

Abstract
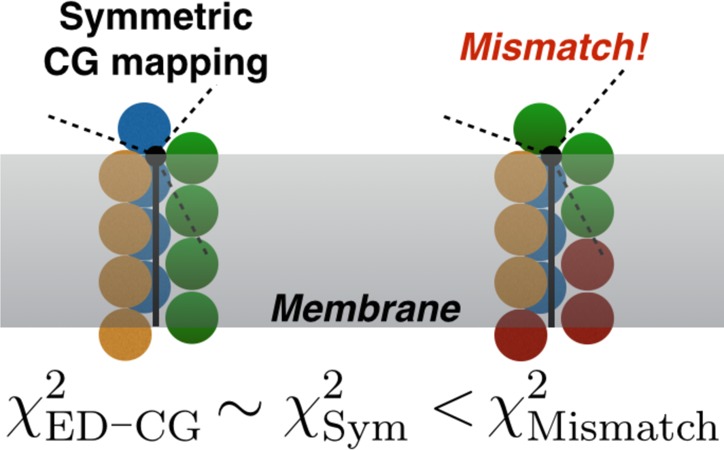
Numerous biomolecules and biomolecular complexes, including transmembrane proteins (TMPs), are symmetric or at least have approximate symmetries. Highly coarse-grained models of such biomolecules, aiming at capturing the essential structural and dynamical properties on resolution levels coarser than the residue scale, must preserve the underlying symmetry. However, making these models obey the correct physics is in general not straightforward, especially at the highly coarse-grained resolution where multiple (∼3–30 in the current study) amino acid residues are represented by a single coarse-grained site. In this paper, we propose a simple and fast method of coarse-graining TMPs obeying this condition. The procedure involves partitioning transmembrane domains into contiguous segments of equal length along the primary sequence. For the coarsest (lowest-resolution) mappings, it turns out to be most important to satisfy the symmetry in a coarse-grained model. As the resolution is increased to capture more detail, however, it becomes gradually more important to match modular repeats in the secondary structure (such as helix-loop repeats) instead. A set of eight TMPs of various complexity, functionality, structural topology, and internal symmetry, representing different classes of TMPs (ion channels, transporters, receptors, adhesion, and invasion proteins), has been examined. The present approach can be generalized to other systems possessing exact or approximate symmetry, allowing for reliable and fast creation of multiscale, highly coarse-grained mappings of large biomolecular assemblies.
I. Introduction
The symmetry of biomolecules originating from gene duplication and consolidated by evolution,1−3 while often only approximate, is intimately linked to functionality.4 For transmembrane proteins (TMPs) in particular, symmetry is one of the common properties shared in their functional states,5,6 and it has been related to dynamics,7 fast folding kinetics,8 high stability,2 and allosteric regulation.9−11 In addition, engineering of proteins with internal symmetry has become an emerging field with a growing body of reported success.12−14
TMPs, such as G protein-coupled receptors and ion channels, are crucial targets in drug discovery due to their physiological roles as direct rectors for drug-like solutes;15,16 it has been suggested that for receptors of neurotransmitters, for instance,17−19 an indirect and less specific mechanism whereby solutes absorbed into the lipid bilayer20−24 can affect the receptor. With around half of current drug targets being these TMPs25 and most of those drugs targeting only a few members,26 it is hardly surprising that the study of TMPs has become an active field of research with an increasing amount of experimental and computational efforts for potential pharmaceutical applications.
Despite advances in both hardware and software for atomistic molecular simulations,27−30 there is still a large gap between the duration of all-atom molecular dynamics (MD) trajectories produced on a routine basis (typically, microseconds) and time scales of biologically relevant events observed in experiments involving TMPs (usually milliseconds to seconds31,32). One fruitful strategy to overcome this gap and better bridge experiments and simulations is to apply a coarse-graining approach. The structure-based coarse-grained (CG) representation encompasses a reduced level of detail of the system, as atoms are grouped into “effective” particles also termed CG sites, and many of the biofunctionally irrelevant degrees of freedom are integrated out. One level of coarse-graining is the “high resolution” level in which each amino acid is represented by several CG sites or “beads”. Another level of coarse-graining is the “low resolution” highly CG level, where each CG site or bead represents some number of amino acids (e.g., tens or more). This paper concerns the latter limit of CG models.
A variety of modern coarse-graining approaches have been developed to define highly CG protein models, including essential dynamics coarse-graining,33−36 topology representing network,37 and rigid unit recognition.38 At the highly coarse-grained level, constructing CG models that satisfy the correct underlying physics is by no means a trivial task and often the resulting models are neither unique nor transferable.39,40 To simulate TMPs at very large spatial and temporal scales relevant for most biological processes, it is both useful and necessary to resort to models of the lowest, such as ultracoarse-grained (UCG) models,41,42 where one CG site represents many amino acid residues and may also have internal “states” to represent the various conformations, chemical forms, etc., of those eliminated amino acids from each CG site. The UCG methodology, often motivated in the context of modeling of the actin filament,41 has only recently been applied to other families of proteins43 but not yet to TMPs.
This work therefore describes our most recent efforts to construct highly CG models for TMPs based on the essential dynamics coarse-graining (ED-CG) method.33−36 The ED-CG method (or a similar approach44) is a systematic variational way of creating CG models that capture the most essential functional motions of biomolecules by a direct mapping of their atomistic motions. In this context, the essential dynamics45 from the atomistic simulations is used as a proxy for the functionally relevant motions. The ED-CG method determines the assignment of atoms to CG sites (the CG mapping) such that the essential dynamics subspace is best preserved at a given resolution.33 The ED-CG method has been applied to a variety of globular proteins and protein complexes, including a ribosome,46 actin filaments,47 and a hydrogenase.48 However, two limitations of the ED-CG method should be taken into consideration. First, the ED-CG approach does not by itself automatically determine the optimal resolution level of a CG model. The total number of CG sites is an externally set parameter by the user. (We note that this issue has been partially resolved in our previous work where we developed a set of criteria to choose optimal numbers of CG sites in different parts of a large biomolecular complex in a self-consistent way.35) Second, there is no guarantee that the ED-CG technique will create the same CG model for a protein in two or more discrete functional states. A previous study from our group shows that the ED-CG models of globular proteins like G-actin only share 60–80% of similarity between the ATP- and ADP-bound states.47 This creates a difficulty in using a CG representation, especially when it is desirable to study effects of transitions between distinct topological conformations. In this work, we have focused on addressing these issues for an important class of proteins, namely, TMPs.
As pointed out in prior work,41 it is important to understand the biomolecular features and peculiarities of the systems in order to construct meaningful CG models. It is generally appreciated in the field of coarse-graining that even small “inadequacies” in the CG mapping can manifest as damage beyond repair when the usual pairwise interaction potentials are used; two-site methanol is a classic example of a problematic CG mapping for a molecular liquid.49,50 For TMPs, the membrane environment imposes particular constraints onto the structure and dynamics of the transmembrane domains inserted into the lipid bilayer51 and differentiates them from extra- or intracellular domains of TMPs or their soluble counterparts. Such constraints give rise to many intriguing structural and dynamic properties of TMPs to account for their functions, such as symmetry. Although TMPs often exist in multimeric symmetric complexes of several repeating subunits with similar tertiary structures (even though the primary sequences of these subunits may be diverse),6 they also frequently possess approximate internal symmetry.
This work is primarily focused on TMPs with approximate internal symmetry, but the findings have the potential to be extended to cases with generalized symmetry; a comparison is made between CG models built using ED-CG methods and ones built on a simple and intuitive heuristic that exploits the molecular symmetry. It is shown that, by exploiting symmetry, we are able to construct CG mappings of TMPs for highly CG simulations consistent with the mappings resulting from the systematic “bottom-up” ED-CG method without the need for fine-grained MD trajectories and complex numerical optimization schemes.
II. Models, Theory, and Methods
In principle, the ED-CG method could be applied to all the atoms in a given protein structure. However, as a matter of practice, we use a residue-based strategy instead, wherein the position of each residue is represented solely by its Cα atom. Given a protein of Nres amino acid residues and NCG CG sites to assign (Nres≫ NCG), we can calculate the ED-CG variational residual χ2 and use it as a measure of the accuracy of a CG mapping to an underlying atomistic MD trajectory with nt frames. As defined in prior work,33 the residual is given by
| 1 |
where ΔriED(t) is the fluctuation of the Cα atom of residue i in the essential subspace at time t, calculated from principal component analysis52 of the atomistic MD simulation. If the Cα atom of another residue j exhibits motion (in the essential subspace) similar to that of the Cα atom of residue i, then it is reasonable to assign residues i and j to the same CG site I. This idea is mirrored in the definition of the χ2 (a “cost function”) by summing terms of fluctuation differences |Δri(t) – ΔrjED(t)|2 over pairs of atoms belonging to the same CG site. In this scheme, the ED-CG method samples a variety of possible ways to group atoms/residues and selects the one with the minimum residual χ2 as the optimal CG model.35
Sequence-Based and Space-Based ED-CG Methods
The ED-CG approach comes in two main variations, namely, sequence-based33 and space-based36 ED-CG. Both methods group the atoms into CG sites based on minimizing intrasite correlated fluctuations, but the different variants of the method applies different rules in sampling to locate the global minimum of χ2. The sequence-based ED-CG method divides the primary sequence of the protein into contiguous CG domains, while the spaced-based ED-CG method favors CG site definitions with atoms/residues close in the three-dimensional space. Provided the contiguous sequence constraint, the sequence-based ED-CG method is less demanding in sampling, but it does not permit nonadjacent domains in the same CG site, even if they are correlated in fluctuation but separated in the sequence (for example, in the case of a disulfide bond). Because of the much greater number of CG mappings allowed by space-based ED-CG, a brute-force search for the global minimum of χ2 would require looking through an exponentially greater number of combinations in comparison to sequence-based ED-CG. The use of simulated annealing and steepest descent techniques significantly decreases the number of combinations to be considered.33 Nevertheless, the amount of computations required to achieve a reasonably low value of χ2 is still greater in the case of the space-based ED-CG, and this gap increases with the number of atoms or residues in the biomolecule under investigation.
Power Law Scaling of the ED-CG Residual χ2
In our prior work,35 it was demonstrated that the ED-CG residual χ2 for the optimal CG map with a given number of CG sites can be approximated by a simple function of the protein size and the number of CG sites,
| 2 |
where the anomalous dimension γ is a protein-specific parameter, δ is a protein-independent coefficient, and C′(T) is a temperature-dependent prefactor. For a wide class of proteins, the value of γ was found35 to range from 0.00 to 0.91 (however, TMPs were not included into the studied set of proteins), while δ ≈ 0.35.
Internal Symmetry, Protein Fluctuation, and Symmetric CG Models
Internal symmetry will provide additional restraints in the coarse-graining of TMPs. In the context of biomolecules, we use the term internal symmetry for symmetry operations obeyed by the three-dimensional structure of the primary polypeptide chain sequence. On the basis of normal-mode analysis of MD simulations and group theory, Matsunaga and co-workers revealed that structural symmetry of homooligomers is a principal determinant of the entire protein complex’s symmetric fluctuation.7 In the same way, TMPs with internal symmetry should also have symmetric thermal fluctuation, which can be captured by ED-CG methods. Mapped onto the CG model, the symmetric domains of a TMP should result in identical CG domains. Directly, this suggests that the CG model should better describe symmetric fluctuation of the target TMP if it is consistent with the protein symmetry. In the simplest case of building a two-site CG model for a protein with perfect S-fold symmetry, we can always obtain the lowest ED-CG residual χ2 when either CG site contains half of the residues.
Our direct method (without ED-CG) of systematically constructing directly comparable CG mappings (of adjustable resolution) that satisfy the three-dimensional structural symmetry of the molecule that it represents is as follows. The contiguous protein sequence is evenly divided into NCG domains, which gives rise to a CG model that has an identical number of residues in each CG site (setting aside rounding errors); we shall refer to this construction as a symmetric model in this present work because these mappings satisfy a modular symmetry in the sense that each CG site is of equal size and separation in sequence space (N.B. only a subset of these mappings will be consistent with the structural symmetry of the molecule). We have collected a representative benchmark data set of eight important TMPs from Protein Data Bank (PDB)53 (Table 1) that all exhibit approximate internal symmetry in order to compare the CG models built by the ED-CG method to these symmetric CG models and thereby elaborate on the necessity of preserving symmetries, exact or approximate, when constructing highly CG mappings for biomolecular systems.
Table 1. Transmembrane Proteins Analyzed in This Work Belong to Different Structural Types and Approximate Symmetry Groups.
| protein | PDB ID code | residues | approximate symmetry point group | number of modular repeats | structure type |
|---|---|---|---|---|---|
| human integral membrane protein (hIMP) TMEM14A | 2LOP(75) | 25–99 | C3 | 3 | α-helical bundle |
| transmembrane domain of N-acetylcholine receptor (nAChR) β2 subunit | 2KSR(76) | 25–164 | C4 | 4 | α-helical bundle |
| human water channel aquaporin-1 (AQP1) | 1H6I(77) | 9–233 | S2 (=Ci) | 8 | α-helical bundle |
| mitochondrial ADP/ATP carrier | 1OKC(78) | 2–293 | C3 | 9 | α-helical bundle |
| ammonia transporter (AMT1) | 2B2F(79) | 1–391 | S2 (=Ci) | 11 | α-helical bundle |
| cytochrome c oxidase subunit 1 (COX1)-β | 1QLE(80) | 17–554 | C3 | 12 | α-helical bundle |
| outer membrane protein X (OmpX) | 1Q9F(81) | 1–148 | C4 | 8 | β-barrel |
| outer membrane protein A (OmpA) | 2GE4(82) | 0–176 | C4 | 8 | β-barrel |
Modeling and Simulations of Transmembrane Proteins
We selected a set of test cases by choosing TMPs with internal symmetry and no missing residues in the sequence. Our set of eight proteins represents TMPs of different size, structure, symmetry, function, and complexity and includes structures of either α-helical bundles or β-barrels (see Table 1 and Figure 1). We note that all of these proteins are folded and fluctuate around the stable equilibrium structure with no large-scale conformational rearrangements.
Figure 1.

Cartoon representations of eight transmembrane proteins studied in this work. Different colors are used to show symmetric units. PDB ID codes are indicated in parentheses.
These protein models were set up in a membrane-bound environment before performing the atomistic MD simulations. With Maestro (Schrödinger, Inc.), each PDB structure was prepared using Protein Preparation Wizard and embedded in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer by the System Builder. The TMP-membrane assemblies were placed in the simulation boxes, which were filled with explicit water (TIP3P water model54) and physiological salt (0.15 M NaCl) on both sides of the membrane. The distance between protein atoms and the box boundaries was at least 12 Å in all directions. CHARMM22/CMAP protein55,56 and CHARMM36 lipid57 force fields were used to assign parameters with the tool Viparr.58 After a 9-step standard relaxation protocol, which has been successfully applied in previous studies,59−61 each atomistic MD simulation was run for 30 ns in the isothermal–isobaric ensemble with constant temperature, T = 310 K, and constant pressure, P = 1 atm, using the Martyna-Tobias-Klein coupling scheme.62 Electrostatic forces were calculated using the particle mesh Ewald technique.63,64 van der Waals and short-range electrostatics were cut off at 9 Å. Long-range electrostatics were updated every third time step. All MD simulations were performed in the Desmond 3.0 simulation package65 with an integration time step of 2 fs. Hereafter, we applied the ED-CG method33 to build the CG models from the simulated all-atom MD trajectories.
Data Analysis
Structure visualization was performed with VMD66 and PyMOL.67 Plots were prepared using Grace (xmgrace; http://plasma-gate.weizmann.ac.il/Grace) and NumPy68/matplotlib.69 For the different sets of CG models for each TMP with the same CG resolution level, we computed and analyzed the naïve model similarity defined as the fraction of residues assigned to the same CG sites in the two compared models,
where Nres is the number of residues and δM(i),N(i) is the Kronecker delta function adding to the similarity whenever residue i is mapped to the same CG site by the two mappings of equal resolution, M and N.
III. Results and Discussion
ED-CG and Symmetric Models of Transmembrane Proteins
Initial tests were performed to compare the CG models built with space- and sequence-based ED-CG methods. We decided to proceed with the sequence-based variant as a suitable representative approach; results were almost identical for the systems and resolutions studied in this paper (in general, though, they will not be), as the space-based method exhibited much slower convergence rates.
To compare the highly CG models built with the ED-CG method to the symmetric CG models, we calculated the value of the residual χ2 (Figure 2) over a range of different numbers of CG sites (i.e., the CG resolution), corresponding to the highly CG mapping regime where multiple amino acid residues are represented by a single CG site. It is seen that the residuals for the symmetric models tend to exhibit an oscillatory behavior compared to ED-CG models and that the period of this oscillation depends on the CG resolution. These oscillatory “footprints” indicate that the collective dynamics, which encompasses symmetric modes for structurally symmetric molecules, is better captured by CG mappings that preserve the dominant symmetries. Since the calculated ED-CG χ2 residuals are a good proxy for the lower bound of the residual χ2 at a certain mapping resolution, we can identify a subset of the symmetric models that is optimal in the sense that the symmetric residual χ2 is almost identical to its lower bound for these models. The error of the symmetric model can be estimated by comparing its residual χ2 to the ED-CG χ2-residual-minimized mapping. While this error tends to be small, it increases systematically whenever the CG mapping does not preserve the structural symmetry of the TMPs, giving rise to what we shall call a symmetry mismatch that appears as an oscillatory difference in χ2 between the symmetric model and the ED-CG model (Figure 2). It can therefore be eliminated to the point where the residual χSym2 ≅ χEDCG2 by appropriately choosing the symmetric model that optimally aligns with the topological features of the TMP. The penalty for a symmetry mismatch follows the same power law relation as the ED-CG χ2 residual, and the relative error is therefore strongly dampened as the resolution of the mapping is increased.
Figure 2.

Plots of the χ2 residuals for the symmetric mappings (squares, green) and the ED-CG method resulting mappings (circles, red) for the eight transmembrane proteins plotted against numbers of CG sites (NCG). The panel with blue dots below each major plot shows the difference in χ2 between the symmetric model and the ED-CG model. Note the logarithmic scale for the y axis in the plotted χ2 residuals.
Optimal Symmetric Models in the CG Regime with ∼10–20 Amino Acid Residues per CG Site Satisfy Symmetry
For all
the test cases (Table 1 and Figure 1), it is observed that, for low
values of NCG (highly
CG models), the optimal subset of symmetric models always contains
models for which the number of CG sites complies with the symmetry
point group in the sense that 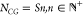 for the TMP with approximate S-fold internal symmetry. When this rule is not obeyed, there will
in general be a penalty in the χ2 residual. Our results
also show a number of differences between small and large TMPs. For
small proteins, such as TMEM14A (75 residues), we observe excellent
agreement between the two CG models when the NCG is a multiple of 3, which can be visually
understood looking at the CG map with 6 sites (Figure 3). The relatively large symmetry-mismatch
penalty observed in the χ2 residual for TMEM14A is
attributed to two factors: (1) the small size of the protein and (2)
the fact that the protein has three modular repeats (α-helices
in this case), which coincides with the approximate 3-fold axis of
symmetry (C3). For the larger TMPs in
our set of test cases, this effect is weaker (Figure 2). Model similarity between the ED-CG mapping
and the symmetric mapping at this level of resolution was very high
(∼80–90%) in all tested cases.
for the TMP with approximate S-fold internal symmetry. When this rule is not obeyed, there will
in general be a penalty in the χ2 residual. Our results
also show a number of differences between small and large TMPs. For
small proteins, such as TMEM14A (75 residues), we observe excellent
agreement between the two CG models when the NCG is a multiple of 3, which can be visually
understood looking at the CG map with 6 sites (Figure 3). The relatively large symmetry-mismatch
penalty observed in the χ2 residual for TMEM14A is
attributed to two factors: (1) the small size of the protein and (2)
the fact that the protein has three modular repeats (α-helices
in this case), which coincides with the approximate 3-fold axis of
symmetry (C3). For the larger TMPs in
our set of test cases, this effect is weaker (Figure 2). Model similarity between the ED-CG mapping
and the symmetric mapping at this level of resolution was very high
(∼80–90%) in all tested cases.
Figure 3.
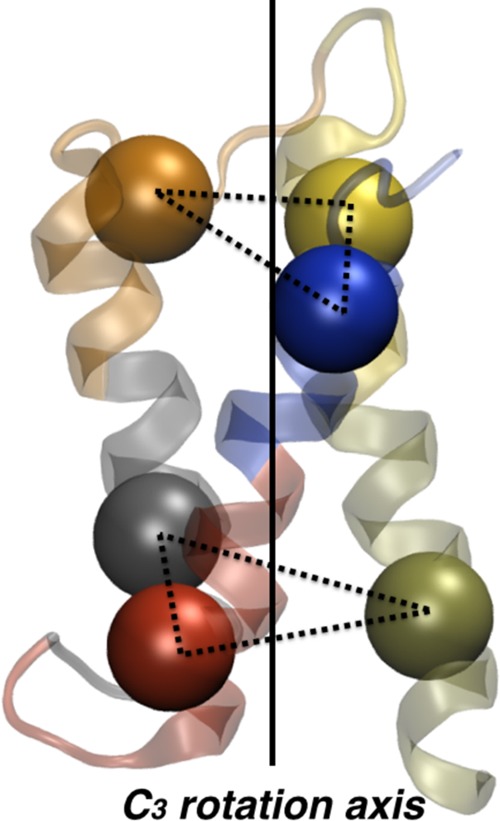
An example of a symmetric CG map for the protein TMEM14A. The backbone of the atomistic X-ray crystal structure is shown as translucent ribbons. The corresponding CG sites of the mapped structure are shown as solid spheres. The approximate C3 symmetry axis is indicated by a vertical solid line. The geometric planes that flank the molecule in the long (transmembrane) dimension are indicated by dashed-line triangles.
Optimal Symmetric Models in the CG Regime with ∼5–10 Amino Acid Residues per CG Site Satisfy Modular Repeats in the Secondary Structure
The symmetry mismatch penalty for the higher resolution models is negligible. While model similarity between the ED-CG mapping and the symmetric mapping at this level of resolution for the tested cases varied somewhat (∼45–75%), the absolute difference in the values of the residual χ2 is subtle (Figure 2, lower panels). This makes physical sense because, at a certain threshold resolution (here, ∼10 amino acids per CG site), there will be enough CG sites in the asymmetric subunit to adequately represent the dynamics of the unit in the essential subspace. However, it turns out that longer-period oscillations appear instead. These oscillations can be interpreted as mismatches (albeit numerically very small compared with the previously described symmetry mismatches) to the modular repeats in the secondary structure of the TMP. For α-helical bundles (β-barrels), the modular repeats are the individual helix-loop (strand-loop) motifs.
Physical Significance of the Anomalous Dimension γ
On the basis of the data plotted in Figure 2, we calculated the values of the anomalous dimension γ and the temperature-dependent prefactor C(T,Nres), as defined by eq 2. As shown in Table 2, γ falls in a small range around 1.0 for α-helical bundles and in another small range around 1.5 for β-barrels. These values are generally higher than the values previously reported for globular proteins like ubiquitin (γ = 0.50) or G-actin (γ = 0.33), implying that χ2 decreases faster for TMPs than for other proteins when the resolution of the CG mapping is increased. Our results also show that the anomalous dimension γ falls within a very well-defined range for specific TMPs with similar topology. In addition, the similar γ values between the sequence-based ED-CG models and the symmetric CG models indicate good agreement with respect to scaling behavior through the whole range of mapping resolutions.
Table 2. Anomalous Dimensions γ of TMPs Are Close to 1, Unlike Those of Globular Proteinsa.
| protein | Nres | ED-CG γ | sym. γ |
|---|---|---|---|
| human integral membrane protein (hIMP) TMEM14A | 75 | 1.10 (0.02) | 0.95 (0.04) |
| transmembrane domain of N-acetylcholine receptor (nAChR) β2 subunit | 140 | 0.96 (0.01) | 0.99 (0.03) |
| human water channel aquaporin-1 (AQP1) | 225 | 0.96 (0.03) | 0.98 (0.01) |
| mitochondrial ADP/ATP carrier | 292 | 1.06 (0.04) | 1.08 (0.05) |
| ammonia transporter (AMT1) | 391 | 1.01 (0.01) | 1.03 (0.02) |
| cytochrome c oxidase subunit 1 (COX1)-β | 538 | 1.15 (0.02) | 1.18 (0.03) |
| outer membrane protein X (OmpX) | 148 | 1.54 (0.05) | 1.57 (0.04) |
| outer membrane protein A (OmpA) | 177 | 1.44 (0.05) | 1.49 (0.03) |
Standard deviations of our estimates of γ are shown in parentheses.
To explain why the values of γ in the case of TMPs are typically higher than in the case of globular proteins studied earlier, we studied two simplified models: one of a solid ball and the other of a straight rod. The anomalous dimensions for these two extreme case model systems are demonstrated to be 0 and 1, respectively (see Appendix A for details). Most proteins considered in the previous work35 are globular; hence, it is reasonable that their anomalous dimensions are typically closer to 0. On the other hand, most TMPs considered in this work are formed by sets of transmembrane α-helices. A set of straight rods, in the approximation of weak interactions between the rods, has the same anomalous dimension as a single rod does (for details, see Appendix B). This analytical result explains why the anomalous dimensions of TMPs are closer to 1 and, therefore, greater than those for globular proteins.
The difference in the anomalous dimensions of the two groups of proteins (or, in general, any biomolecules) leads to an interesting consequence for a multimolecular complex formed by weakly interacting nrod “rod-shaped” components (such as α-helices embedded into a lipid bilayer) and nball “ball-shaped” molecules (such as extra- or intracellular parts of membrane-associated proteins). In this case, an increase in the average resolution level of the CG model of the complex leads to a higher resolution representation of the “ball-shaped” parts in comparison to the “rod-shaped” parts or, in other words, the new CG sites added to the complex upon increasing resolution mainly end up in “ball-shaped” (e.g., extra- or intracellular) components of the complex. In mathematical terms, if the total number of CG sites in the multimolecular complex is denoted NCGtotal, then the ratio of the optimal number of CG sites per each “rod-shaped” component NCG per rod to the optimal number of CG sites per each “ball-shaped” component NCG per ball has the following asymptotic behavior as NCG → ∞:
| 3 |
or, equivalently, the fraction of CG sites within “rod-shaped” components decreases with the increase of the resolution level of a CG model of the complex
| 4 |
Inversely, in coarser CG models, for example, UCG models,41 the optimal distribution of the CG sites implies a more detailed description of “rod-shaped” components (e.g., filamentous proteins or α-helices in a protein).
The oscillatory behavior of the χ2(NCG) curves for TMPs with nsymm-fold rotational or rotoreflection symmetry can be explained on the basis of the universal scaling behavior for χ2 provided by eq 2 and the fact that the anomalous dimension for straight rods equals 1 (see details in Appendix B). The dependence of χ2 on NCG predicted by this simple model is shown in Figure 4 in black solid lines. The behavior of these χ2(NCG) curves is qualitatively similar to those in Figure 2 (especially, TMEM14A, AMT1, and COX1-β) despite the fact that the model of weakly interacting rods provides a simplified representation of dynamical behavior of TMPs.
Figure 4.

A model of nsymm = 3 (left panels) and nsymm = 7 (right panels) weakly interacting straight rods demonstrates an oscillatory behavior of the χ2(NCG) curves (shown with solid lines and circles; the corresponding χlower2(NCG) curves are shown with dashed lines). Therefore, the damped oscillatory behavior of the χ2(NCG) curves for TMPs analyzed in this Article (see Figure 2) is qualitatively captured by the simple model approximating TMPs by several interaction rods. Note the logarithmic scale for the y axis in the top panels.
Connection to Information Content in the CG Model
Very recently, Foley et al.70 investigated the connection between the entropic component of the potential of mean force (PMF) and the CG representation both in general terms and for concrete models, notably the Gaussian linear chain model where an exact explicit PMF could be derived. Their analysis suggests that there are bounds on the resolution range wherein information-efficient CG mappings can found. Our results presented herein add a new perspective by emphasizing that careful consideration of structural symmetries and local modularities in approximately symmetric transmembrane proteins may help to choose between CG mappings that preserve a comparable fraction of nontrivial information.
IV. Conclusions
In this work, we have demonstrated that accurate and precise CG mappings can be generated for a diverse class of TMPs without the use of computationally expensive MD simulations and subsequent global residual χ2 minimization. To investigate the design principle in a general sense, we have studied CG mappings that partition transmembrane domains into contiguous segments of equal length along the primary sequence. The relative error in χ2 resulting from the use of the proposed heuristic rule is oscillatory and strongly damped, which has two practical consequences. First, symmetry mismatch generally decreases for an increasing number of CG sites. Second, it is possible for the heuristic to produce CG mappings with negligible relative difference in χ2 values to ED-CG methods even in the UCG regime, as long as the number of CG sites agrees with the overall symmetry group of the system (most important for low-resolution CG models) and conforms with the modular repeats (most important for medium-resolution CG models). It is likely that this heuristic will be especially useful when used in conjugation with other procedures to select optimal CG mappings on a case-by-case basis. Moreover, from the analysis of simple models, we predict that low resolution UCG models generated with the ED-CG approach should contain more CG sites in “rod-shaped” parts of proteins and protein complexes, such as α-helices immersed into a lipid bilayer, while higher-resolution CG models with more CG sites contain a larger fraction of CG sites in “ball-shaped” parts of the system, such as extra- or intracellular parts. In summary, our study provides new insight into highly CG modeling of TMPs and facilitates CG simulations by demonstrating that simple symmetry-preserving CG mappings are fast and reliable constructions, which have potential applications to future highly CG (or UCG) simulations of large TMPs and TMP assemblies on long time scales.
Acknowledgments
This research was supported by the National Institutes of Health (NIH Grant R01-GM053148) and the National Science Foundation (NSF Grant CHE-1465248). J.J.M. is grateful for support from the Carlsberg Foundation in the form of a postdoctoral fellowship. The authors thank Drs. Jun Fan and Severin T. Schneebeli for helpful discussions. Computation resources were provided by the Texas Advanced Computing Center through the Extreme Science and Engineering Discovery Environment (XSEDE) network (Ranger and Stampede machines) and the Research Computing Center (RCC) at The University of Chicago.
Appendix A
Two simple models are analyzed in this appendix: a solid ball and a straight rod. The solid ball was approximated by a set of Nres points (pseudoatoms) placed at random positions within a sphere of a constant radius with a uniform density distribution. The potential energy V of the system was approximated using an elastic network model (ENM),71−73 namely,
| 5 |
where Δrij = rij – rij0 is the fluctuation of the distance between pseudoatoms i and j, rij is the equilibrium distance between these pseudoatoms, and the spring constants kij were chosen according to the following formula to make the model parameter-free:74
| 6 |
where c is a constant. For various values of Nres, the Hessian matrix and the covariance matrix for the potential energy defined by eq 5 were computed as described by Zhang et al.,34 followed by building CG models for various values of NCG with the use of the space-based ED-CG method.36,72 The space-based version of ED-CG was employed since the primary sequence of pseudoatoms is not defined in this model. The values of the anomalous dimension γ, as well as R2 values characterizing the accuracy of eq 2, were computed by the method of least-squares for the resulting χ2 (NCG) dependencies in the log–log coordinates. Our numerical results indicate (see Table 3) that the value of the anomalous dimension monotonically decreases when the number of pseudoatoms Nres increases. The rate of the decrease in the anomalous dimension suggests that, in the limit of a continuous solid ball, γ = 0.
Table 3. Anomalous Dimensions γ of a Solid Ball and a Straight Rod Converge to 0 and 1, Respectively, in the Continuous Limit of the Number of Pseudo-Atoms Nres → ∞, Confirming the Validity of eq 2a.
| Nres | 500 | 1000 | 5000 | |
|---|---|---|---|---|
| solid ball | γ | 0.063 | 0.041 | 0.020 |
| R2 | 0.99992 | 0.99998 | 0.99999 | |
| straight rod | γ | 1.00005 | 1.00001 | 1.00000 |
| R2 | 1.00000 | 1.00000 | 1.00000 |
The second simple model is a straight rod formed by Nres pseudoatoms equidistantly positioned along the z-axis in three-dimensional space. The potential energy V of this system was defined in the following way:
| 7 |
where k is a constant, and the summation is performed only over all pairs of neighboring pseudoatoms. Expanding the right-hand side of eq 7 in a Taylor series in terms of the fluctuations in the Cartesian coordinates of each pseudoatom dxi, dyi, and dzi and omitting the terms containing the third or higher powers of dxi, dyi, or dzi, one arrives at the expression
| 8 |
suggesting that the problem of a straight rod in a three-dimensional space effectively reduces to the problem in a one-dimensional space. The Hessian matrix for the 1D problem is
 |
9 |
(Note that omitting the third order terms in eq 8 does not affect the accuracy of eq 9, since these terms lead to zero contributions to the second derivatives at the reference geometry with zero displacements.) Using this Hessian, the covariance matrix and, subsequently, the χ2(NCG) dependence can be obtained as described by Zhang et al.34,36 The space-based and the sequence-based variants of the ED-CG method are effectively equivalent in the case of a straight rod, since sets of pseudoatoms located in space in the most compact way correspond to contiguous fragments of the sequence naturally defined in this model.
The values of the anomalous dimension, γ, as well as R2 values, characterizing the goodness-of-fit of eq 2 for the model under consideration, are given in Table 3. They suggest that, in the limit of an infinitely large number of pseudoatoms Nres or, equivalently, for a continuous rod, the value of the anomalous dimension reaches γ = 1.
The two simple models analyzed here can be considered as two extreme cases of possible protein shapes. Globular proteins have shapes closer to a solid ball, and therefore, the values of the anomalous dimension γ are closer to 0, while rod-shaped proteins, as well as α-helices within a single protein in the approximation of weak interhelical interactions, are closer to the simple model of a straight rod, and therefore, their typical values of the anomalous dimension γ are closer to 1.
Appendix B
A symmetric TMP can be approximately represented as a set of nsymm weakly interacting straight rods. The term “weakly interacting” here implies the following. In the absence of interactions between the rods, the potential energy of the system is the sum of the potential energies of all rods, and the Hessian and covariance matrices acquire a block-diagonal form. Since each of these blocks is a positively defined matrix, the minimum of χ2 defined by eq 1 is achieved for a CG mapping with atoms from different rods belonging to different CG domains. In other words, no CG domain in an optimal CG mapping includes atoms from different rods. Now, in the case of interacting rods, the lowest-order nontrivial terms in the expression for the potential energy are linear in terms of deviations of atoms from their reference positions. These terms, however, do not affect the Hessian since it is a matrix of the second order derivatives of the potential energy, and therefore, the linear terms accounting for inter-rod interactions do not change the values of the residual χ2. In physical terms, this means that some attractive or repulsive forces may act between the rods and possibly distort their shapes, but these interactions between the rods and these distortions of the rods do not affect the stiffness of the rods. This approximation of “weak interactions” seems reasonable (at least some of TMPs), since it is well-known that the transmembrane α-helices and β-barrels in TMPs are structurally stable.
Due to the block-diagonal structure of the covariance matrix and therefore separability of the sum of squares of atomistic fluctuations into contributions from individual rods, the total residual χ2 for the whole system of nsymm rods, according to eq 1, can be written as a function of the set of numbers of CG sites placed in the i-th rod NCG,i in the following form:
| 10 |
where χi2 (NCG,i) is the value of the variational residue χ2 for the i-th rod when the number of CG sites placed in that rod equals NCG,i. Taking into consideration the universal scaling law for χ2 provided by eq 2 and the fact that for straight rods the anomalous dimension is 1, eq 10 can be rewritten as
| 11 |
Minimization of χ2({NCG,i}) under the constraints that the total number of CG sites in the system equals NCG
| 12 |
and that each NCG,i is an integer leads to the desired result for χ2 of the system of nsymm rods as a function of the total number of CG sites NCG:
| 13 |
We now define the function χlower2(NCG) in the following way:
| 14 |
This definition differs from eq 13 in that NCG,i′ values are not limited to integers. Since the right-hand sides of both eqs 13 and 14 involve minimization but the latter case is less restricted, the following inequality must be satisfied:
| 15 |
The equality in this expression is achieved only when all NCG,i′ values from eq 14 are integers; otherwise, χ2 > χlower2. The values of NCG,i and χlower2 (NCG) in eq 14 can be found using the technique of Lagrangian multipliers, leading to
| 16 |
Therefore, χlower2 (as a function of NCG) possesses the same power law behavior with γ = 1 as χ2 for each rod. Note that {NCG,i′} values are integers if and only if the total number of CG sites NCG is a multiple of the order of the symmetry axis nsymm. If this condition is not satisfied, then χ2(NCG) > χlower2(NCG). This explains why the χ2(NCG) curve oscillates above χlower2(NCG), only touching it when the CG mapping is consistent with the structural symmetry of the system (Figure 4). The χlower2(NCG) curves in this figure were plotted using eq 16, while the χ2(NCG) values were obtained by numerically solving the minimization problem in eq 13.
Author Present Address
§ A.V.S.: Department of Chemistry, Stanford University, Stanford, CA 94305, United States.
Author Present Address
# J.L.: Department of Chemistry, The University of Vermont, Burlington, VT 05405, United States.
Author Contributions
‡ J.J.M., A.V.S., and J.L.: Authors contributed equally.
The authors declare no competing financial interest.
References
- Saier M. H. Tracing pathways of transport protein evolution. Mol. Microbiol. 2003, 48 (5), 1145–1156. 10.1046/j.1365-2958.2003.03499.x. [DOI] [PubMed] [Google Scholar]
- Goodsell D. S.; Olson A. J. Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 105–153. 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- Hennerdal A.; Falk J.; Lindahl E.; Elofsson A. Internal duplications in alpha-helical membrane protein topologies are common but the nonduplicated forms are rare. Protein Sci. 2010, 19 (12), 2305–2318. 10.1002/pro.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell T. L.; Srinivasan N. Symmetry, stability, and dynamics of multidomain and multicomponent protein systems. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (25), 14243–14248. 10.1073/pnas.93.25.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. Membrane-protein topology. Nat. Rev. Mol. Cell Biol. 2006, 7 (12), 909–918. 10.1038/nrm2063. [DOI] [PubMed] [Google Scholar]
- Choi S.; Jeon J.; Yang J.-S.; Kim S. Common occurrence of internal repeat symmetry in membrane proteins. Proteins: Struct., Funct., Genet. 2008, 71 (1), 68–80. 10.1002/prot.21656. [DOI] [PubMed] [Google Scholar]
- Matsunaga Y.; Koike R.; Ota M.; Tame J. R. H.; Kidera A. Influence of Structural Symmetry on Protein Dynamics. PLoS One 2012, 7 (11), e50011. 10.1371/journal.pone.0050011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy Y.; Cho S. S.; Shen T.; Onuchic J. N.; Wolynes P. G. Symmetry and frustration in protein energy landscapes: A near degeneracy resolves the Rop dimer-folding mystery. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (7), 2373–2378. 10.1073/pnas.0409572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J.; Wyman J.; Changeux J. P. On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol. 1965, 12, 88–118. 10.1016/S0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Changeux J. P.; Edelstein S. J. Allosteric mechanisms of signal transduction. Science 2005, 308 (5727), 1424–1428. 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- Changeux J. P. Allostery and the Monod-Wyman-Changeux model after 50 years. Annu. Rev. Biophys. 2012, 41, 103–133. 10.1146/annurev-biophys-050511-102222. [DOI] [PubMed] [Google Scholar]
- Fortenberry C.; Bowman E. A.; Proffitt W.; Dorr B.; Combs S.; Harp J.; Mizoue L.; Meiler J. Exploring Symmetry as an Avenue to the Computational Design of Large Protein Domains. J. Am. Chem. Soc. 2011, 133 (45), 18026–18029. 10.1021/ja2051217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair J. C.; Davies K. M.; Venien-Bryan C.; Noble M. E. M. Generation of protein lattices by fusing proteins with matching rotational symmetry. Nat. Nanotechnol. 2011, 6 (9), 558–562. 10.1038/nnano.2011.122. [DOI] [PubMed] [Google Scholar]
- Worsdorfer B.; Henning L. M.; Obexer R.; Hilvert D. Harnessing Protein Symmetry for Enzyme Design. ACS Catal. 2012, 2 (6), 982–985. 10.1021/cs300076t. [DOI] [Google Scholar]
- Overington J. P.; Al-Lazikani B.; Hopkins A. L. Opinion - How many drug targets are there?. Nat. Rev. Drug Discovery 2006, 5 (12), 993–996. 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Rask-Andersen M.; Almen M. S.; Schioth H. B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discovery 2011, 10 (8), 579–90. 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- Cantor R. S. The lateral pressure profile in membranes: a physical mechanism of general anesthesia. Biochemistry 1997, 36 (9), 2339–2344. 10.1021/bi9627323. [DOI] [PubMed] [Google Scholar]
- Cantor R. S. Receptor desensitization by neurotransmitters in membranes: are neurotransmitters the endogenous anesthetics?. Biochemistry 2003, 42 (41), 11891–11897. 10.1021/bi034534z. [DOI] [PubMed] [Google Scholar]
- Cantor R. S. The evolutionary origin of the need to sleep: an inevitable consequence of synaptic neurotransmission?. Front. Synaptic Neurosci. 2015, 7, 15. 10.3389/fnsyn.2015.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Ye F.; Velardez G. F.; Peters G. H.; Westh P. Affinity of four polar neurotransmitters for lipid bilayer membranes. J. Phys. Chem. B 2011, 115 (1), 196–203. 10.1021/jp108368w. [DOI] [PubMed] [Google Scholar]
- Orlowski A.; Grzybek M.; Bunker A.; Pasenkiewicz-Gierula M.; Vattulainen I.; Mannisto P. T.; Rog T. Strong preferences of dopamine and l-dopa towards lipid head group: importance of lipid composition and implication for neurotransmitter metabolism. J. Neurochem. 2012, 122 (4), 681–90. 10.1111/j.1471-4159.2012.07813.x. [DOI] [PubMed] [Google Scholar]
- Peters G. H.; Wang C.; Cruys-Bagger N.; Velardez G. F.; Madsen J. J.; Westh P. Binding of serotonin to lipid membranes. J. Am. Chem. Soc. 2013, 135 (6), 2164–2171. 10.1021/ja306681d. [DOI] [PubMed] [Google Scholar]
- Peters G. H.; Werge M.; Elf-Lind M. N.; Madsen J. J.; Velardez G. F.; Westh P. Interaction of neurotransmitters with a phospholipid bilayer: a molecular dynamics study. Chem. Phys. Lipids 2014, 184, 7–17. 10.1016/j.chemphyslip.2014.08.003. [DOI] [PubMed] [Google Scholar]
- Postila P. A.; Vattulainen I.; Rog T. Selective effect of cell membrane on synaptic neurotransmission. Sci. Rep. 2016, 6, 19345. 10.1038/srep19345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundstrom An overview on GPCRs and drug discovery: structure-based drug design and structural biology on GPCRs. Methods Mol. Biol. 2009, 552, 51–66. 10.1007/978-1-60327-317-6_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappano R.; Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat. Rev. Drug Discovery 2011, 10 (1), 47–60. 10.1038/nrd3320. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26 (16), 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Huang C.; Zheng G.; Bohm E.; Bhatele A.; Phillips J. C.; Yu H.; Kale L. V. Scalable molecular dynamics with NAMD on the IBM Blue Gene/L system. IBM J. Res. Dev. 2008, 52 (1–2), 177–188. 10.1147/rd.521.0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror R. O.; Dirks R. M.; Grossman J. P.; Xu H. F.; Shaw D. E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. 10.1146/annurev-biophys-042910-155245. [DOI] [PubMed] [Google Scholar]
- Pronk S.; Pall S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; Hess B.; Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29 (7), 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. X.; Ruta V.; Chen J. Y.; Lee A.; MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature 2003, 423 (6935), 42–48. 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- Vilardaga J.-P. Theme and variations on kinetics of GPCR activation/deactivation. J. Recept. Signal Transduction Res. 2010, 30 (5), 304–312. 10.3109/10799893.2010.509728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Lu L.; Noid W. G.; Krishna V.; Pfaendtner J.; Voth G. A. A systematic methodology for defining coarse-grained sites in large biomolecules. Biophys. J. 2008, 95 (11), 5073–5083. 10.1529/biophysj.108.139626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Pfaendtner J.; Grafmüller A.; Voth G. A. Defining coarse-grained representations of large biomolecules and biomolecular complexes from elastic network models. Biophys. J. 2009, 97 (8), 2327–2337. 10.1016/j.bpj.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinitskiy A. V.; Saunders M. G.; Voth G. A. Optimal Number of Coarse-Grained Sites in Different Components of Large Biomolecular Complexes. J. Phys. Chem. B 2012, 116 (29), 8363–8374. 10.1021/jp2108895. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Y.; Voth G. A. Coarse-Grained Representations of Large Biomolecular Complexes from Low-Resolution Structural Data. J. Chem. Theory Comput. 2010, 6 (9), 2990–3002. 10.1021/ct100374a. [DOI] [PubMed] [Google Scholar]
- Martinetz T.; Schulten K. Topology Representing Networks. Neural Networks 1994, 7 (3), 507–522. 10.1016/0893-6080(94)90109-0. [DOI] [Google Scholar]
- Hespenheide B. M.; Jacobs D. J.; Thorpe M. F. Structural rigidity in the capsid assembly of cowpea chlorotic mottle virus. J. Phys.: Condens. Matter 2004, 16 (44), S5055–S5064. 10.1088/0953-8984/16/44/003. [DOI] [Google Scholar]
- Krishna V.; Noid W. G.; Voth G. A. The multiscale coarse-graining method. IV. Transferring coarse-grained potentials between temperatures. J. Chem. Phys. 2009, 131 (2), 024103. 10.1063/1.3167797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorobyov I.; Kim I.; Chu Z. T.; Warshel A. Refining the treatment of membrane proteins by coarse-grained models. Proteins: Struct., Funct., Genet. 2016, 84 (1), 92–117. 10.1002/prot.24958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dama J. F.; Sinitskiy A. V.; McCullagh M.; Weare J.; Roux B.; Dinner A. R.; Voth G. A. The Theory of Ultra-Coarse-Graining. 1. General Principles. J. Chem. Theory Comput. 2013, 9 (5), 2466–2480. 10.1021/ct4000444. [DOI] [PubMed] [Google Scholar]
- Davtyan A.; Dama J. F.; Sinitskiy A. V.; Voth G. A. The Theory of Ultra-Coarse-Graining. 2. Numerical Implementation. J. Chem. Theory Comput. 2014, 10 (12), 5265–5275. 10.1021/ct500834t. [DOI] [PubMed] [Google Scholar]
- Grime J. M. A.; Dama J. F.; Ganser-Pornillos B. K.; Woodward C. L.; Jensen G. J.; Yeager M.; Voth G. A. Coarse-grained simulation reveals key features of HIV-1 capsid self-assembly. Nat. Commun. 2016, 7, 11568. 10.1038/ncomms11568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Zhang J. Z.; Xia F. Constructing Optimal Coarse-Grained Sites of Huge Biomolecules by Fluctuation Maximization. J. Chem. Theory Comput. 2016, 12 (4), 2091–100. 10.1021/acs.jctc.6b00016. [DOI] [PubMed] [Google Scholar]
- Amadei A.; Linssen A. B.; Berendsen H. J. Essential dynamics of proteins. Proteins: Struct., Funct., Genet. 1993, 17 (4), 412–425. 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Sanbonmatsu K. Y.; Voth G. A. Key Intermolecular Interactions in the E. coli 70S Ribosome Revealed by Coarse-Grained Analysis. J. Am. Chem. Soc. 2011, 133 (42), 16828–16838. 10.1021/ja2028487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J.; Saunders M. G.; Voth G. A. Coarse-Graining Provides Insights on the Essential Nature of Heterogeneity in Actin Filaments. Biophys. J. 2012, 103 (6), 1334–1342. 10.1016/j.bpj.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullagh M.; Voth G. A. Unraveling the Role of the Protein Environment for [FeFe]-Hydrogenase: A New Application of Coarse-Graining. J. Phys. Chem. B 2013, 117 (15), 4062–4071. 10.1021/jp402441s. [DOI] [PubMed] [Google Scholar]
- Izvekov S.; Voth G. A. Modeling real dynamics in the coarse-grained representation of condensed phase systems. J. Chem. Phys. 2006, 125 (15), 151101. 10.1063/1.2360580. [DOI] [PubMed] [Google Scholar]
- Davtyan A.; Dama J. F.; Voth G. A.; Andersen H. C. Dynamic force matching: A method for constructing dynamical coarse-grained models with realistic time dependence. J. Chem. Phys. 2015, 142 (15), 154104. 10.1063/1.4917454. [DOI] [PubMed] [Google Scholar]
- Popot J. L.; Engelman D. M. Membranes Do Not Tell Proteins How To Fold. Biochemistry 2016, 55 (1), 5–18. 10.1021/acs.biochem.5b01134. [DOI] [PubMed] [Google Scholar]
- Jolliffe I. T.Principal component analysis, 2nd ed.; Springer: New York, 2002. [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiorkiewicz-Kuczera J.; Yin D.; Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102 (18), 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D.; Feig M.; Brooks C. L. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conforma- tional distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25 (11), 1400–1415. 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; Lopes P. E.; Mittal J.; Feig M.; Mackerell A. D. Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8 (9), 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmond Molecular Dynamics System, version 3.0; D. E. Shaw Research: New York, NY, 2011.
- Lyman E.; Higgs C.; Kim B.; Lupyan D.; Shelley J. C.; Farid R.; Voth G. A. A role for a specific cholesterol interaction in stabilizing the Apo configuration of the human A(2A) adenosine receptor. Structure 2009, 17 (12), 1660–1668. 10.1016/j.str.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Ziemba B. P.; Falke J. J.; Voth G. A. Interactions of protein kinase C-alpha C1A and C1B domains with membranes: a combined computational and experimental study. J. Am. Chem. Soc. 2014, 136 (33), 11757–66. 10.1021/ja505369r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Jonsson A. L.; Beuming T.; Shelley J. C.; Voth G. A. Ligand-dependent activation and deactivation of the human adenosine A(2A) receptor. J. Am. Chem. Soc. 2013, 135 (23), 8749–59. 10.1021/ja404391q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyna G. J.; Tobias D. J.; Klein M. L. Constant-Pressure Molecular-Dynamics Algorithms. J. Chem. Phys. 1994, 101 (5), 4177–4189. 10.1063/1.467468. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle Mesh Ewald: An N·Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98 (12), 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103 (19), 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Maestro-Desmond Interoperability Tools, version 3.0; Schrödinger: New York, NY, 2011.
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.7.4; Schrödinger, LLC: New York, NY, 2014.
- van der Walt S.; Colbert S. C.; Varoquaux G. The NumPy Array: A Structure for Efficient Numerical Computation. Comput. Sci. Eng. 2011, 13 (2), 22–30. 10.1109/MCSE.2011.37. [DOI] [Google Scholar]
- Hunter J. D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9 (3), 90–95. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- Foley T. T.; Shell M. S.; Noid W. G. The impact of resolution upon entropy and information in coarse-grained models. J. Chem. Phys. 2015, 143 (24), 243104. 10.1063/1.4929836. [DOI] [PubMed] [Google Scholar]
- Tirion M. M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996, 77 (9), 1905–1908. 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- Sinitskiy A. V.; Voth G. A. Coarse-graining of proteins based on elastic network models. Chem. Phys. 2013, 422, 165–174. 10.1016/j.chemphys.2013.01.024. [DOI] [Google Scholar]
- Haliloglu T.; Bahar I.; Erman B. Gaussian dynamics of folded proteins. Phys. Rev. Lett. 1997, 79 (16), 3090–3093. 10.1103/PhysRevLett.79.3090. [DOI] [Google Scholar]
- Yang L.; Song G.; Jernigan R. L. Protein elastic network models and the ranges of cooperativity. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (30), 12347–12352. 10.1073/pnas.0902159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klammt C.; Maslennikov I.; Bayrhuber M.; Eichmann C.; Vajpai N.; Chiu E. J.; Blain K. Y.; Esquivies L.; Kwon J. H.; Balana B.; Pieper U.; Sali A.; Slesinger P. A.; Kwiatkowski W.; Riek R.; Choe S. Facile backbone structure determination of human membrane proteins by NMR spectroscopy. Nat. Methods 2012, 9 (8), 834–839. 10.1038/nmeth.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko V.; Tillman T.; Xu Y.; Tang P. NMR structure of the transmembrane domain of the n-acetylcholine receptor beta2 subunit. Biochim. Biophys. Acta, Biomembr. 2010, 1798 (8), 1608–1614. 10.1016/j.bbamem.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot B. L.; Engel A.; Grubmüller H. A refined structure of human aquaporin-1. FEBS Lett. 2001, 504 (3), 206–211. 10.1016/S0014-5793(01)02743-0. [DOI] [PubMed] [Google Scholar]
- Pebay-Peyroula E.; Dahout-Gonzalez C.; Kahn R.; Trezeguet V.; Lauquin G. J.; Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426 (6962), 39–44. 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- Andrade S. L.; Dickmanns A.; Ficner R.; Einsle O. Crystal structure of the archaeal ammonium transporter Amt-1 from Archaeoglobus fulgidus. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (42), 14994–14999. 10.1073/pnas.0506254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrenga A.; Michel H. The cytochrome c oxidase from Paracoccus denitrificans does not change the metal center ligation upon reduction. J. Biol. Chem. 1999, 274 (47), 33296–33299. 10.1074/jbc.274.47.33296. [DOI] [PubMed] [Google Scholar]
- Fernandez C.; Hilty C.; Wider G.; Guntert P.; Wuthrich K. NMR structure of the integral membrane protein OmpX. J. Mol. Biol. 2004, 336 (5), 1211–1221. 10.1016/j.jmb.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Cierpicki T.; Liang B.; Tamm L. K.; Bushweller J. H. Increasing the accuracy of solution NMR structures of membrane proteins by application of residual dipolar couplings. High-resolution structure of outer membrane protein A. J. Am. Chem. Soc. 2006, 128 (21), 6947–6951. 10.1021/ja0608343. [DOI] [PMC free article] [PubMed] [Google Scholar]