

Abstract
Background
Long QT syndrome (LQTS) confers susceptibility to ventricular arrhythmia, predisposing to syncope, seizures and sudden death. While rare globally, LQTS is ~15 times more common in First Nations of Northern British Columbia largely due to a known mutation in KCNQ1. However, two large multigenerational families were affected, but negative for the known mutation.
Methods and Results
LQTS panel testing was carried out in the index case of each family, and clinical information was collected. Cascade genotyping was performed. Biochemical and myocyte-based assays were performed to evaluate the identified gene variant for loss-of-function activity. Index cases in these two families harbored a novel ANK2 c.1937C>T variant (p.S646F). An additional 16 carriers were identified, including two with structural heart disease: one with cardiomyopathy resulting in sudden death, and the other with congenital heart disease. For all carriers of this variant, the average QTc was 475 ms (±40). While ankyrin-B p.S646F is appropriately folded and expressed in bacteria, the mutant polypeptide displays reduced expression in cultured H9c2 cells and aberrant localization in primary cardiomyocytes. Further, myocytes expressing ankyrin-B p.S646F lack normal membrane targeting of the ankyrin-binding partner, the Na/Ca exchanger. Thus, ankyrin-B p.S646F is a loss-of-function variant.
Conclusions
We identify the first disease-causing ANK2 variant localized to the membrane-binding domain resulting in reduced ankyrin-B expression and abnormal localization. Further study is warranted on the potential association of this variant with structural heart disease given the role of ANK2 in targeting and stabilization of key structural and signaling molecules in cardiac cells.
Keywords: arrhythmia, long QT syndrome, genetic variation, Wolff-Parkinson-White syndrome, congenital heart disease, ANK2, First Nations, membrane binding domain
Introduction
A prolonged QT interval on electrocardiogram (ECG), corrected for heart rate (QTc), is a marker for increased risk for arrhythmia and sudden cardiac death1 and may be a result of genetic and/or non-genetic factors. Congenital long QT syndrome (LQTS types 1–13) is caused by variants in at least 13 known genes that encode for or affect the stability of critical ion channel proteins2. LQTS is characterized by a QTc >450 ms for men and >460 ms for women1,3,4 and can be associated with ventricular arrhythmia, syncope, and seizures. Notably, sudden death can be the first manifestation5. Certain LQTS-associated variants also contribute to broader phenotypes such as heart failure, cardiomyopathy, congenital structural heart disease and non-LQTS arrhythmia6. Although relatively rare (1:2000 people world-wide7), LQTS is at least 15 times more common (~1:125)8 in the Gitxsan First Nation, a remote community in Northern British Columbia (BC) whose ancestors have resided in the region for thousands of years8. The frequency in this community is consistent with one or more ‘founder effects’, whereby rare autosomal dominant conditions not affecting fecundity may become common as rare variants are passed from generation to generation. We have previously identified a pathogenic variant (KCNQ1 p.V205M) which could be traced back at least seven generations, explaining LQTS type 1 in the majority of cases of LQTS9,10 in that region; however, until recently we were unable to identify a potentially causal variant in a second group of kindreds with LQTS but lacking the KCNQ1 variant.
Here, we report the identification of a novel ANK2 variant (ANK2 c.1937C>T resulting in p.Ser646Phe) associated with LQTS and cardiac structural abnormalities in the Gitxsan population. This loss-of-function variant is the first identified human ‘ankyrin-B syndrome’ variant reported in the ankyrin membrane-binding domain (MBD).
Methods
As part of a larger study9, participants were invited to enroll if they had clinical features of LQTS, or were related to an individual with a diagnosis of LQTS. Community approval and individual consents were obtained, following University of British Columbia (H05-70330) and Northern Health Authority (RRC-2007-0038) ethics approval. Referrals for participation were through affected family members and healthcare providers. Health information was recorded upon enrollment through questionnaire and medical records review to confirm clinical diagnosis or suspicion of LQTS. Multigenerational family histories were obtained.
Index case identification
Two apparently unrelated persons from the community, each with a clinical electrophysiology diagnosis of LQTS, were negative for the KCNQ1 p.V205M variant, therefore 12-gene clinical LQTS sequencing panels were carried out revealing the ANK2 c.1937C>T (p.Ser646Phe) variant in each of the cases.
Expanded clinical studies
Twenty one additional close family members (1st and 2nd degree relatives) were tested for the ANK2 c.1937C>T (p.S646F) variant. The majority had been previously ascertained through the original LQTS study. All ECGs on file were reviewed by cardiac electrophysiologists blinded to mutation and clinical status. The QT interval was measured manually using the tangent method11. The reported QT interval was the longest identified within all 12 leads and corrected for heart rate using the Bazett formula. Echocardiograms were performed in carriers when possible.
Animals
All animal studies were performed in accordance with the American Physiological Society Guiding Principles for Research Involving Animals and Human Beings, and approved by the Ohio State University IACUC (2011A00000034-R2). The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the NIH. Mice utilized were post-natal day 1 ankyrin-B+/− mice12.
Neonatal myocyte experiments
Cardiomyocytes from ankyrin-B+/− neonatal mice were isolated and transfected as described13. Neonatal myocytes were transfected with WT GFP-ankyrin-B DNA, p.S646F GFP-ankyrin-B, and GFP 24 hours post-isolation. Constructs were generated from cDNA for canonical 220 kDa ankyrin-B (NP_066187.2) and variants introduced by standard site-directed mutagenesis. Constructs were fully sequenced to ensure no additional variants were present. Cells were allowed to recover from transfection for 48 hours in complete media. Cells were fixed for 15 minutes in 2% PFA and immunolabeled utilizing primary antibodies specific for Na/Ca exchanger (Swant; 1:50) and GFP (1:100)14. Myocytes were co-labeled to mark the nuclei (DAPI, 1.5 µg/mL). Secondary antibodies included Alexa conjugated donkey anti-mouse 568 and donkey anti-rabbit 488 (Invitrogen). Neonatal cardiomyocytes were mounted on Mat-Tek plates and imaged on an LSM780 confocal microscope.
Protein expression and purification
The coding sequences of ankyrin-B MBD (residues 28–873) were PCR amplified using the full-length 220 kDa ankyrin-B as the template. The point mutation was created using the Quick Change site-directed mutagenesis kit and confirmed by DNA sequencing. These coding sequences were cloned into a home-modified pET32a vector for protein expression. The N-terminal thioredoxin-His6-tagged proteins were expressed in Escherichia coli BL21 (DE3) and purified as previously described15. The thioredoxin-His6-tag was removed by incubation with HRV 3C protease and separated by size exclusion columns.
Fast Protein Liquid Chromatography coupled with static light scattering
Protein samples (100 µl at a concentration of 50 µM, pre-equilibrated with corresponding column buffer) were injected into an AKTA Fast Protein Liquid Chromatography (FPLC) system with a Superose 12 10/300 GL column (GE Healthcare) with the column buffer of 50 mM Tris-HCl, 100 mM NaCl, and 1 mM DTT, pH 7.8. The chromatography system was coupled to a static light-scattering detector (MiniDawn, Wyatt) and differential refractive index detector (Optilab, Wyatt). Data were analyzed with ASTRA 6 (Wyatt).
Circular dichroism
Circular Dichroism (CD) spectra of WT ankyrin-B MBD and the p.S646F variant were measured on a JASCO J-815 CD spectropolarimeter at room temperature using a cell path length of 1 mm. Each spectrum was collected with three scans spanning a spectral window of 200–260 nm. The samples were dissolved in 25 mM Tris buffer containing 50mM NaCl, 0.5 mM EDTA and 0.5 mM DTT at pH 7.8 with the increasing concentrations of urea in the same buffer. The protein concentration used in the CD experiment was 10 µM.
Structural modeling
Analysis of the location of p.S646 residue and proposed binding groove of the ANK repeat 17–21 was performed using the high resolution crystal structure of ankyrin-B MBD16. The structural figures were prepared using PyMOL (www.pymol.org).
H9c2 cell studies
H9c2 rat ventricular-derived cardiomyoblasts were purchased from American Type Culture Collection (ATCC CRL-1446). Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 µg/mL streptomycin (37°C, 5% CO2) and passaged at 70–80% confluence. Transfections were performed using jetPEI (Polyplus/VWR) according to the manufacturer’s protocol. The 220 kDa ankyrin-B isoform 2 (NP_066187.2) was commercially synthesized (Bio Basic Inc., Markham, ON) and subcloned into pACGFP1-n1. Ankyrin-B p.S646F-pACGFP1-n1 was constructed using a QuickChange II XL Site-Directed Mutagenesis Kit (Aligent Technologies, Mississauga, ON) following the manufacturer’s protocol. All primers were purchased from Integrated DNA Technologies (Coralville, IA). All plasmids were verified by sequencing prior to use. Cell lysis was performed as previously described17–19. Lysates were heated for 5 minutes (95°C) under reducing conditions, separated by SDS-PAGE and transferred to 0.2 µm pore-size polyvinyldene fluoride (PVDF) membrane for 3 hours at 200mA. Primary antibodies used were anti-GFP polyclonal (1:2000; Life Technologies) and anti-β-actin monoclonal (1:1000; Sigma-Aldrich). Secondary antibody used was horseradish peroxidase (HRP)-conjugated AffiniPure donkey anti-rabbit IgG (1:2000; Jackson ImmunoResearch) and HRP-conjugated AffiniPure donkey anti-mouse IgG (1:2000; Jackson ImmunoResearch). Proteins were detected using Clarity enhanced chemiluminescence reagent (BioRad) and were quantified using Image J. Statistical analyses were performed using Prism for Mac OS X v5.0d software (http://graphpad.com; GraphPad Software). All variances were reported as standard error of the mean.
Results
Identification of novel LQTS variant in the Gitxsan First Nations Community
We identified a novel c.1937 C>T variant in exon 18 (p.S646F) in the ANK2 gene in two Gitxsan multigenerational families with LQTS featured in Figure 1A and B. We first identified ankyrin-B p.S646F in a 28-year-old man with a clinical diagnosis of LQTS who experienced recurrent syncope despite treatment with beta-blockers necessitating insertion of an implantable cardioverter-defibrillator. The KCNQ1 p.V205M mutation was absent and no other potential disease causing mutations were detected on a standard 12 gene LQTS sequencing panel20. Five of his relatives, including two with a previous diagnosis of LQTS, were subsequently confirmed to harbor the ankyrin-B p.S646F variant (Figure 1A and Table 1). The ankyrin-B p.S646F variant was also identified in a second ‘unrelated’ family with LQTS (Figure 1B). The proband for this family had a clinical diagnosis of LQTS provided by her electrophysiologist, prompting the genetic testing. LQTS segregated with the variant in an additional five members of the family, including one who died suddenly with a diagnosis of dilated cardiomyopathy. An additional variant-positive family member had a borderline QTc of 468 ms, but has been noted on repeated ECGs to have Wolf- Parkinson-White syndrome. Her infant daughter inherited the variant and notably was born with a congenital structural heart defect (total anomalous pulmonary venous return-TAPVR). To date, within these two families, a total of thirteen adults and five children have been identified with ankyrin-B p.S646F (Table 1). The LQTS phenotype (Figure 1) segregates with the variant (average QTc in adults =475 ms ± 40; range 430–604 ms), and non-carriers do not have the LQTS phenotype. Of note, there is only minimal evidence of a prolonged QTc interval in the age group under 25 years. Of nine individuals reporting a history of seizures, eight were carriers of the p.S646F variant (44% of carriers). In summary, LQTS, dilated cardiomyopathy with associated sudden death, congenital heart malformation TAPVR, Wolf-Parkinson White (WPW) syndrome, and seizures have been identified in the ankyrin-B p.S646F carriers (Figure 1; Table 1). See also four representative ECGs from carriers shown in Figure 2.
Figure 1.
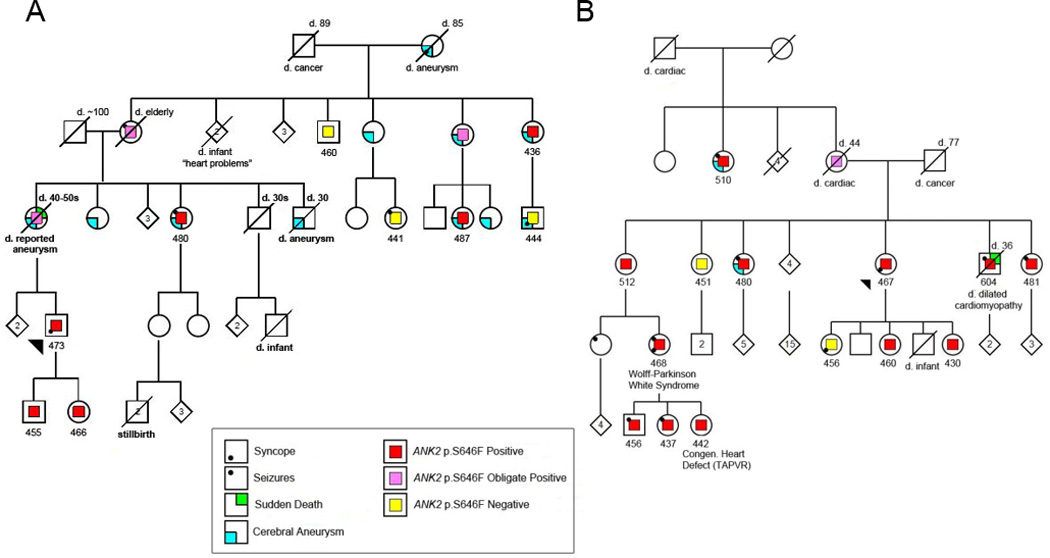
Pedigrees of two Gitxsan families with the p.S646F variant. A) Family 1; B) Family 2. Legend denotes variant status and clinical features. Number below each individual represents manual QTc (ms) read blinded to mutation and clinical status.
Table 1.
Summary of clinical features for two Gitxsan families with the ankyrin-B p.S646F variant
| Family | Indv # | Sex | p.S646F variant status |
Age (yrs) | Highest QTc (ms)† |
ECG findings | Echocardiogram | Clinical Features/Diagnoses | Seizures | Cerebral Aneurysms (age) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | M | pos | 34 | 473 | U-wave, Tachycardia, | Mild mitral and tricuspid insufficiency |
Recurrent pre-syncope, 2X syncope with exercise while on β blockers (prior to ICD implant) |
- | - |
| 2* | F | pos | 2 | 466 | NA | - | - | |||
| 3* | M | pos | 12 | 455 | Notching in V2 & V3 | NA | Pre-syncope with exercise | - | - | |
| 4 | F | pos | 54 | 480 | NA | + | ++(30–40 y) | |||
| 5 | F | pos | 64 | 436 | Mild diastolic dysfunction | Recurrent pre-syncope | - | ++(60 y) | ||
| 6 | F | pos | 40 | 487 | Trace mitral and tricuspid regurgitation |
- | +(39 y) | |||
| N1 | M | neg | 44 | 444 | NA | Syncope prior to aneurysm diagnosis |
- | +(33 y) | ||
| N2 | F | neg | 51 | 441 | NA | + | - | |||
| N3 | M | neg | 67 | 460 | NA | - | - | |||
| 2 | 7 | F | pos | 43 | 467 | Mild U-wave, incomplete Right Bundle Branch Block |
Normal | Implantable loop recorder revealed SVT, recurrent syncope |
- | - |
| 8 | F | pos | 35 | 481 | NA | Recurrent pre-syncope | + | - | ||
| 9 | F | pos | 48 | 480 | Borderline broad-based T-waves in V2 & V3 |
NA | ++ | +(40 y) | ||
| 10 | M | pos | d.36 | 604 | ?Pre-excitation, ?nodal accessory pathway |
Severe dilated left ventricle with ejection fraction 20–25%, diastolic dysfunction, valve regurgitation, pulmonary hypertension, inferior vena cava dilation |
Severe idiopathic dilated cardiomyopathy on autopsy (sudden cardiac death at 36 y) |
++ | - | |
| 11 | F | pos | 51 | 512 | Early precordial transition (R>S in V1), U pattern in V2 |
Mild mitral, tricuspid, and aortic regurgitation. Ascending aorta upper limits of normal (36 mm) |
- | - | ||
| 12 | F | pos | 23 | 460 | Mild mitral and tricuspid regurgitation |
- | - | |||
| 13 | F | pos | 24 | 430 | U-wave, sinus bradycardia |
NA | - | - | ||
| 14 | F | pos | 69 | 510 | Right bundle branch block, notching in V2 |
NA | ++ | + (47 y) | ||
| 15 | F | pos | 23 | 468 | Pre-excitation, AVRT | Trivial mitral and tricuspid regurgitation |
Wolff-Parkinson-White syndrome, recurrent syncope |
+ | - | |
| 16* | M | pos | 8 | 456 | ?Borderline U-wave, notching in V2 |
Normal | + | - | ||
| 17* | F | pos | 6 | 437 | Abnormal T-waves in V1 & V2, T-wave inversion in V1, notching in V2 |
Normal | + | - | ||
| 18* | F | pos | 1 | 442 | Congenital heart defect: Total Anomalous Pulmonary Venous Return (TAPVR) |
TAPVR, surgical repair at 2 m of age |
- | - | ||
| N4 | F | neg | 49 | 451 | NA | - | - | |||
| N5* | F | neg | 17 | 456 | Notching in V2 | NA | Reflex-mediated syncope | - | - |
N1–N5 are negative for the variant, all others are positive,
age<18 years,
manually read, blinded to clinical and mutation status,
++ indicates multiple, NA = Not available.
Figure 2.
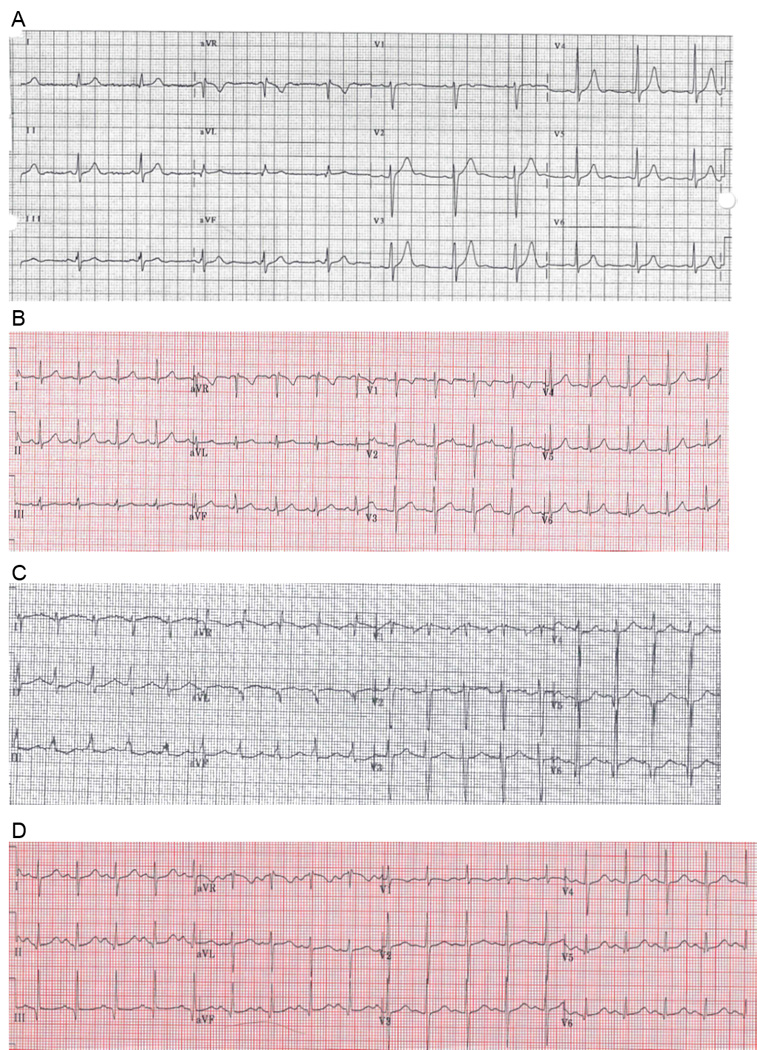
Representative ECGs from four carriers of the p.S646F variant. A) Family 1, Indv 1: note U-waves; B) Family 1, Indv 3: note abnormal notching in V2; C) Family 2, Indv 10: note prolonged QTc; D) Family 2, Indv 11: note prolonged QTc
The ankyrin-B p.S646 residue is highly conserved across evolution, and this variant results in a non-conservative amino acid substitution of a polar amino acid (serine) for a non-polar amino acid (phenylalanine) in the large amino-terminal ankyrin-B membrane-binding domain (Figure 3A–B). In silico analyses using MutationTaster21, Polyphen22, and PROVEAN23 predict the p.S646F variant as disease causing/possibly damaging/deleterious. This variant was absent in more than 6,500 control population samples in the NHLBI Exome Sequencing Project. Further, to date, this variant in ANK2 is not reported in over 120,000 alleles (MAF: Minor Allele Frequency=0.0000 from ExAC: Exome Aggregation Consortium24; NHLBI: National Heart, Lung, and Blood Institute25; 1000 Genomes26) (Table 2).
Figure 3.
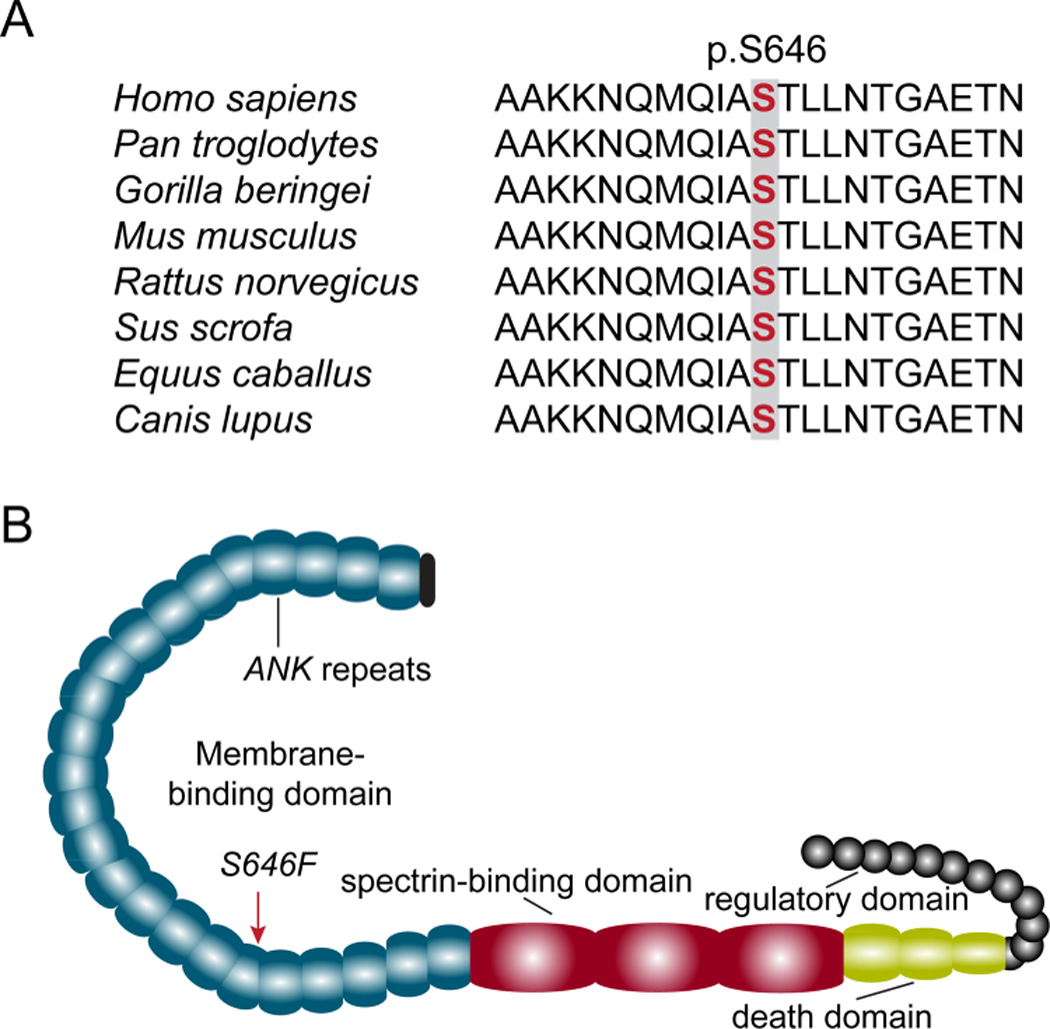
Ankyrin-B p.S646 is located within membrane-binding domain and highly conserved across species. A) Conservation of p.S646 across vertebrates. B) Ankyrin-B domain organization and location of p.S646F variant on membrane-binding domain.
Table 2.
Minor allele frequency data for ANK2 c.1937 C>T
| Database* | Variant Allele Count | Total Allele Count |
|---|---|---|
| ExAC | 0 | 117,264 |
| NHLBI | 0 | 13,004 |
| 1000 Genomes | 0 | 5,008 |
Ankyrin-B p.S646F membrane-binding domain is appropriately expressed and folded
To test the impact of the p.S646F variant on the ankyrin-B polypeptide, we performed biochemical analysis of purified ankyrin-B MBD (residues 28–873) harboring the p.S646F variant in parallel with wild-type ankyrin-B MBD. Following expression of the recombinant proteins in bacteria, polypeptides were purified and subjected to FPLC analytical gel filtration coupled with static light-scattering to detect column behavior and molecular weight, CD to evaluate secondary structure, and CD-based denaturation to analyze protein stability (Figure 4). We observed no significant difference in protein expression between ankyrin-B MBD and ankyrin-B MBD p.S646F (Figure 4A). Specifically, protein quantities and column behavior were nearly identical for the two polypeptides, with appropriate molecular weight mobility (Figure 4B–C). We did however observe that the ankyrin-B p.S646F polypeptide was unable to be concentrated to the same extent as wild-type ankyrin-B MBD, causing protein precipitation at high concentrations. CD spectra showed that both wild-type ankyrin-B MBD and ankyrin-B p.S646F MBD are enriched in helical structures with no significant structural differences (Figure 4D). Finally, using CD-based urea denaturation experiments we observed no differences in protein stability between wild-type ankyrin-B MBD and ankyrin-B p.S646F MBD (Figure 4E). It is worth noting that these biochemical data are in line with prior structural analysis of the ankyrin-B MBD. Ankyrin-B p.S646F resides on the αB of the 19th ANK repeat on the outer surface of the ANK repeat solenoid (Figure 4F–G). While increasing local hydrophobicity, this site is not predicted to directly interfere with binding sites for ankyrin-B membrane protein targets (sites located on inner groove of ANK repeats; Figure 4F–G). In summary, p.S646F does not significantly alter ankyrin-B MBD expression or structural properties.
Figure 4.
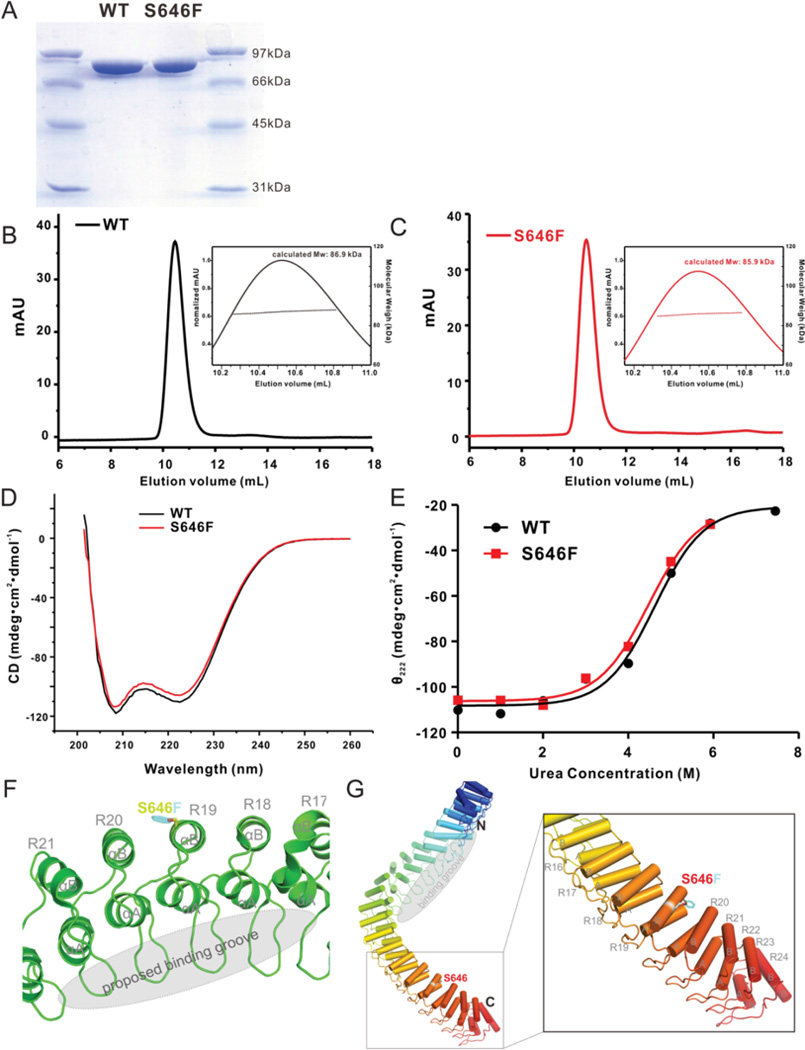
Ankyrin-B MBD p.S646F protein is appropriately expressed and folded in vitro. A) SDS-PAGE shows the purified protein qualities of WT ankyrin-B MBD and p.S646F mutant. The molecular markers are shown on both sides. B-C) FPLC analytical gel filtration coupled with static light scattering profiles of WT and p.S646F show symmetric column behavior and nearly identical molecular weight. D) Circular Dichroism (CD) spectra of WT ankyrin-B MBD and p.S646F show the enrichment of helical structures. E) Urea denaturation-based assay of the stabilities of the WT ankyrin-B MBD and p.S646F mutant shows no obvious difference. F-G) Structural model shows that the p.S646 residue locates on the of the 19th ANK repeat on the outer surface of the ANK repeat solenoid (Protein Data bank ID code: 4 RLV).
Ankyrin-B p.S646F alters polypeptide stability in a model cardiomyocyte cell line
Ankyrin-B p.S646F does not alter expression or folding of the bacterially-expressed purified MBD (Figure 4); however, to interrogate the stability of the full-length ankyrin-B p.S646F in living cells, we transfected H9c2 rat ventricular cardiomyoblasts, a commonly used model cardiomyocyte cell line, with GFP-tagged wild-type ankyrin-B and ankyrin-B harboring the p.S646F variant. While cells transfected at similar efficiencies, subsequent immunoblotting revealed that two days post-transfection, steady-state ankyrin-B p.S646F levels were significantly lower than wild-type ankyrin-B (Figure 5). Together, these data support that while the purified MBD harboring the p.S646F variant displayed normal expression, folding and stability, the full length ankyrin-B p.S646F variant was unstable when expressed in model cardiomyocytes.
Figure 5.
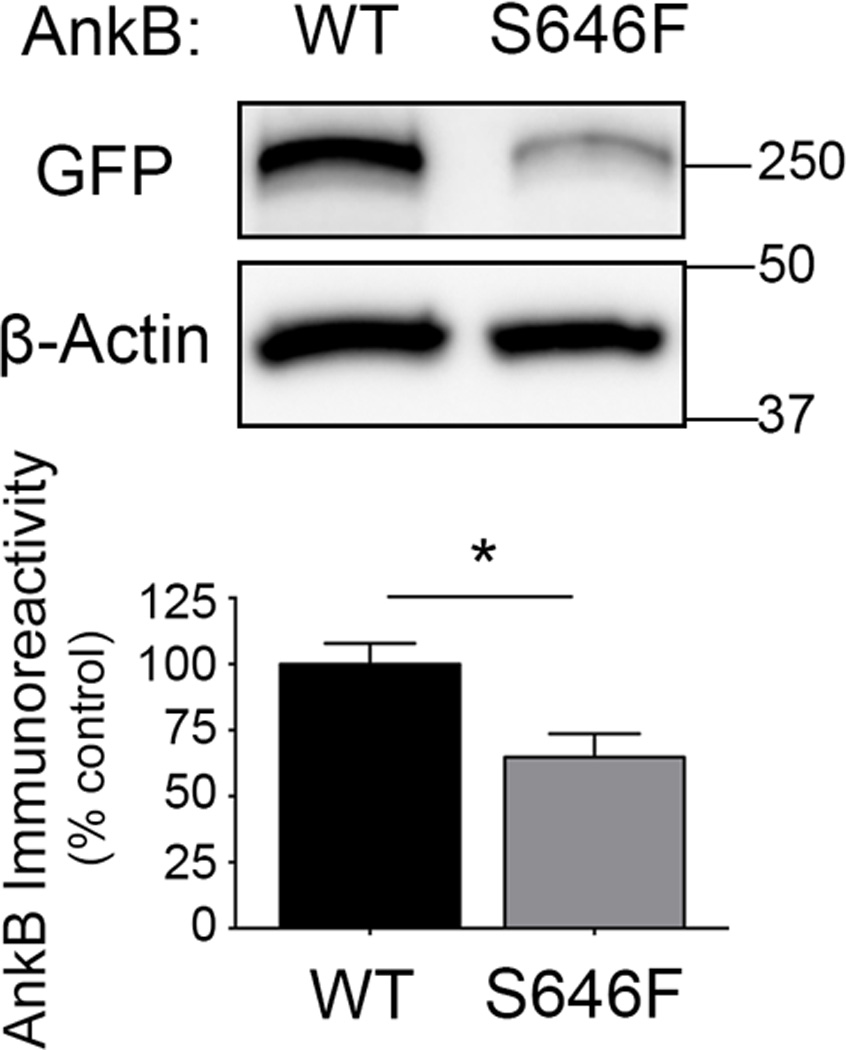
Ankyrin-B p.S646F exhibits decreased post-translational stability in H9c2 cardiomyoblasts. A) Immunoblot of GFP-tagged ankyrin-B (AnkB; wild-type or p.S646F)-expressing H9c2 cell lysates 48 hours post-transfection probed with anti-GFP (upper panel) and anti-β-actin (lower panel). B) Quantification of GFP immunoreactivity revealed that ankyrin-B p.S646F expression is significantly reduced in comparison to the wild-type protein (n=3; *, p = 0.0408 by unpaired t-test). These data are representative of 3 separate independent experiments, each with n ≥ 3 independent biological replicates.
Ankyrin-B p.S646F alters ankyrin-B and Na/Ca exchanger targeting in primary cardiomyocytes
We tested the expression and function of the ankyrin-B p.S646F variant in primary cardiomyocytes. Specifically, we introduced GFP, GFP-ankyrin-B, and GFP-ankyrin-B p.S646F into myocytes derived from mice heterozygous for a null mutation in ankyrin-B (ankyrin-B+/−) to mimic the autosomal dominant background in human carriers. Unlike wild-type GFP-ankyrin-B that localized to the plasma membrane and rescued localization of the Na/Ca exchanger (NCX), ankyrin-B p.S646F was unable to rescue abnormal targeting of NCX (Figure 6). In fact, compared with membrane-associated GFP-ankyrin-B, GFP-ankyrin-B p.S646F was concentrated intracellularly in large cytoplasmic puncta resembling the endosome/lysosome network (Figure 6B). In summary, our data support that ankyrin-B p.S646F displays loss-of-function activity in myocytes resulting in reduced expression and abnormal localization, ultimately affecting both expression and localization of downstream binding partners including NCX.
Figure 6.

Ankyrin-B p.S646F is a loss-of-function variant in myocytes. A) GFP-ankyrin-B localizes to the membrane (arrows) of ankyrin-B+/− neonatal cardiomyocytes. We observe similar localization of Na/Ca exchanger at the membrane of immature primary neonatal cardiomyocytes. Note: GFP localized primarily to nucleus in control experiments as expected (inset). B) GFP-ankyrin-B p.S646F is enriched in the perinuclear region of ankyrin-B+/− neonatal cardiomyocytes. We observed similar localization of the Na/Ca exchanger in these myocytes (arrows). Scale bar equals 20 microns.
Discussion
Long QT syndrome type 4, caused by mutations in ANK2, accounts for less than 1% of all cases of LQTS2. The clinical presentation is well known for its variable phenotype and association with non-LQTS cardiac phenotypes27, necessitating the broader term named for the protein responsible, ‘the ankyrin-B syndrome’. Consistent with previously published reports12,28–30, the QTc range in p.S646F variant carriers was broad (430–604 ms) and only eight of eighteen carriers had a QTc greater than 470 ms. Of note, and also in keeping with previous reports, there was minimal evidence of a prolonged QTc interval in the age group under 25 years. Other cardiac phenotypes, however, were present (see Table 1).
Ankyrin-B dysfunction is associated with acquired cardiovascular disease in humans and cardiovascular disease modeled in animals30–33. Loss-of-function ankyrin-B variants underlie a complex cardiovascular human phenotype. Previously reported cases have displayed ventricular arrhythmias (with or without history of prolonged QTc) as well as sinus node dysfunction and atrial fibrillation12,27,28,30,34–37 depending on the location and severity of the specific variant. Ankyrin-B is comprised of four domains: the MBD required for association with key membrane protein targets and auto-inhibition38,39, a spectrin-binding domain, a death domain and a C-terminal regulatory domain. While human arrhythmia disease variants have previously been localized to both the spectrin-binding domain and C-terminal regulatory domain, ankyrin-B p.S646F is the first identified loss-of-function variant located within the MBD. Based on our findings along with our previous work, we propose that the “ankyrin-B syndrome” phenotype is broader than previously reported, and our study adds to that spectrum. We propose the phenotype (LQTS4, WPW syndrome/tachycardia, congenital heart malformation, and cardiomyopathy with associated sudden death) observed in individuals expressing ankyrin-B p.S646F is caused at the cellular level by impaired Ca2+ clearance (Figure 7), with consequences for the regulation of cardiomyocyte excitability similar to previous ankyrin-B mutations (reviewed in40). Ankyrin-B is responsible for the proper anchoring of the tripartite complex (NCX, Na/K ATPase, inositol 1,4,5-trisphosphate receptor) (Figure 7). The NCX normally extrudes Ca2+, in exchange for three Na+ ions inward, using the Na+ gradient from the Na/K ATPase. Based on our current study together with previous reports on ankyrin-B variants, it is reasonable to postulate that the unchecked accumulation of cytosolic Ca2+ due to loss of NCX localization produces Ca2+ overload in the sarcoplasmic reticulum resulting in uncontrolled leakage and afterdepolarizations, and a prolonged QT interval (Figure 7). Similarly, the occurrence of WPW syndrome (essentially an extra electrical pathway between the atria and ventricles that causes tachycardia) in the ankyrin-B p.S646F population could also be linked to the impaired localization of NCX, with recent work in genetic models demonstrating that NCX plays a critical role in cardiac pacemaking41,42. Furthermore, [Ca2+]i, is a critical determinant of cardiomyocyte development43 regulating the transcription of a number of pivotal developmental genes.
Figure 7.
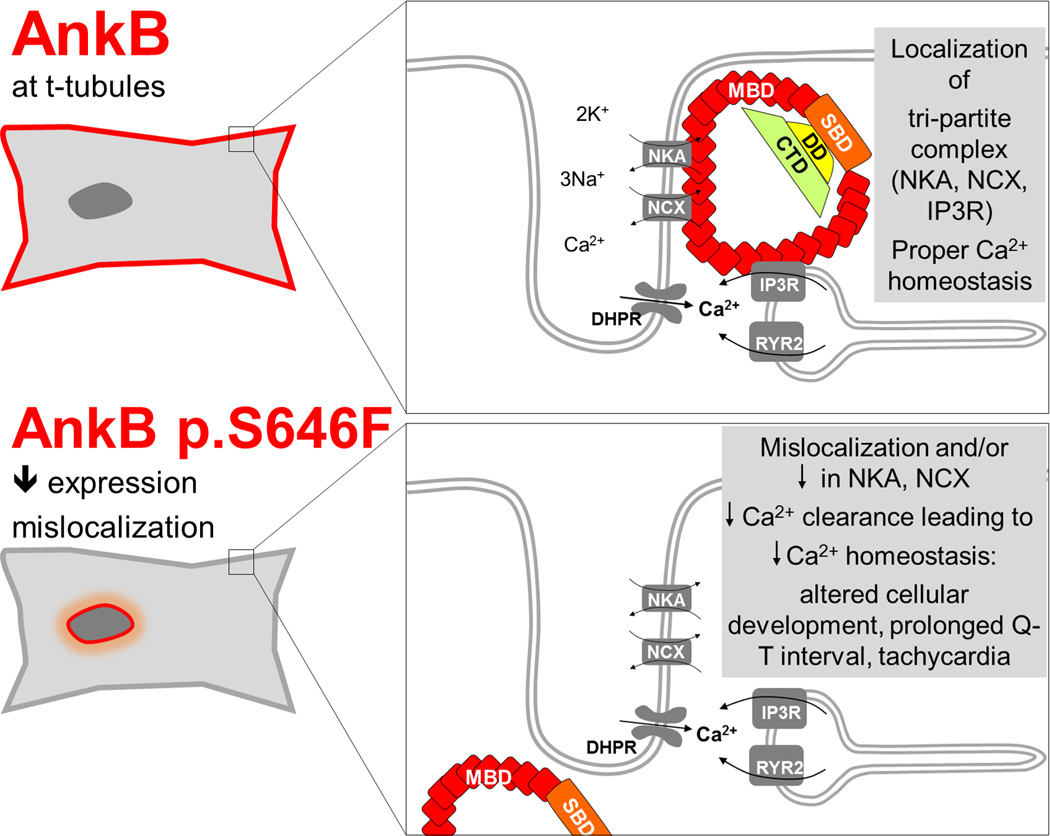
Model for novel disease-causing variant identified in the membrane-binding domain of the Ankyrin B protein (ANK2 c. 1937C>T p.Ser646Phe). NCX indicates Na/Ca exchanger; NKA, Na/K ATPase; IP3R, inositol 1,4,5-trisphosphate receptor; DHPR, dihydropyridine receptor.
In addition to the variable phenotype observed, another notable characteristic of the affected individuals is that the clinical presentation of LQTS4 may be proportional to age27. We suspect this could be due to age-related decreased expression of molecular chaperones (reviewed in44), which normally act to buffer the cardiomyocyte from stress associated with genetic mutations (reviewed in45). For example, with a high level of molecular chaperone co-expression, the catalytic activity of enzymes harboring mutations is preserved to a significantly greater extent46. Therefore, it is reasonable to speculate that the relatively small difference in protein expression and partial mislocalization of AnkB p.S646F with concomitant effects on NCX observed in the in vitro system (Figure 6) over a relatively short duration (days), would likely be exacerbated with age in the context of the living organism, consistent with what is observed in our patient population. Furthermore, heterogeneity in these cellular systems that buffer against genetic mutations and associated cellular dysfunction likely also impacts on the phenotypic variability that manifests with the mutation. Future in vitro work will examine the interplay between AnkB p.S646F and cellular chaperone systems to further explore these ideas and potentially to develop novel therapeutic strategies targeting molecular chaperones.
As noted above, ankyrin-B p.S646F is localized to the MBD, a large protein domain comprised of 24 consecutive ANK repeats. Computational algorithms predict the effect of this non-conservative substitution of a polar residue (serine) for a non-polar amino acid (phenylalanine) is deleterious. Due to the critical role of the ankyrin-B MBD, it was generally viewed that significant variation in this domain would be incompatible with postnatal life. Because the localization of this variant to the 19th ANK repeat is not predicted to directly interfere with the binding site for known ankyrin-B interacting proteins (Figure 3) and the overall structure of the MBD was indistinguishable from wildtype, the variant likely modulates tertiary structure in such a way that determinants of its own localization and stability are disrupted. In fact, we have learned that the ankyrin MBD plays a critical role in regulating binding to interacting proteins through a complex “auto-regulatory” mechanism that involves MBD association with the spectrin-binding domain38. Further, mutations within this region, highly conserved across evolution, may interfere with a critical trafficking molecule required for the localization of ankyrin-B to the myocyte membrane following biosynthesis. The aberrant immunolocalization of GFP-ankyrin-B p.S646F to intracellular clusters supports this hypothesis (Figure 6). Finally, it is worth noting that beyond the fact that this variant was not present in 6,500 control samples at the time of sequencing, this variant has not been identified in any global cohorts regardless of ancestry (minor allele frequency of 0.000 in over 120,000 alleles) (Table 2). Consistent with the category of ‘likely pathogenic’, predicting at least a 90% chance of pathogenicity as per the American College of Medical Genetics and Genomics Standards and Guidelines for Interpretation of Sequence variants47, our studies support that this variant contributes to inherited heart disease. It will be important in future experiments to further define the role of this variant, and this specific ANK repeat in ankyrin-B biosynthesis and regulation in myocytes, both in vitro and in vivo.
An unexpected and intriguing finding here was the presence of seven p.S646F carriers (five established with genetic testing and two obligate carriers), between the two families confirmed to be affected with cerebral aneurysm. One family in particular (Figure 1A) has multiple additional relatives with cerebral aneurysms who have not yet undergone p.S646F testing. Familial cerebral aneurysm is a rare event with only 0.2% of cases reporting more than 1 affected first degree relative48. It is premature, however, to postulate any role of the variant on the cerebral vasculature especially given a paucity of evidence for ankyrin-B expression in endothelial cells49,50. Furthermore, of note, one individual without the p.S646F variant developed a cerebral aneurysm (Figure 1A). Of possible relevance, given the high rate of seizures in p.S646F carriers with and without cerebral aneurysms (Table 1), ankyrin-B has previously been shown to play a critical role in neurite outgrowth, and perhaps may contribute to the genesis of abnormal brain activity, such as that leading to seizures. This hypothesis remains to be tested in the laboratory.
Given its critical roles in targeting and stabilization of key structural and signaling molecules in cardiac cells as well as other organ systems13,51–53, it is not surprising that phenotypes beyond ventricular arrhythmia were observed in the two p.S646F kindreds. The presence of other arrhythmias, as well as structural heart disease and possibly cerebral involvement, underscores the importance of future studies investigating the broad impact of the p.S646F variant as a model for other variants localized to the membrane-binding domain.
Limitations
This study featured a sub population of a cohort being investigated for a high rate of Long QT syndrome in Northern British Columbia leading to ascertainment of these families. Most clinical data collection was by medical record review. Because of the nature of the original study, the delineation of the possible neurological phenotype deserves further and detailed study. Despite these limitations, this study represents an important advance in our understanding of the role of ankyrin-B in cardiac health and disease and lays the groundwork for additional studies.
Clinical Perspective.
Human ANK2 loss-of-function variants are linked with type 4 long QT syndrome. ANK2 encodes ankyrin-B, a critical multi-functional cardiac regulatory “scaffold” protein with key roles in ion channel and transporter membrane targeting and modulation. It is not surprising, therefore, that ANK2 variants contribute to cardiac arrhythmia phenotypes beyond LQTS (sinus node dysfunction, atrial fibrillation), necessitating the broader term, ‘ankyrin-B syndrome’. Here, we report the identification of a novel ANK2 variant (ANK2 c.1937C>T resulting in p.Ser646Phe), and describe the clinical characteristics in 18 carriers, introducing the notion that ANK2 dysfunction causes both cardiac electrical and structural phenotypes. The variant we describe, present in a remote Indigenous community, is uniquely placed in the membrane binding domain of ankyrin-B, supporting that altered protein expression and function, along with the specific location of the variant may influence the clinical presentation. We show that the p.S646F variant alters both ankyrin-B expression as well as activity in in vitro cell lines and primary cardiomyocytes. We propose that the described phenotype in our cohort, (LQTS4, WPW syndrome/tachycardia, congenital heart malformation, and cardiomyopathy) arises at the cellular level resulting from dysregulation of Ca2+ clearance affecting cardiomyocyte excitability and further affects the localization of ankyrin-B to the myocyte membrane, potentially leading to congenital and later life structural heart disease. In summary, our findings represent a critical advance in our understanding of the complexities of heart disease within a defined population, and further underscore the importance of ankyrin-B in cardiac health.
Acknowledgments
We acknowledge our on-going partnership with the Gitxsan Health Society, their role in this research, and extend our gratitude to the participants. An earlier version of this manuscript was reviewed by the Gitxsan Health Society Board.
Sources of Funding: L.A.S. is supported by a Michael Smith Foundation for Health Research & BC Schizophrenia Society Foundation Scholar award. L.C. is supported by a Natural Sciences and Engineering Research Council of Canada CGS-M and University of Victoria graduate scholarships. The clinical cohort work was funded through the Canadian Institutes of Health Research (CIHR), Ottawa, ON, Research grant no. 81197 (to L.A.). Funding for the molecular, cellular and mouse studies was provided by NIH Grants HL084583, HL083422, and HL114383 (to P.J.M.); the American Heart Association (to P.J.M.); the JB Project and the William D. and Jacquelyn L. Wells Fund for Cardiovascular Research at the Ohio State University (to P.J.M); University of Victoria seed funds (to L.A.S.), CIHR bridge funding (to L.A. and L.A.S).
Footnotes
Disclosures: None
References
- 1.Roden DM. Keep the QT interval: it is a reliable predictor of ventricular arrhythmias. Heart Rhythm. 2008;5:1213–1215. doi: 10.1016/j.hrthm.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giudicessi JR, Ackerman MJ. Genotype- and phenotype-guided management of congenital long QT syndrome. Curr Probl Cardiol. 2013;38:417–455. doi: 10.1016/j.cpcardiol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldenberg I, Horr S, Moss AJ, Lopes CM, Barsheshet A, McNitt S, et al. Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J Am Coll Cardiol. 2011;57:51–59. doi: 10.1016/j.jacc.2010.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88:782–784. doi: 10.1161/01.cir.88.2.782. [DOI] [PubMed] [Google Scholar]
- 5.Goldenberg I, Moss AJ. Long QT Syndrome. J Am Coll Cardiol. 2008;51:2291–2300. doi: 10.1016/j.jacc.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 6.Alders M, Christiaans I. Long QT Syndrome [Internet] In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 2003. [Accessed May 2016]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1129/ [Google Scholar]
- 7.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–1767. doi: 10.1161/CIRCULATIONAHA.109.863209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jackson H, Huisman L-A, Sanatani S, Arbour LT. Long QT syndrome. CMAJ. 2011;183:1272–1275. doi: 10.1503/cmaj.100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arbour L, Rezazadeh S, Eldstrom J, Weget-Simms G, Rupps R, Dyer Z, et al. A KCNQ1 V205M missense mutation causes a high rate of long QT syndrome in a First Nations community of northern British Columbia: a community-based approach to understanding the impact. Genet Med. 2008;10:545–550. doi: 10.1097/gim.0b013e31817c6b19. [DOI] [PubMed] [Google Scholar]
- 10.Jackson HA, McIntosh S, Whittome B, Asuri S, Casey B, Kerr C, et al. LQTS in Northern BC: homozygosity for KCNQ1 V205M presents with a more severe cardiac phenotype but with minimal impact on auditory function. Clin Genet. 2014;86:85–90. doi: 10.1111/cge.12235. [DOI] [PubMed] [Google Scholar]
- 11.Postema PG, Wilde AAM. Do patients with long QT syndrome remain at risk for sudden cardiac death after 40 years of age? Nat Clin Pract Cardiovasc Med. 2008;5:602–603. doi: 10.1038/ncpcardio1305. [DOI] [PubMed] [Google Scholar]
- 12.Mohler PJ, Schott J-J, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 13.Cunha SR, Mohler PJ. Obscurin targets ankyrin-B and protein phosphatase 2A to the cardiac M-line. J Biol Chem. 2008;283:31968–31980. doi: 10.1074/jbc.M806050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohler PJ, Davis JQ, Davis LH, Hoffman JA, Michaely P, Bennett V. Inositol 1,4,5-trisphosphate receptor localization and stability in neonatal cardiomyocytes requires interaction with ankyrin-B. J Biol Chem. 2004;279:12980–12987. doi: 10.1074/jbc.M313979200. [DOI] [PubMed] [Google Scholar]
- 15.Wang C, Yu C, Ye F, Wei Z, Zhang M. Structure of the ZU5-ZU5-UPA-DD tandem of ankyrin-B reveals interaction surfaces necessary for ankyrin function. Proc Natl Acad Sci U S A. 2012;109:4822–4827. doi: 10.1073/pnas.1200613109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang C, Wei Z, Chen K, Ye F, Yu C, Bennett V, et al. Structural basis of diverse membrane target recognitions by ankyrins. eLife. 2014;3:e04353. doi: 10.7554/eLife.04353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wicki-Stordeur LE, Boyce AKJ, Swayne LA. Analysis of a pannexin 2-pannexin 1 chimeric protein supports divergent roles for pannexin C-termini in cellular localization. Cell Commun Adhes. 2013;20:73–79. doi: 10.3109/15419061.2013.791681. [DOI] [PubMed] [Google Scholar]
- 18.Wicki-Stordeur LE, Dzugalo AD, Swansburg RM, Suits JM, Swayne LA. Pannexin 1 regulates postnatal neural stem and progenitor cell proliferation. Neural Develop. 2012;7:11. doi: 10.1186/1749-8104-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wicki-Stordeur LE, Swayne LA. Panx1 regulates neural stem and progenitor cell behaviours associated with cytoskeletal dynamics and interacts with multiple cytoskeletal elements. Cell Commun Signal CCS. 2013;11:62. doi: 10.1186/1478-811X-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.GeneDx. Cardiology Genetics: Long QT Syndrome (LQTS) Panel [Internet] [Accessed May 2016]; Available from: http://www.genedx.com/test-catalog/available-tests/lqts-gene-sequencing-deldup-panel/ [Google Scholar]
- 21.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 22.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinforma Oxf Engl. 2015;31:2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.ExAC Browser [Internet] [Accessed May 2016]; Available from: http://exac.broadinstitute.org/ [Google Scholar]
- 25.Exome Variant Server [Internet] [Accessed May 2016]; Available from: http://evs.gs.washington.edu/EVS/ [Google Scholar]
- 26.1000 Genomes | A Deep Catalog of Human Genetic Variation [Internet] [Accessed May 2016]; Available from: http://www.internationalgenome.org/ [Google Scholar]
- 27.Schott JJ, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour JB, et al. Mapping of a gene for long QT syndrome to chromosome 4q25-27. Am J Hum Genet. 1995;57:1114–1122. [PMC free article] [PubMed] [Google Scholar]
- 28.Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K, et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U S A. 2004;101:9137–9142. doi: 10.1073/pnas.0402546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohler PJ, Le Scouarnec S, Denjoy I, Lowe JS, Guicheney P, Caron L, et al. Defining the cellular phenotype of “ankyrin-B syndrome” variants: human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation. 2007;115:432–441. doi: 10.1161/CIRCULATIONAHA.106.656512. [DOI] [PubMed] [Google Scholar]
- 30.Cunha SR, Hund TJ, Hashemi S, Voigt N, Li N, Wright P, et al. Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation. 2011;124:1212–1222. doi: 10.1161/CIRCULATIONAHA.111.023986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hund TJ, Wright PJ, Dun W, Snyder JS, Boyden PA, Mohler PJ. Regulation of the ankyrin-B-based targeting pathway following myocardial infarction. Cardiovasc Res. 2009;81:742–749. doi: 10.1093/cvr/cvn348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kashef F, Li J, Wright P, Snyder J, Suliman F, Kilic A, et al. Ankyrin-B protein in heart failure: identification of a new component of metazoan cardioprotection. J Biol Chem. 2012;287:30268–30281. doi: 10.1074/jbc.M112.368415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith SA, Hughes LD, Kline CF, Kempton AN, Dorn LE, Curran J, et al. Dysfunction of the β2-spectrin-based pathway in human heart failure. Am J Physiol Heart Circ Physiol. 2016;310:H1583–H1591. doi: 10.1152/ajpheart.00875.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohler PJ, Healy JA, Xue H, Puca AA, Kline CF, Allingham RR, et al. Ankyrin-B syndrome: enhanced cardiac function balanced by risk of cardiac death and premature senescence. PLoS One. 2007;2:e1051. doi: 10.1371/journal.pone.0001051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith SA, Sturm AC, Curran J, Kline CF, Little SC, Bonilla IM, et al. Dysfunction in the βII spectrin-dependent cytoskeleton underlies human arrhythmia. Circulation. 2015;131:695–708. doi: 10.1161/CIRCULATIONAHA.114.013708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Scouarnec S, Bhasin N, Vieyres C, Hund TJ, Cunha SR, Koval O, et al. Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc Natl Acad Sci U S A. 2008;105:15617–15622. doi: 10.1073/pnas.0805500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robaei D, Ford T, Ooi S-Y. Ankyrin-B syndrome: a case of sinus node dysfunction, atrial fibrillation and prolonged QT in a young adult. Heart Lung Circ. 2015;24:e31–e34. doi: 10.1016/j.hlc.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 38.He M, Tseng W-C, Bennett V. A single divergent exon inhibits ankyrin-B association with the plasma membrane. J Biol Chem. 2013;288:14769–14779. doi: 10.1074/jbc.M113.465328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mohler PJ, Davis JQ, Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 2005;3:e423. doi: 10.1371/journal.pbio.0030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hashemi SM, Hund TJ, Mohler PJ. Cardiac ankyrins in health and disease. J Mol Cell Cardiol. 2009;47:203–209. doi: 10.1016/j.yjmcc.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herrmann S, Lipp P, Wiesen K, Stieber J, Nguyen H, Kaiser E, et al. The cardiac sodium-calcium exchanger NCX1 is a key player in the initiation and maintenance of a stable heart rhythm. Cardiovasc Res. 2013;99:780–788. doi: 10.1093/cvr/cvt154. [DOI] [PubMed] [Google Scholar]
- 42.Torrente AG, Zhang R, Zaini A, Giani JF, Kang J, Lamp ST, et al. Burst pacemaker activity of the sinoatrial node in sodium-calcium exchanger knockout mice. Proc Natl Acad Sci U S A. 2015;112:9769–9774. doi: 10.1073/pnas.1505670112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Porter GA, Makuck RF, Rivkees SA. Intracellular calcium plays an essential role in cardiac development. Dev Dyn. 2003;227:280–290. doi: 10.1002/dvdy.10307. [DOI] [PubMed] [Google Scholar]
- 44.Calderwood SK, Murshid A, Prince T. The shock of aging: molecular chaperones and the heat shock response in longevity and aging--a mini-review. Gerontology. 2009;55:550–558. doi: 10.1159/000225957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tarone G, Brancaccio M. Keep your heart in shape: molecular chaperone networks for treating heart disease. Cardiovasc Res. 2014;102:346–361. doi: 10.1093/cvr/cvu049. [DOI] [PubMed] [Google Scholar]
- 46.Tokuriki N, Tawfik DS. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature. 2009;459:668–673. doi: 10.1038/nature08009. [DOI] [PubMed] [Google Scholar]
- 47.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Korja M, Kaprio J. Controversies in epidemiology of intracranial aneurysms and SAH. Nat Rev Neurol. 2016;12:50–55. doi: 10.1038/nrneurol.2015.228. [DOI] [PubMed] [Google Scholar]
- 49.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 50.The Human Protein Atlas [Internet] [Accessed May 2016]; Available from: http://www.proteinatlas.org/ [Google Scholar]
- 51.Gudmundsson H, Hund TJ, Wright PJ, Kline CF, Snyder JS, Qian L, et al. EH domain proteins regulate cardiac membrane protein targeting. Circ Res. 2010;107:84–95. doi: 10.1161/CIRCRESAHA.110.216713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kline CF, Kurata HT, Hund TJ, Cunha SR, Koval OM, Wright PJ, et al. Dual role of K ATP channel C-terminal motif in membrane targeting and metabolic regulation. Proc Natl Acad Sci U S A. 2009;106:16669–16674. doi: 10.1073/pnas.0907138106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhasin N, Cunha SR, Mudannayake M, Gigena MS, Rogers TB, Mohler PJ. Molecular basis for PP2A regulatory subunit B56alpha targeting in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2007;293:H109–H119. doi: 10.1152/ajpheart.00059.2007. [DOI] [PubMed] [Google Scholar]