

Abstract
FeIII-(hydro)peroxy intermediates have been isolated in two classes of mononuclear non-heme Fe enzymes that are important in bioremediation: the Rieske dioxygenases and the extradiol dioxygenases. The binding mode and protonation state of the peroxide moieties in these intermediates are not well defined, due to a lack of vibrational structural data. Nuclear resonance vibrational spectroscopy (NRVS) is an important technique for obtaining vibrational information on these and other intermediates, as it is sensitive to all normal modes with Fe displacement. Here, we present the NRVS spectra of side-on FeIII-peroxy and end-on FeIII-hydroperoxy model complexes and assign these spectra using calibrated DFT calculations. We then use DFT calculations to define and understand the changes in the NRVS spectra that arise from protonation and from opening the Fe-O-O angle. This study identifies four spectroscopic handles that will enable definition of the binding mode and protonation state of FeIII-peroxy intermediates in mononuclear non-heme Fe enzymes. These structural differences are important in determining the frontier molecular orbitals available for reactivity.
Graphical abstract
1 Introduction
Mononoclear non-heme iron (NHFe) enzymes are important in many biological catalyzed transformations, including antibiotic and neurotransmitter biosynthesis, bioremediation, hypoxia regulation, and DNA repair.1–6 These enzymes catalyze a wide range of reactions involving hydroxylation, desaturation, ring closure and expansion, or ring cleavage by employing intermediates that include FeIV-oxo and FeIII-(hydro)peroxy species.7–11 FeIII-peroxy intermediates specifically have been invoked in two classes of NHFe enzymes: the Rieske dioxygenases12 and the extradiol dioxygenases,13,14 both important in bioremediation. In the Rieske dioxygenase benzoate 1,2-dioxygenase (BZDO), which catalyzes the cis-dihydroxylation reaction converting benzoate to 1-carboxy-1,2-cis-dihydroxycyclohexa-3,5-diene, a high-spin FeIII-(hydro)peroxy intermediate (BZDOp) has been trapped using a peroxide shunt reaction.12 BZDOp was found to be active in cis-dihydroxylation, making it an important target for further study, since the binding mode and protonation state of the peroxy moiety in this intermediate are unknown. In the extradiol dioxygenase 2,3-homoprotocatechuate dioxygenase (HPCD), which catalyzes the extradiol cleavage of 3,4-dihydroxyphenylacetate (HPCA) to 5-carboxymethyl2-hydroxymuconic semialdehyde, high-spin FeIII peroxy intermediates active in extradiol cleavage have been proposed in H200N13 and H200C14 variants on the basis of EPR and Mössbauer spectroscopies. Again, the structures of the peroxy units in these intermediates are unknown due to a lack of direct structural data.
Resonance Raman (rR) spectroscopy has been an important tool for obtaining vibrational and thus structural data on heme peroxy complexes.15 However, rR requires a chromophore such as heme at the active site of interest, which is often absent in NHFe sites or, in the case of the Rieske dioxygneases, obscured by the strong Rieske chromophore.16 Resonance Raman spectroscopy can also be susceptible to fluorescence overpowering the vibrational data of interest and to photodecay.17 Besides these practical concerns, the types of vibrational data that can be obtained from resonance Raman spectroscopy are quite specific, as the Raman resonance enhances modes must be symmetric and reflect excited state distortions of the associated electronic transition. Nuclear resonance vibrational spectroscopy (NRVS), a third-generation synchrotron technique where one measures vibrational sidebands of the 14.4 keV 57Fe nuclear Mössbauer transition, is a useful complement to rR spectroscopy.18,19 All vibrational modes that involve Fe displacement (and only these modes) have NRVS intensity that is proportional to the amount of Fe displacement in a normal mode of the active site. NRVS provides a wealth of vibrational data selective for the NHFe site, making it useful for structure determination when coupled to DFT calculations that have been calibrated on structurally defined complexes.20–22
We have previously used NRVS to determine the structure and reactivity of activated bleomycin (ABLM), the FeIII-hydroperoxy intermediate in the glycopeptide anticancer drug bleomycin that effects the double-stranded cleavage of DNA.22 However, ABLM is low-spin, unlike the high-spin peroxy intermediates in BZDO and HPCD, and spin state is important in governing the reactivity of FeIII-peroxy complexes.23,24 To be able to define the structure of high-spin FeIII-(hydro)peroxy species using their NRVS spectra, it is first necessary to apply this method to structurally defined models.
In high-spin FeIII-peroxy complexes, the peroxy moiety can be bound in both a side-on (η2) and an end-on (η1) conformations and can be unprotonated or singly-protonated. Of these, models with an unprotonated side-on O22− structure 25–27 and a protonated end-on OOH− structure25,27,28 have been characterized. These respectively perform nucleophilic deformylation reactions and electrophilic H-atom abstraction and hydroxylation reactions. In addition to the above, a single example of a putative end-on O22− has been reported.29 In this study, we use a combined NRVS/DFT methodology to assign the NRVS spectra of two high-spin FeIII-peroxo complexes bearing the TMC (where TMC is 1,4,8,11-tetramethyl1,4,8,11-tetraazacyclotetradecane) ligand: the side-on [FeIII(O2)TMC](CF3SO3) (1) and the end-on [FeIII(OOH)TMC](CF3SO3)+ (2). We further present a systematic correlation between the side-on peroxy and end-on hydroperoxy limits to uncouple the effects of protonation and binding mode on the NRVS spectrum. This study provides spectroscopic handles for determining the structures of high-spin FeIII-peroxy enzyme intermediates. We further consider how the protonation and hydroperoxo coordination mode can impact the reactivity of these intermediates.
2 Methods
2.1 Preparation of 1 and 2
57Fe(CF3SO3)2·2CH3CN was prepared according to the literature method:30 Triflic acid (10 g; 67mmol) in water (20 mL) was added dropwise to finely divided 57Fe powder (1.5 g; 27 mmol) in a round glass bottle flask over 5 min, as the reaction is highly exothermic. The reaction solution was stirred for 1 day. The unreacted 57Fe powder was removed by filtration through a sand core funel. The resulting solution was condensed to dry under vacuum. The 57Fe(CF3SO3)2·2CH3CN product was recrystallized from dried CH3CN/Et2O at 0 °C. 57Fe(CF3SO3)2·2CH3CN was obtained as a white solid power. [(TMC)57FeII(CH3CN)](CF3SO3)2 and 1 were prepared according to methods described in the literature:31 [(TMC)57FeII(CH3CN)](CF3SO3)2 was synthesized by reacting 57Fe(CF3SO3)2·2CH3CN with the tetramethylcyclam ligand (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) under an Ar atmosphere in CH3CN at 25 °C. Then, 1 was generated by reacting [(TMC)57FeII(CH3CN)](CF3SO3)2 (69.3 mg, 0.10 mmol) with 5 equiv. of H2O2 (51 μL, 30% in water, 0.50 mmol) in the presence of 2 equiv. triethylamine (TEA; 28 μL, 0.20 mmol) in CF3CH2OH (2 mL) at −40 °C. Then, Et2O (40 mL) was added to the resulting solution to yield purple precipitates at −40 °C. These purple precipitates, [(TMC)57FeIII(O2)](CF3SO3), were washed with Et2O and dried under Ar atmosphere. 2 was generated from 1 following the procedure in ref 25.
2.2 Collection and Processing of NRVS data
57Fe NRVS spectra of 1 and 2 were collected at BL09XU at SPring-8 in Hyogo, Japan. Samples were maintained at ~60K on a copper block in a liquid helium cryostat. The NRVS energy scale was calibrated to [FeIIICl4][NEt4].32 Scans were added together using the PHOENIX software package33 until a good signal-to-noise ratio was obtained. The elastic peak was subtracted and partial vibrational density of states spectra were generated using PHOENIX. Data were collected on two replicates of each sample, from which final data sets were generated by adding the scans on both replicates and reprocessing after it was confirmed that they reproduced.
2.3 DFT calculations and NRVS simulations
DFT calculations for 1 and 2 were performed using the Gaussian 09 software package.34 The B3LYP functional was used, employing a mixed basis set consisting of the 6-311g* on Fe and 6-311g on all other atoms. This is the same functional and basis set combination that was previously used to evaluate the electrophilic reactivity of 2, giving results consistent with experiment,23 and which had previously given good results on a low-spin end-on FeIII-hydroperoxy and high-spin side-on FeIII-peroxy ligated by N4Py.26 Implicit solvation effects were modeled using the Polarzable Continuum Model (PCM) as implemented in Gaussian 09, with the solvent specified as acetonitrile. The crystal structure of 1 was used at the starting point for its geometry optimization.25 This structure was then protonated as a starting point for the geometry optimization of 2, and this structure was correlated to previous EXAFS data of 2.25 Angle and bond constraints were included using the modredundant keyword in Gaussian. NRVS spectra were simulated from DFT frequency calculations using the gennrvs script.35 NRVS simulations with adjusted force constants were obtained by modifying the Cartesian force constants manually to perform a frequency calculation. Molecular orbital contours were generated using LUMO,36 and Mulliken poplation analyses were performed using QMForge.37
3 Results and Analysis
3.1 NRVS spectra and assignments
3.1.1 Side-on FeIII-OO2− (1)
The partial vibrational density of states (PVDOS) NRVS spectrum taken on a powder sample of 1 is presented in Figure 1A.
Figure 1.

A: NRVS spectrum of 1 divided into four energy regions with a depiction of its structure as an inset. B: DFT-simulated NRVS spectrum of 1, with sticks corresponding to the amount of Fe displacement in each mode and the DFT-optimized structure as an inset. C: Schematic depictions of the vibrations in 1 divided by energy region.
There are three main regions in the spectrum with significant NRVS intensity (the spectrum in Figure 1A is divided into four regions for consistency with data presented below for 2): a single peak at 434 cm−1 (region I) and two pairs of split features in the 210–250 cm−1 (region III) and 145–200 cm−1 (region IV) energy ranges. The integrated intensity ratio of these three regions is 1.2 : 2 : 1 in going from low to high energy. Previous resonance Raman studies on 1 found the symmetric Fe-(O2) stretch at 487 cm−1 in d6-acetone (which shifted to 468 cm−1 with 18O substitution)25 and at 493 cm−1 in MeCN (which shifted to 478 cm−1 with 18O substitution).27 However, those data were taken in solution, where there can be interactions with the solvent. Thus, rR was performed on an equivalent solid sample of 1. The symmetric stretch is now at 432 cm−1 (Figure S1), enabling assignment of the 434 cm−1 peak in Region I of the NRVS spectrum as this symmetric Fe-O stretch (Figure 1C, I ν8) (see SI for the solvent effect on this stretch). DFT calculations were used to simulate the NRVS spectrum of 1 and assign the rest of the spectrum (Figure 1B). The DFT-optimized structure of 1 (Figure 1B, inset and Figure S2) agrees well with the crystal structure (Figure S2, Table S1). The calculated NRVS spectrum is in reasonable agreement with the data), validating this computational modeling of the NRVS data: the symmetric Fe-O stretch is at 429 cm−1 compared to the experimental value of 434 cm−1 (the antisymmetric stretch, ν7 in Figure 1C I, is calculated at 418 cm−1 and is not distinguishable in the NRVS data), and the two pairs of split features in the 210–250 cm−1 and 145–200 cm−1 regions are reproduced. The calculated O-O stretch, which has no Fe displacement and thus does not appear in the NRVS spectrum, is at 850 cm−1 in reasonable agreement with the rR value (825 cm−1).25,27 The intensity ratio of the three regions of the simulated spectrum is 1.2 : 2 : 1 in going from low to high energy, also consistent with experiment.
In 1, which has ~C4v symmetry (actually C2v due to the Fe(O2) plane), there are four equatorial Fe-N stretches that transform as A1,B1, and E. On the basis of the DFT calculations, only the E symmetry vibrations have Fe displacement. The intense peaks at 230 cm−1 and 246 cm−1 (Region III of Figure 1A), calculated at 221 and 244 cm−1, are assigned as this pair of trans antisymmetric equatorial Fe-N stretches of approximately E symmetry. Due to the C2v distortion these split in energy by 16 cm−1 and localize along x and y, respectively (ν4a,4b in Figure 1C), where x is in the Fe(O2) plane, y is perpendicular to the Fe(O2) plane, and z bisects the O-O bond. Also in this region is a high-energy shoulder at 275 cm−1, which can be correlated to a calculated mode at 270 cm−1 that consists of twists of the methyls bound to the nitrogens mixed with the equatorial Fe-N stretch along y. Finally, the Fe(O2) unit vibrates in in-plane rock and out-of-plane wag motions. From the DFT calculations, only the rock has Fe displacement, and the peak at 188 cm−1 in Region IV (calculated at 182 cm−1) is assigned as this rock (ν2 in Figure 1C). The peak at 160 cm−1 in Region IV (calculated at 161 cm−1) is assigned as translation of the Fe-OO unit along the z-axis (ν1 in Figure 1C). This vibration mixes with a TMC ligand breathing mode, distributing some of this z displacement into a mode at 140 cm−1. Adding this intensity into the 161 cm−1 mode gives an intensity ratio of the 161 to 182 cm−1 bands consistent with experiment.
While the calculated vibrational energies and intensity ratios of the three regions are in good agreement with experiment, the relative intensities of the bands in Region III is off. To obtain better agreement, the force constants for the four equatorial nitrogens were increased by 10% and the NRVS data were resimulated (Figure S3, panel B). The relative integrated intensity of the three regions remains 1.2 : 2 : 1, consistent with experiment, and now the relative intensities of the bands in Region III are in better agreement with experiment. The in-plane equatorial Fe-N vibration (split over several modes centered at 226 cm−1) is now more intense than its out-of plane partner at 248 cm−1. In addition, the shoulder at 275 cm−1 is now better modeled, still assigned as the equatorial Fe-N stretch along y mixing into the metyl twists, now at 271 cm−1.
3.1.2 End-on FeIII-OOH (2)
The PVDOS NRVS spectrum taken on 2 in acetonitrile is presented in Figure 2A. Note that 2 has been previously synthesized using both acetonitrile and acetone as the solvent; acetonitrile was chosen for the NRVS experiment due to its higher melting point.
Figure 2.

A: NRVS spectrum of 2 with a depiction of its structure as an inset. B: DFT-simulated NRVS spectrum of 2 using the best structure obtained, with sticks corresponding to the amount of Fe displacement in each mode and the DFT-optimized structure as an inset. C: schematic depictions of relevant vibrations involving Fe displacement.
There are four main regions of the spectrum with significant NRVS intensity: a broad feature centered at 365 cm−1 (region II), an intense feature at 263 cm−1 flanked by shoulders at 290 and 235 cm−1 (region III), and in region IV a single low-energy peak at 179 cm−1. In the higher energy region I, an Fe-O stretch is expected based on rR observations of such a mode at 676 cm−1 in frozen MeCN,27 which shifted to 652 cm−1 with 18O substitution. (For 2 in frozen d6-acetone, this stretch appears at 658 cm−1 and shifts to 633 cm−1 with 18O substitution.)25 The NRVS spectrum contains two weak features in region I, one peak at 507 cm−1 and another at 678 cm−1. The 678 cm−1 peak can be assigned as the Fe-O stretch, as it is in good agreement with the energy from resonance Raman.27 The integrated intensity ratio of the four regions, going from low to high energy, is 3 : 8 : 1.2 : 1.
DFT calculations were again used to assign the spectrum, employing the same methodology validated for 1. Note that although no crystal structure exists for 2, structural information is available from EXAFS (Fe-O bond length) and resonance Raman (Fe-O and O-O stretching frequencies) data, and these serve as benchmarks for the accuracy of the computational structure.25,27 An initial structure was obtained by protonating the DFT optimized structure of 1 and optimizing this without constraints. The Fe-O bond length of 1.84 Å is in good agreement with the EXAFS distance of 1.85 Å, and the average Fe-equatorial ligand distances (2.19 Å) are also consistent with EXAFS (2.16 Å). However, there are two major issues with this structure. First, the calculated Fe-O stretching frequency of 536 cm−1 is in poor agreement with the NRVS frequency of 678 cm−1. Second, there are discrepancies between the experimental and calculated intensities (Figure S4, panel B): the relative intensities of regions IV : III : II : I are (with region III normalized to 8) 5 : 8 : 0.7 : 2.2, with too much intensity in regions I and IV and an incorrect intensity ratio of the two peaks in Region III.
To improve the structural model, explicit solvation by water molecules was evaluated, as 2 is generated by adding an excess of perchloric acid to 1. Inclusion of a single water molecule accepting a hydrogen bond from the proton on the distal O of the hydroperoxy moiety leads to reasonable agreement between the experimental data and the DFT simulation in the low energy region, both in energy and relative peak intensity (Figure S4, panel C); however, the calculated νFe-O (553 cm−1) was still too low relative to experiment (658 cm−1 in d6-acetone and 676 cm−1 in acetonitrile) and too high in intensity. To evaluate whether the energy disagreement was functional-dependent, the same structure was optimized using BP86, which found a lower νFe-O energy (521 cm−1) and longer Fe-O bond (1.85 Å) and gave a NRVS simulation in significantly worse agreement with experiment (Figure S5 and Table S2); thus, B3LYP was used for the analysis. The νFe-O in the B3LYP solvated model was found in the calculations to mix significantly with the higher energy νO-O, offering another possible explanation for its calculated low energy relative to the rR and NRVS data. In a previous study of vibrations in binucler FeIII-peroxy species, mechanical coupling was found between the Fe-O stretch and O-O bend. This coupling is angle dependent, where decreasing the Fe-OO angle led to an increased Fe-O stretching energy.38 This raised the possiblilty that the computationally-observed mixing between νFe-O and νO-O modes could be decreased by decreasing the Fe-OO angle, as the total energy calculated for 2 is quite insensitive to this angle (vide infra). A series of hydroperoxide structures was optimized where the Fe-O distance was fixed at the freely-optimized (and EXAFS) value and the Fe-OO angle was varied over a series of more acute angles relative to the freely optimized angle (121°). A structure with an Fe-OO angle of 104° gave an νFe-O of 619 cm−1, now in better agreement with the solvent-dependent range of 658 to 676 cm−1 for this stretch. Note that, as shown in Figure S6, more acute angles give poor intensity agreement in Region III, while more obtuse angles give similarly good intensities in Region III but worse energy and intensity agreement for the νFe-O (which increases in intensity and decreases in energy as the Fe-OO angle increases from 106°). The DFT calculated value of the νFe-O for this structure is in similar agreement with those previously calculated for high-spin FeIII-alkylperoxy species in two separate studies, where B3LYP predicted a stretching energy 39–45 cm−1 lower than experiment.24,39 One of these studies24 found that this was primarily due to the B3LYP functional underpredicting the Fe-O force constant, and a normal coordinate analysis gave a force constant of 2.8 relative to the B3LYP calculated value of 2.1 mdyn/Å. The νO-O of this fixed-angle structure of 2, 842 cm−1, is also in reasonable agreement with the rR value (868 cm−1). This structure is within 1 kcal/mol of the fully optimized structure, and the NRVS simulation derived from this structure is presented in Figure 2B. The integrated intensity ratios of the four regions of the simulation are 4 : 8 : 0.6 : 2, with the νFe-O region I having double the intensity relative to experiment but reasonable intensity agreements in the other three regions.
To further refine the simulation, the force constant for the proximal O was increased by 25% along the νFe-O vector, and the NRVS spectrum was resimulated. The results are presented in Figure S7, panel B. The integrated intensity ratios of the four regions in this simulation are, from low to high energy, 3 : 8 : 0.7 : 1, in reasonable agreement with experiment. The νFe-O now appears at 678 cm−1 in this model, and the relative intensities of the bands within Region III now more closely mirror experiment.
As the modes in Regions II-IV do not change significantly in energy upon adjusting the proximal O force constant, the simulation in Figure 2B was used to assign the spectrum. Figure 2B also includes an inset depicting the geometry optimized structure of 2, which is expanded in Figure S8. The remaining feature in Region I at 507 cm−1 is assigned as the approximately degenerate pair of Neq-Fe-Neq bends (not observed in 1, one of which is shown schematically as ν8 in Figure 2C). As for 1, four equatorial Fe-N stretches of A1, B1, and E symmetry are expected in 2 (which also has ~C4v symmetry, actually close to Cs due to the Fe(O2H) plane). The features in Region II of Figure 2A centered at 365 cm−1 are assigned as the ~B1 and ~A1 equatorial ligand stretches (ν5 and ν6 in Figure 2C II), which now have Fe displacement and mix into several modes in this energy region. In the most intense region of the spectrum, Region III in Figure 2A, the peak at 263 cm−1 and its shoulder at 290 cm−1 (calculated at 258 and 278 cm−1) are assigned as the equatorial Fe-ligand stretches of ~E symmetry, split to localize in Cs symmetry along y and x, respectively (ν4b and ν4a in Figure 2C III), with y perpendicular to the Fe-OOH plane, x in the Fe-OOH plane, and z along the Fe-O bond. Also in Region III, the 235 cm−1 shoulder (calculated at 227 cm−1) is assigned as the δFe-OOH (ν3 in Figure 2C). Finally, the peak at 179 cm−1 in Region IV (calculated at 180 cm−1) is assigned as translation of the Fe-OOH unit along z (ν1 in Figure 2C).
3.2 Correlation between side-on FeIII-OO2− and end-on FeIII-OOH−
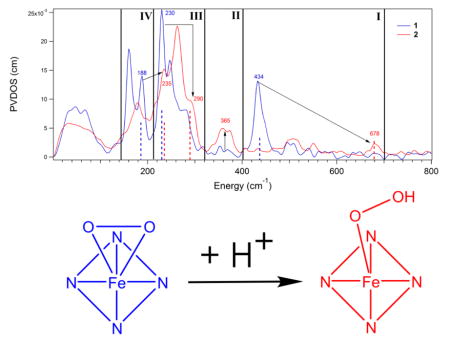
From the above NRVS data, reproduced in DFT simulations, there are four spectral changes that occur upon conversion of the side-on peroxy complex 1 to the end-on hydroperoxy complex 2. These derive from both protonation of the peroxy unit and elimination of one of the Fe-O bonds. These 1 to 2 NRVS spectral changes are highlighted in the NRVS data overlay in Figure 3, depicted schematically for the corresponding normal modes of vibration in Figure 4, and summarized below (with explanations of the origins of these changes to follow):
Figure 3.

Overlay of the NRVS spectra of 1 and 2 divided into the four regions where major differences are observed and highlighted using arrows. Dashed lines of the same height correspond to features that are correlated between 1 and 2.
Figure 4.
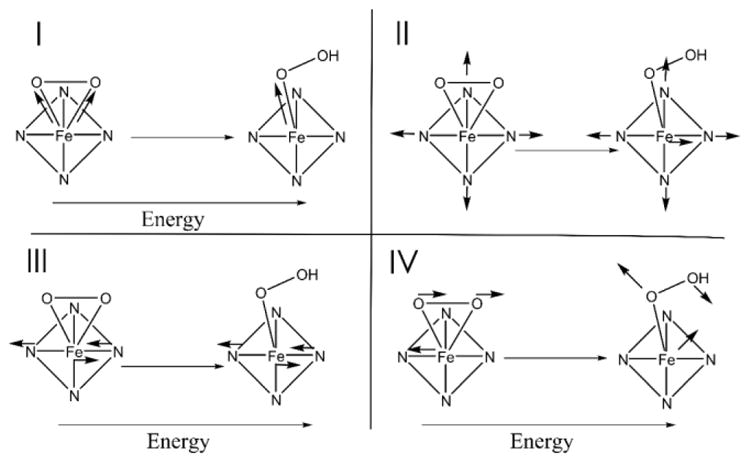
Schematic depiction of the major differences in the vibrations of 1 and 2, labeled according to the energy regions in Figure 3. Note that vibrations in I, III, and IV all involve an increase in the energy of the vibration going from 1 to 2, while II involves a change in the amount of Fe displacement.
Region I of Figure 3, depicted schematically in Figure 4, panel I: The symmetric νFe-(O2) localizes into a single νFe-O and increases in energy from 434 cm−1 to 678 cm−1. The antisymmetric νFe-(O2) (only observed computationally in 1) no longer exists in 2.
Region II in Figure 3 and Figure 4, panel II: The equatorial Fe-ligand stretching modes of ~A1 and ~B1 symmetry centered around 365 cm−1 have no NRVS intensity in 1, but gain Fe displacement and thus NRVS intensity in 2.
Region III in Figure 3 and Figure 4, panel III: The equatorial Fe-ligand ~E vibration in the Fe-OO plane shifts from 230 cm−1 to 290 cm−1, while its perpendicular partner changes less in energy, shifting from 246 cm−1 to 263 cm−1.
Region IV in Figure 3 and Figure 4, panel IV: The in-plane Fe-OO rock at 188 cm−1 disappears, and an in-plane Fe-OOH bend appears at 235 cm−1 in region III.
To systematically define the origins of these changes, NRVS simulations were carried out on a series of hydroperoxy structures optimized with varying fixed Fe-OO angles, beginning with the 66° angle obtained for 1 and ending with the freely-optimized angle of 120°obtained for 2. This series corresponds to protonating the side-on structure followed by opening the Fe-OO angle. These results, summarized in Table 1, define which spectral changes arise from protonation of the peroxy moiety and which arise from opening of the Fe-OO angle. Note that since opening the Fe-OO angle without protonation leads to a change in electronic structure from Fe(III)-OO2− to Fe(II)-OO•− beginning early in the correlation (for all Fe-OO angles ≥ 96°), which is not observed experimentally, this computational series was not considered. Also note that the effect of angle on the FeIII-OOH vibrations was examined for structures both with and without a water accepting a hydrogen bond from the distal hydroperoxy proton. Both sets of calculations gave very similar results. The results without water are presented below (the calculations with water are given in Table S3, and simulations with water are also given in Figure S6).
Table 1.
Structural and vibrational information on the side-on peroxy structure (row 1) and distally protonated structures with varying angles, without water accepting a hydrogen bond from the proton. All protonated structures demonstrated mixing of Fe displacement into the ~A1 and ~B1 equatorial Fe-ligand stretching modes. As above, x is in the FeOO plane and y perpendicular to the plane.
| Fe-Nx distance (Å) | νFe-Nx (~E) (cm−1) | Fe-Ny distance (Å) | νFe-Ny (~E) (cm−1) | Fe-O distance (Å) | νFe-O (cm−1) | Fe-OO rock (cm−1) | Fe-OO bend (cm−1) | |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Side-on | 2.28 | 221 | 2.24 | 244 | 1.92, 1.92 | 430 | 182 | - |
| 66° | 2.22 | 260 | 2.22 | 251 | 2.01, 1.99 | 456 | 186 | - |
| 76° | 2.21 | 269 | 2.22 | 255 | 1.93 | 521 | 195 | - |
| 82° | 2.19 | 273 | 2.22 | 255 | 1.90 | 553 | 197 | - |
| 96° | 2.18 | 276 | 2.22 | 258 | 1.88 | 575 | - | 230 |
| 106° | 2.17 | 281 | 2.22 | 257 | 1.86 | 566 | - | 231 |
| 116° | 2.17 | 283 | 2.22 | 257 | 1.85 | 549 | - | 231 |
| 120° | 2.16 | 283 | 2.22 | 258 | 1.84 | 542 | - | 228 |
The modeling results, summarized in Table 1 (as well as Table S3 and Figure S6), provide insight into the origins of the four spectral changes described above:
Region I in Figure 3: upon protonation, the symmetric Fe-(O2) stretch goes up in energy by 26 cm−1, from 430 to 456 cm−1. As the Fe-OO(H) angle is opened and one of the Fe-O bonds breaks, the symmetric stretch localizes to a single νFe-O and increases in energy until an angle of 96°, where this stretching energy is 575 cm−1. As the angle increases above 96°, the νFe-O begins decreasing, reaching a final value of 542 cm−1 in the freely optimized case (120°). This reflects the fact that increasing the Fe-OO angle above 96 leads the mixing of the νFe-O with the νO-O vibration, as discussed above and in ref 36. The antisymmetric νFe-(O2) (not included in Table 1) decreases in energy from 434 to 323 cm−1 upon protonation and is no longer identifiable when the angle is ≥ 82° and one of the Fe-O bonds is eliminated. Thus protonation and angle both contribute to the difference in the Fe-O stretching energy between 1 and 2 (434 cm−1 vs 678 cm−1).
Region II in Figure 3: in all structures, independent of Fe-OO angle, protonation mixes iron displacement into the ~A1 and ~B1 equatorial Fe-ligand stretches centered at 365 cm−1. Protonation of the OO moiety decreases the symmetry from C2v to Cs. In 2, this decrease in symmetry allows mixing of the Fe displacement present in the ~E symmetry equatorial ligand stretches at 263 and 290 cm−1 into the ~A1 and B1 equatorial ligand stretches, giving them NRVS intensity in region II (Note that for C4v → C2v, A1 → A1, B1 → A1, E →B1 + B2, and there is no allowed mixing; for C4v → Cs, A1 → A′, B1 → A′, E → A′ + A″, thus the mixing is allowed). This mixing further leads to a decrease in Fe displacement and thus a decrease of NRVS PVDOS intensity in the equatorial Fe-N ~E stretches in 2 (at 263 and 290 cm−1) relative to 1 (at 221 and 244 cm−1).
Region III in Figure 3: protonation while maintaining a side-on structure raises the energy of one component of the equatorial Fe-N stretch of ~E symmetry in the Fe-OO plane (along x) from 221 to 260 cm−1, with concomitant lengthening of both Fe-O bonds that leads to shortening of the equatorial Fe-N bonds in the plane from 2.28 to 2.22 Å. With increasing Fe-OO angle, the energy of this vibration continues to increase to a final value of 283 cm−1 as one of the Fe-O bonds is eliminated and the equatorial Fe-Nx bonds continue to shorten to a final value of 2.16 Å. This trend also leads to an increased splitting between the two equatorial Fe-N ~E stretches (9–18 cm−1 for structures with Fe-OO angles ≤ 96°, 24–26 cm−1 for structures with Fe-OO angles > 96°), as the Fe-N bonds and stretch perpendicular to the Fe-OO plane do not change significantly in length or energy with Fe-OO angle because Fe-OO bonding does not change along this axis. This spectral change is dependent on both protonation state and Fe-OO angle, which together shift the energy of the in-plane component of the Fe-N ~E stretch from 230 to 290 cm−1.
Region IV in Figure 3: for Fe-OO angles ≤ 82°, an in-plane Fe-OO rock is present at ~180 cm−1, while for angles greater than 82°, there is no rock (as the bond between Fe and the protonated O is eliminated) and an Fe-OOH bend is instead observed at ~230 cm−1, which shifts this mode into region III. This change is due to opening of the Fe-OO angle and loss of an Fe-O bond and leads to the disappearance of a peak at 188 cm−1 and the growth of a peak 235 cm−1.
In summary, protonation of a side-on Fe-peroxy complex leads to an increase in the Fe-OO(H) stretching energy, mixing of Fe displacement hence NRVS intensity into A1 and B1 equatorial Fe-L stretches in region II due to decreased symmetry, and decreases the splitting between the pair of E symmetry equatorial Fe-L stretches in region III. The opening of the Fe-OOH angle and elimination of an Fe-O bond leads to a further increase in Fe-OOH stretching energy in region I, an increase in the energy of the equatorial component of the Fe-ligand stretch of ~E symmetry in the Fe-OOH plane in region III with a concommittant increase in the splitting between this vibration and its out-of-plane partner, and elimination of an Fe-OOH rock in region IV with appearance of an Fe-OOH bend in region III.
Discussion
From the above analysis, four spectral handles are defined that can be used to determine whether a high-spin FeIII-peroxy complex has a side-on peroxy, side-on hydroperoxy, or an end-on hydroperoxy structure on the basis of its NRVS spectrum. The first is the Fe-O stretch: a side-on FeIII-peroxy will have a relatively low energy stretch (~430 cm−1), a protonated side-on (or nearly side-on) hydroperoxy an intermediate energy stretch (~450 – 520 cm−1), and an end-on hydroperoxy a high energy Fe(O2H) stretch (~540 cm−1 or higher, depending on the Fe-OO angle). The energy actually observed within each region will depend on coordination number, as well as the nature of the other ligands bound to the Fe. The second spectral handle is the splitting of the equatorial Fe-ligand ~E stretches in region III between 205 and 310 cm−1: the experimentally-observed splitting for 1 is 16 cm−1 (Figure 1A), and that for 2 is 27 cm−1 (Figure 2A). This is also observed computationally, with splittings of 9–18 cm−1 calculated for structures with Fe-OO(H) angles ≤ 96° and 24–26 cm−1 for structures with Fe-OO angles > 96°. In a non-heme enzyme system where the Fe is ligated by a 2His/1 carboxylate facial triad, such as BZDO, the equatorial stretches in this region would reflect the cis His ligands, which would also split in energy by an amount determined by the mode of peroxide coordination to the iron. A third handle is the presence of a lower-energy Fe-OO(H) rock as opposed to an Fe-OOH bend: a rock is observed for the side-on 1 at 188 cm−1 and predicted computationally for Fe-OOH structures with Fe-OO angles between 66 and 82°, while a bend is observed at higher energy for the end-on 2 at 235 cm−1 and is predicted for structures with Fe-OO angles greater than 82°; these can be distinguished on the basis of their energy. Finally, a fourth spectral handle arising from protonation is the presence of Fe mixing into the equatorial Fe-N stretching modes of A1 and B1 symmetry associated with the decrease in symmetry at the Fe due to protonation, which is distinguishable by the prescence of NRVS intensity around 365 cm−1.
Definining the structure of an FeIII-peroxy enzyme intermediate using the four spectral handles determined above provides insight into its reactivity, since side-on peroxy, side-on hydroperoxy, and end-on hydroperoxy species involve different modes of peroxide activation (Figure 5).
Figure 5.

FMOs and reactivities of side-on FeIII-peroxy species 1 (left) and end-on FeIII-hydroperoxy species 2 (right), as well as the FMO and potential reactivity of a putative side-on FeIII-hydroperoxy species. The character for each O in the FMO is given. The side-on hydroperoxy structure has a fixed Fe-OO angle of 76°, with other structural details given in Table 1. Arrows indicate direction of electron flow.
High-spin NHFe side-on peroxy intermediates exhibit nucleophilic reactivities such as deformylation25,40 and being protonated.41 This involves attack on an unoccupied orbital by their occupied peroxide π*v frontier molecular orbital. High-spin NHFe end-on hydroperoxy intermediates primarily exhibit electrophilic reactivity, in particular H-atom abstraction23 and sulfoxidation42 using a LUMO with hydroperoxy σ* character where only the distal O is accessible for reaction with substrate. Note that 2 also performs nucleophilic reactivity which is complicated by the presence of excess acid.25 Though there is not yet definitive evidence of the existence of a side-on high-spin Fe(III)-hydroperoxy species in the literature for either model or enzyme systems, such a species has been proposed as an intermediate prior to aromatic ring hydroxylation in the peroxide shunt reaction of BZDO and in computational studies.12,43 A side-on hydroperoxy intermediate could have additional channels of electrophilic reactivity available relative to the end-on conformer. In particular this could utilize the now accessible unprotonated oxygen through the hydroperoxy σ* orbital (middle of Figure 5; note the greater unprotonated O character in this FMO). It has also been proposed that a side-on FeIII-OOH species could undergo heterolytic O-O bond cleavage to form an FeV(O)(OH) species prior to attack on the aromatic ring.44 Because of this availability of different reaction pathways for side-on peroxy, side-on hydroperoxy, and end-on hydroperoxy complexes, structural definition of high-spin Fe(III) peroxy intermediates in enzymes and models is crucial, and as presented above, NRVS is a powerful tool for providing such structural definition.
Supplementary Material
Acknowledgments
Funding for this work was provided by the National Institutes of Health (GM-40392 to E.I.S.), the NRF of Korea through CRI (NRF-2012R1A3A2048842 to W.N.) and GRL (NRF-2010-00353 to W.N.), JSPS KAKENHI (Grant No. 24221005 to M. Seto), and the U.S. National Science Foundation (CHE-1361773 to L.Q.). Synchrotron experiments at SPring-8 were performed with the approval of the Japan Synchrotron Radiation Research Institute (JASRI; proposal no. 2013B0105).
Footnotes
Supporting information. Resonance Raman method; discussion of solvent effects for 1; resonance Raman spectrum of solid 1; overlay of the crystal and DFT structures of 1; table comparing select geometric parameters for crystal and DFT structures of 1; NRVS simulation of 1 with adjusted force constants; NRVS simulations of 2 for models without discrete water and freely optimized with discrete water; NRVS simulation using the BP86 functional for 2 with discrete water; table comparing BP86 and B3LYP calculated structural and vibrational parameters for 2; NRVS simulations for a series of Fe-hydroperoxy structures with varying fixed Fe-OO angles; NRVS simulation for 2 with adjusted force constant; depiction of final DFT structure of 2; table comparing calculated structural and vibrational parameters for a series of Fe-hydroperoxy structures with varying Fe-OO angles, including water hydrogen bonding; supplementary references.
References
- 1.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 2.Baldwin JE, Abraham E. Nat Prod Reports. 1988;5:129–145. doi: 10.1039/np9880500129. [DOI] [PubMed] [Google Scholar]
- 3.Fitzpatrick PF. Adv Enzymol. 2000;74:235–294. doi: 10.1002/9780470123201.ch6. [DOI] [PubMed] [Google Scholar]
- 4.Hrywna Y, Tsoi TV, Maltseva OV, Quensen JF, Tiedje JM. Appl Environ Microbiol. 1999;65:2163–2169. doi: 10.1128/aem.65.5.2163-2169.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahon PC, Hirota K, Semenza GL. Genes & Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kataoka H, Yamamoto Y, Sekiguchi M. J Bacteriol. 1983;153:1301–1307. doi: 10.1128/jb.153.3.1301-1307.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Solomon EI, Light KM, Liu LV, Srnec M, Wong SD. Accounts Chem Res. 2013;46:2725–2739. doi: 10.1021/ar400149m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hausinger RP. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 9.Busby RW, Townsend CA. Bioorganic & Med Chem. 1996;4:1059–1064. doi: 10.1016/0968-0896(96)00088-0. [DOI] [PubMed] [Google Scholar]
- 10.Lloyd MD, Lee HJ, Harlos K, Zhang ZH, Baldwin JE, Schofield CJ, Charnock JM, Garner CD, Hara T, van Scheltinga ACT, Valegard K, Viklund JAC, Hajdu J, Andersson I, Danielsson A, Bhikhabhai R. J Mol Biol. 1999;287:943–960. doi: 10.1006/jmbi.1999.2594. [DOI] [PubMed] [Google Scholar]
- 11.Vaillancourt FH, Bolin JT, Eltis LD. Crit Rev Biochem Mol Biol. 2006;41:241–267. doi: 10.1080/10409230600817422. [DOI] [PubMed] [Google Scholar]
- 12.Neibergall MB, Stubna A, Mekmouche Y, Munck E, Lipscomb JD. Biochemistry. 2007;46:8004–8016. doi: 10.1021/bi700120j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mbughuni MM, Chakrabarti M, Hayden JA, Meier KK, Dalluge JJ, Hendrich MP, Muenck E, Lipscomb JD. Biochemistry. 2011;50:10262–10274. doi: 10.1021/bi201436n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meier KK, Rogers MS, Kovaleva EG, Mbughuni MM, Bominaar EL, Lipscomb JD, Münck E. Inorg Chem. 2015;54:10269–10280. doi: 10.1021/acs.inorgchem.5b01576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Proshlyakov DA, Ogura T, Shinzawa-Itoh K, Yoshikawa S, Appelman EH, Kitagawa T. J Biol Chem. 1994;269:29385–29388. [PubMed] [Google Scholar]
- 16.Kuila D, Fee JA, Schoonover JR, Woodruff WH, Batie CJ, Ballou DP. J Am Chem Soc. 1987;109:1559–1561. [Google Scholar]
- 17.Que L. Physical methods in bioinorganic chemistry: spectroscopy and magnetism. Sterling Publishing Company; 2000. [Google Scholar]
- 18.Seto M, Yoda Y, Kikuta S, Zhang XW, Ando M. Phys Rev Lett. 1995;74:3828–3831. doi: 10.1103/PhysRevLett.74.3828. [DOI] [PubMed] [Google Scholar]
- 19.Sturhahn W, Toellner TS, Alp EE, Zhang X, Ando M, Yoda Y, Kikuta S, Seto M, Kimball CW, Dabrowski B. Phys Rev Lett. 1995;74:3832–3835. doi: 10.1103/PhysRevLett.74.3832. [DOI] [PubMed] [Google Scholar]
- 20.Bell CB, III, Wong SD, Xiao Y, Klinker EJ, Tenderholt AL, Smith MC, Rohde J-U, Que L, Jr, Cramer SP, Solomon EI. Angew Chemie-International Ed. 2008;47:9071–9074. doi: 10.1002/anie.200803740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong SD, Srnec M, Matthews ML, Liu LV, Kwak Y, Park K, Bell CB, III, Alp EE, Zhao J, Yoda Y, Kitao S, Seto M, Krebs C, Bollinger JM, Jr, Solomon EI. Nature. 2013;499:320–323. doi: 10.1038/nature12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu LV, Bell CB, III, Wong SD, Wilson SA, Kwak Y, Chow MS, Zhao J, Hodgson KO, Hedman B, Solomon EI. Proc Natl Acad Sci United States Am. 2010;107:22419–22424. doi: 10.1073/pnas.1016323107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu LV, Hong S, Cho J, Nam W, Solomon EI. J Am Chem Soc. 2013;135:3286–3299. doi: 10.1021/ja400183g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehnert N, Ho RYN, Que L, Solomon EI. J Am Chem Soc. 2001;123:12802–12816. doi: 10.1021/ja011450+. [DOI] [PubMed] [Google Scholar]
- 25.Cho J, Jeon S, Wilson SA, Liu LV, Kang EA, Braymer JJ, Lim MH, Hedman B, Hodgson KO, Valentine JS, Solomon EI, Nam W. Nature. 2011;478:502–505. doi: 10.1038/nature10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roelfes G, Vrajmasu V, Chen K, Ho RY, Rohde JU, Zondervan C, la Crois RM, Schudde EP, Lutz M, Spek AL. Inorg Chem. 2003;42:2639–2653. doi: 10.1021/ic034065p. [DOI] [PubMed] [Google Scholar]
- 27.Li F, Meier KK, Cranswick MA, Chakrabarti M, Van Heuvelen KM, Münck E, Que L., Jr J Am Chem Soc. 2011;133:7256–7259. doi: 10.1021/ja111742z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wada A, Ogo S, Nagatomo S, Kitagawa T, Watanabe Y, Jitsukawa K, Masuda H. Inorg Chem. 2002;41:616–618. doi: 10.1021/ic001058h. [DOI] [PubMed] [Google Scholar]
- 29.McDonald AR, Van Heuvelen KM, Guo Y, Li F, Bominaar EL, Münck E, Que L. Angew Chem Int Ed. 2012;51:9132–9136. doi: 10.1002/anie.201203602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hagen KS. Inorg Chem. 2000;39:5867–5869. doi: 10.1021/ic000444w. [DOI] [PubMed] [Google Scholar]
- 31.Bang S, Lee YM, Hong S, Cho KB, Nishida Y, Seo MS, Sarangi R, Fukuzumi S, Nam W. Nat Chem. 2014;6:934–940. doi: 10.1038/nchem.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith MC, Xiao Y, Wang H, George SJ, Coucouvanis D, Koutmos M, Sturhahn W, Alp EE, Zhao J, Cramer SP. Inorg Chem. 2005;44:5562–5570. doi: 10.1021/ic0482584. [DOI] [PubMed] [Google Scholar]
- 33.Sturhahn W. Hyperfine Interactions. 2000;125:149–172. [Google Scholar]
- 34.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA. Inc; Wallingford, CT: 2009. [Google Scholar]
- 35.Tenderholt A. gennrvs. 2009 http://www.stanford.edu/group/solomon/gennrvs/gennrvs.py.txt.
- 36.Kieber-Emmons MT. Lumo, version 1.0.1. 2012 http://www.kieber-emmons.com/Lumo/
- 37.Tenderholt AT. QMForge, version 2.1. 2007 http://qmforge.sourceforge.net/
- 38.Brunold TC, Tamura N, Kitajima N, Moro-oka Y, Solomon EI. J Am Chem Soc. 1998;120:5674–5690. [Google Scholar]
- 39.Bautz J, Comba P, Que L. Inorg Chem. 2006;45:7077–7082. doi: 10.1021/ic0602401. [DOI] [PubMed] [Google Scholar]
- 40.Shokri A, Que L., Jr J Am Chem Soc. 2015;137:7686–7691. doi: 10.1021/jacs.5b01053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neese F, Solomon EI. J Am Chem Soc. 1998;120:12829–12848. [Google Scholar]
- 42.Kim YM, Cho KB, Cho J, Wang B, Li C, Shaik S, Nam W. J Am Chem Soc. 2013;135:8838–8841. doi: 10.1021/ja404152q. [DOI] [PubMed] [Google Scholar]
- 43.Bassan A, Blomberg MRA, Siegbahn PEM. J Biol Inorg Chem. 2004;9:439–452. doi: 10.1007/s00775-004-0537-0. [DOI] [PubMed] [Google Scholar]
- 44.Wolfe MD, Lipscomb JD. J Biol Chem. 2003;278:829–835. doi: 10.1074/jbc.M209604200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.