

Abstract
Disease Overview
Ring sideroblasts (RS) are erythroid precursors with abnormal perinuclear mitochondrial iron accumulation. Two myeloid neoplasms defined by the presence of RS, include refractory anemia with ring sideroblasts (RARS), now classified under myelodysplastic syndromes with RS (MDS-RS) and RARS with thrombocytosis (RARS-T); now called myelodysplastic/myeloproliferative neoplasm with RS and thrombocytosis (MDS/MPN-RS-T).
Diagnosis
MDS-RS is a lower risk MDS, with single or multilineage dysplasia (SLD/MLD), <5% bone marrow (BM) blasts and ≥15% BM RS (≥5% in the presence of SF3B1 mutations). MDS/MPN-RS-T, now a formal entity in the MDS/MPN overlap syndromes, has diagnostic features of MDS-RS-SLD, along with a platelet count ≥ 450 × 10(9)/L and large atypical megakaryocytes (similar to BCR-ABL1 negative MPN).
Mutations and Karyotype
Mutations in SF3B1 are seen in ≥80% of patients with MDS-RS-SLD and MDS/MPN-RS-T, and strongly correlate with the presence of BM RS; MDS/MPN-RS-T patients also demonstrate JAK2V617F, ASXL1, DNMT3A, SETBP1, and TET2 mutations; with ASXL1/SETBP1 mutations adversely impacting survival. Cytogenetic abnormalities are uncommon in both diseases.
Risk stratification
Most patients with MDS-RS-SLD are stratified into lower risk groups by the revised-International Prognostic Scoring System (R-IPSS). Disease outcome in MDS/MPN-RS-T is better than that of MDS-RS-SLD, but worse than that of essential thrombocythemia. Both diseases have a low risk of leukemic transformation.
Treatment
Anemia and iron overload are complications seen in both and are managed similar to lower risk MDS and MPN. Aspirin therapy is reasonable in MDS/MPN-RS-T, especially in the presence of JAK2V617F, but the value of platelet-lowering drugs is uncertain.
Keywords: Myelodysplastic, Myeloproliferative, Overlap, SF3B1, JAK2, Review
DISEASE OVERVIEW
Ring sideroblasts (RS) are erythroid precursors in which after Prussian blue staining (Perls reaction) there are a minimum of five siderotic granules covering at least a third of the nuclear circumference (figure 1).[1] The iron deposited in the perinuclear mitochondria of RS is present in the form of mitochondrial ferritin. The detection of bone marrow RS can be seen in a variety of clonal hematological and non-clonal disorders (table 1). Clonal conditions associated with RS include myeloid neoplasms such as, myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPN) and MDS/MPN overlap syndromes. Amongst these, two myeloid neoplasms synonymous with the presence of bone marrow (BM) RS include: refractory anemia with ring sideroblasts (RARS), now classified under MDS with ring sideroblasts (MDS-RS) and RARS with thrombocytosis (RARS-T); now called MDS/MPN with RS and thrombocytosis (MDS/MPN-RS-T).[2] Non-clonal conditions associated with RS include excess alcohol use, lead toxicity, copper or pyridoxine deficiency, isoniazid therapy and congenital sideroblastic anemias (CSA).[3, 4]
Figure 1.
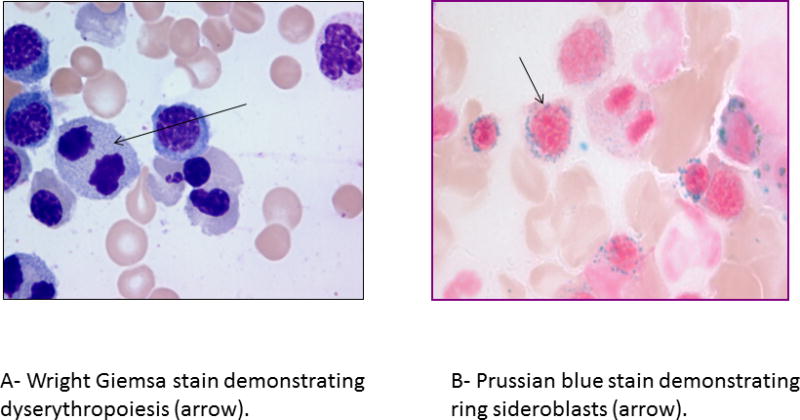
Bone marrow aspirate findings in patients with myelodysplastic syndrome with ring sideroblasts and single lineage dysplasia (MDS-RS-SLD).
Table 1.
Differential diagnosis of bone marrow ring sideroblasts
| Clonal – Myeloid Neoplasms | Non Clonal Causes |
|---|---|
1. Myelodysplastic syndrome (MDS)
|
1. Hereditary sideroblastic anemia
|
2. Myeloproliferative neoplasms (MPN)
|
2. Alcoholism |
3. MDS/MPN overlap syndromes
|
4. Drug induced sideroblastic anemia
|
| 5. Copper deficiency | |
| 6. Lead poisoning | |
| 7. Zinc toxicity |
Regardless of the etiology, the presence of RS usually signifies ineffective erythropoiesis and mitochondrial iron overload. Mitochondrial ferritin, which is a ferroxidase enzyme, is encoded by FTMT, an intronless gene located on chromosome 5q23.1.[5] Mitochondrial ferritin is normally undetectable in normal erythropoiesis.[6, 7] The differential diagnosis of disorders associated with RS, can broadly be divided into clonal hematological and non-clonal conditions (figure 2), summarized as follows.
Figure 2.

A schematic approach to the differential diagnosis of bone marrow ring sideroblasts.
Key- BM- bone marrow, RS- ring sideroblasts, MDS-RS-SLD- myelodysplastic syndrome with ring sideroblasts and single lineage dysplasia (previously called RARS), MDS-RS-MLD- myelodysplastic syndromes with ring sideroblasts and multilineage dysplasia (previously called RCMD-RS), MDS-EB-myelodysplastic syndrome with excess blasts (previously called RAEB), PMF- primary myelofibrosis, CMML- chronic myelomonocytic leukemia, PB- peripheral blood, XLSA- X linked sideroblastic anemia, XLSA/A- XLSA with ataxia, SA- sideroblastic anemia.
A. Non-clonal sideroblastic anemias
These are inherited or acquired, non-clonal conditions that give rise to BM RS. Acquired conditions include excess alcohol use, copper deficiency, lead and zinc toxicity, and drugs such as isoniazid, chloramphenicol, penicillamine and linezolid (table 1).[3, 4]
Genetically defined congenital sideroblastic anemias can be attributed to mutations in 1 of 3 mitochondrial pathways: heme synthesis, iron-sulfur cluster biogenesis and protein synthesis, or, rarely, a mutation in a protein involved in mitochondrial respiration itself.[8] The most common CSA is the X-linked sideroblastic anemia (XLSA) occurring secondary to heterogenous, usually missense substitutions of conserved amino acids in the delta-aminolevulinate synthase 2 gene (ALAS2)[9]; a gene encoding the first enzyme of the heme biosynthetic pathway. The activity of ALAS2 is pyridoxal 5-phosphate-dependant, with mutations that decrease the binding of the pyridoxal phosphate cofactor accounting for the pyridoxine responsiveness seen in some patients. Affected individuals usually have a microcytic hypochromic anemia and then eventually go on to develop signs and symptoms of iron overload (hemosiderosis).[3] Loss of function mutations in SLC25A38 have been shown to cause a sideroblastic anemia (SA) similar to XLSA, but with an autosomal recessive inheritance.[10] XLSA with ataxia is an inherited disorder associated with SA and non-progressive spinocerebellar ataxia (cerebellar hypoplasia), secondary to mutations involving the ATP-binding cassette sub-family B member 7 (ABCB7).[11] Similarly, mutations involving glutaredoxin-5 (GLRX5), a gene that encodes a mitochondrial protein involved in iron-sulfur cluster biogenesis can give rise to CSA.[12] Recently, a recurring mutation in NDUFB11 (nuclear-encoded mitochondrial complex I protein) essential for mitochondrial oxidative phosphorylation was identified in 5 individuals from 4 families with variably syndromic, normocytic CSA.[8] Additional rare causes of CSA have been outlined in table one and include mitochondrial deletion syndromes (more common in pediatric patients). Gene sequencing, including mitochondrial DNA sequencing and the establishment of heteroplasmy are becoming more accessible, leading to the availability of better diagnostic tests for these conditions.
B. Clonal Sideroblastic Anemias
Two clonal myeloid disorders characteristically associated with anemia and BM RS include RARS (now referred to as MDS-RS-single lineage dysplasia) and RARS-T (now referred to as MDS/MPN-RS-T). The World Health Organization (WHO) criteria for the diagnosis of MDS-RS used to necessitate the presence of ≥15% BM RS[1], a criterion that was thought to be arbitrary and one without prognostic relevance.[13] In a study with 200 MDS patients without excess blasts and with ≥ 1% RS, the impact of RS% was assessed both as a continuous and categorical variable: <5% (n = 56), 5%-14% (n = 32), 15%-50% (n = 79), and >50% (n = 33).[13] Ring sideroblast % correlated (p<.05) directly with age, platelet count, transfusion dependency, BM cellularity, and mutant SF3B1 and inversely with hemoglobin level, multilineage dysplasia, and high-risk karyotype. Neither univariate nor multivariable analysis showed significant effect for RS% on overall (OS) or leukemia-free survival (LFS), suggesting the limited prognostic value of quantifying BM RS.[13, 14] Based on this, the revised 2016 WHO criteria now allow for the diagnosis of MDS-RS with ≥5% BM RS, in the presence of SF3B1 mutations.[2] The number of RS required for a diagnosis of MDS/MPN-RS-T (≥15%) is not altered by the presence or absence of SF3B1 mutations.[2]
1. Myelodysplastic syndromes with ring sideroblasts
Disease overview
MDS-RS is usually a lower risk MDS characterized by anemia, morphologic dysplasia involving one or more lineages and BM RS comprising ≥ 15% of the BM erythroid progenitors (≥5% in the presence of SF3B1 mutations).[1, 2] The BM myeloblast content should be <5%, without any circulating blasts and secondary causes of RS need to be excluded. MDS-RS is further sub-classified into two groups; MDS-RS with single lineage dysplasia (SL D; formerly RARS) and MDS-RS with multilineage dysplasia (MLD; formerly RCMD-RS).[2] MDS-RS-SLD constitutes approximately 3–10% of all cases of MDS and has a median age of presentation of 71 years, with a slight male preponderance.[14–16] Clonal cytogenetic changes can be seen in 5–20% of cases, and when present usually involve a single chromosome.[16]
MDS-RS-MLD is characterized by bicytopenia or pancytopenia, and dysplasia affecting >10% of the cells in two or more myeloid cell lineages.[1, 2] Clinically these patients have a shorter OS with a higher risk for leukemic transformation (LT), in comparison to patients with MDS-RS-SLD.[14]
Mutations and Prognosis in MDS-RS
In 2011, with the advent of next generation sequencing (NGS) technology, somatic spliceosome component mutations (SF3B1-2q33.1, SRSF2-17q25.1, U2AF1-21q22.3, ZRSR2-Xp22.1, SF3A1-22q12.2 and U2AF2-19q13.42) were first described in patients with MDS (table 2).[17, 18] Amongst these, SF3B1 (splicing factor 3B subunit 1) mutations were most common in patients with MDS-RS.[14, 17, 18] The SF3B1 protein is a core component of the U2 snRNP, which binds to the branch site, thereby base pairing with the intron RNA; a critical process during RNA splicing (figure 3). Most mutations in SF3B1 are heterozygous substitutions and tend to cluster in exons 12–16 of the gene (chromosome 2q33.1). The SF3B1 K700E mutation usually accounts for 50% of the variants, with additional codons such as 666, 662, 622 and 625 acting as hot spot sites.[14, 17, 18]
Table 2.
Spectrum of molecular abnormalities in myelodysplastic syndromes with ring sideroblasts and single lineage dysplasia (RARS) and MDS/MPN with ring sideroblasts and thrombocytosis (RARS-T)
| Mutations and Study | RARS | RARS-T | Additional Myeloid neoplasms evaluated | Prognostic Impact |
|---|---|---|---|---|
|
| ||||
| JAK2V617F mutation: | ||||
| 1. Szpurka et al. Blood 2006[76] | 0/13 (0%) | 6/9 (67%) | RCMD-RS -0/16 (0%) RAEB-1/25 (4%) MDS with isolated del (5q) - 0/4 (0%) MDS-U-0/7 (0%) CMML-2/22 (9%) MDS/MPN-U – 3/26 (12%) Secondary AML-1/24 (4%) CML-0/21 (0%) PV-36/40 (90%) ET-17/30 (27%) PMF-18/33 (55%) |
|
| 2. Renneville et al. Leukemia 2006[77] | 0/7 (0%) | 5/7 (71%) | RAEB – 1/28 (4%) AML/MDS-1/9 (11%) CMML-2/15 (13%) ET-78/148 (53%) PV-86/92 (93%) PMF-30/50 (60%) |
|
| 3. Malcovati et al. Blood 2009[78] | 0/24 (0%) | 10/19 (53%) | ||
| 4. Broseus et al. Leukemia 2014[64] | 47/95 (49.4%) | |||
| 5. Jeromin et al. Haematologica 2014 [61] | 54/92 (58.7%) | |||
|
| ||||
| CALR Exon 9 mutation: | ||||
| 1. Klampf et al NEJM 2013[62] | 0% | 3/24 (12.5%) | MDS-0% AML-0% CMML-0% ET-25% PMF-35% PV-0% |
In PMF CALR mutations associated with better OS. |
| 2. Nangalia et al. NEJM 2013[63] | 3/27(11%) | 0/6 (0%) | Refractory anemia-9% RAEB-2/17-12% RA (5q−)-0% RCMD-0% RCMD-RS-0% ET-80/250 (32%) PMF-18/129 (14%) |
CALR mutations associated with higher platelet counts and lower hemoglobin |
| 3. Patnaik et al. Leukemia 2014[32] | 0/53 (0%) | |||
| 4. Broseus et al. Leukemia 2014[64] | 1/95 (1%) | |||
|
| ||||
| MPL mutations: | ||||
| 1. Malcovati et al. Blood 2009[78] | 1/19 (5%) | |||
| 2. Broseus et al. Leukemia 2014[64] | 1.1% (n=88) | |||
| 3. Jeromin et al. Haematologica 2014[61] | 2/92 (2.2%) | |||
|
| ||||
| SF3B1 mutations | ||||
| 1. Yoshida et al., Nature 2011 [17] | n=23 19/23 (82.6%) |
MDS without RS-6.5% RCMD-RS-38/50 (76% CMML -4.5% AML/MDS-4.8% De Novo AML-2.6% MPN-0% |
||
| 2. Papaemmanuil et al. NJEM 2011 [16] | 9/91 (10%) | RCMD-RS-13/23(56.5%) RCMD-3/53(6%) RAEB-6/110 (5%) PV-0% ET-6/189 (3%) PMF-6/136 (4.4%) CMML-5/106 (4.7%) |
Better OS- (p=0.01) Longer EFS (p=0.008) Longer LFS- (p=0.05) |
|
| 3. Patnaik et al. Blood 2012 [15] | 35 (66%) | RCMD (30%) RAEB-1 (4%) RAEB-2(0%) |
No benefit with regard to OS or LFS (p=0.6 and 0.2 respectively) | |
| 4. Visconte et al. Leukemia 2012 [79] | 9/14 (64%) | 13/18 (72%) | ||
| 5. Broseus et al. Leukemia 2014 [64] | 88.3% (n=94) | |||
| 6. Malcovati et al. Blood 2014[80] | 31/34 (91.2%) | NA | RCMD-RS - 16/28 (57.1%) RCMD-1 RA-3 |
SF3B1 mutations were associated with better survival (OS and LFS). |
| 7. Jeromin et al. Haematologica 2014[61] | 83/92 (90%) | |||
| 8. Patnaik et al Leukemia 2016 [23] | 41/48 (85%) | SF3B1 mutations were associated with inferior TFS. | ||
|
| ||||
| SRSF2 mutations: | ||||
| 1. Yoshida et al., Leukemia 2011[17] | 5.5% | NA | MDS with our RS-11.6% CMML-28.4% AML/MDS-6.5% De novo AML-2.6% MPN-1.9% |
|
| 2. Jeromin et al. Haematologica 2014[61] | 5/88 (5.7%) | |||
| 3. Patnaik et al. Leukemia 2016 [23] | 2/48 (4%) | |||
|
| ||||
| U2AF1 mutations: | ||||
| 1. Yoshida et al., Leukemia 2011[17] | 0% | NA | MDS without RS-11.6% CMML-8.0% AML/MDS-9.7% De novo AML-1.3% MPN-1.3% |
|
| 2. Jeromin et al. Haematologica 2014[61] | 4/89 (4.5%) | |||
|
| ||||
| TET2 mutations: | ||||
| 1. Jeromin et al. Haematologica 2014[61] | 21/90 (23.3%) | |||
| 2. Patnaik et al. Leukemia 2016 [23] | 5/48 (10%) | |||
|
| ||||
| DNMT3A mutations: | ||||
| 1. Jeromin et al. Haematologica 2014[61] | 13/78 (16.7%) | |||
| 2. Patnaik et al. Leukemia 2016 [23] | 6/48 (13%) | |||
|
| ||||
| ASXL1 mutations: | ||||
| 1. Jeromin et al. Haematologica 2014[61] | 13/91 (14.3%) | |||
| 2. Patnaik et al. Leukemia 2016 [23] | 14/42 (29%) | |||
Key: RARS, Refractory anemia with ringed sideroblasts; RARS-T, Refractory anemia with ringed sideroblasts and thrombocytosis, MDS, Myelodysplastic syndrome; RAEB, refractory anemia with excess blasts, CMML, Chronic myelomonocytic anemia; MPN, Myeloproliferative neoplasms; PV, polycythemia vera; PMF, primary myelofibrosis; ET, essential thrombocythemia; RCMD, refractory cytopenia with multilineage dysplasia; AML, acute myeloid leukemia; RA, refractory anemia; RS, ringed sideroblasts; OS, Overall survival; LFS, Leukemia free survival; TFS- Thrombosis free survival; WHO- World Health Organization.
Figure 3.
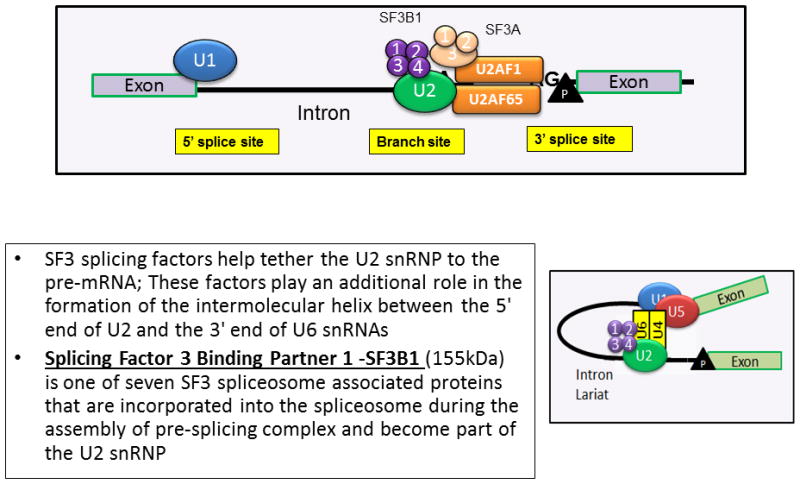
Physiological role of Splicing Factor 3 Binding Partner 1 (SF3B1).
SF3B1 mutations can be seen in ~80% of patients with MDS-RS-SLD, ~40% of patients with MDS-RS-MLD, with the percentage of BM RS often correlating directly with the SF3B1 mutant allele burden.[14, 17–19] Meayamycin, a pharmacological inhibitor of SF3B1, can induce RS in healthy in vitro BM cells, and BM RS can be seen in sf3b1-heterozygous-knockout mice, supporting the thought that SF3B1 haploinsufficiency strongly correlates with the development of BM RS.[20] The molecular mechanism behind the development of RS in relation to SF3B1 mutations is unclear. One hypothesis is that SF3B1 mutations could alter ABCB7 gene expression, dysregulating mitochondrial iron homeostasis, resulting in the formation of RS. [21] Currently, SF3B1 mutations have a positive predictive value for disease phenotype with RS of 97.7% (95% CI, 93.5–99.5%), and conversely the absence of these mutations, have a negative predictive value of 97.8% (95% CI, 93.8–99.5%).[22] It is important to note that SF3B1 mutations can be seen in a variety of myeloid neoplasms with BM RS such as; MDS/MPN-RS-T (~80%), primary myelofibrosis (PMF~7%) and chronic myelomonocytic leukemia (CMML~6%).[23–26] They have also been described in non-myeloid cancers such as, chronic lymphocytic leukemia (CLL~15%, enriched in patients with del11q), where they are associated with an adverse prognostic impact.[27]
The prognostic impact of SF3B1 mutations in MDS-RS is somewhat controversial, with a few reports demonstrating a favorable independent prognostic impact,[22] with most others not confirming the same.[14, 28, 29] In the Mayo Clinic study, we studied 107 patients with MDS-RS, including 48 with MDS-RS-SLD (RARS), 43 with MDS-RS-MLD (RCMD-RS), 11 with refractory anemia with excess blasts-1 (RAEB1)-RS, and 5 with RAEB2-RS.[14] SF3B1 mutations were detected in 53 (~50%) patients: 35 MDS-RS-SLD (73%), 16 MDS-RS-MLD (37%), and 2 RAEB1-RS (18%). In univariate analysis, the presence of SF3B1 mutations was associated with better OS (p<.01) and LFS (p<.01); however, in both instances, significance was completely accounted for by WHO morphologic risk categorization.[14] In other words, SF3B1 mutations have a useful phenotypic correlation with BM RS; however in the setting of an accurate morphological diagnosis (SLD versus MLD) they do not offer any additional prognostic value. Similarly, amongst 288 low risk MDS patients (MD Anderson lower-risk prognostic scoring system-LR-PSS), SF3B1 mutations were seen in 78% of patients with BM RS, they frequently co-occurred with DNMT3A mutations (p<0.001) and did not impact OS or LFS.[29] In this study, gene mutations associated with an independent prognostic impact in patients with MDS included; EZH2, RUNX1, Tp53 and ASXL1. Of these, only EZH2 mutations retained significance in a multivariable model that included LR-PSS and the aforementioned mutations.[29]
Thus conventional prognostication of patients with MDS-RS is usually carried out using MDS based risk stratification systems such as the IPSS (international prognostic scoring system)[30], revised IPSS (R-IPSS)[31], MD LR-PSS[32] and the WPSS (WHO classification based prognostic scoring system)[33]. Additional prognostic strategies incorporating molecular aberrations with clinical parameters are currently evolving.[34]
Clinical Outcomes
Patients with MDS-RS are generally stratified into lower risk categories using the aforementioned MDS prognostic models. In the Italian WPSS cohort there were 43 (10%) patients with RARS (MDS-RS-SLD) with their IPSS cytogenetic stratification being; good-82%, intermediate-15% and poor-3%, respectively.[33] Thirty % were red cell transfusion dependent. Similarly in the German (Dusseldorf) WPSS validation cohort there were 28 (4%) patients with RARS (MDS-RS-SLD) with their IPSS cytogenetic stratification being; good-86%, intermediate-10% and poor risk 4%, respectively. Twenty five % were red cell transfusion dependant.[33] In the Mayo Clinic cohort there were 48 patients with RARS (MDS-RS-SLD) and their risk stratification was as follows; IPSS- low-10% and intermediate-1–90%, R-IPSS- very low-34%, low-64% and intermediate-2%, respectively.[14] In this cohort, 17% were red cell transfusion dependant, 81% had SF3B1 mutations, 4% had the JAK2V617F mutation, 2% had IDH2 mutations and there were no patients with IDH1, MPL and CALR mutations, respectively.[14, 35] The median OS for patients with MDS-RS-SLD ranges from 69–108 months, with a very low risk for LT (<2%).[14, 35, 36] The survival of patients with MDS-RS-MLD is inferior to that of MDS-RS-SLD, but better than that of patients with MDS-MLD without BM RS or SF3B1 mutations.[14]
Risk Adapted Therapy
The management of patients with RARS involves supportive care strategies outlined for lower risk MDS patients and includes the use of erythropoiesis stimulating agents (ESA), transfusional supportive care and iron chelation therapy where applicable (figure 4).[37, 38]
Figure 4.

Schematic approach for the management of myelodysplastic syndrome with ring sideroblasts and single lineage dysplasia.
Key- MDS-RS- myelodysplastic syndrome with ring sideroblasts, EPO- erythropoietin, rH- recombinant human, G-CSF- granulocyte colony stimulating factor, ESA- erythropoiesis stimulating agents.
* Immunosuppressive therapy could be considered in individuals with hypoplastic bone marrows and a HLA-DR-15 phenotype.
i) Management of anemia
Commercially available ESA include recombinant human erythropoietin (rh-EPO) and darbepoetin. Response rates in lower risk MDS patients range from 30–60%, with the median duration of response being ~ 24 months.[39–41] Parameters predictive of ESA response include; low transfusion burden (<2 units a month), diagnosis of RA/RARS, use of a fixed-dose versus weight-based EPO regimen, shorter time from diagnosis to starting treatment, and a lower baseline serum EPO level (<500 IU/ml).[39, 40] Most responses to ESA occur within 8 weeks of treatment and close monitoring of hemoglobin level is required to avoid increases to >12 g/dL, which is associated with a potential risk of systemic hypertension and thrombosis.[42] Weekly doses of 40 000 units of EPO-α, 30 000 units of EPO-β, or 150 to 300 microgram of darbepoetin yield ~ 50–60% of erythroid responses.[39, 40, 42, 43] Meta-analyses have not demonstrated any significant difference between rH-EPO versus darbepoetin, while there might be a benefit to the addition of G-CSF (granulocyte- colony stimulating factor) to ESA.[39, 40, 42] Interestingly, ~ 70% of the relapses of anemia after initial response to ESA are not associated with progression to higher-risk MDS. [44]
Lenalidomide an immunomodulatory agent with preferential activity in MDS patients with isolated del(5q), can alleviate transfusional requirements in approximately 26–57% of lower risk MDS patients with a normal karyotype.[45–47] In a phase II study, for transfusion dependant (TD) lower risk MDS patients, 30 (35%) of 86 patients with MDS-RS-SLD achieved transfusion independence (TI).[46] In a recent, pivotal, randomized, phase III clinical trial, using lenalidomide or placebo (2:1) in TD, lower-risk MDS patients with non del(5q), RBC-TI ≥ 8 weeks was achieved in 26.9% and 2.5% of patients in the lenalidomide and placebo groups, respectively (n=239, ~8% with MDS-RS-SLD).[47] Median onset of RBC-TI was at 10.1 weeks and 90% responded within 16 weeks. In multivariate analysis, prior ESA use and transfusion burden were independent predictors of response, whereas EPO level was not.[47] Peripheral blood cytopenias, skin rash and pruritus were the most common adverse events. Only 3% of patients receiving lenalidomide developed venous thrombosis.[47] Lenalidomide can also be used in combination with ESA in the setting of clinical trials.[38]
Sotatercept (ACE-011) and Luspatercept (ACE-536) are soluble fusion proteins that inhibit molecules in the TGF-β (transforming growth factor) superfamily, increasing hemoglobin levels by targeting a pathway fundamentally distinct from EPO.[48] Preliminary results from MDS trials are encouraging (NCT01736683 & NCT02268383-www.clinicaltrials.gov). Similarly, eltrombopag, a small molecule agonist of c-mpl (megakaryocyte receptor) has shown efficacy in improving cytopenias, including red cell TD in patients with MDS (NCT01772420 and NCT01584531-www.clinicaltrials.gov).[49, 50]
ii) Iron chelation therapy
The indolent nature of MDS-RS, coupled with infective erythropoiesis and red cell TD, can result in iron overload.[51] The iron status can be monitored non-invasively by measuring serum ferritin levels, transferrin saturation and by the utilization of T2* MRI (magnetic resonance imaging) scans.[51, 52] Increased serum ferritin levels in MDS, can be secondary to TD, cancer associated inflammation (acute phase reactant) and liver disease.[53–55] Each of these comorbidities are prognostically detrimental and it is unclear if iron overload, or for that matter an elevated ferritin level, per se contribute to mortality and morbidity.[53–55] Data suggesting hemosiderosis related causes of death in MDS are scant and are limited to case reports. In a retrospective study of 126 MDS-RS patients, independent prognosticators for survival included, IPSS (p<0.0001) categories and red cell TD at diagnosis (p=0.001), but not the number of red cell units transfused during the disease course (p =0.17).[53] There were no correlations between survival and serum ferritin level, measured either at diagnosis or during follow-up. Similarly, there was no difference in survival when patients were stratified by serum ferritin levels of < or ≥1,000 ng/mL at diagnosis or peak serum ferritin levels of <1,000, 1,000–5,000, or >5,000 ng/mL during follow-up.[53] Similarly, in 88 patients with WHO defined MDS with isolated del(5q), serum ferritin levels did not impact survival and with a median OS of 66 months, with 66% deaths documented at last follow up, there were no deaths attributed to iron overload.[56] A recent prospective study also found no impact of an elevated liver iron content, as measured by MRI scans, on post allogeneic stem cell transplant (HSCT) survival and outcomes.[57] Additionally, iron chelation agents such as desferrioxamine (parenteral), deferiprone and deferasirox (both oral) are expensive and are associated with side effects.[58] Desferrioxamine has been associated with deafness and visual side effects,[52] deferiprone with gastrointestinal (GI) side effects and agranulocytosis, [59] and deferasirox with renal failure, liver failure, cytopenias and GI side effects.[60] The use of iron chelation therapy in MDS-RS awaits rigorous prospective evaluation and until this can be established, its routine use cannot be recommended.
iii) Hypomethylating agent use
The upfront use of hypomethylating agents (HMA) such as 5-Azacitidine and decitabine in patients with MDS-RS is uncommon; exceptions being patients that have failed ESA or have developed additional cytopenias/disease progression.[38] HMA have been reported to yield red cell TI in ~ 40% of patients and may also be effective for other cytopenias in lower-risk MDS.[61, 62] In a phase II trial randomizing AZA and AZA + EPO in red cell TD lower-risk MDS patients clearly identified as ESA resistant, the red cell TI rate was 17%, without difference between the 2 treatment arms.[63] Ongoing studies are exploring the safety and efficacy of low dose HMA in TD lower-risk MDS. CC-486 is an oral formulation of azacitidine. In an expanded phase 1 trial, CC-486 demonstrated clinical and biological activity in lower-risk MDS with poor prognostic features including anemia and/or thrombocytopenia. The overall response rate was 40 %, including HI in 28 % and RBC TI sustained for 56 days in 47 % of patients with baseline transfusion dependence.[64] It is important to note that to date, HMA have not demonstrated a survival advantage in patients with lower risk MDS.
iv) Imetelstat
The telomerase inhibitor imetelstat is an oligonucleotide that targets the RNA template of human telomerase reverse transcriptase. In a recent study, imetelstat produced clinical and molecular remissions in patients with myelofibrosis with SF3B1 or U2AF1 mutations; thus leading to its use in a pilot study for patients with MDS-RS (n=5) and MDS/MPN-RS-T (n=3).[65, 66] Three (38%) of 8 TD patients became TI at a median time of 11 weeks (range, 9–14) and their response lasted for a median of 28 weeks.[65, 66] Treatment emergent myelosuppression and liver function test abnormalities were the most common side effects seen. The drug is currently being evaluated in a larger phase II clinical study in patients with TD lower risk MDS (NCT02598661- www.clinicaltrials.gov).
v) Allogeneic stem cell transplantation
Allogeneic hematopoietic stem cell transplantation (HSCT) is the only curative strategy for patients with MDS. This modality however is fraught with complications such as acute and chronic graft versus host disease (GVHD) and non-relapse mortality (NRM). Seminal studies have shown no benefit for upfront allo-HSCT for lower risk MDS patients, regardless of a myeloablative [67] or a reduced intensity conditioning (RIC) strategy.[68]
Cutler et al. demonstrated that for patients with lower risk MDS, delayed myeloablative allo-HSCT was associated with maximum life expectancy.[67] In this study, the median OS of patients with IPSS low and intermediate-1 MDS were 141.1 and 62.9 months with non-transplant management, while these numbers decreased to 40.2 and 20.5 months with early transplant strategies.[67] In addition, adjustment for quality of life did not change the preferred treatment strategy. In this study 16% of patients in the non-transplant group and approximately 5% in the transplant group had a morphological diagnosis of MDS-RS-SLD.[67] Similarly, in de novo lower risk MDS patients with ages between 60–70 years, OS was better with non-transplant strategies (77 months), in comparison to a RIC allo-HSCT (38 months).[68] Once again in this cohort quality-adjusted life expectancy did not alter preferred treatment strategy. In the non-transplant group, the lower risk MDS patients received ESA and supportive care, while the higher risk patients received HMA.[68]
2. Myelodysplastic/Myeloproliferative neoplasm with ring sideroblasts and thrombocytosis
Disease overview
MDS/MPN-RS-T, formerly RARS-T, was previously considered a provisional entity with overlapping features of MDS and MPN.[1, 69] Based on comprehensive clinical and molecular data, the WHO in their 2016 revision recommendations, formally classified RARS-T as a MDS/MPN overlap subtype and renamed it MDS/MPN-RS-T.[1, 2] Patients with MDS/MPN-RS-T have features of MDS-RS-SLD and thrombocytosis (platelet count ≥ 450 × 10(9)/L) with proliferation of large atypical megakaryocytes similar to those observed in BCR-ABL1 negative MPN. In 2008 the minimum platelet count required for inclusion was lowered from 600 to 450 × 10(9)/L for consistency with the definition criteria for essential thrombocythemia (ET).[1, 2] In the differential diagnosis, it is important to exclude patients with ET or reactive thrombocytosis associated with BM RS. The median age for presentation usually ranges from 71–75 years and approximately 80% of patients do not have detectable clonal cytogenetic abnormalities.[69–71] Other entities included in the MDS/MPN overlap syndromes are; CMML, juvenile myelomonocytic leukemia (JMML), atypical CML and MDS/MPN- unclassifiable.[72]
Mutations and Prognosis in MDS/MPN-RS-T
Gene mutations encountered in patients with MDS/MPN-RS-T include; SF3B1 (~85%), JAK2V617F (~50%), TET2 (~25%), ASXL1 (~20%), DNMT3A (~15%) and SETBP1 (~10%).[70, 71] In a recent European study (n=92), gene mutations detected included; spliceosome components - SF3B1-90%, SRSF2-6.7%, U2AF1-5.3%, ZRSR2-2.7%, signalling pathways- JAK2V617F- 57%, MPL-2.7%, CBL-4%, epigenetic regulators- ASXL1-15%, TET2-25%, EZH2-7%, DNMT3A-15%, IDH2-4%, and transcription regulators- ETV6-2.7%, RUNX1-1.3%.[71] Approximately 50% of patients harbored both the JAK2V617F and SF3B1 mutations. Conversely, most of the SF3B1wt patients had JAK2V617F and ASXL1 mutations.[71]
In a Mayo Clinic study, 94% of patients had ≥1 mutations; common mutations being: SF3B1 85%, JAK2V617F 33%, ASXL1 29%, DNMT3A 13%, SETBP1 13% and TET2 10%.[70] In a multivariable survival analysis (n = 82), anemia (P = 0.02) and abnormal karyotype (P =.01) were independently prognostic for inferior survival. In patients with NGS information (n = 48), univariate analysis showed association between poor survival and presence of SETBP1 (P = 0.04) or ASXL1 (P = 0.08) mutations whereas the absence of these mutations (ASXL1wt/SETBP1wt) was favorable (P = 0.04); the number of concurrent mutations did not provide additional prognostication (P = 0.3).[70] This resulted in a hazard ratio-weighted prognostic model, with 2 points for an abnormal karyotype, 1 point for either ASXL1 and/or SETBP1 mutations, and 1 point for a HB level < 10 gm/dl, which effectively stratified patients into three risk categories; low (0 points), intermediate (1 point) and high (≥2 points), with median survivals of 80, 42 and 11 months respectively (P = 0.01) (figure 5A).[70] Mutations in exon 9 of the calreticulin gene (CALR) have been reported as mutually exclusive of JAK2 and MPL mutations and can be seen in 67–71% of ET and 56–88% of PMF patients with wild-type JAK2 or MPL.[73] However, unlike in MPN, MPL (1–3%) and CALR (0–3%) mutations are infrequent in MDS/MPN-RS-T.[70, 71, 73]
Figure 5.
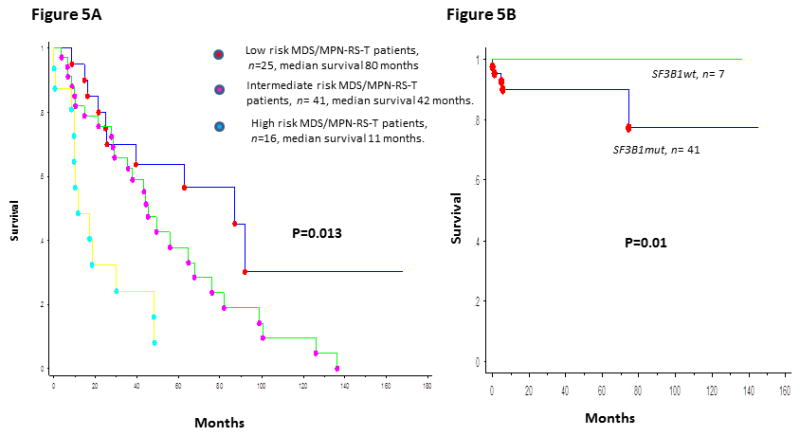
5A: Over-all survival of 82 patients with MDS/MPN-RS-T, stratified by the Mayo Clinic prognostic model.
5B: Thrombosis free survival in 48 patients with MDS/MPN-RS-T, stratified by their SF3B1 mutation status.
In a large study comparing patients with MDS-RS, MDS/MPN-RS-T and ET, patients with MDS/MPN-RS-T were found to have a better median OS than MDS-RS-SLD (76 months versus 63 months), and an inferior OS in comparison to patients with ET (76 months versus 117 months).[25] Importantly there was no difference in survival noted regardless of the JAK2 mutation status or the platelet threshold (> or < 600 × 10(9)/L).[25] There was no difference in supportive care, including the use of transfusions and ESA between MDS-RS and MDS/MPN-RS-T patients, with approximately 50% receiving transfusions and ESA respectively. However, MDS/MPN-RS-T patients were more frequently treated with antiplatelet agents (51.5%) and cytoreductive therapies such as hydroxyurea (32%).[25] The leukemic transformation rate per 100 years was similar in MDS/MPN-RS-T (1.8) and MDS-RS-SLD (2.4) and higher in MDS/MPN-RS-T in comparison to ET (0.7).
Thrombosis in MDS/MPN-RS-T
The frequency of thrombotic events in MDS/MPN-RS-T is thought to be similar to that of ET (3.6 vs 3.9/100 patient-years), but more frequent than MDS-RS (3.6 vs 0.9/100 patient-years).[25] In a prior study (n=15), 6 of 10 (60%) patients with SF3B1mut RARS-T had thrombotic events; whereas there were no thrombotic events in SF3B1wt patients.[19] In a recent Mayo Clinic study, at a median follow-up of 27 months, 48 (59%) deaths and 16 (20%) thrombotic events were documented (median OS 44 months).[23] Cardiovascular (CV) risk factors were present in 52 (63%) patients. Eight (10%) patients had a thrombotic event prior to or at the time of diagnosis of MDS/MPN-RS-T (venous-8, arterial-0).[23] Nine (11%) patients developed subsequent thrombotic events (venous-7, arterial-2) with no fatalities. Six (85%) of seven subsequent venous events were unprovoked deep vein thromboses (DVT), with two patients having concomitant pulmonary emboli; whereas one patient developed portal vein thrombosis.[23] In univariate analysis, hemoglobin <10 g/dl (P=0.008), BM RS % (P=0.04), history of thrombosis (P=0.02) and presence of SF3B1 mutations (P=0.017) were associated with an inferior thrombosis free survival (TFS). Age (P=0.07), JAK2 mutation status (P=0.56) and CV risk factors (P=0.95) did not have prognostic impact. Given the association between SF3B1 mutations and BM RS, in a multivariable model that included these two as covariates, only the presence of SF3B1 mutations retained prognostic significance (P=0.02) (figure 5B). The presence of SF3B1 mutation remained significant (P=0.04) (hazard ratio—8.9; 95% confidence interval 1.1–4.5) when history of thrombosis (P=0.12) or hemoglobin <10 g/dl (P=0.14) was introduced into the multivariable model. Thrombotic events occurring at or prior to diagnosis (P=0.53), thrombotic events occurring after diagnosis (P=0.69), SF3B1 (P=0.83) and JAK2V617F (P=0.4) mutational status, and CV risk factors (P=0.05) did not impact OS.[23] Thus, unlike in ET, prior thrombotic events in MDS/MPN-RS-T do not impact OS.[23] The mechanism by which SF3B1 mutations might increase thrombotic predisposition in patients with MDS/MPN-RS-T requires further study.
Risk adapted therapy
Given that, until recently, RARS-T was considered a provisional entity in the MDS/MPN overlap syndromes, no formal guidelines for the management of this disease exist. Current management strategies, including response criteria are extrapolated from related diseases such as low risk MDS-RS and MPN (ET). Management is individualized to address presenting problems (figure 6).
Figure 6.

Management of thrombocytosis in patients with MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T).
In patients with anemia, management is very similar to lower risk MDS, where the use of ESA and transfusional supportive care is instituted early on. There are case reports documenting the efficacy of lenalidomide in alleviating red cell transfusion need in patients with MDS/MPN-RS-T.[74] In one particular case report, of 2 patients with JAK2V617mut MDS/MPN-RS-T, lenalidomide was able to achieve a complete molecular remission in one patient.[74] Clinical trials with newer agents such as sotatercept/luspatercept, eltrombopag and the novel telomerase inhibitor imetelstat are either in conception phase or are currently ongoing.[66]
Similar to ET, patients with MDS/MPN-RS-T are at risk for vasomotor symptoms such as, migraine headaches, palpitations, acral paresthesias, atypical chest pain and erythromelalgia along with arterial and venous thrombosis.[23, 25] Some patients may also develop an acquired von Willebrand’s disease, particularly in the presence of extreme thrombocytosis (> 1000 × 10(9)/L), and are at risk for aspirin related bleeding.[75]
Similar to ET, we risk stratify patients with MDS/MPN-RS-T (especially those with SF3B1 mutations) into two categories; low risk (0 factors) and high risk (1 or 2 factors), based on age >60 years and a prior thrombotic event (figure 6).[76] We believe (in the absence of extreme thrombocytosis) that low risk patients with MDS/MPN-RS-T benefit from low dose aspirin therapy (81–100 mg), with a reduction in the risk for arterial thrombosis and a significant reduction in microvascular/vasomotor symptoms.[77] Recent data, suggests an added benefit for twice daily aspirin dosing in patients with ET, a finding that needs to be explored in MDS/MPN-RS-T.[78] Alternative antiplatelet agents such as clopidogrel, prasugrel and ticagrelor are considered in the setting of aspirin resistance, aspirin hypersensitivity or the need for dual antiplatelet therapy.
The value of cytoreductive therapy in MDS/MPN-RS-T is uncertain and its use might exacerbate anemia, which is the most frequent disease complication in MDS/MPN-RS-T. We therefore avoid the use of cytoreductive drugs in patients with anemia unless compelled by the presence of multiple risk factors for thrombosis.[76] Our drug of choice, if cytoreductive therapy is indicated, is hydroxyurea, which has been the mainstay of cytoreductive therapy, with randomized data in patients with ET demonstrating a significant reduction in thrombotic events. In a randomized study, after 27 months follow up, thrombotic events were seen in 3.6% of ET patients on hydroxyurea and in 24% of patients in the control arm.[79] Alternate cytoreductive agents that we consider in the setting of hydroxyurea failure, refractoriness or intolerance, include; lenalidomide, interferon alpha or busulfan.[76] Two recent studies of pegylated INF-α (~90 μg SC weekly) in PV and ET reported hematologic remissions of ~80% accompanied by decreases in JAK2V617F allele burden (complete molecular remission rate of 5–10%).[80, 81] In one of the two studies, 77 cases were evaluable after a median follow up of 21 months and 76% and 70% of patients with ET or PV, respectively, achieved a complete hematologic remission, mostly in the first 3 months; side effects were recorded in 96% of the patients and 22% had discontinued treatment.[81] A small study of busulfan in 36 patients with ET demonstrated safety and efficacy of the agent, without an increase in LT rates.[82] In the setting of extreme thrombocytosis, the diagnosis of acquired von Willebrand’s disease should be evaluated for, using the ristocetin cofactor activity and the von Willebrand’s factor multimer analysis.[76] Affected patients demonstrate an improvement in bleeding parameters after a reduction in the platelet count is achieved with cytoreductive agents.
Similar to lower risk MDS and MPN patients, allo-HSCT is reserved for patients with refractory cytopenias or progressive disease. The risks of allo-HSCT namely, acute and chronic GVHD and the NRM currently limit the upfront use of this strategy for affected patients.[83]
CONCLUSION
MDS-RS and MDS/MPN-RS-T are myeloid neoplasms synonymous with the presence of BM RS and the presence of SF3B1 mutations. MDS-RS is further subclassified based on the extent of BM dysplasia into MDS-RS-SLD and MDS-RS-MLD. MDS/MPN-RS-T shares features of both MDS and MPN and is now formally classified as a MDS/MPN overlap neoplasm (2016 WHO revision recommendations). SF3B1 mutations are seen in ~ 80% of patients with MDS-RS-SLD and MDS/MPN-RS-T. Additional mutations seen in MDS/MPN-RS-T include; JAK2V617F (~50%), TET2 (~25%), ASXL1 (~20%), DNMT3A (~15%) and SETBP1 (~10%). Most patients with MDS-RS, especially those with SLD prognosticate into lower risk categories of the IPSS and R-IPSS, indicative of favorable outcomes. Managements strategies for MDS-RS are similar to that of lower risk MDS. The prognosis of patients with MDS/MPN-RS-T is better than that of MDS-RS, but inferior to that of ET. Survival in MDS/MPN-RS-T is adversely impacted by low hemoglobin levels, abnormal karyotype and the presence of ASXL1/SETBP1 mutations; with SF3B1 mutations adversely impacting TFS. However, unlike in ET, prior thrombotic events in MDS/MPN-RS-T do not impact either the OS or TFS. While symptom management is similar to patients with low risk MDS and MPN, patients with MDS/MPN-RS-T have a higher incidence of thrombosis and often need antiplatelet and/or cytoreductive therapy. The recent classification of MDS/MPN-RS-T as a formal MDS/MPN overlap neoplasm is a much needed step, one that will hopefully result in the development of disease specific response criteria and clinical trials.
Acknowledgments
The authors would like to acknowledge Dr. Emnet Wassie, Dr. Curtis Hanson and Dr. Terra Lasho for their help in the preparation of the manuscript.
Current publication is supported in part by grants from the “The Henry J. Predolin Foundation for Research in Leukemia, Mayo Clinic, Rochester, MN, USA”.
This publication was supported by CTSA Grant Number KL2 TR000136 from the National Center for Advancing Translational Science (NCATS). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
References
- 1.Swerdlow S, Camp E, Harris NL, Jaffe ES, Stefano PA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon: International Agency for Research on Cancer; 2008. [Google Scholar]
- 2.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 3.Camaschella C. Hereditary sideroblastic anemias: pathophysiology, diagnosis, and treatment. Semin Hematol. 2009;46:371–377. doi: 10.1053/j.seminhematol.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Willekens C, Dumezy F, Boyer T, et al. Linezolid induces ring sideroblasts. Haematologica. 2013;98:e138–140. doi: 10.3324/haematol.2013.092395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levi S, Corsi B, Bosisio M, et al. A human mitochondrial ferritin encoded by an intronless gene. J Biol Chem. 2001;276:24437–24440. doi: 10.1074/jbc.C100141200. [DOI] [PubMed] [Google Scholar]
- 6.Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood. 2013;122:4021–4034. doi: 10.1182/blood-2013-09-381665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cazzola M, Invernizzi R, Bergamaschi G, et al. Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia. Blood. 2003;101:1996–2000. doi: 10.1182/blood-2002-07-2006. [DOI] [PubMed] [Google Scholar]
- 8.Lichtenstein DA, Crispin AW, Sendamarai AK, et al. A recurring mutation in the respiratory complex 1 protein NDUFB11 is responsible for a novel form of X-linked sideroblastic anemia. Blood. 2016;128:1913–1917. doi: 10.1182/blood-2016-05-719062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cotter PD, Baumann M, Bishop DF. Enzymatic defect in “X-linked” sideroblastic anemia: molecular evidence for erythroid delta-aminolevulinate synthase deficiency. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:4028–4032. doi: 10.1073/pnas.89.9.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guernsey DL, Jiang H, Campagna DR, et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat Genet. 2009;41:651–653. doi: 10.1038/ng.359. [DOI] [PubMed] [Google Scholar]
- 11.Allikmets R, Raskind WH, Hutchinson A, et al. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A) Human molecular genetics. 1999;8:743–749. doi: 10.1093/hmg/8.5.743. [DOI] [PubMed] [Google Scholar]
- 12.Rouault TA, Tong WH. Iron-sulfur cluster biogenesis and human disease. Trends in genetics: TIG. 2008;24:398–407. doi: 10.1016/j.tig.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patnaik MM, Hanson CA, Sulai NH, et al. Prognostic irrelevance of ring sideroblast percentage in World Health Organization-defined myelodysplastic syndromes without excess blasts. Blood. 2012;119:5674–5677. doi: 10.1182/blood-2012-03-415356. [DOI] [PubMed] [Google Scholar]
- 14.Patnaik MM, Lasho TL, Hodnefield JM, et al. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood. 2012;119:569–572. doi: 10.1182/blood-2011-09-377994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Germing U, Gattermann N, Strupp C, et al. Validation of the WHO proposals for a new classification of primary myelodysplastic syndromes: a retrospective analysis of 1600 patients. Leuk Res. 2000;24:983–992. doi: 10.1016/s0145-2126(00)00088-6. [DOI] [PubMed] [Google Scholar]
- 16.Germing U, Gattermann N, Aivado M, et al. Two types of acquired idiopathic sideroblastic anaemia (AISA): a time-tested distinction. Br J Haematol. 2000;108:724–728. doi: 10.1046/j.1365-2141.2000.01940.x. [DOI] [PubMed] [Google Scholar]
- 17.Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–1395. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 19.Visconte V, Makishima H, Jankowska A, et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia. 2012;26:542–545. doi: 10.1038/leu.2011.232. [DOI] [PubMed] [Google Scholar]
- 20.Visconte V, Rogers HJ, Singh J, et al. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood. 2012;120:3173–3186. doi: 10.1182/blood-2012-05-430876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nikpour M, Scharenberg C, Liu A, et al. The transporter ABCB7 is a mediator of the phenotype of acquired refractory anemia with ring sideroblasts. Leukemia. 2013;27:889–896. doi: 10.1038/leu.2012.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malcovati L, Papaemmanuil E, Bowen DT, et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood. 2011;118:6239–6246. doi: 10.1182/blood-2011-09-377275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patnaik MM, Lasho TL, Finke CM, et al. Vascular events and risk factors for thrombosis in refractory anemia with ring sideroblasts and thrombocytosis. Leukemia. 2016;30:2273–2275. doi: 10.1038/leu.2016.216. [DOI] [PubMed] [Google Scholar]
- 24.Lasho TL, Finke CM, Hanson CA, et al. SF3B1 mutations in primary myelofibrosis: clinical, histopathology and genetic correlates among 155 patients. Leukemia. 2012;26:1135–1137. doi: 10.1038/leu.2011.320. [DOI] [PubMed] [Google Scholar]
- 25.Broseus J, Florensa L, Zipperer E, et al. Clinical features and course of refractory anemia with ring sideroblasts associated with marked thrombocytosis. Haematologica. 2012;97:1036–1041. doi: 10.3324/haematol.2011.053918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patnaik MM, Lasho TL, Finke CM, et al. Spliceosome mutations involving SRSF2, SF3B1, and U2AF35 in chronic myelomonocytic leukemia: prevalence, clinical correlates, and prognostic relevance. Am J Hematol. 2013;88:201–206. doi: 10.1002/ajh.23373. [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497–2506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Damm F, Kosmider O, Gelsi-Boyer V, et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood. 2012;119:3211–3218. doi: 10.1182/blood-2011-12-400994. [DOI] [PubMed] [Google Scholar]
- 29.Bejar R, Stevenson KE, Caughey BA, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. 2012;30:3376–3382. doi: 10.1200/JCO.2011.40.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–2088. [PubMed] [Google Scholar]
- 31.Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–2465. doi: 10.1182/blood-2012-03-420489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kantarjian H, O’Brien S, Ravandi F, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. 2008;113:1351–1361. doi: 10.1002/cncr.23697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malcovati L, Germing U, Kuendgen A, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol. 2007;25:3503–3510. doi: 10.1200/JCO.2006.08.5696. [DOI] [PubMed] [Google Scholar]
- 34.Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–2506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patnaik MM, Belachew A, Finke C, et al. CALR mutations are infrequent in WHO-defined refractory anemia with ring sideroblasts. Leukemia. 2014;28:1370–1371. doi: 10.1038/leu.2014.47. [DOI] [PubMed] [Google Scholar]
- 36.Germing U, Strupp C, Kuendgen A, et al. Prospective validation of the WHO proposals for the classification of myelodysplastic syndromes. Haematologica. 2006;91:1596–1604. [PubMed] [Google Scholar]
- 37.Malcovati L, Hellstrom-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122:2943–2964. doi: 10.1182/blood-2013-03-492884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia-Manero G. Myelodysplastic syndromes: 2014 update on diagnosis, risk-stratification, and management. Am J Hematol. 2014;89:97–108. doi: 10.1002/ajh.23642. [DOI] [PubMed] [Google Scholar]
- 39.Hellstrom-Lindberg E. Efficacy of erythropoietin in the myelodysplastic syndromes: a meta-analysis of 205 patients from 17 studies. Br J Haematol. 1995;89:67–71. doi: 10.1111/j.1365-2141.1995.tb08909.x. [DOI] [PubMed] [Google Scholar]
- 40.Moyo V, Lefebvre P, Duh MS, et al. Erythropoiesis-stimulating agents in the treatment of anemia in myelodysplastic syndromes: a meta-analysis. Ann Hematol. 2008;87:527–536. doi: 10.1007/s00277-008-0450-7. [DOI] [PubMed] [Google Scholar]
- 41.Jadersten M, Montgomery SM, Dybedal I, et al. Long-term outcome of treatment of anemia in MDS with erythropoietin and G-CSF. Blood. 2005;106:803–811. doi: 10.1182/blood-2004-10-3872. [DOI] [PubMed] [Google Scholar]
- 42.Santini V. Clinical use of erythropoietic stimulating agents in myelodysplastic syndromes. Oncologist. 2011;16(Suppl 3):35–42. doi: 10.1634/theoncologist.2011-S3-35. [DOI] [PubMed] [Google Scholar]
- 43.Gabrilove J, Paquette R, Lyons RM, et al. Phase 2, single-arm trial to evaluate the effectiveness of darbepoetin alfa for correcting anaemia in patients with myelodysplastic syndromes. Br J Haematol. 2008;142:379–393. doi: 10.1111/j.1365-2141.2008.07181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park S, Grabar S, Kelaidi C, et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood. 2008;111:574–582. doi: 10.1182/blood-2007-06-096370. [DOI] [PubMed] [Google Scholar]
- 45.List A, Kurtin S, Roe DJ, et al. Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med. 2005;352:549–557. doi: 10.1056/NEJMoa041668. [DOI] [PubMed] [Google Scholar]
- 46.Raza A, Reeves JA, Feldman EJ, et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1 risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood. 2008;111:86–93. doi: 10.1182/blood-2007-01-068833. [DOI] [PubMed] [Google Scholar]
- 47.Santini V, Almeida A, Giagounidis A, et al. Randomized Phase III Study of Lenalidomide Versus Placebo in RBC Transfusion-Dependent Patients With Lower-Risk Non-del(5q) Myelodysplastic Syndromes and Ineligible for or Refractory to Erythropoiesis-Stimulating Agents. J Clin Oncol. 2016;34:2988–2996. doi: 10.1200/JCO.2015.66.0118. [DOI] [PubMed] [Google Scholar]
- 48.Raje N, Vallet S. Sotatercept, a soluble activin receptor type 2A IgG-Fc fusion protein for the treatment of anemia and bone loss. Curr Opin Mol Ther. 2010;12:586–597. [PubMed] [Google Scholar]
- 49.Roth M, Will B, Simkin G, et al. Eltrombopag inhibits the proliferation of leukemia cells via reduction of intracellular iron and induction of differentiation. Blood. 2012;120:386–394. doi: 10.1182/blood-2011-12-399667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prica A, Sholzberg M, Buckstein R. Safety and efficacy of thrombopoietin-receptor agonists in myelodysplastic syndromes: a systematic review and meta-analysis of randomized controlled trials. Br J Haematol. 2014;167:626–638. doi: 10.1111/bjh.13088. [DOI] [PubMed] [Google Scholar]
- 51.Valent P, Krieger O, Stauder R, et al. Iron overload in myelodysplastic syndromes (MDS) - diagnosis, management, and response criteria: a proposal of the Austrian MDS platform. Eur J Clin Invest. 2008;38:143–149. doi: 10.1111/j.1365-2362.2007.01915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greenberg PL, Rigsby CK, Stone RM, et al. NCCN Task Force: Transfusion and iron overload in patients with myelodysplastic syndromes. J Natl Compr Canc Netw. 2009;7(Suppl 9):S1–16. doi: 10.6004/jnccn.2009.0082. [DOI] [PubMed] [Google Scholar]
- 53.Chee CE, Steensma DP, Wu W, et al. Neither serum ferritin nor the number of red blood cell transfusions affect overall survival in refractory anemia with ringed sideroblasts. Am J Hematol. 2008;83:611–613. doi: 10.1002/ajh.21192. [DOI] [PubMed] [Google Scholar]
- 54.Tefferi A, Mesa RA, Pardanani A, et al. Red blood cell transfusion need at diagnosis adversely affects survival in primary myelofibrosis-increased serum ferritin or transfusion load does not. Am J Hematol. 2009;84:265–267. doi: 10.1002/ajh.21391. [DOI] [PubMed] [Google Scholar]
- 55.Tefferi A, Stone RM. Iron chelation therapy in myelodysplastic syndrome - Cui bono? Leukemia. 2009;23:1373. doi: 10.1038/leu.2009.39. [DOI] [PubMed] [Google Scholar]
- 56.Patnaik M, Lasho T, Finke C, et al. WHO-defined ‘myelodysplastic syndrome with isolated del (5q)’in 88 consecutive patients: survival data, leukemic transformation rates and prevalence of JAK2, MPL and IDH mutations. Leukemia. 2010;24:1283–1289. doi: 10.1038/leu.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trottier BJ, Burns LJ, DeFor TE, et al. Association of iron overload with allogeneic hematopoietic cell transplantation outcomes: a prospective cohort study using R2-MRI-measured liver iron content. Blood. 2013;122:1678–1684. doi: 10.1182/blood-2013-04-499772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Durairaj S, Chew S, Hyslop A, et al. Predicted costs of iron-chelators in myelodysplastic syndromes: a 10-year analysis based on actual prevalence and red cell transfusion rates. Am J Hematol. 2011;86:406–410. doi: 10.1002/ajh.22001. [DOI] [PubMed] [Google Scholar]
- 59.Cermak J, Jonasova A, Vondrakova J, et al. Efficacy and safety of administration of oral iron chelator deferiprone in patients with early myelodysplastic syndrome. Hemoglobin. 2011;35:217–227. doi: 10.3109/03630269.2011.578515. [DOI] [PubMed] [Google Scholar]
- 60.Vichinsky E. Clinical application of deferasirox: practical patient management. Am J Hematol. 2008;83:398–402. doi: 10.1002/ajh.21119. [DOI] [PubMed] [Google Scholar]
- 61.Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429–2440. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- 62.Silverman LR, McKenzie DR, Peterson BL, et al. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin Oncol. 2006;24:3895–3903. doi: 10.1200/JCO.2005.05.4346. [DOI] [PubMed] [Google Scholar]
- 63.Itzykson R, Kosmider O, Renneville A, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31:2428–2436. doi: 10.1200/JCO.2012.47.3314. [DOI] [PubMed] [Google Scholar]
- 64.Garcia-Manero G, Gore SD, Cogle C, et al. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J Clin Oncol. 2011;29:2521–2527. doi: 10.1200/JCO.2010.34.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tefferi A, Lasho TL, Begna KH, et al. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N Engl J Med. 2015;373:908–919. doi: 10.1056/NEJMoa1310523. [DOI] [PubMed] [Google Scholar]
- 66.Tefferi A, Al-Kali A, Begna KH, et al. Imetelstat therapy in refractory anemia with ring sideroblasts with or without thrombocytosis. Blood Cancer J. 2016;6:e405. doi: 10.1038/bcj.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cutler CS, Lee SJ, Greenberg P, et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood. 2004;104:579–585. doi: 10.1182/blood-2004-01-0338. [DOI] [PubMed] [Google Scholar]
- 68.Koreth J, Pidala J, Perez WS, et al. Role of reduced-intensity conditioning allogeneic hematopoietic stem-cell transplantation in older patients with de novo myelodysplastic syndromes: an international collaborative decision analysis. J Clin Oncol. 2013;31:2662–2670. doi: 10.1200/JCO.2012.46.8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patnaik MM, Tefferi A. Refractory anemia with ring sideroblasts and RARS with thrombocytosis. Am J Hematol. 2015;90:549–559. doi: 10.1002/ajh.24038. [DOI] [PubMed] [Google Scholar]
- 70.Patnaik MM, Lasho TL, Finke CM, et al. Predictors of survival in refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) and the role of next-generation sequencing. Am J Hematol. 2016;91:492–498. doi: 10.1002/ajh.24332. [DOI] [PubMed] [Google Scholar]
- 71.Jeromin S, Haferlach T, Weissmann S, et al. Refractory anemia with ring sideroblasts and marked thrombocytosis cases harbor mutations in SF3B1 or other spliceosome genes accompanied by JAK2V617F and ASXL1 mutations. Haematologica. 2015;100:e125–127. doi: 10.3324/haematol.2014.119032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Patnaik MM, Tefferi A. Chronic Myelomonocytic Leukemia: Focus on Clinical Practice. Mayo Clinic proceedings. 2016;91:259–272. doi: 10.1016/j.mayocp.2015.11.011. [DOI] [PubMed] [Google Scholar]
- 73.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391–2405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huls G, Mulder AB, Rosati S, et al. Efficacy of single-agent lenalidomide in patients with JAK2 (V617F) mutated refractory anemia with ring sideroblasts and thrombocytosis. Blood. 2010;116:180–182. doi: 10.1182/blood-2010-01-263087. [DOI] [PubMed] [Google Scholar]
- 75.Michiels JJ, Berneman Z, Schroyens W, et al. The paradox of platelet activation and impaired function: platelet-von Willebrand factor interactions, and the etiology of thrombotic and hemorrhagic manifestations in essential thrombocythemia and polycythemia vera. Semin Thromb Hemost. 2006;32:589–604. doi: 10.1055/s-2006-949664. [DOI] [PubMed] [Google Scholar]
- 76.Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2015 update on diagnosis, risk-stratification and management. Am J Hematol. 2015;90:162–173. doi: 10.1002/ajh.23895. [DOI] [PubMed] [Google Scholar]
- 77.Alvarez-Larran A, Cervantes F, Pereira A, et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood. 2010;116:1205–1210. doi: 10.1182/blood-2010-01-263319. quiz 1387. [DOI] [PubMed] [Google Scholar]
- 78.Pascale S, Petrucci G, Dragani A, et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood. 2012;119:3595–3603. doi: 10.1182/blood-2011-06-359224. [DOI] [PubMed] [Google Scholar]
- 79.Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med. 1995;332:1132–1136. doi: 10.1056/NEJM199504273321704. [DOI] [PubMed] [Google Scholar]
- 80.Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. 2008;112:3065–3072. doi: 10.1182/blood-2008-03-143537. [DOI] [PubMed] [Google Scholar]
- 81.Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol. 2009;27:5418–5424. doi: 10.1200/JCO.2009.23.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shvidel L, Sigler E, Haran M, et al. Busulphan is safe and efficient treatment in elderly patients with essential thrombocythemia. Leukemia. 2007;21:2071–2072. doi: 10.1038/sj.leu.2404743. [DOI] [PubMed] [Google Scholar]
- 83.Sharma P, Shinde SS, Damlaj M, et al. Allogeneic hematopoietic stem cell transplant in adult patients with myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN) overlap syndromes. Leuk Lymphoma. 2016:1–10. doi: 10.1080/10428194.2016.1217529. [DOI] [PubMed] [Google Scholar]