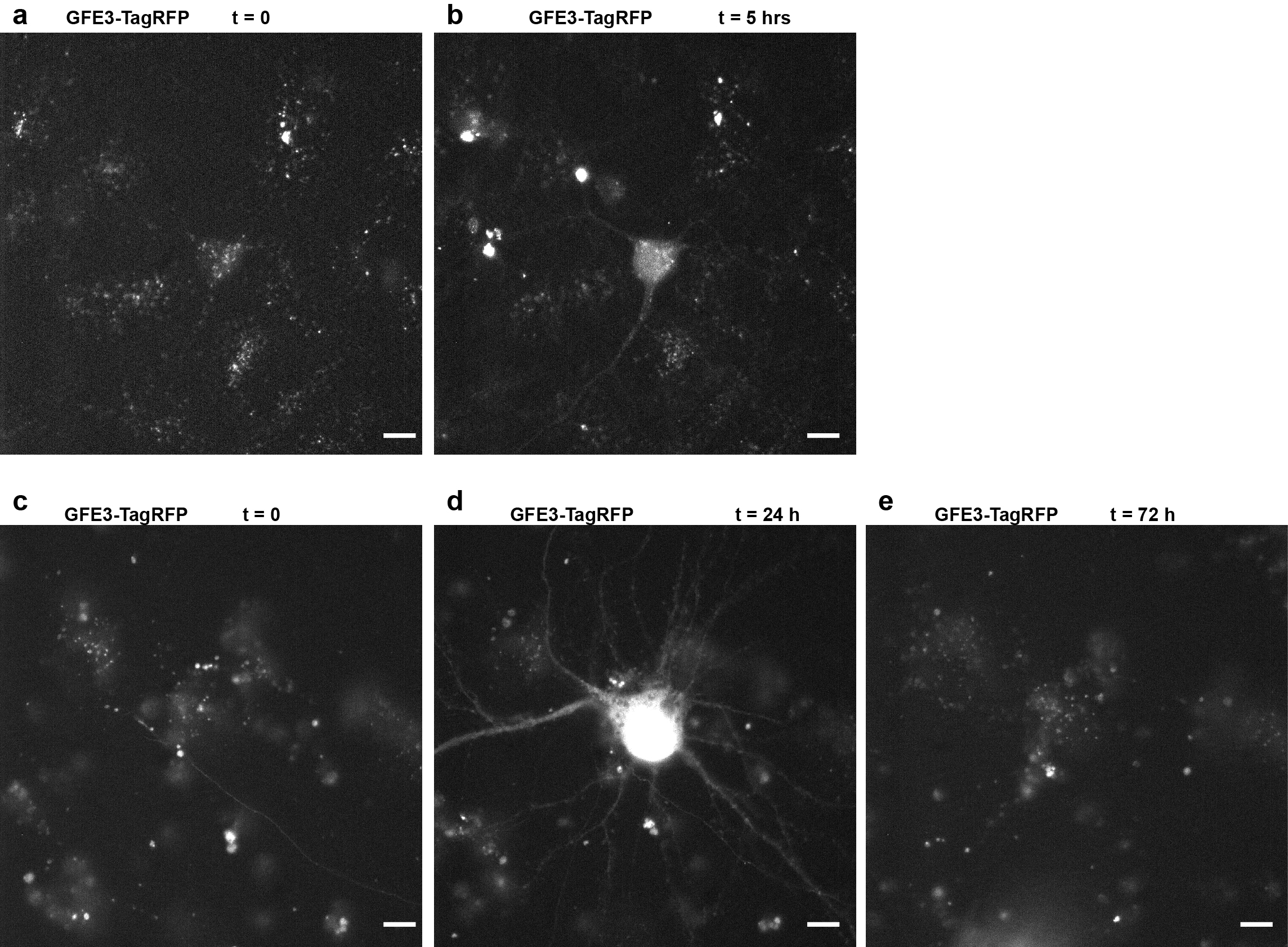
An advantage of the GFE3 approach is that Gephyrin expression is modified at the level of protein degradation, and thus the level of Gephyrin is expected to respond quickly to the expression or elimination of GFE3. To test this hypothesis, we expressed GPHN.FingR-GFP along with doxycycline(dox)-dependent GFE3-TagRFP in cortical neurons in culture and incubated for 4–7 days without dox, labeling inhibitory synapses with GPHN.FingR-GFP. We then induced expression of GFE3-TagRFP using dox (Fig. 5a, Supplementary Fig. 9), and found efficient elimination of Gephyrin in less than 5 hours as indicated by the absence of GPHN.FingR-GFP labeling (Fig 5a, b). Fixing the same cells and immunostaining for endogenous Gephyrin (green) and GFE3-TagRFP (red) confirmed that endogenous Gephyrin had been eliminated (Fig. 5c). In contrast, transfection of siRNA led to an 80% reduction in the number of Gephyrin puncta after 5 days in dissociated hippocampal cultures26.
Figure 5. GFE3 mediates transient and reversible ablation of Gephyrin.

(a,b) GPHN.FingR-GFP localization in a rat cortical neuron co-expressing GPHN.FingR-GFP and doxycycline(dox)-inducible GFE3-TagRFP before (a) and after (b) addition of dox (1 µg/ml) for 5 hours. (c) Immunostaining of GFE3-TagRFP (red) and Gephyrin (green) in the same cell as in (a) and (b). (d) GPHN.FingR-GFP localization in a rat cortical neuron co-expressing GPHN.FingR-GFP and dox-inducible GFE3-TagRFP before (d) and after addition of dox (1 µg/ml) for 24 hours (e) as well as after subsequent removal of dox for 48 hours (f). (g) An overlay of time points t = 0 (d) and t = 72 hrs (f). (h) Immunostaining of the neuron in (f) for GPHN.FingR-GFP (green), Gephyrin (red), and GAD-65 (blue) at t = 72 hrs. Inset: arrowheads point to puncta where all three proteins are colocalized. Scale bars represent 10 µm. (i) Quantitation of relative synaptic strength, by measuring total intensity of puncta labeled with GPHN.FingR-GFP, indicates that no significant difference exists between synapses at 0 hrs and at 72 hrs in neurons treated with dox or with vehicle (P > 0.2, Mann-Whitney; n = 7 cells, 2 independent experiments).
To demonstrate reversible elimination of Gephyrin expression, we added dox to neurons transfected with GPHN.FingR-GFP and dox-dependent GFE3-TagRFP for 24 hours, and then removed dox and waited 48 hours to determine whether the Gephyrin puncta would return (Supplementary Fig. 9). As in the previous experiment, Gephyrin puncta labeled with GPHN.FingR-GFP disappeared following 24 hours of dox-induced GFE3-TagRFP expression (Fig. 5d, e; Supplementary Fig. 9). Removal of dox for 48 hours resulted in a reappearance of Gephyrin puncta (Fig. 5f), many in places that are close to their original locations (Fig. 5g). Gephyrin immunostaining at 48 hours after removal of Dox showed that newly formed Gephyrin puncta co-localized with staining for presynaptic GAD-65 (Fig. 5h), suggesting that the recovered Gephyrin puncta have corresponding presynaptic sites, and thus are likely part of reconstituted synapses. Finally, quantitation of GPHN.FingR-GFP shows that following recovery total FingR staining in puncta is present at levels that are comparable to initial levels (Fig. 5i). Thus, by controlling the expression of GFE3 it is possible to induce the reversible ablation of Gephyrin.
Discussion
FingRs can target fluorescent proteins to endogenous target proteins, allowing the targets and the macromolecular structures where they reside to be visualized in vivo11,27. Here we show that an E3 ligase can also be targeted specifically using a FingR, causing expression of its target protein to be quickly and reversibly eliminated. Although this is not the first method developed for direct elimination of protein expression, it differs in important respects from previously described methods. Many such methods are specific to exogenously expressed proteins9,28, or are not genetically encoded29. Furthermore, FingRs bind to their targets without affecting target function or expression11, which means that protein elimination is entirely due to increased degradation through the E3 ligase and the Ubiquitin proteasome system. This is distinct from scFv-based proteins that reduce Gephyrin expression through an unknown mechanism, with unknown off-target effects30. In addition, because the Gephyrin FingR was specifically engineered to bind Gephyrin with very high affinity and specificity, the fidelity with which it degrades its target is likely to be much higher than that of a system that depends on naturally occurring protein-protein interactions, such as those involving PDZ domains, which have relatively low affinity and specificity31,32. Furthermore, positively charged cell-permeant peptides such as Tat, upon which systems mediating degradation with peptides rely, are inefficient, unstable, and induce cytotoxicity33.
E3 ligases can be fused to FingRs targeted against other proteins, suggesting that our approach could be generalizable for reversibly ablating a wide range of endogenous targets. In addition, because of its modular nature, we envision that GFE3 function could be engineered to rely on light dependent association of the two domains, making the complex active in response to light.
Modulating the excitability of neurons using optogenetics has revealed the electrophysiological properties of specific neuronal cell types and their roles in circuits and functional networks, which was not possible with previous pharmacological approaches. Like optogenetic proteins, GFE3 can be expressed in specific cell types. However, it acts at a qualitatively different level than optogenetics - modifying endogenous synaptic connectivity rather than directly inducing the opening of exogenous ion channels or the activation of pumps34. GFE3 also differs from AS-PaRac-1, which can inducibly reduce the strength of excitatory synapses, in that GFE3 eliminates Gephyrin, a component of the synapse, whereas AS-PaRac1 causes changes in the actin cytoskeleton supporting dendritic spines by modulating function of the G protein Rac135.
By targeting expression of GFE3 to excitatory cells it should increase excitability within a specific brain region, whereas expressing it in inhibitory cells should decrease excitability. Thus, it should be possible to probe the effects of systematically modulating the excitability of specific regions of the brain, both in wild-type animals and in disease models in a way that does not directly interfere with the firing patterns of cells.
In conclusion, GFE3 provides a fast, specific and reversible method for modulating inhibition onto defined cells. Furthermore, it provides a framework for generating a diverse array of genetically encoded modulators of expression and function that act specifically on virtually any target protein.
Online Methods
cDNA Constructs
The Gephyrin (GFE3) and Random (RandE3) ablating FingRs were constructed by fusing a DNA sequence encoding three consecutive HA-tags followed by the amino acids 440–496 of the RING domain of rat XIAP 3’ to the Gephyrin FingR gene11 or to the Random FingR gene FN0436 (Supplementary Fig. 1). In addition, the gene for EGFP or TagRFP (Evrogen, cat. # FP141) was linked 3’ to a DNA spacer encoding the amino acid sequence GGGS repeated four times, which was then fused 5’ to the Gephyrin FingR to allow for direct visualization of the GFE3 in living cells. Expression constructs were made by ligating these genes into the pCAG, pFUW or pAAV37 expression vector. The construction of transcriptionally controlled Gephyrin.FingR-GFP-CCR5ZFL-KRAB(A) (Addgene, cat. # 46296) and Gephyrin-GFP were described previously11. Note that in COS-7 cell experiments a non-transcriptionally controlled version of GPHN.FingR was used, but transcriptionally controlled GPHN.FingR was used in all other contexts. GHA-tagged Human Ubiquitin inserted into the constitutive mammalian expression vector pcDNA3 (Addgene plasmid # 18712)38. Plasmids for expression of RandE3 or GFE3 in zebrafish were created by ligating the assembled gene (RandE3 or GFE3) into the modified expression vector pMT(3mnx1), which contains a triplicated enhancer element from the mnx1 gene upstream of the MCS that drives expression of the ablating FingR in spinal cord neurons20. A vector to mediate inducible expression of GFE3 (GFE3-TagRFPind) was generated by subcloning tagRFP-4(GGGS)-GFE3 into the pTRE-3G vector (Tet-On 3G, Clontech, cat. # 631337). GFE3, RandE3, or GPHN.FingR was inserted into the MCS of the pFUW vector for production of lentivirus at the USC School of Pharmacy Lentiviral Laboratory. Similarly, GFE3 or GPHN.FingR was subcloned into the ‘cre-on’ pAAV-EF1a-DIO vector for production of AAV at the University of Pennsylvania School of Medicine Vector Core. The sequences for all constructs were verified by DNA sequencing.
Expression of Gephyrin Ablating FingRs in Cells
Constitutive expression of GPHN.FingR, GFE3 or RandE3 in mammalian cells was achieved through CalPhos (Clontech, cat. # 631312) or Lipofectamine 2000 (Thermo Fisher Scientific, cat. # 11668019) transfection, lentiviral transduction or adeno-associated virus (AAV) transduction of the construct into cells. Inducible expression of GFE3 was achieved by CalPhos-mediated co-transfection of GFE3-TagRFPind with the doxycycline-dependent Tet-On expression vector pCMV-Tet-3G (Tet-On 3G, Clontech, cat. # 631337) into cells. In experiments where the transcriptionally controlled Gephyrin FingR was co-expressed with doxycycline-induced GFE3, 2 µg of Gephyrin.FingR-GFP-CCR5ZFL-KRAB(A) was co-transfected with 1 µg each of GFE3-TagRFPind and pCMV-Tet-3G into cells. 1 µg of doxycycline (EMD Millipore, cat. # 631311) in DMSO was added per mL of medium to cells to activate the Tet-On 3G expression system, and was removed by washing the cells six times with conditioned medium. Expression time of constructs ranged from 5 hours to 10 days, as specified. For lentiviral infection 4 infectious units (IFU) of lentivirus at a titer of 3.0×107 IFU/mL was used per cell by adding concentrated virus to the medium for 16 to 24 hours. Transfection or transduction of cells was performed according to the manufacturer’s suggested protocol. Experimental protocols were conducted according to the US National Institutes of Health guidelines for animal research and were approved by the Institutional Animal Care and Use Committee at the University of Southern California, the Animal Care Committee at Laval University and the Institutional Animal Care and Use Committee at Harvard Medical School.
Ubiquitination and Proteasome Dependency
COS-7 cells (ATCC, cat. # CRL-1651) were cultured in Dulbecco’s modified eagle’s medium (ATCC, cat. # 30-2002) supplemented with 10% fetal bovine serum in 5% CO2 to a confluency of 80% on a 100 × 20 mm tissue culture dish before being co-transfected with constructs constitutively expressing the RandE3, GFE3 or the Gephyrin ablating FingR control along with Gephyrin-GFP and HA-Ubiquitin. 24 hours after transfection, cells were lysed in 1 mL of lysis buffer (150 mM NaCl, 1 mM EDTA, 10 mM Tris, pH 8.0, 1% NP40, 0.12 mg/mL PMSF, 2 µg/mL Leupeptin, 1 µg/mL Aprotinin, 10 mM NaF, 1 µg/mL Pepstatin). Proteasome activity in cells was arrested by adding the highly specific and irreversible inhibitor clasto-Lactacystin-[beta]-lactone (10 µM, VWR, cat. # 80052-808) in DMSO to the culture medium. To examine Gephyrin ubiquitination in cells, 500 µl of lysate was pre-cleared with agarose beads for 2 hours at 4°C and then incubated overnight at 4°C with a rabbit anti-Gephyrin antibody (Bethyl Laboratories, cat. # A304-245A) diluted 1:100. The next day, 50 µl of protein A/G agarose beads (Pierce, cat. # 20421) were added to the lysate. The beads were then nutated at 4°C for 8 hours to immunoprecipitate (IP) Gephyrin, washed six times in wash buffer (as lysis buffer, except with 0.1% NP40) and finally boiled for 5 minutes in Laemmli sample buffer (62.5 mM Tris-HCl, pH 6.8, 2.5% SDS, 10% Glycerol, 0.02% bromophenol blue, 5% 2-mercaptoethanol). Cell lysate and eluate from the Gephyrin IP were run on pre-cast Any kD SDS-PAGE gels (Bio-Rad, cat. # 4569033) and transferred to a low fluorescence PVDF membrane (Bio-Rad, cat. # 1620264). The blots were then probed with a mouse anti-Gephyrin antibody (Synaptic Systems, cat. # 147 111), 1:1000, a mouse anti-beta-tubulin antibody (Sigma, cat. # T4026), 1:5000, or a mouse anti-HA antibody (Covance/BioLegend, cat. # 901513), 1:500. Secondary antibody used was goat anti-mouse Alexa Fluor 750 (Thermo Fisher Scientific, cat. # A-21037), 1:1000. Blots were imaged with the Odyssey Infrared Imaging System (LI-COR Biosciences). Steady-state protein levels were determined using ImageJ software (US National Institute of Health). Prior to analyses of data, Gephyrin-GFP levels were normalized to beta-tubulin levels. Data are represented as mean ± SEM and were compared using the non-parametric Kruskal-Wallis test. A P value < 0.05 was considered significant. The supplier (ATCC) tested the cells for mycoplasma and confirmed their species of origin by COI assay.
Lentiviral Transduction Efficiency
Efficiency of lentiviral transduction with GFE3, RandE3 or GPHN.FingR was assessed by counting the number of GFP positive and negative cells in five 133.3 µm2 images taken from infected Delta T dishes. In brief, live cell morphology and GFP expression were imaged and analyzed first with brightfield illumination and FITC fluorescence. Cells were then fixed and stained for GFP and Gephyrin, and mounted in Fluoromount-G with DAPI (Electron Microscopy Sciences, cat. # 17984-24). A GFP positive cell was identified by colocalization of GFP with DAPI in the cell body, whereas a GFP negative cell had no colocalization of DAPI with GFP. Image analyses were done blinded with ImageJ software. Data are represented as mean ± SEM and were subjected to a Kruskal-Wallis test. A P value < 0.05 was considered significant.
Quantitation of knockdown of synaptic protein levels following expression of GFE3
Dissociated rat cortical neurons seeded at a density of 7.5 × 105 on 100 × 20 mm tissue culture dishes were transduced with 3.0 × 106 IFUs of lentivirus with the RandE3, GFE3 or GPHN.FingR. After 16 hours of expression, cells were lysed in 1 mL of lysis buffer with 1% Triton X-100. Samples were then run on Any kD SDS-PAGE gels and transferred to PVDF membrane. The blots were probed with a mouse anti-Gephyrin antibody, 1:1000, a rabbit anti-GABAA receptor alpha1 antibody (EMD Millipore, cat. # 06-868), 1:1000, a mouse anti-PSD-95 antibody (NeuroMab, cat. # 75-028), 1:1000, a mouse anti-GluA1 antibody (NeuroMab, cat. # 75-327), 1:1000, a mouse anti-GluN2B antibody (NeuroMab, cat. # 75-097), 1:1000, a mouse anti-Collybistin II antibody (Santa Cruz Biotechnology, cat. # sc-136393), 1:1000, a rabbit anti-GABARAP antibody (Origene, cat. # TA307845), 1:1000, or a mouse anti-beta-tubulin antibody, 1:5000. Secondary antibody used was goat anti-mouse Alexa Fluor 750, 1:1000, or goat anti-rabbit Alexa Fluor 750, 1:1000 (Thermo Fisher Scientific, cat. # A-21039). The blots were imaged at multiple intensities using the Odyssey Infrared Imaging System. Measurements of synaptic protein steady-state levels from four independent experiments were obtained using ImageJ software. This data was normalized to beta-tubulin levels and subjected to a Kruskal-Wallis test. All data are expressed as mean ± SEM. A P value < 0.05 was considered significant.
Preparation of Cortical Neurons
Cortices from E17 Sprague-Dawley rat embryos were dissected in 0.1 mM HEPES (Thermo Fisher Scientific, cat. # 15630-080) supplemented Hank’s balanced salt solution (HBSS, Thermo Fisher Scientific, cat. # 14025076), HEPES-HBSS. The dissected cortices were trypsinized in 0.25 % trypsin, 0.1 mM HEPES-HBSS for 15 min at 37°C; the tissue was then washed 3 times in fresh HEPES-HBSS. Afterwards, the cortices were triturated to dissociate the cells. The neurons were then plated in poly-D-lysine and laminin pre-treated 22 × 22 mm coverslips (Thermo Fisher Scientific, cat. # 12-541B) in 6 well plates (VWR International, cat. # 29442-036) at a density of 5.0×104, 0.17 mm Delta T culture dishes (Bioptechs, cat. # 04200417C) at a density of 3.5×104, 100 × 20 mm tissue culture dishes (VWR, cat. # 25382-166) at a density of 7.5×105, and 96 well plates (Sigma, cat. # CLS3610) at a density of 2.5×103 cells per well in supplemented Neurobasal medium (NBM, Thermo Fisher Scientific, cat. # 21103-049). The NBM was supplemented with 10 mL/L Glutamax (Thermo Fisher Scientific, cat. # 35050-061), 1 mg/L gentamicin solution (Thermo Fisher Scientific, cat. # 15750-078), 20 mL/L B-27 supplement (Thermo Fisher Scientific, cat. # 17504-044), and 50 mL/L fetal bovine serum (Biowest, cat. # S162H). Four hours after the neurons were plated, the medium was diluted 1:3 with serum-free supplemented NBM. After 7 days in vitro, the medium in the wells was diluted 1:2 with fresh supplemented NBM. Neurons were cultured 12–16 days in vitro (DIV) before transfection with CalPhos or 16–20 DIV prior to transduction with lentivirus.
Immunocytochemistry of Cortical Neurons
Cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences, cat. # 15714) for 5 minutes, and then subjected to three 5 minute washes with PBS. Prior to staining with primary antibody, cells were blocked with blocking solution (1% bovine serum albumin, 5% normal goat serum, 0.1% Triton X-100 in PBS) for 30 minutes. After blocking, primary antibody was diluted in blocking solution and added to cells for 1 hour. This was followed by three 5 minute washes of cells in PBS. Secondary antibody was diluted in blocking solution and added to cells for 30 minutes in the dark. After staining, cells were washed for 5 minutes three times with PBS and then mounted onto 75 × 25 mm glass microscope slides (VWR, cat. # 48300-025) in Fluoromount-G (Electron Microscopy Sciences, cat. # 17984-25) or Fluoromount-G with DAPI. All steps were performed at room temperature. Primary antibody concentrations were as follows: chicken anti-GFP (Aves, cat. # GFP-1020), 1:4000, mouse anti-Gephyrin (Synaptic Systems, cat. # 147 008), 1:300, rabbit anti-tRFP (Evrogen, cat. # AB234), 1:5000, mouse anti-GAD-65 (Synaptic Systems, cat. # 198 111), 1:500, mouse anti-PSD-95 (NeuroMab, cat. # 75-028), 1:1000, and rabbit anti-GABAA receptor alpha1 (EMD Millipore, cat. # 06-868), 1:1000. Secondary antibodies used were: goat anti-chicken Alexa Fluor 488, goat anti-mouse Alexa Fluor 594, goat anti-rabbit Alexa Fluor 594, goat anti-rabbit Alexa Fluor 647, goat anti-mouse IgG2a Alexa Fluor 594, goat anti-mouse IgG3 Alexa Fluor 594, goat anti-mouse IgG1 Alexa Fluor 594 and goat anti-mouse IgG1 Alexa Fluor 647 (Thermo Fisher Scientific, cat. # A-11039, A-11005, R-37117, A-27040, A-21135, A-21155, A-21125, A-21240), all at dilutions of 1:1000.
Image Capture and Analysis of Cortical Neurons
Imaging of fixed, immunostained cells was done on a Bio-Rad MRC-1024 confocal microscope. Each cell was imaged as a single optical section with either an ×20 objective at 1.0× zoom or an ×40 objective at 2.0× zoom, unless otherwise noted. Stained levels of Gephyrin, PSD95 and GABAAR were determined by measuring mean pixel intensity on dendrites from neurons with similar morphology that expressed GFE3, RandE3 or GPHN.FingR. Image analyses were performed blinded with ImageJ software. Data are represented as mean ± SEM and were compared using a Kruskal-Wallis or Mann-Whitney test. A P value < 0.05 was considered significant. Living cells on Delta T culture dishes were imaged in HBSS buffer, supplemented with 10mM HEPES and pre-warmed to 37°C, with an ×20 air objective or ×60 water objective at 1.0× zoom on an Olympus X81 inverted microscope equipped with the Delta T system (Bioptechs, cat. # 0420-4), an EM-CCD digital camera (Hamamatsu, cat. # C9100-02), DAPI, GFP/mCherry and Cy5.5 filter cubes (Chroma Technology, cat. # 49000, cat. # 59022 and # SP105), an MS-2000 XYZ automated stage (Applied Scientific Instrumentation), an X-cite exacte mercury lamp (Excelitas Technologies) and Metamorph software (Molecular Devices). Imaging of GFP and TagRFP was done at 25% intensity with a 400 ms exposure to minimize bleaching and phototoxicity affects. Intensity values of puncta labeled by transcriptionally controlled GPHN.FingR-GFP in images taken at different time points were determined with ImageJ software. All analyses of cells were performed blinded. Only neurons with healthy morphology were selected for quantification, and none of the images used contained saturated pixels in the axon or dendrites of cells. Data are represented as mean pixel intensities ± SEM and were compared using the Mann-Whitney test. A P value < 0.05 was considered significant.
Cell Viability, Cytotoxicity, ATP Levels and Proteasome Activity
2.5 × 103 rat cortical neurons/well in 96 well white-walled plates were transduced with 9×103 IFUs of lentivirus with GFE3, RandE3 or GPHN.FingR for 16 hours. Cell viability and cytotoxicity were assessed using the MultiTox-Glo Multiplex Cytotoxicity Assay kit (Promega, cat. # G9270) that measures: 1) fluorescence generated in living cells from selective uptake of a protease substrate conjugated to a fluorophore (AFC) that fluoresces on proteolytic cleavage, and 2) light produced by Luciferase from a substrate (aminoluciferin) that is activated by proteases released into the medium from dead cells. The ATP levels of neurons were measured using the CellTiter-Glo Luminescent Cell Viability Assay kit (Promega, cat. # G7570), which uses ATP released from lysed cells and Luciferase to generate light. As a positive control for cell viability and cytotoxicity assays, the mitochondrial toxin 3-nitropropionic acid (3-NP, Sigma, cat. # N5636) was applied to cells at a final concentration of 15 mM for 48 hours39. Proteasome activity in transduced cells was determined using the Proteasome-Glo 3-Substrate Cell-Based Assay kit (Promega, cat. #G1180), which uses proteasome caspase-, trypsin- and chymotrypsin-like activity to produce light via aminoluciferin-based substrates and Luciferase. As a positive control for proteasome activity, the highly specific proteasome inhibitor clasto-Lactacystin-[beta]-lactone was added to the medium of cells at a final concentration of 10 µM for 3 hours before obtaining measurements. All assays were performed according to the instructions provided by the manufacturer. Fluorometric and luminometric measurements were taken with a Fluoroskan Ascent FL (Thermo Fisher Scientific) microplate reader. 24 wells were measured per condition. Data are represented as measurements of transduced (or positive control) cells divided by the mean of non-transduced (or vehicle control) cells ± SEM and were compared using the Kruskal-Wallis test. A P value < 0.05 was considered significant.
Preparation and Transfection of Hippocampal Neurons
Dissociated hippocampal neurons were prepared as described previously40. Briefly, dissected hippocampi from P1–P3 Sprague Dawley rats of both sexes were dissociated enzymatically (papain, 12 U/mL; Worthington) and mechanically (trituration with a Pasteur pipette). After dissociation, the cells were washed, centrifuged and plated on poly-D-lysine-coated Aclar (12 mm) coverslips at a density of 350–450 cells/mm2. Growth medium consisted of NBM and B27 (50:1), supplemented with penicillin/streptomycin (50 U/mL; 50 µg/mL) and 0.5 mM Glutamax. Fetal bovine serum (2%, Hyclone, GE Healthcare, cat. # SH30088.02) was added at the time of plating. After 5 days, half the medium was changed to a medium without serum, but with Ara-C added (5 µM; Sigma-Aldrich; cat. # C1768) to limit proliferation of non-neuronal cells. Twice a week, half of the growth medium was replaced with serum and Ara-C free medium. Neurons were transfected at 11–13 DIV using Lipofectamine 2000, as previously described40. To avoid synaptic scaling after gephyrin ablation, cultures were treated with TTX (1 µM) 24 h before recording. By that time, expression of FingR constructs was observed
Electrophysiology of Hippocampal Neurons
Coverslips containing transfected neurons were transferred to a recording chamber and perfused with a recording solution which consisted of HBSS containing: 1.2 mM CaCl2, and 1.0 mM MgCl2,10 mM Hepes, 2.0 mM glucose, pH 7.3. The osmolarity of the solution was adjusted to 290 mOsm/kg. Whole-cell voltage clamp recordings were obtained with borosilicate glass pipettes (4–6 MΩ). For mIPSC recordings, pipettes were filled with an internal solution containing: 100 mM CsMeSO3, 20 mM CsCl, 2.5 MgCl2, 70 mM Hepes, 0.6 mM Cs-EGTA, 4 mM Mg-ATP, 0.4 mM Tris-GTP, pH 7.3 and clamped at 0 mV in the presence of TTX (1 µM). Evoked GABA currents were recorded with an internal solution containing: 120 mM CsCl, 2.0 mM MgCl2, 1.0 mM CaCl2, 10 mM Hepes, 11 mM Cs-EGTA, 4 mM Mg-ATP, 1 mM TEA, pH 7.3. After a stable baseline, a high concentration of GABA (1 mM) was added to the bath during 1 minute and then washed out. For mEPSC recordings pipettes were filled with an internal solution containing: 100 mM CsMeSO3, 20 mM CsCl, 2.5 MgCl2, 70 mM Hepes, 0.6 mM Cs-EGTA, 4 mM Mg-ATP, 0.4 mM Tris-GTP, pH 7.3. Miniature excitatory and inhibitory post-synaptic currents (mEPSCs and mIPSCS) were recorded at holding potentials of −70 mV and 0 mV, respectively, in the presence of TTX (1 µM).
All recordings were performed at room temperature. Signals were low-pass filtered at 2 kHz and digitized at 10 kHz using a Multiclamp 700B amplifier, Digidata 1440 digitizer and pClamp 10 software (Molecular Devices). Comparisons of parameters for mIPSCs and mEPSCs were made using a Kruskal-Wallis test followed by a Dunn's multiple parameter comparison. Responses to GABA applications were analyzed using 2-tailed unpaired Mann-Whitney test. A P value < 0.05 was considered significant. All data are expressed as mean ± SEM.
Noise analysis
To see if unclustering of GABAA receptors affects their kinetic properties, we performed stationary noise analysis of GABAA receptors activated by the exogenous application of GABA (1mM) as previously described41. Briefly, a region of 50 s after peak GABA activation was high pass filtered at 1 Hz and a power spectrum was derived. This spectrum was fitted with the sum of three Laurentzian functions of the form:
where L(f) is the spectral density at frequency f. Si is the power of the spectrum at f = 0 and fci is the cutoff frequency at which the spectral power is half. The corresponding time constants (τi) are derived by the relationship τi = 1/(2πfci)41.
Zebrafish Husbandry and Experimentation
Wild-type AB and AB/TL zebrafish (Danio rerio) were raised and nucleic acid injections were performed as described previously42. Embryos were maintained at 28°C for up to 5 days post fertilization (dpf) in egg water (1.19 g NaCl, 0.377 g CaSO4·2H2O, 265 µL methylene blue, filled to 5 L with ddH2O). Within the 28°C incubator, the fish were exposed to a 13/11 hour day/night light cycle. At the zygote stage, zebrafish were left uninjected (control) or were injected with 45–70 pg of DNA, along with 35 pg of Tol2 Transposase43 mRNA27 S to facilitate mosaic integration of the pMT(3mnx1) plasmid DNA into the zebrafish genome. Before behavior experiments (i.e. spontaneous tail flick), 17–22 hour post fertilization zebrafish embryos in egg water were carefully dechorionated with a pair of #5 forceps (Fine Science Tools). 200 µL of egg water supplemented with 0.003% 1-phenyl 2-thiourea (PTU, Sigma-Aldrich, cat. # P7629) – to inhibit pigment formation – was added to individual wells of a 96-well plate, and individual zebrafish were placed into each of the filled wells with a glass pipette. Gender could not be determined due to the age of the fish44. Zebrafish tail flicking behavior was recorded (40 frames per second) from below the well plate using a compact USB 3.0 CMOS camera (Cat #DCC1545M, Thorlabs). After tail flick imaging, zebrafish were kept overnight at 28°C within the 96-well plate. At 2 dpf, zebrafish injected with the spinal cord neuron expressing GFE construct were anesthetized with 0.015–0.03% MS-222 (Argent Laboratories), embedded in 1–1.5% SeaPlaque agarose within a 30X Danieau’s solution (17.4 mM NaCl, 0.21 mM KCl, 0.12 mM MgSO4, 0.18 mM Ca(NO3)2, 1.5 mM HEPES), and imaged using a Plan Apochromat ×20 objective on a commercial LSM 700 confocal microscope (Carl Zeiss AG), as similarly described45. Mosaic-labeled GFP positive cells were visualized throughout the zebrafish spinal cord using the tiled confocal stack feature of the microscope. For immunostaining of zebrafish, 1–2 dpf zebrafish were anesthetized in 0.03% MS-222 and were fixed in 2% Trichoroacetic acid for 3 hours at room temperature. After 5 washes in PBS pH 7.4 with 0.5% Triton X-100 (PBT), fish were blocked overnight in blocking buffer (PBS pH 7.4, 1% bovine serum albumin, 1% DMSO, 0.1% Triton X-100, 2% normal goat serum) at 4 °C on an orbital shaker. Mouse anti-Gephyrin antibody (Synaptic Systems, cat. # 147 111) was applied at a 1:250 dilution in blocking buffer and the embryos were left at 4 °C on an orbital shaker for 72–96 hours. After 5 washes in PBT, goat anti-mouse Alexa Fluor 594 antibody (Thermo Fisher Scientific, cat. #A-11005) was applied at a 1:2000 dilution in blocking buffer, and the sample was left on an orbital shaker at 4 °C for 14–18 hours. Following 5 washes in PBT, samples were washed in PBS pH 7.4. For clearing, samples were progressively dehydrated in glycerol, ultimately ending in a solution of 75% glycerol / 25% PBS pH 7.4. Samples were placed on #1.5 coverslips and were imaged with a commercial LSM 780 confocal microscope (Carl Zeiss AG) using an ×63 water objective. Analyses of Gephyrin levels in confocal images were performed blinded with ImageJ software. All data are expressed as mean ± SEM. Zebrafish were included in analysis that had at least 20 labeled motoneurons. No randomization was used.
Stereotaxic Intracranial Injections
To examine inhibitory synaptic inputs, a total of 1 µl AAV-DIO-GFE3-GFP (‘cre-on’) mixed at a 2:1 ratio with AAV-FAS-ChR2-mCherry (‘cre-off’37) was delivered by intracranial injection into the striatum of Adora2a-cre mice (GENSAT: B6.FVB(Cg)-Tg(Adora2acre)KG139Gsat/Mmucd) (P18-P22) of both sexes maintained under deep isoflurane anesthesia. The non-ablating gephyrin intrabody (AAV-DIO-GPHN.FingR-GFP11) was used as an internal control in the other hemisphere of each mouse. Note that AAV-EF1a-DIO-GFE3 and AAV-EF1a-DIO-GPHN.FingR are AAV serotype 9, AAV-EF1a-FAS-ChR2-mCherry is serotype 8 and 3.8×1010 genome copies (GC) at a titer of 3.8×1013 GC/mL were used. To test excitatory inputs, AAV-DIO-GFE3-GFP or AAV-DIO-GPHN.FingR-GFP mixed with AAV-hSyn-cre at a 100:1 GC-ratio was injected into the striatum of age-matched wildtype mice of both sexes (C57Bl/6, Charles River). Injection coordinates were 0.7 mm anterior from Bregma, 1.8 mm lateral, and 2.45 mm below pia, and expression time was at 12–14 days.
Slice Preparation and Electrophysiology
Acute sagittal brain slices were prepared, and electrophysiological recordings obtained, as described before46, with the following variations: Spiny projection neurons of the indirect pathway were identified by GFP expression and their electrical properties, and synaptic currents were recorded using a high-chloride pipette solution containing (in mM) 125 CsCl, 10 TEA-Cl, 0.1 EGTA (CsOH), 10 HEPES, 3.3 QX-314 (Cl-), 1.8 MgCl2, 4 Na2-ATP, 0.3 Na-GTP, 8 Na2-phosphocreatine (pH 7.3 adjusted with CsOH; 295 mOsm·kg−1). GABAA receptor-mediated currents were recorded as inward currents in voltage-clamp mode at Vhold = −70mV in the presence of the AMPA and NMDA-type glutamate receptor inhibitors NBQX and (R)-CPP (10 µm each). Excitatory currents were isolated by adding SR95331 (10 µM) to the bath solution. Tonic GABAA receptor mediated currents were defined as the difference in holding current before and after bath application of SR95331 (10 µM). Pipette resistance was 2.5–3 MΩ, and all recordings were performed at 33–34°C. Errors due to the voltage drop across the series resistance (< 15 MΩ) were left uncompensated. ChR2 was activated by a single 1 ms pulse of light delivered by a 473 nm laser using full-field illumination through the objective (~3 mW·mm−2) at 30 s intervals. All pharmacological reagents were obtained from Tocris.
Data Acquisition and Analysis
Membrane currents were acquired as described previously46, and analyzed in Igor Pro (Wavemetrics). For display and analysis of optically evoked synaptic membrane currents, 10 consecutive traces were averaged for each cell. Data are represented as mean ± SEM (bars) overlaid over individual data points (gray circles) and were compared using the non-parametric Mann-Whitney test. P values are reported directly, and values smaller than 0.05 were considered statistically significant.
Statistics
No statistical methods were used to predetermine sample size. Nonparametric tests were used throughout, as the data are not normally distributed. Data are represented as mean ± SEM. Statistical comparisons are between groups with similar variances.
Supplementary Material
Supplementary Figure 1, related to Figure 1. Generation of a synthetic E3 ligase that targets Gephyrin to proteasomes for degradation. (a) RING domain of X-linked Inhibitor of Apoptosis Protein (XIAP) is fused to the C-terminus of GPHN.FingR with a triplicated HA-tag (3HA), and GFP or a red fluorescent protein (TagRFP) is fused to the GPHN.FingR N-terminus through a GGGS linker (4GGGS) to give GFE3. A FingR containing randomized residues within its binding interface (Random FingR) is exchanged for GPHN.FingR to give RandE3. GPHN.FingR with GFP fused to the N-terminus is used as a control. (b) Gephyrin and PSD-95 immunostaining in cortical neurons indicates a 93 ± 2% (P < 0.0001, Kruskal-Wallis) reduction in Gephyrin levels in the presence of GFE3 versus RandE3 or GPHN.FingR, and no change in PSD-95 levels (P > 0.97, Kruskal-Wallis). n = 10 neurons per condition. (c) Lysates from COS-7 cells expressing GFE3, HA-tagged Ubiquitin (HA-Ubiquitin), and Gephyrin-GFP for 24 hours labeled with an anti-Gephyrin antibody. GFE3 strongly reduced Gephyrin-GFP (80 ± 2%, P = 0.01, Kruskal-Wallis) compared to RandE3 or GPHN.FingR, but not in the presence of 10 µM Lactacystin (P > 0.78, Kruskal-Wallis). Beta-tubulin is the loading control. The band below Gephyrin-GFP represents an endogenous COS-7 cell protein recognized by the anti-Gephyrin antibody, but not by GPHN.FingR. n = 4 replicate blots. Bars indicate means ± SEM. (d) Immunoprecipitation of Gephyrin-GFP from COS cell lysate using an anti-Gephyrin antibody, and staining for HA-Ubiquitin. GFE3 co-expression results in Gephyrin ubiquitination, whereas RandE3 or Gephyrin.FingR co-expression does not. High molecular weight bands (>150 kD) in the GFE3 lane likely correspond to poly-ubiquitinated Gephyrin that accumulates due to proteasome inhibition by Lactacystin. Elevated HA-Ubiquitin in lysate from cells expressing GFE3 or RandE3 vs. GPHN.FingR can be attributed to auto-ubiquitination by the overexpressed FingR-E3 ligase.
{kind=link}
Supplementary Figure 2, related to Figure 1. Efficacy of lentiviral mediated expression of GFE3 in cortical neurons. (a–l) Cultured cortical neurons infected with approximately 4 infectious units (IFU) of lentivirus per cell. Cells were fixed and stained for GFP (green), Gephyrin (red) and DAPI, to label nuclei, (blue). Scale bar represents 10 µm. (m) Quantification of DAPI labeled, GFP positive, and negative, cells suggests a transduction efficiency of 84 ± 2% of GFE3, 85 ± 4% of RandE3 and 84 ± 2% of GPHN.FingR (P > 0.93, Kruskal-Wallis). Bars indicate means ± SEM.
{kind=link}
Supplementary Figure 3, related to Figure 1. Expression of GFE3 in cortical neurons results in specific and rapid degradation of endogenous Gephyrin. (a–i) Western blotting for endogenous Gephyrin (a, arrow), GABAA receptor alpha1 (b, arrow), the Gephyrin binding protein Collybistin II (c, arrow), GluA1 (d, arrow), GluN2B (e, arrow), PSD-95 (f, arrow), and the Gephyrin and GABAA receptor binding protein GABARAP (h, arrow) in cells expressing GFE3, RandE3, GPHN.FingR or in untransduced (Control) cells. (g) Beta-tubulin levels that serve as a loading control for a–f. (i) Beta-tubulin levels that serve as a loading control for h.
{kind=link}
Supplementary Figure 4, related to Figure 1. Quantification of endogenous synaptic protein levels in cortical neurons expressing GFE3. (a–g) Western blotting for synaptic proteins in GFE3 transduced dissociated cortical neurons suggests that GFE3 specifically reduces endogenous Gephyrin levels. A marked decrease in Gephyrin (a; 80 ± 3%, P = 0.01, Kruskal-Wallis) occurs in neurons expressing GFE3 compared to neurons not transduced (Control) or transduced with RandE3 or Gephyrin.FingR. In contrast, no significant difference is observed for the steady-state levels of the alpha1 subunit of the GABAA receptor, PSD-95, the GluA1 subunit of the AMPA receptor, the GluN2B subunit of the NMDA receptor, or the Gephyrin interacting proteins Collybistin II and GABARAP (b–g; P > 0.8, Kruskal-Wallis). n = 4 replicate blots. (h, i) Quantification of endogenous Gephyrin and GABAA receptor immunostaining in cortical neurons showed a 93 ± 1% (P < 0.0001, Mann-Whitney) reduction in Gephyrin levels in cells transfected with GFE3 compared to cells with RandE3, and no change in GABAA receptor levels (P > 0.99). Sample size is n = 10 neurons per condition. Bars indicate means ± SEM.
{kind=link}
Supplementary Figure 5, related to Figure 1. Expression of GFE3 in cortical neurons does not affect cell viability or proteasome activity. (a, b) Lentiviral mediated expression of GFE3, RandE3 or GPHN.FingR in cortical neurons for 16 hours had a similar effect on cell viability (P > 0.96, Kruskal-Wallis) and cytotoxicity (P > 0.98), based on a multiplexed assay of protease activity in live cells and from dead cells. In contrast, addition of the mitochondrial toxin 3-nitropropionic acid (3-NP, 15 mM) to the medium for 48 hours induced an 80 ± 1% decrease in cell viability (P < 0.0001) and a 5-fold increase in cytotoxicity (P < 0.0001). (c) ATP levels in cells expressing GFE3, RandE3 or GPHN.FingR for 16 hours were unchanged (P > 0.96, Kruskal-Wallis), whereas cells that were treated with 3-NP for 48 hours exhibited an 80 ± 1% decrease in ATP levels (P < 0.0001). (d–f) Measurement of proteasome caspase- (d; P > 0.96, Kruskal-Wallis), chymotrypsin- (e; P > 0.87, Kruskal-Wallis) and trypsin-like (f; P > 0.86, Kruskal-Wallis) activity in cells transduced with GFE3, RandE3 or GPHN.FingR for 16 hours indicates that GFE3 does not alter proteasome function. In contrast, addition of the highly specific proteasome inhibitor clasto-Lactacystin-[beta]-lactone (Lactacystin, 10 µM) to the medium of cells for 3 hours resulted in the inhibition of all three types of proteasome activity (P < 0.0001). Data is shown as experimental (transduced or drug-treated) cell measurements normalized to the mean of Control (not transduced or vehicle-treated) cell measurements. Bars indicate means ± SEM. Sample size is n = 24 wells of cells in a 96 well plate per condition.
Supplementary Figure 6, related to Figure 2. Expression of GFE3 does not affect frequency or amplitude of EPSCs. (a, b) GFE3-mediated ablation of Gephyrin does not affect mEPSCs. Representative traces of mEPSCs obtained from dissociated hippocampal neurons expressing GFE3 or non-transfected controls (a). No significant change in mEPSC amplitude or frequency was observed in GFE3 expressing cells compared to controls (b; P > 0.75, Mann-Whitney). n = 5 independent experiments for controls and GFE3. Bars indicate means ± SEM.
{kind=link}
Supplementary Figure 7, related to Figure 2. Noise analysis of GABA evoked currents. (a) Power spectrum of a continuous recording after GABA (1 mM) application was fitted with the sum (solid lines) of three Laurentzian functions (dotted lines). Spectral analysis of GABA-induced noise in control (non-transfected) cells revealed three average corner frequencies at 3.2 ± 0.6, 31.6 ± 4.2, 380.0 ± 75.4 Hz (n = 5) corresponding to time constants of 53.7, 4.9, 0.3 ms, respectively. (b) In GFE3 expressing cells the three corner frequencies obtained were 1.7 ± 0.3, 36.7 ± 10.6, 316.9 ± 129.7 Hz (n = 6), corresponding to time constants of 105.4, 6.9, 0.9 ms, respectively. (c) No statistical difference was observed between these measurements (P > 0.05, Mann-Whitney) indicating that uncoupling the GABAA receptors from Gephyrin did not cause an observable change in channel gating. Data are expressed as means ± SEM.
{kind=link}
Supplementary Figure 8, related to Figure 4. GFE3 expression in spinal cord neurons of zebrafish impairs tail flick behavior. (a) Time-lapse images of a zebrafish age 24 hours post fertilization with mosaic expression of RandE3 in spinal motoneurons showed stereotyped tail flick behavior, which is comprised of a coordinated curling of the tail that brings the tip to the base. (b) In contrast, an age-matched sibling that expresses GFE3 in spinal motoneurons exhibits abnormal tail flick behavior, being unable to curl its tail sufficiently to touch the tip to the base.
{kind=link}
Supplementary Figure 9, related to Figure 5. Transient expression of GFE3-TagRFP in cortical neurons. (a) A rat cortical neuron transfected with doxycycline (dox)-inducible GFE3-TagRFP, before addition of dox to the culture medium (t = 0 hrs). (b) Following incubation with 1 µg/mL of dox for 5 hours, GFE3-TagRFP expression is visible (t = 5 hrs). (c) A rat cortical neuron transfected with dox-inducible GFE3-TagRFP prior to incubation with dox (t = 0 hrs). (d) High levels of GFE3-TagRFP are present in the neuron 24 hours after adding 1 µg/mL of dox (t = 24 hrs). (e) Dox was removed by washing six times with conditioned medium. No GFE3-TagRFP is detectable in the neuron following 48 hours of recovery. Scale bar represents 10 µm.
{kind=link}
Supplementary Video 1, related to Figure 4. Zebrafish expressing GFE3.
Zebrafish expressing GFE3-GFP in motoneurons in the spinal cord displays an abnormal tail flick.
Supplementary Video 2, related to Figure 4. Zebrafish expressing GFE3.
Zebrafish expressing GFE3-GFP in motoneurons in the spinal cord displays an abnormal tail flick.
Supplementary Video 3, related to Figure 4. Zebrafish expressing RandE3.
Zebrafish expressing RandE3-GFP in motoneurons in the spinal cord displays a normal tail flick.
Supplementary Video 4, related to Figure 4. Zebrafish expressing RandE3.
Zebrafish expressing RandE3-GFP in motoneurons in the spinal cord displays a normal tail flick.
Acknowledgments
We thank M. Roth for technical support, Y. Davis and C. Tian for assistance in analyzing the data, L. Chen for use of his microplate reader, and E. Liman for helpful suggestions on the manuscript. Lentivirus was created by L. Asatryan at USC School of Pharmacy Lentiviral Core Laboratory. This work was supported by NIH (grants AI085583 and GM083898 to R.W.R., NS046579 to B.L.S. and NS081687 to D.B.A.), by the Human Frontier Science Program (to Y.D.K and D.B.A), by the Canadian Institutes of Health Research (MOP12942 to Y.D.K. and MOP286161 to P.D.K) and by the McKnight foundation (to D.B.A).
Footnotes
Author Contributions:
G.G.G. and D.B.A conceived of the project and designed the imaging, cell biology and biochemistry experiments, which were performed by G.G.G. C.S. and B.L.S. designed and C.S. performed electrophysiology experiments in slices. J.P.S., P.D.K, and Y.D.K. designed and J.P.S. performed electrophysiology experiments on dissociated neurons. W.P.D and D.B.A designed and W.P.D. performed experiments on zebrafish assisted by L.A.T and S.E.F. J.A.J. and R.W.R. contributed to the design of constructs. D.B.A. and G.G.G wrote the paper with contributions from all authors.
Competing Financial Interest Statement
The authors declare no competing financial interests.
References
- 1.Cabot JB, Bushnell A, Alessi V, Mendell NR. Postsynaptic gephyrin immunoreactivity exhibits a nearly one-to-one correspondence with gamma-aminobutyric acid-like immunogold-labeled synaptic inputs to sympathetic preganglionic neurons. J Comp Neurol. 1995;356:418–432. doi: 10.1002/cne.903560309. [DOI] [PubMed] [Google Scholar]
- 2.Craig AM, Banker G, Chang W, McGrath ME, Serpinskaya AS. Clustering of gephyrin at GABAergic but not glutamatergic synapses in cultured rat hippocampal neurons. J Neurosci. 1996;16:3166–3177. doi: 10.1523/JNEUROSCI.16-10-03166.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng G, et al. Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity. Science. 1998;282:1321–1324. doi: 10.1126/science.282.5392.1321. [DOI] [PubMed] [Google Scholar]
- 4.Capecchi MR. Altering the genome by homologous recombination. Science. 1989;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- 5.McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nature reviews. Genetics. 2002;3:737–747. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]
- 6.Hedstrom KL, Ogawa Y, Rasband MN. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183(4):635–640. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Incontro S, Asensio CS, Edwards RH, Nicoll RA. Efficient, complete deletion of synaptic proteins using CRISPR. Neuron. 2014;83:1051–1057. doi: 10.1016/j.neuron.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 9.Caussinus E, Kanca O, Affolter M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat Struct Mol Biol. 2012;19:117–121. doi: 10.1038/nsmb.2180. [DOI] [PubMed] [Google Scholar]
- 10.Yeh JT, Binari R, Gocha T, Dasgupta R, Perrimon N. PAPTi: a peptide aptamer interference toolkit for perturbation of protein-protein interaction networks. Sci Rep. 2013;3:1156. doi: 10.1038/srep01156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gross GG, et al. Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron. 2013;78:971–985. doi: 10.1016/j.neuron.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galban S, Duckett CS. XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ. 2010;17:54–60. doi: 10.1038/cdd.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kins S, Betz H, Kirsch J. Collybistin, a newly identified brain-specific GEF, induces submembrane clustering of gephyrin. Nat Neurosci. 2000;3:22–29. doi: 10.1038/71096. [DOI] [PubMed] [Google Scholar]
- 14.Chen L, Wang H, Vicini S, Olsen RW. The gamma-aminobutyric acid type A (GABAA) receptor-associated protein (GABARAP) promotes GABAA receptor clustering and modulates the channel kinetics. Proc Natl Acad Sci U S A. 2000;97:11557–11562. doi: 10.1073/pnas.190133497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacob TC, et al. Gephyrin regulates the cell surface dynamics of synaptic GABAA receptors. J Neurosci. 2005;25:10469–10478. doi: 10.1523/JNEUROSCI.2267-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson CJ. GABAergic inhibition in the neostriatum. Prog Brain Res. 2007;160:91–110. doi: 10.1016/S0079-6123(06)60006-X. [DOI] [PubMed] [Google Scholar]
- 17.Ade KK, Janssen MJ, Ortinski PI, Vicini S. Differential tonic GABA conductances in striatal medium spiny neurons. J Neurosci. 2008;28:1185–1197. doi: 10.1523/JNEUROSCI.3908-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santhakumar V, Jones RT, Mody I. Developmental regulation and neuroprotective effects of striatal tonic GABAA currents. Neuroscience. 2010;167:644–655. doi: 10.1016/j.neuroscience.2010.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tritsch NX, Oh WJ, Gu C, Sabatini BL. Midbrain dopamine neurons sustain inhibitory transmission using plasma membrane uptake of GABA, not synthesis. eLife. 2014;3:e01936. doi: 10.7554/eLife.01936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zelenchuk TA, Bruses JL. In vivo labeling of zebrafish motor neurons using an mnx1 enhancer and Gal4/UAS. Genesis. 2011;49:546–554. doi: 10.1002/dvg.20766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirata H, Takahashi M, Yamada K, Ogino K. The biological role of the glycinergic synapse in early zebrafish motility. Neuroscience research. 2011;71:1–11. doi: 10.1016/j.neures.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Cui WW, et al. The zebrafish shocked gene encodes a glycine transporter and is essential for the function of early neural circuits in the CNS. J Neurosci. 2005;25:6610–6620. doi: 10.1523/JNEUROSCI.5009-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirata H, et al. Zebrafish bandoneon mutants display behavioral defects due to a mutation in the glycine receptor beta-subunit. Proc Natl Acad Sci U S A. 2005;102:8345–8350. doi: 10.1073/pnas.0500862102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirata H, Carta E, Yamanaka I, Harvey RJ, Kuwada JY. Defective glycinergic synaptic transmission in zebrafish motility mutants. Frontiers in molecular neuroscience. 2009;2:26. doi: 10.3389/neuro.02.026.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ganser LR, et al. Distinct phenotypes in zebrafish models of human startle disease. Neurobiol Dis. 2013;60:139–151. doi: 10.1016/j.nbd.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu W, et al. Gephyrin clustering is required for the stability of GABAergic synapses. Mol Cell Neurosci. 2007;36:484–500. doi: 10.1016/j.mcn.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mora RJ, Roberts RW, Arnold DB. Recombinant probes reveal dynamic localization of CaMKIIalpha within somata of cortical neurons. J Neurosci. 2013;33:14579–14590. doi: 10.1523/JNEUROSCI.2108-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banaszynski LA, Wandless TJ. Conditional control of protein function. Chem Biol. 2006;13:11–21. doi: 10.1016/j.chembiol.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Sakamoto KM, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varley ZK, et al. Gephyrin regulates GABAergic and glutamatergic synaptic transmission in hippocampal cell cultures. J Biol Chem. 2011;286:20942–20951. doi: 10.1074/jbc.M111.234641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan X, Jin WY, Lu J, Wang J, Wang YT. Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation. Nat Neurosci. 2014;17:471–480. doi: 10.1038/nn.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dev KK. PDZ domain protein-protein interactions: a case study with PICK1. Curr Top Med Chem. 2007;7:3–20. doi: 10.2174/156802607779318343. [DOI] [PubMed] [Google Scholar]
- 33.Young Kim H, Young Yum S, Jang G, Ahn D-R. Discovery of a non-cationic cell penetrating peptide derived from membrane-interacting human proteins and its potential as a protein delivery carrier. Scientific Reports. 2015;5:11719. doi: 10.1038/srep11719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang F, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- 35.Hayashi-Takagi A, et al. Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature. 2015;525:333–338. doi: 10.1038/nature15257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 36.Olson CA, Roberts RW. Design, expression, and stability of a diverse protein library based on the tenth fibronectin type III domain. Protein Science. 2007;16:476–484. doi: 10.1110/ps.062498407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saunders A, Johnson CA, Sabatini BL. Novel recombinant adeno-associated viruses for Cre activated and inactivated transgene expression in neurons. Frontiers in neural circuits. 2012;6:47. doi: 10.3389/fncir.2012.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blobel G, Sabatini DD. Controlled Proteolysis of Nascent Polypeptides in Rat Liver Cell Fractions. Journal of Cell Biology. 1970;45:130–145. doi: 10.1083/jcb.45.1.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pang Z, Geddes JW. Mechanisms of cell death induced by the mitochondrial toxin 3-nitropropionic acid: acute excitotoxic necrosis and delayed apoptosis. J Neurosci. 1997;17:3064–3073. doi: 10.1523/JNEUROSCI.17-09-03064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hudmon A, et al. A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. J Neurosci. 2005;25:6971–6983. doi: 10.1523/JNEUROSCI.4698-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Koninck Y, Mody I. Noise analysis of miniature IPSCs in adult rat brain slices: properties and modulation of synaptic GABAA receptor channels. Journal of Neurophysiology. 1994;71:1318–1335. doi: 10.1152/jn.1994.71.4.1318. [DOI] [PubMed] [Google Scholar]
- 42.Detrich HW, Westerfield M, Zon LI. Essential zebrafish methods : cell and developmental biology. Elsevier: 2009. [Google Scholar]
- 43.Kawakami K, Shima A. Identification of the Tol2 transposase of the medaka fish Oryzias latipes that catalyzes excision of a nonautonomous Tol2 element in zebrafish Danio rerio. Gene. 1999;240:239–244. doi: 10.1016/s0378-1119(99)00444-8. [DOI] [PubMed] [Google Scholar]
- 44.Uchida D, Yamashita M, Kitano T, Iguchi T. Oocyte apoptosis during the transition from ovary-like tissue to testes during sex differentiation of juvenile zebrafish. J Exp Biol. 2002;205:711–718. doi: 10.1242/jeb.205.6.711. [DOI] [PubMed] [Google Scholar]
- 45.Dempsey WP, et al. In vivo single-cell labeling by confined primed conversion. Nat Methods. 2015;12:645–648. doi: 10.1038/nmeth.3405. [DOI] [PubMed] [Google Scholar]
- 46.Straub C, Tritsch NX, Hagan NA, Gu C, Sabatini BL. Multiphasic modulation of cholinergic interneurons by nigrostriatal afferents. J Neurosci. 2014;34:8557–8569. doi: 10.1523/JNEUROSCI.0589-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1, related to Figure 1. Generation of a synthetic E3 ligase that targets Gephyrin to proteasomes for degradation. (a) RING domain of X-linked Inhibitor of Apoptosis Protein (XIAP) is fused to the C-terminus of GPHN.FingR with a triplicated HA-tag (3HA), and GFP or a red fluorescent protein (TagRFP) is fused to the GPHN.FingR N-terminus through a GGGS linker (4GGGS) to give GFE3. A FingR containing randomized residues within its binding interface (Random FingR) is exchanged for GPHN.FingR to give RandE3. GPHN.FingR with GFP fused to the N-terminus is used as a control. (b) Gephyrin and PSD-95 immunostaining in cortical neurons indicates a 93 ± 2% (P < 0.0001, Kruskal-Wallis) reduction in Gephyrin levels in the presence of GFE3 versus RandE3 or GPHN.FingR, and no change in PSD-95 levels (P > 0.97, Kruskal-Wallis). n = 10 neurons per condition. (c) Lysates from COS-7 cells expressing GFE3, HA-tagged Ubiquitin (HA-Ubiquitin), and Gephyrin-GFP for 24 hours labeled with an anti-Gephyrin antibody. GFE3 strongly reduced Gephyrin-GFP (80 ± 2%, P = 0.01, Kruskal-Wallis) compared to RandE3 or GPHN.FingR, but not in the presence of 10 µM Lactacystin (P > 0.78, Kruskal-Wallis). Beta-tubulin is the loading control. The band below Gephyrin-GFP represents an endogenous COS-7 cell protein recognized by the anti-Gephyrin antibody, but not by GPHN.FingR. n = 4 replicate blots. Bars indicate means ± SEM. (d) Immunoprecipitation of Gephyrin-GFP from COS cell lysate using an anti-Gephyrin antibody, and staining for HA-Ubiquitin. GFE3 co-expression results in Gephyrin ubiquitination, whereas RandE3 or Gephyrin.FingR co-expression does not. High molecular weight bands (>150 kD) in the GFE3 lane likely correspond to poly-ubiquitinated Gephyrin that accumulates due to proteasome inhibition by Lactacystin. Elevated HA-Ubiquitin in lysate from cells expressing GFE3 or RandE3 vs. GPHN.FingR can be attributed to auto-ubiquitination by the overexpressed FingR-E3 ligase.
Supplementary Figure 2, related to Figure 1. Efficacy of lentiviral mediated expression of GFE3 in cortical neurons. (a–l) Cultured cortical neurons infected with approximately 4 infectious units (IFU) of lentivirus per cell. Cells were fixed and stained for GFP (green), Gephyrin (red) and DAPI, to label nuclei, (blue). Scale bar represents 10 µm. (m) Quantification of DAPI labeled, GFP positive, and negative, cells suggests a transduction efficiency of 84 ± 2% of GFE3, 85 ± 4% of RandE3 and 84 ± 2% of GPHN.FingR (P > 0.93, Kruskal-Wallis). Bars indicate means ± SEM.
Supplementary Figure 3, related to Figure 1. Expression of GFE3 in cortical neurons results in specific and rapid degradation of endogenous Gephyrin. (a–i) Western blotting for endogenous Gephyrin (a, arrow), GABAA receptor alpha1 (b, arrow), the Gephyrin binding protein Collybistin II (c, arrow), GluA1 (d, arrow), GluN2B (e, arrow), PSD-95 (f, arrow), and the Gephyrin and GABAA receptor binding protein GABARAP (h, arrow) in cells expressing GFE3, RandE3, GPHN.FingR or in untransduced (Control) cells. (g) Beta-tubulin levels that serve as a loading control for a–f. (i) Beta-tubulin levels that serve as a loading control for h.
Supplementary Figure 4, related to Figure 1. Quantification of endogenous synaptic protein levels in cortical neurons expressing GFE3. (a–g) Western blotting for synaptic proteins in GFE3 transduced dissociated cortical neurons suggests that GFE3 specifically reduces endogenous Gephyrin levels. A marked decrease in Gephyrin (a; 80 ± 3%, P = 0.01, Kruskal-Wallis) occurs in neurons expressing GFE3 compared to neurons not transduced (Control) or transduced with RandE3 or Gephyrin.FingR. In contrast, no significant difference is observed for the steady-state levels of the alpha1 subunit of the GABAA receptor, PSD-95, the GluA1 subunit of the AMPA receptor, the GluN2B subunit of the NMDA receptor, or the Gephyrin interacting proteins Collybistin II and GABARAP (b–g; P > 0.8, Kruskal-Wallis). n = 4 replicate blots. (h, i) Quantification of endogenous Gephyrin and GABAA receptor immunostaining in cortical neurons showed a 93 ± 1% (P < 0.0001, Mann-Whitney) reduction in Gephyrin levels in cells transfected with GFE3 compared to cells with RandE3, and no change in GABAA receptor levels (P > 0.99). Sample size is n = 10 neurons per condition. Bars indicate means ± SEM.
Supplementary Figure 5, related to Figure 1. Expression of GFE3 in cortical neurons does not affect cell viability or proteasome activity. (a, b) Lentiviral mediated expression of GFE3, RandE3 or GPHN.FingR in cortical neurons for 16 hours had a similar effect on cell viability (P > 0.96, Kruskal-Wallis) and cytotoxicity (P > 0.98), based on a multiplexed assay of protease activity in live cells and from dead cells. In contrast, addition of the mitochondrial toxin 3-nitropropionic acid (3-NP, 15 mM) to the medium for 48 hours induced an 80 ± 1% decrease in cell viability (P < 0.0001) and a 5-fold increase in cytotoxicity (P < 0.0001). (c) ATP levels in cells expressing GFE3, RandE3 or GPHN.FingR for 16 hours were unchanged (P > 0.96, Kruskal-Wallis), whereas cells that were treated with 3-NP for 48 hours exhibited an 80 ± 1% decrease in ATP levels (P < 0.0001). (d–f) Measurement of proteasome caspase- (d; P > 0.96, Kruskal-Wallis), chymotrypsin- (e; P > 0.87, Kruskal-Wallis) and trypsin-like (f; P > 0.86, Kruskal-Wallis) activity in cells transduced with GFE3, RandE3 or GPHN.FingR for 16 hours indicates that GFE3 does not alter proteasome function. In contrast, addition of the highly specific proteasome inhibitor clasto-Lactacystin-[beta]-lactone (Lactacystin, 10 µM) to the medium of cells for 3 hours resulted in the inhibition of all three types of proteasome activity (P < 0.0001). Data is shown as experimental (transduced or drug-treated) cell measurements normalized to the mean of Control (not transduced or vehicle-treated) cell measurements. Bars indicate means ± SEM. Sample size is n = 24 wells of cells in a 96 well plate per condition.
Supplementary Figure 6, related to Figure 2. Expression of GFE3 does not affect frequency or amplitude of EPSCs. (a, b) GFE3-mediated ablation of Gephyrin does not affect mEPSCs. Representative traces of mEPSCs obtained from dissociated hippocampal neurons expressing GFE3 or non-transfected controls (a). No significant change in mEPSC amplitude or frequency was observed in GFE3 expressing cells compared to controls (b; P > 0.75, Mann-Whitney). n = 5 independent experiments for controls and GFE3. Bars indicate means ± SEM.
Supplementary Figure 7, related to Figure 2. Noise analysis of GABA evoked currents. (a) Power spectrum of a continuous recording after GABA (1 mM) application was fitted with the sum (solid lines) of three Laurentzian functions (dotted lines). Spectral analysis of GABA-induced noise in control (non-transfected) cells revealed three average corner frequencies at 3.2 ± 0.6, 31.6 ± 4.2, 380.0 ± 75.4 Hz (n = 5) corresponding to time constants of 53.7, 4.9, 0.3 ms, respectively. (b) In GFE3 expressing cells the three corner frequencies obtained were 1.7 ± 0.3, 36.7 ± 10.6, 316.9 ± 129.7 Hz (n = 6), corresponding to time constants of 105.4, 6.9, 0.9 ms, respectively. (c) No statistical difference was observed between these measurements (P > 0.05, Mann-Whitney) indicating that uncoupling the GABAA receptors from Gephyrin did not cause an observable change in channel gating. Data are expressed as means ± SEM.
Supplementary Figure 8, related to Figure 4. GFE3 expression in spinal cord neurons of zebrafish impairs tail flick behavior. (a) Time-lapse images of a zebrafish age 24 hours post fertilization with mosaic expression of RandE3 in spinal motoneurons showed stereotyped tail flick behavior, which is comprised of a coordinated curling of the tail that brings the tip to the base. (b) In contrast, an age-matched sibling that expresses GFE3 in spinal motoneurons exhibits abnormal tail flick behavior, being unable to curl its tail sufficiently to touch the tip to the base.
Supplementary Figure 9, related to Figure 5. Transient expression of GFE3-TagRFP in cortical neurons. (a) A rat cortical neuron transfected with doxycycline (dox)-inducible GFE3-TagRFP, before addition of dox to the culture medium (t = 0 hrs). (b) Following incubation with 1 µg/mL of dox for 5 hours, GFE3-TagRFP expression is visible (t = 5 hrs). (c) A rat cortical neuron transfected with dox-inducible GFE3-TagRFP prior to incubation with dox (t = 0 hrs). (d) High levels of GFE3-TagRFP are present in the neuron 24 hours after adding 1 µg/mL of dox (t = 24 hrs). (e) Dox was removed by washing six times with conditioned medium. No GFE3-TagRFP is detectable in the neuron following 48 hours of recovery. Scale bar represents 10 µm.
Supplementary Video 1, related to Figure 4. Zebrafish expressing GFE3.
Zebrafish expressing GFE3-GFP in motoneurons in the spinal cord displays an abnormal tail flick.
Supplementary Video 2, related to Figure 4. Zebrafish expressing GFE3.
Zebrafish expressing GFE3-GFP in motoneurons in the spinal cord displays an abnormal tail flick.
Supplementary Video 3, related to Figure 4. Zebrafish expressing RandE3.
Zebrafish expressing RandE3-GFP in motoneurons in the spinal cord displays a normal tail flick.
Supplementary Video 4, related to Figure 4. Zebrafish expressing RandE3.
Zebrafish expressing RandE3-GFP in motoneurons in the spinal cord displays a normal tail flick.