

SUMMARY
The mammalian sperm genome is thought to lack substantial information to regulate future expression after fertilization. Here we show that most promoters in mouse sperm are flanked by well-positioned nucleosomes marked by active histone modifications. Analysis of these modifications suggests that many enhancers and super-enhancers functional in embryonic and adult tissues are already specified in sperm. The sperm genome is bound by CTCF and cohesin at sites that are also present in round spermatids and embryonic stem cells (ESCs). These sites mediate interactions that organize the sperm genome into domains and compartments that overlap extensively with those found in mESCs. These results suggest that sperm carry a rich source of regulatory information, encoded in part by its three-dimensional folding specified by CTCF and cohesin. This information may contribute to future expression during embryonic and adult life, suggesting mechanisms by which environmental effects on the paternal germline are transmitted trans-generationally.
Keywords: chromatin, transcription, CTCF, TAD, pluripotency, stem cell
Graphical Abstract
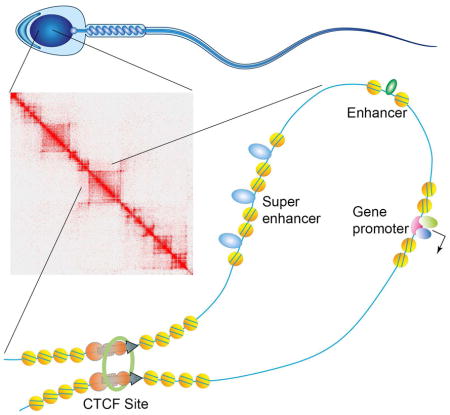
INTRODUCTION
Evidence suggesting the inheritance of epigenetically-mediated phenotypes inter- and transgenerationally in mammals is accumulating rapidly (Rando, 2016; Lim and Brunet, 2013). Here we use the term epigenetics in a broad sense to describe the self-propagation of a transient event, including chromatin-based mechanisms by which cells retain memories of past states (Henikoff and Greally, 2016). The mechanisms by which this information can be passed from parent to offspring through the male germline remain difficult to rationalize (Heard and Martienssen, 2014). DNA methylation is erased, although not completely, and re-established during germ cell differentiation. In addition, this information is rapidly but not fully erased from the paternal chromosomes after fertilization (Hackett and Surani, 2013). Although some sites in the genome maintain their methylation status, this epigenetic modification may not be the primary causal carrier of information accounting for transgenerational inheritance of epiphenotypes without some additional information that specifies the re-establishment of methylation patterns after each erasure.
Covalent modifications of histones may explain, at least in part, the inheritance of epigenetic information by affecting the demethylation and remethylation of the paternal chromosomes after fertilization. Canonical histones in mammals are exchanged for protamines during spermiogenesis, the transition from round spermatids to mature sperm. Pioneering work from several laboratories during the last few years has mapped the distribution of retained nucleosomes using micrococcal nuclease digestion of chromatin in the mouse or human genomes (Carone, et al., 2014; Samans, et al., 2014; Erkek, et al., 2013; Brykczynska, et al., 2010; Hammoud, et al., 2009). Some studies have suggested that nucleosomes are enriched at promoters of microRNAs, imprinted genes, and particular genes with functions in early mammalian development such as HOX genes (Hammoud, et al., 2009). Other studies have found nucleosomes along the entire genome, with only a minor enrichment at TSSs (Carone, et al., 2014; Brykczynska, et al., 2010), whereas other results suggest that nucleosomes are depleted at HOX gene clusters but enriched at repetitive regions of the genome (Samans, et al., 2014). Some of these discrepancies have been attributed to experimental differences in the digestion of chromatin by micrococcal nuclease (MNase). Only after extensive digestion can nucleosomes be detected over promoters of developmental genes, suggesting that nucleosomes at these promoters are less accessible or more resistant to digestion (Carone, et al., 2014). Sperm nucleosomes contain H3K4me3 and/or H3K27me3, with partial overlap between bivalent promoters in sperm and hESCs (Hammoud, et al., 2009). Analysis of the distribution of H3K4me2 suggests that a large fraction of genes containing this histone modification in human sperm were expressed during the prior spermatid stages or at later stages during embryogenesis (Brykczynska, et al., 2010). However, most genes containing H3K27me3 in sperm were not expressed during spermatogenesis or early embryogenesis. Non-coding RNAs present in sperm have also been shown to mediate the transfer of epigenetic information between generations (Sharma, et al., 2016).
Transcription factors may be retained in sperm bound to specific regulatory sequences, and these proteins may carry information to the next generation, perhaps by regulating patterns of demethylation and remethylation of the paternal chromosomes during early zygote development. However, the distribution of DNA-binding proteins on the mammalian sperm has not been analyzed using approaches such as ChIP-seq, perhaps due to technical difficulties owing to the highly condensed sperm chromatin. Binding motifs for CTCF can be detected at sites resistant to digestion by MNase, suggesting that this protein may be retained in the sperm nucleus (Carone, et al., 2014). CTCF and cohesin are responsible, at least in part, for the establishment of the three-dimensional organization of the genome, which in turn can elicit specific patterns of gene expression by mediating interactions between specific enhancers and promoters (Ong and Corces, 2014). To obtain a comprehensive view of the mouse sperm epigenome and the type of information present in mammalian sperm that can be transmitted to the next generation, we used ATAC-seq to map the location of DNA-binding proteins and nucleosomes at high resolution in live mouse sperm. We also used ChIP-seq to map the distribution of various histone modifications, CTCF, and cohesin. Integration of this information with HiC data suggests that the sperm epigenome is in part a consequence of the transcriptional state of the preceding round spermatid stage as well as a prelude to expression patterns observed in ESCs and adult tissues. The results suggest that mammalian sperm chromatin carries an expansive amount of epigenetic information, partly encoded in the 3D organization of the genome, which could be modulated by the paternal environment and transmitted to subsequent generations to elicit phenotypic changes in the progeny.
RESULTS
Promoters of sperm genes contain positioned nucleosomes with active histone modifications
Sperm used in all experiments were isolated from the cauda epididymis using the swim up assay and were at least 99.9% pure (less than one contaminating cell per 1000 sperm, Figure S1A). Only 1% of the mouse sperm genome is thought to be associated with histones based on electrophoretic analyses of HCl-extracted proteins from sperm chromatin (Balhorn, et al., 1977). To more accurately determine the amount of histones present in the nuclei of mature mouse sperm, we used Western blot analysis of intact sperm following two different strategies. First, we compared the amount of histones H3 and H4 in mouse sperm to the amount of these histones in the nuclei of two somatic tissues, liver and brain. We assumed that the mass of the base pairs (ca. 200 bp/Mr= 132,600) covered by the histone octamer including histone H1 (Mr=130,500) are approximately identical (Van Holde, 1989). In the second strategy, the amount of H3, H3.3, and H4 present in sperm were directly determined by titration using recombinant histone counterparts (Figure S2A). In both instances, the two approaches gave the same result and allowed us to determine the amount of histones present in mouse sperm nuclei as 8.5% (with histone H1) and 6.4% (without H1) (see Extended Experimental Procedures for a detailed explanation of the calculations employed). Therefore, the overall amount of histones in mouse sperm nuclei could be considered to be 7.5 ± 1% of that present in one genome equivalent of normal diploid cells. Using a similar approach, the amount of H3.3 was determined to be 24% of the total H3 present in sperm nuclei (Figure S2A). To further explore the issue of what fraction of nucleosomes persist in mouse sperm, we performed MNase digestion experiments carried out in parallel with 780,000 sperm and 42,500 (85,000 genome equivalents) mESCs. Results suggest a similar accessibility to MNase in the two cell types (Figure S2B). Furthermore, quantification of the mono- and di-nucleosome bands in the gel shown in Figure S2B indicates an 8-fold difference between the number of nucleosomes present in sperm and mESCs after taking into account the difference in ploidy between the two cell types. The combination of results from quantitation of histones and nucleosomes suggests the presence of more nucleosomes in the mouse sperm genome than previously thought.
To analyze the distribution of nucleosomes in mouse sperm, we then used ATAC-seq on live sperm permeabilized with digitonin using the new Fast-ATAC protocol (Corces, et al., 2016). Experiments performed in parallel with sperm and mESCs suggest that sperm chromatin is accessible to Tn5 transposase to the same degree as chromatin from mESCs (Figure S2B). We obtained three independent biological replicates of sperm ATAC-seq with high correlation values (Figure S1B). We combined the three replicates and separately analyzed reads in the 50–115 bp range and reads with lengths of 180–247 bp (Figure S1C). The former corresponds to sites depleted of nucleosomes where bound proteins protect the DNA from the Tn5 transposase (Tn5 hypersensitive sites, THSSs) whereas the latter corresponds to mono-nucleosomes (Schep, et al., 2015). We then used DANPOS2 (Chen, et al., 2013) to map nucleosomes using the 180–247 bp nucleosomal reads. Figure 1A shows the distribution of THSSs and nucleosomes in a region of the mouse genome surrounding the Jarid2 gene. As a comparison, we show previously published MNase data for mouse sperm (Brykczynska, et al., 2010). ATAC-seq detects the presence of several well-positioned nucleosomes surrounding the transcription start site (TSS), with a strong THSSs region between the +1 and −1 nucleosomes. Genome-wide analyses of the distribution of THSSs and nucleosomes with respect to various gene features indicate that most are present in genes or their promoters, with approximately 25% present in intergenic regions (Figure 1B). To determine the nature of the sequences corresponding the THSSs, we first used all mouse TSSs as anchors and carried out k-means clustering using the nucleosomal and non-nucleosomal reads. Of the 24,643 TSSs examined, 16,003 are sensitive to Tn5 transposition and these TSSs are flanked by nucleosomes (clusters 1–4), whereas the remaining ca. 8,640 (cluster 5) are not (Figure 1C). Results from MNase-seq (Brykczynska, et al., 2010) agree with the information obtained by ATAC-seq, but the resolution of the data does not allow the mapping of individual nucleosomes (Figure 1C). Results from ATAC-seq indicate that the +1 nucleosome is well positioned and appears to be placed at the same location in all accessible TSSs, whereas the signal corresponding to the −1 nucleosome appears more intense in clusters 1–3. In these three clusters, the −1 nucleosome has a broad signal with a shoulder that partially overlaps the THSS regions (Figure 1D). In addition to the +1 nucleosome, weaker signals can be detected in the downstream region. The lower intensity of these signals with respect to the +1 nucleosome may be a consequence of the preference of the Tn5 transposase for the two nucleosomes flanking the TSS (Schep, et al., 2015) or may correspond to nucleosomes that are not well-positioned (Figure 1D, arrowheads).
Figure 1. Epigenetic profiles of sperm chromatin at transcription start sites (TSS).

(A) Track view of THSS and nucleosome reads obtained using ATAC-seq, as well as ChIP-seq signal for H3K4me3 and RNAPII-Ser2ph around the Jarid2 TSS. MNase-seq data (Brykczynska, et al., 2010) is shown for comparison.
(B) Genome-wide distribution of THSSs and nucleosomes identified from sperm ATAC-seq. TSS ± 2 kb are listed as promoters.
(C) Heatmaps showing chromatin features around TSSs (± 1.5 kb). Sites are ordered by nucleosome and THSS signal from ATAC-seq.
(D) Average THSS (red) and nucleosome (green) ATAC-seq enrichment profiles relative to TSSs. Arrow heads indicate the position of nucleosomes downstream of the TSS.
(E) Average profiles of H3K9ac ChIP-seq for each cluster shown in panel C.
(F) Comparison of sperm nucleosome-occupied TSSs with gene ontology terms obtained using the GREAT tool, gene expression (RPKM from RNA-seq), and round spermatid chromatin state.
RS=round spermatid; SP=sperm; ZY=zygote; 2C= 2 cell embryo; 4C= 4 cell embryo; ICM= inner cell mass; Tro= trophectoderm.
See also Figures S1, S2 and S3
To gain insights into the epigenetic state of sperm promoters and its possible relationship to the patterns of nucleosomes observed by ATAC-seq, we performed native ChIP-seq using micrococcal nuclease (MNase) in non-fixed nuclei and antibodies against H3K4me3, H3K27ac, H3K9ac, H3K36me3, H3K9me3, and H3K27me3. Western analyses indicate that these modifications are present in sperm histones (Figure S2C). Under the digestion conditions employed (see Materials and Methods), most of the chromatin is digested to mono nucleosome size (Brykczynska, et al., 2010). We also carried out whole genome DNA methylation analysis using bisulfite sequencing (BS-seq). We then aligned the reads to the clustering matrix obtained with ATAC-seq data (Figure 1C). We find that the +1 nucleosome contains all three active marks, and that, in agreement with ATAC-seq results, there appears to be several additional nucleosomes containing these histone modifications upstream and throughout the coding region of these genes (Figures 1C and 1E). All promoters flanked by nucleosomes and containing active histone modifications are devoid of 5mC (Figure 1C). All other promoters lacking clear ATAC-seq signal and active histone modifications (cluster 5) appear to be embedded in regions containing H3K9me3, H3K27me3, and higher levels of 5mC (Figure 1C). Furthermore, analysis of H3K36me3 by ChIP-seq suggests that this modification, which is normally present throughout the coding region and 3′ end of transcribed genes and correlates with active elongation (Bannister, et al., 2005), is present at these promoters, although it does not appear to extend far through the coding region of genes (Figure 1C).
The active epigenetic state of a subset of sperm promoters correlates with their previous activity in round spermatids
The transcriptional state of sperm promoters may be, at least in part, a consequence of transcription during previous stages of spermatogenesis. Alternatively, the epigenetic state of these promoters may prepare the genes for expression at later stages of development after fertilization. Round spermatids represent the last stage of spermatogenesis that is easily accessible for genome-wide analysis. The transition from this state to mature sperm takes approximately 13.5 days (Oakberg, 1957). We therefore examined RNA levels from published RNA-seq data obtained in round spermatids, sperm, zygote, 2 and 4 cell stages, inner cell mass (ICM), and trophoblasts (Hammoud, et al., 2014), and arranged RNA levels according to the order of genes present in the 5 clusters shown in Figure 1C. We also compared this information with the distribution of histone modifications in round spermatids (Hammoud, et al., 2014). Results show that clusters 1 and 2, marked in orange and yellow in Figure 1F, correspond to genes with high RNA levels in both round spermatids and sperm. These genes contain active histone modifications in round spermatids (Figure 1F) and sperm (Figure 1C). RNA levels corresponding to these genes are lower in zygotes and early stages of embryogenesis, although some appear to be transcribed in ICM cells. Gene ontology analyses using GREAT suggest that genes in these two clusters are enriched for functions related to spermatogenesis and male gamete generation (Figure 1F). The presence of these RNAs in the sperm may be a consequence of their expression in round spermatids or during late stages of spermiogenesis.
A subset of accessible promoters containing active histone modifications in sperm are transcribed in cells of the ICM
RNA levels for genes in clusters 3 and 4 are relatively high in the zygote and ICM stages compared to round spermatids and sperm (Figure 1F). These sperm promoters could be in an active epigenetic state in preparation for future transcription in the zygote. On the other hand, promoters in cluster 5, which are enriched in GO terms related to immune system function and lack both a nucleosome free region and well-positioned flanking nucleosomes, do not appear to be transcribed during embryonic development. These promoters may correspond to genes that only become active in adult tissues. However, some of the genes whose promoters are not accessible in sperm appear to be transcribed in the one-cell stage, possibly from the maternal chromosome. To gain further insights into the biological significance of the epigenetic states of promoters in clusters 3–5, we compared accessibility to Tn5 transposase and nucleosome positioning around these TSSs using ATAC-seq data obtained in sperm (this study), mouse embryonic stem cells (mESCs), and mouse embryonic fibroblasts (MEFs) (Maza, et al., 2015). These sites were clustered using TSSs as anchors and mESC ATAC-seq data. The results suggest that promoters present in an epigenetically active state in sperm are also in the same state in mESCs and MEFs, where they contain strong THSSs and are flanked by well-positioned nucleosomes (Figure 2A, clusters 1–3). These genes are highly expressed in cells of the ICM (Figure 2A). However, a subset of non-accessible or with low accessibility sperm promoters become accessible in mESCs and MEFs (cluster 4). Non-accessible sperm promoters in cluster 5 are also in this state in mESCs and MEFs. Presumably, these promoters become accessible in other adult cell types. It should be noted that the observation of a subset of promoters in an epigenetically active state in sperm and mESCs cannot be taken as evidence for the maintenance of this state during embryonic development and up to the blastocyst stage. In fact, it appears that many of these genes are not expressed during the two- and four-cell stages (Figure 1F). Instead, we favor the hypothesis that the open chromatin state of sperm promoters may function to guide the re-methylation of the sperm genome early in embryogenesis, thus ensuring that these promoters are not permanently silenced by 5mC.
Figure 2. Comparison of THSSs and nucleosome positioning around TSSs among sperm, mESCs, and MEFs.

(A) Heatmaps representing THSSs and nucleosomes occupancy between cell types after re-clustering TSSs present in cluster 3–5 of figure 1C by H3K4me3 and H3K27me3 of sperm and mESCs. Occupancy of protamine 1, H3.3, and RPKM value in round spermatids, sperm, ICM cells, and trophoblasts is also shown. Each panel represents 1.5 kb upstream and downstream of the TSS.
(B) Heatmaps indicating THSSs and nuclesomes from ATAC-seq in sperm, mESCs, and MEFs at summit of sperm THSSs located at TSSs (± 1.5 kb).
(C) Track view of protamine 1 and input enrichment in a typical region of the genome.
(D) Average profile of THSSs, nucleosomes, H3.3, and H3K4me3 around THSSs in cluster 2 of panel B.
(E) Average profile of THSSs, nucleosomes, and protamine 1 around TSSs in panel A.
See also Figure S2
Promoters of developmental genes are in a poised state in ESCs, containing both H3K4me3 and H3K27me3 (Mikkelsen, et al., 2007). The distribution of these two histone modifications at sperm promoters based on our ChIP-seq experiments correlates well with results from previous work (Erkek, et al., 2013) (Figure S2D). To explore the epigenetic state of these promoters in sperm, we examined the distribution of these two histone modifications in mESCs, and found that genes in cluster 4 contain H3K27me3 and H3K4me3 (Figure 2A). We then examined the distribution of these two histone modifications in sperm promoters. Results show that the same cluster 4 genes that are in a poised state in mESCs are already in a similar state in sperm (Figure 2A).
Sperm promoters in an epigenetically active state show an unusual chromatin structure
Most active promoters in mESCs and MEFs have an open chromatin organization in the sperm. However, the distribution of ATAC-seq nucleosomal signal around these active TSSs is different in sperm versus mESCs or MEFs (Figure 2A, clusters 1–3). The latter two cell types show a well-positioned +1 nucleosome and a weaker signal at the −1 nucleosome, whereas the same promoters in sperm display a broader and stronger signal at the location of the −1 versus the +1 nucleosomes. To explore the significance of this difference, we used sperm promoters with strong nucleosomal ATAC-seq signal and we re-clustered the data using the summits of the THSSs as anchors instead of TSSs. Results from this analysis indicate that THSSs detected by ATAC-seq are flanked by two nucleosomes in mESCs and MEFs. However, the same promoters in sperm contain a strong signal in the nucleosomal ATAC-seq reads (size range 180–247 bp) located between the −1 and +1 nucleosomes (Figures 2B and 2D), suggesting that the labile nucleosome present in this region is more stable in sperm than in normal cells. The same result was observed when using THSSs from mESCs or MEFs as anchors (Figure S3A). Sperm chromatin is mostly devoid of histones and it contains protamines instead. To gain additional insights into the reasons for the unusual chromatin organization of sperm TSSs, we carried out ChIP-seq with antibodies to protamin 1 (PRM1) and examined the distribution of histone H3.3 (Brykczynska, et al., 2010). Results show a broad and persistent occupancy of PRM1 throughout the sperm genome, with an enrichment around TSSs and other specific sites (Figure 2C). PRM1 and H3.3 are only present in sperm promoters that contain strong THSSs, well-positioned flanking nucleosomes and active histone modifications. Promoters that lack THSSs and well-positioned nucleosomes do not contain PRM1 or H3.3, whereas poised promoters contain intermediate levels (Figure 2A). Therefore, the enrichment of PRM1 and H3.3 correlates with an active chromatin state of sperm gene promoters, but their location with respect to TSSs follows different patterns. H3.3 is enriched in the “nucleosome-free” region overlapping the THSSs (Figure 2D). PRM1, on the other hand, is enriched on both sides of the nucleosome-free region, with preferential accumulation around the −1 and +1 nucleosomes (Figure 2E). Therefore, the ATAC-seq nucleosomal signal observed between the −1 and +1 nucleosomes of sperm TSSs could be due to the presence of a nucleosome containing H3.3 that is more stable than in somatic cells. Interestingly, H3.3 present between the −1 and +1 nucleosomes is not methylated in Lys4, in contrast with the histone H3 present in the flanking nucleosomes (Figure 2D). This unusual promoter organization is not observed in mESCs or MEFs (Figures 2B and S3A). The same organization can be detected at a subset of non-TSS THSSs in sperm but not mESCs or MEFs (cluster 3, Figure S3B)
Sperm contain enhancers and super-enhancers that normally function in ESCs or adult tissues
The finding of H3K4me1 in sperm chromatin, a histone modification characteristic of enhancers, raises the question of whether these regulatory sequences are already defined in sperm. To investigate this issue, we used ROSE (Whyte, et al., 2013) to define enhancers and super-enhancers utilizing ChIP-seq signal for H3K4me1 and H3K27ac, in both sperm and mESCs (Figure 3A). Using these criteria, we find 4,678 enhancers in sperm, of which 1337 overlap with those previously defined in embryonic or adult mouse tissues (Shen, et al., 2012). Interestingly, a subset of these enhancers also appears to contain H3K27me3, suggesting a bivalent state (cluster 2 in Figure 3A). These enhancers are associated with GO terms related to cell fate commitment, pattern specification processes, and embryo development. We also carried out a parallel analysis by assuming that the ca. 58,000 THSSs detected by ATAC-seq that do not correspond to TSSs or CTCF sites (see below) must correspond to transcription factors bound to regulatory sequences. Using these criteria, we find 10,240 THSSs that correspond to previously characterized enhancers in embryonic or adult tissues (Figure 3B) (Shen, et al., 2012).
Figure 3. Putative enhancers and super-enhancers identified in sperm.

(A) Sperm enhancers defined by the presence of H3K4me1 and H3K27ac. A subset in cluster 2 also contains H3K27me3.
(B) Overlap of enhancers between sperm and other tissues.
(C) Identification of super-enhancers by H3K27ac signal in sperm (left) and mESCs (right).
(D) Overlap of super-enhancers between sperm and other tissues.
(E) Track view of ChIP-seq binding profiles at a super enhancer present in sperm and mESCs.
(F) Track view of ChIP-seq binding profiles at a super enhancer present in sperm and heart.
See also Figure S2
Using ROSE, we also find 645 super-enhancers present in the sperm genome with a median size of 22 kb; of these, 109 overlap with those defined in mESC by the same criteria (Figure 3C). We then used ROSE to define super-enhancers in various adult tissues using published data for H3K27ac, and found that many super-enhancers found in sperm overlap with those present in adult tissues (Figure 3D). An example of a super-enhancer located between the Mreg and Pecr genes in sperm and mESCs is shown in Figure 3E. A super-enhancer located adjacent to Mir3089 in both sperm and heart is shown in Figure 3F. These results suggest that regulatory elements critical for the differentiation of specific adult cell types are already specified in the sperm.
CTCF and cohesin are present at new as well as persistent sites in the sperm genome
THSSs accessible to Tn5 transposase represent open regions of chromatin where proteins are bound. Results from the ATAC-seq analysis suggest the existence of ca. 97,000 of these sites in the sperm genome. As described above, ca. 16,000 of these THSSs correspond to promoters of genes in an active epigenetic state and an additional 10,240 may correspond to proteins bound to putative enhancer sequences. The remaining THSSs may correspond to sites bound by other proteins in the mouse sperm genome. One strong candidate protein to be present at sperm THSSs is CTCF, since the consensus motif for CTCF is present at MNase sites in sperm (Carone, et al., 2014). To determine whether CTCF is present in mouse sperm we carried out western and immunofluorescence analyses. Antibodies to CTCF detect the presence of a band of the appropriate size in western blots, although this band is less intense in sperm than mESCs (Figure S2E). Both sperm and mESCs show additional bands that may correspond to degradation products of CTCF or, in the case of sperm, fragments of the BORIS protein likely recognized by the same antibodies. Experiments using immunofluorescence confocal microscopy confirm the presence of CTCF in mouse sperm nuclei (Figure S2G). We thus carried out ChIP-seq with antibodies to CTCF. During the ChIP procedure, sperm DNA is broken down by sonication to the same extend than that of somatic cells (Figure S2F). We find 5,325 CTCF peaks distributed throughout the sperm genome that contain the consensus CTCF motif (Figure 4A). Of these, 3,827 are also present in round spermatids and in mESCs, whereas 841 are specific for sperm (Figure 4A). CTCF requires the cohesin complex for function and, therefore, we also examined the presence of this protein in mouse sperm. Western blot and immunofluorescence analysis confirm the presence of Smc1 in sperm (Figures S2E and S2G). We then determined the distribution of Smc1 in the sperm genome using ChIP-seq and compared its distribution with respect to that found in round spermatids and mESCs (Figure 4B). We used sperm CTCF peaks as anchors to cluster previously published CTCF and cohesin sites from round spermatids and mESCs (Pugacheva, et al., 2015; Nitzsche, et al., 2011; Kagey, et al., 2010). Results show that most sperm CTCF sites are also present in round spermatids and mESCs but a subset are specific for sperm and are absent in these two other cell types (Figures 4B and 4C, cluster 1). Interestingly, these sperm-specific CTCF sites lack Smc1. Alignment of ATAC-seq reads to the same clustering matrix suggests that these sites have weaker THSS signal than those containing Smc1. Furthermore, although the rest of sperm CTCF sites are flanked by positioned nucleosomes, sperm-specific CTCF sites are not (Figure 4C, cluster 1). A subset of 2,549 sites in sperm correspond to 2X sites found in round spermatids that contain both CTCF and BORIS in adjacent sites separated by 30–50 bp (Figures 4B and 4C, cluster 2) (Pugacheva, et al., 2015). These CTCF sites are present in unmethylated regions of the sperm genome (Figure 4C). Although all sperm CTCF/cohesin sites are flanked by nucleosomes containing H3K4me1, only those adjacent to BORIS in round spermatids are also marked by H3K27ac and H3K9ac. Sperm CTCF sites lacking BORIS in round spermatids also lack these histone modifications and are located in regions containing 5mC (cluster 3, Figures 4B and 4C). Therefore, it appears that sperm CTCF sites can be classified in three different groups depending on the presence of cohesin and various histone modifications. These groups correlate with the previous co-occupancy of CTCF with BORIS in round spermatids, suggesting that BORIS may help mark different CTCF sites for future function. A similar analysis comparing CTCF and cohesin sites in sperm, mESCs, and round spermatids but using the summits of CTCF peaks defined in round spermatids as anchors gives additional information on the dynamics of CTCF localization during the transitions between these cell types (Figure 4D). A total of 35,978 CTCF sites have been mapped in round spermatids (Pugacheva, et al., 2015). Of these, 23,679 also contain BORIS, and are maintained in sperm and mESCs. In addition, 12,299 sites present in round spermatids are lost in sperm and re-gained in mESCs. These sites are present in methylated regions of sperm DNA (Figure 4D).
Figure 4. Genomic profiling of CTCF occupancy in sperm.

(A) CTCF consensus motif found in CTCF occupied site in sperm (left). Venn diagram showing the number of sperm CTCF occupied sites in round spermatids (RS) and mESCs.
(B) Representative examples of CTCF and Smc1 occupancy in round spermatids (RS), sperm (S), and mESCs (ES) in each cluster shown in panel C.
(C) Heatmaps showing a comparison of CTCF, BORIS and cohesin (Smc1) occupancy in round spermatids (RS), sperm, and mESCs at all CTCF occupied sites of sperm (± 1.5 kb)
(D) Heatmaps showing a comparison of CTCF, BORIS, and cohesin (Smc1) occupancy in round spermatids (RS), sperm, and mESCs at all CTCF occupied sites of RS (± 1.5 kb)
(E) Average profiles of THSSs and nuclesomes from ATAC-seq (left), and H3K4me1 (right) around CTCF binding sites from panel D.
See also Figures S2, S4, and S6
Analysis of ChIP-seq data for CTCF using MACS predicts the existence of 5,325 peaks of this protein in the sperm genome. However, the clustering analysis of sperm, round spermatid and mESC ChIP-seq data suggests the existence of 23,679 sites in the sperm genome where CTCF ChIP-seq signal is enriched above background (Figure 4D). These sites also contain Smc1, suggesting that they are bona fide CTCF sites. One possible explanation for the discrepancy between this information and that obtained from ChIP-seq analysis of CTCF in sperm is that, due to technical limitations because of the compact state of sperm chromatin, the signal to noise ratio of the sperm CTCF ChIP-seq data is lower than that of round spermatids and mESCs. The differential occupancy of CTCF sites in clusters 1–2 versus 3 in Figure 4D was further explored by mapping the CTCF footprint at these two groups of sites. CTCF sites present in clusters 1–2 show the characteristic CTCF footprint (Figure S4A) whereas those in cluster 3 do not show a discernible footprint (Figure S4B), underscoring the conclusion that these sites are not occupied by CTCF in sperm. To further investigate whether all possible 23,679 CTCF sites correspond to genuine signal, we examined nucleosomal and THSSs ATAC-seq data around these sites. Results show that indeed all 23,679 CTCF sites are sensitive to Tn5 transposition and are flanked by 5–7 well-positioned nucleosomes marked by H3K4me1 on either side (Figures 4D and 4E). These nucleosomes appear to contain low levels of H3K27ac and H3K9ac and are depleted of H3K4me3 (Figure 4D). Similar results were obtained when using CTCF peaks mapped in mESCs as anchors to carry out the clustering analysis, showing that all but 10,702 sites occupied by CTCF in mESCs are already occupied in sperm (Figure S4C). Interestingly, CTCF sites present in round spermatids and sperm that persist in mESCs are embedded in regions of chromatin containing H3K4m1, H3K4me3 and H3K27ac in these cells, whereas sites present in round spermatids that disappear in sperm, but are regained in mESCs, are present in regions that lack these marks (Figure S4D and S4E).
Global features of chromatin 3D organization are similar between sperm and ESCs
Chromatin is considered to be highly packed in the sperm nucleus due to the presence of protamines and absence of nucleosomes in large regions of the genome. To study this largely unexplored area of sperm chromosome biology, we used HiC to map genome wide interactions in the sperm genome. The 3D organization of the mouse sperm nucleus using HiC has been described previously but the sparseness of the data (272 million sequenced reads, of which only 8.6 million intra-chromosomal reads passed QC using the same pipeline employed to analyze our HiC data) did not allow a high resolution analysis of interactions (Battulin, et al., 2015). We prepared HiC libraries from two biological replicates using HindIII and obtained 199 million high quality mapped reads after various quality control steps (Tables S1 and S2). We then compared our results with HiC data previously obtained using Patski (Darrow, et al., 2016), mESCs (Selvaraj, et al., 2013) and CH12-LX B-cell lymphoma cells (Rao, et al., 2014). At a global level, contact domains observed in sperm HiC data appear similar to those previously characterized in normal mammalian diploid cells. Sperm contain either one X or one Y chromosomes. It has been previously shown that the active (Xa) and inactive (Xi) chromosomes of mouse and human cells adopt very different 3D organization (Darrow, et al., 2016). To analyze the 3D organization of the X chromosome present in sperm, we compare results from HiC analyses with those previously obtained in the Patski cell line, which was obtained in a cross between M. musculus and M. spretus (Darrow, et al., 2016). Results indicate that the sperm X chromosome lacks the two superdomains separated by the DXZ4 locus characteristic of Xi. Instead, the sperm X chromosome shows the typical multi-domain organization observed in Xa (Figure 5A).
Figure 5. Comparison of 3D organization between sperm, mESCs and B-cell lymphoma cells.

(A) Organization of the X chromosome in sperm. Contact HiC maps are shown for the inactive, active, and sperm X chromosomes.
(B)Compartment organization in sperm. Eigen vector and visualization of HiC maps suggest that while some compartment interactions are maintained among all three cell lines, sperm compartments are more closely conserved in mESCs. Figures correspond to mouse chromosome 17.
(C) TAD organization in sperm. Figure shows a region of mouse chromosome 1 containing several TADs. Two of them are demarcated by yellow squares with sub-domains indicated by blue squares. Domains in the top left quadrant are established by interactions between CTCF sites and are conserved between sperm and mESCs but not in CH12-LX cells. Domains present in the bottom right quadrant are conserved in all three cell types.
See also Figure S6
Studies using ESCs or various cell lines have shown that, at a coarse level, chromatin in mammals is partitioned into two compartments, the gene-rich and accessible A-compartment and the repressive B-compartment (Lieberman-Aiden, et al., 2009). Sperm HiC data shows this same type of compartmental signal, and the sperm compartments at 500 kb resolution appear more similar to those present in mESCs than in CH12-LX cells (Figure 5B). Compartment calls at 10 kb resolution in sperm and mESC HiC data showed considerable similarity (Figure 6A) with 87.4% of the genome sharing the same compartment state (42.2% A, 45.1% B). The remainder of the genome is split between transitioning from the A-compartment in sperm to the B-compartment in mESCs (5.4%) or from B to A (7.3%). To determine the characteristics of compartment boundaries, we profiled CTCF, protamine 1, and several histone modifications anchored at boundaries and extending up- and down-stream one Mb in sperm (this study) and mESCs cells (Selvaraj, et al., 2013) (Figure 6B). Because there are specific enrichments associated with TSSs, we partitioned the profiles based on the presence or absence of TSSs in each 20 kb bin. Overall, sperm and mESC compartment boundaries show highly consistent signal across CTCF, protamine 1, THSSs, and active histone mark signals with enrichment in signal in A-compartments peaking just inside the boundary and depletion in B-compartments (Figures 6B and 6C). This pattern is much stronger in TSS-occupied regions, although the same pattern is observed in non-TSS profiles as well. Repressive histone marks H3K27me3, H3K9me3, and H3K36me3 show a reversal of this pattern in sperm cells, with depletion in A-compartments and enrichment in B-compartments regardless of the presence of TSSs. These findings suggest that sperm already are physically configured at a higher order into a state highly consistent with that seen in mESCs, although there are some clear sperm-specific features in the organization of compartments (Figure S5A).
Figure 6. Comparison of genes, histone modifications and proteins across sperm and mESC chromatin compartments.

(A) Chromatin compartment calls for sperm and ES cells overlaid on a heatmap of 10 kb bin pairwise correlations.
(B) Average ChIP-seq signal centered on compartment boundaries +/− 1 Mb. Signals were binned in 20 kb bins and were truncated to at the 95th percentile of signal prior to finding means to remove outliers. All signals were oriented to go from an A-compartment (left) to a B-compartment (right) prior to finding the mean signal. Left-side plots show mean signal for bins containing at least one TSS while right-side plots show mean signal for all non-TSS containing bins.
(C) Log-enrichments of mean ChIP-seq signals for each compartment class compared to the A/A TSS-free signal for each factor. Mean signals were found across all bases within a class that fell within one kb of a TSS within that class (top) and for all other bases in each class (bottom).
See also Figures S5
Histone marks of transcriptional state indicate global chromatin organization in a sperm-specific fashion
Sperm cells demonstrate a pattern of histone enrichment and modification in compartments that is quite different from that observed in mESCs (Figure 6B and 6C). In mESCs, ATAC-seq data show a small depletion of nucleosomes in the B-compartment compared to the A compartment at locations not within 1 kb of a TSS and a two-fold depletion of H3.3 in these same regions. Sperm show higher levels of nucleosomes across the B-compartment, especially relative to sites surrounding TSSs. In non-TSS regions, the active histone marks H3K9ac, H3K27ac, and H3K4me1 show patterns consistent with a sperm-like state, with enrichment in the sperm A-compartment and depletion in the B-compartment including transitional regions overlapping the mESC A-compartment. At TSSs, H3K27ac and H3K4me1 show enrichment patterns more consistent with a transitional state, highest in the constitutive A-compartment, lowest in the constitutive B-compartment, and middle-level and equal between the two transitional compartment types. H3K4me3 shows a pattern more consistent with mESCs, regardless of the presence of TSSs (Figures 6B and S5).
The traditionally repressive histone marks show a pattern that is inconsistent with what we observe in mESCs. H3K27me3 shows a stronger enrichment in B-compartment TSSs compared to mESCs. Further, in non-TSS regions, sperm cells show an enrichment of this mark in regions transitioning from the B-compartment to the A-compartment in mESCs and a strong depletion in regions transitioning in the other direction. This is in contrast to mESCs, were H3K27me3 depletion is strongest across the B-compartment. More striking are the other repressive marks, H3K9me3 and H3K36me3, which show enrichment across the sperm B-compartment. This pattern is completely inconsistent with that seen in mESCs cells (Figure S5A). The strength of the enrichment of H3K36me3, particularly at non-TSS regions is of particular importance as this mark has been shown to be absent from paternally derived chromatin in very early embryogenesis (Boskovic, et al., 2012). ChIP-seq data in round spermatids (Erkek, et al., 2013) show that H3K27me3 has a profile across sperm compartment boundaries consistent with mESCs (Figure S5B), meaning that this pattern of histone methylation is established later in spermiogenesis either by targeting histones in the B-compartment or demethylating histones in the A-compartment. H3K9me3 showed the same pattern in round spermatid as in sperm cells, showing that this pattern is already being established in early spermiogenesis.
Sperm domains already reflect mESC chromatin architecture physically but contain key epigenetic differences
TADs found in sperm are conserved with respect to those found in mESCs and differentiated CH12-LX cells, but whereas the sub-domain organization is very similar between sperm and mESCs, these sub-domains are very different in differentiated cells (Figure 5C). In order to examine domain-level chromatin architecture in sperm, we used a divisive clustering domain-calling approach and compared domain structure and characteristics between sperm and mESCs (Figure 7A). Using 10 kb bins, this approach yielded 6,868 and 5,177 domains in sperm and mESCs, respectively. More than 50% of domain boundaries called in mESCs occurred within 20 kb, or two bins away, from sperm domain boundaries, suggesting a strong overlap between cell types. We also used a boundary metric that measures correlations and signal differences for groups of interactions straddling potential boundary points. When scores from this metric profiles were anchored at domain boundaries, we found a strong negative correlation and large signal difference across shared boundaries (Figure S6A). We also observed cell type-specific bias associated with sperm and mESC-specific domain boundaries, suggesting that at least some portion of these domain differences is biologically driven.
Figure 7. Domain boundary comparison and associations in sperm and ES cells.

(A) Domain calls and boundary metric scores for sperm and ES HiC data. Domain calls (black, center) are overlaid on fend-corrected HiC enrichment data, binned at 100 kb resolution. Bins with no observations are in gray.
(B) Average ChIP-seq signal centered on domain boundaries +/− 500 kb. A union set of sperm and ES boundaries was used, collapsing boundaries occurring within 40 kb or each other together. Boundaries that did not overlap and were not collapsed across cell-types were considered cell-type specific. Signals were binned in 10 kb bins and were truncated at the 95th percentile of signal prior to finding means to remove outliers.
(C) Enrichment of HiC signal at intersecting CTCF sites less than 500 kb apart. Interactions were partitioned based on CTCF motif orientation and the presence of a Smc1 peak within 500 bases of CTCF peak calls. CTCF sites less than 4 kb apart were excluded to avoid false signals. HiC signal was binned at 2 kb intervals and signal +/− 100 kb from site intersections were found.
(D) The joint domain boundary set for ES and sperm cells was used to partition the genome and find ChIP-seq and RNA-seq mean signals for each interval. Intervals were sorted first by compartment class and then by mean ES expression level.
(E) Correlations between mean ChIP-seq domain signals and mean RNA-seq expression levels for each pairwise combination.
To determine the relationship of histone modifications and CTCF to domain boundary calls, we determined ChIP-seq signal profiles anchored at domain boundaries and partitioned by whether the boundaries were shared between cell-types (Figure 7B). As expected, there is an enrichment of CTCF at domain boundaries in both sperm and mESCs. Although smaller than at shared boundaries, both cell-types show enrichment for CTCF signal at non-shared boundaries, regardless of which cell-type the boundaries are called in. The active histone marks H3K4me3, H3K9ac, and H3K27ac all show enrichment at boundaries. Like CTCF, shared boundaries exhibit the strongest enrichment and sperm-specific boundaries show enrichment in both cell-types. H3K4me1 at shared and sperm-specific boundaries is slightly depleted in sperm cells but slightly enriched in mESCs. Also of note is that THSSs, which show some enrichment in shared boundaries in sperm, is narrowly enriched at shared boundaries and shows weak enrichment at both cell-type specific boundaries (Figure 7B). This suggests that additional factors are being recruited to genomic positions already established as architecturally important in sperm.
We then examined both boundary metrics and long-range interaction enrichments to determine whether the domain association with CTCF is reflective of physical interactions in the sperm or proteins poised to establish loops after fertilization. Interactions show a very strong negative correlation across CTCF-bound sites in sperm, with no difference in magnitude to that seen in mESCs (Figure S6B). We also examined HiC signal enrichment for all CTCF pairs within 500 kb of each other and containing the CTCF motif (Figure 7C). These were partitioned both by CTCF motif orientation and the presence of Smc1. Only convergent pairs of CTCF with cohesin at both sites show a strong enrichment peak, although weaker peaks are apparent for pairs with cohesin present on one site. CTCF pairs in the same orientation and with cohesin at both sites show evidence of enrichment though far weaker than inward facing CTCF pairs. This lends weight to the idea that not only is CTCF positioned on the sperm chromatin, but also establishing long-range interactions that underpin the observed domain structure. Interestingly, CTCF occupancy at domain boundaries is tied to the presence of super-enhancers. When compared to randomly distributed positions, super-enhancers in sperm are significantly enriched in domains bound on both sides by CTCF (Table S4).
To assess how domain partitioning varies across cell-types and expression levels, we intersected all domain boundaries, collapsing boundaries within 40 kb of each other, to produce a common set of domains/sub-domains and calculated mean ChIP-seq and expression levels for each interval (Figure 7D). This partitioning shows some variation across both cell-type and expression level, but confirms trends observed at the compartment level with respect to the flipping of H3K9me3 and H3K36me3 modifications between sperm and mESCs. In round spermatids, it is clear that strong marking of the B-compartment with H3K9me3 is already established prior to the completion of spermatogenesis. We also examined correlations between mean ChIP-seq signals and expression levels across round spermatid, sperm, and ES cells (Figure 7E). In all three cell-types, the active histone marks H3K4me3 and H3K9ac are reflective of the current (or most recent) transcriptionally-active state with strong positive correlations to their own cell-type expression levels, although both round spermatid and mESC marks also show increased correlation with sperm expression levels in the A/A compartment. The marks H3K4me1 and H3K27ac show this same pattern of correlation in sperm and mESCs, although no data was available for round spermatids.
To determine what, if any, specific categories of TSSs are enriched at domain boundaries, we examined TSS density with TSSs parsed using the clustering presented in Figures 1 and 2 (Figure S6C). All of the clusters from Figure 1 associated with nucleosomes and active histone modifications (clusters 1–4) show enrichment at shared and sperm-specific domain boundaries, although cluster 1 was less enriched than the others. mESC-specific domain boundaries only show enrichment of clusters 3 and 4, and TSSs not associated with round spermatid or sperm RNA-seq signal. Clusters from Figure 2 show enrichment only for clusters 1, 4, and 5, all TSSs associated with strong expression signal at the inner cell mass stage and mixed signal in round spermatid and sperm cells. No non-TSS clusters show enrichment at domain boundaries. Taken together, the results suggest that CTCF and cohesin are responsible for the establishment of domains in mouse sperm by forming loops between CTCF sites oriented in convergent orientation, and these domains are very like those present in mESCs.
DISCUSSION
The sperm nucleus has been classically considered a passive vessel whose role is to deliver the highly condensed paternal DNA complexed with protamines to the egg during fertilization. Recent work from several laboratories has resulted in the mapping of the distribution of H3K4me3, H3K27me3, and H3.3, in the mouse and human sperm genomes (Carone, et al., 2014; Samans, et al., 2014; Brykczynska, et al., 2010; Hammoud, et al., 2009). To better understand the informational content of the sperm epigenome, we have used ATAC-seq to map the location of nucleosomes and native ChIP-seq to map the location of different active and repressive histone modifications. In addition, we have mapped the location of the architectural proteins CTCF and cohesin, and determined the three-dimensional arrangement of the chromatin fiber using HiC. The results of this analysis offer an unprecedented view of the epigenomic content of the sperm nucleus.
Approximately 60% of sperm promoters are in an active epigenetic state. The TSSs of these promoters are flanked by 3–4 nucleosomes upstream and 5–6 nucleosomes downstream, and this number could actually be higher since these techniques only detect nucleosomes when they are positioned in the same location in different cells of a population. These promoters are flanked by nucleosomes containing histones H3K4me3, H3K27ac, H3K9ac, and H3K36me3, all modifications that are deposited during the initiation or elongation of the transcription process. A subset of these promoters belong to genes known to be active in round spermatids but RNAs for these genes are also found in sperm. Many of these sperm mRNAs are truncated (Soumillon, et al., 2013), and it is possible that they were made prior to sperm differentiation, since new RNA synthesis was not observed in human sperm when examining incorporation of radioactive UTP in total RNA by gel electrophoresis (Grunewald, et al., 2005). The remaining sperm TSSs containing active histone modifications correspond to genes for which RNA is found at low levels in the sperm. These promoters are active in mESCs or in differentiated cells of the embryo such as MEFs, implying that the sperm epigenome is primed for future gene expression. Interestingly, although protamine 1 is broadly distributed throughout sperm DNA, it appears to be enriched at the locations of the −1 and +1 nucleosomes flanking these TSSs. It is possible that protamine 1 preferentially interacts with canonical nucleosomes or, alternatively, that some sperm in a population contain protamine 1 at this location whereas others contain nucleosomes. The two nucleosomes flanking sperm TSSs border a putative nucleosome free region that contains the histone variant H3.3. This is unlike what we observe in mESCs and MEFs, where ATAC-seq fails to detect this large, nucleosome-sized, structure resistant to Tn5 transposition.
Sperm not only have promoters in a primed state that correlates with expression in mESCs but they also appear to have enhancers in a similar state. Results from ATAC-seq experiments suggest the presence of around 58,000 THSSs that may be bound by specific transcription factors, 10,240 of which correspond to enhancers previously identified in embryonic or adult tissues. In addition to typical enhancers, the sperm genome also contains around 645 super-enhancers, most of them in common with mESCs or specific cell lineages found in the adult organism. Interestingly, super-enhancers are present in loops formed by CTCF and cohesin to create insulated neighborhoods as previously found in mESCs (Dowen, et al., 2014). These results suggest that enhancer elements are already specified in the sperm and may be primed for subsequent function during embryogenesis and in the establishment of specific cell fates during the formation of differentiated tissues.
The three-dimensional organization of the genetic material has come into focus in recent years as an essential player in the establishment of specific transcription patterns. This organization is mediated by CTCF and cohesin, which establish point-to-point interactions between CTCF binding sites, preferentially when arranged in a forward-reverse orientation (Guo, et al., 2015; Rao, et al., 2014). Sperm contain around 23,000 CTCF sites also present in mESCs. These sites also have cohesin, suggesting that they are involved in productive long-range interactions that contribute to the organization of the sperm genome in a particular 3D architecture. Analysis of interactions determined by HiC suggests that sperm and mESCs share many features in the 3D organization of their genomes. The sperm genome is organized into A and B compartments that overlap extensively with those found in mESCs. The distribution of various DNA binding proteins and histone modifications along these compartments is similar between the two cell types, with the exception of the unusual enrichment of H3K9me3 and H3K36me3 observed in the sperm B compartment. Furthermore, at a lower level of organization, sperm DNA is organized into domains, TADs and sub-TADs, that overlap considerably with those found in mESCs. The borders of these domains are preferentially established at sites containing CTCF and Smc1, with the CTCF binding motifs preferentially arranged in a convergent orientation. These results indicate that the 3D organization of the sperm genome follows the same principles described for other cell types. This organization is closely related to that observed in mESCs, suggesting that the architecture of the sperm nucleus may be programed for future transcription in the embryo.
Our results indicate that mammalian sperm contain a rich and complex palette of epigenetic information. It is possible that the role of this information is to directly instruct transcription patterns during early embryonic development. Alternatively, the presence of DNA-bound transcription factors and histone modifications in specific regions of the genome may serve to guide re-methylation of the DNA after the extensive demethylation of the paternal chromosomes that takes place immediately after fertilization. Under this model, although epigenetic information specified by DNA methylation of the paternal chromosomes may be erased after fertilization, this information may be maintained by the interaction of specific DNA-bound transcription factors that preserve a memory of critical regulatory functions encoded by enhancers, super-enhancers, and 3D organization. Environmental factors that alter transcription during the development of the paternal germline may result in the persistence of DNA-binding proteins at new sites in the genome, which in turn would alter DNA re-methylation after fertilization and elicit novel phenotypic outcomes in the next generation.
EXPERIMENTAL PROCEDURES
Assay for transposase-accessible chromatin using sequencing (ATAC-seq)
Mouse sperm was isolated from the cauda epididymis as described in the Extended Experimental Procedures with a purity of more than 99.9% as determined by microscopy. ATAC-seq was carried out using the Fast-ATAC protocol (Corces, et al., 2016). We first determined the appropriate concentration of digitonin to ensure sperm permeabilization by examining uptake of trypan blue. The purified sperm pellet was then resuspended in the transposase reaction mix containing 0.05% digitonin and incubated for 30 min at 37°C. Following incubation, sperm were treated with Proteinase K at 55°C for 2 hr, and gDNA was isolated by phenol:chloroform:isoamyl alcohol and EtOH precipitation. Library amplification was done with 2x KAPA HiFi mix and 1.25 μM indexed primers using the following PCR conditions: 72°C for 5 min; 98°C for 30 s; and 10–11 cycles at 98°C for 10 s, 63°C for 30 s, and 72°C for 1 min. ATAC-seq experiments were performed on three biological replicates and each one contained at least 80 million reads after all the quality control steps.
ChIP-seq in sperm cells
Chromatin immunoprecipitation to detect the localization of CTCF and Smc1 in sperm was performed using the standard ChIP-seq protocol with some modifications. In brief, 10–20 million sperm cells were crosslinked with 1% formaldehyde in 1x PBS for 10 min at RT, and the reaction was quenched with 125 mM glycine for 10 min at RT. After washing with PBS, crosslinked sperm were lysed with 5 mM PIPES, 85 mM KCl, 0.5% NP40 and 1x proteinase inhibitor on ice for 10 min. Then we performed a second fixation step by directly adding 1% formaldehyde to the lysis buffer on ice. After 10 min sperm were homogenized with a glass dounce homogenizer. Following quenching and washing, sperm cells were resuspended in RIPA buffer (1x PBS; 1% NP40; 0.5% sodium deoxycholate; 0.1% SDS; 1x proteinase inhibitor) and incubated on ice for 20 min. The purified sperm chromatin was sonicated to 300–1000 bp using a Diagenode Bioruptor and 25 cycles of 30 s on and 30 s off and the supernatant was collected. Immunoprecipitation steps are described in the Extended Experimental Procedures.
Analysis of ChIP-seq and ATAC-seq data
All libraries were sequenced using an Illumina Hiseq2500 v4 sequencer and 50 bp paired-end format. Paired reads were aligned to the mouse reference genome mm9 using Bowtie 0.12.7 for ATAC-seq and ChIP-seq, and Bismark for BS-seq. ATAC-seq reads were aligned using default parameters except -X 2000-m 1. Duplicates were removed using Samtools. To adjust for fragment size, we aligned all reads as + strands offset by +4 bp and − strands offset by −5 bp (Schep, et al., 2015). The nucleosome-free and mono-nucleosome fractions were separated by choosing reads 50–115 bp and 180–247 bp in length, respectively. Mono-nucleosome reads were analyzed using DANPOS2 (Chen, et al., 2013). MACS2 was used for peak calling with nucleosome-free reads (Liu, 2014).
Preparation of HiC libraries
HiC was performed using the in situ protocol as described (Rao, et al., 2014) using two biological replicates, and each was sequenced twice. Information on the different quality control steps performed during read mapping is summarized in Tables S1–S3. Subsequent normalization steps are described in detail in the Extended Experimental Procedures.
Supplementary Material
Acknowledgments
We would like to thank the Genomic Services Lab at the HudsonAlpha Institute for Biotechnology, and specially Drs. Braden Boone, Angela Jones and Terri Pointer, for their help in performing Illumina sequencing of samples. This work was supported by U.S. Public Health Service Award R01 GM035463 from the National Institutes of Health to VGC and Natural Sciences and Engineering Research Council of Canada (NSERC) 46399-2012 grant to JA. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
ACCESSION NUMBERS
ChIP-seq, ATAC-seq and BS-seq data are available from NCBI’s Gene Expression Omnibus (GEO) under accession number GSE79230.
Supplemental Information includes Supplemental Experimental Procedures, six figures, and four tables and can be found with this article online at
AUTHOR CONTRIBUTIONS
YHJ and VGC conceived the project; YHJ, XL, and MSC performed experiments; YHJ and MEGS analyzed the data; JA, JT, and VGC designed experiments and analyzed results; YHJ, MEGS, JA, JT, and VGC wrote the manuscript with input from all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balhorn R, Gledhill BL, Wyrobek AJ. Mouse sperm chromatin proteins: quantitative isolation and partial characterization. Biochemistry. 1977;16:4074–80. doi: 10.1021/bi00637a021. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Schneider R, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. The Journal of biological chemistry. 2005;280:17732–6. doi: 10.1074/jbc.M500796200. [DOI] [PubMed] [Google Scholar]
- Battulin N, Fishman VS, Mazur AM, Pomaznoy M, Khabarova AA, Afonnikov DA, Prokhortchouk EB, Serov OL. Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome biology. 2015;16:77. doi: 10.1186/s13059-015-0642-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boskovic A, Bender A, Gall L, Ziegler-Birling C, Beaujean N, Torres-Padilla ME. Analysis of active chromatin modifications in early mammalian embryos reveals uncoupling of H2A.Z acetylation and H3K36 trimethylation from embryonic genome activation. Epigenetics. 2012;7:747–57. doi: 10.4161/epi.20584. [DOI] [PubMed] [Google Scholar]
- Brykczynska U, Hisano M, Erkek S, Ramos L, Oakeley EJ, Roloff TC, Beisel C, Schubeler D, Stadler MB, Peters AH. Repressive and active histone methylation mark distinct promoters in human and mouse spermatozoa. Nature structural & molecular biology. 2010;17:679–87. doi: 10.1038/nsmb.1821. [DOI] [PubMed] [Google Scholar]
- Carone BR, Hung JH, Hainer SJ, Chou MT, Carone DM, Weng Z, Fazzio TG, Rando OJ. High-resolution mapping of chromatin packaging in mouse embryonic stem cells and sperm. Developmental cell. 2014;30:11–22. doi: 10.1016/j.devcel.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KF, Xi YX, Pan XW, Li ZY, Kaestner K, Tyler J, Dent S, He XW, Li W. DANPOS: Dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2013;23:341–351. doi: 10.1101/gr.142067.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet. 2016 doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrow EM, Huntley MH, Dudchenko O, Stamenova EK, Durand NC, Sun Z, Huang SC, Sanborn AL, Machol I, Shamim M, et al. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E4504–12. doi: 10.1073/pnas.1609643113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen JM, Fan ZP, Hnisz D, Ren G, Abraham BJ, Zhang LN, Weintraub AS, Schuijers J, Lee TI, Zhao K, et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159:374–87. doi: 10.1016/j.cell.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkek S, Hisano M, Liang CY, Gill M, Murr R, Dieker J, Schubeler D, van der Vlag J, Stadler MB, Peters AH. Molecular determinants of nucleosome retention at CpG-rich sequences in mouse spermatozoa. Nature structural & molecular biology. 2013;20:868–75. doi: 10.1038/nsmb.2599. [DOI] [PubMed] [Google Scholar]
- Grunewald S, Paasch U, Glander HJ, Anderegg U. Mature human spermatozoa do not transcribe novel RNA. Andrologia. 2005;37:69–71. doi: 10.1111/j.1439-0272.2005.00656.x. [DOI] [PubMed] [Google Scholar]
- Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell. 2015;162:900–10. doi: 10.1016/j.cell.2015.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett JA, Surani MA. Beyond DNA: programming and inheritance of parental methylomes. Cell. 2013;153:737–9. doi: 10.1016/j.cell.2013.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoud SS, Low DHP, Yi CI, Carrell DT, Guccione E, Cairns BR. Chromatin and Transcription Transitions of Mammalian Adult Germline Stem Cells and Spermatogenesis. Cell Stem Cell. 2014;15:239–253. doi: 10.1016/j.stem.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, Cairns BR. Distinctive chromatin in human sperm packages genes for embryo development. Nature. 2009;460:473–8. doi: 10.1038/nature08162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell. 2014;157:95–109. doi: 10.1016/j.cell.2014.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Greally JM. Epigenetics, cellular memory and gene regulation. Curr Biol. 2016;26:R644–8. doi: 10.1016/j.cub.2016.06.011. [DOI] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–5. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JP, Brunet A. Bridging the transgenerational gap with epigenetic memory. Trends in genetics : TIG. 2013;29:176–86. doi: 10.1016/j.tig.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. Use model-based Analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol Biol. 2014;1150:81–95. doi: 10.1007/978-1-4939-0512-6_4. [DOI] [PubMed] [Google Scholar]
- Maza I, Caspi I, Zviran A, Chomsky E, Rais Y, Viukov S, Geula S, Buenrostro JD, Weinberger L, Krupalnik V, et al. Transient acquisition of pluripotency during somatic cell transdifferentiation with iPSC reprogramming factors. Nature biotechnology. 2015;33:769–74. doi: 10.1038/nbt.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–60. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitzsche A, Paszkowski-Rogacz M, Matarese F, Janssen-Megens EM, Hubner NC, Schulz H, de Vries I, Ding L, Huebner N, Mann M, et al. RAD21 cooperates with pluripotency transcription factors in the maintenance of embryonic stem cell identity. PloS one. 2011;6:e19470. doi: 10.1371/journal.pone.0019470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakberg EF. Duration of Spermatogenesis in the Mouse. Nature. 1957;180:1137–1138. doi: 10.1038/1801137a0. [DOI] [PubMed] [Google Scholar]
- Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nature Reviews Genetics. 2014;15:234–246. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugacheva EM, Rivero-Hinojosa S, Espinoza CA, Mendez-Catala CF, Kang S, Suzuki T, Kosaka-Suzuki N, Robinson S, Nagarajan V, Ye Z, et al. Comparative analyses of CTCF and BORIS occupancies uncover two distinct classes of CTCF binding genomic regions. Genome biology. 2015;16:161. doi: 10.1186/s13059-015-0736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ. Intergenerational Transfer of Epigenetic Information in Sperm. Cold Spring Harbor perspectives in medicine. 2016 doi: 10.1101/cshperspect.a022988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–80. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samans B, Yang Y, Krebs S, Sarode GV, Blum H, Reichenbach M, Wolf E, Steger K, Dansranjavin T, Schagdarsurengin U. Uniformity of nucleosome preservation pattern in Mammalian sperm and its connection to repetitive DNA elements. Developmental cell. 2014;30:23–35. doi: 10.1016/j.devcel.2014.05.023. [DOI] [PubMed] [Google Scholar]
- Schep AN, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, Greenleaf WJ. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res. 2015;25:1757–1770. doi: 10.1101/gr.192294.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj S, JRD, Bansal V, Ren B. Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing. Nature biotechnology. 2013;31:1111–8. doi: 10.1038/nbt.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma U, Conine CC, Shea JM, Boskovic A, Derr AG, Bing XY, Belleannee C, Kucukural A, Serra RW, Sun F, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351:391–6. doi: 10.1126/science.aad6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–20. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, Gu H, Barthes P, Kokkinaki M, Nef S, Gnirke A, et al. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell reports. 2013;3:2179–90. doi: 10.1016/j.celrep.2013.05.031. [DOI] [PubMed] [Google Scholar]
- Van Holde KE. Chromatin. New York: Springer-Verlag; 1989. [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–19. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.