

ABSTRACT
In skeletal tissue, loss or mutation of the gap junction protein connexin 43 (Cx43, also known as GJA1) in cells of the osteoblast lineage leads to a profound cortical bone phenotype and defective tissue remodeling. There is mounting evidence in bone cells that the C-terminus (CT) of Cx43 is a docking platform for signaling effectors and is required for efficient downstream signaling. Here, we examined this function, using a mouse model of Cx43 CT-truncation (Gja1 K258Stop). Relative to Gja1+/− controls, male Gja1−/K258Stop mice have a cortical bone phenotype that is remarkably similar to those reported for deletion of the entire Cx43 gene in osteoblasts. Furthermore, we show that the Cx43 CT binds several signaling proteins that are required for optimal osteoblast function, including PKCδ, ERK1 and ERK2 (ERK1/2, also known as MAPK3 and MAPK1, respectively) and β-catenin. Deletion of the Cx43 CT domain affects these signaling cascades, impacting osteoblast proliferation, differentiation, and collagen processing and organization. These data imply that, at least in bone, Cx43 gap junctions not only exchange signals, but also recruit the appropriate effector molecules to the Cx43 CT in order to efficiently activate signaling cascades that affect cell function and bone acquisition.
KEY WORDS: Gap junction, Osteoblast, Osteocyte, Signal transduction, Intercellular communication, Cx43 K258Stop
Highlighted Article: The C-terminal domain of Cx43 in skeletal tissue affects signaling cascades and osteoblast function. Truncation of this domain recapitulates key aspects of Cx43 conditional deletion in bone.
INTRODUCTION
Gap junctions are transmembrane pores formed between two cells that allow for the direct passage of small molecules and second messengers, typically ∼1 kDa or smaller. Intercellular communication through gap junctions is a broadly conserved mechanism for the coordination of tissue function. Gap junction communication is important to physiological function and changes in gap junctions underpin many diseases (Kelly et al., 2015; Laird, 2014). The gap junction protein, connexin 43 (Cx43, also known as GJA1) is broadly expressed in many tissues of the body. Indeed, global Cx43 gene deletion (Gja1−/−) is perinatally lethal due to a cardiac defect (Reaume et al., 1995; Ya et al., 1998). Additionally, the skeleton of these mice exhibit delayed mineralization and a cell autonomous osteoblast dysfunction, including a failure to progress through differentiation (Lecanda et al., 2000). Similarly, conditional deletion of the Gja1 allele in cells of the osteoblast lineage also leads to a skeletal phenotype with cortical thinning, increased marrow cavity area, disproportionate collagen network and an imbalance in bone remodeling due to abnormal osteoblast differentiation (Bivi et al., 2012a,b; Chung et al., 2006; Watkins et al., 2011; Zhang et al., 2011). These data indicate an important role for intercellular communication by Cx43-containing gap junctions among the extensive interconnected network of cells (i.e. osteoblast, osteocytes and osteoprogenitor cells). However, details of the molecular mechanisms by which Cx43 affects osteoblast function and bone remodeling are unclear.
Connexin proteins have a conserved structure with four transmembrane domains and intracellular N- and C-termini. Twenty-one genes encoding connexin proteins are present in humans (Söhl and Willecke, 2004). The need for so many genes expressing proteins of largely similar function is unclear. While the biophysical properties of the permeability of each of these connexins often functionally overlap, the C-terminal (CT) domain of these connexins is the least conserved region within the protein family (Palatinus et al., 2012). This domain is the target of many signaling pathways that regulate the gap junction channel open or closed states (Palatinus et al., 2012; Solan and Lampe, 2009). Furthermore, numerous signaling proteins exist in a complex with the Cx43 CT (Hervé and Derangeon, 2013; Hervé et al., 2012; Palatinus et al., 2012). In osteoblasts, a growing body of evidence suggests that protein–protein interactions with the Cx43 CT domain are important for the spatio-temporal recruitment of signaling proteins to gap junctions for efficient downstream signaling (Batra et al., 2012; Bivi et al., 2011; Hebert and Stains, 2013; Niger et al., 2010). In vitro, we have shown that Cx43 interacts with PKCδ in osteoblasts (Hebert and Stains, 2013; Niger et al., 2010) and that the Cx43 CT domain is necessary, but not sufficient, to allow optimal signaling through PKCδ and ERK1 and ERK2 (ERK1/2, also known as MAPK3 and MAPK1, respectively), despite supporting similar levels of gap junctional intercellular communication (Hebert and Stains, 2013). Cx43 regulation of these signaling pathways converge on essential osteoblast transcription factors, like Runx2 (Lima et al., 2009; Niger et al., 2012, 2013). In addition, the Cx43 CT domain is needed for the Cx43-dependent activation of osteoblast gene expression, as a full-length Cx43, but not CT-truncated Cx43 protein, stimulates the expression of osteocalcin (encoded by Bglap), type I collagen (encoded by Col1a1), osterix (encoded by Sp7) and osteoprotegerin (OPG; encoded by Tnfrsf11b) (Hebert and Stains, 2013). In this regard, we speculated that signaling proteins, such as ERK1/2, PKCδ and β-catenin, are locally recruited to the gap junction plaque via their interaction with the Cx43 CT domain. Accordingly, as ions and second messengers are communicated through the gap junction pore, the locally assembled signaling machinery can be efficiently activated to affect bone cell function.
In the present study, we tested the hypothesis that the Cx43 CT is a docking platform for signaling proteins and is required for efficient downstream signaling in bone, using a mouse model of Cx43 CT truncation (Gja1 K258Stop). In this mouse model, the endogenous Gja1 allele was replaced with a truncation mutant, lacking much of the CT domain (amino acids 258–382 removed) downstream of the microtubule interaction domain (Maass et al., 2004). The resultant truncated Cx43 traffics to the plasma membrane and forms functional gap junctions with single channel properties similar to intact Cx43 gap junctions (Maass et al., 2007). However, the Cx43 K258Stop gap junction plaques tend to be larger in size and fewer in number (Maass et al., 2007). In contrast to Gja1−/− mice (Reaume et al., 1995; Ya et al., 1998), there is no lethal obstruction of the cardiac ventricular outflow tract in homozygous Gja1K258Stop/K258Stop mice, although these mice die as a result of a postnatal epidermal barrier defect (Maass et al., 2004). In contrast, Gja1−/K258Stop mice, which have just a single copy of the truncated Cx43 allele, are viable and healthy with no overt phenotype (Maass et al., 2007). Here, we show that 6-week-old Gja1−/K258Stop male mice have an obvious skeletal phenotype that includes increased femoral diaphyseal marrow cavity area, enhanced periosteal apposition and endosteal resorption, increased cortical porosity and cortical thinning relative to Gja1+/− controls. Truncation of Cx43 reduced the mRNA levels of genes involved in osteoblast proliferation, differentiation and collagen processing. Furthermore, truncation of the Cx43 CT resulted in reduced levels of phosphorylated (p)PKCδ, pERK1/2 and active β-catenin, signaling effectors that bind the Cx43 CT and play roles in osteoblast function. These data suggest that the Cx43 CT serves as a docking platform for signaling proteins and is required for efficient signal pathway activation to affect cell proliferation, differentiation and function.
RESULTS
The Cx43 CT interacts with signaling proteins
In osteoblasts, Cx43 is in a complex with several proteins, including PKCδ, β-arrestin, ZO-1 (also known as TJP1), and α5β1 integrin (Batra et al., 2012; Bivi et al., 2011; Hervé and Derangeon, 2013; Niger et al., 2010). Furthermore, alteration of Cx43 abundance regulates the activation of ERK1/2 and PKCδ signaling cascades (Lima et al., 2009; Niger et al., 2012, 2013). In vitro, overexpression of a full-length, intact Cx43, but not a Cx43 S244Stop CT truncation mutant, increases signal transduction through the PKCδ and ERK1/2 pathways and elevates the mRNA levels of osteoblast-associated genes, even though both constructs support similar levels of intercellular communication (Hebert and Stains, 2013).
In order to establish that the Cx43 CT interacts with signaling proteins that are known to be affected by Cx43 abundance and are relevant to bone biology, we performed co-immunoprecipitations in UMR106 osteoblast-like cells (Fig. 1). Immunoprecipitation of ERK1/2, PKCδ and β-catenin resulted in the co-precipitation of Cx43 (Fig. 1A). Conversely, overexpression of just a FLAG-tagged Cx43 CT domain (amino acids 236–382) reduced the binding of the full-length Cx43 to these signaling proteins. When the reverse co-immunoprecipitation was performed, these signaling effectors were found in complex with the overexpressed FLAG–Cx43 CT construct (Fig. 1B). These data show that PKCδ, ERK1/2 and β-catenin are in a complex with Cx43 and suggest that the overexpressed Cx43 CT competes for binding of the signaling proteins with the endogenous intact Cx43.
Fig. 1.

Overexpression of the Cx43 CT domain competes for binding of signaling proteins with the intact Cx43 protein. Co-immunoprecipitations (co-IPs) were performed to assess protein–protein interactions between the indicated signaling proteins and the Cx43 C-terminus. UMR106 cells were transfected with a construct encoding the CT domain of Cx43 (FLAG-tagged) or with an empty vector control. (A) Input and bead fractions are shown for co-IPs performed with anti-ERK1/2, anti-PKCδ and anti β-catenin antibodies and blotted with anti-Cx43 antibodies. A negative control co-IP with an antibody to a protein unlikely to be found at the plasma membrane (anti-Sp1) was performed and blotted with anti-Cx43 antibodies and is shown below. (B) IPs were performed for the Cx43 C-terminus (with anti-FLAG antibody), and then blotted for the indicated signaling proteins.
Male Gja1−/K258Stop mice have no overt morphological phenotype
Given the accumulated in vitro data showing that the Cx43 CT binds numerous signaling proteins to affect signaling and cell function, we analyzed the consequence of Cx43 CT truncation in vivo. Specifically, we examined the skeletal phenotype of 6-week-old male Gja1−/K258Stop mice expressing a single allele of the Cx43 gene that lacks the majority of the Cx43 CT domain, relative to Gja1+/− mice expressing a single allele of the intact Cx43 gene (Fig. 2). X-rays showed no gross skeletal differences between male Gja1−/K258Stop mice and Gja1+/− littermate controls (Fig. 2A). Furthermore, there were no differences in body length or body weight between genotypes (Fig. 2B). Using an antibody recognizing a C-terminal epitope of Cx43, we confirmed absence of the Cx43 CT in the Gja1−/K258Stop mice. Quantitative real-time PCR (qRT-PCR) performed on cDNA from tibial extracts confirmed a loss of Gja1 mRNA in the Gja1−/K258Stop mice as compared to the heterozygous controls (Fig. 2C). In contrast, the mRNA expression of Gjc1 (Cx45), the second most abundant connexin expressed in bone, was unchanged. A prior report had shown a compensatory upregulation of Cx45 in Gja1−/− animals (Lecanda et al., 2000).
Fig. 2.
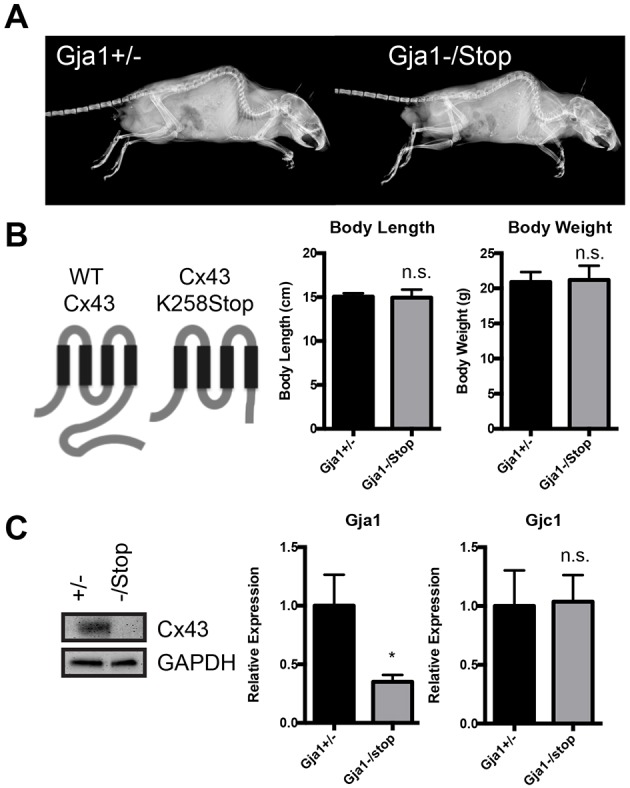
No apparent morphological defects are observed in mice with truncation of the Cx43 CT. (A) Digital X-rays, and (B) body length and weight measurements in 6-week-old male Gja1+/− (n=11) and Gja1−/K258Stop (n=8) mice. (B) Schematic of full-length (WT Cx43) and truncated Cx43 (Cx43 K258Stop) proteins, showing deletion of most of the C-terminus. (C) Western blot performed on tibial extracts (marrow flushed) for Cx43 (C-terminal epitope). qRT-PCR from RNA isolated from tibia (marrow flushed) of the indicated genotype was performed for Gja1 (Cx43; 3′ primers to the C-terminal encoding region) and Gjc1 (Cx45) mRNA expression. (n=7 for Gja1+/− and n=5 for Gja1−/K258Stop). Graphs depict mean±s.d. *P<0.05; n.s., not significant (two-tailed t-test).
Altered cortical bone in male Gja1−/K258Stop mice
Despite gross morphological similarity between Gja1−/K258Stop and Gja1+/− mice, microcomputed tomography (microCT) revealed a cortical bone phenotype in the femurs of male Gja1−/K258Stop mice (Fig. 3). At the femoral mid-diaphysis, there was a pronounced expansion of the cross-sectional area, with an increase in the periosteal (15%) and endosteal (26%) bone perimeter in Gja1−/K258Stop mice relative to age- and gender-matched littermate Gja1+/− controls. In addition, Gja1−/K258Stop mice demonstrated cortical thinning and a 3.7-fold increase in cortical porosity. The changes in cortical bone geometry resulted in a 48% increase in the mean polar moment of inertia. In contrast, there was no trabecular bone phenotype detected in 6-week-old male Gja1−/K258Stop mice relative to controls, despite an overall increase in cross-sectional volume in the distal femur (Table S1).
Fig. 3.

Truncation of Cx43 results in a cortical phenotype with increased cross-sectional area, increased cortical porosity, cortical thinning and marrow cavity expansion in 6-week-old male Gja1−/K258Stop mice. (A) Top, cross section through the femoral mid-diaphysis from a representative microCT slice from Gja1+/− and Gja1−/K258Stop mice. Bottom, 3D reconstruction of intact femurs. (B) Quantification of the cortical phenotype at the femoral mid-diaphysis by microCT (n=11 for Gja1+/− and n=8 for Gja1−/K258Stop); see Materials and Methods for the definitions of the parameters. Graphs depict mean±s.d. *P<0.05 (two-tailed t-test).
Altered bone formation rates in male Gja1−/K258Stop mice
Dynamic histomorphometric analysis of the femoral mid-diaphyseal cortical bone showed a nearly 2-fold increase in periosteal bone formation rate without a concomitant change in mineral apposition rate (P=0.24) in mice expressing the truncated Cx43 (Fig. 4A). By contrast, on the endosteal surface the bone formation rate was diminished but did not reach statistical significance (P=0.10) in the Gja1−/K258Stop mice compared to control mice (Fig. 4B). Analysis of serum type I collagen N-propeptide (P1NP), a marker of bone formation, was performed using a commercial ELISA. P1NP levels were increased in the Gja1−/K258Stop samples (Fig. 4C). Static histomorphometry showed no difference in the osteoblast number normalized to the bone surface area between groups, although it was slightly elevated in the Gja1−/K258Stop samples (Fig. 4D). As osteocyte apoptosis has been implicated as one of the mechanisms of the effects of Cx43 on bone (Bivi et al., 2012a; Xu et al., 2015), empty and occupied osteocyte lacunae were counted in cortical bone for mice of each genotype. The percentage of occupied osteocyte lacunae was unaffected by Cx43 CT truncation (Fig. 4E).
Fig. 4.

Substantial periosteal bone apposition contributes to the cortical expansion of the mid-diaphysis in 6-week-old male Gja1−/K258Stop mice. Calcein and Alizarin dual labeling and quantification of the mineral apposition rate (MAR) and bone formation rate (BFR) is shown for each genotype at the (A) periosteal (Ps) and (B) endosteal (Es) surface of the femoral diaphysis (n=3 animals per genotype, 3 sections per animal). (C) Quantification of serum levels of P1NP (n=4 per genotype). (D) Static histomorphometry of osteoblast number normalized to the bone surface area (n=5 per genotype). (E) Quantification of the percentage of occupied osteocyte lacunae observed in cortical bone from the indicated genotype (n=3 animals and >1000 lacunae/genotype). A representative hematoxylin-stained image of cortical bone is shown. White arrowheads point to occupied lacunae. Graphs depict mean±s.d. *P<0.05; n.s., not significant (two-tailed t-test).
Enhanced bone resorption in male Gja1−/K258Stop mice
Osteoblast-lineage-specific Cx43 deletion has been shown to indirectly affect osteoclast-mediated bone resorption via the RANKL and OPG pathway, an effect that contributes to the expansion of the marrow cavity in those models (Bivi et al., 2012a; Lloyd et al., 2013; Watkins et al., 2011, 2012; Xu et al., 2015; Zhang et al., 2011). Accordingly, we looked at RANKL and OPG mRNA levels in RNA extracts from tibial extracts by qRT-PCR (Fig. 5A). In Gja1−/K258Stop mice, the relative expression of the gene encoding OPG was 68% of the levels in controls. Relative expression of the gene encoding RANKL was statistically unchanged between genotypes. As predicted by the increased RANKL-to-OPG ratio, the number of TRAP-positive osteoclasts normalized to the bone surface area was increased 2.8-fold in the mice expressing the truncated Cx43 (Fig. 5B). Similarly, serum markers of bone turnover as determined with a commercial ELISA for bone resorption (type I collagen C-telopeptide, CTX) were markedly increased in Gja1−/K258Stop mice (Fig. 5C).
Fig. 5.
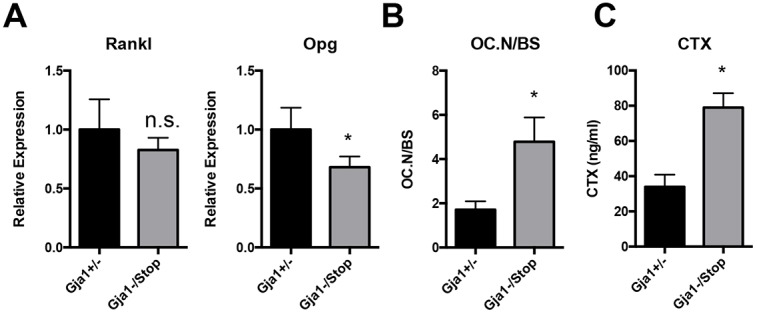
Cx43 truncation affects regulators of osteoclastogenesis, leading to increased osteoclast number and increased bone resorption. (A) qRT-PCR from RNA isolated from tibia (marrow flushed) of the indicated genotype was performed for RANKL and OPG mRNA expression (n=7 for Gja1+/− and n=5 for Gja1−/K258Stop). (B) Static histomorphometry of osteoclast number normalized to the bone surface area (OC.N/BS; n=5 per genotype). (C) Quantification of serum levels of CTX (n=3 for Gja1+/− and n=4 for Gja1−/K258Stop). Graphs depict mean±s.d. *P<0.05; n.s., not significant (two-tailed t-test).
Signal transduction pathway activation is reduced in the Gja1−/K258Stop mice
To glean insights into the molecular mechanisms by which the Cx43 CT domain influences bone acquisition and remodeling, we examined the activation of the signaling proteins (i.e. PKCδ, ERK1/2 and β-catenin) that physically associated with the Cx43 CT and have been shown to be impacted by Cx43 levels in vitro. As predicted, the levels of pPKCδ, pERK1/2 and active β-catenin were reduced in protein extracts from Gja1−/K258Stop tibiae (Fig. 6A). Consistent with decreased β-catenin activity, Axin2 mRNA expression was decreased in the tibiae of Gja1−/K258Stop mice as compared to Gja1+/− controls (Fig. 6B). The mRNA expression of the Wnt/β-catenin antagonist sclerostin (Sost) was unexpectedly decreased in these samples as well (Fig. 6B). Next, we examined the expression of key transcriptional regulators of the osteoblast-lineage, Twist1, Runx2 and Osterix (Sp7). The relative mRNA expression of Twist1 and Osterix (Sp7) were reduced, while Runx2 levels were unaffected by truncation of the Cx43 CT domain (Fig. 6C). Western blotting confirmed a downregulation of Osterix, whereas Runx2 was only slightly reduced, in primary bone marrow stromal cells (BMSCs) isolated from Gja1−/K258Stop mice (Fig. 6C). We were unable to detect Twist1 protein in these extracts (data not shown). Similarly, markers of osteoblast differentiation were uniformly reduced in the tibiae of Gja1−/K258Stop mice (Fig. 6D). In total, these data confirm that Cx43 and the Cx43 CT domain play a role throughout the osteoblast lineage, including in the early stages of osteoblastogenesis.
Fig. 6.

Cx43 truncation impairs signal cascade activation and osteoblast differentiation. (A) Western blots were probed for indicated signaling proteins in tibial (marrow flushed) extracts of the indicated genotypes. The GAPDH blot shown in Fig. 6A (right panel) is the same as in Fig. 8B, as the same membrane was re-probed for the indicated factors. qRT-PCR from RNA isolated from tibia (marrow flushed) of the indicated genotype was performed for (B) Wnt/β-catenin-related genes, (C) transcriptional regulators of osteoblastogenesis, and (D) osteoblast differentiation markers (n=7 for Gja1+/− and n=5 for Gja1−/K258Stop). (C) Western blots on extracts from BMSCs cultured for 7 days in mineralization medium were probed for Runx2, Osterix and GAPDH (loading control). Graphs depict mean±s.d. *P<0.05; n.s., not significant (two-tailed t-test).
Collagen processing and matrix maturation is disrupted in Gja1−/K258Stop mice
Cx43 gene deletion in cells of the osteoblast lineage has been shown to affect the expression of lysyl oxidase (encoded by Lox), an enzyme involved in collagen fiber cross-linking and collagen organization (Bivi et al., 2012b; Watkins et al., 2011). Similarly, Lox mRNA is reduced in tibial extracts from Gja1−/K258Stop mice (Fig. 7A). In addition, the gene expression of the collagen-specific chaperone Serpinh1 (also known as Hsp47) is also reduced in these mice (Fig. 7A). Western blotting of primary BMSCs isolated from Gja1−/K258Stop and Gja1+/− mice confirmed the downregulation of Lox (proform and mature isoforms) and SerpinH1 proteins upon truncation of the Cx43 CT domain (Fig. 7A). Using Picrosirius Red staining in conjunction with polarized light microscopy to look at collagen fiber thickness and orientation, the collagen at the cortical bone in the femoral diaphysis of Gja1−/K258Stop had thinner fibers with a less-organized orientation in comparison to the same anatomical region in the Gja1+/− mice (Fig. 7B). This was confirmed by quantification of the birefringence hue distribution, with red indicating thicker more cross-linked fibers and green indicating thinner, less cross-linked fibers (Bauman et al., 2014; Whittaker and Rich, 2005). Orange and yellow colors indicate intermediate levels of thickness and orientation.
Fig. 7.
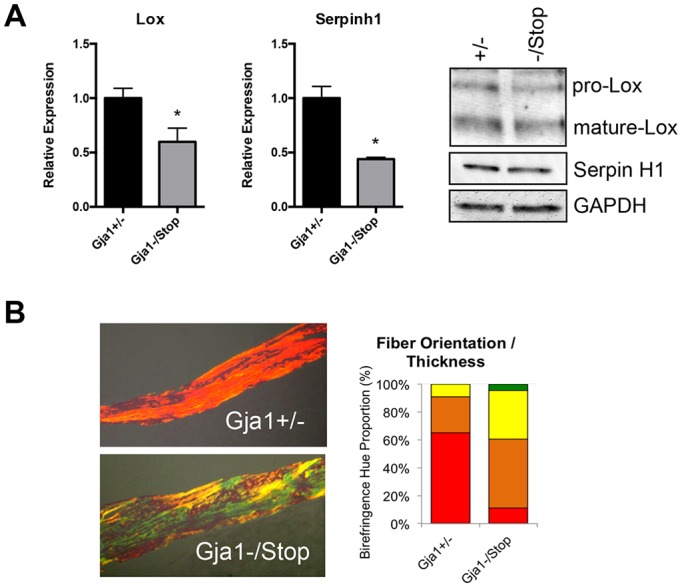
Cx43 truncation reduces mRNA expression of collagen processing factors and results in a disorganized and less mature collagen network in cortical bone. (A) qRT-PCR from RNA isolated from tibia (marrow flushed) of the indicated genotype was performed for expression of collagen-processing factors. Graphs depict mean±s.d. (n=7 for Gja1+/− and n=5 for Gja1−/K258Stop). *P<0.05 (two-tailed t-test). Western blot on extracts from BMSCs cultured for 7 days in mineralization medium and immunoblotted for Lox, Hsp47 and GAPDH (load control) are also shown. Bands for the preform (pro) and mature isoform of Lox are detected. (B) Picrosirius Red staining was performed and quantified at the mid-diaphyseal cortical bone, using a hue-range distribution method. A representative image of mid-diaphyseal cortical bone is shown. Quantification of fiber orientation and length is shown as a percentage of composition of the birefringent hue (red, orange, yellow and green).
A cell autonomous defect in proliferation and signaling in osteoblasts from Gja1−/K258Stop mice
In order to establish whether a cell autonomous defect in the osteoblast-lineage underlies the skeletal phenotype, bone marrow stromal cells (BMSCs) were isolated from mice of each genotype. Consistent with the increased osteoblast activity as determined by P1NP and bone formation rate findings, cultured BMSCs from Gja1−/K258Stop mice exhibited an accelerated rate of proliferation relative to controls (Fig. 8A). Indeed, cyclin D1 expression was increased in tibial extracts from the Gja1−/K258Stop mice (Fig. 8B). As was observed in vivo, the levels of pPKCδ, pERK, and active β-catenin were reduced in Gja1−/K258Stop BMSCs in culture (Fig. 8C). Finally, we determined mineralization capacity of primary cells from both genotypes. Increased Alizarin Red staining from Gja1−/K258Stop BMSCs is apparent after 14 days under mineralizing conditions (Fig. 8D).
Fig. 8.

Truncation of Cx43 leads to cell autonomous defects in osteoblast proliferation, signaling and mineralization. (A) Cell proliferation on BMSCs after 48 h in culture (n=6 wells per genotype). (B) Western blot on extracts from BMSCs cultured for 7 days in mineralization medium and immunoblotted for indicated signaling proteins. The blot for GAPDH shown in Fig. 8B is the same as in Fig. 6A (right panel), as the same membrane was re-probed for the indicated factors. (C) Alizarin Red S staining in replicate wells (n>4 wells per genotype) in BMSCs cultured for 14 days in mineralization medium. Graphs depict mean±s.d. *P<0.05 (two-tailed t-test).
DISCUSSION
This study demonstrates that the Cx43 CT binds signaling proteins and is required for efficient signaling and osteoblast differentiation in vitro and in vivo. We show, for the first time, that truncation of just the C-terminus of Cx43 in 6-week-old male mice results in a skeletal phenotype, primarily in cortical bone, that closely copies the phenotype of osteoblast-lineage-specific deletion of full-length Cx43 (Bivi et al., 2012a,b; Chung et al., 2006; Watkins et al., 2011; Zhang et al., 2011). This strongly suggests that, at least in skeletal tissue, the Cx43 CT domain is functionally as important as the entire intact Cx43 protein. Among the shared features between the Gja1−/K258Stop mice and most of the osteoblast-lineage-specific Cx43 conditional knockout mice are the increased marrow cavity area, due to enhanced periosteal bone apposition and endosteal bone resorption; alterations in RANKL and OPG ratio; increased osteoclast number; and changes in Lox gene expression that affect collagen processing. The reduction in Serpinh1 mRNA levels observed in Gja1−/K258Stop mice have not been reported previously in Cx43-knockout models. Decreased expression of this important collagen chaperone, which is implicated in osteogenesis imperfecta (Christiansen et al., 2010; Drögemuller et al., 2009; Kojima et al., 1998), may also contribute to the defective extracellular matrix and inferior material properties reported in other models of Cx43 deficiency (Bivi et al., 2012b; Lecanda et al., 2000; Watkins et al., 2011). These data confirm a role for Cx43 and the Cx43 CT in the organization and maturation of the collagenous extracellular matrix of bone (Bivi et al., 2012b; Hammond et al., 2016; Pacheco-Costa et al., 2016; Watkins et al., 2011).
Additionally, we observed decreased expression of osteoblast differentiation markers in vivo, and increased proliferation and mineralization capacity in in vitro cultures from Gja1−/K258Stop mice. While the increases in mineralization and concomitant decreases in expression of differentiation markers seem paradoxical, these findings are consistent with previous reports (Watkins et al., 2011). We speculate this may be due to a reciprocal role for Cx43 (and the Cx43 CT) in regulating early osteoprogenitor commitment and osteoblast differentiation. The control of progenitor cell commitment by Cx43 is supported by increases in BMSC proliferation rates, increases in cyclin D1 and a loss of Twist1 in Gja1−/K258Stop cells. Twist1 maintains the osteoprogenitor pool in part by inhibiting the activity of Runx2, a key transcriptional regulator of early osteoblast differentiation (Miraoui and Marie, 2010). Thus, a reduction in Twist1 mRNA could cause an expansion of the committed osteoblast progenitor population. However, we and others have shown here and elsewhere (Hebert and Stains, 2013; Niger et al., 2011; Watkins et al., 2011) that Cx43 and the Cx43 CT domain influence the expression of Osterix (Sp7), a transcription factor required for osteoblast differentiation downstream of Runx2. We think this reciprocal regulation could lead to a bottleneck of osteoprogenitors that are unable to efficiently pass through terminal osteoblast differentiation. Such an expansion of the osteoprogenitor pool could account for the substantial increase in periosteal bone formation observed here and in Cx43 conditional knockout models. Of course there are limitations to extrapolating in vitro proliferation data to the in vivo context. Careful dissection of the osteoprogenitor pool by lineage tracking or other means in vivo will be required to confirm the details of this apparent bottleneck of early osteogenic differentiation.
A key aspect of these findings is that, while loss of the Cx43 CT nearly perfectly phenocopies the loss of the intact Cx43 in bone cells, these mice do not exhibit the developmental cardiovascular defect observed in the Cx43 global null (Reaume et al., 1995). This implies that whatever signaling function the Cx43 CT plays, it is not universally required in all tissues or throughout development. Unlike the perinatal lethality observed upon global Cx43 gene deletion (Gja1−/−), Gja1−/K258Stop mice show no significant overt phenotype, but present with increased myocardial infarct size and susceptibility to arrhythmias following experimentally induced coronary occlusion (Maass et al., 2009). Notably, we cannot exclude the possibility that the Gja1 K258Stop allele might function as a neomorph. Gja1K258Stop/K258Stop mice exhibit an unexpected epidermal barrier defect that is present only in the homozygous mutants but not in Gja1−/K258Stop or Gja1+/K258Stop mice (Maass et al., 2004). However, the striking phenotypic overlap with osteoblast-lineage Cx43 conditional knockouts and the Gja1−/K258Stop mice makes it unlikely that this is a unique neomorphic function of the Cx43 K258Stop protein. Similar to our findings in bone, Gja1−/K258Stop phenocopies key aspects of Cx43 conditional knockout in neuronal tissue (Cina et al., 2009), indicating that the function of the Cx43 CT is important in a subset of tissues and is not exclusive to bone.
Several mechanisms have been proposed for how Cx43 deletion affects skeletal tissue, including signaling defects and disruption of osteocyte viability (Buo and Stains, 2014; Plotkin, 2014; Plotkin and Bellido, 2013; Stains and Civitelli, 2016). Here, we report the impact of Cx43 CT truncation on signaling via ERK1/2, PKCδ and β-catenin. Tissue extracts from cortical bone of male Gja1−/K258Stop mice showed that Cx43 truncation resulted in reduced levels of pPKCδ, pERK and active β-catenin, signaling effectors that bind to the Cx43 CT and play a role in osteoblast differentiation and function (Ge et al., 2007; Kobayashi et al., 2015; Tu et al., 2007). While in vitro data suggested such a link (Batra et al., 2012; Bivi et al., 2011, 2013; Hebert and Stains, 2013; Niger et al., 2010; Plotkin et al., 2002), this is the first report of altered signaling in these pathways downstream of Cx43 CT truncation in vivo. These results are consistent with the requirement of the Cx43 CT as a docking platform for efficient signaling within interconnected bone cells. Cx43-containing gap junctions are present throughout bone, forming a functional syncytium of interconnected osteoblasts, osteocytes and progenitor cells. This network of cells is believed to rapidly and efficiently propagate signals among the coupled cells. Accordingly, it is intuitive that truncation of the Cx43 CT, which binds and locally recruits signaling proteins, would lead to reduced signaling. Furthermore, the signals being passed among these cells are controlling the fate of signaling cascades known to be involved in both proliferation and maintenance of progenitors as well as differentiation through the osteoblast lineage, depending on the context of the cells and the signals passed.
Another mechanism contributing to the cortical phenotype of Cx43 deficiency is osteocyte apoptosis (Plotkin, 2014). Up to 30% unoccupied osteocyte lacunae have been observed in cortical bone of Cx43 conditional knockout mice (Bivi et al., 2012a; Lloyd et al., 2014; Xu et al., 2015). Despite the nearly identical cortical bone phenotype observed in the Gja1−/K258Stop and Cx43 conditional knockout models, we did not detect any difference in empty lacunae between groups, suggesting that osteocytic apoptosis does not precede the phenotype, at least in male Gja1−/K258Stop mice. Of course, this does not preclude a key role for osteocytes (viable or apoptotic) in the intercellular communication of bone remodeling signals. Indeed, osteocytes, osteoblasts and likely osteoprogenitors all form a functional syncytium of shared signals via intercellular communication through Cx43 (Stains and Civitelli, 2016).
Recently, a similar Cx43 truncation model was reported, in which the same Gja1 K258Stop allele was combined with osteocyte-specific deletion (Dmp1-Cre) of a floxed Gja1 allele and performed in 4.5-month-old female mice (Pacheco-Costa et al., 2016). These female Gja1flox/K258Stop; Dmp1-Cre mice had a strong trabecular phenotype (decreased bone volume fraction, trabecular thickness, number and separation), but no statistically significant cortical phenotype; male mice were not examined in that study (Pacheco-Costa et al., 2016). On the surface, these data appear in direct contrast with our findings, as 6-week-old male Gja1−/K258Stop mice had no change in trabecular parameters and had striking changes in the cortical envelope in our study. However, based on this recent publication (Pacheco-Costa et al., 2016), we conducted a screen of 6-week-old female Gja1+/− (n=3) and Gja1−/K258Stop (n=4) mice. Similar to their findings, we found comparable trends in 6-week-old female Gja1−/K258Stop mice in the trabecular and cortical compartments, where trabecular bone volume fraction, trabecular number and trabecular thickness approached significance (Table S2). These findings suggest the intriguing possibility that, beyond the disparate roles of Cx43 in the cortical and trabecular bone compartments, the effects of Cx43 on bone are sexually dimorphic. In female mice, estrogens are known to inhibit periosteal apposition and stimulate endocortical bone apposition; these effects may counteract the action of Cx43 to stimulate periosteal apposition and endocortical resorption (Callewaert et al., 2010b). However, to systematically address the sexually dimorphic skeletal phenotype in these mice, studies should be conducted comparing estrogen-replete and estrogen-deficient mice. Importantly, gender-specific differences in bone are a result of numerous systemic factors, thus estrogen signaling alone is unlikely to fully account for this discrepancy (Callewaert et al., 2010a; Manolagas et al., 2013).
In total, these data suggest that the Cx43 CT is required for optimal osteoblast signaling and gene expression, and that absence of the Cx43 CT in male mice results in a skeletal phenotype analogous to deletion of the entire Cx43 gene in osteoblasts. These data also imply that Cx43-containing gap junctions not only exchange signals, but also must recruit the appropriate effector molecules to the Cx43 CT in order to optimally modulate cell function and bone acquisition.
MATERIALS AND METHODS
Materials
All chemicals and reagents were purchased from Sigma (St Louis, MO) unless indicated otherwise. The antibodies use in this study were: Sigma, anti-Cx43 (C6219); Cell Signaling Technology, total ERK1/2 (9102L), phospho-ERK1/2 (9101L), total PKCδ (2058S), phospho-PKCδ Thr505 (9374S), active β-catenin (8814S), CyclinD1 (2978S), Runx2 (8486S), anti-rabbit HRP (7074S), anti-mouse HRP (7076S); BD Biosciences, total β-catenin (6310153); Agilent, FLAG (200470-2); Millipore, Osterix/Sp7 (AB3743), GAPDH (MAB374); Enzo Life Sciences, HSP47/SerpinH1 (ADI-SPA-470); Invitrogen, Lox (PA1-16953); Santa Cruz, Sp1 (sc-59). The FLAG-tagged Cx43 CT construct, pCMV3XFLAGm43CT (amino acids 236–382), was a gift from Cecilia Lo (Addgene plasmid # 17664). Primary antibodies were used at a 1:1000 dilution, except for anti-Cx43, which was used at 1:8000. Secondary antibodies were used at 1:2000.
Cell culture
UMR106 cells were obtained from the ATCC and were cultured as described previously (Gupta et al., 2016). Primary BMSCs were prepared from the long bones of mice of the indicated genotype as described (Buo et al., 2016). Briefly, bone marrow was flushed with sterile saline from long bones, and pelleted at 500 g for 10 min. The pellet was resuspended in αMEM containing 10% fetal bovine serum, penicillin (50 IU/ml), streptomycin (50 μg/ml) and gentamycin (50 μg/ml) and filtered through a 70 μm cell strainer. Cells were plated and grown to confluence. Cells were kept at 37°C in a humidified atmosphere of 95% air and 5% CO2. Medium was replaced every 2–3 days. For Alizarin Red S staining, confluent cultures of BMSCs were maintained for 14 days in mineralizing medium, which contains ascorbic acid-2-phosphate (50 ng/ml) and glycerol-2-phosphate (10 μM). For western blotting, confluent cultures of BMSCs were maintained for 7 days in mineralizing medium, which contains ascorbic acid-2-phosphate (50 ng/ml) and glycerol-2-phosphate (10 μM). Subsequently, the cells were rinsed three times in HBSS and fixed in 10% neutral buffered formalin prior to staining mineralized nodules with 1 mg/ml Alizarin Red S (pH 4.2) solution, as described previously (Mbalaviele et al., 2005). For cell proliferation assays, freshly isolated BMSCs isolated from mice of each genotype were seeded at 5000 cells/well in a 48-well plate and cultured in complete medium for 48 h. Cell number was quantitated using a CCK-8 colorimetric assay (Enzo Life Sciences, Farmingdale, NY) according to the manufacturer's specifications in comparison to parallel wells of known cell numbers.
Transient transfections, co-immunoprecipitations and western blotting
UMR106 cells were transiently transfected using the Jetprime transfection reagent (Polyplus, New York, NY), as described previously (Gupta et al., 2016). Endotoxin-free plasmid DNA was prepped using a Qiagen HI-Speed Maxi prep system, according to the manufacturer's specifications. Cells were lysed at 48 h post transfection with 1× cell lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100 and 1× HALT phosphatase and protease inhibitor cocktail). Whole-cell lysates (500 μg total proteins) were pre-cleared with protein A/G agarose beads at 4°C for 30 min. The cleared supernatants were incubated overnight at 4°C with 2 μg of the indicated antibodies (anti-ERK1/2, anti-PKCδ, anti-β-catenin, anti-FLAG and anti-Cx43 antibodies), followed by a 30 min incubation at 4°C with protein A/G agarose beads. After five washes in ice-cold PBS-T (PBS + 0.1% Tween 20), the proteins were eluted from the beads by heating the samples for 5 min at 95°C in Laemmli 3× SDS buffer [62.5 mM Tris-HCl, 2% (w/v) SDS, 10% glycerol, 50 mM DTT, 0.01% (w/v) Bromophenol Blue]. A fraction of the eluted proteins (bead fraction) and input fraction were analyzed by western blotting (as described below) with the indicated antibodies. Briefly, equal amounts of proteins were electrophoresed on 10% SDS-PAGE gels, blotted to PVDF membranes and probed with the indicated antibodies. Membranes were stripped and re-probed with anti-GAPDH antibodies to ensure equal loading of proteins among lanes. All blots were repeated at least three times with independent cell cultures or tibial extracts. Representative data are shown for each.
Mouse colony
All animal studies were performed with approval by the Animal Care and Use Committee at the University of Maryland School of Medicine. Gja1−/K258Stop mice on a C57BL/6 background were provided by Mario Delmar (School of Medicine, New York University, NY). Gja1+/− mice on a mixed (C57BL/6-C129/J) background were provided by Roberto Civitelli (Division of Endocrinology, Metabolism and Lipid Research, Washington University in St Louis, St Louis, MO). Breeding colonies were maintained at the University of Maryland School of Medicine. Littermates were used for all experiments. Mice were given ad libitum access to standard chow and water and were maintained on a 12-h-light–12-h-dark cycle. For genotyping, DNA was extracted from tail snips prior to weaning, using an Accustart genotyping mix (Quanta Bio, Gaithersburg, MD). PCR genotyping for the Gja1+ and Gja1− alleles was carried out using allele-specific PCR primers (primer 1, 5′-CACGTGAGCCAAGTACAGGA-3′; primer 2, 5′-AGGTGGTGTCCAGAGCCTTA-3′; primer 3, 5′-AATCCATCTTGTTCAATGGCCGATC-3′), as provided by the Jackson laboratory (stock number 002201). PCR genotyping for the Gja1+ and Gja1K258Stop alleles was carried out using allele-specific PCR primers (primer 1, 5′-GCATCCTCTTCAAGTCTGTCTTCG-3′; primer 2, 5′-CAAAACACCCCCCAACAAGGAACCTAG-3′) as described previously (Maass et al., 2004).
X-ray imaging
Lateral and AP radiographs (35 kV, 10 s) were taken of the mice, post euthanasia using a Faxitron digital x-ray system, as described (Gupta et al., 2015).
Protein extracts
For long-bone extracts, tibiae were dissected from both genotypes and cleaned of soft tissue. Subsequently, the epiphyses were removed and the marrow cavity flushed with sterile saline, prior to lysis in a modified RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 mM Na4P2O7, 10 mM 2-glycerolphosphate, 10 mM NaF, 10 mM EDTA, 1 mM EGTA, 1× HALT phosphatase and protease inhibitor cocktail). Sample homogenization was performed using 5 mm-diameter stainless steel beads and a Qiagen tissue lyser LT at 50 MHz for up to 10 min or until the sample was completely homogenized, as described (Buo et al., 2016). Samples were then centrifuged at maximum speed for 10 min at 4°C. The supernatant was used for protein expression analysis by western blotting. For BMSCs, cells were lysed in modified RIPA, as described (Gupta et al., 2014).
Quantitative real-time RT-PCR
Total RNA was isolated from tibial extracts (flushed of marrow) using TRIzol reagent and homogenizing the sample with 5-mm-diameter stainless steel beads and a Qiagen tissue lyzer LT at 50 MHz for up to 10 min. RNA (1 μg) was reverse transcribed and used for qRT-PCR as described previously (Hebert and Stains, 2013). The levels of the genes were simultaneously normalized to expression levels for Gapdh, Rpl13 and Hprt. PCR primer sequences are as published and/or are available upon request (Hebert and Stains, 2013; Niger et al., 2013).
MicroCT
Femurs were dissected from 6-week-old male Gja1+/− and Gja1−/K258Stop mice and fixed in 4% PFA for 2–4 days and then transferred to 70% ethanol. Three-dimensional microCT was performed on the femurs of each genotype using SkyScan 1172 (Bruker, Kontich, Belgium). The skeletal parameters assessed by microCT followed published nomenclature guidelines (Bouxsein et al., 2010). Bone morphology and microstructure were assessed at the mid-diaphysis for cortical parameters, including cortical bone volume (Ct.TV), periosteal perimeter (Ct.Peri.Pm), endosteal perimeter (Ct.Endo.Pm), cortical thickness (Ct.Th), cortical porosity (Ct.Po) and mean polar moment of inertia (MMI). Trabecular parameters were assessed at the distal femoral metaphysis for trabecular parameters, including the trabecular bone volume fraction (BV/TV), trabecular bone thickness (Tb.Th), trabecular number (Tb.N) and trabecular separation (Tb.Sp). Analysis was completed on 11 (Gja1+/−) and 8 (Gja1−/K258Stop) animals for each genotype, respectively. Femurs were scanned with 2K resolution, a 10 μm3 voxel size, and 0.5 mm Al filter at 60 kV and 167 µA. Trabecular bone was delineated manually in a region of interest 0.2 mm to 2.0 mm proximal to the distal femoral growth plate. For cortical bone parameters, transverse microCT scans were performed at the femoral diaphysis beginning at 56% of the femoral length (measured from the head of the femur) extending 0.6 mm distally.
ELISA
Serum was collected from animals, and serum CTX and P1NP levels were quantified using an IDS assay for P1NP production and RatLaps for CTX production, according to the manufacturer's specifications (Immunodiagnostic Systems Inc., Gaithersburg, MD).
Bone histomorphometry
Fixed femurs were decalcified in 14% EDTA (pH 8.0). Subsequently, the tissue was embedded in paraffin and 5 μm serial longitudinal sections were prepared. Sections were stained for tartrate-resistant acid phosphate using a leukocyte acid phosphatases (TRAP) kit (Sigma), as described previously (Nanjundaiah et al., 2012). Osteoblasts and osteoclasts adjacent to the bone surface were quantified using the Bioquant Osteo measure histology software, as described previously (Castro et al., 2004; Chung et al., 2006). Sections were stained with hematoxylin and cortical occupied and empty lacunae were counted [n=3 animals per genotype; 1002 (Gja1+/−) and 1015 (Gja1−/K258Stop) lacunae, respectively]. Picrosirius Red staining was performed on sections, which were allowed to air dry before imaging. Quantification of fiber thickness and orientation was completed using a hue range method (Bauman et al., 2014; Whittaker and Rich, 2005). For dynamic parameters, 6-week-old mice of each genotype were injected with calcein (7.5 mg/kg body weight) and alizarin 3-methyl iminodiacetic acid (30 mg/kg body weight) at 7 and 2 days before euthanasia, respectively. Femoral bone formation rates were quantified by dynamic histomorphometry, as described previously (Tomlinson and Silva, 2015). Following fixation, each femur was embedded in poly-(methyl methacrylate) and 100 μm thick transverse sections were cut (Leco VC50) at the femoral midpoint, then polished to 30 μm and mounted on glass slides. Images of each section was generated for analysis using fluorescence microscopy (Olympus IX-71). Images were analyzed for endosteal (Es) and periosteal (Ps) bone formation rate normalized to bone surface area (BFR/BS) and mineral apposition rate (MAR) as defined by the ASBMR Committee for Histomorphometry Nomenclature (Dempster et al., 2013).
Statistics
All data are displayed as mean±s.d. Unless stated otherwise, experiments were repeated at least three times (n≥3). For microCT and histological analysis, the reviewer was blinded to the genotype of the mouse. Mean expression was compared between samples by a two-tailed t-test for unpaired samples. Data were statistically analyzed using GraphPad Prism 6 software. P<0.05 was taken as being statistically significant.
Acknowledgements
We thank Mario Delmar (NYU) and Roberto Civitelli (Washington University St Louis) for providing the Gja1−/K258Stop and Gja1+/− mice, respectively. We thank Christopher Ward and Ramzi Khairallah for their assistance with the preparation of the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization, M.C.M. and J.P.S.; Investigation, M.C.M., C.H., R.E.T., S.R.I., M.C.; Writing, M.C.M., J.P.S.; Supervision, J.P.S.; Project administration, J.P.S.; Funding acquisition, J.P.S.
Funding
This work was supported by grants the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01-AR063631 to J.P.S.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.197285.supplemental
References
- Batra N., Burra S., Siller-Jackson A. J., Gu S., Xia X., Weber G. F., DeSimone D., Bonewald L. F., Lafer E. M., Sprague E. et al. (2012). Mechanical stress-activated integrin alpha5beta1 induces opening of connexin 43 hemichannels. Proc. Natl. Acad. Sci. USA 109, 3359-3364. 10.1073/pnas.1115967109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman T. M., Nicholson T. M., Abler L. L., Eliceiri K. W., Huang W., Vezina C. M. and Ricke W. A. (2014). Characterization of fibrillar collagens and extracellular matrix of glandular benign prostatic hyperplasia nodules. PLoS ONE 9, e109102 10.1371/journal.pone.0109102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivi N., Lezcano V., Romanello M., Bellido T. and Plotkin L. I. (2011). Connexin43 interacts with betaarrestin: a pre-requisite for osteoblast survival induced by parathyroid hormone. J. Cell. Biochem. 112, 2920-2930. 10.1002/jcb.23208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivi N., Condon K. W., Allen M. R., Farlow N., Passeri G., Brun L. R., Rhee Y., Bellido T. and Plotkin L. I. (2012a). Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J. Bone Miner. Res. 27, 374-389. 10.1002/jbmr.548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivi N., Nelson M. T., Faillace M. E., Li J., Miller L. M. and Plotkin L. I. (2012b). Deletion of Cx43 from osteocytes results in defective bone material properties but does not decrease extrinsic strength in cortical bone. Calcif. Tissue Int. 91, 215-224. 10.1007/s00223-012-9628-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivi N., Pacheco-Costa R., Brun L. R., Murphy T. R., Farlow N. R., Robling A. G., Bellido T. and Plotkin L. I. (2013). Absence of Cx43 selectively from osteocytes enhances responsiveness to mechanical force in mice. J. Orthop. Res. 31, 1075-1081. 10.1002/jor.22341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouxsein M. L., Boyd S. K., Christiansen B. A., Guldberg R. E., Jepsen K. J. and Müller R. (2010). Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res. 25, 1468-1486. 10.1002/jbmr.141 [DOI] [PubMed] [Google Scholar]
- Buo A. M. and Stains J. P. (2014). Gap junctional regulation of signal transduction in bone cells. FEBS Lett. 588, 1315-1321. 10.1016/j.febslet.2014.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buo A. M., Williams M. S., Kerr J. P. and Stains J. P. (2016). A cost-effective method to enhance adenoviral transduction of primary murine osteoblasts and bone marrow stromal cells. Bone Res. 4, 16021 10.1038/boneres.2016.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callewaert F., Sinnesael M., Gielen E., Boonen S. and Vanderschueren D. (2010a). Skeletal sexual dimorphism: relative contribution of sex steroids, GH-IGF1, and mechanical loading. J. Endocrinol. 207, 127-134. 10.1677/JOE-10-0209 [DOI] [PubMed] [Google Scholar]
- Callewaert F., Venken K., Kopchick J. J., Torcasio A., van Lenthe G. H., Boonen S. and Vanderschueren D. (2010b). Sexual dimorphism in cortical bone size and strength but not density is determined by independent and time-specific actions of sex steroids and IGF-1: evidence from pubertal mouse models. J. Bone Miner. Res. 25, 617-626. 10.1359/jbmr.090828 [DOI] [PubMed] [Google Scholar]
- Castro C. H. M., Shin C. S., Stains J. P., Cheng S.-L., Sheikh S., Mbalaviele G., Szejnfeld V. L. and Civitelli R. (2004). Targeted expression of a dominant-negative N-cadherin in vivo delays peak bone mass and increases adipogenesis. J. Cell Sci. 117, 2853-2864. 10.1242/jcs.01133 [DOI] [PubMed] [Google Scholar]
- Christiansen H. E., Schwarze U., Pyott S. M., AlSwaid A., Al Balwi M., Alrasheed S., Pepin M. G., Weis M. A., Eyre D. R. and Byers P. H. (2010). Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 86, 389-398. 10.1016/j.ajhg.2010.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung D. J., Castro C. H. M., Watkins M., Stains J. P., Chung M. Y., Szejnfeld V. L., Willecke K., Theis M. and Civitelli R. (2006). Low peak bone mass and attenuated anabolic response to parathyroid hormone in mice with an osteoblast-specific deletion of connexin43. J. Cell Sci. 119, 4187-4198. 10.1242/jcs.03162 [DOI] [PubMed] [Google Scholar]
- Cina C., Maass K., Theis M., Willecke K., Bechberger J. F. and Naus C. C. (2009). Involvement of the cytoplasmic C-terminal domain of connexin43 in neuronal migration. J. Neurosci. 29, 2009-2021. 10.1523/JNEUROSCI.5025-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster D. W., Compston J. E., Drezner M. K., Glorieux F. H., Kanis J. A., Malluche H., Meunier P. J., Ott S. M., Recker R. R. and Parfitt A. M. (2013). Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 28, 2-17. 10.1002/jbmr.1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drögemuller C., Becker D., Brunner A., Haase B., Kircher P., Seeliger F., Fehr M., Baumann U., Lindblad-Toh K. and Leeb T. (2009). A missense mutation in the SERPINH1 gene in Dachshunds with osteogenesis imperfecta. PLoS Genet. 5, e1000579 10.1371/journal.pgen.1000579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge C., Xiao G., Jiang D. and Franceschi R. T. (2007). Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J. Cell Biol. 176, 709-718. 10.1083/jcb.200610046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A., Niger C., Buo A. M., Eidelman E. R., Chen R. J. and Stains J. P. (2014). Connexin43 enhances the expression of osteoarthritis-associated genes in synovial fibroblasts in culture. BMC Musculoskelet. Disord. 15, 425 10.1186/1471-2474-15-425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R. R., Kim H., Chan Y.-K., Hebert C., Gitajn L., Yoo D. J., O'Toole R. V., Hsieh A. H. and Stains J. P. (2015). Axial strain enhances osteotomy repair with a concomitant increase in connexin43 expression. Bone Res. 3, 15007 10.1038/boneres.2015.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A., Anderson H., Buo A. M., Moorer M. C., Ren M. and Stains J. P. (2016). Communication of cAMP by connexin43 gap junctions regulates osteoblast signaling and gene expression. Cell. Signal. 28, 1048-1057. 10.1016/j.cellsig.2016.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond M. A., Berman A. G., Pacheco-Costa R., Davis H. M., Plotkin L. I. and Wallace J. M. (2016). Removing or truncating connexin 43 in murine osteocytes alters cortical geometry, nanoscale morphology, and tissue mechanics in the tibia. Bone 88, 85-91. 10.1016/j.bone.2016.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert C. and Stains J. P. (2013). An intact connexin43 is required to enhance signaling and gene expression in osteoblast-like cells. J. Cell. Biochem. 114, 2542-2550. 10.1002/jcb.24603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervé J.-C. and Derangeon M. (2013). Gap-junction-mediated cell-to-cell communication. Cell Tissue Res. 352, 21-31. 10.1007/s00441-012-1485-6 [DOI] [PubMed] [Google Scholar]
- Hervé J.-C., Derangeon M., Sarrouilhe D., Giepmans B. N. G. and Bourmeyster N. (2012). Gap junctional channels are parts of multiprotein complexes. Biochim. Biophys. Acta 1818, 1844-1865. 10.1016/j.bbamem.2011.12.009 [DOI] [PubMed] [Google Scholar]
- Kelly J. J., Simek J. and Laird D. W. (2015). Mechanisms linking connexin mutations to human diseases. Cell Tissue Res. 360, 701-721. 10.1007/s00441-014-2024-4 [DOI] [PubMed] [Google Scholar]
- Kobayashi Y., Uehara S., Koide M. and Takahashi N. (2015). The regulation of osteoclast differentiation by Wnt signals. Bonekey Rep. 4, 713 10.1038/bonekey.2015.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima T., Miyaishi O., Saga S., Ishiguro N., Tsutsui Y. and Iwata H. (1998). The retention of abnormal type I procollagen and correlated expression of HSP 47 in fibroblasts from a patient with lethal osteogenesis imperfecta. J. Pathol. 184, 212-218. [DOI] [PubMed] [Google Scholar]
- Laird D. W. (2014). Syndromic and non-syndromic disease-linked Cx43 mutations. FEBS Lett. 588, 1339-1348. 10.1016/j.febslet.2013.12.022 [DOI] [PubMed] [Google Scholar]
- Lecanda F., Warlow P. M., Sheikh S., Furlan F., Steinberg T. H. and Civitelli R. (2000). Connexin43 deficiency causes delayed ossification, craniofacial abnormalities, and osteoblast dysfunction. J. Cell Biol. 151, 931-944. 10.1083/jcb.151.4.931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima F., Niger C., Hebert C. and Stains J. P. (2009). Connexin43 potentiates osteoblast responsiveness to fibroblast growth factor 2 via a protein kinase C-delta/Runx2-dependent mechanism. Mol. Biol. Cell 20, 2697-2708. 10.1091/mbc.E08-10-1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd S. A., Loiselle A. E., Zhang Y. and Donahue H. J. (2013). Connexin 43 deficiency desensitizes bone to the effects of mechanical unloading through modulation of both arms of bone remodeling. Bone 57, 76-83. 10.1016/j.bone.2013.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd S. A., Loiselle A. E., Zhang Y. and Donahue H. J. (2014). Evidence for the role of connexin 43-mediated intercellular communication in the process of intracortical bone resorption via osteocytic osteolysis. BMC Musculoskelet. Disord. 15, 122 10.1186/1471-2474-15-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass K., Ghanem A., Kim J.-S., Saathoff M., Urschel S., Kirfel G., Grümmer R., Kretz M., Lewalter T., Tiemann K. et al. (2004). Defective epidermal barrier in neonatal mice lacking the C-terminal region of connexin43. Mol. Biol. Cell 15, 4597-4608. 10.1091/mbc.E04-04-0324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass K., Shibayama J., Chase S. E., Willecke K. and Delmar M. (2007). C-terminal truncation of connexin43 changes number, size, and localization of cardiac gap junction plaques. Circ. Res. 101, 1283-1291. 10.1161/CIRCRESAHA.107.162818 [DOI] [PubMed] [Google Scholar]
- Maass K., Chase S. E., Lin X. and Delmar M. (2009). Cx43 CT domain influences infarct size and susceptibility to ventricular tachyarrhythmias in acute myocardial infarction. Cardiovasc. Res. 84, 361-367. 10.1093/cvr/cvp250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolagas S. C., O'Brien C. A. and Almeida M. (2013). The role of estrogen and androgen receptors in bone health and disease. Nat. Rev. Endocrinol. 9, 699-712. 10.1038/nrendo.2013.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbalaviele G., Sheikh S., Stains J. P., Salazar V. S., Cheng S.-L., Chen D. and Civitelli R. (2005). Beta-catenin and BMP-2 synergize to promote osteoblast differentiation and new bone formation. J. Cell. Biochem. 94, 403-418. 10.1002/jcb.20253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miraoui H. and Marie P. J. (2010). Pivotal role of Twist in skeletal biology and pathology. Gene 468, 1-7. 10.1016/j.gene.2010.07.013 [DOI] [PubMed] [Google Scholar]
- Nanjundaiah S. M., Venkatesha S. H., Yu H., Tong L., Stains J. P. and Moudgil K. D. (2012). Celastrus and its bioactive celastrol protect against bone damage in autoimmune arthritis by modulating osteoimmune cross-talk. J. Biol. Chem. 287, 22216-22226. 10.1074/jbc.M112.356816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niger C., Hebert C. and Stains J. P. (2010). Interaction of connexin43 and protein kinase C-delta during FGF2 signaling. BMC Biochem. 11, 14 10.1186/1471-2091-11-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niger C., Lima F., Yoo D. J., Gupta R. R., Buo A. M., Hebert C. and Stains J. P. (2011). The transcriptional activity of osterix requires the recruitment of Sp1 to the osteocalcin proximal promoter. Bone 49, 683-692. 10.1016/j.bone.2011.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niger C., Buo A. M., Hebert C., Duggan B. T., Williams M. S. and Stains J. P. (2012). ERK acts in parallel to PKCdelta to mediate the connexin43-dependent potentiation of Runx2 activity by FGF2 in MC3T3 osteoblasts. Am. J. Physiol. Cell Physiol. 302, C1035-C1044. 10.1152/ajpcell.00262.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niger C., Luciotti M. A., Buo A. M., Hebert C., Ma V. and Stains J. P. (2013). The regulation of runt-related transcription factor 2 by fibroblast growth factor-2 and connexin43 requires the inositol polyphosphate/protein kinase Cdelta cascade. J. Bone Miner. Res. 28, 1468-1477. 10.1002/jbmr.1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco-Costa R., Davis H. M., Atkinson E. G., Katchburian E., Plotkin L. I. and Reginato R. D. (2016). Osteocytic connexin 43 is not required for the increase in bone mass induced by intermittent PTH administration in male mice. J. Musculoskelet. Neuronal. Interact. 16, 45-57. [PMC free article] [PubMed] [Google Scholar]
- Palatinus J. A., Rhett J. M. and Gourdie R. G. (2012). The connexin43 carboxyl terminus and cardiac gap junction organization. Biochim. Biophys. Acta 1818, 1831-1843. 10.1016/j.bbamem.2011.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin L. I. (2014). Connexin 43 hemichannels and intracellular signaling in bone cells. Front. Physiol. 5, 131 10.3389/fphys.2014.00131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin L. I. and Bellido T. (2013). Beyond gap junctions: Connexin43 and bone cell signaling. Bone 52, 157-166. 10.1016/j.bone.2012.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin L. I., Manolagas S. C. and Bellido T. (2002). Transduction of cell survival signals by connexin-43 hemichannels. J. Biol. Chem. 277, 8648-8657. 10.1074/jbc.M108625200 [DOI] [PubMed] [Google Scholar]
- Reaume A. G., de Sousa P. A., Kulkarni S., Langille B. L., Zhu D., Davies T. C., Juneja S. C., Kidder G. M. and Rossant J. (1995). Cardiac malformation in neonatal mice lacking connexin43. Science 267, 1831-1834. 10.1126/science.7892609 [DOI] [PubMed] [Google Scholar]
- Söhl G. and Willecke K. (2004). Gap junctions and the connexin protein family. Cardiovasc. Res. 62, 228-232. 10.1016/j.cardiores.2003.11.013 [DOI] [PubMed] [Google Scholar]
- Solan J. L. and Lampe P. D. (2009). Connexin43 phosphorylation: structural changes and biological effects. Biochem. J. 419, 261-272. 10.1042/BJ20082319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stains J. P. and Civitelli R. (2016). Connexins in the skeleton. Semin. Cell Dev. Biol. 50, 31-39. 10.1016/j.semcdb.2015.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson R. E. and Silva M. J. (2015). HIF-1alpha regulates bone formation after osteogenic mechanical loading. Bone 73, 98-104. 10.1016/j.bone.2014.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu X., Joeng K. S., Nakayama K. I., Nakayama K., Rajagopal J., Carroll T. J., McMahon A. P. and Long F. (2007). Noncanonical Wnt signaling through G protein-linked PKCdelta activation promotes bone formation. Dev. Cell 12, 113-127. 10.1016/j.devcel.2006.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins M., Grimston S. K., Norris J. Y., Guillotin B., Shaw A., Beniash E. and Civitelli R. (2011). Osteoblast connexin43 modulates skeletal architecture by regulating both arms of bone remodeling. Mol. Biol. Cell 22, 1240-1251. 10.1091/mbc.E10-07-0571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins M. P., Norris J. Y., Grimston S. K., Zhang X., Phipps R. J., Ebetino F. H. and Civitelli R. (2012). Bisphosphonates improve trabecular bone mass and normalize cortical thickness in ovariectomized, osteoblast connexin43 deficient mice. Bone 51, 787-794. 10.1016/j.bone.2012.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker P. and Rich L. (2005). Collagen and picrosirius red staining: A polarized light assessment of fibrillar hue and spatial distribution. Braz J. Morphol. Sci. 22, 97-104. [Google Scholar]
- Xu H., Gu S., Riquelme M. A., Burra S., Callaway D., Cheng H., Guda T., Schmitz J., Fajardo R. J., Werner S. L. et al. (2015). Connexin 43 channels are essential for normal bone structure and osteocyte viability. J. Bone Miner. Res. 30, 436-448. 10.1002/jbmr.2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ya J., Erdtsieck-Ernste E. B. H. W., de Boer P. A. J., van Kempen M. J. A., Jongsma H., Gros D., Moorman A. F. M. and Lamers W. H. (1998). Heart defects in connexin43-deficient mice. Circ. Res. 82, 360-366. 10.1161/01.RES.82.3.360 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Paul E. M., Sathyendra V., Davison A., Sharkey N., Bronson S., Srinivasan S., Gross T. S. and Donahue H. J. (2011). Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS ONE 6, e23516 10.1371/journal.pone.0023516 [DOI] [PMC free article] [PubMed] [Google Scholar]