

Abstract
Autophagy is an evolutionarily conserved intracellular degradation/recycling system that is essential for cellular homeostasis but is dysregulated in a number of diseases, including myocardial hypertrophy. Although it is clear that limiting or accelerating autophagic flux can result in pathological cardiac remodeling, the physiological signaling pathways that fine-tune cardiac autophagy are poorly understood. Herein, we demonstrated that stimulation of cardiomyocytes with phenylephrine (PE), a well known hypertrophic agonist, suppresses autophagy and that activation of focal adhesion kinase (FAK) is necessary for PE-stimulated autophagy suppression and subsequent initiation of hypertrophic growth. Mechanistically, we showed that FAK phosphorylates Beclin1, a core autophagy protein, on Tyr-233 and that this post-translational modification limits Beclin1 association with Atg14L and reduces Beclin1-dependent autophagosome formation. Remarkably, although ectopic expression of wild-type Beclin1 promoted cardiomyocyte atrophy, expression of a Y233E phosphomimetic variant of Beclin1 failed to affect cardiomyocyte size. Moreover, genetic depletion of Beclin1 attenuated PE-mediated/FAK-dependent initiation of myocyte hypertrophy in vivo. Collectively, these findings identify FAK as a novel negative regulator of Beclin1-mediated autophagy and indicate that this pathway can facilitate the promotion of compensatory hypertrophic growth. This novel mechanism to limit Beclin1 activity has important implications for treating a variety of pathologies associated with altered autophagic flux.
Keywords: autophagy, Beclin-1 (BECN1), cardiac hypertrophy, muscle atrophy, PTK2 protein tyrosine kinase 2 (PTK2) (focal adhesion kinase) (FAK), atrophy
Introduction
Macroautophagy is an evolutionarily conserved intracellular degradation/recycling process in which cytoplasmic cargo is non-selectively enveloped within double-membraned vesicles called autophagosomes that are transported to and fuse with lysosomes. Within these so-called autolysosomes, cytoplasmic cargo and the inner membrane are degraded so that the amino acids, fatty acids, and glucose released can be used to support cellular metabolism or to synthesize new proteins. Cardiomyocytes are both highly metabolically active cells and long-lived cells and as such are particularly dependent on macroautophagy (hereafter referred to as autophagy) for energy production and removal of damaged organelles or misfolded proteins. However, in some cases up-regulation of autophagy (or failure of autophagy suppression) can lead to detrimental cardiac remodeling.
Autophagy is regulated in a stepwise fashion that involves the formation of distinct protein complexes composed of autophagy-related genes (Atg proteins) and their enzymatic binding partners. Nutrient deprivation-induced activation of the AMP kinase/Unc-51-like autophagy-activating kinase 1 (ULK) kinase cascade leads to the recruitment of core proteins VPS34, VPS15, and Beclin1 (complex I) to endoplasmic reticulum exit sites to form phosphatidylinositol 1,4,5-phosphate 3-kinase-dependent double membrane autophagosome precursors. Complex I then facilitates the recruitment of additional proteins, including the phosphatidylinositol 3-phosphate-binding WIPI/ATG18-ATG2 complex and the ubiquitin-like ATG8/LC3-phospatidylethanol conjugation system, to drive membrane expansion and the formation of a phagophore and eventually to promote phagophore closure to form an autophagosome. The final step of autophagosome-lysosome fusion is mediated by a second Beclin1-containing complex (complex II) in which UVRAG bridges Beclin1 with Vps34/Vps15 on the surface of the lysosome (1).
Numerous studies indicate that tight spatial and temporal regulation of autophagy is necessary for both basal cardiac function and for protection from pathological remodeling following ischemic or hemodynamic stress (2, 3). The finding that cardiac-restricted depletion of the autophagy related gene 5 (Atg5; a protein essential for the capture of ubiquitinated proteins and LC3-dependent expansion of autophagosomes) led to progressive contractile dysfunction in adult mice in the absence of a pathological insult indicates that a basal level of autophagy is necessary for cardiac function (4). Autophagy is elevated following ischemia/reperfusion (I/R)3 injury, and numerous studies have revealed that autophagy plays an important cardioprotective role in this setting. For example, treatment with bafilomycin A (which blocks lysosomal degradation of autophagosomes) or expression of a dominant-inhibitory variant of Atg5 worsened I/R-mediated injury and promoted the accumulation of damaged mitochondria in this setting (5, 6). However, the finding that Beclin1+/− mice (that exhibit decreased autophagy) were protected from I/R damage indicates that in some cases suppression of autophagy (or at least limiting Beclin1-dependent induction of autophagy) can be beneficial (7, 8). Interestingly, genetic deficiency in Beclin1 impaired the regression of cardiac hypertrophy induced by mechanical or neurohumoral unloading (9, 10) indicating that Beclin1-dependent autophagy might also play an important role in myocyte atrophy. Notably, although cardiac autophagy is increased following I/R, autophagy is suppressed during compensatory hypertrophic growth in response to moderate hemodynamic stress (4) or stimulation with the β-adrenergic agonist isoproterenol (11). However, the molecular mechanisms underlying autophagy suppression and its consequences with respect to adrenergic signaling-dependent hypertrophic growth remain unclear.
As noted above, Beclin1 plays a critical role in both autophagosome formation and autophagosome-lysosome fusion, and recent studies reveal that Beclin1 serves as a nexus for autophagy regulation in response to various signaling pathways. For example, Beclin1 ubiquitination is promoted by tumor necrosis factor receptor-associated factor 6 (TRAF6) (12), neural precursor cell-expressed developmentally down-regulated 4 (Nedd4) (13), and Ambra1 (14). Beclin1 is also phosphorylated by serine/threonine kinases Akt (15), Mst1 (16), Ulk1 (17), ROCK1 (18), and MAPKAPK2/3 (19) as well as the receptor tyrosine kinase epidermal growth factor receptor (EGFR) (20). Each of these post-translational modifications alters the ability of Beclin1 to associate with complex I and II proteins or with the autophagy inhibitors Bcl2 and Rubicon. However, despite findings that limiting Beclin1 activity is markedly cardioprotective and may govern compensatory cardiac growth, we know very little about the molecular mechanisms that regulate Beclin1 function in the heart.
We and others have reported that α1-adrenergic stimulation of cardiac hypertrophy was augmented by integrin-dependent activation of focal adhesion kinase (FAK) (21, 22), and mice deficient in FAK were resistant to pressure overload-induced hypertrophy (23, 24). Because FAK is a critical downstream regulator of cardiac integrins and growth factor receptors that are known to inhibit autophagy (25), we explored the possibility that FAK might promote the initiation of hypertrophic growth, at least in part by suppressing autophagy-induced myocyte atrophy. Indeed, we found that FAK is both sufficient and necessary for α-adrenergic signaling-dependent autophagic suppression in vitro and in vivo. Collectively, our studies described herein identify FAK as a novel negative regulator of Beclin1-mediated autophagy and indicate that repression of Beclin1-dependent autophagy can facilitate the initiation of hypertrophic cardiac growth.
Results
Activation of FAK Suppresses Starvation-induced Cardiomyocyte Autophagy
We previously showed that elevated FAK activity limits cardiomyocyte apoptosis and provides remarkable cardioprotection in the face of various pathological stressors (26, 27). Because recent evidence indicates that apoptotic and autophagic signaling pathways are intimately interlinked and because activation of integrins (the major upstream regulators of FAK) is known to limit autophagy (25), we reasoned that levels of cardiac FAK activity might also impart control over autophagic flux. To explore a putative role for FAK in cardiomyocyte autophagy, we evaluated levels of canonical autophagic markers in nutrient-deprived primary neonatal rat cardiomyocytes (NRCMs) infected with adenoviruses expressing green fluorescent protein (GFP, control) or an active FAK variant (K578E/K581E) termed SuperFAK that exhibited enhanced catalytic activity (28). As shown in Fig. 1A, SuperFAK expression induced a robust increase in protein levels of p62 (also known as Sequestosome 1, or SQSTM1, a substrate degraded by autophagy) and a decrease in the conversion of the cytosolic precursor form of microtubule-associated protein light chain 3 (LC3-I) to the autophagosome-localized, cleaved, and lipidated form (LC3-II) as assessed by both quantification of LC3-II/LC3-I ratios and total LC3-II levels. Autophagic vacuoles can be visualized and quantified by ectopic expression of epitope-tagged LC3, and as shown in Fig. 1B, ectopic expression of SuperFAK in HL-1 atrial cardiomyocytes (29) significantly reduced the number of RFP-LC3 puncta, providing further support that FAK activity suppresses autophagy. Suppression of autophagic flux was confirmed in a separate set of experiments using the lysosome inhibitor, chloroquine (CQ) to block autophagy at the substrate degradation step. As expected, incubation with CQ led to a robust increase in LC3-II levels in all samples, but ectopic expression of SuperFAK significantly reduced LC3 turnover (i.e. the difference of LC3-II levels in the presence and absence of CQ as quantified using previously reported methods (Fig. 1C) (30). Notably, SuperFAK-dependent autophagic suppression was abolished by treatment with PF-573228 (PF-228), an inhibitor of FAK catalytic activity (Fig. 1D). Collectively, these studies indicate that elevating FAK activity might provide an effective means to dampen autophagic flux.
FIGURE 1.

Activation of FAK limits cardiomyocyte autophagy. A, NRCMs were infected with GFP or SuperFAK adenoviruses for 72 h, and cell lysates were blotted with indicated antibodies with GAPDH as a loading control. Quantification shown on right, n = 3; *, p < 0.05; **, p < 0.01 versus GFP. B, HL-1 cardiomyocytes were co-transfected with RFP-LC3 or HA-Beclin1 for 30 h, followed by infection with GFP or SuperFAK adenoviruses for 18 h and starvation for 4 h in the presence of CQ. Cells were then fixed with 4% paraformaldehyde and stained to detect HA (purple) and nuclei (DAPI, blue). GFP- (asterisk) or SuperFAK-infected cells appear green. Overexpression of SuperFAK reduced the number of RFP-LC3 puncta (autophagosomes, arrow). ***, p < 0.001 versus GFP. Quantification represents mean ± S.E. of three independent experiments (at least 50 cells positive for RFP-LC3, GFP, and HA-Beclin1 were analyzed in each group). Scale bar, 10 μm. C, NRCMs were infected with GFP or SuperFAK adenoviruses for 48 h. The lysosomal inhibitor CQ (10 μm) was added to half of the samples to assess autophagic flux. LC3-II turnover is defined as the amount of LC3-II delivered to lysosomes for degradation within a certain period of time and calculated by subtracting LC3-II band intensity in the presence of CQ with that in the absence of CQ. *, p < 0.05 versus GFP. D, NRCMs were infected with GFP or SuperFAK adenoviruses for 48 h with or without the potent and selective FAK inhibitor PF-228 in the last 24 h. *, p < 0.05; **, p < 0.01. NS, not significant. Data are mean ± S.E. of three independent experiments. All data were analyzed using two-tailed Student's t tests.
α-Adrenergic Signaling Limits Cardiac Autophagy via FAK Activation
We have shown that phenylephrine (PE)-dependent activation of myocardial α1-adrenergic receptor induces FAK activity, and others have shown that compensatory hypertrophic signaling induced by related Gq-coupled GPCRs is associated with a blunted autophagic response (4, 11, 31). Although a recent report indicated that long term treatment with PE led to adrenergic stress-induced autophagy (32), we reasoned that PE might limit autophagy during the initial compensatory phase when FAK is active. To test this postulate, we first treated C57Bl/6 mice with a single injection of PE at a concentration previously shown to induce compensatory hypertrophy (20 mg/kg, s.c.) (33) and evaluated myocardial p62 and LC3-I and -II levels 24 h following treatment. At this time point PE induced a marked increase in p62 protein levels and a concomitant decrease in the LC3-II/LC3-I ratio and total LC3-II levels but did not alter heart weight (Fig. 2A, data not shown), indicating that PE treatment suppresses autophagy prior to the promotion of hypertrophy. Although Beclin1 levels have been shown to positively correlate with autophagy in severe pressure overload-induced cardiac hypertrophy (34), no change in Beclin1 protein levels was observed following 24 h of PE treatment (Fig. 2A). Importantly, treatment of serum-starved NRCM with PE (50 μm) induced a time-dependent increase in p62 protein and a decrease in the LC3-II/LC3-I ratio and total LC3-II levels (Fig. 2B), indicating that PE-mediated autophagic suppression is autonomous to cardiomyocytes. Moreover, LC3 turnover was reduced in cells treated with PE and CQ, indicating that PE treatment inhibited autophagic flux in cardiac myocytes (Fig. 2C). Accordingly, PE treatment significantly attenuated serum starvation-induced accumulation of autophagic puncta in NRCMs (Fig. 2D) and in intact hearts as assessed using fasted transgenic RFP-GFP-LC3 mice treated with bafilomycin A1 (Fig. 2E) (35). Together, these results indicate that adrenergic signaling and elevated signaling through FAK lead to a similar transient suppression of cardiac autophagy.
FIGURE 2.

Activation of α-adrenergic signaling suppresses cardiac autophagy. A, C57BL/6 mice received a single injection of PE (20 mg/kg, s.c.) or saline control (n = 4 in each group). Hearts were harvested. and protein lysates were blotted using indicated antibodies with GAPDH as a loading control. B, NRCMs were treated with PE (50 μm) for various times from 2 to 24 h. Cell lysates were blotted with indicated antibodies. Quantification of p62 and LC3-II/LC3-I ratio and LC3-II levels are shown on right in A and B. *, p < 0.05; **, p < 0.01 versus non-treated control. Results are mean ± S.E. of three independent experiments. C, NRCMs were incubated with PE (50 μm) for 24 h with or without CQ (10 μm) in the last 4 h of treatment. LC3-II turnover was calculated by subtracting LC3-II band intensity in the presence of CQ with that in the absence of CQ. *, p < 0.05 versus vehicle. D, NRCMs were treated with PE (50 μm) or vehicle for 4 h in the presence of CQ (10 μm). Cells were subsequently stained with the CYTO-ID® autophagy detection kit was used to visualize autophagic vacuoles (green puncta). Treatment with PE significantly reduced puncta formation. *, p < 0.05 versus vehicle. At least 50 cells were examined per sample. Data are mean ± S.E. of three independent experiments. Scale bar, 20 μm. **, p < 0.01. E, CAG-RFP-EGFP-LC3 mice were transferred to food-free cages and received a single injection of PE (20 mg/kg, s.c.) or saline control (n = 3 in each group), and hearts were collected at 24 h. The lysosomal acidification inhibitor bafilomycin A1 was injected 2 h before euthanization. Autophagosomes appear orange, and autolysosomes appear red. Number of puncta was quantified from at least five different high power fields. *, p < 0.05. NS, not significant. Data were analyzed using two-tailed Student's t tests.
We next sought to determine whether PE-inhibited autophagy in a FAK-dependent fashion. Importantly, treatment of NRCM with PE induced a robust increase in FAK activity as assessed by increased auto-phosphorylation at Tyr-397 and increased Src-dependent phosphorylation of FAK at Tyr-576/577 and Tyr-925, collective indicators of full FAK catalytic activation (Fig. 3A). Importantly, siRNA-mediated knockdown of FAK fully attenuated PE-induced changes in p62 levels and LC3 modification, whereas knockdown of FAK only modestly increased basal autophagy (Fig. 3B) and autophagic flux (data not shown). Pre-treatment with the selective FAK inhibitor, PF-228, also abolished the PE-induced increase in p62 and blunted PE-induced changes in LC3-II (Fig. 3C). Moreover, PE failed to reduce the number of autophagic puncta induced by serum starvation when cultured primary cardiomyocytes were pre-treated with PF-228 (Fig. 3D). We next used our cardiac-restricted FAK gain- or loss-of-function mouse models to assess the extent that FAK contributes to PE-mediated autophagy suppression in vivo. Although fed/unstimulated cardiac-specific SuperFAK transgenic (SF2) mice tended to have lower levels of LC3-II than WT littermates, this effect was not significantly different. Similarly, basal autophagy was not altered in hearts from myocyte-restricted FAK knock-out mice when compared with WT hearts (Fig. 3E). This lack of effect could be due to compensation over time as these models exhibit early post-natal changes in cardiac FAK activity (23). However, the ability of PE to suppress basal autophagy was significantly blunted in the hearts from MFKO mice and significantly augmented in the hearts from SF2 mice relative to littermate controls (Fig. 3, F–H). These data indicate that PE signals through FAK to limit cardiac autophagy. We next determined whether elevated FAK activity can limit cardiac autophagy following mechanical overload by subjecting SF2 mice and littermates to either moderate or severe aortic constriction (induced by ligating the aorta to the width of a 27- or 28-gauge needle, respectively) for 3 days (34). SuperFAK was capable of suppressing cardiac autophagy-induced moderate hemodynamic overload (Fig. 3I). However, although SuperFAK was also clearly capable of limiting autophagy following severe pressure overload as assessed by elevated p62 levels and reduced LC3II/LC3I ratio, total LC3II levels were increased in these mice (Fig. 3J). Although future studies will be important to fully understand this finding, it is formally possible that accumulation of LC3II in this model results from a combination of a compensatory induction of autophagy by endoplasmic reticulum or mitochondrial stress pathways combined with a reduction in autophagy-dependent LC3II clearance. Collectively, these studies indicate that modulation of FAK activity can provide a means to control excessive autophagy in pathological settings.
FIGURE 3.

α-Adrenergic-dependent autophagy suppression requires FAK. A, NRCMs were treated with PE (50 μm) for various times from 2 to 24 h. Cell lysates were blotted with indicated antibodies. Identical lysates were used in Fig. 2B. Results are representative of three independent experiments. Quantification of FAK phosphorylation/total FAK is indicated (relative to the 0 time point) for three separate experiments. B, NRCMs were transfected with control or FAK siRNA (25 nm) for 72 h prior to treatment with PE (50 μm) for 24 h. Cell lysates were blotted with indicated antibodies (right panel). Data were quantified by densitometry and graphed as mean ± S.E. of three independent experiments; *, p < 0.05; **, p < 0.01. C, NRCMs were pre-treated with the pharmacological FAK inhibitor PF-228 (1 μm) for 1 h prior to the addition of PE (50 μm) for 24 h. Pre-treatment with FAK inhibitor blocked PE-mediated autophagic suppression (right panel). Data were quantified by densitometry and graphed as mean ± S.E. of three independent experiments; *, p < 0.05; **, p < 0.01. D, NRCMs were pre-treated with FAK inhibitor PF-228 (1 μm) for 1 h prior to incubation with PE (50 μm) for 4 h in the presence of CQ (10 μm). Autophagic vesicles were detected by staining with the CYTO-ID® autophagy detection kit. ***, p < 0.001. Scale bar, 20 μm. E, quantification of LC3-II levels in basal WT, MFKO myocyte-restricted FAK knock-out, and SF2 hearts (NS, not significant; n = 3–5 per group). F–H, MFKO, SF2 mice and WT littermates received a single injection of PE (20 mg/kg, s.c.). Hearts were harvested at 24 h, and protein lysates were blotted using indicated antibodies. Blots are representative of 3–5 hearts per group. H, quantification of LC3-II levels in PE-treated WT, MFKO, and SF2 hearts (**, p < 0.01; ***, p < 0.001). I and J, SF2 mice were subjected to moderate (I) or severe (J) TAC. Hearts were isolated at day 3, and protein lysates were blotted with indicated antibodies. Data are representative of three hearts per group.
FAK Phosphorylates the Core Autophagy Protein Beclin1 at Tyr-233
Although FAK can associate with p85PI3K and promote p85PI3K/AKT signaling to limit autophagy in some cancer cell types (36), we previously reported that FAK does not regulate AKT activity in cardiomyocytes (37), and our subsequent findings that phospho-AKT was not elevated in SF2 hearts either at baseline (data not shown) or following challenge (Fig. 3J) indicates that FAK-dependent autophagy suppression is likely not mediated by AKT signaling in these cells. Because the catalytic activity of FAK was necessary to suppress autophagy, we reasoned that FAK might phosphorylate a key substrate of the core autophagy initiation complex. As noted above, Beclin1 has been previously identified as a substrate for both serine/threonine and tyrosine kinases (15, 20) and through the use of the Group-based Prediction System algorithm (38), we identified Tyr-233 (a site highly conserved from lower vertebrates to mammals; Table 1) as a putative FAK phosphorylation site. To determine whether Beclin1 is a bona fide FAK substrate, COS cells were co-transfected with HA-tagged Beclin1 and FAK catalytic variants that included either wild-type FAK, a kinase deficient non-phosphorylatable mutant FAK (Y397F), or SuperFAK. Immunoprecipitation of HA-Beclin1 followed by Western blotting with a pan-Tyr(P) antibody revealed that Beclin1 tyrosine phosphorylation was markedly induced by expression of SuperFAK and to a lesser extent by wild-type FAK but not by the Y397F-FAK variant (Fig. 4A). To test the hypothesis that FAK phosphorylates Beclin1 at Tyr-233, a similar experiment was performed, and the blot was probed with a specific phospho-Beclin1 (Tyr-233) antibody. As shown in Fig. 4B, the level of phospho-Beclin1 (Tyr-233) was dramatically increased by expression of SuperFAK but not by FAK (Y397F). To test our postulate that Tyr-233 of Beclin1 is a major FAK phosphorylation site, we tested the ability of SuperFAK to induce phosphorylation of a Y233F Beclin1 variant. As shown in Fig. 4C, mutation of Tyr-233 to Phe-233 completely abolished SuperFAK-induced Beclin1 tyrosine phosphorylation, indicating that Tyr-233 is the sole FAK phosphorylation site on Beclin1. Because FAK acts in a bipartite complex with the tyrosine kinase pp60Src, we next explored whether FAK kinase activity per se was necessary for Beclin1 Tyr-233 phosphorylation. Indeed, we found that treatment with the FAK-specific catalytic inhibitor PF-228 attenuated SuperFAK-dependent Beclin1 phosphorylation without influencing total protein expression (Fig. 4D) indicating that Beclin1 is a substrate for FAK. Notably, in vitro kinase assays revealed that Beclin1 is also a substrate for Src; however, active Src induced comparable phosphorylation of WT and Y233F-Beclin1, indicating that Src and FAK likely phosphorylate distinct tyrosines (Fig. 4E). Importantly, phosphorylation of endogenous Beclin1 on Tyr-233 is evident in isolated cardiomyocytes following treatment with PE underscoring the importance of this post-translational modification in a physiological setting (Fig. 4F). Moreover, PE-induced phosphorylation of Tyr-233 Beclin is induced in vivo and is elevated in hearts from SuperFAK mice (Fig. 4G). The subsequent finding that siRNA-mediated knockdown of Beclin1 attenuated starvation-induced autophagic flux (data not shown) and SuperFAK-dependent increases in p62 protein levels (Fig. 5A) highlights the importance of Beclin1 in mediating FAK-dependent autophagic suppression.
TABLE 1.
Protein sequence of Beclin1 surrounding Tyr-233 in different species
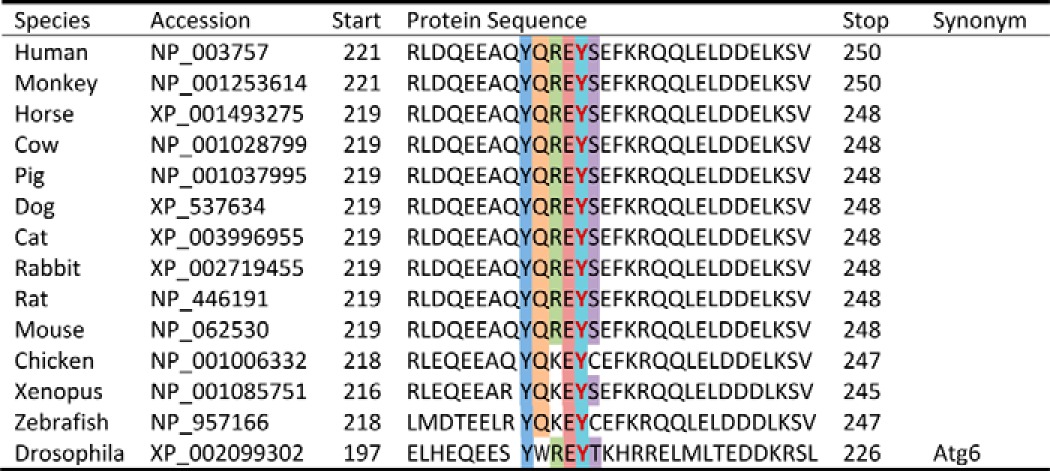
FIGURE 4.

FAK induces Beclin1 phosphorylation on Tyr-233. A, COS cells were co-transfected with HA-Beclin1 and wild-type FAK, non-phosphorylatable FAK mutant (Y397F), or SuperFAK. At 24 h, cell lysates were subjected to immunoprecipitation (IP) with anti-HA followed by Western blotting with indicated antibodies, including an anti-phosphotyrosine antibody (Tyr(P), 4G10). WCL, whole cell lysate. B, protein lysates from COS cells co-transfected with HA-Beclin1 and non-phosphorylatable FAK mutant (Y397F) or SuperFAK were immunoprecipitated with an anti-HA antibody followed by Western blotting analysis with anti-phospho-Beclin1 (Tyr-233). C, COS cells were co-transfected with SuperFAK and HA-Beclin1 or a non-phosphorylatable Beclin1 mutant (Y233F). Cell lysates were immunoprecipitated with an anti-HA antibody followed by Western blotting analysis with an anti-phosphotyrosine antibody (Tyr(P), 4G10). D, COS cells were co-transfected with SuperFAK and HA-Beclin1 prior to treatment with FAK inhibitor PF-228 for 16 h. Cell lysates were immunoprecipitated with an anti-HA antibody followed by Western blotting analysis with anti-phospho-Beclin1 (Tyr-233). E, in vitro Src kinase assay. Purified GST-Beclin1 and GST-Beclin1-Y233F MAPK were incubated with GST-Src and [γ-32P]ATP. Samples were analyzed by SDS-PAGE, and the gel was dried and exposed to Kodak XAR film for 2 h, followed by Coomassie Blue staining. Gel is representative of four separate experiments. F, NRCMs were treated with PE (50 μm) for 8 h. Duplicate cell lysates were blotted with indicated antibodies. G, cardiac-specific SuperFAK transgenic (SF2) mice and littermates received a single injection of PE (20 mg/kg, s.c.). Hearts were harvested at 24 h, and protein lysates were immunoprecipitated with Beclin1 antibody followed by immunoblotting using indicated antibodies. Blots represent three hearts per group, and data shown are mean ± S.E.; p < 0.05.
FIGURE 5.

Phosphorylation of Tyr-233 by FAK limits Beclin1-dependent autophagy. A, Beclin1 is required for SuperFAK-mediated autophagy suppression. NRCMs were infected with GFP or SuperFAK adenoviruses and transfected with control or Beclin1 siRNAs for 48 h prior to treatment with PE (50 μm) for 24 h. Cell lysates were blotted with indicated antibodies with GAPDH as a loading control. Quantification of mean ± S.E. of three independent experiments revealed that knockdown of Beclin1 reduced SuperFAK-induced increase in p62 protein levels when compared with si-Control (right panel). **, p < 0.01; ***, p < 0.001. B, FAK activity and FAK-dependent phosphorylation of Beclin1 alters the Beclin1 interactome. HL-1 cells were transfected with HA-Beclin1 or a phosphomimetic Beclin1 mutant (Y233E) for 48 h prior to starvation in the presence of FAK inhibitor PF-228 (1 μm) or vehicle control for 22 h. Lysates were immunoprecipitated with anti-HA and subjected to Western blotting with indicated antibodies. C, co-immunoprecipitation of endogenous Beclin1 with Atg14L in starved HL-1 cardiomyocytes treated with PF-228 (1 μm) or vehicle control for 22 h. D, Beclin Y233E phosphomimetic has limited capacity to induce autophagy. HL-1 cells were co-transfected with GFP-LC3 and HA-Beclin1 or a phosphomimetic Beclin1 variant (Y233E) for 48 h prior to treatment with starvation in the presence of FAK inhibitor PF-228 (1 μm) and CQ (10 μm) for 4 h. Cells were fixed and stained for GFP (green), HA (red), and nuclei (DAPI, blue). Beclin1(Y233E)-expressing cells exhibited less GFP-LC3 puncta (autophagosomes) than wild-type Beclin1-expressing cells. ***, p < 0.001 versus wild-type Beclin1. Results are mean ± S.E. of three independent experiments (>50 cells were analyzed each group). Scale bar, 10 μm. All data were analyzed using two-tailed Student's t tests.
FAK-mediated Phosphorylation of Beclin1 at Tyr-233 Compromised Beclin1-dependent Autophagy
Because FAK suppressed autophagy in a Beclin1dependent fashion, we reasoned that phosphorylation of Tyr-233 might either enhance Beclin1's association with autophagy inhibitors (i.e. Bcl2 and rubicon) or limit its association with complex I (Atg14L/Vps15/Vps34) or complex II (UVRAG/Vps15/Vps34) binding partners that are necessary for autophagosome formation or autophagosome-lysosomal fusion, respectively (15, 16, 20, 39). As shown in Fig. 5B, FAK inhibition markedly enhanced the association of HA-Beclin1 with Atg14L (i.e. complex I formation) but did not influence UVRAG binding (i.e. complex II formation). FAK inhibition induced a similar increase in Atg14L association with endogenous Beclin1 in HL-1 cells (Fig. 5C). Conversely, co-immunoprecipitation assays revealed that a phosphomimetic Y233E Beclin1 variant exhibited a weaker interaction with Atg14L and slightly stronger interaction with UVRAG when compared with WT Beclin1 (Fig. 5B, right panels). Because Tyr-233 lies within the coiled-coil domain that is known to mediate Atg14L association (40), it is likely that phosphorylation of this site directly interferes with Atg14L binding. Although the coiled-coil domain of Beclin1 is also responsible for Beclin1 homodimerization and inactivation, Tyr-233 phosphorylation did not alter Beclin1 self-association as assessed by co-immunoprecipitation of FLAG- and HA-tagged WT or Y233E variants (data not shown). We next assessed a functional role for Tyr-233 phosphorylation in Beclin1-dependent autophagosome formation. To this end, we co-expressed GFP-LC3 with HA-tagged Beclin1 variants in HL-1 cells and subjected the cells to nutrient depletion for 4 h. As shown in Fig. 5D, WT Beclin1 induced numerous GFP-LC3 puncta in starved HL-1 cells, whereas ectopic expression of the phosphomimetic variant Y233E induced significantly fewer puncta. Thus, FAK-dependent phosphorylation of Beclin1 on Tyr-233 suppressed autophagy by reducing Atg14L binding and limiting autophagosome formation.
To further evaluate the regulation of Beclin1 by FAK, we next investigated whether FAK and Beclin1 interact. Indeed, as shown in Fig. 6A, WT Beclin1 co-immunoprecipitated with WT FAK when co-expressed in COS cells. To map the interaction site(s) of FAK on Beclin1, similar co-immunoprecipitation experiments were performed in COS cells co-transfected with Myc-FAK and various Beclin1 fragments. Amino acids 1–242 but not 1–150 of Beclin1 co-immunoprecipitated with FAK, indicating that amino acids 151–242 (containing the CCD domain) contribute to the interaction between Beclin1 and FAK. In addition, the Beclin1 fragment 243–450 containing the evolutionarily conserved domain (ECD) co-precipitated with FAK. Thus, both the CCD and the ECD domain of Beclin1 are capable of binding to FAK. The finding that FAK associates more strongly with full-length Beclin1 than it does with either individual domain suggests that these sites might act in a cooperative fashion to promote FAK binding. We next tested the extent to which FAK activity might influence FAK-Beclin1 interactions. Interestingly, we found that inactive Y397F-FAK interacted more strongly with Beclin1 than did WT FAK (Figs. 4A and 6B). Accordingly, short term treatment with the FAK inhibitor (FAKi) significantly decreased FAK phosphorylation at Tyr-397 and enhanced the interaction between WT FAK and WT Beclin1 (Fig. 6C). Although FAK inhibition may also lead to de-phosphorylation of Beclin1 over time, the increased FAK-Beclin1 association observed in the previous experiment was not likely due to this occurrence because our subsequent studies revealed that phospho-deficient Y233F-Beclin1 actually associated slightly less avidly with Y397F-FAK than did WT Beclin1 (Fig. 6D). Collectively, these findings indicate that nutrient starvation promotes Beclin1-Atg14L containing complex formation and subsequent autophagosome formation. In turn, FAK activation promotes Beclin1 Tyr-233 phosphorylation, reduces FAK-Beclin1 interactions, attenuates complex I formation, and limits autophagy initiation (Fig. 6E). Thus, FAK regulates Beclin1-dependent autophagy in an activation-state specific manner.
FIGURE 6.

FAK associates with Beclin1 in an activation-specific fashion. A, top, schematic representation of three functional domains of Beclin1 (the N-terminal Bcl-2 homology 3 domain (BH3), the central coiled-coil domain (CCD), and the C-terminal ECD), and FLAG-tagged (FL) Beclin1 deletion constructs. Bottom, FAK interaction sites on Beclin1 were mapped by co-immunoprecipitation of Beclin1 with FAK in COS cells co-transfected with Myc-FAK and indicated FLAG-Beclin1 constructs. B, immunoprecipitation of Beclin1 with FAK in COS cells co-transfected with HA-Beclin1 and wild-type FAK or the non-phosphorylatable FAK mutant (Y397F). C, immunoprecipitation of Beclin1 with FAK in COS cells co-transfected with HA-Beclin1 and wild-type FAK and incubated ± FAK inhibitor 14 (50 μm for 1 h) prior to lysis. D, immunoprecipitation of Beclin1 with FAK in COS cells co-transfected with FAK(Y397F) and wild-type Beclin1 or the non-phosphorylatable Beclin1 variant (Y233F). E, model depicting the effect of FAK-mediated Beclin1 phosphorylation on autophagy initiation. Under conditions of nutrient starvation, Beclin1 forms a complex with Atg14L to promote autophagy. In response to neurohormone stimulation or mechanical stretch, FAK-mediated phosphorylation of Tyr-233 Beclin1 disrupts the Beclin1-Atg14L interaction and suppresses autophagy.
FAK-dependent Suppression of Autophagy Promotes Cardiomyocyte Hypertrophy
As we and others previously showed that adrenergic signaling-induced cardiac hypertrophy was mediated by activation of FAK (21, 22), we reasoned that FAK-dependent autophagy suppression might underlie this growth response. To test this possibility, we first sought to determine whether Beclin1 was required for PE-induced cardiac hypertrophy in SF2 mice. To this end, we crossed SF2 mice to available Beclin1+/− mice to obtain SF2/Beclin1+/− and SF2/Beclin1+/+ littermates, treated mice with PE for 24 h, and measured heart weight and cardiomyocyte cross-sectional area. Remarkably, we found that haplo-insufficiency of Beclin1 significantly attenuated the PE-induced cardiomyocyte hypertrophy that is apparent in SF2 (but not WT) mice at this time point (Fig. 7, A–C). When coupled with our observation that siRNA-mediated knockdown of Beclin1 prevented SuperFAK's ability to inhibit autophagy in NRCMs (Fig. 5A), these data suggest that limiting Beclin1-dependent autophagy contributes to the adaptive increase in myocardial mass induced by PE and FAK. Accordingly, pre-treatment of NRCMs with the FAK selective inhibitor, PF-228 not only blocked the PE-mediated autophagy suppression (refer to Fig. 3, C and D) but also abrogated PE-induced hypertrophic growth (Fig. 7, D–F). Finally, we explored a specific role for Tyr(P)-233 Beclin1 in the regulation of myocyte growth. As shown in Fig. 8, ectopic expression of WT Beclin1 induced marked atrophy in starved HL-1 cells, whereas ectopic expression of Y233E-Beclin1 did not affect cell size. Collectively, these studies indicate that FAK-dependent phosphorylation of Beclin1 facilitates PE-induced compensatory hypertrophic growth by suppressing autophagy-dependent myocyte atrophy.
FIGURE 7.

FAK-mediated cardiomyocyte hypertrophy requires Beclin1 and FAK activity. A–C, cardiac-specific SuperFAK transgenic (SF2) or non-transgenic (NTG) mice on a Beclin1+/+ or Beclin1+/− background received a single injection of PE (20 mg/kg, s.c.), and hearts were harvested 24 h following treatment. A, heart weight normalized to tibia length. B and C, myocyte cross-sectional area as assessed by staining with wheat germ agglutinin (red), cardiac troponin T (green), and DAPI (blue). At least 200 cells from five different fields were analyzed in each heart. *, p < 0.05; **, p < 0.01. Data are mean ± S.E. from 3 to 4 animals per group. D–F, pre-treatment with FAK inhibitor abrogated PE-induced cardiomyocyte hypertrophy. NRCMs were treated with PE (50 μm) with or without FAK inhibitor PF-228 (1 μm) for 48 h. D, cardiac myocytes were labeled with anti-cardiac troponin T (red), and cell surface area was quantified using ImageJ software (National Institutes of Health). **, p < 0.01. Data are mean ± S.E. of four independent experiments (>200 cells were analyzed each group). Scale bar, 20 μm. E, quantitative RT-PCR analysis of hypertrophy markers ANP (left) and BNP (right). *, p < 0.05; **, p < 0.01; ***, p < 0.001. Data are mean ± S.E. of three independent experiments and were analyzed using two-tailed Student's t tests. ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide.
FIGURE 8.

Beclin1 but not Y233E Beclin1 induces cardiomyocyte atrophy. HL-1 cells were transfected with HA-Beclin1 WT, HA-Beclin1 Y233E variant, or empty vector for 48 h. Cells were then fixed in 2% paraformaldehyde and stained with HA (red), phalloidin (green), and DAPI (blue). HL-1 cell size was measured using ImageJ software. Results are mean ± S.E. of three independent experiments (>50 cells were analyzed each group). Scale bar, 10 μm. NS, not significant. Data were analyzed by one-way analysis of variance with Dunnett's post hoc test.
Discussion
Autophagy promotes the clearance of toxic misfolded proteins and damaged organelles and is necessary to maintain cellular homeostasis, particularly for post-mitotic cells such as cardiac myocytes that may live for decades. Autophagy is a highly controlled and precise metabolic function, and dysregulation of autophagy has been implicated in a number of cardiovascular disorders such as cardiac hypertrophy, myocardial infarction, and doxorubicin-induced cardiomyopathy (4, 34, 41). Although it is clear that too much or too little autophagy can be detrimental, the mechanisms that underlie the tight spatio-temporal control of autophagy following cardiac stress have yet to be fully elucidated. Here, we showed that stimulation of α1-adrenergic receptor, a major GPCR family member essential for the development of cardiac hypertrophy, transiently suppressed autophagy by activation of FAK. Mechanistically, we demonstrated that direct phosphorylation of Beclin1 at Tyr-233 by FAK altered Beclin1's interactome, leading to diminished autophagic initiation. Moreover, we identified Beclin1 as a critical mediator of FAK-induced induction of myocyte hypertrophy, as depletion of Beclin1 blunted the FAK-dependent hypertrophic response to PE.
Despite mounting evidence for a causal role of autophagy suppression in the promotion of compensatory hypertrophy (4, 11, 31, 42–46), the mechanistic links between pressure overload and adrenergic signaling-induced autophagic suppression have remained largely unsolved. A prior report indicated that endothelin-1 can suppress autophagy by inducing ubiquitination and degradation of Atg14L through ZBTB16-Cullin3-Rock1 E3 ubiquitin ligase complex (47). Interestingly, we found that PE treatment also impaired Atg14L function, although through a distinct mechanism. In response to PE stimulation, FAK-mediated phosphorylation of Beclin1 at Tyr-233 disrupted the Beclin1-Atg14L interaction and suppressed autophagy initiation. Notably, we and others previously reported that α1-adrenergic stimulation of cardiac hypertrophy was augmented by integrin-dependent activation of FAK (21, 22), and mice deficient in FAK were resistant to pressure overload-induced hypertrophy (23, 24). Although global Beclin1 depletion did not affect basal anabolic heart growth, we showed that Beclin1 was required for the initial hypertrophic response that occurs in a PE/FAK-dependent fashion in un-starved mice. When coupled with our findings that phosphorylation of Tyr-233 Beclin1 not only suppressed autophagy, but also limited Beclin1-dependent cardiomyocyte atrophy in vitro, these data suggest that repression of Beclin1-dependent atrophy is necessary for FAK-dependent compensatory hypertrophic growth. The blockade of this basal bulk catabolic program likely primes cardiomyocytes for ensuing hypertrophic growth that occurs via de novo synthesis of proteins. Interestingly, very recent studies indicate that autophagy may compete with endocytic recycling as these pathways share common vesicle trafficking machinery (48, 49). Thus, it is also formally possible that a temporary block of autophagy in the face of continued secretory/endocytic recycling vesicle trafficking could promote plasma membrane expansion, an event that could ultimately prevent growth-induced stress on the plasma membrane and/or de-repress a growth-inhibitory negative feedback system. As FAK is known to be activated by TAC and other hypertrophic agonists, it will be future interest to determine whether this is a general mechanism by which these agents lead to suppression of myocyte autophagy-dependent atrophy.
As noted above, Beclin1 has previously been shown to be phosphorylated by the serine/threonine kinases Mst1 (16), ROCK1 (18), DAPK and Akt (15) among others (50), and these post-translational modifications have been shown to either promote or inhibit Beclin1 autophagy initiation complex formation. Beclin1 protein has a BH3 domain (amino acids 114–123) that interacts with the autophagy inhibitor Bcl-2/Bcl-xl, a CCD domain (amino acids 144–269) that binds autophagy promoting proteins Atg14L, UVRAG, Ambra1, and VPS34 and mediates Beclin1 homodimer formation, and an ECD domain (amino acids 244–337) that interacts with VPS34 and the anti-autophagic protein Rubicon (51). ROCK and DAPK phosphorylate Thr-119 within the BH3 (Bcl-2 binding) domain of Beclin1, and this phosphorylation limits Bcl-2/Bcl-xl binding and promotes autophagy (18, 52). In contrast, Mst1 phosphorylation of Beclin1 on a nearby residue (Thr-108) enhances the binding affinity for Bcl-2/Bcl-xl and limits autophagy (16). AKT-dependent phosphorylation of Beclin1 also limits autophagy, but in this case the mechanism involves phospho-Ser-234- and Ser-295-induced formation of a distinct autophagy-inhibitory Beclin1/14-3-3·vimentin intermediate filament complex (15). Of these serine/threonine Beclin1 phosphosites, the MST-1 target Thr-108 is the only one to date that has been studied in the context of cardiac biology, and these studies revealed that the Asp-108 phosphomimetic Beclin1 variant reduced cardiomyocyte autophagy and simultaneously promoted cardiomyocyte apoptosis by promoting the sequestration of Bcl-2/Bcl-xl from Bax (16).
Recently, the Levine laboratory (20) reported that Beclin1 is also a target for tyrosine phosphorylation-dependent regulation. In fact their studies showed that Tyr-233 was one of multiple tyrosine phosphorylation sites induced by EGFR signaling that collectively inhibited Beclin1-dependent autophagy. Mechanistically, EGFR-dependent tyrosine phosphorylation increased Beclin1 dimerization, promoted Beclin1 binding with Bcl-2 and Rubicon, and decreased Beclin1·VPS34 complex formation (20). However the specific function of Tyr-233 was not evaluated in their studies. Our studies are the first to show that Tyr(P)-233 is a major phospho-site in cardiomyocytes in vitro and in vivo and is the first to describe the consequence of this phosphosite on Beclin1 activity. We identified Tyr-233 within the CCD domain of Beclin1 as the sole site of phosphorylation by FAK. Moreover, we demonstrated that phosphorylation of Tyr-233 limits association of Beclin1 with the CCD-binding partner, Atg14L, indicating Tyr-233 is a critical site in defining the Beclin1 interactome and in modulating Beclin1-mediated autophagy and cardiomyocyte size. Because Tyr-233 is located at the antiparallel dimer packing interface of the metastable (imperfect) Beclin1 homodimer (53), we tested the possibility that the Y233E mutation might promote homodimer stability and thus limit the monomeric “pool” available for Atg14L binding. However, we found that the Y233E mutation did not have a major effect on Beclin1 dimerization as assessed by co-immunoprecipitation nor did it promote inhibitory (Bcl2/Beclin1 or rubicon/Beclin1) complex formation. This finding is consistent with the lack of effect of the phosphomimetic Y233E Beclin1 on cardiomyocyte apoptosis (data not shown) and our previous findings that FAK is a cardioprotective kinase (26, 27). Thus, although we cannot exclude that Tyr-233 phosphorylation results in subtle changes in dimer stabilization, the studies above combined with knowledge that only a small fraction of endogenous Beclin1 exists in a homodimeric form indicate that phosphorylation of Tyr-233 may alter Atg14L association by a distinct mechanism (54). In this regard, it is noteworthy that the nearby Glu-224 is a critical residue in the electrostatic pairing of Beclin1 with Atg14L, and thus it will be of interest to determine whether Tyr(P)-233 interferes with this charge pair (53, 54). In sum, although the precise mechanism by which Tyr(P)-233 regulates Atg14L binding remains to be identified, our study provides important insight into a novel regulatory pathway that controls the formation of various Beclin1-VPS34 sub-complexes to modulate autophagic flux. It will be of future interest to study the differential regulation and consequences of Beclin1 tyrosine and serine/threonine phosphorylations as coordinated phosphorylation of these sites will likely have a major impact on the regulation of Beclin1 function.
Because FAK is activated by EGFR signaling (55), it is likely that EGFR-induced Beclin1 phosphorylation at Tyr-233 is mediated, at least in part, by FAK, that phosphorylation of this site is sufficient to limit complex 1 formation, but that additional FAK-independent EGFR tyrosine phosphorylations are necessary to fully attenuate Beclin1-dependent autophagy. EGFR is known to be transactivated by numerous GPCRs, and EGFR function is critical for GPCR-stimulated mitogenic responses; however, further studies are necessary to determine the extent to which EGFR transactivation regulates PE-mediated autophagy suppression. Nevertheless, given that EGF signaling promotes mitogenic responses in many cell types and that EGFR-mediated autophagy suppression facilitates tumor initiation, progression, and acquired resistance to tyrosine kinase inhibitors (11), the consequence of FAK-dependent tyrosine phosphorylation of Beclin1 likely reaches beyond the physiological initiation of cardiac hypertrophy defined herein.
Autophagy is known to be enhanced following cardiac I/R, but studies targeting various components of this pathway have led to different conclusions as to whether autophagy is adaptive or maladaptive in the context of ischemic heart disease (2, 3). Although treatment with bafilomycin A (which blocks lysosomal degradation of autophagosomes) or inhibition of Atg5 (a protein essential for the capture of ubiquitinated proteins and LC3-dependent expansion of autophagosomes) worsened I/R injury and promoted the accumulation of damaged mitochondria in this setting (5, 6), mice haplo-insufficient for Beclin1 that exhibited decreased autophagy were protected from I/R damage (7, 8). Similar discordant results were observed in pressure overload models of cardiac hypertrophy. Indeed, inactivation of Atg5 was detrimental, whereas inactivation of Beclin1 was protective (2, 4, 34). Moreover, very recent studies indicate that the partial suppression of autophagy initiation conferred by Beclin1 haplo-insufficiency also protected mice from doxorubicin-induced cardiotoxicity (35). The latter studies showed that doxorubicin blocks autophagic flux by alkalinizing the lysosome and that the resultant build-up of autophagosomes contributes to cardiotoxicity. Thus, decreased autophagy initiation was cardioprotective in this setting as it resulted in a diminished demand on the lysosomal system. We reason that this mechanism may also contribute to the cardioprotection conferred by Beclin1 haplo-insufficiency in I/R and TAC models and posit that FAK-dependent phosphorylation of Beclin1 may act in a similar protective fashion. Indeed, we found that FAK-dependent Beclin1 phosphorylation attenuates complex I formation and autophagosome formation but permits complex II formation and the fusion of pre-existing autophagosomes with lysosomes thus limiting the build-up of autophagic vesicle intermediates. Interestingly, there are striking parallels between challenged Beclin1+/− mice and SF2 mice. Both lines of mice exhibit preserved systolic function and reduced perivascular and interstitial fibrosis following I/R or doxorubicin treatment (7, 26, 27, 35). Moreover, myocardial infarct size and myocyte apoptosis following I/R was significantly reduced in both models (8, 26). Thus, it will be of future interest to determine the extent that FAK-dependent phosphorylation of Beclin1 regulates the pathological progression of ischemic heart disease.
Although the mechanisms were previously unclear, integrin-dependent signals have also long been known to limit autophagy. We now show that FAK suppresses autophagy by phosphorylation of Beclin1 at Tyr-233 and that this response is causal for the initiation of PE/FAK-dependent myocyte hypertrophy. Tyr-233 lies within the CCD domain of Beclin1, and FAK-dependent phosphorylation of this site limits Beclin1·Atg14L complex formation, reduces bulk autophagy, and limits myocyte atrophy. This novel mechanism to limit Beclin1 activity has important implications for future treatment of a variety of pathophysiological processes associated with altered autophagic flux.
Experimental Procedures
Animals
C57BL/6 mice were purchased from Charles River Laboratories. MFKO mice and cardiac-specific SuperFAK (SF) transgenic mice (SF2) were generated on a C57BL/6 background as described previously (23, 26). CAG-RFP-EGFP-LC3 mice were kindly provided by Dr. Thomas G. Gillette (University of Texas Southwestern Medical Center). Beclin1+/− mice were obtained from The Jackson Laboratory. Mice were injected with phenylephrine (Sigma) at a dose (20 mg/kg, s.c.) that has previously been shown to induce cardiac hypertrophy (33) or 0.9% NaCl as a control. In some experiments, cardiac hypertrophy was induced by pressure overload using the TAC surgery as described previously (23).
NRCM Isolation and Treatments
NRCMs were isolated from 2- to 3-day-old Wistar rats using the Neonatal Cardiomyocyte Isolation System (Worthington) as described previously (27). Cells were plated on fibronectin-coated dishes and cultured in Media 199 supplemented with 15% FBS, 1% penicillin/streptomycin, and 100 μm BrdU for 24 h and then switched to serum-free Medium 199 with 1% penicillin/streptomycin. The following drug concentrations were used: phenylephrine (50 μm), PF-228 (1 μm), chloroquine (Sigma, 10 μm) or as indicated.
Adenoviral Infection and siRNA Transfection
Adenoviral infection was performed at a multiplicity of infection of 50 using adenoviruses for GFP or SuperFAK containing an IRES-GFP cassette. Transfection with siRNA was carried out using HiPerfect (Qiagen) following the manufacturer's protocol. The following siRNAs were used: rat FAK siRNA (sc-156037, Santa Cruz Biotechnology), Beclin1 siRNA-1 (CUCAGGAGAGGAGCCAUUU(dT)(dT)), Beclin1 siRNA-2 (GGGUAAUAUUAAACCACAU(dT)(dT)), and control scrambled siRNA (Life Technologies, Inc.).
Cell Lines and Plasmid Transfection
HL-1 cells (mouse atrial cardiomyocyte cell line, a generous gift from Dr. William C. Claycomb) were cultured in Claycomb Medium supplemented with 10% FBS, 1% penicillin/streptomycin, 0.1 mm norepinephrine, and 2 mm l-Glutamine. COS cells (monkey kidney fibroblast-like cell line) were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Plasmid transfection was performed using Lipofectamine 2000 (Life Technologies, Inc.) for HL-1 cells or TransIT-LT1 (Mirus Bio) for COS cells as per manufacturer's instructions.
Immunoprecipitation and Immunoblotting
Protein extracts were prepared from tissue and cultured cells using RIPA lysis buffer supplemented with HaltTM protease and phosphatase inhibitor mixtures (Thermo Scientific), and protein concentration was measured using the Pierce BCA protein assay kit (Thermo Scientific). Protein lysates (500 μg to 1 mg) were incubated with mouse anti-HA (sc-7392, Santa Cruz Biotechnology) or mouse anti-Myc (catalog no. 2276, Cell Signaling) bound with Dynabeads® Protein G (Life Technologies, Inc.) at 4 °C for 1 h. Precipitated protein complexes were eluted by boiling in sample buffer for 5 min. For immunoblotting, protein lysates (10–50 μg of protein) were separated by SDS-PAGE, transferred to PVDF membranes, and blotted with the following antibodies: rabbit anti-pFAK (Y397) (44624G, Invitrogen, 1:1000); rabbit anti-pFAK (Y576/577) (catalog no. 3281, Cell Signaling, 1:1000); rabbit anti-pFAK (Y925) (catalog no. 3284, Cell Signaling, 1:1000); mouse anti-FAK (clone 4.47, Millipore, 1:1000); mouse anti-phosphotyrosine (4G10, Millipore, 1:1000); rabbit anti-HA (sc-805, Santa Cruz Biotechnology, 1:1000); rabbit anti-Beclin1 (sc-11427, Santa Cruz Biotechnology, 1:1000); rabbit anti-phospho-Beclin1(Y233) (kind gift from Dr. Beth Levine, custom-produced by Phosphosolutions Inc., 1:400); mouse anti-SQSTM1/p62 (ab56416, Abcam, 1:2000); rabbit anti-LC3A/B (catalog no. 12741, Cell Signaling, 1:1000); rabbit anti-VPS34 (catalog no. 4263, Cell Signaling, 1:1000); rabbit anti-Atg14L (PD026, MBL, 1:500); mouse anti-UVRAG (M160–3, MBL, 1:1000); rabbit anti-FLAG (catalog no. 2368, Cell Signaling, 1:1000); and rabbit anti-GAPDH (sc-25778, Santa Cruz Biotechnology, 1:1000).
In Vitro Phosphorylation of Beclin1
Recombinant active GST-Src (100 ng, Abcam ab60884) was combined with equal amounts of GST-Beclin1 fusion proteins (∼1 μg each) and [γ-32P]ATP (1 μCi) in kinase reaction buffer (10 mm MOPS, pH 7.2, 5 mm glycerol 2-phosphate, 10 mm MgCl2, 2 mm EGTA, 0.8 mm EDTA, 1 μm DTT, and 1 μm ATP). After incubation for 15 min at 30 °C, the reaction mixtures were boiled in SDS sample buffer, resolved by 7.5% SDS-PAGE, and visualized by autoradiography. Total protein was measured by Coomassie staining.
Autophagic Activity and Flux Assays
HL-1 cells were transfected with GFP-LC3 (kind gift from Dr. Douglas Cyr, University of North Carolina, Chapel Hill) followed by treatment with FAK inhibitor PF-573228 (Sigma, 1 μm) in serum-free media for 4 h or as indicated. Autophagy was assessed by quantitation of GFP-LC3 puncta numbers per cell blindly using the ImageJ software or by immunoblotting for LC3-I/LC3-II and SQSTM1/p62. In cultured NRCMs, autophagic vacuoles were stained by using the Cyto-IDTM autophagy detection kit (Enzo Life Sciences). To monitor autophagic flux in vitro, cells were treated with the lysosomal inhibitor chloroquine (10 μm) for 4–24 h. For assessment of autophagic flux in vivo, fasted CAG-RFP-EGFP-LC3 mice received a single injection of PE (20 mg/kg, s.c.) at 0 h, an i.p. injection of the lysosomal inhibitor bafilomycin A1 (1.5 mg/kg) at 22 h, and mice were euthanized at 24 h based on a recently published protocol (35). Hearts were then isolated and processed for frozen sectioning using Tissue-Tek O.C.T. Compound. During the study period, mice have free access to water but were deprived of food. Tissue sections were observed under a Zeiss 700 confocal microscope using the ×63 oil lens. Maturation of autophagosome (neural pH) into autolysosome (acidic pH) is visualized by the loss of the acid-sensitive EGFP signal.
Immunofluorescence Staining
NRCMs or HL-1 cells cultured in chamber slides were fixed with 4% paraformaldehyde, permeabilized with PBS containing 0.1% Triton X-100, and incubated with mouse anti-cardiac troponin T (MS-295-P, Thermo Scientific, 1:100), mouse anti-HA (catalog no. 2367, Cell Signaling, 1:50), and Acti-stain 488 fluorescent phalloidin (PHDG1, Cytoskeleton, Inc., 100 nm). Paraffin-embedded tissue sections were subjected to deparaffinization, rehydration, and antigen retrieval as described previously (26). Sections were then incubated with mouse anti-cardiac troponin T (MS-295-P, Thermo Scientific, 1:100) and wheat germ agglutinin (W32466, Invitrogen, 1:100). Immunofluorescence signals were visualized by incubation with appropriated Alexa Fluor® fluorescent-conjugated secondary antibodies (Invitrogen) and then observed under a Zeiss LSM 710 confocal laser-scanning microscope (Zeiss, Germany). Cross-sectional area and surface area of cardiomyocytes were measured by tracing cell peripheries using ImageJ software (National Institutes of Health).
Quantitative Reverse Transcription-PCR
Total RNA was isolated from cells using the Quick-RNATM MiniPrep kit (Zymo Research), and cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad). Quantitative real time PCR was performed using the Maxima SYBR Green/ROX quantitative PCR master mix (Thermo Scientific) in an Applied Biosystems 7500 real time PCR system with GAPDH as an internal control. Primer pairs used were as follows: atrial natriuretic peptide, 5′-CCAGGCCATATTGGAGCAAATCC-3′ and 5′-CATGACCTCATCTTCTACCGGCAT-3′; brain natriuretic peptide, 5′-TGGGAAGTCCTAGCCAGTCTCC-3′ and 5′-CTTTTCTCTTATCAGCTCCAGCAGC-3′; and GAPDH, 5′-GCTGGCATTGCTCTCAATGACAA-3′ and 5′-GTCCACCACCCTGTTGCTGTA-3′.
Statistics
Results are presented as mean ± S.E. Statistical comparisons between two groups were performed using two-tailed unpaired Student's t test. Multigroup comparisons were performed using one-way analysis of variance followed by post hoc tests to calculate pairwise differences. A p value of less than 0.05 was considered significant.
Study Approval
All procedures were performed in accordance with National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the IACUC at the University of North Carolina, Chapel Hill.
Author Contributions
J. M. T. conceived the hypothesis and supervised the project; Z. C. and J. M. T. designed the research, conducted the experiments, analyzed the data, and wrote the paper; R. D., Q. Z., and Z. O. conducted the experiments and analyzed the data; C. P. M. provided intellectual input; D. M. C provided intellectual input and reagents.
Acknowledgments
We thank Dr. Beth Levine (University of Texas Southwestern Medical Center) and Phosphosolutions, Inc., for providing critical reagents.
This work was supported in part by National Institutes of Health Grants HL-081844 and HL-130367 from NHLBI and American Heart Association Grant 0355776U (to J. M. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
- I/R
- ischemia/reperfusion
- FAK
- focal adhesion kinase
- PE
- phenylephrine
- NRCM
- neonatal rat cardiomyocyte
- GPCR
- G-protein-coupled receptor
- EGFR
- epidermal growth factor receptor
- CQ
- chloroquine
- PF-228
- PF-573228
- TAC
- transverse aortic constriction
- EGFP
- enhanced GFP
- ECD
- evolutionarily conserved domain
- MFKO
- myocyte-restricted FAK knock-out
- SF
- SuperFAK.
References
- 1. Wesselborg S., and Stork B. (2015) Autophagy signal transduction by atg proteins: From hierarchies to networks. Cell. Mol. Life Sci. 72, 4721–4757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rothermel B. A., and Hill J. A. (2008) Autophagy in load-induced heart disease. Circ. Res. 103, 1363–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gustafsson A. B., and Gottlieb R. A. (2009) Autophagy in ischemic heart disease. Circ. Res. 104, 150–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nakai A., Yamaguchi O., Takeda T., Higuchi Y., Hikoso S., Taniike M., Omiya S., Mizote I., Matsumura Y., Asahi M., Nishida K., Hori M., Mizushima N., and Otsu K. (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 13, 619–624 [DOI] [PubMed] [Google Scholar]
- 5. Kanamori H., Takemura G., Goto K., Maruyama R., Tsujimoto A., Ogino A., Takeyama T., Kawaguchi T., Watanabe T., Fujiwara T., Fujiwara H., Seishima M., and Minatoguchi S. (2011) The role of autophagy emerging in postinfarction cardiac remodelling. Cardiovasc. Res. 91, 330–339 [DOI] [PubMed] [Google Scholar]
- 6. Hamacher-Brady A., Brady N. R., and Gottlieb R. A. (2006) Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 281, 29776–29787 [DOI] [PubMed] [Google Scholar]
- 7. Hariharan N., Zhai P., and Sadoshima J. (2011) Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid. Redox Signal. 14, 2179–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matsui Y., Takagi H., Qu X., Abdellatif M., Sakoda H., Asano T., Levine B., and Sadoshima J. (2007) Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and beclin 1 in mediating autophagy. Circ. Res. 100, 914–922 [DOI] [PubMed] [Google Scholar]
- 9. Hariharan N., Ikeda Y., Hong C., Alcendor R. R., Usui S., Gao S., Maejima Y., and Sadoshima J. (2013) Autophagy plays an essential role in mediating regression of hypertrophy during unloading of the heart. PLoS ONE 8, e51632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oyabu J., Yamaguchi O., Hikoso S., Takeda T., Oka T., Murakawa T., Yasui H., Ueda H., Nakayama H., Taneike M., Omiya S., Shah A. M., Nishida K., and Otsu K. (2013) Autophagy-mediated degradation is necessary for regression of cardiac hypertrophy during ventricular unloading. Biochem. Biophys. Res. Commun. 441, 787–792 [DOI] [PubMed] [Google Scholar]
- 11. Pfeifer U., Föhr J., Wilhelm W., and Dämmrich J. (1987) Short-term inhibition of cardiac cellular autophagy by isoproterenol. J. Mol. Cell. Cardiol. 19, 1179–1184 [DOI] [PubMed] [Google Scholar]
- 12. Shi C. S., and Kehrl J. H. (2010) Traf6 and a20 regulate lysine 63-linked ubiquitination of beclin-1 to control tlr4-induced autophagy. Sci. Signal. 3, ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Platta H. W., Abrahamsen H., Thoresen S. B., and Stenmark H. (2012) Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor beclin 1. Biochem. J. 441, 399–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xia P., Wang S., Du Y., Zhao Z., Shi L., Sun L., Huang G., Ye B., Li C., Dai Z., Hou N., Cheng X., Sun Q., Li L., Yang X., and Fan Z. (2013) Wash inhibits autophagy through suppression of beclin 1 ubiquitination. EMBO J. 32, 2685–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang R. C., Wei Y., An Z., Zou Z., Xiao G., Bhagat G., White M., Reichelt J., and Levine B. (2012) Akt-mediated regulation of autophagy and tumorigenesis through beclin 1 phosphorylation. Science 338, 956–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maejima Y., Kyoi S., Zhai P., Liu T., Li H., Ivessa A., Sciarretta S., Del Re D. P., Zablocki D. K., Hsu C. P., Lim D. S., Isobe M., and Sadoshima J. (2013) Mst1 inhibits autophagy by promoting the interaction between beclin1 and bcl-2. Nat. Med. 19, 1478–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Russell R. C., Tian Y., Yuan H., Park H. W., Chang Y. Y., Kim J., Kim H., Neufeld T. P., Dillin A., and Guan K. L. (2013) Ulk1 induces autophagy by phosphorylating beclin-1 and activating vps34 lipid kinase. Nat. Cell Biol. 15, 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gurkar A. U., Chu K., Raj L., Bouley R., Lee S. H., Kim Y. B., Dunn S. E., Mandinova A., and Lee S. W. (2013) Identification of rock1 kinase as a critical regulator of beclin1-mediated autophagy during metabolic stress. Nat. Commun. 4, 2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wei Y., An Z., Zou Z., Sumpter R., Su M., Zang X., Sinha S., Gaestel M., and Levine B. (2015) The stress-responsive kinases mapkapk2/mapkapk3 activate starvation-induced autophagy through beclin 1 phosphorylation. eLife 10.7554/eLife.05289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei Y., Zou Z., Becker N., Anderson M., Sumpter R., Xiao G., Kinch L., Koduru P., Christudass C. S., Veltri R. W., Grishin N. V., Peyton M., Minna J., Bhagat G., and Levine B. (2013) Egfr-mediated beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 154, 1269–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Taylor J. M., Rovin J. D., and Parsons J. T. (2000) A role for focal adhesion kinase in phenylephrine-induced hypertrophy of rat ventricular cardiomyocytes. J. Biol. Chem. 275, 19250–19257 [DOI] [PubMed] [Google Scholar]
- 22. Pham C. G., Harpf A. E., Keller R. S., Vu H. T., Shai S. Y., Loftus J. C., and Ross R. S. (2000) Striated muscle-specific β(1d)-integrin and fak are involved in cardiac myocyte hypertrophic response pathway. Am. J. Physiol. Heart Circ. Physiol. 279, H2916–H2926 [DOI] [PubMed] [Google Scholar]
- 23. DiMichele L. A., Doherty J. T., Rojas M., Beggs H. E., Reichardt L. F., Mack C. P., and Taylor J. M. (2006) Myocyte-restricted focal adhesion kinase deletion attenuates pressure overload-induced hypertrophy. Circ. Res. 99, 636–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clemente C. F., Tornatore T. F., Theizen T. H., Deckmann A. C., Pereira T. C., Lopes-Cendes I., Souza J. R., and Franchini K. G. (2007) Targeting focal adhesion kinase with small interfering RNA prevents and reverses load-induced cardiac hypertrophy in mice. Circ. Res. 101, 1339–1348 [DOI] [PubMed] [Google Scholar]
- 25. Lock R., and Debnath J. (2008) Extracellular matrix regulation of autophagy. Curr. Opin. Cell Biol. 20, 583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheng Z., DiMichele L. A., Hakim Z. S., Rojas M., Mack C. P., and Taylor J. M. (2012) Targeted focal adhesion kinase activation in cardiomyocytes protects the heart from ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 32, 924–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheng Z., DiMichele L. A., Rojas M., Vaziri C., Mack C. P., and Taylor J. M. (2014) Focal adhesion kinase antagonizes doxorubicin cardiotoxicity via p21(cip1.). J. Mol. Cell. Cardiol. 67, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gabarra-Niecko V., Keely P. J., and Schaller M. D. (2002) Characterization of an activated mutant of focal adhesion kinase: ‘Superfak’. Biochem. J. 365, 591–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Claycomb W. C., Lanson N. A. Jr, Stallworth B. S., Egeland D. B., Delcarpio J. B., Bahinski A., and Izzo N. J. Jr. (1998) Hl-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U.S.A. 95, 2979–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mizushima N., Yoshimori T., and Levine B. (2010) Methods in mammalian autophagy research. Cell 140, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ceylan-Isik A. F., Dong M., Zhang Y., Dong F., Turdi S., Nair S., Yanagisawa M., and Ren J. (2013) Cardiomyocyte-specific deletion of endothelin receptor a rescues aging-associated cardiac hypertrophy and contractile dysfunction: role of autophagy. Basic Res. Cardiol. 108, 335. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Nakaoka M., Iwai-Kanai E., Katamura M., Okawa Y., Mita Y., and Matoba S. (2015) An α-adrenergic agonist protects hearts by inducing akt1-mediated autophagy. Biochem. Biophys. Res. Commun. 456, 250–256 [DOI] [PubMed] [Google Scholar]
- 33. Huang S., Zou X., Zhu J. N., Fu Y. H., Lin Q. X., Liang Y. Y., Deng C. Y., Kuang S. J., Zhang M. Z., Liao Y. L., Zheng X. L., Yu X. Y., and Shan Z. X. (2015) Attenuation of microrna-16 derepresses the cyclins d1, d2 and e1 to provoke cardiomyocyte hypertrophy. J. Cell. Mol. Med. 19, 608–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhu H., Tannous P., Johnstone J. L., Kong Y., Shelton J. M., Richardson J. A., Le V., Levine B., Rothermel B. A., and Hill J. A. (2007) Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest. 117, 1782–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li D. L., Wang Z. V., Ding G., Tan W., Luo X., Criollo A., Xie M., Jiang N., May H., Kyrychenko V., Schneider J. W., Gillette T. G., and Hill J. A. (2016) Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation 133, 1668–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sandilands E., Schoenherr C., and Frame M. C. (2015) P70s6k is regulated by focal adhesion kinase and is required for src-selective autophagy. Cell. Signal. 27, 1816–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DiMichele L. A., Hakim Z. S., Sayers R. L., Rojas M., Schwartz R. J., Mack C. P., and Taylor J. M. (2009) Transient expression of frnk reveals stage-specific requirement for focal adhesion kinase activity in cardiac growth. Circ. Res. 104, 1201–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xue Y., Ren J., Gao X., Jin C., Wen L., and Yao X. (2008) Gps 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics. 7, 1598–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Funderburk S. F., Wang Q. J., and Yue Z. (2010) The beclin 1-vps34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 20, 355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Itakura E., Kishi C., Inoue K., and Mizushima N. (2008) Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian atg14 and uvrag. Mol. Biol. Cell 19, 5360–5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nishida K., and Otsu K. (2016) Autophagy during cardiac remodeling. J. Mol. Cell. Cardiol. 10.1016/j.yjmcc.2015.12.003 [DOI] [PubMed] [Google Scholar]
- 42. Nishida K., Kyoi S., Yamaguchi O., Sadoshima J., and Otsu K. (2009) The role of autophagy in the heart. Cell Death Differ. 16, 31–38 [DOI] [PubMed] [Google Scholar]
- 43. Dämmrich J., and Pfeifer U. (1983) Cardiac hypertrophy in rats after supravalvular aortic constriction. I. Size and number of cardiomyocytes, endothelial and interstitial cells. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 43, 265–286 [DOI] [PubMed] [Google Scholar]
- 44. Dämmrich J., and Pfeifer U. (1983) Cardiac hypertrophy in rats after supravalvular aortic constriction. Ii. Inhibition of cellular autophagy in hypertrophying cardiomyocytes. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 43, 287–307 [DOI] [PubMed] [Google Scholar]
- 45. Wauson E. M., Dbouk H. A., Ghosh A. B., and Cobb M. H. (2014) G protein-coupled receptors and the regulation of autophagy. Trends Endocrinol. Metab. 25, 274–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hathaway C. K., Grant R., Hagaman J. R., Hiller S., Li F., Xu L., Chang A. S., Madden V. J., Bagnell C. R., Rojas M., Kim H. S., Wu B., Zhou B., Smithies O., and Kakoki M. (2015) Endothelin-1 critically influences cardiac function via superoxide-mmp9 cascade. Proc. Natl. Acad. Sci. U.S.A. 112, 5141–5146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang T., Dong K., Liang W., Xu D., Xia H., Geng J., Najafov A., Liu M., Li Y., Han X., Xiao J., Jin Z., Peng T., Gao Y., Cai Y., et al. (2015) G-protein-coupled receptors regulate autophagy by zbtb16-mediated ubiquitination and proteasomal degradation of atg14l. Elife 4, e06734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim H. J., Zhong Q., Sheng Z. H., Yoshimori T., Liang C., and Jung J. U. (2012) Beclin-1-interacting autophagy protein atg14l targets the snare-associated protein snapin to coordinate endocytic trafficking. J. Cell Sci. 125, 4740–4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Farré J. C., and Subramani S. (2011) Rallying the exocyst as an autophagy scaffold. Cell 144, 172–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Maejima Y., Isobe M., and Sadoshima J. (2016) Regulation of autophagy by beclin 1 in the heart. J. Mol. Cell. Cardiol. 95, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kang R., Zeh H. J., Lotze M. T., and Tang D. (2011) The beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zalckvar E., Berissi H., Mizrachy L., Idelchuk Y., Koren I., Eisenstein M., Sabanay H., Pinkas-Kramarski R., and Kimchi A. (2009) Dap-kinase-mediated phosphorylation on the bh3 domain of beclin 1 promotes dissociation of beclin 1 from bcl-xl and induction of autophagy. EMBO Rep. 10, 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li X., He L., Che K. H., Funderburk S. F., Pan L., Pan N., Zhang M., Yue Z., and Zhao Y. (2012) Imperfect interface of beclin1 coiled-coil domain regulates homodimer and heterodimer formation with atg14l and uvrag. Nat. Commun. 3, 662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hurley J. H., and Schulman B. A. (2014) Atomistic autophagy: the structures of cellular self-digestion. Cell 157, 300–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Long W., Yi P., Amazit L., LaMarca H. L., Ashcroft F., Kumar R., Mancini M. A., Tsai S. Y., Tsai M. J., and O'Malley B. W. (2010) Src-3delta4 mediates the interaction of egfr with fak to promote cell migration. Mol. Cell 37, 321–332 [DOI] [PMC free article] [PubMed] [Google Scholar]