

Abstract
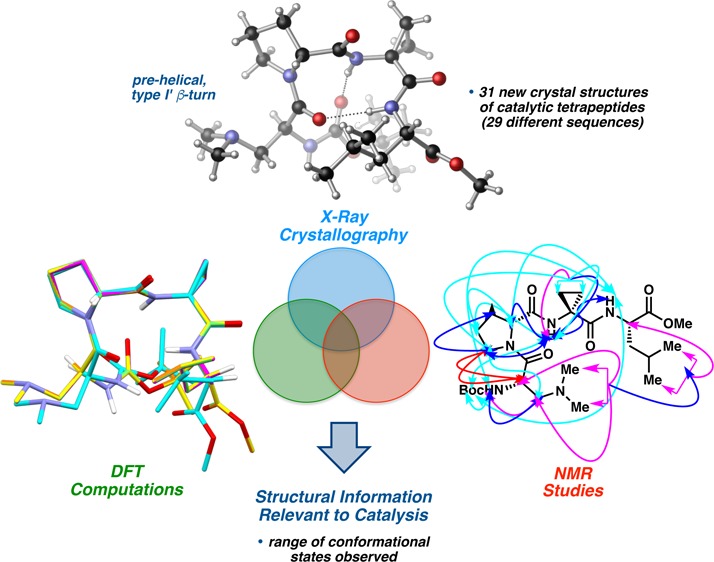
X-ray crystallography has been applied to the structural analysis of a series of tetrapeptides that were previously assessed for catalytic activity in an atroposelective bromination reaction. Common to the series is a central Pro-Xaa sequence, where Pro is either l- or d-proline, which was chosen to favor nucleation of canonical β-turn secondary structures. Crystallographic analysis of 35 different peptide sequences revealed a range of conformational states. The observed differences appear not only in cases where the Pro-Xaa loop-region is altered, but also when seemingly subtle alterations to the flanking residues are introduced. In many instances, distinct conformers of the same sequence were observed, either as symmetry-independent molecules within the same unit cell or as polymorphs. Computational studies using DFT provided additional insight into the analysis of solid-state structural features. Select X-ray crystal structures were compared to the corresponding solution structures derived from measured proton chemical shifts, 3J-values, and 1H–1H-NOESY contacts. These findings imply that the conformational space available to simple peptide-based catalysts is more diverse than precedent might suggest. The direct observation of multiple ground state conformations for peptides of this family, as well as the dynamic processes associated with conformational equilibria, underscore not only the challenge of designing peptide-based catalysts, but also the difficulty in predicting their accessible transition states. These findings implicate the advantages of low-barrier interconversions between conformations of peptide-based catalysts for multistep, enantioselective reactions.
Introduction
Nature has evolved the capacity to catalyze chemical reactions with remarkable rate acceleration and selectivity. The catalytic efficacy of enzymes stems, in part, from their well defined, folded structures. Within the active site, catalytic residues are precisely oriented in space to accommodate well orchestrated, and often highly stabilized, transition states. Thus, a significant kinetic advantage is provided by active site preorganization, which itself is enforced and buttressed by the folded structure of the protein.1 An ongoing goal of structural biology has been to develop a means to predict the folding patterns of proteins based solely on their primary amino acid sequences.2 Endeavors to this end have produced important advances in the field, including analyses of structural trends from the protein data bank.3 These studies have not only increased our collective understanding of protein secondary structures, but they have also provided a platform for the design of other peptidic systems, such as synthetic foldamers4 and molecular devices.5
One interdisciplinary approach in the field of asymmetric catalysis for organic synthesis involves the design of minimal peptides that aim to capture key features of enzymatic active sites within a substantially simplified chiral environment. We,6 and others,7 have pursued this goal in recent years, which has led to the development of a variety of peptide-based catalysts that mediate asymmetric reactions. In general, catalysts of this type have been identified by implementation of either high-throughput screening8 or hypothesis-driven design.9 Whereas on-bead screening has delivered highly effective catalyst sequences that may not have been predicted on the basis of structural attributes, efforts to design peptide-based catalysts rationally have largely focused on sequences that are predisposed to certain secondary structures. The β-turn motif10 has proven particularly fruitful in this regard. Inspired by comprehensive analyses of trends from the protein data bank,3,10 as well as pioneering studies of low-molecular-weight peptides in organic solution,11 we sought to take advantage of predictable β-turn geometry in our catalyst design. In many cases, the tendency of certain β-turn-containing peptides to adopt hairpin structures provides further conformational support through an additional interstrand hydrogen bond (H-bond).12 This approach allows for the possibility of positioning catalytically active amino acid side-chains in close proximity to other functional groups, such as backbone amides, that might also interact with substrates through H-bonding.13Figure 1 highlights three representative asymmetric reactions developed in our laboratory that are catalyzed by such β-turn-containing peptides.14 In each case, experimental data support the designation of a β-turn secondary structure.
Figure 1.
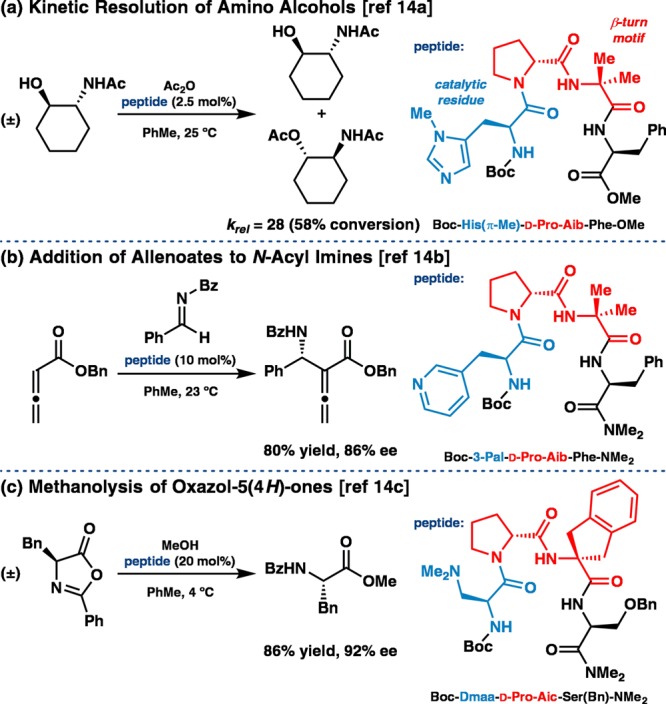
Examples of asymmetric reactions catalyzed by β-turn-containing peptides, including (a) a kinetic resolution of amino alcohols,14a (b) an addition of allenoates to N-acyl imines,14b and (c) a methanolytic dynamic kinetic resolution of oxazol-5(4H)-ones.14c
The potential homology among substructure elements in proteins and much smaller peptide-based catalysts has consistently influenced our studies. For context in terms of structure,15 it has been estimated that up to 25% of all residues in proteins are involved in β-turns.3,10 Accordingly, this scaffold has been studied in numerous contexts spanning biology and chemistry.16 In fact, a major focus in the field of peptidomimetics has been to develop β-turn mimics, or compounds that exhibit turn-like conformations but whose biological functions are modulated in some way.17 β-Turns have been historically classified according to the ϕ and ψ dihedral angles of the loop-region (ϕ,ψ(i+1) and ϕ,ψ(i+2), Figure 2a).3,10 The most common β-turn motifs in proteins are types I and II, though the so-called “mirror image” turns, types I′ and II′, predominate in β-hairpin substructures (Figure 2b).3 β-Turns of types I/I′ are related to types II/II′ by a plane-flip of the loop-region amide, a relatively low-barrier process that is known to interconvert turn-types in proteins.18 Among all of the common turn-types, the possibility exists for a ten-membered ring H-bond between N–H(i+3) and O(i). While the folding of a peptide into a β-turn is a multifaceted process driven in part by local torsional preferences, interstrand H-bonding can offer additional stabilization of the turn.3,10,11 This is especially apparent in studies of β-hairpins, wherein the additional 14-membered ring H-bond between N–H(i) and O(i+3) reinforces close interstrand distances, often less than 7 Å between the α-carbons of the i and i+3 residues, and perpetuate β-sheet-type structures in proteins.12,19
Figure 2.

(a) Pertinent features of a β-turn-containing peptide. (b) Canonical β-turn-types most frequently observed in protein crystal structures.3 (c) The work of Gellman and co-workers showed that heterochiral β-turn sequences are conformationally more apt to adopt β-hairpin structures relative to homochiral sequences.11a
All of the β-turn-containing peptide catalysts shown in Figure 1, and many others not depicted,20 possess a two-residue Pro-Xaa loop-region, where Pro is d- or l-proline, and Xaa is a locally achiral, α,α-disubstituted amino acid. Our initial decision to explore Pro-containing catalysts was built upon the established propensity of Pro to restrict the conformational space available to the peptide by virtue of its pyrrolidine ring.21 In fact, Pro is the most frequent residue to occur at the i+1 position of type I and II β-turns in proteins.3 The potential for control of conformation through defined stereochemical alteration of the i+1 Pro residue, as explored by Gellman and others, has proven particularly powerful in catalyst design.11a,22 For example, replacement of l-Pro with d-Pro in an otherwise l-homochiral turn sequence often changes not only the turn sense from type II to type II′, but also shifts the conformational equilibrium to favor the β-hairpin form (Figure 2c). The hairpin conformer is not typically favored in homochiral oligopeptides, which instead tend to equilibrate between β- and γ-turn forms.11a,23 In addition, Toniolo and co-workers have thoroughly investigated the use of achiral, α,α-disubstituted amino acids in short peptide sequences.24 These residues are more conformationally restricted than glycine, the most frequent residue found at the i+2 position of β-turns in proteins,3 and they have been found to bias Pro-Xaa β-turns into the type II/II′ regime when incorporated at the i+2 position.25
Application of these important principles allowed the development of highly enantioselective peptide-based catalysts biased toward type II (Pro-Xaa) or type II′ (d-Pro-Xaa) β-turns with seemingly limited conformational flexibility. The large number of effective catalysts that have been developed within this motif has also raised questions regarding the possibly “privileged” nature of the Pro-Xaa turn sequence for asymmetric catalysis (Figure 1).14,20,26 Additional catalysts based upon this structural motif are emerging with some regularity.27 Even so, our resolution in catalyst design has remained relatively low, as the connection between subtle changes in peptide sequence and their effect on enantioselectivity has rarely been clear. Moreover, as we will detail below, the conformational mobility of intentionally biased sequence space has also proven greater than our initial intuition suggested.
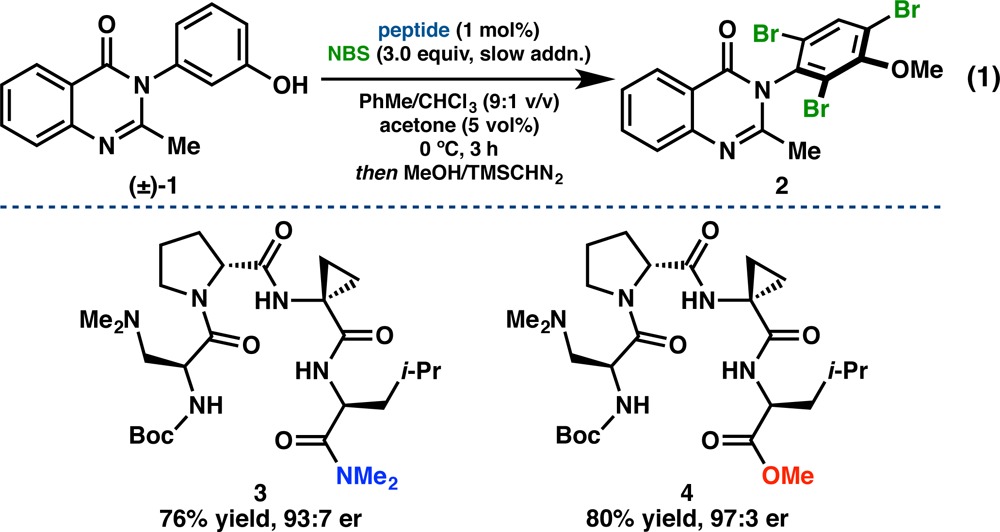
For context in terms of function, we recently developed a catalytic, atroposelective bromination of 3-arylquinazolin-4(3H)-ones (1), providing access to highly enantioenriched tribromides (2, eq 1).28a Peptide 3, which possesses a tertiary amine-containing l-β-dimethylaminoalanine (Dmaa) residue at the i-position and a d-Pro-Acpc turn-motif, emerged as the lead catalyst for this transformation from an initial library of 24 sequences biased toward presumed type II′ β-turns. Upon expanding our catalyst library to 54 total peptides, we discovered an improved catalyst (4) that differed from 3 only in the C-terminal cap, and yet it delivered 2 in 97:3 er at only 1 mol % loading (eq 1).28b Motivated by a desire to better understand the delicate interplay between peptide structure and enantioselectivity in this reaction, we sought to study structural aspects of our bromination catalyst library using X-ray crystallography and NMR spectroscopy. It is this library that most comprehensively revealed the structural heterogeneity available to peptide-based catalysts of this type.
 |
1 |
We report herein a detailed structural study of 35 Pro-Xaa-containing peptides, each of which proved suitable for analysis in the solid state using single crystal X-ray diffraction. Crystallographic data on such catalytic tetrapeptides have been limited in previous years. Of the sequences examined, X-ray crystal structures of only six have been reported previously (3, 4, 34–37, Chart 1).9,28,29 The 29 newly reported peptides (5–33, Chart 1) represent a substantial increase in the number of small molecule, β-turn-containing crystal structures in the Cambridge Structural Database (CSD).30 Furthermore, we have observed an unexpected frequency of polymorphism and symmetry-independent conformational isomerism. Thus, the total number of peptide conformational states we describe sums to 53. In Chart 1, we denote in red the number of discrete states observed for a given sequence. The combination of X-ray crystallography, DFT computational studies, and solution-phase NMR spectroscopy has also helped us to understand better the conformational effects associated with changes to the primary sequence. Of particular interest were the effects of various i+2 substitutions on secondary structure, since changes to this particular residue often perturb the enantioselectivity significantly in peptide-catalyzed reactions. In general, a wider range of ground state structures was observed than might have been expected based on standard design principles.3,11a Focusing on ground state structural issues alone, the conformational diversity that may be reasonably populated within the generic β-turn framework is striking.31 Conformations that span the canonical β-turn classifications, and even populate their boundaries, have been observed. These findings are consistent with multiple accessible states, which intersects with ongoing studies regarding the importance of catalyst dynamics.32 Critically, the results of this study offer opportunities to expand our understanding of the connection between catalyst structure and enantioselectivity outcomes. As is well appreciated, these issues present tremendous challenges for combined experimental and computational approaches that, together, promise to advance our understanding of the structure–function continuum for catalytic reactions.33
Chart 1. Peptides Analyzed by X-ray Crystallography.

Results and Discussion
Conformational Polymorphs and Pseudopolymorphs
Crystallographic Analysis of Peptide 3
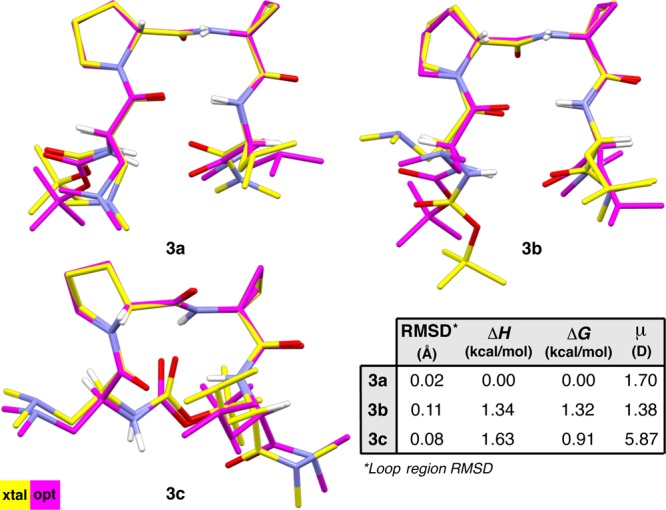
The observation of three distinct conformations of peptide 3 in the solid state (Figure 3) stimulated an aggressive inquiry into the potential generality of the phenomenon. Conformers 3a and 3b were identified within the same asymmetric unit,28 and both are characterized as type II′ β-hairpins on the basis of their loop-region dihedrals, ϕ,ψ(i+1) and ϕ,ψ(i+2), and the presence of N–H(i+3)···O(i) and N–H(i)···O(i+3) intramolecular H-bonds. Conformer 3c, on the other hand, was identified within a polymorphic crystal grown under nearly identical conditions, and yet its structure most closely resembles a type I′ β-turn when considering the ϕ and ψ dihedrals of the i+1 and i+2 positions and the N–H(i+3)···O(i) H-bond. Another ten-membered ring N–H(i+2)···O(i–1) H-bond delineates a second β-turn within the same tetrameric framework. The i and i+1 residues comprise the loop-region of this N-terminal β-turn, which is characterized by type II ϕ and ψ dihedrals. This sequential, II/I′ double β-turn motif provides a nascent 310′-helical structure to 3c, although the sequence is too short to constitute a true helix (Tables S4.04 vs S4.05).34 Nonetheless, the observation of 3c was initially unexpected for a d-Pro-Xaa-containing sequence, which is often presumed to nucleate a type II′ β-hairpin geometry.3,11 A structural overlay of the conformers of 3 highlights the differences between the prehelical 3c and 3a,b (Figure 3).
Figure 3.
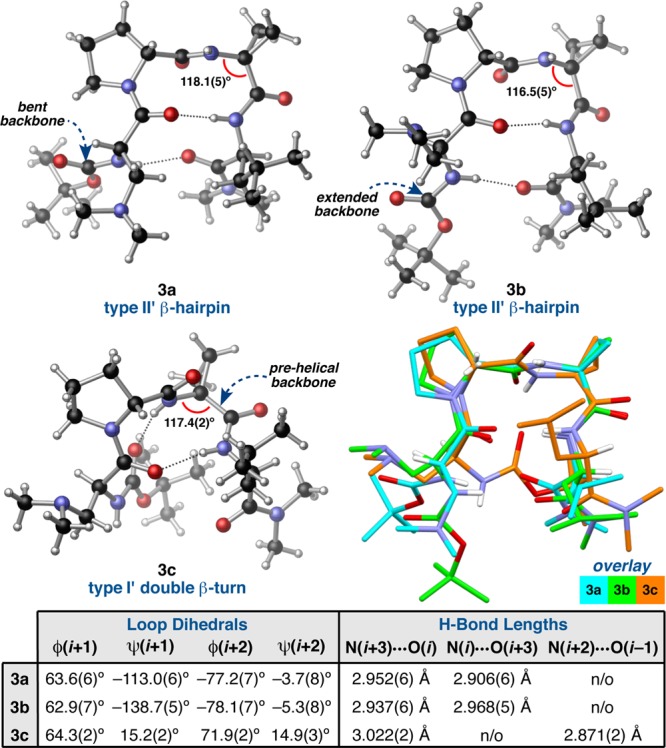
Three distinct conformations of peptide 3 with relevant crystallographic measurements. A structural overlay highlights the differences among the conformers (loop RMSD (3a/3b) = 0.14 Å, loop RMSD (3a/3c) = 0.74 Å).
Although 3a and 3b broadly classify into the same structural motif, there are a number of differences between the two structures. For instance, the ψ(i+1) value of 3a is nearly 26° contracted relative to that of 3b. This is possibly a manifestation of the incipient N–H(i+2)···O(i) intramolecular H-bond in 3a. This type of H-bond is characteristic of a γ′-turn,23 though the comparatively long N···O length of 3.029(6) Å and the acute N–H···O angle of 96(4)° suggest that the β-turn geometry is favored. Perhaps the most prominent difference, aside from the orientation of the i and i+3 side-chains, is the degree of backbone bending at the C- and N-terminal residues. The termini of conformer 3a bend approximately 66° from the plane defined by the α-carbons of the i, i+1, i+2, and i+3 residues, while that of 3b bends only 41° (Table S4.12 and associated figure). Thus, conformer 3a is more bent than 3b, resulting in a more compact structure. Another feature that differentiates these two conformers is the degree of twisting, as measured by the virtual dihedral angle (ϖ) defined by the four α-carbons (Table S4.14 and associated figure).35 Conformers 3a and 3b twist in opposite directions, with ϖ measuring 9.4° and −21.3°, respectively, suggesting that both twist-senses are accessible to peptide 3. These details provide insight into the range of conformations available to short peptides of the same overall structural class.
A closer analysis of conformer 3c also reveals some structural nuances (Figure 3). Though ϕ,ψ(i+1) and ϕ,ψ(i+2) classify the central turn-motif as a type I′ turn, the dihedral values are distorted from their canonical values.3 For instance, ψ(i+1) is nearly 15° contracted, while ϕ(i+2) and ψ(i+2) are wider than predicted by 19° and 15°, respectively. It is possible that these deviations may be caused by the N–H(i+2)···O(i–1) β-turn H-bond, which is significantly shorter than the central N–H(i+3)···O(i) H-bond of 3c (as well as both of the H-bonds in 3a and 3b). Formation of this particularly favorable H-bond likely counterbalances the torsional distortions in the backbone. The i and i+1 residues define the loop-region of the N-terminal β-turn. In this case, the N–H(i+3)···O(i) H-bond likely causes some deviations from the ideal type II torsional potentials. The second residue of a type II β-turn typically possesses ϕ values of 80°. However, this value is contracted by nearly 16° in 3c, a direct consequence of the pyrrolidine ring of d-Pro, which typically locks ϕ at 60 ± 10°. The pronounced twist of 3c (ϖ = −71.5°) reflects the prehelical nature of the sequential double β-turn motif (Table S4.16).
In order to gain insight into the degree to which crystal packing forces might affect the geometries and to assess the energy differences (ΔH° and ΔG°) between the conformers, we optimized the crystallographic coordinates of 3a–c using DFT (see the Supporting Information for details).36 We found that the loop-regions of the DFT-optimized structures are nearly coincident with their respective X-ray crystal structures. However, the computed structures showed deviations in the i and i+3 residues, especially with respect to the side-chains (Figure 4). In the case of 3b, geometry optimization causes the backbone to bend such that it more closely resembles that of 3a. In fact, the optimized structures of 3a and 3b overlay with a backbone RMSD of only 0.24 Å; the only major differences occur in the side-chains. Optimization of conformer 3c produces a structure in which the loop-region dihedrals of both β-turns have approached their canonical values, and the N-terminal carbamate has torqued about ϕ(i) to lengthen the N–H(i+2)···O(i–1) β-turn H-bond. This result is consistent with our hypothesis that formation of the N–H(i+2)···O(i–1) H-bond is coupled to the conformational distortion of the backbone. It is notable that 3c does not converge to a type II′ β-hairpin, which solidifies its characterization as a conformational polymorph of 3a,b.37 In terms of relative energies, conformer 3a was found to be the lowest-energy conformer, while the prehelical 3c was higher by only 0.91 kcal/mol. Conformer 3c has the highest relative enthalpy, which suggests that there is some entropic benefit to occupying the prehelical geometry. It also has a significantly larger dipole moment (μ) than either of the β-hairpin conformers, suggesting that its population might be sensitive to solvent. Surprisingly, conformer 3b was 0.41 kcal/mol higher in energy than 3c, despite the tight overlay between 3a and 3b. This is possibly due to the orientation of the i and i+3 side-chains in 3b.
Figure 4.
Three solid-state conformations of peptide 3 were optimized using DFT at the M06-2X/6-311++G(2d,3p)//B3LYP/6-31+G(d,p) level of theory.
NMR Analysis of Peptide 3
We next sought to compare the solution conformational profile of 3 to its observed solid-state structures. The solution structure of peptide 3 has been studied extensively in our laboratory. For example, we have recently reported an NMR-derived structure of 3 that was computed using a NOE-restrained simulated annealing/DFT protocol.28b This treatment produced a solution structure of 3 that was a hybrid of its solid-state conformers, 3a–c. A more comprehensive analysis presented herein supports these findings and is predicated on a NMR-based, three-pronged approach: (1) three-bond couplings between the amide- and Cα-protons (3JNH–Hα) of the i and i+3 residues were extracted from 1H NMR spectra and used to calculate ϕ(i) and ϕ(i+3) (Table S5.40 and Figure S5.15);38 (2) the relative chemical shifts and peak widths of the NH(i), NH(i+2), and NH(i+3) signals were compared to assign their H-bonded state (Tables S5.41–S5.43);39 and (3) NOE contacts from two-dimensional 1H–1H-NOESY spectra were analyzed for information about through-space interactions.40 All NMR analyses were performed using benzene-d6 (C6D6) as the solvent in order to simulate the reaction conditions (9:1 PhMe/CHCl3) with fewer residual solvent peaks that could complicate analysis. Comparable enantioselectivities were obtained upon substitution of benzene for toluene in the catalytic bromination of 1 (eq 1).28 Concentrations of 0.01 M with respect to peptide were used, which is 10 times more concentrated than the reaction conditions. However, we previously demonstrated that this concentration is still below the aggregation limit for peptides of this class.
Our investigation of 3 in solution (0.01 M, C6D6, 25 °C) provided data consistent with ϕ(i) = −86° or −154° and ϕ(i+3) = −95° or −145° based on the 3JNH–Hα (Table S5.40). Based on the empirically derived cutoffs, these dihedral values do not preclude any specific secondary structure. Relative to other peptides with similar primary sequences, the NH(Leu) and NH(Dmaa) signals appear at downfield chemical shifts, while that of NH(Acpc) appears at a relatively upfield shift (Table S5.41). These data suggest that NH(Leu) and NH(Dmaa) are both involved in intramolecular H-bonds, while NH(Acpc) is exposed to the solvent. Five especially interesting cross-peaks in the NOESY spectrum of 3 are indicative of the following through-space interactions: NH(Dmaa) ↔ NH(Leu), NH(Acpc) ↔ NH(Leu), NH(Dmaa) ↔ NMetrans(Leu), NH(Leu) ↔ α(d-Pro), and β(Dmaa) ↔ β(Leu). A particularly strong NH(Acpc) ↔ α(d-Pro) NOE was also observed (Figure S5.01). These interactions, in conjunction with the proposed intramolecular H-bonds, point to a type II′ β-hairpin structure in solution, much like 3a/3b. However, the presence of NH(Acpc) ↔ δ(d-Pro) and Boc(Dmaa) ↔ β(Acpc) NOE correlations provides evidence that a prehelical conformer much like 3c is populated to a lesser extent in solution. Moreover, the calculated ϕ(i) and ϕ(i+3) dihedrals are also consistent with nonhairpin β-turn and prehelical conformations, further supporting our assertion that 3, and other peptides of this type, may populate multiple conformations in solution.
Crystallographic Analysis of Peptide 16
We also observed three, distinct, solid-state conformations of peptide 16, a homologue of 3 in which the i+2 position is substituted with an Acbc residue (Figure 5). Type II′ β-hairpins 16a and 16b were identified within the same unit cell, while type I′ double β-turn 16c was found in a pseudopolymorphic crystal. Despite the fact that the crystallization samples were prepared under nearly identical conditions, the single crystals that gave rise to 16a,b and 16c are not truly polymorphic due to the presence of 2 equiv of HCl in the unit cell of the former.41 In both 16a and 16b, the Dmaa residues appear to be protonated at the tertiary amine moiety, and the resulting ammonium ions are engaged in intermolecular H-bonds with each of the two chloride counterions. Conformer 16a, however, is also associated with the other chloride via an intermolecular H-bond donated from the N–H(i+2). It is possible that this intermolecular H-bond accounts for the wider ψ(i+1) in 16a, though the sampling of an N–H(i+2)···O(i) H-bond in 16b may also contribute to the difference.
Figure 5.
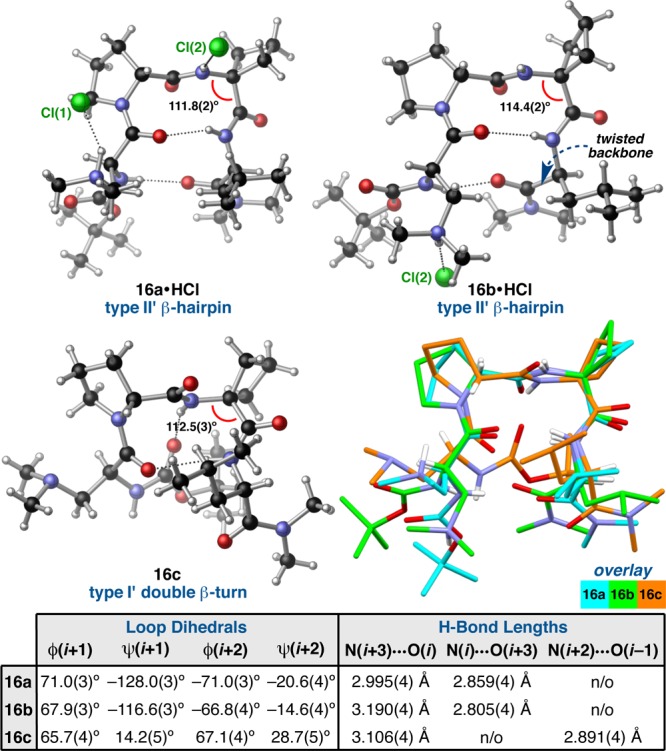
Three distinct conformations of peptide 16 with relevant crystallographic measurements. A structural overlay highlights the differences among the conformers (loop RMSD (16a/16b) = 0.11 Å, loop RMSD (16a/16c) = 0.74 Å).
As with 3a and 3b, type II′ β-hairpins 16a and 16b exhibit significant variation despite sharing the same secondary structural motif. For example, the i+2 main-chain angles (τ) differ substantially between 16a and 16b, with that of the former measuring 111.8(2)° compared to 114.4(2)° in that of the latter (Figure 5). We initially wondered if this difference might be related to the orientation of the cyclobutane ring.42 In 16a, the i+2 cyclobutane ring puckers away from the d-Pro residue, whereas it puckers toward the d-Pro residue in 16b. Structure 16c shares the same pucker direction as 16a and has a similar τ(i+2) of 112.5(3)°. However, the difference in τ(i+2) may also be coupled to the incipient N–H(i+2)···O(i) γ′-turn H-bond in 16b, an orientation that might exacerbate the electrostatic repulsion between the N–H(i+2) and N–H(i+3) σ-bonds, and thereby widen τ(i+2) relative to 16a.43 This hypothesis was further substantiated by DFT optimization of 16a and 16b without the associated HCl equivalents. The optimized structures both exhibit τ(i+2) values >114°, and while the loop-region of 16b remains nearly constant, that of 16a becomes more like 16b and begins to adopt a γ′-turn H-bond (Figure 6). Another distinguishing feature between 16a and 16b is the backbone twist. Much like 3a and 3b, these two conformers twist in opposite directions, with ϖ measuring −7.9° and 17.8°, respectively (Table S4.14). The i and i+3 side-chains of 16a and 16b are also oriented differently, with 16a adopting a more compact arrangement.
Figure 6.
Three solid-state conformations of peptide 16 were optimized using DFT at the M06-2X/6-311++G(2d,3p)//B3LYP/6-31+G(d,p) level of theory.
Much like 3c, conformer 16c is characterized by a prehelical, type I′ β-turn secondary structure that is underdocumented for a d-Pro-Xaa sequence (Figure 5). Many of the structural characteristics of 3c are mirrored in 16c; the two structures overlay with a backbone RMSD of only 0.12 Å. The primary difference between the two structures is the ψ(i+2) value, which measures nearly twice as wide in 16c at 28.7(5)°. It is possible that this difference is related to the torquing of the cyclobutane ring away from the central β-turn. The 23° deviation of ϕ(i+2) from the ideal values may also stem from this torquing effect. These values of ϕ,ψ(i+2) are at the very boundary of what would canonically be considered a type I′ β-turn,3 though ϕ,ψ(i+1) are in accordance with this assignment.
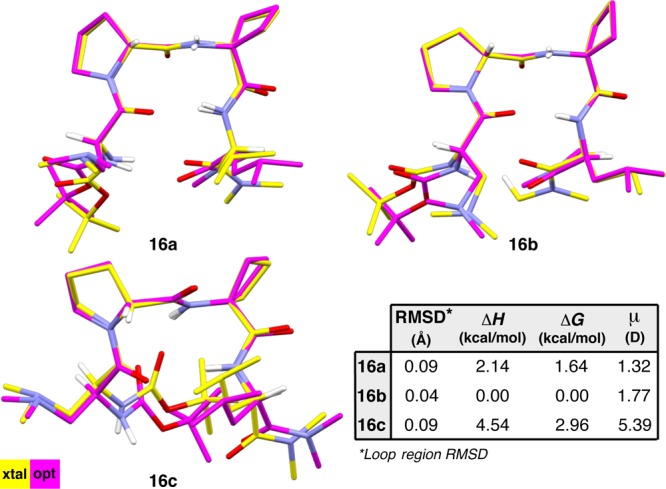
It is interesting to note that the comparisons among 3a–c and 16a–c appear to be analogous, despite the presence of HCl salts for 16a and 16b. While this is certainly a caveat worth considering, we do not expect that protonation of the peptides and association of the chloride counterion necessarily affects the structure in a way that might devalue these comparisons. Wennemers and co-workers recently reported that separate crystal structures of a short peptide and its corresponding TFA salt did not deviate to any significant extent.44 To probe these issues further, we omitted the HCl equivalents and optimized structures 16a and 16b using DFT. The optimized geometries overlaid closely with the corresponding X-ray crystal structures (loop RMSDs < 0.10 Å, Figure 6). Thus, the presence of the HCl appears not to alter the structures significantly. Furthermore, DFT optimization of 16a–c also allowed us to assess the relative energies of the three structures. In this case, conformer 16b was found to be significantly lower in energy than the others. Prehelical 16c is significantly more disfavored relative to the β-hairpin geometries in the context of peptide 16 than it is in peptide 3 (Figure 4). This underscores an influence of the i+2 residue on the conformational energy landscape of these peptides.
NMR Analysis of Peptide 16
Solution NMR data were acquired for peptide 16 to provide insight into its conformational profile (0.01 M, C6D6, 20 °C). In the 1H NMR spectrum of 16, the NH(Dmaa) signal appears as a broad singlet, implying a ϕ(i) dihedral of approximately −30°. It is difficult to draw definitive conclusions based on this ϕ(i), as the broadness of the resonance might point to conformational averaging at this residue. On the other hand, the 3JNH–Hα of NH(Leu) is consistent with ϕ(i+3) values of either −96° or −143° (Table S5.40), which are most in-line with the β-turn or hairpin conformations (i.e., 16a/16b) and are on the upper limit of what might be expected for a prehelical, type I′ β-turn assignment (i.e., 16c). Compared to peptide 3, the chemical shifts of the NH(Leu) and NH(Dmaa) signals are upfield-shifted, while that of the NH(Acbc) occurs significantly further downfield (Table S5.41). Taken together, these relative chemical shift data provide evidence for a conformation in which: (1) NH(Acbc) is involved in a strong, intramolecular H-bond; (2) NH(Dmaa) is mostly solvent-exposed; and (3) NH(Leu) samples multiple H-bonded states on the NMR time-scale. In terms of through-space interactions, a strong NH(Acbc) ↔ α(d-Pro) NOE is present in the NOESY spectrum of 16. This correlation typically supports type II′ loop dihedrals, much like 16a/16b. However, the absence of cross-strand NOEs perhaps intimates that the structure is in-flux between multiple conformers (Figure S5.05). These cumulative data provide evidence that supports a γ′-turn that is perhaps in equilibrium with the nonhairpin β-turn form. Conformer 16b shows some characteristics of an incipient γ′-turn conformation in the solid state (Figure 5).
Packing Polymorphs
Crystallographic Analysis of Peptide 4
While peptides 3 and 16 exhibit multiple solid-state conformations that are significantly different from one another in their polymorphic or pseudopolymorphic crystal structures, we have also observed cases in which the degree of structural variation is subtler. For example, two polymorphic crystal structures of peptide 4 give rise to five distinct states, all of which are similar to one another (Figure 7). The initial crystal structure provided 4a, a type I′ double β-turn similar to both 3c and 16c. Upon recrystallization of peptide 4 under nearly identical conditions, a polymorphic unit cell was found to contain four symmetry-independent molecules (4b–e) that adopt secondary structures similar to that of 4a. As such, crystal structures 4b–e may be described as a packing polymorph of 4a.37 An overlay of 4a–e reveals that the five states are remarkably consistent across the i+1 and i+2 residues, with loop-region RMSDs (relative to 4a) of <0.03 Å (Figure 7a). The loop dihedral with the most variation is ψ(i+1), which varies from 12.6(6)° to 21.8(6)° across the set. Unlike the loop-regions, the termini of 4a–e vary more significantly. The C-terminus, in particular, shows a range of conformations in both the backbone and the Leu side-chain. The principle metric of this variation is ϕ(i+3),15 which ranges from −65.1(6)° in 4c to −135.7(4)° in 4e with an average of −101.4 ± 27.2° (Table S4.03). The high standard deviation of the mean ϕ(i+3) value is a reflection of the conformational heterogeneity at the C-terminus. We note that ψ(i+3) also varies across 4a–e, but to a lesser extent, ranging from 150.4(4)° in 4b to 178.3(4)° in 4e. The variation observed in the i+3 side-chain appears to be coupled to the backbone deviations, as the side-chain dihedral χ1(i+3) remains fairly constant across 4a–d,15 only ranging from −58.3(5)° in 4d to −67.2(5)° in 4c (Table S4.32). The exception is conformer 4e, in which χ1(i+3) measures 57.3(5)°. Positive gauche (g+) values of χ1 are quite infrequent at the i+3 position of these β-turn-containing tetrapeptides, since side-chains oriented in this way incur two destabilizing gauche interactions from the main-chain.
Figure 7.
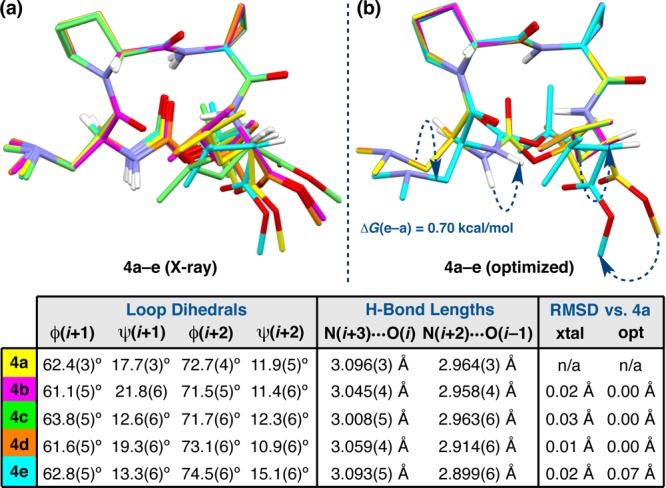
(a) Overlay of the five solid-state conformations of peptide 4 that were identified within two polymorphic unit cells. (b) DFT Optimization of 4a–e at the M06-2X/6-311++G(2d,3p)//B3LYP/6-31+G(d,p) level of theory led to two distinct families of conformers, shown overlaid. The optimized structures of 4a–d were degenerate, while 4e was found to be 0.70 kcal/mol higher energy than 4a–d. Loop RMSDs are reported in the table.
Given the subtlety of the structural differences among 4a–e, a pertinent question is whether each might converge to the same conformation upon minimization. As such, we optimized the crystallographic geometries of 4a–e using DFT and found that 4a–d converge to a single structure (Figure 7b). The optimized structure of 4e, however, is distinct, with a small, yet nonzero, loop-region RMSD of 0.07 Å relative to 4a (Figure S6.02) and some significant structural deviations in the termini (Figure 7b, arrows). As we observed with the DFT-optimized structures of 3c and 16c above, the N-terminal carbamates of 4a–e torque about ϕ(i), which orients the N–H(i)s toward O(i+3) on the opposite strands; this may be coupled to the lengthening of the N–H(i+2)···O(i–1) H-bonds and the minor differences in the Dmaa side-chain of 4e relative to 4a–d. This effect is more prominent in 4e, wherein N–H(i) is in close enough range for a bona fide β-hairpin H-bond to O(i+3). Minor differences in the Dmaa side-chain of 4e relative to 4a–d may be related to this pronounced torquing. The values of ϕ(i+3) and χ1(i+3) observed in the crystal structure of 4e are largely maintained after optimization, measuring −128.0° and 56.4°, respectively. The unusual gauche χ1(i+3) of 4e may be the source of the difference between the optimized structures of 4e and 4a–d, since the ϕ(i+3) dihedrals of 4a–d were quite disparate, and yet they still converged during optimization. Conformer 4e was found to be 0.70 kcal/mol higher in energy than 4a–d, which suggests that its distorted β-hairpin geometry may be accessible to some extent at ambient temperature (Table S6.10). This energy difference is consistent with a gauche interaction.
NMR Analysis of Peptide 4
We also analyzed the structure of peptide 4 in solution (0.01 M, C6D6, 20 °C). Using the extracted 3JNH–Hα values, we calculated ϕ(i) and ϕ(i+3) to be −30° and either −89° or −151°, respectively (Table S5.40). Based on these criteria, no specific conformations may be excluded, but the data are most consistent with either a prehelical or nonhairpin β-turn. The relative chemical shifts of the NH(Leu), NH(Acpc), and NH(Dmaa) resonances point to a scenario in which all three are involved in intramolecular H-bonds (Table S5.41 and S5.42). However, the very broad peak width of the NH(Dmaa) signal suggests that the carbamate is experiencing different H-bonded states on the NMR time-scale (Table S5.43). The presence of NH(Dmaa) ↔ NH(Leu), Boc(Dmaa) ↔ NH(Leu), and Boc(Dmaa) ↔ NH(Acpc) NOE contacts, as well as a particularly weak NH(Acpc) ↔ α(d-Pro) correlation, is consistent with a prehelical, type I′ β-turn as the predominant solution conformation (Figure S5.02). In keeping with the downfield-shifted NH(Dmaa) signal, a type I′ β-hairpin might also be sampled to some extent.28b This assignment is in agreement with the turn-type observed for all five solid-state conformers 4a–e (Figure 7).
Symmetry-Independent Conformers
Among the 38 different crystal structures examined in this study, we have observed multiple, distinct peptide molecules within the same unit cell in 13 instances, representing 34% of our peptide structure library (Table 1). The observation of symmetry-independent molecules is relatively uncommon among crystal structures of small organic molecules. According to Steiner’s 2000 study of the CSD, 73.3% of organic crystal structures have only one formula unit per unit cell, while 15.9% have fewer and 10.8% have more than one molecule per unit cell.45 Broken down further into compound classes, it was found that 81.6% of peptidic crystal structures have only one molecule per unit cell, while 5.2% have fewer and 13.2% have more than one molecule in the unit cell. Compared to organic molecules on the whole, peptides appear slightly more prone to this phenomenon. For example, symmetry-independent molecules have been observed in crystal structures of helical peptides acquired by Gellman and co-workers,46 as well as in examples of β-hairpins reported by Balaram.19 It has been proposed that flexible molecules, such as peptides, may crystallize with more than one molecule in the unit cell to achieve better packing than would be possible with only one, “awkwardly shaped” formula unit.37 Another likely possibility is aggregation through N–H···O intermolecular H-bonding.
Table 1. Symmetry-Independent Molecules Observed.
In light of these findings, we were curious about the degree to which the symmetry-independent molecules we observed differ from one another and whether or not they could be considered conformers.47 Cruz-Cabeza and Bernstein studied the correlation between crystallographic and DFT-optimized RMSDs in pairs of conformers and developed a cutoff all-atom RMSD value of 0.375 Å, above which polymorphic or symmetry-independent structures are often true conformers.37aTable 1 shows the 13 cases in which we observed symmetry-independent molecules and the degree to which these peptides differ from one another using three different RMSD metrics (loop, backbone, and all-atom; Tables S4.49 and S4.50). In general, loop RMSD values were found to be lower than both backbone and all-atom RMSDs between the pairs of symmetry-independent molecules. Given that all of these peptides contain Pro-Xaa β-turn motifs, it is unsurprising that the well-defined loop-regions are more coincident than the peripheral residues. Backbone RMSDs are lower than the corresponding all-atom values for similar reasons. Eight of the symmetry-independent molecules qualify as distinct conformers based on their all-atom RMSD values. This includes the cases 3a,b and 16a,b discussed previously, which have all-atom RMSDs of 1.45 and 1.10 Å, respectively. Similarly, structures 4b–e are symmetry-independent, yet computation reveals that only 4e may be considered a distinct conformer. Two of the remaining five instances, those of peptides 7 and 19, are discussed below as illustrative examples of symmetry-independent conformers.
Symmetry-Independent Conformers with Different Turn-Motifs
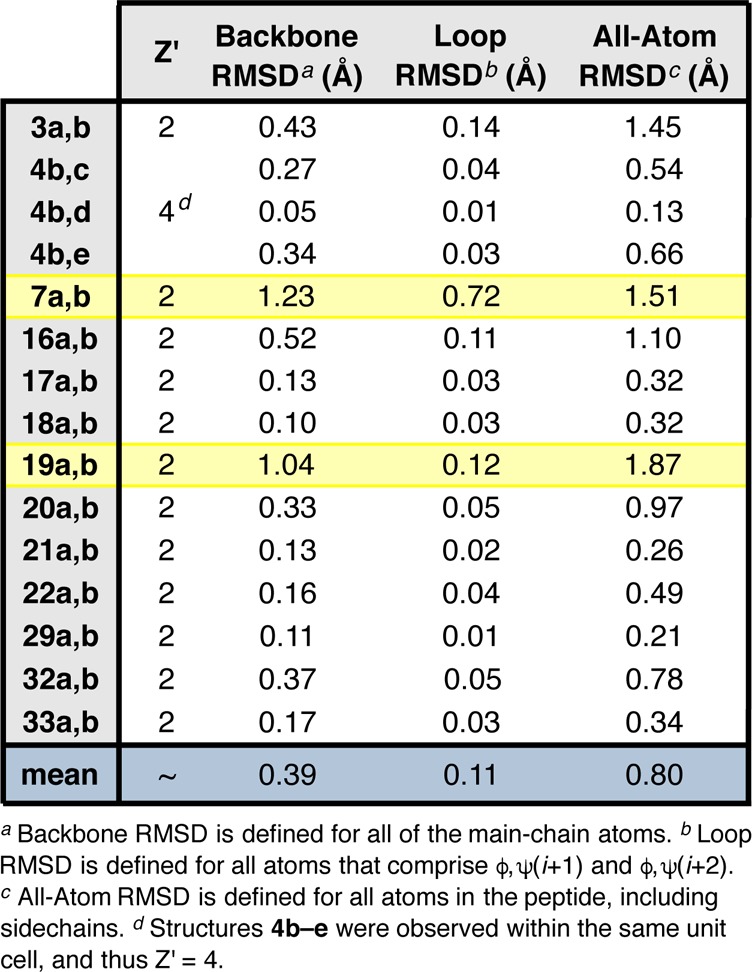
The symmetry-independent molecules of peptide 7 show significant deviations from one another according to all three RMSD metrics (Table 1). These differences are sufficient to warrant classification as discrete conformers. Within the unit cell, conformers 7a and 7b interact with one another via reciprocal N–H(i)···NMe2(i′) intermolecular H-bonds (Figure 8a). Analyzing the conformers separately, it is clear that 7a and 7b are not only conformers of one another, but they are different β-turn-types all together. This observation is supported by the high loop RMSD of 0.72 Å between the two conformers. The loop-region ϕ and ψ dihedrals of conformer 7a are quite close to the canonical values of a type II′ β-turn, while conformer 7b exhibits the loop dihedrals and intramolecular H-bonding network characteristic of the prehelical, type I′ double β-turn we have observed in 3c, 4a–e, and 16c (Figure 8b). Analogously, the deviations from the canonical type I′ β-turn observed in 7b are attributed to the presence of the additional N–H(i+2)···O(i–1) β-turn H-bond that gives the conformer its prehelical structure. However, this H-bond is much longer and more acute than previously observed, measuring 3.256(5) Å from N(i+2)-to-O(i–1) and 127(4)° in the N–H···O angle. This nonideal H-bond may be coupled to the pronounced pyramidalization of the i+2 amide nitrogen-atom; N(i+2) deviates 0.14 Å from the plane defined by the N-substituents, which is quite significant compared to the 0.48 Å deviation in the corresponding tertiary amine moiety of the Dmaa residue.48 On the other hand, conformer 7a does not exist as a β-hairpin in the solid state, even though this might be expected based on its near-canonical type II′ turn-motif and stereochemical array. It is possible that the i+3 Gly residue may provide less of a bias in favor of the β-hairpin than an α-substituted residue during crystal seeding.3 Perhaps a more likely cause of the distortions in both 7a and 7b is the involvement of N–H(i) of each in intermolecular H-bonds (Figure 8a). In other words, systematic effects may favor the observed conformations to maximize packing efficiency at the expense of conformational strain.37
Figure 8.
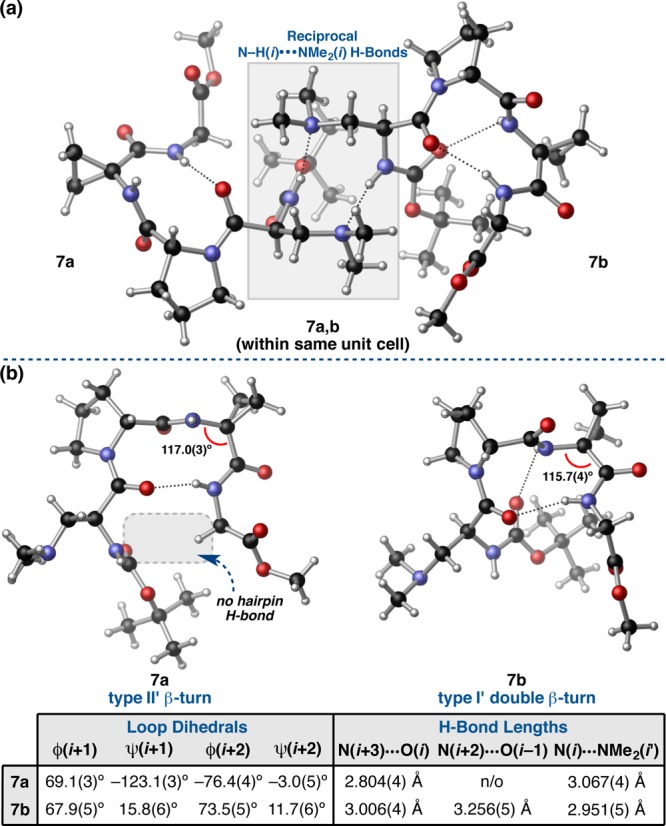
(a) The two symmetry-independent conformers of peptide 7 within the unit cell. (b) Analysis of the conformers revealed two different turn-motifs, a type II′ β-turn in 7a and a type I′ double β-turn in 7b.
DFT optimization of 7a and 7b provided gas-phase geometries that coincided well with the corresponding crystal structures, especially in the loop-regions (Figure S6.03). The optimized structure of 7a differs substantially from the crystallographic coordinates only at the i-position. The ϕ,ψ(i) values are, nonetheless, still atypical compared to the rest of the structure library. Optimization of 7b results in the planarization of N(i+2) and a concomitant shortening and widening of the N–H(i+2)···O(i–1) H-bond to 3.125 Å (N···O) and 167° (N–H···O), respectively. These results lend credence to the hypothesis that the intermolecular interactions between 7a and 7b distort the solid-state geometries. Conformer 7b was found to be 4.22 kcal/mol lower in energy than 7a, reflecting both the torsional strain in the peripheral residues and the absence of a second intramolecular H-bond (Table S6.13).
In order to assess the relevance of these solid-state conformers in solution, peptide 7 was subjected to NMR analysis under the typical conditions (0.01 M, C6D6, 25 °C). The absence of J-coupling in the NH(Dmaa) signal suggests that the time-averaged value of ϕ(i) is approximately −30° (Table S5.40). However, the 3JNH–Hα of the i+3 Gly residue gives rise to ϕ(i+3) dihedrals of either −74° or −166°, values that are most consistent with a nonhairpin or prehelical β-turn. In addition, the downfield-shifted 1H NMR resonances corresponding to NH(Gly) and NH(Acpc) provide evidence that these amides are engaged in intramolecular H-bonds; NH(Gly) most likely donates an NH(i+3)···O(i) β-turn H-bond, though the nature of the H-bond involving NH(Acpc) is more ambiguous (Tables S5.41–S5.43). The relatively upfield chemical shift of the NH(Dmaa) resonance is consistent with a solvent-exposed carbamate that is not involved in an intramolecular H-bond. A number of long-range NOEs are observed in the NOESY spectrum of 7, including Boc(Dmaa) ↔ α(Gly), NH(Dmaa) ↔ NH(Gly), Boc(Dmaa) ↔ β(Acpc), and NH(Gly) ↔ α(d-Pro) correlations (Figure S5.03). Much like peptides 3 and 16, a strong NH(Acpc) ↔ α(d-Pro) NOE provides evidence for type II′ loop dihedrals, although the moderately strong δ(d-Pro) ↔ NH(Acpc) and NH(Dmaa) ↔ NH(Acpc) contacts are typically associated with the prehelical, type I′ β-turn structure. Based on these data, it is likely that peptide 7 samples multiple conformations in solution, perhaps favoring a γ′-turn or a prehelical β-turn similar to 7b.
Symmetry-Independent Conformers with the Same Turn-Motif
The symmetry-independent molecules of Aic-containing peptide 19 also differ from one another significantly, as evidenced by the all-atom RMSD of 1.87 Å between 19a and 19b. Unlike in the case of peptide 7, this large global disparity stems mostly from the peripheral i and i+3 residues, as the loop RMSD is only 0.12 Å between the two conformers (Table 1). Both 19a and 19b are broadly characterized as type I′ double β-turns, but they do differ from the canonical geometries (Figure 9). The loop dihedrals of 19a deviate substantially from the canonical values; ϕ(i+1) is over 11° wider than might be expected, and ϕ,ψ(i+2) are both more than 30° distorted. The value of ψ(i+1) also deviates from the ideal value of 30°, but this distortion is consistent across many of the type I′ double β-turns we have studied and presumably relates to the overall prehelical geometry afforded by the N–H(i+2)···O(i–1) β-turn H-bond. However, the degree of distortion in ϕ,ψ(i+2) is quite significant, extending beyond the classically defined boundaries (±30°) for the various turn-types (Figure 2b).3 The N–H(i+3)···O(i–1) H-bond lengths of 19a are also quite long, both of which measure >3.19 Å, though the N–H···O angles are both directional in favor of H-bonding (>150°). Despite these significant departures from the canonical type I′ β-turn, which likely contribute a non-negligible amount of strain in the backbone, overall 19a resembles the other double β-turn structures we have examined (e.g., 3c, 4a–e, 16c, and 7b).
Figure 9.
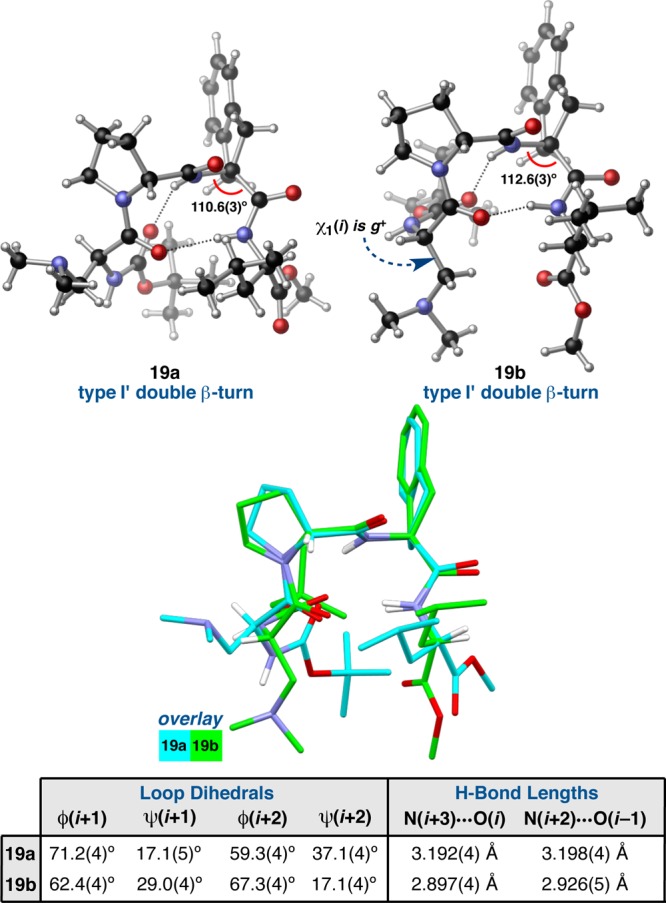
Two symmetry-independent conformers of peptide 19, both of which are classified as type I′ double β-turns. A structural overlay highlights the differences between the conformers (loop RMSD (19a/19b) = 0.12 Å).
On the other hand, conformer 19b has loop-region dihedrals that are in-line with the canonical values; the ϕ,ψ(i+1) values are very close to the established potentials, and ϕ,ψ(i+2) deviate to the degree we have observed previously. The two β-turn H-bonds are also much shorter than in 19a and equally directional in terms of the N–H···O angles. However, 19b has a rather different overall geometry owing to an unusual orientation of the C- and N-terminal residues, as shown in an overlay of 19b with 19a (Figure 9). We can quantify these differences by analyzing the ϕ and ψ dihedral angles of the i and i+3 positions.15 In 19a, ϕ(i) and ψ(i) measure −49.2(4)° and 133.4(3)°, respectively, in good accordance with most of the other type I′ double β-turns examined in this study (Table S4.03). The same dihedrals measure 50.3(4)° and 46.3(4)°, respectively, in 19b. Thus, there is a difference of 99.5° in ϕ(i) and 87.1° in ψ(i) between the two structures, with 19b exhibiting the only positive ϕ(i) value and the most acute ψ(i) value in the entire data set. Perhaps coupled to the unusual main-chain dihedrals of 19b is the gauche χ1(i) value of −51.3(4)°. In 19a, χ1(i) measures −175.0(3)°, which is much more reflective of the remainder of the type I′ β-turn structures that tend to have anti-periplanar χ1(i) values (Table S4.32). At the C-terminus of 19b, ϕ(i+3) and ψ(i+3) measure −117.8(4)° and −175.9(3)°, respectively, compared to −99.8(4)° and 41.4(4)° in 19a (Table S4.03). These differences reflect the more extended backbone of 19b. In this case, perceived deviations in the Leu side-chain orientation are almost entirely due to differences in the backbone, as the χ1(i+3) values of 19a and 19b are approximately equal at −63.9(4)° and −64.4(4)°, respectively (Table S4.32).
DFT Optimization of the crystallographic coordinates provided energy minimized structures of 19a and 19b that overlay with the corresponding X-ray crystal structures with loop RMSDs of 0.13 and 0.06 Å, respectively (Figure 10). The optimized structure of 19a has loop dihedrals that are much more similar to the canonical values than was observed in the solid state, hence the relatively high loop RMSD. Furthermore, the central β-turn H-bond shortens and becomes more directional, but the N–H(i+3)···O(i–1) β-turn H-bond breaks down completely upon geometry optimization. Instead, the N-terminal carbamate torques about ϕ(i) in such a way as to orient N–H(i) toward the opposite strand. The C-terminal ester, however, remains oriented away from the N-terminal strand, and a hairpin H-bond is not present. Only minor adjustments to the geometry of 19b are observed after optimization, with an attendant lengthening of both β-turn H-bonds to values above 3.0 Å. An overlay of the two optimized geometries reveals that the primary deviation is in the N-terminal residue–the orientation of the backbone and Dmaa side-chain (Figure S6.05). These differences amount to 2.07 kcal/mol in free energy in favor of conformer 19b, which benefits from two H-bonding interactions relative to one in 19a.
Figure 10.
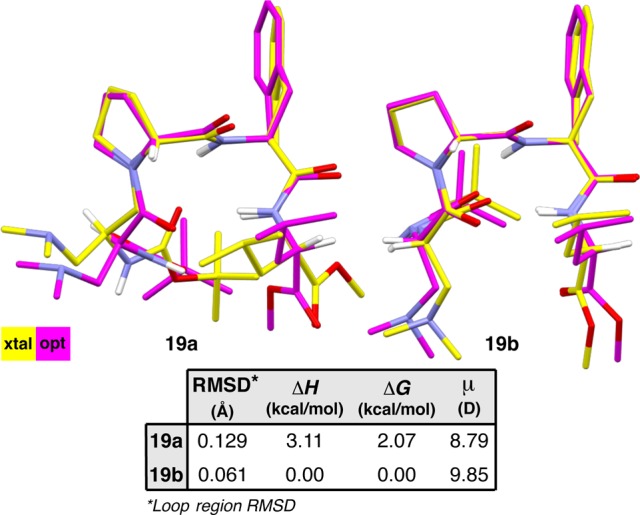
Two solid-state conformations of peptide 19 were optimized using DFT at the M06-2X/6-311++G(2d,3p)//B3LYP/6-31+G(d,p) level of theory.
Crystal Structure Library Analysis
Thus far, the discussion has primarily focused on the difference among discrete conformers of a given peptide. The degree of variation that we observed motivated an analysis of the peptide X-ray crystal structure library as a whole (Chart 1), in search of trends that might provide insight into (1) the effect of primary sequence modifications on secondary structure and (2) the source of differences between and within structural archetypes.15 While crystal packing forces and systematic effects certainly influence the solid-state structures to some degree, the primary goal of this study has been to observe, document, and quantify the conformational space available to β-turn-containing tetrapeptides. In a way, deviations caused by packing forces may help us achieve this goal, as systematic effects are unlikely to give rise to a truly inaccessible geometry.49 Furthermore, analysis of the structure library as a whole has shown that similar structures tend to deviate from the canonical turn-potentials in similar ways, allowing us to draw conclusions based on average values within of a given structural motif. It is useful to consider the possible sources of these differences, as they often provide insight into the steric and stereoelectronic effects that govern secondary structure. A summary of our findings is presented in Figure 11, and additional details can be found in the Supporting Information. These structural considerations may prove relevant to the design and study of future peptide-based catalysts.
Figure 11.

Structural attributes of the peptide X-ray crystal library. Ramachandran plots show the ϕ,ψ distribution at the (a) i, (b) i+1, (c) i+2, and (d) i+3 positions. Groupings of similar structural types are labeled in each plot. (e) A plot of χ1(i+3) as a function of χ1(i) shows the distribution of side-chain dihedrals. Histograms of H-bond (f) lengths and (g) angles show the distribution of H-bond geometries. Histograms of main-chain angles (τ) in (h) type II/II′ β-turns and (i) type I/I′ β-turns show the differences between the two secondary structural types. (j) A loose correlation between O(i)···C′(i+1) length and O(i)···C′=O(i+1) angle is observed for the n → π* stereoelectronic interaction that stabilizes the turn.
Backbone Dihedrals
Inspired by the work of Ramachandran and co-workers,50 we assessed the conformational space occupied by each residue of our peptides using ϕ,ψ dihedral plots (Figure 11a–d). It is instantly clear from these Ramachandran plots that the i+1 and i+2 residues are more tightly grouped than the i and i+3 residues. This is perhaps unsurprising given that the i+1 and i+2 residues comprise the loop-region of these peptides, which is biased to favor nucleation of a β-turn in every case by taking advantage of the Pro-Xaa sequence. Though the loop residues tend to cluster more tightly than the peripheral residues, they still show a number of interesting deviations from the canonical values (Figure 2b).3
i+1 Position
The Ramachandran plot of the i+1 residue shows four relatively tight groupings corresponding to the four common β-turn motifs (i–iv, Figure 11b). Because the i+1 residue is either d- or l-Pro in all of the peptides analyzed, the clusters tend to center on ±60° in ϕ, the value set by the pyrrolidine ring.3,21 The type II cluster (i, Figure 11b) consists of six peptides with average ϕ and ψ values of −53.4 ± 3.7° and 135.8 ± 4.7°, respectively (Table S4.02). Approximately equal and opposite values are observed for the type II′ cluster (ii, Figure 11b), with average ϕ = 60.6 ± 5.0° and average ψ = −130.2 ± 6.6° based on 23 peptides (Table S4.01). In both cases, the ψ value is noticeably wider than the ideal value of 120°, which may be related to repulsive interactions between the N–H(i+2) and N–H(i+3) σ-bonds, as discussed previously. The wider ψ value also permits a more geometrically favorable nO(i) → π*C′=O(i+1) interaction, which is known to stabilize β-turns.51 The type I′ cluster (iii, Figure 11b) consists of 21 peptides, significantly more than might have been expected from this library given the bias toward type II/II′ β-turns. It is perhaps noteworthy to mention that all of the type I′ β-turns observed in the solid state exist in the prehelical geometry described above by virtue of the N–H(i+2)···O(i–1) intramolecular H-bond. The average value of ϕ for this cluster is 65.3 ± 4.3°, and the average ψ value is 14° contracted relative to the ideal at 16.0 ± 5.1° (Table S4.03). These departures are consistent with an orientation of the loop amide that both promotes N–H(i+2)···O(i–1) H-bonding and minimizes nO(i)-nO(i+1) Pauli repulsion. The lone type I β-turn (iv, Figure 11b) is peptide 37, a structure we reported previously.29c The distorted type I β-turn is characterized by ϕ and ψ dihedrals measuring −47.7(3)° and −37.8(4)°, respectively. These values are perhaps buttressed by a 10-membered ring, side-chain-to-main-chain H-bond between N–H(i+2) and the free imidazole nitrogen-atom of the His(τ-Bn) side-chain, reminiscent of an ASX-turn.52 The two outlying points centered around ϕ,ψ = 81°, –170° correspond to the symmetry-independent conformers of peptide 32 (32a and 32b), both of which were largely unfolded in the solid state despite being biased toward a type II′ β-turn using a d-Pro-Phe central sequence (Figure S3.33). We also note parenthetically that we were able to rule out the Cγ-pucker of Pro as a relevant source of differences in the ϕ,ψ dihedrals of the peptides (Table S4.20–S4.24).53
i+2 Position
Unlike in the i+1 position, many different residues are found at the i+2 position in the structure library, ranging from Gly to a variety of α,α-disubstituted residues (Chart 1). Nevertheless, only two clusters are observed in the Ramachandran plot (Figure 11c), because ψ = 0° in all of the common turn-motifs, and the ideal ϕ values of types I/II′ (−90°/–80°) and types I′/II (90°/80°) are too proximal to distinguish them (Figure 2b).3 The first cluster (i, Figure 11c) is composed of one type I and 23 type II′ β-turns. Again, the lone type I turn (37) is distorted in i+2 owing to its ASX-like turn geometry, with ϕ,ψ = −63.5(4)°, – 21.8(4)°. The average ϕ,ψ(i+2) values for the type II′ β-turns are −69.9 ± 9.1° in ϕ and −11.7 ± 12.0° in ψ (Table S4.01). Moreover, a wide span of ψ values were observed, ranging from −31.4° to 22.5°, and giving rise to a standard deviation greater than the mean. The ψ = 22.5(2)° data point corresponds to Ala-containing peptide 31 and might be an outlier of the cluster, the average of which lies below the negative ψ region. The second grouping (ii, Figure 11c) includes six type II and 21 type I′ β-turns. The average dihedrals for the type II β-turns measure 67.6 ± 8.3° in ϕ and 17.8 ± 12.6° in ψ (Table S4.02). The 28.7° range in ψ, as well as the large standard deviation of 12.6°, suggests that a variety of torsions are accessible within this archetype. The type I′ β-turns differ only slightly from the type II, with average ϕ and ψ of 69.1 ± 7.3° and 17.1 ± 9.3°, respectively (Table S4.03). In this case, wide ranges of 27.0° in ϕ and 33.0° ψ are observed. It is perhaps noteworthy that, within each grouping, the Acpc-containing peptides tend to have values closer to ψ = 0° than other i+2 variants. This may be related to the fact that Acpc tends to occupy the so-called “bridge region” of the Ramachandran plot, as highlighted by Toniolo and co-workers,24a owing in part to the wide main-chain angle that relieves the repulsive interaction between N–H(i+2) and N–H(i+3) to some degree.43 The unfolded peptides 32a,b are outliers (ϕ,ψ = −135°, – 147°) at this position, as well (Figure S3.33).
It is noteworthy that all four turn-types tend to deviate from their canonical ϕ,ψ(i+2) values in the same overall direction, although the degree varies from type to type (Figure 11c). On average, ϕ(i+2) is 10–27° more acute than anticipated, and ψ(i+2) is 12–22° wider. The deviations in ϕ are coupled to the pitch of the loop amide; the established potentials require a relatively close distance between N–H(i+2) and N–H(i+3) in the type II/II′ β-turns and between O(i+2) and O(i) in the type I/I′ turns, and contraction of the ϕ value may alleviate these strains to some degree. The average ϕ(i+2) among the type I′ turns is nearly two-times more contracted relative to the type II/II′ turns, a possible manifestation of the N–H(i+2)···O(i–1) H-bonds that stabilize the prehelical, double β-turn structures. In terms of ψ(i+2), the canonical value of 0° requires the i+2 carbonyl to perfectly bisect the α-substituents (Figure 12a). The fact that ψ(i+2) is universally wider than typical suggests that a combination of steric and stereoelectronic factors may cause the i+2 α-substituents to torque either forward or backward, providing a skew conformation (Figure 12b,c). In the type I/II′ cluster, the α-substituents tend to pivot forward (toward the front/endo face of the β-turn) on average, while they tend to pivot backward (toward the back/exo face of the β-turn) in the type II/I′ cluster.
Figure 12.
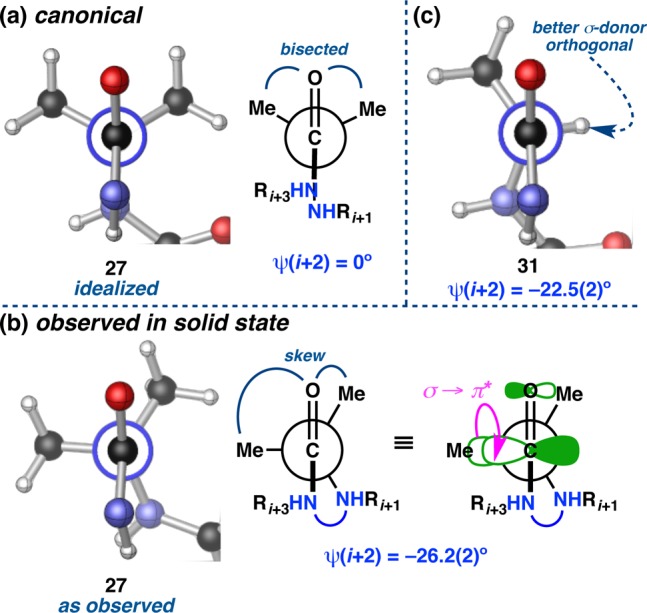
Deviations from canonical ψ(i+2) values, as exemplified by peptides 27 and 31. (a) The canonical value of ψ(i+2) requires a bisected geometry. (b) In the solid state, however, a skew geometry is observed, which may be the result of an incipient σ → π* stereoelectronic effect. (c) Ala-containing peptide 31 is skewed in the opposite direction relative to the other members of its type II′ β-turn class (e.g., 27), possibly to achieve orthogonality between the C–H bond and the adjacent π-system.
These observations appear consistent with an incipient σ → π* stereoelectronic effect, wherein torquing about ψ(i+2) orients one α-substituent orthogonal to the π-system of the i+2 carbonyl (Figure 12b). This hypothesis is supported by the following observations: (1) Acpc-containing peptides tend closer to the ideal ψ(i+2) of 0°, as the sp2 character of the cyclopropyl C–C bonds would make them poor σ-donors; and (2) Ala-containing peptide 31 is oriented such that its Cα–H bond, a better σ-donor than the C–Me bond, is orthogonal to the adjacent π-system, producing a ψ-deviation in the opposite direction of the other type II′ β-turns (Figure 12c). A stereoelectronic effect of this type would typically be associated with a shortening of the Cα–C′ bond. The lack of an appropriate zero-point has made this difficult to assess, but the directionality of the interaction, as governed by the relative orientation of the donor and acceptor, is favorable in each case. Both symmetry-independent molecules of Gly-containing peptide 33 are nearly ideal in ϕ(i+2) and ψ(i+2), suggesting that a steric effect, such as allylic strain minimization about the loop amide, may be coupled to these stereoelectronic effects.
i Position
The ϕ,ψ plot for the i position is more diffuse than either of the two loop positions, but some grouping of similar structural types is observed nonetheless (Figure 11a). For example, most of the type I′ double β-turns tend to tightly cluster together due to the fact that the i position of these prehelical structures also serves as the first loop residue in the N-terminal, type II β-turn of the double turn-motif (i, Figure 11a). The ASX-turn-containing peptide 37 also appears in this cluster, but outside of the main grouping. The average ϕ(i) and ψ(i) values of −56.3 ± 4.6° and 131.7 ± 3.5°, respectively, closely align with those measured for the type II cluster in the ϕ,ψ plot of the i+1 position (i, Figure 11b, Table S4.06). Deviations from the canonical type II potentials are likely due to the N–H(i+3)···O(i) H-bond, which affects the N-terminal β-turn in much the same way as the N–H(i+2)···O(i–1) H-bond affects the central β-turn. The single outlier at ϕ,ψ = 50.3°, 46.3° corresponds to 19b, which belongs in this cluster based on its overall topology but has quite different i dihedrals. All of the type II and II′ β-turns split into two relatively loose clusters based on the degree of backbone bending observed. Type II/II′ β-turns and hairpins with extended backbones are characterized by anticlinal ϕ and ψ values (ii, Figure 11a), with a mean ϕ of −133.8 ± 23.0° and a mean ψ of 145.9 ± 13.6° (Table S4.07). On the other hand, type II′ β-hairpins with bent backbones also have anticlinal ϕ values but are more gauche in ψ (iii, Figure 11a), with mean ϕ and ψ values of −146.0 ± 11.2° and 79.8 ± 12.4°, respectively (Table S4.08). These data show that the directionality of N–H(i), and thus the ability to engage in an intramolecular hairpin H-bond, is related to ϕ(i), while the degree of backbone bending relates to ψ(i) (Table S4.13 and Figure S4.06).
i+3 Position
Within the crystal structure library, the i+3 position shows the most residue variation. Unsubstituted and alkyl-substituted residues, such as Gly, Leu, Nle, Val, and Chg, as well as benzylic residues, including Phe, d-Phe, 3-Pal, and 2-Thi, are found at the i+3 position. As such, it is not necessarily surprising that there is a lack of obvious trend in the i+3 Ramachandran plot (Figure 11d). Of the 50 peptides possessing i+3 residues, 41 (82%) possess negative values of ϕ(i+3) and positive values of ψ(i+3) (i, Figure 11d), the averages being −93.7 ± 25.2° and 128.6 ± 32.1°, respectively (Table S4.09). The large standard deviations reflect the diffuseness of the cluster, though there does seem to be some preference for orthogonal ϕ(i+3) and anticlinal ψ(i+3). The three d-Phe-containing peptides (14 and 21a,b) are found in the bottom-right quadrant, as expected for an inversion of the α-stereocenter. Peptides 7a and 7b are also outliers at i+3 for reasons previously discussed. It is difficult to say whether or not the cluster centered on ϕ,ψ = −115°,–168° (ii, Figure 11d) is a group of outliers or a continuation of the main cluster (i, Figure 11d). Interestingly, the symmetry-independent conformers of peptide 33 are outliers (iii, Figure 11d). As discussed previously, the unusually acute (and negative) ψ-values orient the i+3 carbonyl away from the N-terminal strand, whereas the carbonyl typically points toward the opposite strand.
Side-Chain Dihedrals
Since the pioneering work of Dunbrack and co-workers,15,54 the interplay of the side-chain dihedrals with those of the backbone, as well as their overall influence on protein structure, have become well appreciated. The steric and electronic profiles of side-chains have informed studies that aim to define “allowed” and “forbidden” χ-dihedral space for combinations of amino acids and to apply these parameters in a predictive manner.55 In an effort to contribute to this growing literature, we analyzed the χ1 dihedrals at the i and i+3 positions of our peptide crystal structure library and plotted χ1(i+3) against χ1(i) in search of structural trends (Figure 11e). To avoid ambiguity, Val-containing peptides 9, 10, and 26 and Chg-containing peptide 11 were removed from this analysis, as were the peptides without i+3 residues or side-chains (7a,b and 35–37).
Perhaps the most obvious characteristic of this plot is that no positive χ1 values were observed at the i position (Table S4.32). Negative χ1(i) values orient the side-chain away from the peptide scaffold so as to minimize unfavorable steric interactions. Positive values of χ1(i+3) are relatively uncommon for similar reasons, with over 82% of the peptides analyzed having negative χ1(i+3) values. Moreover, the side-chains at both the i and i+3 positions tend to cluster around gauche (g) and anti (a) values of χ1, the expected minima on the torsional potential energy surface of amino acid–based systems (Tables S4.33–S4.37). About 75% of the peptides are gauche at the i+3 position, while the i position is more evenly split between gauche and anti (40:60). The two major clusters are ag– (i.e., anti at i, negative gauche at i+3), consisting of 21 peptides, and g–g–, comprising 11. The type I′ double β-turn structures demonstrate a strong preference for the ag– arrangement of side-chains, as 17 out of the 19 such peptides analyzed are found in the ag– cluster. Further, all but one of the type I′ peptides have anti χ1(i) values, and all of them have gauche χ1(i+3) values (Table S4.38). The only type I′ peptide to have a gauche χ1(i) is 19b, and the lone ag+ peptide is 4e, also a prehelical, type I′ β-turn, both of which were discussed previously. It is plausible that anti χ1(i) values are favored because they orient the side-chain as far away as possible from both the backbone and the C-terminal side-chain. It seems that the preference for gauche χ1(i+3) values avoids destabilizing syn-pentane-type strain with the C-terminus, while also promoting a more compact structure. The side-chains of the type II/II′ β-turns are significantly less directional and appear in all of the clusters. Of the 23 examined, about 35% have anti χ1(i) values, and 61% have gauche values of χ1(i+3) (Table S4.38). This suggests that gauche/anti differential is less energetically significant at both the i and i+3 positions among the type II/II′ β-turns.
H-Bond Geometries
The ability to form intra- and intermolecular H-bonds is a fundamental feature of peptides that is essential to their higher order structures and functions. In the context of β-turns, intramolecular H-bonds stabilize the folded structure, and intermolecular H-bonds play a role in association with molecular guests or other peptides.14,20 The H-bonding patterns of our peptide X-ray crystal structure library were analyzed and compared with known data. Only amide–amide H-bonds are considered in our analysis to ensure a proper reference point. We found the H-bonding patterns of our crystal structures to be typical overall.56
The N···O distances (r) and N–H···O angles (θ) for 94 intramolecular, amide–amide H-bonds were measured and analyzed. Four different types of intramolecular H-bonds are observed across our library: (1) N–H(i+3)···O(i) β-turn H-bonds found in every member of the library except unfolded peptides 32a and 32b, (2) N–H(i)···O(i+3) hairpin H-bonds found in types II′ and II β-turns, (3) N–H(i+2)···O(i–1) β-turn H-bonds found exclusively in the type I′ double β-turns, and (4) N–H(i+2)···O(i) γ-turn H-bonds observed sporadically. While the distances were often favorable for γ-turn H-bonding, the angles were often too acute to be relevant, and thus we excluded the N–H(i+2)···O(i) metrics from this analysis. There are only minor differences among the other three types of H-bonds. The average value of r across the whole set measures 3.005 ± 0.124 Å, with a median of 2.979 Å. A range of 2.804–3.358 Å was observed, spanning from strong to very weak intramolecular H-bonds.56 A histogram showing the distribution of r values possesses two local maxima, one at 2.85–2.89 Å and the other at 2.95–2.99 (Figure 11f). Another interesting feature of the distribution is the relatively high proportion of H-bonds with r greater than 3.20 Å. Above this length, H-bonds tend to be quite weak. It may even be tempting to discount these interactions as proper H-bonds. However, we included them in the analysis if the N–H···O angles were appropriately directional. The average value of θ measures 160 ± 8°, which is typical for amide–amide H-bonds in the solid state. A range of 127° to 175° was observed, and the distribution is skewed toward the higher end of the range (Figure 11g). The most favorable H-bonds have a near linear N–H···O angle, but this is difficult to achieve given the geometric constraints of the β-turn structure. The values at the lower end of this distribution may be questionable as to whether they constitute a true H-bond or not, but they were included on the basis of their lengths.
Main-Chain Angles
The ideal value of a main-chain angle (τ) can be considered to be 109.5°, in accord with a perfectly tetrahedral α-carbon.15 It is not uncommon, however, to observe a range of τ values, especially for those residues involved in secondary structures.43 The main-chain angles of each residue in our peptide library were measured, providing 208 τ values for analysis. We observe a distribution of τ values ranging from 105.9° to 118.1°, with a mean of 111.5 ± 2.8° and a median of 110.8° (Table S4.25). The average τ is likely skewed larger than tetrahedral due to the prevalence of Acpc residues in our library, which have wide τ values on average (117.2 ± 0.6°). A τ histogram shows a clear maximum at tetrahedral values (109–110°) and perhaps more wide-angle counts than might be expected (Figure S4.08). When analyzed on a position-by-position basis, some interesting trends are observed (Figure S4.09). The peripheral residues tend to be tetrahedral on average, with averages for τ(i) and τ(i+3) measuring 109.5 ± 1.7° and 110.3 ± 1.7°, respectively. However, the loop-region angles are significantly wider. The mean τ(i+2) measures 113.9 ± 2.9°, which is perhaps skewed wide due to the 20 peptides that have an Acpc residue at the i+2 position. As such, the τ(i+2) histogram is bimodal, with maxima at 111–112° and 117–118° (Figure S4.08). Though all of the peptides possess a d- or l-Pro residue at the i+1 position, the average value of τ(i+1) is also quite wide at 112.2 ± 2.5°.57 As there are no strained rings at i+1, this widening must be a result of the conformational stresses imposed by the well-defined β-turn loop-region. The τ(i+1) histogram also shows two maxima, one at 109–110° and the other at 113–114°; there is a comparatively large proportion of τ(i+1) values in the 115–116° range, as well.
Analyzing the τ positional distributions as a function of turn-motif provided some additional insight. In the type II/II′ β-turns, the τ(i+1) distribution is centered at approximately tetrahedral (Figure 11h), with a mean value of 110.1 ± 1.0° (Table S4.27). Among the type I/I′ β-turns, however, τ(i+1) averages 114.9 ± 1.0°, nearly equal to the 114.2 ± 2.9° mean τ(i+2) value (Table S4.29). The distribution is skewed quite wide, as well; no τ(i+1) values are found below 113–114°, which is also the bin with the highest count (Figure 11i). These data suggest that the observed widening of τ(i+1) across the whole library is due to the presence of the 22 type I/I′ β-turns, which have significantly wider τ(i+1) values on average than their type II/II′ counterparts. In fact, the entire distribution is shifted wide compared to that of the type II/II′ turns, including the peripheral positions. From a conformational standpoint, we attribute this widening to the prehelical geometry of the type I/I′ β-turns. This is especially evident at the i+1 position, wherein the proximal and codirectional i and i+1 carbonyls may induce nO–nO Pauli repulsion that widens τ(i+1) angle (Figure 13a).58 This proposed interaction is absent in the type II/II′ β-turn structures, in which the carbonyls are oriented in opposite directions.
Figure 13.
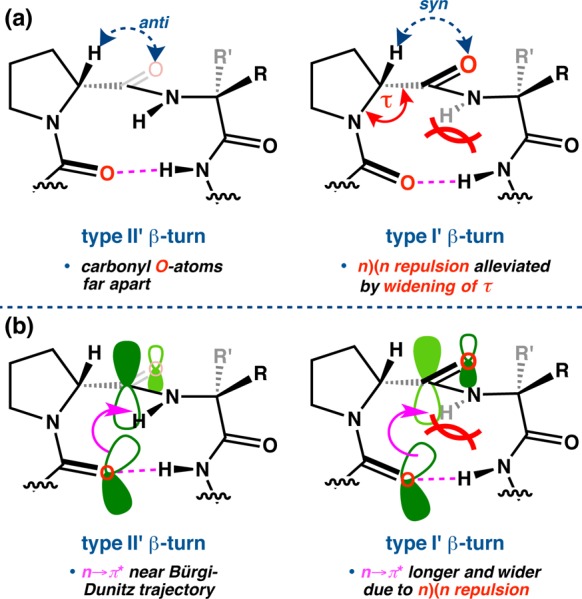
Some differences between type II′ and I′ β-turns. (a) Widening of τ(i+1) as a consequence of Pauli repulsion in type I′ β-turns. (b) As a consequence of this repulsion in type I/I′ turns, types II/II′ are able to access a more favorable n → π* geometry, with shorter interaction O···C′ distances and near Bürgi–Dunitz trajectories.
n → π* Geometries
We also examined the peptide library for its exhibition of the n→π* stereoelectronic effect, wherein lone pair electron density localized on the carbonyl oxygen-atom of the i residue donates into the antibonding π-orbital of the proceeding i+1 carbonyl.51,58,59 This type of delocalization stabilizes the β-turn via a partial bonding interaction that tethers O(i) to C′(i+1). The most favorable n→π* interactions are typically shorter than the sum of the van der Waals radii of the two atoms (r < 3.2 Å) and have O···C=O angles (θ) near the Bürgi-Dunitz trajectory (θ = 107–110°).60 In order to assess the degree to which n→π* effects are present in this structure library, we measured the O(i)···C′(i+1) lengths and O(i)···C=O(i+1) angles for each peptide.
The average O(i)···C′(i+1) distance measures 2.893 ± 0.098 Å, with a range from 2.716 to 3.185 Å (Table S4.44), consistent with the operation of n→π* effects. However, the average O(i)···C′=O(i+1) angle is wider than the Bürgi-Dunitz trajectory at θ = 113.6 ± 11.3°. Plotting θ against r reveals a loose correlation between length and angle (R2 = 0.76) with a positive slope; longer distances tend to accommodate wider angles and vice versa (Figure 11j). These n→π* effects are thus perturbed by additional local structural contexts.
When analyzed as a function of β-turn motif, the data again show differences between the canonical turn-types. The six type II β-turns are characterized by average r and θ values of 2.763 ± 0.031 Å and 99.2 ± 1.6°, respectively, the shortest and most acute n→π* geometries observed for the set (Table S4.45), whereas the average of the 23 type II′ β-turns measures 2.857 ± 0.071 Å in r and 106.8 ± 6.7° in θ (Table S4.46). The more favorable interaction geometries in the type II turns may be coupled to their ϕ,ψ(i+1) dihedrals, which are more distorted on average relative to the diastereomeric type II′ turns (i vs ii, Figure 11b). The widening of ψ(i+1), especially, accommodates a more acute O(i)···C′=O(i+1) angle, and therefore allows for a shorter O(i)···C′(i+1) distance. However, the 21 type I′ double β-turns exhibited longer O(i)···C′(i+1) distances and wider O(i)···C′=O(i+1) angles, with an average r and θ values of 2.974 ± 0.067 Å and 125.2 ± 3.5°, respectively (Table S4.47). It is possible that this may be coupled to the wider τ(i+1) observed for this turn-motif, as the structures must balance the effects of destabilizing nO-nO Pauli repulsion, N–H(i+2)···O(i–1) H-bonding, and favorable n→π* overlap (Figure 13b).58 Thus, the n→π* geometries may tend toward a less favorable donor–acceptor orientation to maximize the O(i)···O(i+1) distance. Though it has been shown that the Cγ-pucker of Pro can affect n→π* geometry in model systems,51 this issue is not obviously a source of deviation among the β-turn-types in this library.
Stereoelectronic effects of this type might be expected to produce concomitant lengthening of C=O(i+1) and pyramidalization of C′(i+1). Possibly due to the lack of a proper zero-point, we were unable to observe any significant lengthening of C=O(i+1); the average length across the entire library measures 1.228 ± 0.006 Å, which is identical to the often quoted value of 1.23 Å. However, the pyramidalization of C′(i+1), as measured by ΔC′, is non-negligible, with a mean ΔC′ of 0.10 ± 0.08 Å (Table S4.44).44 The large standard deviation reflects significant variation within the data set. Interestingly, the type I′ β-turns show the most significant pyramidalization of C′(i+1) despite their nonideal interaction geometry (average ΔC′ = 0.014 ± 0.007 Å). This is likely a function of the inherent strains in this turn-motif.
Distribution of Turn-Types and Homologous Structure Series
We now discuss structural data for three series of homologous peptides, including those that vary only at the i+2 position, those that differ at the C-terminal cap, and those containing l-Pro instead of d-Pro. These data enable a systematic comparison of structures that differ by a single substitution. Discussion of the relationship between peptide structure and catalytic activity in the bromination of quinazolinone 1, namely enantioselectivity, are discussed where appropriate.61
Effects of the i+2 Residue
The i+2 residue plays an important role in the nucleation and conformational stabilization of a β-turn.3,11 We intentionally biased all of the peptides in this structure library to adopt either type II′ or type II β-turns depending on whether d- or l-Pro was incorporated at the i+1 position. However, we observed a wider range of turn-motifs in the solid state than initially expected, and we wondered how the nature of the i+2 residue might influence this distribution. Figure 14 presents the incidence of canonical β-turn-types as a function of i+2 residue.62
Figure 14.
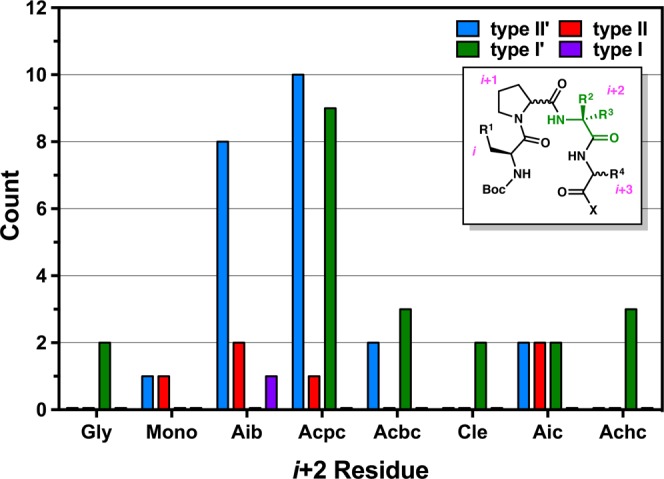
Occurrence of canonical turn-motifs as a function of i+2 residue. Peptide 32 was excluded from this analysis.
Over half of the library (nearly 57%) consists of Aib- and Acpc-containing peptides, and the data show a significant difference between these two subsets. While the Aib-containing peptides largely tend to nucleate the anticipated type II/II′ turns depending on the Pro stereochemistry, those with Acpc residues at the i+2 are more bimodal—that is, they are distributed more evenly among the type II/II′ and prehelical, type I′ turns. The lone type I turn (peptide 37) contains a central l-Pro-Aib sequence, which suggests circumstantially that the type I′ conformation should also be accessible for Aib-containing peptides. Even so, none were observed in this library. Despite the fact that Aib and Acpc only differ by an equivalent of molecular hydrogen, they seem to behave differently in the context of β-turn structure. These data suggest that Acpc may allow for more conformational freedom than Aib in these particular sequences, possibly as a consequence of the strained cyclopropyl ring that widens τ(i+2) and allows access to the bridge-region of Ramachandran space.63 The Acbc-containing peptides are also more evenly divided between types II′ and I′ turns, perhaps for similar reasons. It is interesting to note that the seemingly “Aib-like” residues, such as Cle, Aic, and Achc,24 behave differently than Aib itself and are observed to promote the nucleation of prehelical type I′ β-turns in this context (Figure 14).
Crystallographic Analysis of Homologous i+2 Series
We were able to obtain X-ray crystallographic data for a homologous series of seven peptides that differ from one another only at the i+2 residue (Figure 15a). All of the peptides possess a common Boc-Dmaa-d-Pro-Xaa-Leu-OMe sequence that was designed to nucleate a type II′ β-hairpin. Of the seven homologues, only the Aib-containing peptide 25 exhibits the targeted secondary structure. All of the others (4, 17–19, 22, and 33) present prehelical, type I′ β-turn structures in the solid state. An overlay of these six structures shows a tight correspondence in the loop-region (RMSD ≤ 0.11 Å) with more variability at the peripheral positions (Figure 15b). The N-terminal residues are more cohesive than those of the C-terminus. The Gly-containing peptide 33 is particularly noteworthy for its prehelical structure, as Pro-Gly sequences were previously studied as archetypal β-hairpin inducers.19,22 The average value of τ(i+2) in 33 measures 115.5 ± 0.1° for the two symmetry-independent molecules 33a and 33b. Notably, the wide τ(i+2) observed for Gly is reminiscent of Acpc, and perhaps this signifies that Gly is similar to Acpc in terms of its influence over the conformational space available to β-turn-biased structures.
Figure 15.

(a) Homologous series of peptides differing from one another only in the i+2 residue. For sequences with more than one symmetry-independent structure, only one is shown. (b) Overlay of the type I′ β-turns from the homologous series showing tight overlap in the loop-region and more deviations at the peripheral residues. All loop RMSDs are less than 0.11 Å. For more structural information, see Figure S4.21.
NMR Analysis of Homologous i+2 Series
To investigate whether or not the secondary structures observed in the solid state were populated in solution, we acquired NMR data for each member of the i+2 homologous series under identical conditions (600 MHz, 0.01 M, C6D6, 20 °C). The same protocol that was applied to peptide 4 (vide supra) was also used to analyze peptides 17–19, 22, 25, and 33. Additional details, including stacked 1H NMR spectra (Figure S5.16), are provided in the Supporting Information. In general, as with compounds discussed earlier, our data are consistent with a variety of populated states for these peptides in solution.
The NMR data for Acbc-containing peptide 17 are quite similar to those of 4. The 3JNH–Hα values extracted from the 1H NMR spectrum are consistent with a ϕ(i) of approximately −30° and ϕ(i+3) values of either −89° or −151° (Table S5.40). As for 4, these torsions can accommodate a variety of conformations, but they are perhaps most consistent with the prehelical and nonhairpin β-turns. The chemical shifts of the NH(Leu) and NH(Acbc) resonances suggest that these amides are involved in intramolecular bonds, while that of NH(Dmaa) is in-line with a non-H-bonded state (Table S5.41). It is notable that the NH(Leu) signal of 17 is 0.58 ppm more upfield than that of 4; in fact it is the most upfield-shifted NH(Leu) signal of the entire homologous series (Figure S5.16), which intimates that the NH(i+3)···O(i) β-turn H-bond could be particularly weak in the case of 17. Moreover, the following structure-suggestive NOE contacts are observed in the NOESY spectrum: Boc(Dmaa) ↔ NH(Acbc), Boc(Dmaa) ↔ NH(Leu), and δ(d-Pro) ↔ NH(Acbc). Although these correlations are supportive of a prehelical, type I′ β-turn structure, the presence of a moderate strength α(d-Pro) ↔ NH(Acbc) NOE indicates that 17 may sample type II′ loop dihedrals in solution (Figure S5.06). As such, these data support either a γ′-turn conformation similar to that proposed for homologue 16 or a prehelical structure mirroring the solid-state observations (17a,b) for this sequence.
Data gleaned from the NMR spectra of Cle-containing peptide 18 indicate that its most populated conformer in the solution phase differs from the type I′ double β-turn it shows in the solid state (18a,b). The ϕ(i) and ϕ(i+3) dihedral angles of 18 are calculated to be −63° or −177° and −89° or −142°, respectively, using the corresponding 3JNH–Hα values (Table S5.40). These dihedrals are most in-line with those expected for a β-turn or hairpin. The relative chemical shifts of the amide resonances suggest that NH(Leu) is likely engaged in an NH(i+3)···O(i) H-bond, while NH(Cle) is largely solvent-accessible (Table S5.41). Compared to those of peptides 4 and 17, the NH(Dmaa) signal of 18 occurs at a downfield shift and is significantly sharper in terms of peak width (Table S5.43). These chemical shift data are in-line with a hairpin conformation. Moreover, a number of long- and short-range NOE correlations are apparent in the NOESY spectrum of 18 (Figure S5.07), including the following structure-suggestive contacts: NH(Leu) ↔ NH(Cle), β(Dmaa) ↔ NH(Leu), δ(d-Pro) ↔ NH(Cle), and Boc(Dmaa) ↔ NH(Leu). A strong α(d-Pro) ↔ NH(Cle) NOE is also observed, providing evidence in support of type II′ loop dihedrals despite the NOE-contacts (e.g., the latter two) that point to a prehelical, type I′ structure. Overall, these NMR data are consistent with a type II′ β-hairpin solution structure that may also be in equilibrium with nonhairpin conformational states.
The solution structure of Aic-containing peptide 19 presents an intermediate case. The 3JNH–Hα value of the i position is consistent with ϕ(i) dihedrals of either −89° or −142°, and that of the i+3 position yields ϕ(i+3) values of either −89° or −151° (Table S5.40). These calculated torsions are most representative of the β-turn and hairpin conformations, though the prehelical form cannot be rigorously excluded. The 1H NMR chemical shifts for the NH(Leu) and NH(Dmaa) signals indicate that both are engaged in intramolecular H-bonds. The NH(Aic) resonance is less diagnostic; it is moderately downfield-shifted, which might suggest that it samples H-bonded and solvent-exposed states on the NMR time-scale (Table S5.41), although it could also be attributed to deshielding anisotropic effects stemming from the indane ring. A strong α(d-Pro) ↔ NH(Aic) correlation observed in the NOESY spectrum of 19 is consistent with a type II′ loop-region (Figure S5.08). Additional long- and short-range NOEs are also apparent, including fairly strong NH(Leu) ↔ NH(Aic) and NMe2(Dmaa) ↔ δ(Leu) contacts, as well as weak δ(Leu) ↔ α(d-Pro), Boc(Dmaa) ↔ NH(Leu), and Boc(Dmaa) ↔ β(Aic) correlations. In the solid state, peptide 19 was found to exhibit distorted, prehelical, type I′ β-turn structures in both symmetry-independent conformers (Figure 9). In contrast, these NMR data suggest that 19 likely populates both type II′ β-hairpin and γ′-turn forms.
Peptides 22 and 25, which contain Achc and Aib residues at the i+2 position, respectively, provided similar NMR data profiles. In both cases, the 3JNH–Hα of the i position gives rise to calculated ϕ(i) values of either −67° or −172° (Table S5.40). The calculated ϕ(i+3) values differ only slightly between 22 and 25, with those of the former being either −90° or −150° and those of the latter being −94° or −151°. These dihedrals are able to accommodate all of the proposed conformations. The 1H NMR spectra of both peptides show downfield-shifted NH(Leu) and NH(Dmaa) resonances and particularly upfield-shifted NH(i+2) signals. These chemical shift data support conformations wherein NH(Leu) and NH(Dmaa) are engaged in intramolecular H-bonds, perhaps NH(i+3)···O(i) and NH(i)···O(i+3) H-bonds, while NH(i+2) is free (Table S5.41). The NOESY spectra of peptides 22 and 25 peptide differ slightly. Peptide 22 exhibits multiple long-range contacts that imply interstrand proximity (Figure S5.10). For example, the following NOEs are observed: Boc(Dmaa) ↔ β(Achc), α(Dmaa) ↔ NH(Leu), δ(Leu) ↔ γ(d-Pro), and a fairly strong α(d-Pro) ↔ NH(Achc) suggestive of type II′ loop dihedrals. Peptide 25 shares many of the short-range contacts exhibited by 22 (Figure S5.11). Strong α(d-Pro) ↔ NH(Aib) and NH(Leu) ↔ NH(Aib) are consistent with type II′ loop dihedrals, but the absence of long-range NOEs might reflect a conformationally dynamic solution structure. Taken together, the data for peptides 22 and 25 are well aligned with type II′ β-hairpin conformations in solution, although it is important to acknowledge the possibility that nonhairpin conformations, such as a γ′-turn, may also be populated. This assignment is in agreement with the crystal structure of 25, but for 22 the solution phase and solid-state structures point to different conformations, as its crystal structure presents a prehelical, type I′ conformation (Figure 15).
The solution NMR data of Gly-containing peptide 33 accommodate multiple conformational states. The 3JNH–Hα values of the peripheral residues are consistent with ϕ(i) dihedrals of either −119° or −180° and ϕ(i+3) dihedrals of either −89° or −151° (Table S5.40). These calculated torsions are most representative of the β-turn and hairpin conformations. The 1H NMR resonances of NH(Leu), NH(Gly), and NH(Dmaa) occur at relatively downfield frequencies, suggesting that all three are involved in, or at least sampling, intramolecular H-bonds (Table S5.41). The longer-range NOE contacts observed in the NOESY spectrum of 33 provide evidence for interstrand proximity, but are not hallmarks of any specific conformation (Figure S5.14). These contacts are as follows: α(Dmaa) ↔ δ(Leu), γ(d-Pro) ↔ NH(Leu), γ(d-Pro) ↔ NH(Gly), and NMe2(Dmaa) ↔ NH(Leu). Furthermore, a moderate-weak α(d-Pro) ↔ NH(Acbc) correlation perhaps demonstrate that type II′ turn dihedrals are favored to a lesser extent. This might be coupled to the weak NH(Leu) ↔ NH(Gly) NOE, which implies that a β-turn may not be the most populated state in solution. Overall, these data could signal a type II′ β-hairpin/γ′-turn equilibrium favoring the γ′-form. In the solid state, peptide 33 adopts a prehelical geometry in both symmetry-independent molecules (Figure 15). As these data do not necessarily preclude this conformation, it is possible that the prehelical form is also populated to some degree in solution.
It is interesting to note that, while six of the seven peptides in the homologous i+2 series can exhibit the prehelical, type I′ β-turn structure in the solid state (Figure 15), only one peptide definitively populates this conformation in solution (i.e., 4). The solution NMR data presented above capture the dynamic behavior of these β-turn biased sequences in conditions relevant to catalysis.
Connections to Enantioselective Catalysis in the Homologous i+2 Series
Table 2 summarizes the observed solid-state and solution-phase conformations of the homologous i+2 peptides and presents the enantioselectivity exhibited in the bromination of quinazolinone 1 under the conditions of eq 2. These data highlight the importance of the i+2 position in enantioselective catalysis, as the er of tribromide 2 ranges from 61:39 to 95:5 depending on the identity of this residue. We note that conversion is complete in every case. Interestingly, the enantioselectivity decreases as a function of ring size in the nominally spirocyclic i+2 residues, with strained rings (e.g., Acpc and Acbc) providing higher selectivities. The only exception is Aic-containing 19, which is the least selective catalyst and provides 2 in only 61:39 er. Although Aic is related to Cle, the benzo-ring of Aic could either (a) induce conformational changes in the catalyst structure that render it less selective or (b) interact with the substrate via π-stacking interactions that are deleterious to enantioinduction. Moreover, Achc-containing 22 performs comparably to Aib-containing 25, which might be expected given that they both adopt type II′ β-hairpins in solution. What is surprising, however, is the low er of 68:32 observed for both 22 and 25, as our hypothesized model for enantioinduction invokes docking of 1 to a type II′ β-hairpin conformer of peptide 3.28a Indeed, the best catalysts for this reaction are found to adopt nonhairpin conformations in the solid state and in solution. Acpc-containing peptide 4 significantly outperforms 3, yet it adopts a prehelical, type I′ β-turn in its crystal and NMR structures. Additionally, Gly-containing 33 is nearly as selective as 3, delivering 2 in 91:9 er despite the lack of α-substituents at the i+2. Catalyst 33 was also found to adopt a prehelical, type I′ β-turn in the solid state, and it is most likely in fast-exchange equilibrium between multiple conformers in solution.
Table 2. Enantioselectivity Observed for Homologous i+2 Seriesa.
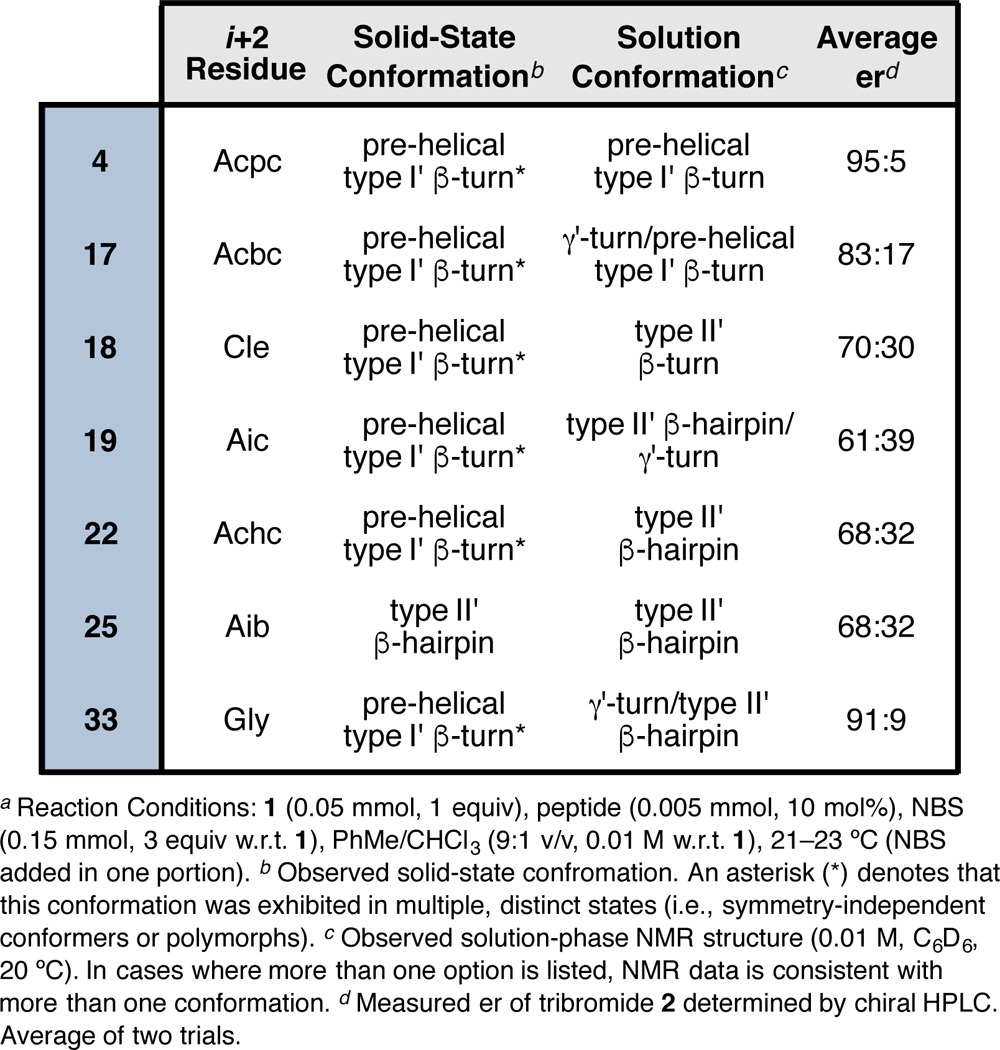
Taken together, these results suggest that peptidic catalysts that nucleate rigid β-turns are not necessarily the most effective in the context of this atroposelective bromination reaction. In fact, there seems to be some benefit to catalyst flexibility that is not yet fully understood. There are a number of caveats associated with making enantioselectivity predictions based on structurally dynamic catalysts.33 In this particular reaction, however, both highly strained (e.g., Acpc) and unstrained (e.g., Gly) i+2 residues provide highly selective catalysts. This unusual dichotomy suggests that more global conformational effects are likely operative along the selective reaction coordinate.
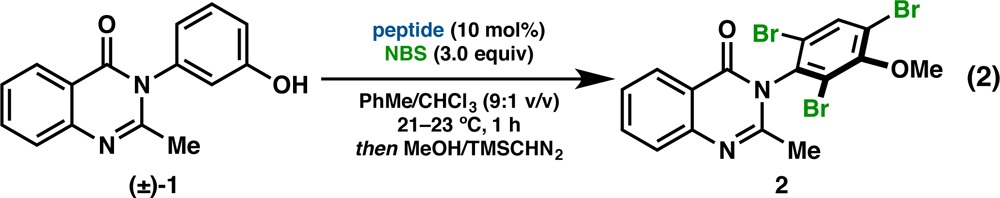 |
2 |
Role of Peripheral Residues
While the peptide library analyzed in this study is highly populated with sequences containing a Dmaa residue at the i-position, peptides with other residues in this position show similar properties. For example, the ability to access a type I′ double β-turn geometry does not seem to require a Dmaa residue at the i position. Peptide 5, a homologue of peptide 4 in which the Dmaa residue has been substituted with Cys(Ph), also adopts the prehelical geometry (Figure 16a). The same is true of peptide 23, the Cys(Ph) variant of 22 (Figure S3.24). However, peptide 6, the carbon-isostere of 4 in which the i residue is Leu instead of Dmaa, adopts a typical type II′ β-hairpin geometry (Figure 16a). We do not expect that this difference is due to the Leu residue specifically, but rather it reflects the bimodal distribution of Acpc-containing peptides.
Figure 16.
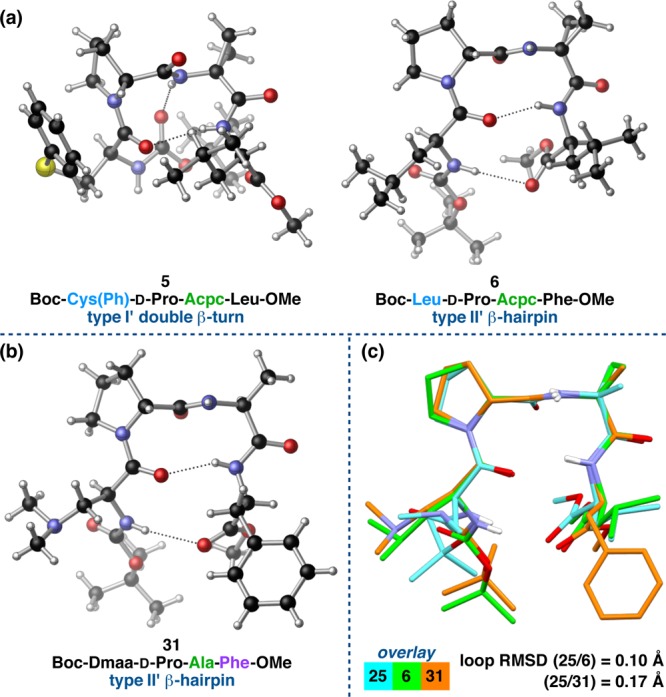
(a) Two i position homologues of peptide 4, Cys(Ph)-containing 5 and Leu-containing 6. (b) The Ala-containing peptide 31 is not quite homologous to the series presented in Figure 15 due to its i+3 Phe residue. (c) An overlay shows the structural similarity between type II′ β-hairpins 25, 6, and 31.
The Ala-containing peptide 31 is not a perfect structural homologue of 6 owing to the i+3 Phe residue, but its type II′ β-hairpin structure closely resembles that of 6 (Figure 16b). Additionally, the structures of both 6 and 31 are quite coincident with Aib-containing peptide 25, with loop RMSDs of 0.10 and 0.17 Å, respectively (Figure 16c). The primary differences among the members of this series are localized to the peripheral residues. It is not entirely clear that a fair comparison can be drawn between 31 and the other members of the homologous i+2 series, all of which are Leu-containing at the i+3 position. The i+3 Phe residue of 31 may not be structurally innocent, as we have not observed prehelical secondary structures for peptides with benzylic i+3 residues. It is possible that the type I′ double β-turn geometry is more favorable when the i+3 residue is alkyl-substituted.
Amide plane flipping is known to interconvert types II and I β-turns in proteins via a relatively low-barrier, concerted rotation about ψ(i+1) and ϕ(i+2).20 However, the type I′ β-turns we have observed would require a more global conformational change to access the observed, prehelical structures from the intended type II′ β-hairpins. This includes significant reorganization of the peripheral residues and side-chains. Only a few similar structures are reported in the CSD, most of which are found within cyclic peptides.64 Furthermore, the documentation of this unexpected structure in solution provides evidence that this conformation is important to consider, at least for this class of peptide-based catalysts.
C-Terminal Cap
An often-overlooked structural attribute of these peptides is the C-terminal protecting group (cap). In the course of optimizing peptide-based catalysts for a wide variety of enantioselective reactions, we have encountered multiple scenarios in which the choice of C-terminal cap proved essential to achieve high levels of enantioinduction.9,14,20,28 In cases where N,N-dimethyl amides outperformed the corresponding methyl esters, the enhanced selectivity has been ascribed to a stabilization of the β-hairpin structure via a more favorable N–H(i)···O(i+3) H-bond, since amides tend to be better H-bond acceptors than esters.65 As such, we wondered if the C-terminal cap might influence the distribution of secondary structures within our library. Figure 17a summarizes these data.62 Methyl esters comprise nearly 65% of the tetrapeptides analyzed, and N,N-dimethyl amides make up the remaining 35%. Despite the population imbalance, there seems to be a difference between the two types of C-terminal caps. While the N,N-dimethyl amides contain a high type II′/I′ ratio, the conformational landscape of the methyl esters is more even, yet still skewed in favor of the prehelical, type I′ β-turn structures. Thus, it seems that the dimethyl amide cap does indeed stabilize type II′ β-turns to some degree. Of the 15 type II′ amides, 13 (87%) are β-hairpins. As a side note, no l-Pro-containing peptides with dimethyl amide caps were analyzed, hence the lack of type II β-turns in amide bin.
Figure 17.
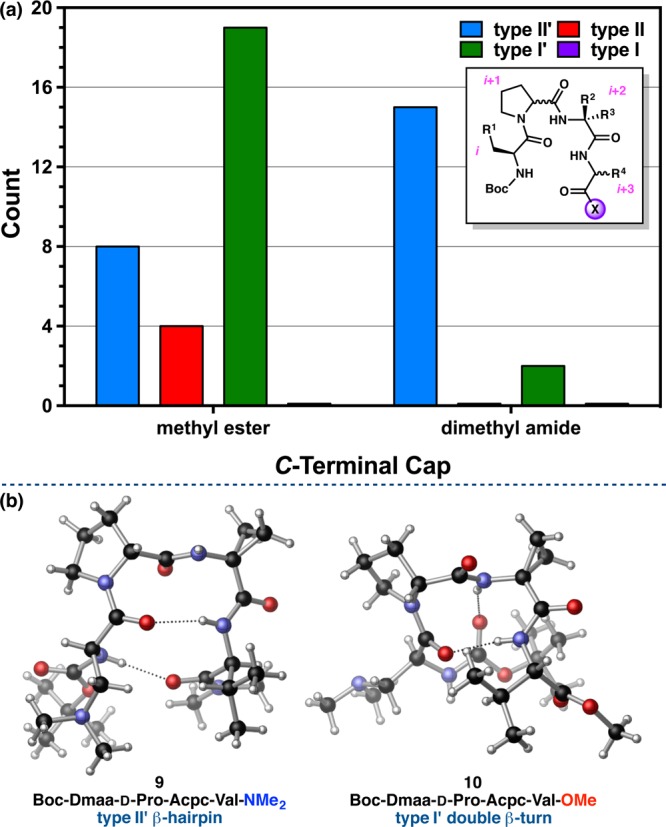
(a) Occurrence of canonical turn-motifs as a function of the C-terminal cap. Peptides 32 and 35–37 were excluded from this analysis. (b) Homologues 9 and 10 differ only in their C-terminal cap, and yet they exhibit different secondary structures.
Additional insight may be gleaned from structural comparisons of homologous pairs. The example of peptides 3 and 4, for instance, suggests that the change from N,N-dimethyl amide to methyl ester within an otherwise identical sequence changes the conformational landscape in favor of the prehelical geometry. The N,N-dimethyl amide variant (3) exists as both type II′ β-hairpins (3a,b) and type I′ double β-turns (3c) in the solid state, while methyl ester-containing 4 only exhibits the type I′ structure across the five symmetry-independent molecules examined. These solid-state observations are mirrored in the NMR solution structures, as well. The same trend was observed in Acbc-containing peptides 16 and 17. In the case of peptides 9 and 10, only one conformer of each was observed in the solid state, with N,N-dimethyl amide 9 adopting a type II′ β-hairpin and methyl ester 10 as a prehelical type I′ β-turn (Figure 17b). These data provide evidence in support of our previous hypothesis that N,N-dimethyl amide caps tend to favor nucleation of β-hairpin structures. Nevertheless, it is important to acknowledge the exceptions that we observed. The crystal structures of homologues 12 and 13 do not support this hypothesis, nor do those of peptides 24 and 25 (Chart 1). All of these crystal structures exhibit type II′ β-hairpin geometries despite the differences in C-terminal cap (Figures S3.12–S3.13 and S3.25–S3.26).
In terms of the influence of the C-terminal cap on the observed enantioselectivity in the bromination of quinazolinone 1 (eq 2), the effects appear to be subtle and context-dependent. While the N,N-dimethyl amides indeed prove to be superior to the methyl esters in lower-selectivity regimes (e.g., 75:25 vs 67:33 er with 24 and 25, respectively),28a this trend did not necessarily hold within the more selective catalyst series.28b In fact, examination of the methyl ester variant of 3 led to the discovery of 4, which was both more reactive and selective. However, N,N-dimethyl amide-containing 16 was modestly more selective than methyl ester-containing 17 (85:15 vs 83:17 er), suggesting that the effect of the N-terminal cap is modulated by the i+2 residue. The i+3 residue also seems to influence the end-cap effects, even when Acpc is incorporated at the i+2 position. For example, Val-containing amide 9 is more selective than its ester variant 10 (94:6 vs 91:9), and Phe-containing amide 12 is more selective than ester 13 (88:12 vs 86:14 er). Thus, it seems that the methyl esters may be beneficial above some selectivity threshold, especially when paired with Leu at the i+3 position. Structurally, it seems plausible that a weakened N–H(i)···O(i+3) β-hairpin H-bond65 could produce a significant geometric change, especially when paired with backbone conformational driving forces, such as the more helical tendencies of Acpc. Another factor that should be considered in this context is that tertiary amides are known to catalyze electrophilic bromination reactions.66 Thus, depending on the disposition of the other amides within the peptide scaffold, the enantioselectivity could be altered by the presence or absence of a C-terminal N,N-dimethyl amide; one might envision this to be either beneficial or deleterious to enantioselectivity depending on the overall conformation of the catalyst.
l-Pro Series
Thus far, the discussion has primarily focused on the d-Pro-containing sequences that comprise a majority (83%) of this structure library. These sequences have been observed to nucleate “mirror image” β-turns (types I′ and II′) in the solid state and in solution. However, the remaining 17% of this library are l-Pro-containing peptides, which are predisposed toward types I and II β-turns. Of the six entries, five are characterized as type II β-turns (15, 21, 30, 35–36), while only a single type I turn (37) is observed (Chart 1). According to the work of Gellman and co-workers, β-hairpin structures are generally disfavored in this class of homochiral (i.e., all l-containing) peptides relative to the corresponding heterochiral (e.g., d-Pro-containing at i+1) peptides.11a This is often attributed to incompatibility between the twist of the antiparallel strands with that of the type I or II loop-region.3,35 The result is that l-homochiral peptides equilibrate between β- and γ-turn geometries (Figure 2c). Our findings in the solid state and in solution support these observations.
Crystallographic Analysis of Homologous l-Pro Series
We were able to obtain X-ray data for a homologous series of l-Pro-containing peptides with the sequence Boc-Dmaa-Pro-Xaa-Leu-OMe (Figure 18), allowing further comparisons to be drawn to the d-Pro variants discussed previously (Figure 15a). The Aib-containing peptide 30 exhibits a type II β-turn in the solid state (Figure 18a). The i+3 residue is oriented such that a β-hairpin H-bond is not possible, in keeping with the previous studies of homochiral sequences.11a,35 The d-Pro variant of 30, peptide 25, does adopt a β-hairpin structure in the solid state (Figure 15a). The Aic-containing peptide 21 demonstrates these same overall structural features as 30 in both symmetry-independent molecules (21a, Figure 18a). This is in direct contrast to the d-Pro variants, peptides 19a and 19b, which nucleate prehelical, type I′ β-turns in the solid state (Figure 9). It is notable that no intermolecular H-bonds to the “free” methyl ester moiety are observed in 30 or 21 (Table S4.41). This suggests that the absence of a hairpin is not the result of lattice effects, but rather it reflects the conformational preferences of this class of peptides.
Figure 18.
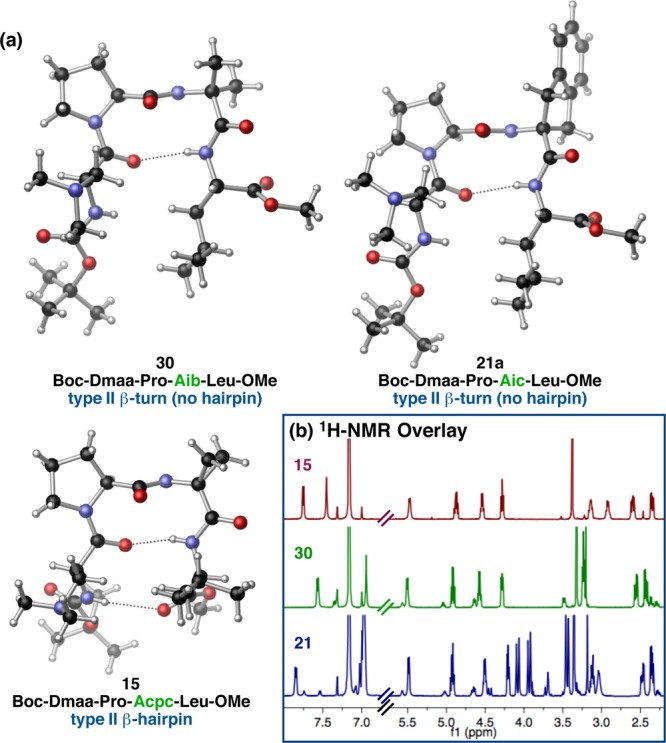
(a) Homologous series of l-Pro-containing peptides differing from one another only in the i+2 residue. Only one symmetry-independent conformer of 21 is shown (21a). (b) Stacked 1H NMR spectra of the peptides shown in (a) acquired under identical conditions (600 MHz, 0.01 M in C6D6, 20–24 °C).
Surprisingly, a type II β-hairpin structure is observed for the l-Pro-Acpc-containing peptide 15 (Figure 18a). The N–H(i)···O(i+3) hairpin H-bond is directional, with an N–H(i)···O(i+3) angle of 165(6)°, but the N(i)···O(i+3) distance is at the upper limit of what might be considered an H-bonding interaction, measuring 3.243(6) Å. For context, the average hairpin N···O length and N–H···O angle are 2.986 ± 0.148 Å and 162 ± 9°, respectively, over all hairpins in the library. DFT optimization of 15 did not shorten the N(i)···O(i+3) distance appreciably (Figure S6.07). These data suggest that the N–H(i)···O(i+3) H-bond of 15 is likely quite weak owing to the conformational pressures that tend to destabilize such interactions in short sequence β-hairpins. The d-Pro variant of 15, peptide 4, was found to exhibit the prehelical, type I′ β-turn structure in the solid state and in solution (Figure 7). This observation further underscores the uniqueness of Acpc, in that it has been found to exist within conformations, such as the type II β-hairpin of 15 and the prehelical, type I′ β-turn of 4, that have been observed less frequently. Overall, compensatory interactions may lead to the accommodation of otherwise unusual conformational profiles.
NMR Analysis of Homologous l-Pro Series
The solid-state observations of this l-Pro-series are largely recapitulated in solution. The 1H NMR spectra of 30 and 21 show some conformational heterogeneity; a set of minor (∼20%) peaks is observed for both peptides, which is consistent with a slow-exchange conformational equilibrium on the NMR time-scale (Figures 19b). While it is possible that this heterogeneity describes the equilibrium between β- and γ-turn structures, the interconversion of these forms is expected to be a fast exchange phenomenon in the context of Pro-Xaa turns,11a in which both H-bonded states are readily accessible and can interconvert with low barriers. Perhaps a more likely scenario is Pro trans/cis isomerization, which could be sufficiently slow on the NMR time scale to allow for the observation of discrete signals.67 This would intimate that peptides 30 and 21 equilibrate between well folded (trans Pro) and unfolded or loosely folded (cis Pro) states in solution as a function of their homochiral sequences. Although a heterochiral sequence, the X-ray crystal structure of peptide 32 provides an example of an unfolded cis-Pro rotamer (Figure S3.33). The spectrum of peptide 15, on the other hand, shows only one set of peaks, which may be attributed to either fast exchange of conformers or stabilization of a single, low-energy conformer. In light of our findings for similar peptides, especially the d-Pro-Acpc variants (e.g., 4), the former is a more plausible explanation. The relatively downfield chemical shift of the NH(Acpc) signal suggests that 15 samples β- and γ-turn conformations in solution.
Figure 19.

Measured NOE correlations for (a) peptide 3 alone, (b) a 1:1 mixture of 3 + 1, and (c) a 1:1 mixture of 3 + 38 (600 MHz, 0.01 M, C6D6, 25 °C). The NOE maps show the inter-residue NOE’s of peptide 3 and the intermolecular NOE’s between 3 and the titrated substrate. The Δ maps show how the NOE contacts change as a function of presumed substrate binding. The data are consistent with a more defined complex 3 + 1, while 3 + 38 is less rigid.
Based on additional NMR analyses, the data for peptide 15 and the major rotamers of peptides 21 and 30 are consistent with type II β-turn conformations in solution (0.01 M, C6D6, 20–24 °C). The ϕ(i) values calculated from 3JNH–Hα for all three peptides are tightly clustered: – 86° or −154° for 15, – 82° or −158° for 21, and −89° or −151° for 30 (Table S5.40). On the other hand, the calculated ϕ(i+3) dihedrals of 15 and 21 are both of −89° or −142°, while the those of 30 are −89° or −151°. These ϕ(i) and ϕ(i+3) values fit within the ranges expected in the proposed type II β-turns (Figure S5.15). Comparing the 1H NMR spectra, the l-Pro-containing peptides are different from the corresponding d-Pro-containing variants (Figure S5.17). For example, the NH(Leu), NH(Acpc), and NH(Dmaa) resonances are all noticeably upfield-shifted in 15 relative to 4, especially with regard to the NH(Dmaa) signal (Table S5.41). In peptide 21, the NH(Leu) signals are essentially equal to those of d-Pro-containing 19, whereas those of NH(Aic) and NH(Dmaa) are both nearly 0.3 ppm upfield-shifted. On the other hand, only the NH(Dmaa) resonance of peptide 30 is upfield-shifted, while the NH(Leu) and NH(Aib) chemical shifts are essentially equal to those of 25. These differences represent changes in the intramolecular H-bonding network that disfavor β-hairpin formation via the N–H(i)···O(i+3) H-bond in the l-Pro-containing peptides. Additionally, the upfield chemical shifts observed for the NH(i+2) signals in peptides 21 and 30 likely signify a solvent-exposed state, while the same signal remains moderately downfield-shifted in 15. The NOESY spectrum of peptide 15 shows NH(Dmaa) ↔ δ(Leu), NH(Acpc) ↔ NH(Leu), NH(Leu) ↔ α(Pro), and δ(Leu) ↔ α(Pro) NOE contacts, in addition to a very strong α(Pro) ↔ NH(Acpc) correlation (Figure S5.04). All of these data align with a type II β-turn/γ-turn conformational equilibrium in solution. Interestingly, neither gas phase DFT optimization nor solution phase NMR analysis provide evidence for the N–H(i)···O(i+3) hairpin H-bond observed in the crystal structure of 15. In peptide 21, the NOESY spectrum reveals a few relevant NOEs, including NH(Dmaa) ↔ β(Leu) and NMe2(Dmaa) ↔ β(Aic) correlations (Figure S5.09). Despite the absence of an α(Pro) ↔ NH(Aic) crosspeak, the cumulative NMR data support a nonhairpin type II β-turn conformation in solution, similar to the crystal structure of 21. Peptide 30 also shares this assignment, which is supported by OMe(Leu) ↔ NH(Aib) and NH(Aib) ↔ β(Dmaa) NOE contacts, as well as a very strong α(Pro) ↔ NH(Aib) correlation (Figure S5.12).
Connection to Enantioselective Catalysis in the Homologous l-Pro Series
Peptide 15 is a highly enantioselective catalyst for the bromination reaction presented in eq 2, delivering tribromide 2 in 11:89 er in favor of the opposite enantiomer provided by peptide 4, its Pro-Cα epimer.28b In other reactions we have studied employing Pro-Xaa-type catalysts, changes from d- to l-Pro (or vice versa) within an otherwise identical catalyst sequence typically provide lower enantioselectivity in favor of the opposite enantiomer.9a Indeed, peptides 21 and 30 follow this trend relative to their d-Pro variants; both 21 and 30 provide tribromide ent-2 in 45:55 er. Thus, the high level of enantiodivergence observed using Acpc-containing catalyst 15 is rather unusual, and it suggests that enantioinduction is primarily dictated by the turn sense. These findings are particularly intriguing in light of the structural observations of this homologous series. Of the three l-Pro-containing peptides examined, only peptide 15 adopts a well-folded conformation in the solid-state and in solution, while 21 and 30 are partially unfolded in solution owing to the minor population of cis-Pro rotamer (Figure 18b). Perhaps this apparent structural homogeneity is partially responsible for the enhanced selectivity of 15 relative to its i+2 homologues. While more folded than 21 and 30 on average, 15 also shows NOEs consistent with more than one folded state, notably β- and γ-turn conformations. Thus, conformational dynamics are possibly involved in the enhanced selectivity of 15.
Further Connections to Enantioselective Catalysis—Catalyst–Substrate Titration Studies
It is important to emphasize that the specific mechanism of Lewis base- and Brønsted base-catalyzed bromination of arenes is not definitively known in terms of the bond forming steps.66 Nevertheless, we are able to glean additional information relevant to catalysis by analyzing the solution structures of peptides in the presence of substrates.32a We thus examined catalyst-substrate mixtures employing NMR techniques. In particular, we compared the solution structure of catalyst 3 to those of 1:1 mixtures that also contained either a very good substrate (3 + 1) or a poor substrate (3 + 38) for the atroposelective bromination reaction described in eq 1.28a Each sample was prepared and analyzed using the same approach employed for peptide 3 alone (600 MHz, 0.01 M in C6D6, 25 °C). However, due to the difficulty in extracting 3JNH–Hα values for the i and i+3 residues of 3 in mixtures 3 + 1 and 3 + 38,68 as well as the differential impacts of substrate anisotropy on the chemical shits of 3, only the NOE data provide a straightforward interpretation. Comparison of the data acquired for 3 + 1 and 3 + 38 to each other and to that of peptide 3 alone reveals significant deviations in catalyst conformation as a function of association. The NOE contacts from the 1H–1H-NOESY spectra of 3, 3 + 1, and 3 + 38 are presented in Figure 19.
As discussed previously, the solution NMR data acquired for peptide 3 are most consistent with a type II′ β-hairpin structure that is possibly in flux with prehelical, type I′ β-turn forms. This assignment was made on the basis of 18 inter-residue NOE correlations (Figure 19a). Upon titration of quinazolinone 1, we observe a change in the NOESY spectrum of 3 that is consistent with a conformational change induced by substrate association. Mixture 3 + 1 exhibits 19 inter-residue NOEs, 13 of which are unique to 3 + 1 and are not observed in 3 alone. In addition, 15 intermolecular contacts between 3 and 1 are observed (Figure 19b). We also note that the 1H-resonance corresponding to NH(Acpc) shifts downfield by 0.93 ppm, which may implicate this amide in substrate docking.28a These through-space interactions suggest that 3 associates with quinazolinone 1 in a rigid type II′ β-hairpin conformation. This assignment is significantly less ambiguous than that of 3 alone, and is supported by α(d-Pro) ↔ NH(Acpc), β(Dmaa) ↔ β(Leu), and NH(Dmaa) ↔ δ(Leu) NOEs, as well as the α(d-Pro) ↔ δ(Leu), Boc(Dmaa) ↔ NMe2(Leu), and NH(Acpc) ↔ β(Acpc) NOEs unique to 3 + 1. In contrast, titration of quinazolinone 38 produces changes in the NOESY spectrum of 3 that are consistent with both a less defined folded state and a looser catalyst-substrate association (Figure 19c). The NOESY spectrum of 3 + 38 shows only nine inter-residue correlations, five of which are unique to the mixture. The strong α(d-Pro) ↔ NH(Acpc) and long-range NMe2(Dmaa) ↔ δ(Leu) NOEs support a type II′ β-hairpin conformation of 3 + 38, although the notably reduced quantity of through-space interactions relative to 3 + 1 suggest that the structure less rigid and possibly fluxional. Moreover, fewer intermolecular NOE contacts are observed between 3 and 38, and these nine correlations provide less coverage of the whole quinazolinone scaffold than in 3 + 1. Although NH(Acpc) also shifts significantly downfield (by 0.88 ppm) in 3 + 38, the data are consistent with a weaker interaction between 3 and 38 or perhaps multiple, interconverting binding orientations.
The emergent picture is one that suggests a higher degree of structural organization, and perhaps a more homogeneous conformation, for catalyst 3 as it associates with a good substrate (1). On the other hand, in the presence of a poor substrate (38), the conformational profile of 3 becomes even more ambiguous than 3 alone. These observations are consistent with the induced fit model that is well characterized for allosteric enzymes as they associate with their substrates69 and underscores the importance of dynamics in peptide-catalyzed, enantioselective reactions.
Conclusions
Our data show that a wide range of conformational states is available to low-molecular-weight tetrapeptides. In the solid state, multiple conformations of the same peptide sequence are manifest in a high incidence of symmetry-independent conformers and polymorphs within this structure library. Interpretations of NMR data are repeatedly compatible with various populated states in solution. On its own, this situation could be claimed to be unsurprising—short peptides are flexible and occupy shallow conformational energy landscapes. Yet, the direct experimental observation of allowable conformations in the solid state and in solution is rare. Moreover, the discovery and application of residue-specific conformational biases for nucleation of secondary structures remain an important tenet of peptide and protein design. These preferences are, of course, context dependent. The results reported herein, including redundant structural analyses of identical sequences and similar analyses of many related analogs employing both X-ray diffraction and NMR methods, culminate in the direct observation of multiple conformational states, and provide an aggregate of data that reveal a high level of variation. We find the level of diversity among peptides biased toward similar structural motifs to be striking. In response, computational analysis has allowed an assessment of relative energies for experimentally observed states, expanding intuition about the complex equilibria available to simple, folded peptides.
What emerges is a database of structural information that illuminates the conformational space available to these peptides, many of which are known to be catalysts for enantioselective reactions. What are the key structural elements that allow these low-molecular-weight, acyclic peptides to function as enantioselective catalysts? It seems that there are likely many, and the importance of catalyst dynamics is often one.32 The analysis of structural information from peptide-based catalyst libraries may also allow for the identification of correlations between structural features and enantioselectivity.70 For example, Figure 20 shows a rather good correlation (R2 = 0.89) between enantioselectivity (ee) in the atroposelective bromination of quinazolinone 1 (eq 2) and the crystallographic τ-angle of the i+2 position in a particular subset of this catalyst library.28 This factor was not among the design considerations when the catalyst library was conceived, and its connection to the observed enantioselectivity is not fully understood. Is it causative or coincidental? The recognition of this provocative trend from the structural database, combined with an improved, experimentally and computationally grounded appreciation for the catalyst conformational profiles, better positions future studies of flexible, peptide-based catalysts to be hypothesis driven.
Figure 20.
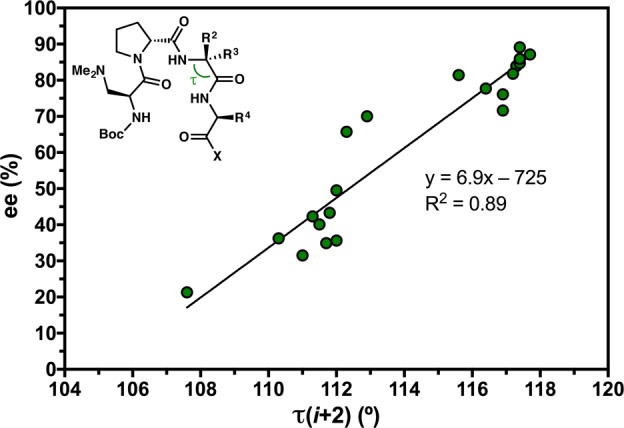
Linear correlation between τ(i+2) and ee of tribromide 2 observed for the reaction shown in eq 2. For structures with more than one symmetry-independent conformer, the average τ(i+2) value was used. Additional details are provided in the Supporting Information.
Acknowledgments
We would like to thank Byoungmoo Kim, Michael Giuliano, Alex Chinn, Christopher Shugrue, David Romney, William Jorgensen, Julian Tirado-Rives, and Matthew Diener for helpful discussions and insightful comments, as well as Guillaume Pelletier, Kimberly Barrett, and Newlyn Joseph for materials and data. A.J.M. gratefully acknowledges early support from the National Science Foundation Graduate Research Fellowship Program. All computational work was supported by the facilities and staff of the Yale University Faculty of Arts and Sciences High Performance Computing Center, and by the National Science Foundation under grant number CNS 08-21132, that partially funded acquisition of the facilities. We would also like to thank Simon Teat of the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory, for acquiring some X-ray crystal data presented herein. The ALS is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract Number DE-AC02-05CH11231. We are grateful to the National Institute of General Medical Sciences of the NIH (GM-068649 and GM-096403) for support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b11348.
Synthetic methods, characterization, X-ray crystallographic data, and solution NMR data for peptides 3–37, as well as additional analyses of structural trends and DFT computational data (PDF)
CIF data for 3a,b (1412920), 3c (1453125), 4a (1453124), 4b–e, 5, 6, 7a,b, 8, 9, 10, 11, 12, 13, 14, 15, 16a,b, 16c, 17a,b, 18a,b, 19a,b, 20a,b, 21a,b, 22a,b, 23, 24, 25, 26, 27, 28, 29a,b, 30, 31, 32a,b, and 33a,b (ZIP)
The authors declare no competing financial interest.
Notes
Crystallographic data are deposited with the Cambridge Crystallographic Data Centre (CCDC) under the following accession numbers: 4b–e (1510507), 5 (1510503), 6 (1510502), 7a,b (1510506), 8 (1510504), 9 (1510505), 10 (1510508), 11 (1510516), 12 (1510513), 13 (1510509), 14 (1510514), 15 (1510510), 16a,b (1510512), 16c (1510511), 17a,b (1510515), 18a,b (1510517), 19a,b (1510525), 20a,b (1510518), 21a,b (1510519), 22a,b (1510522), 23 (1510526), 24 (1510521), 25 (1510520), 26 (1510524), 27 (1510523), 28 (1510530), 29a,b (1510527), 30 (1510531), 31 (1510528), 32a,b (1510529), and 33a,b (1510532).
Supplementary Material
References
- a Knowles J. R. Nature 1991, 350, 121–124. 10.1038/350121a0. [DOI] [PubMed] [Google Scholar]; b Kirby A. J. Angew. Chem., Int. Ed. Engl. 1996, 35, 706–724. 10.1002/anie.199607061. [DOI] [Google Scholar]; c Smith A. J. T.; Müller R.; Toscano M. D.; Kast P.; Hellinga H. W.; Hilvert D.; Houk K. N. J. Am. Chem. Soc. 2008, 130, 15361–15373. 10.1021/ja803213p. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Creighton T. E.Proteins: Structures and Molecular Properties, 2nd ed.; W. H. Freeman & Co.: New York, 1993. [Google Scholar]
- a Baker D. Nature 2000, 405, 39–42. 10.1038/35011000. [DOI] [PubMed] [Google Scholar]; b Dobson C. M.; Šali A.; Karplus M. Angew. Chem., Int. Ed. 1998, 37, 868–893. 10.1002/(SICI)1521-3773(19980420)37:7<868::AID-ANIE868>3.3.CO;2-8. [DOI] [PubMed] [Google Scholar]; c Valentine J. S., Ed. Acc. Chem. Res. 1998, 31, 697 ( (Protein Folding Special Issue), ). [Google Scholar]
- a Wilmot C. M.; Thornton J. M. J. Mol. Biol. 1988, 203, 221–232. 10.1016/0022-2836(88)90103-9. [DOI] [PubMed] [Google Scholar]; b Hutchinson E. G.; Thornton J. M. Protein Sci. 1994, 3, 2207–2216. 10.1002/pro.5560031206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews on synthetic foldamers, see:; a Guichard G.; Huc I. Chem. Commun. 2011, 47, 5933–5941. 10.1039/c1cc11137j. [DOI] [PubMed] [Google Scholar]; b Goodman C. M.; Choi S.; Shandler S.; DeGrado W. F. Nat. Chem. Biol. 2007, 3, 252–262. 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Venkatraman J.; Shankarama S. C.; Balaram P. Chem. Rev. 2001, 101, 3131–3152. 10.1021/cr000053z. [DOI] [PubMed] [Google Scholar]; d Hill D. J.; Mio M. J.; Prince R. B.; Hughes T. S.; Moore J. S. Chem. Rev. 2001, 101, 3893–4012. 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]; e Gellman S. H. Curr. Opin. Chem. Biol. 1998, 2, 717–725. 10.1016/S1367-5931(98)80109-9. [DOI] [PubMed] [Google Scholar]
- For select examples of peptide-based dynamic foldamers and molecular devices, see:; a Le Bailly B. A. F.; Clayden J. Chem. Commun. 2016, 52, 4852–4863. 10.1039/C6CC00788K. [DOI] [PubMed] [Google Scholar]; b Mazzier D.; Crisma M.; De Poli M.; Marafon G.; Peggion C.; Clayden J.; Moretto A. J. Am. Chem. Soc. 2016, 138, 8007–8018. 10.1021/jacs.6b04435. [DOI] [PubMed] [Google Scholar]; c Brown R. A.; Diemer V.; Webb S. J.; Clayden J. Nat. Chem. 2013, 5, 853–860. 10.1038/nchem.1747. [DOI] [PubMed] [Google Scholar]; d Lewandowski B.; De Bo G.; Ward J. W.; Papmeyer M.; Kuschel S.; Aldegunde M. J.; Gramlich P. M.; Heckmann D.; Goldup S. M.; D’Douza D. M.; Fernandes A. E.; Leigh D. A. Science 2013, 339, 189–193. 10.1126/science.1229753. [DOI] [PubMed] [Google Scholar]; e Meillon J.-C.; Voyer N.; Biron É.; Sanschagrin F.; Stoddart J. F. Angew. Chem., Int. Ed. 2000, 39, 143–145. 10.1002/(SICI)1521-3773(20000103)39:1<143::AID-ANIE143>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]; f Dado G. P.; Gellman S. H. J. Am. Chem. Soc. 1993, 115, 12609–12610. 10.1021/ja00079a060. [DOI] [Google Scholar]
- Miller S. J. Acc. Chem. Res. 2004, 37, 601–610. 10.1021/ar030061c. [DOI] [PubMed] [Google Scholar]
- For reviews, see:; a Lewandowski B.; Wennemers H. Curr. Opin. Chem. Biol. 2014, 22, 40–46. 10.1016/j.cbpa.2014.09.011. [DOI] [PubMed] [Google Scholar]; b Wennemers H. Chem. Commun. 2011, 47, 12036–12041. 10.1039/c1cc15237h. [DOI] [PubMed] [Google Scholar]; c Colby Davie E. A.; Mennen S. M.; Xu Y.; Miller S. J. Chem. Rev. 2007, 107, 5759–5812. 10.1021/cr068377w. [DOI] [PubMed] [Google Scholar]
- For a review, see:; a Revell J. D.; Wennemers H. Curr. Opin. Chem. Biol. 2007, 11, 269–278. 10.1016/j.cbpa.2007.05.015. [DOI] [PubMed] [Google Scholar]; For select examples, see:; b Lichtor P. A.; Miller S. J. ACS Comb. Sci. 2011, 13, 321–326. 10.1021/co200010v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Copeland G. T.; Miller S. J. J. Am. Chem. Soc. 1999, 121, 4306–4307. 10.1021/ja984139+. [DOI] [Google Scholar]
- For select examples, see:; a Jarvo E. R.; Copeland G. T.; Papaioannou N.; Bonitatebus P. J.; Miller S. J. J. Am. Chem. Soc. 1999, 121, 11638–11643. 10.1021/ja9931776. [DOI] [Google Scholar]; b Peris G.; Jakobsche C. E.; Miller S. J. J. Am. Chem. Soc. 2007, 129, 8710–8711. 10.1021/ja073055a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For early studies of β-turns in proteins, see:; a Rose G. D.; Gierasch L. M.; Smith J. A. Adv. Protein Chem. 1985, 37, 1–109. 10.1016/S0065-3233(08)60063-7. [DOI] [PubMed] [Google Scholar]; b Richardson J. S. Adv. Protein Chem. 1981, 34, 167–339. 10.1016/S0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]; c Wilmot C. M.; Thornton J. M. Protein Eng., Des. Sel. 1990, 3, 479–493. 10.1093/protein/3.6.479. [DOI] [PubMed] [Google Scholar]; d Lewis P. N.; Momany F. A.; Scheraga H. A. Biochim. Biophys. Acta, Protein Struct. 1973, 303, 211–229. 10.1016/0005-2795(73)90350-4. [DOI] [PubMed] [Google Scholar]
- a Haque T. S.; Little J. C.; Gellman S. H. J. Am. Chem. Soc. 1996, 118, 6975–6985. 10.1021/ja960429j. [DOI] [Google Scholar]; b Liang G.-B.; Rito C. J.; Gellman S. H. J. Am. Chem. Soc. 1992, 114, 4440–4442. 10.1021/ja00037a083. [DOI] [Google Scholar]; c Jiménez A. I.; Cativiela C.; Aubry A.; Marraud M. J. Am. Chem. Soc. 1998, 120, 9452–9459. 10.1021/ja9807439. [DOI] [Google Scholar]; d Boussard G.; Marraud M. J. Am. Chem. Soc. 1985, 107, 1825–1828. 10.1021/ja00293a004. [DOI] [Google Scholar]; e Ravi A.; Venkataram Prasad B. V.; Balaram P. J. Am. Chem. Soc. 1983, 105, 105–109. 10.1021/ja00339a019. [DOI] [Google Scholar]
- a Sibanda B. L.; Thornton J. M. Nature 1985, 316, 170–174. 10.1038/316170a0. [DOI] [PubMed] [Google Scholar]; b Sibanda B. L.; Blundell T. L.; Thornton J. M. J. Mol. Biol. 1989, 206, 759–777. 10.1016/0022-2836(89)90583-4. [DOI] [PubMed] [Google Scholar]; c Blanco F. J.; Jiménez Angeles M.; Herranz J.; Rico M.; Santoro J.; Nieto J. L. J. Am. Chem. Soc. 1993, 115, 5887–5888. 10.1021/ja00066a092. [DOI] [Google Scholar]
- For discussions of H-bonding as it pertains to catalysis, see:; a Knowles R. R.; Jacobsen E. N. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20678–20685. 10.1073/pnas.1006402107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Simón L.; Goodman J. M. J. Org. Chem. 2010, 75, 1831–1840. 10.1021/jo901503d. [DOI] [PubMed] [Google Scholar]; c Bernardi L.; Fochi M.; Comes Franchini M.; Ricci A. Org. Biomol. Chem. 2012, 10, 2911–2922. 10.1039/c2ob07037e. [DOI] [PubMed] [Google Scholar]; d Mader M. M.; Bartlett P. A. Chem. Rev. 1997, 97, 1281–1301. 10.1021/cr960435y. [DOI] [PubMed] [Google Scholar]
- a Copeland G. T.; Jarvo E. R.; Miller S. J. J. Org. Chem. 1998, 63, 6784–6785. 10.1021/jo981642w. [DOI] [PubMed] [Google Scholar]; b Cowen B. J.; Saunders L. B.; Miller S. J. J. Am. Chem. Soc. 2009, 131, 6105–6107. 10.1021/ja901279m. [DOI] [PubMed] [Google Scholar]; c Metrano A. J.; Miller S. J. J. Org. Chem. 2014, 79, 1542–1554. 10.1021/jo402828f. [DOI] [PubMed] [Google Scholar]
- For ease of reference,
we provide definitions
of the following structural parameters discussed throughout the manuscript:
ϕ, the backbone dihedral defined for rotation about C′–N–Cα–C′;
ψ, the backbone dihedral defined for rotation about N–Cα–C′–N;
ω, the backbone dihedral defined for rotation about Cα–C′–N–Cα,
which, due to the planarity of the amide (C′–N) bond,
is typically either 0° or 180°; χ1, the
side-chain dihedral defined for rotation about N–Cα–Cβ–Cγ;
and τ, the N–Cα–C′ main-chain angle.
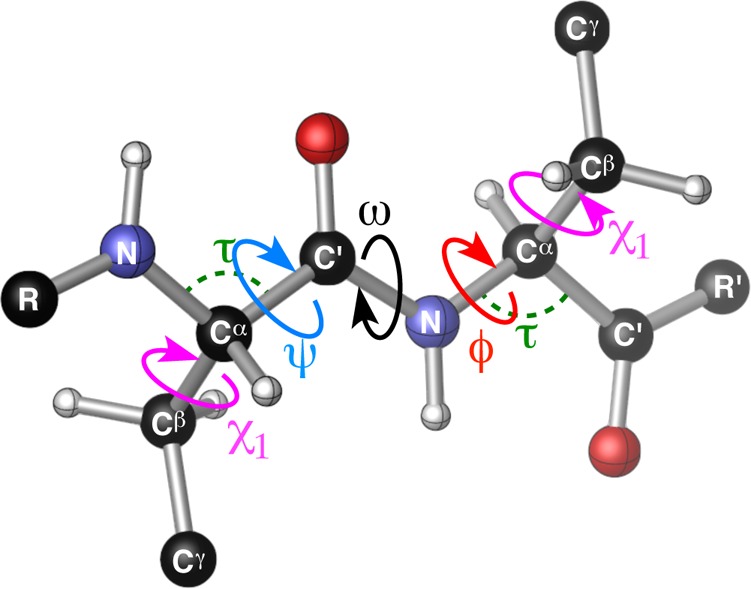
- For reviews on β-turns in biological and medicinal contexts, see:; a Craik D. J.; Fairlie D. P.; Liras S.; Price D. Chem. Biol. Drug Des. 2013, 81, 136–147. 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]; b Robinson J. A. Acc. Chem. Res. 2008, 41, 1278–1288. 10.1021/ar700259k. [DOI] [PubMed] [Google Scholar]; c Che Y.; Books B. R.; Marshall G. R. J. Comput.-Aided Mol. Des. 2006, 20, 109–130. 10.1007/s10822-006-9040-8. [DOI] [PubMed] [Google Scholar]; d Suat K.; Jois S. D. S. Curr. Pharm. Des. 2003, 9, 1209–1224. 10.2174/1381612033454900. [DOI] [PubMed] [Google Scholar]; e Grauer A.; König B. Eur. J. Org. Chem. 2009, 2009, 5099–5111. 10.1002/ejoc.200900599. [DOI] [Google Scholar]
- For some examples of β-turn peptidomimetics, see:; a Souers A. J.; Ellman J. A. Tetrahedron 2001, 57, 7431–7448. 10.1016/S0040-4020(01)00680-9. [DOI] [Google Scholar]; b Whitby L. R.; Ando Y.; Setola V.; Vogt P. K.; Roth B. L.; Boger D. L. J. Am. Chem. Soc. 2011, 133, 10184–10194. 10.1021/ja201878v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Low K. E.; Ler S.; Chen K. J.; Campbell R. L.; Hickey J. L.; Tan J.; Scully C. C. G.; Davies P. L.; Yudin A. K.; Zaretsky S. J. Med. Chem. 2016, 59, 5403–5415. 10.1021/acs.jmedchem.6b00267. [DOI] [PubMed] [Google Scholar]; d Eckhardt B.; Grosse W.; Essen L.-O.; Geyer A. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 18336–18341. 10.1073/pnas.1004187107. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Van der Poorten O.; Knuhtsen A.; Pedersen D. S.; Battet S.; Tourwé D.. J. Med. Chem. 2016, ASAP, DOI: 10.1021/acs.jmedchem.6b01029. [DOI]
- a Gunasekaran K.; Gomathi L.; Ramakrishnan C.; Chandrasekhar J.; Balaram P. J. Mol. Biol. 1998, 284, 1505–1516. 10.1006/jmbi.1998.2154. [DOI] [PubMed] [Google Scholar]; b Hayward S. Protein Sci. 2001, 10, 2219–2227. 10.1110/ps.23101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of extended β-hairpin structures in short peptides, see:; a Karle I. L.; Awasthi S. K.; Balaram P. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 8189–8193. 10.1073/pnas.93.16.8189. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Aravinda S.; Harini V. V.; Shamala N.; Das C.; Balaram P. Biochemistry 2004, 43, 1832–1846. 10.1021/bi035522g. [DOI] [PubMed] [Google Scholar]; c Aravinda S.; Raghavender U. S.; Rai R.; Harini V. V.; Shamala N.; Balaram P. Org. Biomol. Chem. 2013, 11, 4220–4231. 10.1039/c3ob25777k. [DOI] [PubMed] [Google Scholar]
- d-Pro-Aib examples:; a Barrett K. T.; Miller S. J. J. Am. Chem. Soc. 2013, 135, 2963–2966. 10.1021/ja400082x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Han S.; Miller S. J. J. Am. Chem. Soc. 2013, 135, 12414–12421. 10.1021/ja406067v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fowler B. S.; Mikochik P. J.; Miller S. J. J. Am. Chem. Soc. 2010, 132, 2870–2871. 10.1021/ja9107897. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Saunders L. B.; Cowen B. J.; Miller S. J. Org. Lett. 2010, 12, 4800–4803. 10.1021/ol101947s. [DOI] [PubMed] [Google Scholar]; e Lewis C. A.; Miller S. J. Angew. Chem., Int. Ed. 2006, 45, 5616–5619. 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]; d-Pro-Acpc example:; f Shugrue C. R.; Miller S. J. Angew. Chem., Int. Ed. 2015, 54, 11173–11176. 10.1002/anie.201505898. [DOI] [PMC free article] [PubMed] [Google Scholar]; d-Pro-Aic example:; g Mbofana C. T.; Miller S. J. J. Am. Chem. Soc. 2014, 136, 3285–3292. 10.1021/ja412996f. [DOI] [PubMed] [Google Scholar]; l-Pro-Cle examples:; h Fiori K. W.; Puchlopek A. L. A.; Miller S. J. Nat. Chem. 2009, 1, 630–634. 10.1038/nchem.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur M. W.; Thornton J. M. J. Mol. Biol. 1991, 218, 397–412. 10.1016/0022-2836(91)90721-H. [DOI] [PubMed] [Google Scholar]
- a Haque T. S.; Little J. C.; Gellman S. H. J. Am. Chem. Soc. 1994, 116, 4105–4106. 10.1021/ja00088a067. [DOI] [Google Scholar]; b Stanger H. E.; Gellman S. H. J. Am. Chem. Soc. 1998, 120, 4236–4237. 10.1021/ja973704q. [DOI] [Google Scholar]; c Raghothama S. R.; Awasthi S. K.; Balaram P. J. Chem. Soc., Perkin Trans. 2 1998, 137–143. 10.1039/a703331a. [DOI] [Google Scholar]; d Yan Y.; Tropsha A.; Hermans J.; Erickson B. W. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 7898–7902. 10.1073/pnas.90.16.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews on γ-turns in proteins, see:; a Crisma M.; De Zotti M.; Moretto A.; Peggion C.; Drouillat B.; Wright K.; Couty F.; Toniolo C.; Formaggio F. New J. Chem. 2015, 39, 3208–3216. 10.1039/C4NJ01564A. [DOI] [Google Scholar]; b Milner-White E. J. J. Mol. Biol. 1990, 216, 385–397. 10.1016/S0022-2836(05)80329-8. [DOI] [PubMed] [Google Scholar]
- a Toniolo C.; Crisma M.; Formaggio F.; Peggion C. Biopolymers 2001, 60, 396–419. 10.1002/1097-0282(2001)60:6<396::AID-BIP10184>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]; b Toniolo C.; Benedetti E. Macromolecules 1991, 24, 4004–4009. 10.1021/ma00014a006. [DOI] [Google Scholar]; c Fabiano N.; Valle G.; Crisma M.; Toniolo C.; Saviano M.; Lombardi A.; Isernia C.; Pavone V.; Di Blasio B.; Pedone C.; Benedetti E. Int. J. Pept. Protein Res. 1993, 42, 459–465. 10.1111/j.1399-3011.1993.tb00155.x. [DOI] [PubMed] [Google Scholar]; d Toniolo C.; Crisma M.; Formaggio F.; Benedetti E.; Santini A.; Iacovino R.; Saviano M.; Di Blasio B.; Pedone C.; Kamphuis J. Pept. Sci. 1996, 40, 519–527. [Google Scholar]
- a Prasad B. V. V; Balaram H.; Balaram P. Biopolymers 1982, 21, 1261–1273. 10.1002/bip.360210702. [DOI] [Google Scholar]; b Formaggio F.; Barazza A.; Bertocco A.; Toniolo C.; Broxterman Q. B.; Kaptein B.; Brasola E.; Pengo P.; Pasquato L.; Scrimin P. J. Org. Chem. 2004, 69, 3849–3856. 10.1021/jo040107v. [DOI] [PubMed] [Google Scholar]
- For discussions of “privileged” chiral scaffolds, see:; a Yoon T. P.; Jacobsen E. N. Science 2003, 299, 1691–1693. 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]; b Karukurichi K. R.; Fei X.; Swyka R. A.; Broussey S.; Shen W.; Dey S.; Roy S. K.; Berkowitz D. B. Sci. Adv. 2015, 1, e1500066–20. 10.1126/sciadv.1500066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For other examples of Pro-Xaa β-turns in asymmetric catalysts, see:; a Greenfield S. J.; Agarkov A.; Gilbertson S. R. Org. Lett. 2003, 5, 3069–3072. 10.1021/ol035097j. [DOI] [PubMed] [Google Scholar]; b Krattiger P.; Kovàsy R.; Revell J. D.; Ivan S.; Wennemers H. Org. Lett. 2005, 7, 1101–1103. 10.1021/ol0500259. [DOI] [PubMed] [Google Scholar]; c Agarkov A.; Greenfield S. J.; Xie D.; Pawlick R.; Starkey G.; Gilbertson S. R. Biopolymers 2006, 84, 48–73. 10.1002/bip.20395. [DOI] [PubMed] [Google Scholar]; d Revell J. D.; Wennemers H. Adv. Synth. Catal. 2008, 350, 1046–1052. 10.1002/adsc.200800053. [DOI] [Google Scholar]; e Akagawa K.; Akabane H.; Sakamoto S.; Kudo K. Org. Lett. 2008, 10, 2035–2037. 10.1021/ol800031p. [DOI] [PubMed] [Google Scholar]; f Wiesner M.; Revell J. D.; Wennemers H. Angew. Chem., Int. Ed. 2008, 47, 1871–1874. 10.1002/anie.200704972. [DOI] [PubMed] [Google Scholar]; g Wiesner M.; Revell J. D.; Tonazzi S.; Wennemers H. J. Am. Chem. Soc. 2008, 130, 5610–5611. 10.1021/ja801027s. [DOI] [PubMed] [Google Scholar]; h Wiesner M.; Neuburger M.; Wennemers H. Chem.−Eur. J. 2009, 15, 10103–10109. 10.1002/chem.200901021. [DOI] [PubMed] [Google Scholar]; i Chen P.; Qu J. J. Org. Chem. 2011, 76, 2994–3004. 10.1021/jo200403g. [DOI] [PubMed] [Google Scholar]; j Wiesner M.; Revell J. D.; Wennemers H. Angew. Chem., Int. Ed. 2013, 52, 7228–7232. [DOI] [PubMed] [Google Scholar]; k Liao R.-Z.; Santoro S.; Gotsev M.; Marcelli T.; Himo F. ACS Catal. 2016, 6, 1165–1171. 10.1021/acscatal.5b02131. [DOI] [Google Scholar]; l Grünenfelder C. E.; Kisunzu J. K.; Wennemers H. Angew. Chem., Int. Ed. 2016, 55, 8571–8574. 10.1002/anie.201602230. [DOI] [PubMed] [Google Scholar]; m Ueda A.; Umeno T.; Doi M.; Akagawa K.; Kudo K.; Tanaka M. J. Org. Chem. 2016, 81, 6343–6356. 10.1021/acs.joc.6b00982. [DOI] [PubMed] [Google Scholar]
- a Diener M. E.; Metrano A. J.; Kusano S.; Miller S. J. J. Am. Chem. Soc. 2015, 137, 12369–12377. 10.1021/jacs.5b07726. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Metrano A. J.; Abascal N. C.; Mercado B. Q.; Paulson E. K.; Miller S. J. Chem. Commun. 2016, 52, 4816–4819. 10.1039/C6CC01428C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For peptide 34:; a Blank J. T.; Miller S. J. Biopolymers 2006, 84, 38–47. 10.1002/bip.20392. [DOI] [PubMed] [Google Scholar]; For peptide 35, see ref (9a). For peptide 36:; b Jakobsche C. E.; Peris G.; Miller S. J. Angew. Chem., Int. Ed. 2008, 47, 6707–6711. 10.1002/anie.200802223. [DOI] [PMC free article] [PubMed] [Google Scholar]; For peptide 37:; c Blank J. T.; Guerin D. J.; Miller S. J. Org. Lett. 2000, 2, 1247–1249. 10.1021/ol005700n. [DOI] [PubMed] [Google Scholar]
- A search of the Cambridge Structural Database (CSD) on 17 October 2016 returned 424 Pro-Xaa-containing peptides, about half of which were cyclic. Of the 191 noncyclic entries: (1) 54 contained α,α-disubstituted residues as Xaa; (2) 12 contained Acpc as Xaa; and (3) 19 were tetramers.
- For examples in the context of β-turns, see:; a Karnes M. A.; Schettler S. L.; Werner H. M.; Kurz A. F.; Horne W. S.; Lengyel G. A. Org. Lett. 2016, 18, 3902–3905. 10.1021/acs.orglett.6b01936. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Burke N. L.; DeBlase A. F.; Redwine J. G.; Hopkins J. R.; McLuckey S. A.; Zwier T. S. J. Am. Chem. Soc. 2016, 138, 2849–2857. 10.1021/jacs.6b00093. [DOI] [PubMed] [Google Scholar]; For related examples, see:; c Fierman M. B.; O’Leary D. J.; Steinmetz W. E.; Miller S. J. J. Am. Chem. Soc. 2004, 126, 6967–6971. 10.1021/ja049661c. [DOI] [PubMed] [Google Scholar]; d Lawson K. V.; Rose T. E.; Harran P. G. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E3753–E3760. 10.1073/pnas.1311706110. [DOI] [PMC free article] [PubMed] [Google Scholar]; See also ref (22d).
- a Abascal N. C.; Miller S. J. Org. Lett. 2016, 18, 4646–4649. 10.1021/acs.orglett.6b02282. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Alford J. S.; Abascal N. C.; Shugrue C. R.; Colvin S. M.; Romney D. K.; Miller S. J. ACS Cent. Sci. 2016, 2, 733–739. 10.1021/acscentsci.6b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Abascal N. C.; Lichtor P. A.; Giuliano M. W.; Miller S. J. Chem. Sci. 2014, 5, 4504–4511. 10.1039/C4SC01440E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel W. Angew. Chem., Int. Ed. 2014, 53, 8605–8613. 10.1002/anie.201402118. [DOI] [PubMed] [Google Scholar]; Section 4.4 is particularly relevant.
- For discussion of 310-helices, see:Tonlolo C.; Benedetti E. Trends Biochem. Sci. 1991, 16, 350–353. 10.1016/0968-0004(91)90142-I. [DOI] [PubMed] [Google Scholar]
- This virtual dihedral (ϖ) was previously used to describe the twist of an oligopeptide biased toward a β-turn geometry. See:Gunasekaran K.; Ramakrishnan C.; Balaram P. Protein Eng., Des. Sel. 1997, 10, 1131–1141. 10.1093/protein/10.10.1131. [DOI] [PubMed] [Google Scholar]
- For reviews on DFT:; a Pople J. A.; Gill P. M. W.; Johnson B. G. Chem. Phys. Lett. 1992, 199, 557–560. 10.1016/0009-2614(92)85009-Y. [DOI] [Google Scholar]; b Parr R. G.; Yang W.. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, 1989. [Google Scholar]; For M06-2X:; c Zhao Y.; Truhlar D. G. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- For reviews on polymorphism, see:; a Cruz-Cabeza A. J.; Bernstein J. Chem. Rev. 2014, 114, 2170–2191. 10.1021/cr400249d. [DOI] [PubMed] [Google Scholar]; b Nangia A. Acc. Chem. Res. 2008, 41, 595–604. 10.1021/ar700203k. [DOI] [PubMed] [Google Scholar]
- Wang A. C.; Bax A. J. Am. Chem. Soc. 1996, 118, 2483–2494. 10.1021/ja9535524. [DOI] [Google Scholar]
- a Gardner R. R.; Gellman S. H. J. Am. Chem. Soc. 1995, 117, 10411–10412. 10.1021/ja00146a038. [DOI] [Google Scholar]; b Gellman S. H.; Dado G. P.; Liang G.-B.; Adams B. R. J. Am. Chem. Soc. 1991, 113, 1164–1173. 10.1021/ja00004a016. [DOI] [Google Scholar]
- a Wider G.; Macura S.; Kumar A.; Ernst R. R.; Wüthrich K. J. Magn. Reson. 1984, 56, 207–234. 10.1016/0022-2364(84)90099-4. [DOI] [Google Scholar]; b Jeener J.; Meier B. H.; Bachmann P.; Ernst R. R. J. Chem. Phys. 1979, 71, 4546–4553. 10.1063/1.438208. [DOI] [Google Scholar]; c Awasthi S. K.; Raghothama S.; Balaram P. Biochem. Biophys. Res. Commun. 1995, 216, 375–381. 10.1006/bbrc.1995.2634. [DOI] [PubMed] [Google Scholar]
- The batch of peptide 16 that gave rise to polymorphs 16a,b and 16c was catalytically competent in the atroposelective bromination of 1 described in eq 1. Additionally, the NMR spectrum of 16 is consistent with protonated catalyst (see the Supporting Information). As such, it is unlikely that our stock of 16c is protonated. The source of the HCl equivalents remains unclear, but we speculate that the 16a,b crystal may have nucleated on a grain of adventitious, wet NaCl.
- For a computational study of cyclobutane puckering, see:Glendening E. D.; Halpern A. M. J. Phys. Chem. A 2005, 109, 635–642. 10.1021/jp0405097. [DOI] [PubMed] [Google Scholar]
- Zhou A. Q.; O’Hern C. S.; Regan L. Protein Sci. 2011, 20, 1166–1171. 10.1002/pro.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünenfelder C. E.; Kisunzu J. K.; Trapp N.; Kastl R.; Wennemers H.. Biopolymers (Pept. Sci.) 2016, ASAP, DOI: 10.1002/bip.22912. [DOI] [PubMed]
- Steiner T. Acta Crystallogr., Sect. B: Struct. Sci. 2000, B56, 673–676. 10.1107/S0108768100002652. [DOI] [PubMed] [Google Scholar]
- a Choi S. H.; Guzei I. A.; Spencer L. C.; Gellman S. H. J. Am. Chem. Soc. 2010, 132, 13879–13885. 10.1021/ja1062532. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Choi S. H.; Guzei I. A.; Spencer L. C.; Gellman S. H. J. Am. Chem. Soc. 2009, 131, 2917–2924. 10.1021/ja808168y. [DOI] [PubMed] [Google Scholar]
- Thompson H. P. G.; Day G. M. Chem. Sci. 2014, 5, 3173–3182. 10.1039/c4sc01132e. [DOI] [Google Scholar]
- For a discussion of amide pyramidalization in proteins, see:; a MacArthur M. W.; Thornton J. M. J. Mol. Biol. 1996, 264, 1180–1195. 10.1006/jmbi.1996.0705. [DOI] [PubMed] [Google Scholar]; For related examples, see:; b Cruz-Cabeza A. J.; Day G. M.; Motherwell W. D. S.; Jones W. Cryst. Growth Des. 2006, 6, 1858–1866. 10.1021/cg0601756. [DOI] [Google Scholar]; c Alemán C.; Jiménez A. I.; Cativiela C.; Pérez J. J.; Casanovas J. J. Phys. Chem. B 2005, 109, 11836–11841. 10.1021/jp050141t. [DOI] [PubMed] [Google Scholar]
- For example, see:; a Brock C. P.; Minton R. P. J. Am. Chem. Soc. 1989, 111, 4586–4593. 10.1021/ja00195a010. [DOI] [Google Scholar]; b Pearson A. D.; Mills J. H.; Song Y.; Nasertorabi F.; Han G. W.; Baker D.; Stevens R. C.; Schultz P. G. Science 2015, 347, 863–867. 10.1126/science.aaa2424. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Romney D. K.; Miller S. J. Science 2015, 347, 829. 10.1126/science.aaa5623. [DOI] [PubMed] [Google Scholar]; See also ref (47).
- Ramachandran G. N.; Ramakrishnan C.; Sasisekharan V. J. Mol. Biol. 1963, 7, 95–99. 10.1016/S0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- a Hinderaker M. P.; Raines R. T. Protein Sci. 2003, 12, 1188–1194. 10.1110/ps.0241903. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Choudhary A.; Gandla D.; Krow G. R.; Raines R. T. J. Am. Chem. Soc. 2009, 131, 7244–7246. 10.1021/ja901188y. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bartlett G. J.; Choudhary A.; Raines R. T.; Woolfson D. N. Nat. Chem. Biol. 2010, 6, 615–620. 10.1038/nchembio.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Duddy W. J.; Nissink J. W. M.; Allen F. H.; Milner-White E. J. Protein Sci. 2004, 13, 3051–3055. 10.1110/ps.04920904. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Abbadi A.; Mcharfi M.; Aubry A.; Premilat S.; Boussard G.; Marraud M. J. Am. Chem. Soc. 1991, 113, 2729–2735. 10.1021/ja00007a056. [DOI] [Google Scholar]; c Marraud M.; Aubry A. Biopolymers 1996, 40, 45–83. 10.1002/(SICI)1097-0282(1996)40:1<45::AID-BIP3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; See also ref (29c).
- For a discussion of Cγ-puckering in Pro, see:Shoulders M. D.; Kotch F. W.; Choudhary A.; Guzei I. A.; Raines R. T. J. Am. Chem. Soc. 2010, 132, 10857–10865. 10.1021/ja103082y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapovalov M. V.; Dunbrack R. L. Proteins: Struct., Funct., Genet. 2007, 66, 279–303. 10.1002/prot.21150. [DOI] [PubMed] [Google Scholar]
- a Zhou A. Q.; O’Hern C. S.; Regan L. Biophys. J. 2012, 102, 2345–2352. 10.1016/j.bpj.2012.01.061. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhou A. Q.; Caballero D.; O’Hern C. S.; Regan L. Biophys. J. 2013, 105, 2403–2411. 10.1016/j.bpj.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Caballero D.; Smith W. W.; O’Hern C. S.; Regan L. Proteins: Struct., Funct., Genet. 2015, 83, 1488–1499. 10.1002/prot.24837. [DOI] [PubMed] [Google Scholar]; d Caballero D.; Virrueta A.; O’Hern C. S.; Regan L. Protein Eng., Des. Sel. 2016, 29, 367–376. 10.1093/protein/gzw027. [DOI] [PubMed] [Google Scholar]
- For a review of H-bonding in the solid state, see:Steiner T. Angew. Chem., Int. Ed. 2002, 41, 48–76. 10.1002/1521-3773(20020104)41:1<48::AID-ANIE48>3.0.CO;2-U. [DOI] [Google Scholar]
- For a discussion of τ-angle distributions, see:; a Malathy Sony S. M.; Saraboji K.; Sukumar N.; Ponnuswamy M. N. Biophys. Chem. 2006, 120, 24–31. 10.1016/j.bpc.2005.07.012. [DOI] [PubMed] [Google Scholar]; See also ref (43). For a discussion of τ-angles in Pro-residues, see:; b Karplus P. A. Protein Sci. 1996, 5, 1406–1420. 10.1002/pro.5560050719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a related example, see:Jakobsche C. E.; Choudhary A.; Miller S. J.; Raines R. T. J. Am. Chem. Soc. 2010, 132, 6651–6653. 10.1021/ja100931y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Newberry R. W.; Orke S. J.; Raines R. T. Org. Lett. 2016, 18, 3614–3617. 10.1021/acs.orglett.6b01655. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Siebler C.; Maryasin B.; Kuemin M.; Erdmann R. S.; Rigling C.; Günenfelder C. E.; Ochsenfeld C.; Wennemers H. Chem. Sci. 2015, 6, 6725–6730. 10.1039/C5SC02211H. [DOI] [PMC free article] [PubMed] [Google Scholar]; See also ref (44).
- Bürgi H. B.; Dunitz J. D. Acc. Chem. Res. 1983, 16, 153–161. 10.1021/ar00089a002. [DOI] [Google Scholar]
- We acknowledge the caveats associated with drawing comparisons between X-ray crystal structures, NMR solution conformations, and enantioselectivity values that were obtained in different solvents. However, we do not anticipate this to be a source of significant deviation in the present context. Significant changes in peptide conformation have been observed upon switching from nonpolar to strongly H-bonding solvents. For example, see:Awasthi S. K.; Shankaramma S. C.; Raghothama S.; Balaram P. Biopolymers 2001, 58, 465–476. 10.1002/1097-0282(20010415)58:5<465::AID-BIP1022>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]; The solvents examined (EtOAc, PhMe, PhH, and THF) span a narrow dielectric range and are generally considered to be nonpolar solvents. Furthermore, in the rare cases in which we have observed solvent molecules in the crystallographic unit cells, they are not directly interacting with the peptides.
- This chart is not intended to be a frequency histogram, but rather it is meant to highlight the diversity we observed within this crystal structure library.
- For discussions of Acpc (and derivatives) in peptides, see:; a Crisma M.; Borggraeve W. M.; Peggion C.; Formaggio F.; Royo S.; Jiménez A. I.; Cativiela C.; Toniolo C. Chem.−Eur. J. 2006, 12, 251–260. 10.1002/chem.200500865. [DOI] [PubMed] [Google Scholar]; b Casanovas J.; Jiménez A. I.; Cativiela C.; Pérez J. J.; Alemán C. J. Phys. Chem. B 2006, 110, 5762–5766. 10.1021/jp0542569. [DOI] [PubMed] [Google Scholar]; c Zanuy D.; Ballano G.; Jiménez A. I.; Casanovas J.; Haspel N.; Cativiela C.; Curcóm D.; Nussinov R.; Alemán C. J. Chem. Inf. Model. 2009, 49, 1623–1629. 10.1021/ci9001487. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Fabiano N.; Valle G.; Crisma M.; Toniolo C.; Saviano M.; Lombardi A.; Isernia C.; Pavone V.; Di Blasio B.; Pedone C.; Benedetti E. Int. J. Pept. Protein Res. 1993, 42, 459–465. 10.1111/j.1399-3011.1993.tb00155.x. [DOI] [PubMed] [Google Scholar]; e Benedetti E.; Di Blasio B.; Pavone V.; Pedone C.; Santini A.; Barone V.; Fraternali F.; Lelj F.; Bavoso A.; Crisma M.; Toniolo C. Int. J. Biol. Macromol. 1989, 11, 353–360. 10.1016/0141-8130(89)90007-X. [DOI] [PubMed] [Google Scholar]; f Valle G.; Crisma M.; Toniolo C.; Holt E. M.; Tamura M.; Bland J.; Stammer C. H. Int. J. Pept. Protein Res. 1989, 34, 56–65. 10.1111/j.1399-3011.1989.tb01009.x. [DOI] [PubMed] [Google Scholar]; g Varughese K. I.; Srinivasan A. R.; Stammer C. H. Int. J. Pept. Protein Res. 1985, 26, 242–251. 10.1111/j.1399-3011.1985.tb03202.x. [DOI] [PubMed] [Google Scholar]; See also refs (11c and 24a).
- Some type I/I′ β-turns similar to those we have observed were located in the CSD. This substructure was most often found in longer, helical sequences. For example, see:; a Bunkóczi G.; Schiell M.; Vértesy L.; Sheldrick G. M. J. Pept. Sci. 2003, 9, 745–752. 10.1002/psc.496. [DOI] [PubMed] [Google Scholar]; b Karle I. L.; Flippen-Anderson J.; Sukumar M.; Balaram P. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 5087–5091. 10.1073/pnas.84.15.5087. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Karle I. L.; Gurunath R.; Prasad S.; Kaul R.; Rao R. B.; Balaram P. J. Am. Chem. Soc. 1995, 117, 9632. 10.1021/ja00143a003. [DOI] [Google Scholar]; For some examples in shorter peptides, see:; d Francis A. K.; Iqbal M.; Balaram P.; Vijayan M. J. Chem. Soc., Perkin Trans. 2 1982, 1235–1239. 10.1039/p29820001235. [DOI] [Google Scholar]; e De Zotti M.; Ballano G.; Jost M.; Salnikov E. S.; Bechinger B.; Oancea S.; Crisma M.; Toniolo C.; Formaggio F. Chem. Biodiversity 2014, 11, 1163–1191. 10.1002/cbdv.201300394. [DOI] [PubMed] [Google Scholar]; f Ghasparian A.; Moehle K.; Linden A.; Robinson J. A. Chem. Commun. 2006, 174–176. 10.1039/B510812H. [DOI] [PubMed] [Google Scholar]; For some examples of type I′ hairpins that are not helical, see:; g Karle I. L.; Gopi H. N.; Balaram P. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 3716–3719. 10.1073/pnas.071050198. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Bandyopadhyay A.; Misra R.; Gopi H. N. Chem. Commun. 2016, 52, 4938–4941. 10.1039/C6CC00127K. [DOI] [PubMed] [Google Scholar]
- Gilli P.; Pretto L.; Bertolasi V.; Gilli G. Acc. Chem. Res. 2009, 42, 33–44. 10.1021/ar800001k. [DOI] [PubMed] [Google Scholar]
- For example, see:; a Denmark S. E.; Burk M. T. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20655–20660. 10.1073/pnas.1005296107. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a review on Lewis base catalysis, see:; b Denmark S. E.; Beutner G. L. Angew. Chem., Int. Ed. 2008, 47, 1560–1638. 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- For a review on trans/cis isomerization in peptides, see:; a Fischer G. Chem. Soc. Rev. 2000, 29, 119–127. 10.1039/a803742f. [DOI] [Google Scholar]; For an example involving Pro, see:; b Fischer S.; Dunbrack R. L. Jr.; Karplus M. J. Am. Chem. Soc. 1994, 116, 11931–11937. 10.1021/ja00105a036. [DOI] [Google Scholar]; c Wedemeyer W. J.; Welker E.; Scheraga H. A. Biochemistry 2002, 41, 14637–14644. 10.1021/bi020574b. [DOI] [PubMed] [Google Scholar]
- It is difficult to extract 3JNH–Hα values for the i and i + 3 residues of 3 + 1 and 3 + 2, owing to the lower peak resolution due to the presumed catalyst–substrate complexation.
- a Koshland D. E. Jr. J. Cell. Comp. Physiol. 1959, 54, 245–258. 10.1002/jcp.1030540420. [DOI] [PubMed] [Google Scholar]; b Koshland D. E. Jr.; Mémethy G.; Filmer D. Biochemistry 1966, 5, 365–385. 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]; For reviews, see:; c Changeux J.-P. Annu. Rev. Biophys. 2012, 41, 103–133. 10.1146/annurev-biophys-050511-102222. [DOI] [PubMed] [Google Scholar]; d Johnson K. A. J. Biol. Chem. 2008, 283, 26297–26301. 10.1074/jbc.R800034200. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Herschlag D. Bioorg. Chem. 1988, 16, 62–96. 10.1016/0045-2068(88)90038-7. [DOI] [Google Scholar]
- For related examples, see:; a Milo A.; Bess E. N.; Sigman M. S. Nature 2014, 507, 210–214. 10.1038/nature13019. [DOI] [PubMed] [Google Scholar]; b Gustafson J. L.; Sigman M. S.; Miller S. J. Org. Lett. 2010, 12, 2794–2797. 10.1021/ol100927m. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sigman M. S.; Harper K. C.; Milo A. Acc. Chem. Res. 2016, 49, 1292–1301. 10.1021/acs.accounts.6b00194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.