

Supplemental Digital Content is available in the text
Keywords: C1 inhibitor, C4, emergency center, hereditary angioedema, screening
Abstract
Hereditary angioedema (HAE) with deficiency of C1 inhibitor (C1-INH) is an autosomal-dominant disease characterized by recurrent episodes of potentially life-threatening angioedema. The objective is to study the incidence of HAE among patients who visit the emergency department.
This was a 3-year prospective observational screening study involving 13 urban tertiary emergency centers in Osaka prefecture, Japan. Patients were included if they met the following criteria: unexplained edema of the body, upper airway obstruction accompanied by edema, anaphylaxis, acute abdomen with intestinal edema (including ileus and acute pancreatitis), or asthma attack. C1-INH activity and C4 level were measured at the time of emergency department admission during the period between July 2011 and June 2014.
This study comprised 66 patients with a median age of 54.0 (IQR: 37.5–68.3) years. Three patients were newly diagnosed as having HAE, and 1 patient had already been diagnosed as having HAE. C1-INH activity levels of the patients with HAE were below the detection limit (<25%), whereas those of non-HAE patients (n = 62) were 106% (IQR: 85.5%–127.0%) (normal range, 70%–130%). The median level of C4 was significantly lower in the patients with HAE compared with those without HAE (1.2 [IQR: 1–3] mg/dL vs 22 [IQR: 16.5–29.5] mg/dL, P < 0.01) (normal range, 17–45 mg/dL).
Three patients with undiagnosed HAE were diagnosed as having HAE in the emergency department during the 3-year period. If patients have signs and symptoms suspicious of HAE, the levels of C1-INH activity and C4 should be measured.
1. Introduction
Hereditary angioedema (HAE) with deficiency of C1 inhibitor (C1-INH) is a rare but medically important disease characterized by recurrent episodes of potentially life-threatening angioedema.[1] HAE was initially described clinically by Quincke and Osler.[2] HAE is characterized clinically by edema of the subcutaneous or submucosal tissue, usually affecting the face, extremities, upper airway, and gastrointestinal tract.[2] The edema causing upper airway obstruction often leads to life-threating laryngeal attacks.[3] The incidence of HAE is estimated to be approximately 1 in 10,000 to 1 in 150,000 persons.[4,5] A survey of HAE in emergency departments in the UK was conducted, and recommendations for the management of HAE attacks were discussed.[6] In Japan, however, HAE itself is generally less well known among both physicians and the public. It is also reported that HAE shows no predilection as to sex or ethnic background.[4] Therefore, we thought that patients with HAE could have visited the emergency department unnoticed in Japan.
HAE results from a mutation in the C1-INH gene and is inherited via an autosomal-dominant pattern.[1,7] If we could detect patients with undiagnosed HAE in the emergency department, we could potentially save the lives of patients and other undiagnosed family members. In this study, therefore, we examined the incidence of HAE among patients who visited 13 emergency centers in Osaka, Japan.
2. Methods
2.1. Patients and setting
This was a 3-year prospective observational screening study involving 13 urban tertiary emergency centers in Osaka prefecture, Japan. There are 15 urban tertiary emergency centers in Osaka prefecture. Therefore, we covered 86.7% (13/15) of the centers in the prefecture, which has a population of 8.8 million as of October 1, 2014. This study was approved by the Ethics Committee of Osaka University Graduate School of Medicine. The institutional review board waived the need for informed consent because the methods of HAE diagnosis according to the Guideline for HAE 2010 by the Japanese Association for Complement Research[8] include only normal medical examination and tests. Patients were included if they met the following criteria: unexplained edema of the body, upper airway obstruction accompanied by edema, anaphylaxis, acute abdomen with intestinal edema (including ileus and acute pancreatitis), or asthma attack. C1-INH activity and C4 level were measured at the time of emergency department admission during the period between July 2011 and June 2014. Informed consent for genetic analysis was obtained only from patients with newly diagnosed HAE in this study. The flowchart of the recruitment of patients is shown in Supplemental Figure 1.
2.2. Diagnosis of HAE
We diagnosed HAE according to the Guideline for HAE 2010 by the Japanese Association for Complement Research: points for diagnosis and treatment.[8] This guideline was modified from the 2010 International consensus algorithm for diagnosis[9] in accordance with the circumstances in Japan. Because evaluation of the antigenic level of C1-INH is not covered by the Japanese health insurance system, the HAE diagnosis was obtained based on the clinical symptoms, clinical histories, and laboratory data (antigenic levels of C4 and functional levels of C1-INH) of the study patients. If the values of C1-INH activity and C4 were low and patients were diagnosed as having HAE, we informed them of this fact. If they and their families agreed to genetic analysis, they underwent genetic analysis of the C1-INH gene at Kyushu University. The genetic analysis method was as described previously by Yamamoto et al.[10] If family members also desired to have their C1-INH activity and C4 level evaluated, then we measured those values as well.
2.3. Evaluation of clinical background
Blood samples were obtained at the admitting emergency center. We analyzed white blood cells (WBC), C-reactive protein (CRP), glutamic oxaloacetic transaminase (GOT), glutamic pyruvic transaminase (GPT), lactate dehydrogenase (LDH), blood urea nitrogen (BUN), creatinine (CRE), total protein (TP), and functional levels of C1-INH and C4. C1-INH activity values were measured using the Berichrom C1 inhibitor kit (Siemens Healthcare Diagnostics, Deerfield, IL) according to the manufacturer's instructions. Age, sex, and the medical histories of the patients and their families were recorded at the time of admission.
2.4. Statistical analysis
Continuous variables are presented as the median and interquartile range (IQR). The Wilcoxon rank-sum test and Pearson χ2 test were used to compare 2 patient groups. A P value of <0.05 was considered significant. All statistical analyses were performed using JMP Pro 11.2.0 (SAS Institute Inc, Cary, NC).
3. Results
3.1. Patient characteristics
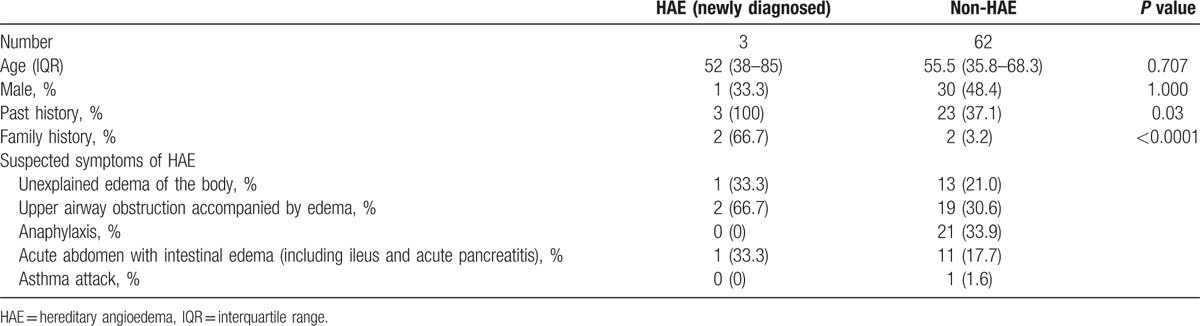
We included 66 patients with a median age of 54.0 (IQR: 37.5–68.3) years in this study. Three patients were newly diagnosed as having HAE, and 1 patient had already been diagnosed as having HAE. Patient characteristics, except for the patient already diagnosed as having HAE, are shown in Table 1. All patients with HAE had a past history of suspected symptoms of HAE. In addition, 2 of the 3 patients with HAE had a family history of suspected symptoms of HAE.
Table 1.
Patient characteristics.
3.2. Evaluation of C1-INH activity, C4, and clinical background
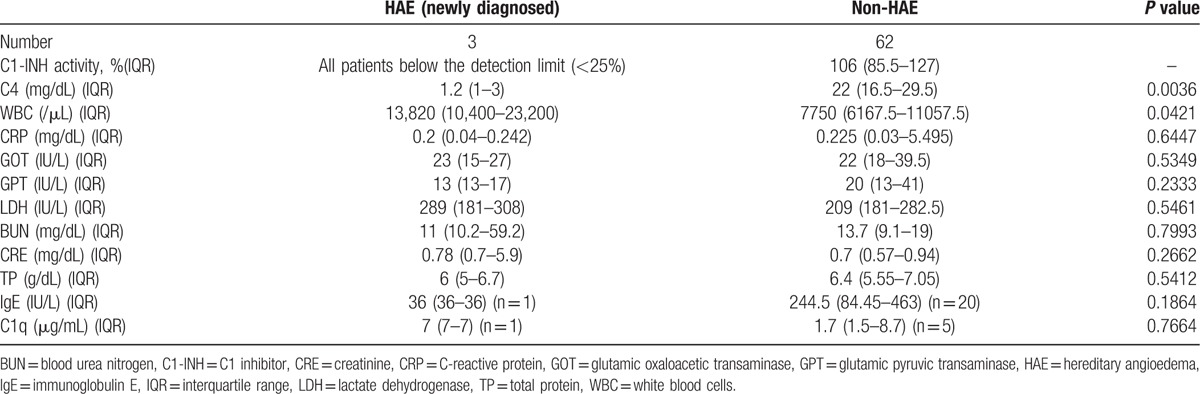
Evaluations of C1-INH activity, C4, and the clinical background of HAE and non-HAE patients are shown in Table 2, and those of each HAE patient are shown in Supplemental Table 1. C1-INH activity levels of the patients with HAE were below the detection limit (<25%), whereas those of the non-HAE patients (n = 62) were 106% (IQR: 85.5%–127.0%) (normal range 70%–130%). The median levels of C4 in the HAE patients were significantly lower than those in the non-HAE patients (1.2 [IQR: 1–3] vs 22 [IQR: 16.5–29.5] mg/dL, P < 0.01) (normal range 17–45 mg/dL). The WBC count was significantly higher in the patients with HAE than in the non-HAE patients (13,820 [IQR: 10,400–23,200] vs 7750 [IQR: 6167.5–11,057.5]/μL, P = 0.04). IgE and C1q were voluntarily measured in some patients depending on the physician's decision. There were no significant differences in levels of CRP, GOT, GPT, LDH, BUN, CRE, TP, IgE, and C1q between the patients with and without HAE. We also measured C1-INH activity levels and C4 in some of the families of the patients with HAE and detected low values of C1-INH activity and C4 in some of them. We could not clarify the exact incidence of HAE in the patients’ families because we could not obtain informed consent from all of the patients with HAE to investigate their families in detail.
Table 2.
Evaluation of C1-INH activity, C4, and other clinical parameters.
3.3. Genetic analysis of the C1-INH gene
Two of the newly diagnosed patients with HAE and some of their family members underwent genetic analysis, whereas the other newly diagnosed patient did not. The C1-INH gene mutation was detected in these 2 patients and in some of their family members.
3.4. Case reports of HAE patients
Case 1: A 52-year-old woman was transferred to the emergency center because of facial edema (Fig. 1A and B). An H1 blocker, H2 blocker, and steroid were not effective in reducing her facial edema. It worsened and speech became difficult. Computed tomography (CT) examination of her face and neck revealed laryngeal edema. She was intubated because of the risk of airway obstruction (Fig. 2). Complement blood tests revealed a low value of C4. On day 4 after intubation, her facial edema had improved naturally without specific treatment, and she was successfully extubated. Intravenous human C1-INH was not administered because the treating physicians were not confident that she had HAE. By day 8, her facial edema had completely resolved (Fig. 1C and D). During the hospital stay, complement blood tests revealed a C1-INH deficiency (below the detection limit [<25%]). On day 10, she was discharged from hospital.
Figure 1.
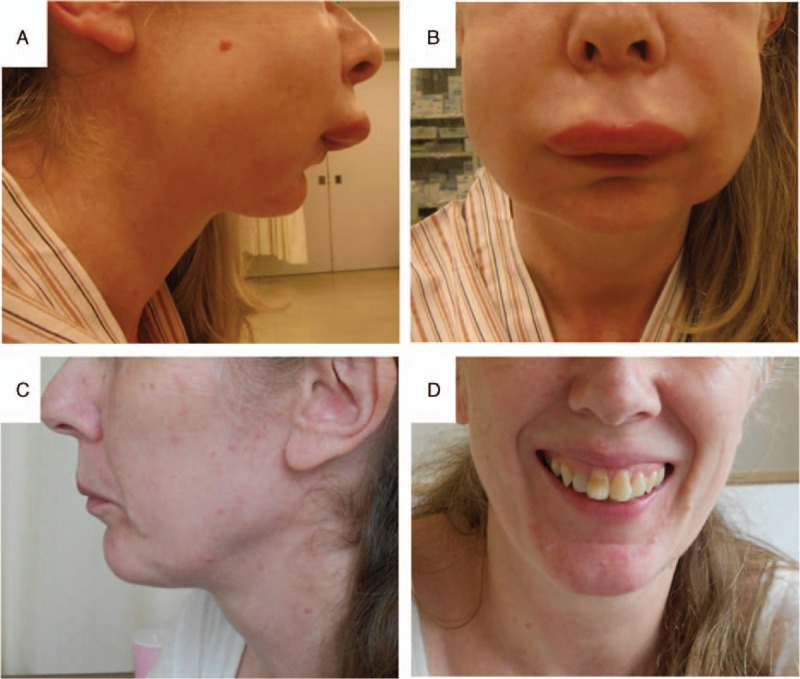
Photographs of a patient with hereditary angioedema (Case 1). Facial edema was apparent on admission (side view, A; front view, B). Facial edema had completely resolved by day 8 (side view, C; front view, D).
Figure 2.
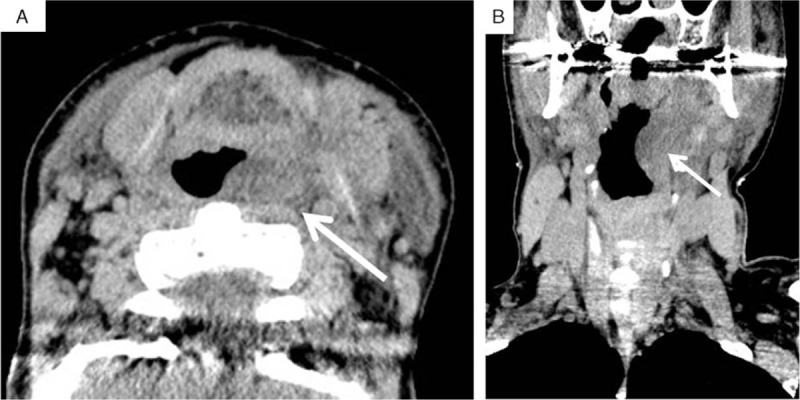
Neck CT images during an attack of hereditary angioedema (Case 1). On admission, facial edema was apparent, and she had difficulty in speaking. CT examination revealed laryngeal edema (transverse plane, A; coronal plane, B), and the risk of airway obstruction was present. Arrow, laryngeal edema. CT = computed tomography.
She had a past history of infectious enteritis involving abdominal pain, intestinal edema, and ascites (Supplemental Figure 2), facial, hand and leg edema, and asthma attacks. The patient presented with abdominal attacks at a hospital 2 or 3 times a year, and she was misdiagnosed as having enteritis and endometriosis. She received hormone treatment for endometriosis over a period of 1 year, and this exacerbated her condition. Prior to diagnosis, she had been experiencing 2 or 3 abdominal attacks monthly and after hormone treatment, sometimes twice a week. She experienced an increase in facial, leg, and hand edema attack. This was not the first time she had experienced life-threatening laryngeal edema. The patient reported 3 instances of life-threating edema episodes, each time with massive facial swelling. On 2 occasions she presented to a doctor and was given steroids and antihistamines, which did not work. On 1 occasion, she was told by the doctor that she was very lucky to have survived the night as the laryngeal edema was so extensive. Nevertheless, there was a delay of 39 years between the onset of symptoms and an accurate diagnosis.
Case 2: A 38-year-old woman was transferred to the emergency center because of abdominal pain and vomiting. Her WBC count was 23,200/μL and her CRP value was 0.242 mg/dL. Her C4 value was low. An abdominal CT examination showed intestinal edema and ascites (Fig. 3). Physicians strongly suspected that this patient had HAE from her medical history, symptoms, and CT findings. Intravenous tranexamic acid was administered, and the abdominal attack improved. She was discharged the next day. A few days later, C1-INH deficiency (below the detection limit [<25%]) was detected. She had a past history of limb edema from age 25, abdominal pain and vomiting from age 32 and occasional facial edema diagnosed as Quincke edema at another hospital.
Figure 3.
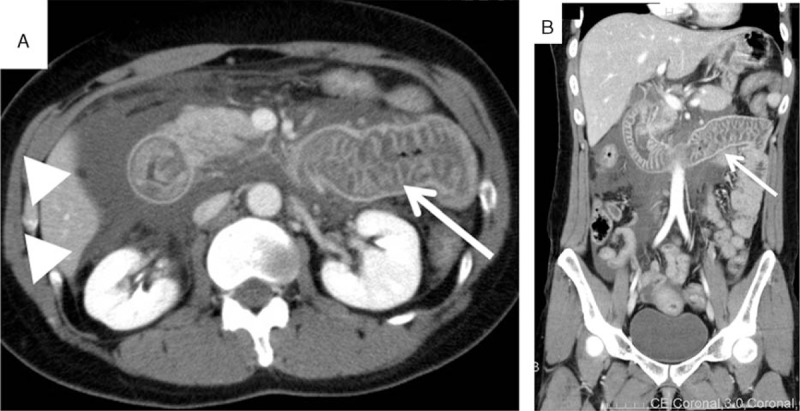
Abdominal CT images during an attack of hereditary angioedema (Case 2). On admission, the patient had abdominal pain and vomiting. Abdominal CT examination revealed intestinal edema (arrow) and ascites (arrowheads). The physician suspected an attack of HAE on the basis of these images and the patient's medical history. CT = computed tomography, HAE = hereditary angioedema.
Case 3: An 85-year-old man was transferred to the emergency center because of out-of-hospital cardiac arrest (OHCA). This case was reported previously as a case report by the attending physicians.[11] The earliest symptoms of facial swelling emerged about 20 hours before the OHCA. The etiology of cardiac arrest was most likely asphyxiation due to upper airway obstruction. A photograph of his facial edema on day 2 can be seen in Supplemental Figure 3. The facial edema persisted on day 3, and a 1500 U dose of human C1-INH (Berinert; CSL Behring) was administered intravenously before an accurate diagnosis of HAE was made, which resulted in rapid improvement of the edema. Complement blood tests revealed a deficiency in C1-INH activity and a low C4 level. The patient was successfully extubated on day 4. He was discharged from the hospital on day 33 with complete neurologic recovery. His past medical history included multiple episodes of facial swelling and an episode of OHCA of unknown cause with facial edema at the age of 76 years.
Case 4: A 45-year-old man was transferred to the emergency center because of facial edema and dyspnea. He had already been diagnosed as having HAE about 10 years before. Human C1-INH was not available in the hospital. An H1 blocker, H2 blocker, steroid, and adrenaline were not effective in improving his edema and dyspnea. About 2 hours later, the attending physician was able to obtain human C1-INH (Berinert; CSL Behring) and administered it to him. His facial edema and dyspnea gradually improved, and he was discharged the next day.
4. Discussion
In this study, we clarified that patients with HAE appear to go unnoticed in emergency departments in Japan. Based on the estimated incidence of HAE patients of approximately 1 in 10,000 to 1 in 150,000 persons,[4,5] it is estimated that about 60 to 880 patients with HAE may live in Osaka prefecture. However, the disease is generally not well known throughout Japan.[12] We believe that the results of this study will contribute to the awareness of HAE in Japan.
We concluded that identifying patients with HAE in the emergency department is especially important for these patients, who have recurrent episodes of various clinical symptoms such as edema of the body (nonpruritic, nonpitting, subcutaneous, or submucosal edema typically involving the arms, legs, hands, feet, bowels, genitalia, trunk, face, tongue, or larynx), abdominal pain, nausea and vomiting, and dyspnea.[1] Currently, they are often misdiagnosed. The average time to diagnosis after the initial presentation of symptoms is 8.3 years, and patients report visiting an average of 4.4 physicians before receiving an accurate diagnosis.[13,14] We think the average time to diagnosis in Japan is much longer. Although the number of patients with HAE who visited emergency departments has not been reported epidemiologically, an online survey of the clinical status of 63 patients with HAE reported an average of 4.7 emergency department visits annually and that nearly one-quarter of the patients were treated for anaphylaxis in the emergency room.[15] The 3 patients with newly diagnosed HAE in this study had not been diagnosed as having HAE although they had visited the emergency department multiple times by that time.
A laryngeal attack in an HAE patient may cause fatal airway obstruction if not diagnosed and treated promptly.[3,16] The patient in Case 1 was at risk of fatal airway obstruction (Figs. 1 and 2), and the patient in Case 3 had suffered asphyxia twice in his life (Supplemental Figure 3).[11] Bork et al[16] reported that most of the patients experience asphyxiation between their 20th and 50th years of life, but asphyxiation can occur even in children. Thus, the possibility that the first episode of laryngeal edema may be fatal must be emphasized to the patient's relatives, and attending physicians must have a high degree of awareness. We think that the accurate diagnosis of HAE can lessen the risk of asphyxia due to a laryngeal attack not only for the patients with HAE themselves, but also for their family members who potentially might become HAE patients.
Abdominal attacks in patients with HAE can cause abdominal pain, vomiting, and diarrhea and can mimic surgical emergencies.[4,17] The patients in Cases 1 and 2 were misdiagnosed as having infectious enteritis many times. Abdominal attacks with HAE are also similar to acute abdominal disease such as perforation or ileus, so unnecessary surgery might be performed.[4] Therefore, physicians working in emergency departments and surgeons performing emergency abdominal surgery need to be aware of HAE. Ohsawa et al[18] reported that in patients with acute abdominal pain and leukocytosis without CRP elevation, attending physicians should consider the possibility of an attack of HAE. In the present study, the WBC count was significantly higher in the patients with HAE than in the non-HAE patients, and elevation of the CRP level could not be detected in all patients with HAE in the emergency department. Thus, the likelihood of an attack of HAE should be considered when patients with abdominal pain, intestinal edema, and a suspected past medical and family history of HAE are examined.
We diagnosed HAE according to the 2010 guidelines published by the Japanese Association for Complement Research if the values of C1-INH activity and C4 were low.[19] However, it takes several weeks to obtain the results of C1-INH activity in Japan; they are not available immediately. Thus, the detection of low levels of C4 and an accurate clinical history are the key to the screening of HAE patients in the emergency department. Ohsawa et al[18] reported that their examination confirmed the diagnosis of HAE in 100% of patients by measuring functional levels of C1-INH and serum levels of C4 and that normal levels of C4 virtually exclude HAE.[8,9] Therefore, if patients have signs and symptoms suspicious of HAE, C4 and C1-INH activity levels should be measured.
The first-line treatment of HAE attacks is plasma-derived C1-INH.[9,20] In the present study, human C1-INH (Berinert; CSL Behring) was administered in Cases 3 and 4. Recently, a bradykinin B2 receptor antagonist or a plasma kallikrein inhibitor for HAE patients was administered for HAE attacks,[21,22] but human C1-INH is the only plasma-derived C1-INH product approved in Japan by the Japanese health insurance system. However, many hospitals do not stock plasma-derived C1-INH due to the low recognition of HAE by physicians.[12] In Case 4, a patient already diagnosed as having HAE could not receive the immediate administration of plasma-derived C1-INH. We think that making both physicians and the public aware of HAE and the establishment of a regional medical system that includes placement of plasma-derived C1-INH and a system to transport the drug to hospitals in which HAE patients can be treated are very important.
Patients with HAE have to endure various burdens including a chronic disease course with early onset, delayed diagnosis, and misdiagnosis; the nature of attacks, which are unpredictable, prolonged, frequent, disfiguring, painful, debilitating, and life threatening; treatment with limited options, accessibility, and side effects; a quality of life that includes depression, anxiety, limitations on social activities, travel and work/school, and hospitalization; and high cost.[23] Some Japanese people have a fear of genetic discrimination.[24] In the present study, 1 newly diagnosed HAE patient did not undergo genetic analysis. Patients suspected of having HAE and their families may continue to miss beneficial opportunities for the diagnosis of HAE, appropriate prophylaxis of HAE attacks and prevention of unnecessary tests and treatments. Accurate recognition of HAE is required for physicians and the public.
There are a few limitations in the present study. First, the enrolled sample size was small due to a lack of recognition of HAE by the emergency department physicians. Therefore, we could not include all patients who might have met the inclusion criteria for this study. Second, there was a difference in the number of cases registered by each institution. A lack of recognition of HAE was present at those institutions. However, the physicians at some institutions were able to recognize HAE.
5. Conclusions
Patients with previously undiagnosed HAE were diagnosed as having HAE in the emergency department. If patients have signs and symptoms that are suspicious of HAE, levels of C1-INH activity and C4 should be measured.
Supplementary Material
Footnotes
Abbreviations: BUN = blood urea nitrogen, C1-INH = C1 inhibitor, CRE = creatinine, CRP = C-reactive protein, GOT = glutamic oxaloacetic transaminase, GPT = glutamic pyruvic transaminase, HAE = hereditary angioedema, IgE = immunoglobulin E, IQR = interquartile range, LDH = lactate dehydrogenase, TP = total protein, WBC = white blood cells.
TomH, TakH, and TS designed the study. TomH, FK, MS, YM, HY, MK, TK, SS, MN, AF, HA, TK, and SK collected and generated the data. TomH wrote the first draft. TomH, TakH, and TS analyzed the data. TakH and TS helped to draft the manuscript. All of the authors read and approved the final manuscript.
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med 2008;359:1027–36. [DOI] [PubMed] [Google Scholar]
- [2].Bork K, Meng G, Staubach P, et al. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med 2006;119:267–74. [DOI] [PubMed] [Google Scholar]
- [3].Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol 2012;130:692–7. [DOI] [PubMed] [Google Scholar]
- [4].Nzeako UC, Frigas E, Tremaine WJ. Hereditary angioedema: a broad review for clinicians. Arch Intern Med 2001;161:2417–29. [DOI] [PubMed] [Google Scholar]
- [5].Cicardi M, Agostoni A. Hereditary angioedema. N Engl J Med 1996;334:1666–7. [DOI] [PubMed] [Google Scholar]
- [6].Jaiganesh T, Hughan C, Webster A, et al. Hereditary angioedema: a survey of UK emergency departments and recommendations for management. Eur J Emerg Med 2012;19:271–4. [DOI] [PubMed] [Google Scholar]
- [7].Davis AE., 3rd C1 inhibitor and hereditary angioneurotic edema. Annu Rev Immunol 1988;6:595–628. [DOI] [PubMed] [Google Scholar]
- [8].Horiuchi T, Ohi H, Ohsawa I, et al. Guideline for hereditary angioedema (HAE) 2010 by the Japanese Association for Complement Research—secondary publication. Allergol Int 2012;61:559–62. [DOI] [PubMed] [Google Scholar]
- [9].Bowen T, Cicardi M, Farkas H, et al. 2010 International consensus algorithm for the diagnosis, therapy and management of hereditary angioedema. Allergy Asthma Clin Immunol 2010;6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yamamoto T, Horiuchi T, Miyahara H, et al. Hereditary angioedema in Japan: genetic analysis of 13 unrelated cases. Am J Med Sci 2012;343:210–4. [DOI] [PubMed] [Google Scholar]
- [11].Fuse T, Nakada TA, Taniguchi M, et al. Cardiac arrest due to airway obstruction in hereditary angioedema. Am J Emerg Med 2015;33:1840.e1–2. [DOI] [PubMed] [Google Scholar]
- [12].Ohsawa I, Nagamachi S, Kusaba G, et al. Hereditary angioedema recognition survey in Japan. Pharma Medica 2011;29:109–18. [Google Scholar]
- [13].Bork K, Davis-Lorton M. Overview of hereditary angioedema caused by C1-inhibitor deficiency: assessment and clinical management. Eur Ann Allergy Clin Immunol 2013;45:7–16. [PubMed] [Google Scholar]
- [14].Lunn ML, Santos CB, Craig TJ. Is there a need for clinical guidelines in the United States for the diagnosis of hereditary angioedema and the screening of family members of affected patients? Ann Allergy Asthma Immunol 2010;104:211–4. [DOI] [PubMed] [Google Scholar]
- [15].Huang SW. Results of an on-line survey of patients with hereditary angioedema. Allergy Asthma Proc 2004;25:127–31. [PubMed] [Google Scholar]
- [16].Bork K, Siedlecki K, Bosch S, et al. Asphyxiation by laryngeal edema in patients with hereditary angioedema. Mayo Clin Proc 2000;75:349–54. [DOI] [PubMed] [Google Scholar]
- [17].Agostoni A, Aygören-Pürsün E, Binkley KE, et al. Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol 2004;114(3 suppl):S51–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ohsawa I, Nagamachi S, Suzuki H, et al. Leukocytosis and high hematocrit levels during abdominal attacks of hereditary angioedema. BMC Gastroenterol 2013;13:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Horiuchi T. [Guideline for hereditary angioedema (HAE) 2010 by the Japanese Association for Complement Research: points for diagnosis and treatment]. Arerugi 2014;63:749–53. in Japanese. [PubMed] [Google Scholar]
- [20].Bhardwaj N, Craig TJ. Treatment of hereditary angioedema: a review (CME). Transfusion 2014;54:2989–96. [DOI] [PubMed] [Google Scholar]
- [21].Bork K. A decade of change: recent developments in pharmacotherapy of hereditary angioedema (HAE). Clin Rev Allergy Immunol 2016;51:183–92. [DOI] [PubMed] [Google Scholar]
- [22].Betschel S, Badiou J, Binkley K, et al. Canadian hereditary angioedema guideline. Allergy Asthma Clin Immunol 2014;10:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Banerji A. The burden of illness in patients with hereditary angioedema. Ann Allergy Asthma Immunol 2013;111:329–36. [DOI] [PubMed] [Google Scholar]
- [24].Murashige N, Tanimoto T, Kusumi E. Fear of genetic discrimination in Japan. Lancet 2012;380:730. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.