

Abstract
DOCK8 is an atypical guanine nucleotide exchange factor for Cdc42, and its mutations cause combined immunodeficiency in humans. Accumulating evidence indicates that DOCK8 regulates the migration and activation of various subsets of leukocytes, but its regulatory mechanism is poorly understood. We here report that DOCK8-deficient macrophages exhibit a migration defect in a 2D setting. Although DOCK8 deficiency in macrophages did not affect the global Cdc42 activation induced by chemokine stimulation, rescue experiments revealed that the guanine nucleotide exchange factor activity of DOCK8 was required for macrophage migration. We found that DOCK8 associated with LRAP35a, an adaptor molecule that binds to the Cdc42 effector myotonic dystrophy kinase-related Cdc42-binding kinase, and facilitated its activity to phosphorylate myosin II regulatory light chain. When this interaction was disrupted in WT macrophages, they showed a migration defect, as seen in DOCK8-deficient macrophages. These results suggest that, during macrophage migration, DOCK8 links Cdc42 activation to actomyosin dynamics through the association with LRAP35a.
Keywords: Cdc42, cell migration, guanine nucleotide exchange factor (GEF), macrophage, signal transduction, actomyosin dynamics
Introduction
Cdc42 is a member of the small GTPases that function as molecule switches by cycling between GDP-bound inactive and GTP-bound active states (1). The stimulus-induced formation of active Cdc42 is mediated by guanine nucleotide exchange factors (GEFs),2 and, when activated, Cdc42 binds to multiple effector molecules (1). One such effector molecule is myotonic dystrophy kinase-related Cdc42-binding kinase (MRCK), which stimulates the phosphorylation of myosin II regulatory light chain (MLC2) and controls actomyosin dynamics (2–5). Cdc42 is known to act as a master regulator of cell polarity in eukaryotic organisms ranging from yeasts to humans (1). In addition, it has been shown that Cdc42-deficient dendritic cells (DCs), a subset of antigen-presenting leukocytes, fail to migrate effectively within 3D extracellular matrix scaffolds, whereas they exhibit only limited defects in a 2D setting (6). Thus, Cdc42 plays a unique role in leukocyte migration.
DOCK8 is a member of the evolutionarily conserved DOCK family proteins that function as GEFs for the Rho family of GTPases (7, 8). Although DOCK8 does not contain the Dbl homology domain typically found in GEFs, it does bind to Cdc42 and mediates the GTP-GDP exchange reaction through the DOCK homology region 2 (DHR2) domain (9). Recently, substantial attention has been paid to the signaling and functions of DOCK8, as it has been reported that Dock8 mutations cause combined immunodeficiency syndrome in humans, with morphological and functional abnormalities of leukocytes (10–15). The role of DOCK8 in leukocytes was also demonstrated using animal models. For example, the N-ethyl-N-nitrosourea-mediated mutagenesis in mice has shown that DOCK8 plays an important role in B cell immunological synapse and long-lasting humoral immunity and also contributes to the development or survival of memory CD8+ T cells (15–17). Furthermore, by generating DOCK8-deficient (Dock8−/−) mice, we and others have revealed that DOCK8 is required for DCs to migrate effectively within 3D, but not 2D, microenvironments (9, 18). However, DOCK8 deficiency in DCs did not affect the global Cdc42 activation induced by chemokine stimulation (9). Therefore, how DOCK8 regulates leukocyte migration remains unclear.
Macrophages are a subset of leukocytes involved in innate immune responses, and they play important roles in the induction of protective immunity to invading pathogens (19). We found that, even in a 2D setting, Dock8−/− macrophages exhibited reduced motility with a defect in nuclear positioning. Although DOCK8 deficiency in macrophages also did not affect global Cdc42 activation, rescue experiments revealed that the GEF activity of DOCK8 was required for macrophage migration. In this study, we show that DOCK8 associates with LRAP35a, an adaptor molecule that binds to MRCK (4), and facilitates MLC2 phosphorylation. Our findings suggest that, during leukocyte migration, DOCK8 links Cdc42 activation to actomyosin dynamics through its association with LRAP35a.
Results
DOCK8 Regulates Macrophage Migration in 2D Environments
To examine the role of DOCK8 in macrophages, we first compared the recruitment of macrophages to inflamed lungs between Dock8+/− and Dock8−/− littermate mice. In response to intratracheal LPS challenge, macrophages rapidly accumulated in the alveolar cavity of Dock8+/− mice (Fig. 1A). However, such accumulation was impaired in Dock8−/− mice (Fig. 1A). It is known that this recruitment of macrophages critically depends on the chemokine CCL2 in alveolar tissues (20). To compare the chemotactic response to CCL2 in vitro, we differentiated macrophages from Dock8+/− and Dock8−/− bone marrow (BM) cells in the presence of M-CSF. Irrespective of DOCK8 expression, these BM-derived macrophages exhibited an adherent morphology (Fig. 1B), and the majority were CD11clowCD11bhigh cells (Fig. 1C). These phenotypes were different from the DCs generated from BM cells with GM-CSF (Fig. 1C). When Dock8+/− macrophages were stimulated with CCL2 in an EZ-TAXIScan chamber, their leading edges efficiently moved toward the chemokine source at a speed of 1.77 μm/min (Fig. 1D). Although DOCK8 deficiency did not affect the directionality and y-forward migration index during macrophage migration (Fig. 1E), the average speed of Dock8−/− BM-derived macrophages was reduced to 61.0% of the Dock8+/− speed (Fig. 1D). Similar results were obtained when thioglycollate (TG)-elicited peritoneal macrophages were isolated from Dock8+/− and Dock8−/− mice and analyzed for migration speed (1.11 versus 0.61 μm/min, Fig. 1F). However, no such defect was observed with Dock8−/− BM-derived DCs in this 2D setting (Fig. 1G), as reported previously (9).
FIGURE 1.

DOCK8 regulates macrophage migration in 2D environments. A, comparison of alveolar macrophage accumulation between Dock8+/− and Dock8−/− mice. The central lines and error bars on the scatterplots represent the mean and S.D., respectively. Data are collected from 5 mice/group. *, p < 0.05 (two-tailed Mann-Whitney test). B, phase-contrast images of Dock8+/− and Dock8−/− BM-derived macrophages migrating along the CCL2 gradient. The blue arrow and red arrowhead indicate the leading edge and the nucleus, respectively. Scale bar = 10 μm. Data are representative of four independent experiments. C, BM-derived macrophages or DCs from Dock8+/− and Dock8−/− mice were analyzed for CD11c and CD11b expression. The numbers in the quadrants indicate the percentage of cells in each quadrant. Data are representative of three independent experiments. D and E, comparison of migration of Dock8+/− (n = 34) and Dock8−/− (n = 30) BM-derived macrophages in response to CCL2 stimulation on a 2D surface. The migration speed (D), directionality (E), and y-forward migration index (E) were calculated based on the movement of the leading edge. The central lines and error bars on the scatterplots represent the mean and S.D., respectively. **, p < 0.01 (two-tailed Student's t test). Data are representative of four independent experiments. F, comparison of migration of Dock8+/− (n = 30) and Dock8−/− (n = 24) peritoneal macrophages in response to CCL2 stimulation on a 2D surface. The migration speed was calculated based on the movement of the leading edge. The central lines and error bars on the scatterplots represent the mean and S.D., respectively. **, p < 0.01 (two-tailed Mann-Whitney test). Data are representative of three independent experiments. G, comparison of the migration of Dock8+/− (n = 30) and Dock8−/− (n = 28) BM-derived DCs in response to CCL21 stimulation on a 2D surface. The migration speed, directionality, and y-forward migration index were calculated based on the movement of the leading edge. The central lines and error bars on the scatterplots represent the mean and S.D., respectively. Data are representative of three independent experiments. H, immunofluorescent staining of chemotaxing Dock8+/− and Dock8−/− BM-derived macrophages with DAPI (1:5000) and phalloidin (1:500). Scale bar = 10 μm. Data are representative of three independent experiments. I, comparison of the relative position of the nucleus between Dock8+/− (n = 33) and Dock8−/− (n = 30) BM-derived macrophages 60 min after migration. The central lines and error bars on the scatterplots represent the mean and S.D., respectively. **, p < 0.01 (two-tailed Student's t test). Data are representative of four independent experiments. J, comparison of the relative position of the nucleus between Dock8+/− (n = 30) and Dock8−/− (n = 24) peritoneal macrophages 60 min after migration. The central lines and error bars on the scatterplots represent the mean and S.D., respectively. **, p < 0.01 (two-tailed Mann-Whitney test). Data are representative of three independent experiments.
During the in vitro chemotaxis assay, we found that nuclear positioning was different between Dock8+/− and Dock8−/− macrophages. Both Dock8+/− and Dock8−/− BM-derived macrophages undergoing chemotaxis actively formed ring-like structures at the leading lamella (Fig. 1B and supplemental Movie 1). This was more clearly observed when cells were stained with phalloidin (Fig. 1H). However, although the nucleus was located immediately below the ring-like structure in Dock8+/− BM-derived macrophages, such localization of the nucleus was hardly detected in Dock8−/− macrophages (Fig. 1, B and H, and supplemental Movie 1). Indeed, when the relative positions of the nucleus within the cell (the distance from the nucleus to the rear per whole cell length) were compared 60 min after migration, the values of Dock8−/− BM-derived macrophages were significantly lower than those of Dock8+/− BM-derived macrophages (0.68 versus 0.44) (Fig. 1I). Similar results were obtained when TG-elicited peritoneal macrophages were analyzed (0.62 versus 0.47, Fig. 1J).
DOCK8 Regulates Macrophage Migration through Cdc42 Activation
Having found that DOCK8 regulates macrophage migration, we next examined whether the GEF activity of DOCK8 is required for this process. By analogy of the DOCK9 DHR2 domain, which is also known to act as a Cdc42 GEF (21), the valine residue at position 1986 of DOCK8 was expected to function as a nucleotide sensor. Indeed, biochemical analyses revealed that the Cdc42 GEF activity of the recombinant DOCK8 DHR2 protein was completely abolished by mutating this valine residue to alanine (VA mutant) despite the quality of the purified proteins being unaffected. (Fig. 2, A–C). When the GFP-tagged WT and VA mutant were expressed in Dock8−/− BM-derived macrophages using Amaxa NucleofectorTM II, they showed comparable expression (Fig. 2D). However, although the migration speed was restored in Dock8−/− BM-derived macrophages expressing WT DOCK8 (Fig. 2E), the expression of the GEF-dead VA mutant failed to rescue the migration defect (Fig. 2E). These results indicate that DOCK8 regulates macrophage migration through Cdc42 activation. However, as seen in Dock8−/− DCs (9), DOCK8 deficiency in BM-derived macrophages did not affect the global Cdc42 activation induced by CCL2 stimulation (Fig. 2F), probably because of functional compensation by other Cdc42 GEFs.
FIGURE 2.

DOCK8 regulates macrophage migration through Cdc42 activation. A, proteins used for the assay. GST fusion Cdc42 and the His-SUMO-tagged DOCK8 DHR2 domain (5 μg each, WT and VA mutant) were separated on a 10% SDS-polyacrylamide gel and stained with Coomassie Brilliant Blue. The position and size (in kilodaltons) of the molecular weight markers (MW) are indicated. B, recombinant WT or mutant (VA) DOCK8 DHR2 proteins at final concentrations of 0.015–1.5 μm were analyzed for their GEF activities toward Cdc42 (10 μm) using BODIPY-FL-GTP. RFU, relative fluorescence units. Data are representative of four independent experiments. C, comparison of the GEF activities of WT and mutant (VA) DOCK8 DHR2 proteins. The initial slope during the first 10 s (in relative fluorescence units/second) was calculated and plotted against the concentrations of DOCK8 DHR2 proteins. *, p < 0.05 (two-tailed Mann Whitney test). D, flow cytometric analyses for BM-derived macrophages transfected with the pCI-GFP vector encoding WT or mutant (VA) DOCK8. The green lines indicate the profiles of cells transfected with an empty vector. E, comparison of the migration speed of Dock8−/− BM-derived macrophages with or without the expression of WT or mutant (VA) DOCK8. As controls, Dock8+/− and Dock8−/− BM-derived macrophages transfected with pCI-GFP vector were simultaneously analyzed and are indicated as (−). At least 20 cells were analyzed per category. The central lines and error bars on the scatterplots represent the mean and S.D., respectively. **, p < 0.01 (two-tailed Mann-Whitney test). Data were collected from five to nine separate experiments. F, activation of Cdc42 in Dock8+/− and Dock8−/− BM-derived macrophages stimulated with CCL2 (100 ng/ml) for 10 s. Cell lysates were subjected to pulldown assays using the GST-fusion Cdc42-binding domain of PAK1 before immunoblotting with anti-Cdc42 antibody. Results were quantified by densitometry and are expressed as the ratio of the GTP-bound form to the total protein after the normalization of the 10-s value of Dock8+/− macrophages to an arbitrary value of 1. Data are indicated as the mean ± S.D. of four independent experiments.
DOCK8 Regulates Actomyosin Dynamics at the Leading Lamella during Macrophage Migration
Nonmuscle myosin II (NMII) is composed of two heavy chains, two essential light chains, and two regulatory light chains (22). Although it is well known that NMII mediates retraction of the rear during cell migration (22, 23), several studies have indicated that NMII also regulates leading edge motion (24–27). To examine the localization of NMII, we stained Dock8+/− and Dock8−/− BM-derived macrophages with antibody for NMIIA. Although the staining pattern of NMIIA at the cell rear was unchanged between Dock8+/− and Dock8−/− BM-derived macrophages, the localization and accumulation of NMIIA at the leading lamella were less evident in Dock8−/− BM-derived macrophages (Fig. 3, A and B). When GFP-tagged MLC2 was retrovirally expressed in Dock8+/− and Dock8−/− BM-derived macrophages (Fig. 3C), the migration speed was reduced, but a difference still existed between Dock8+/− and Dock8−/− macrophages (1.43 versus 0.92 μm/min, Fig. 3D). More importantly, the dynamic movement of MLC2 was found at the leading lamella of Dock8+/− but not Dock8−/− BM-derived macrophages (Fig. 3C). The dynamics of actomyosin critically depend on the phosphorylation status of MLC2 (22, 23). In response to CCL2 stimulation, MLC2 was phosphorylated at the serine residue of position 19 (Ser-19) in Dock8+/− BM-derived macrophages (Fig. 3E). Immunofluorescence staining revealed that phosphorylated MLC2 was localized at the leading lamella and subnuclear region (Fig. 3F). However, such phosphorylation was significantly reduced in Dock8−/− macrophages (Fig. 3, E and F). These results suggest that DOCK8 regulates the actomyosin dynamics during macrophage migration through MLC2 phosphorylation.
FIGURE 3.

DOCK8 regulates actomyosin dynamics at the leading lamella during macrophage migration. A, immunofluorescent staining of chemotaxing Dock8+/− and Dock8−/− BM-derived macrophages with DAPI, phalloidin (1:500), and anti-NMIIA antibody (1:400). Scale bar = 10 μm. Data are representative of three independent experiments. B, fluorescence intensity of NMIIA and phalloidin staining in chemotaxing Dock8+/− and Dock8−/− BM-derived macrophages. The intensity was measured from the cell edge to the cell center. a.u., arbitrary unit. Data are the average of 10 cells with 3 measurements/cell. C, MLC2 dynamics at the leading lamella in chemotaxing Dock8+/− and Dock8−/− BM-derived macrophages after retroviral expression of MLC2-GFP. The white lines indicate the leading edge. Scale bar = 10 μm. As controls, Dock8+/− and Dock8−/− BM-derived macrophages were analyzed after expression of GFP alone. Data are representative of three independent experiments. D, comparison of the migration speed of Dock8+/− (n = 20) and Dock8−/− (n = 20) BM-derived macrophages after retroviral expression of MLC2-GFP. The central lines and error bars on the scatterplots represent the mean and S.D., respectively. **, p < 0.01 (two-tailed Student's t test). Data were collected from three separate experiments. E, comparison of CCL2-induced MLC2 phosphorylation at Ser-19 (phospho-MLC2) between Dock8+/− and Dock8−/− BM-derived macrophages. Results were quantified by densitometry and are expressed as the ratio of the phosphorylated form to the total protein after normalization of the 2 min-value of control samples to an arbitrary value of 1. Data are indicated as the mean ± S.D. of four independent experiments. *, p < 0.05 (two-tailed Mann-Whitney test). MW, molecular weight. F, localization of phosphorylated MLC2 at Ser-19 (red, 1:400) and phalloidin (green, 1:500) in chemotaxing Dock8+/− and Dock8−/− BM-derived macrophages. Scale bar = 10 μm. Data are representative of three independent experiments.
DOCK8 Links Cdc42 Activation to Actomyosin Dynamics through LRAP35a Interaction
MRCKα is a Cdc42 effector that mediates MLC2 phosphorylation at Ser-19 in collaboration with the leucine-rich adaptor protein LRAP35a (2–5). During the search for binding partners of DOCK8, we found that DOCK8 and LRAP35a were coimmunoprecipitated when expressed in HEK-293T cells (Fig. 4A). Similar results were obtained when cell extracts of BM-derived macrophages were pulled down with GST fusion LRAP35a (Fig. 4B). On the other hand, βPIX (28), another Cdc42 GEF, did not show any binding to LRAP35a (Fig. 4A). A previous report indicated that the N-terminal region of LRAP35a containing leucine repeats is important for MRCK binding (4). In contrast, pulldown experiments using recombinant proteins revealed that LRAP35a associates with DOCK8 through the C-terminal region (Fig. 4C). When LRAP35a expression was knocked down in Dock8+/− BM-derived macrophages using siRNA (Fig. 4D), the migration speed was reduced to the level of Dock8−/− macrophages (Fig. 4E). Similar results were obtained by overexpressing an LRAP35a N mutant that failed to bind to DOCK8 (Fig. 4F). In addition, knockdown of either LRAP35a or MRCKα suppressed MLC2 phosphorylation at Ser-19 (Fig. 4, G and H). These results suggest that, during macrophage migration, DOCK8 acts as a signaling molecule to integrate Cdc42 activation and actomyosin dynamics through its interaction with LRAP35a.
FIGURE 4.

DOCK8 regulates macrophage migration through LRAP35a interaction. A, following the expression of DOCK8-HA, βPIX-HA, MRCKα-GFP, and/or LRAP35a-V5 in HEK-293T cells, cell extracts were immunoprecipitated (IP) with anti-HA antibody or anti-GFP antibody for immunoblotting. Data are representative of at least three independent experiments. MW, molecular weight. B, cell extracts from BM-derived macrophages were pulled down with GST alone or GST-fusion LRAP35a, and the bound proteins and total lysate control were probed by immunoblot with anti-DOCK8 antibody. The recombinant proteins used in this assay were detected with Coomassie Brilliant Blue (CBB) staining. Data are representative of three independent experiments. C, following the expression of DOCK8-HA or MRCKα-GFP in HEK-293T cells, cell extracts were pulled down with GST fusion LRAP35a or its mutants. Data are representative of three independent experiments. D, RT-PCR analysis for the expression of Lurap1 encoding LRAP35a in BM-derived macrophages transfected with siRNA or control oligonucleotide. E, effect of LRAP35a knockdown on the migration speed of Dock8+/− BM-derived macrophages. As controls, Dock8+/− and Dock8−/− BM-derived macrophages transfected with control oligonucleotide were analyzed simultaneously. At least 40 cells were analyzed per category. The central lines and error bars on the scatterplots represent the mean and S.D., respectively. **, p < 0.01 (two-tailed Mann-Whitney test). Data were collected from three separate experiments. F, effect of expression of the LRAP35a N mutant on the migration speed of Dock8+/− BM-derived macrophages. As controls, Dock8+/− and Dock8−/− BM-derived macrophages transfected with GFP alone were analyzed simultaneously. At least 20 cells were analyzed per category. The central lines and error bars on the scatterplots represent the mean and S.D., respectively. **, p < 0.01 (two-tailed Student's t test). Data were collected from five separate experiments. G, effect of LRAP35a knockdown on CCL2-induced MLC2 phosphorylation at Ser-19 (phospho-MLC) in Dock8+/− BM-derived macrophages. As controls, Dock8+/− and Dock8−/− BM-derived macrophages transfected with control oligonucleotide were analyzed simultaneously. Results were quantified by densitometry and are expressed as the ratio of the phosphorylated form to total protein after normalization of the 2-min value of control samples to an arbitrary value of 1. Data are indicated as the mean ± S.D. of four independent experiments. *, p < 0.05 (two-tailed Mann-Whitney test). H, effect of MRCKα knockdown on CCL2-induced MLC2 phosphorylation at Ser-19 (phospho-MLC) in Dock8+/− BM-derived macrophages. As controls, Dock8+/− and Dock8−/− BM-derived macrophages transfected with control oligonucleotide were analyzed simultaneously. Results were quantified by densitometry and are expressed as the ratio of the phosphorylated form to total protein after normalization of the 2-min value of control samples to an arbitrary value of 1. Data are indicated as the mean ± S.D. of four independent experiments. *, p < 0.05 (two-tailed Mann-Whitney test).
Role of LRAP35a in DC Migration in 2D and 3D Environments
Although Dock8−/− BM-derived DCs migrate normally on 2D surfaces, they fail to migrate effectively within 3D extracellular matrix scaffolds such as collagen gels (9, 18). To examine the role of LRAP35a in DC migration, we also knocked down LRAP35a expression in Dock8+/− BM-derived DCs (Fig. 5A). Although knockdown of LRAP35a did not affect DC migration in a 2D setting, it resulted in reduced motility of Dock8+/− BM-derived DCs in collagen gels (Fig. 5B). Similar results were obtained by overexpressing LRAP35a N mutant in Dock8+/− BM-derived DCs (Fig. 5C). These results indicate that the DOCK8-LRAP35a signaling axis also acts in DCs to promote migration within 3D environments.
FIGURE 5.

The role of LRAP35a in DC migration in 2D and 3D environments. A, RT-PCR analysis for the expression of Lurap1 encoding LRAP35a in BM-derived DCs transfected with siRNA or control oligonucleotide. B, effect of LRAP35a knockdown on the migration speed of Dock8+/− BM-derived DCs on a 2D surface (left panel) or in 3D collagen gels (right panel). As controls, Dock8+/− and Dock8−/− BM-derived DCs transfected with control oligonucleotide were analyzed simultaneously. At least 30 cells were analyzed per category. The central lines and error bars on the scatterplots represent the mean and S.D., respectively. **, p < 0.01 (two-tailed Student's t test). Data were collected from three separate experiments. C, effect of overexpression of the GFP fusion LRAP35a N mutant on the migration speed of Dock8+/− BM-derived DCs in 3D collagen gels. As controls, Dock8+/− and Dock8−/− BM-derived DCs transfected with GFP alone were simultaneously analyzed. At least 20 cells were analyzed per category. The central lines and error bars on the scatterplots represent the mean and S.D., respectively. **, p < 0.01 (two-tailed Student's t test). Data were collected from three separate experiments.
Identification of the DOCK8 Region Critical for LRAP35a Association
To determine the region of DOCK8 critical for LRAP35a binding, we expressed WT or mutant DOCK8 in HEK-293T cells with LRAP35a and analyzed their interaction. Although WT DOCK8 and its mutant lacking the N-terminal 730 amino acid residues (DOCK8-ΔNΔDHR1) comparably bound to LRAP35a, such interaction was hardly detected when the DOCK8 DHR2 domain was deleted (DOCK8-ΔDHR2, Fig. 6A). Consistent with this, an association between DOCK8 and LRAP35a was observed when the DOCK8 DHR2 domain alone was co-expressed with LRAP35a (Fig. 6B), suggesting that DOCK8 associates with LRAP35a through the DHR2 domain. This association seemed to be direct, as recombinant protein encoding the His-SUMO-tagged DOCK8 DHR2 domain exhibited definite binding to GST fusion LRAP35a but not the LRAP35a N mutant in vitro (Fig. 6C).
FIGURE 6.

DOCK8 associates with LRAP35a through lobe B of the DOCK8 DHR2 domain. A, following the expression of WT DOCK8-HA or its mutants (DOCK8-ΔNΔDHR1-HA or DOCK8-ΔDHR2-HA) with LRAP35a-V5 in HEK-293T cells, cell extracts were immunoprecipitated with anti-HA antibody for immunoblotting. Data are representative of three independent experiments. B, following expression of GFP-tagged DOCK8-DHR2 (amino acid residues 1633–2071), DOCK8-DHR1 (amino acid residues 561–730), or GFP alone with LRAP35a-V5 in HEK-293T cells, cell extracts were immunoprecipitated (IP) with anti-GFP antibody for immunoblotting. Data are representative of three independent experiments. MW, molecular weight. C, recombinant protein encoding the His-SUMO-tagged DOCK8 DHR2 domain (20 μg) was incubated with GST alone, GST fusion LRAP35a, or its mutant (10 μg each) for pulldown assays. Data are representative of three independent experiments. CBB, Coomassie Brilliant Blue. D, following expression of GFP-tagged lobe A (amino acid residues 1633–1808), lobe B (amino acid residues 1764–1976), or lobe C (amino acid residues 1936–2076) of the DOCK8 DHR-2 domain with LRAP35a-V5 in HEK-293T cells, cell extracts were immunoprecipitated with anti-GFP antibody for immunoblotting. Data are representative of three independent experiments. E, following expression of GFP-tagged DOCK8-DHR2-lobe B, DOCK8-DHR1, or DOCK7-DHR2-lobe B (amino acid residues 1769–1981) with LRAP35a-V5 in HEK-293T cells, cell extracts were immunoprecipitated with anti-GFP antibody for immunoblotting. Data are representative of three independent experiments. F, following expression of WT DOCK8-HA or its VA mutant with LRAP35a-V5 in HEK-293T cells, cell extracts were immunoprecipitated with anti-HA antibody for immunoblotting. Data are representative of three independent experiments. G, following expression of WT DOCK8-HA or its VA mutant with LRAP35a-V5 in HEK-293T cells, cell extracts were pulled down with GST fusion Cdc42 or GST alone. Data are representative of three independent experiments.
The DHR2 domain is composed of three lobes: lobe A, lobe B, and lobe C (21). Of these, lobe B of the DHR2 domain was found to be important for LRAP35a association (Fig. 6, D and E). Lobes B and C of DOCK8 are known to mediate Cdc42 binding (9). To examine whether Cdc42 binding affects the interaction between DOCK8 and LRAP35a, we expressed full-length DOCK8 with a VA mutation in the DHR2 domain. Although this mutant failed to bind to Cdc42, the VA mutation did not affect the interaction of DOCK8 with LRAP35a (Fig. 6F). In addition, pulldown experiments using GST fusion Cdc42 showed that DOCK8, LRAP35a, and Cdc42 were able to form a trimolecular complex (Fig. 6G). These results indicate that DOCK8 associates with LRAP35a through lobe B of the DHR2 domain, independent of Cdc42 binding. However, no association with LRAP35a was found when lobe B of the DOCK7 DHR2 domain was analyzed (Fig. 6E).
Discussion
Accumulating evidence indicates that DOCK8 regulates the migration and activation of various subsets of leukocytes (9–18). However, how DOCK8 regulates these processes has been unclear. In this study, we showed that, in the absence of DOCK8, macrophages exhibit a migration defect even in a 2D setting. Although DOCK8 deficiency did not affect global Cdc42 activation, rescue experiments revealed that the Cdc42 GEF activity of DOCK8 is required for macrophage migration. We found that DOCK8 regulates the phosphorylation of MLC2 in chemotaxing macrophages. Mechanistically, DOCK8 associates with LRAP35a and facilitates MLC2 phosphorylation by MRCKα. Interestingly, the association with LRAP35a was not found with other Cdc42 GEFs, such as βPIX and DOCK7 (28, 29). Therefore, it is likely that the interaction with LRAP35a makes DOCK8 a special GEF that links Cdc42 activation to actomyosin dynamics during macrophage migration (Fig. 7).
FIGURE 7.
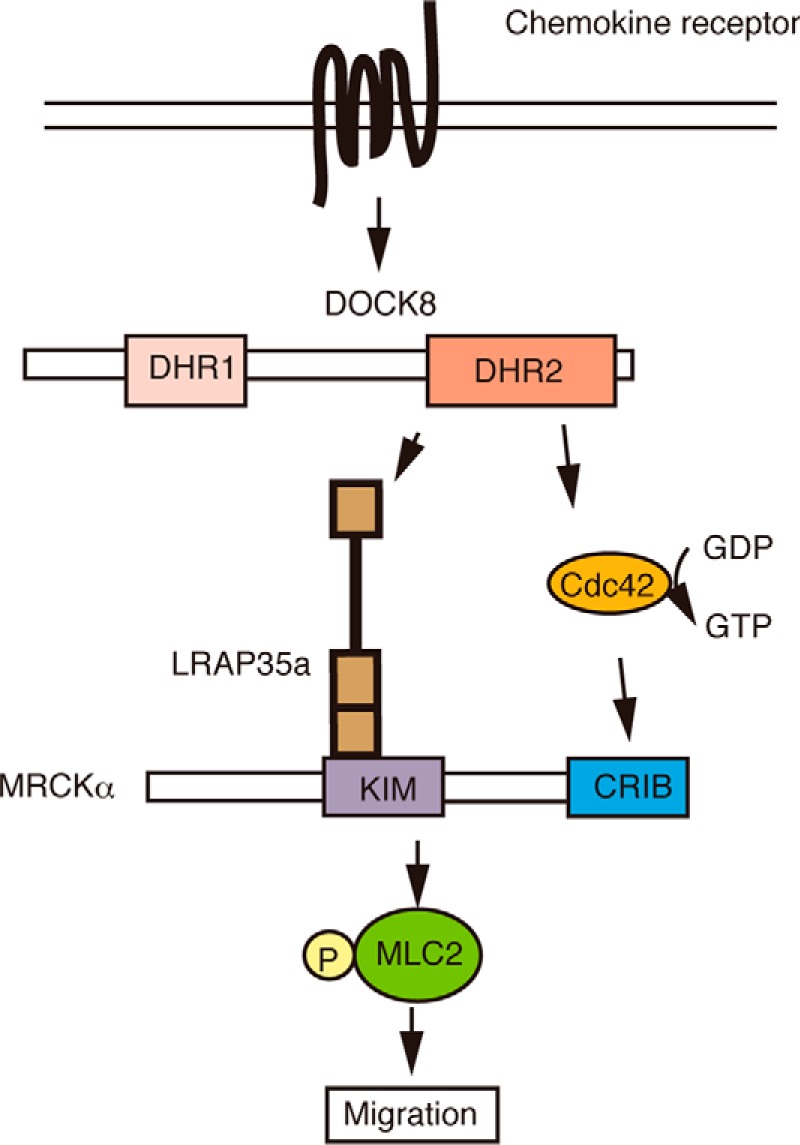
Schematic of the role of DOCK8 in leukocyte migration. In this model, DOCK8 functions not only as a Cdc42 GEF but also as a scaffold protein that links Cdc42 activation to actomyosin dynamics through its interaction with LRAP35a. KIM, kinase inhibitory motif; CRIB, Cdc42/Rac-interactive binding site.
The role of the DOCK8-LRAP35a signaling axis in other leukocyte subsets is also important. In this study, we showed that neither DOCK8 nor LRAP35a deficiency affected the DC migration on 2D surfaces. Although the precise reason is currently unknown, this discrepancy is likely to result from differences in the adhesiveness between macrophages and DCs. However, we found that the migration speed of Dock8+/− DCs in collagen gels was also reduced to the level of Dock8−/− DCs when LRAP35a expression was knocked down. Recent evidence indicates that NMII-mediated contractility is required for leukocytes to squeeze their nucleus through narrow gaps such as endothelial cell-cell junctions during extravasation and dense extracellular matrix scaffold during interstitial migration (30, 31). Therefore, our results suggest that the DOCK8-LRAP35a axis also regulates interstitial DC migration by controlling actomyosin dynamics.
In conclusion, we showed here that DOCK8 regulates leukocyte migration by linking Cdc42 activation to actomyosin dynamics through LRAP35a interaction. Our findings provide novel insights into the mechanism of how DOCK8 regulates leukocyte migration.
Experimental Procedures
Mice
Dock8−/− mice have been described previously (9). Mice were maintained under specific pathogen-free conditions in the animal facility of Kyushu University. All experiments were performed in accordance with the guidelines of the Committee of Ethics of Animal Experiments, Kyushu University.
Cell Preparation
For preparation of BM-derived macrophages, BM cells were cultured for 7 days in complete RPMI medium containing 10% heat-inactivated FCS (Nichirei Bioscience), 50 μm 2-mercaptoethanol (Nacalai Tesque), 2 mm l-glutamine (Life Technologies), 100 units/ml penicillin (Life Technologies), 100 μg/ml streptomycin (Life Technologies), 1 mm sodium pyruvate (Life Technologies), and MEM non-essential amino acids (Life Technologies) plus M-CSF (20 ng/ml, PeproTech). To generate BM-derived DCs, BM cells were cultured for 7 days in complete RPMI medium supplemented with GM-CSF (10 ng/ml, PeproTech) and purified with anti-CD11c antibody-conjugated microbeads (Miltenyi Biotec). To isolate alveolar macrophages, mice were challenged with intratracheal instillations of Escherichia coli LPS (20 μg/mouse). Mice were then killed 96 h later, and the bronchoalveolar lavage fluid was collected as described previously (32). For preparation of TG-elicited peritoneal macrophages, Dock8+/− or Dock8−/− mice were injected intraperitoneally with 200 μl of sterile 3% TG broth solution (Nissui) to elicit an inflammatory response and recruit the peritoneal macrophages. Mice were sacrificed 96 h later by cervical dislocation, and peritoneal exudate cells were harvested and maintained in complete RPMI medium at 37 °C. Three hours later, the adherent cells were collected and used as peritoneal macrophages.
Chemotaxis Assay
BM-derived macrophages or peritoneal macrophages were allowed to migrate under a CCL2 (10 μg/ml, Wako) gradient in an EZ-TAXIScan chamber (Effector Cell Institute). Phase-contrast images of chemotaxing cells were acquired at 1-min intervals over 60 min. To assess the migration of BM-derived DCs, phase-contrast images of cells migrating toward CCL21 (250 μg/ml, R&D Systems) were acquired at 30-s intervals over 30 min. EZ-TAXIScan chemotaxis assays were performed as described previously (9, 33). TAXIScan-FL (Effector Cell Institute) was used when cells were transfected with a plasmid encoding the GFP fusion protein or Block-iT fluorescent oligo (Invitrogen). Images were imported as stacks to the ImageJ software program (National Institutes of Health). The velocity, directionality, and y-forward migration index were analyzed using manual tracking and the chemotaxis and migration tools. 3D collagen gel chemotaxis assays were performed with μ-Slide Chemotaxis3D (Ibidi) as described previously (9). Briefly, cells were mixed with 1.65 mg/ml of collagen type I (Corning) in RPMI medium supplemented with 4% FCS and cast in the chamber before assays. After polymerization of the lattice, images of cells migrating toward CCL21 (10 μg/ml) were taken in a heated chamber every 2 min using an IX-81 inverted microscope (Olympus) equipped with a cooled charge-coupled device camera (CoolSNAP HQ, Roper Scientific), an IX2-ZDC laser-based autofocusing system (Olympus), and an MD-XY60100T-Meta automatically programmable XY stage (Sigma KOKI). Images were taken, and velocities were calculated with the manual tracking feature in the MetaMorph software program.
Immunofluorescence Microscopy
BM-derived macrophages migrating in an EZ-TAXIScan chamber were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 in PBS. Cells were then stained with Alexa Fluor 546-conjugated phalloidin (1:500, Invitrogen), DAPI (1:5000, Dojindo), and anti-NMIIA antibody (1:400, PRB-440P, Covance). To stain activated myosin, cells were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and incubated with the phosphorylation-specific antibody against Ser-19 of MLC2 (1:400, 3671, Cell Signaling Technology) and Alexa Fluor 488-conjugated phalloidin (1:500, Invitrogen) for 1 h. After being washed with PBS, sections were incubated with appropriately labeled secondary antibodies. A microscopic analysis was performed with a laser-scanning confocal microscope (LSM 510 Meta, Carl Zeiss) with Plan-Apochromat ×63/1.4 oil objective at room temperature. The acquisition software program AIM (Carl Zeiss) was used. In some experiments, fluorescence intensity was measured from the cell edge to the cell center using the ImageJ software program. The averages from multiple cells were calculated and plotted against distance from the leading edge.
FACS Analysis
Before staining, cells were preincubated for 10 min at 4 °C with anti-Fcγ III/II receptor (BD Biosciences) to block Fc receptors. Single cell suspensions were stained with biotinylated anti-F4/80 (TONBO Biosciences), FITC-conjugated anti-CD11b (BD Biosciences), and phycoerythrin-conjugated anti-CD11c (BD Biosciences) antibodies, followed by allophycocyanin-conjugated streptavidin (BD Biosciences). Flow cytometric analyses were performed on a FACSCalibur (BD Biosciences).
Plasmids and Transfection
The genes encoding DOCK8, DOCK7, LRAP35a, MRCKα, and βPIX and their mutants with appropriate tags were created by PCR and subcloned into the pcDNA (Invitrogen) or pCI (Promega) vector. These plasmids were transfected into HEK-293T cells with polyethylenimine. The pMX vector was used to generate the plasmid encoding GFP or GFP-tagged MLC2. The pMX-MLC2-GFP or pMXs-GFP vector was transfected into platinum-E packaging cells with FuGENE 6 transfection reagent (Promega). 48 h after transfection, the cell culture supernatants were harvested and supplemented with Polybrene (5 μg/ml) and M-CSF (20 ng/ml) to infect macrophages. The pCI-GFP vector encoding DOCK8, LRAP35a, and/or their mutants was transfected into macrophages or DCs by using Amaxa NucleofectorTM II (Lonza) in accordance with the instructions of the manufacturer. The genes encoding the DOCK8 DHR2 domain (residues 1633–2071) and its mutant were subcloned into the pET-SUMO vector to express fusion proteins with the His tag at their N termini (9). The GST fusion recombinant Cdc42, LRAP35a, and its mutants were expressed with the pGEX6P-1 vector (GE Healthcare).
Immunoprecipitation, Pulldown Assays, and Immunoblotting
The following antibodies were used to examine the association of DOCK8 (WT or its mutant) or βPIX with LRAP35a or MRCKα: anti-GFP antibody (sc-9996, Santa Cruz Biotechnology), anti-HA antibody (11867423001, Roche), anti-V5 antibody (R961-25, Invitrogen), and anti-His antibody (CP15165, Thermo Fisher Scientific). After expression of DOCK8, βPIX, MRCKα, and/or LRAP35a with the appropriate tags in HEK-293T cells, cell extracts were immunoprecipitated with the relevant antibodies and analyzed by SDS-PAGE for immunoblotting. For pulldown assays, cell extracts from BM-derived macrophages and HEK-293T cells expressing HA-tagged DOCK8 or GFP-tagged MRCKα and recombinant protein encoding His-SUMO-tagged DOCK8 DHR2 domain were incubated with GST-fusion recombinant protein encoding LRAP35a or its mutants. In some experiments, HEK-293T cell extracts expressing HA-tagged WT or mutant DOCK8 and V5-tagged LRAP35a were subjected to pulldown assays using GST fusion recombinant Cdc42. The bound proteins were analyzed by SDS-PAGE. The blots were probed with anti-HA antibody (1:1000), anti-GFP antibody (1:1000), anti-V5 antibody (1:4000), or anti-His antibody (1:1000). The polyclonal antibody against DOCK8 was produced by immunizing rabbits with keyhole limpet hemocyanin-coupled synthetic peptide corresponding to the C-terminal sequence (RDSFHRSSFRKCETQLSQGS, residues 2081–2100; 1.88 μg/ml) of human and mouse DOCK8. To assess the phosphorylation status of MLC2, BM-derived macrophages were lysed on ice in lysis buffer containing PhosSTOP phosphatase inhibitor mixture (Roche). Cell extracts were then analyzed by SDS-PAGE, and blots were probed with phosphorylation-specific antibody against Ser-19 of MLC2 (1:1000, 3671, Cell Signaling Technology) and anti-myosin light chain antibody (1:1000, M4401, Sigma-Aldrich) as a control.
RT-PCR
The cells were homogenized in Isogen (Nippon Gene). The RNA was extracted with chloroform, precipitated with isopropanol, and washed with 75% ethanol. The resulting RNA was resolved in diethyl pyrocarbonate-treated water. RNA samples were treated with RNase-free DNase I (Invitrogen), reverse-transcribed with oligo(dT) and SuperScript III reverse transcriptase (Invitrogen), and then amplified by PCR with specific primers.
Knockdown of LRAP35a and MRCKα
The siRNA Mm_1520402A15Rik_1 (Qiagen, SI00791763) was transfected into BM-derived macrophages or DCs to knock down LRAP35a expression in accordance with the instructions of the manufacturer. The irrelevant oligonucleotide SI03650318 was used as a control. 48 h after transfection, cells were harvested for a chemotaxis assay and/or immunoblotting. The knockdown efficiency was checked by RT-PCR. The following primers were used: LRAP35a, 5′-ATGGAGGGGACCGCGGAGT-3′ and 5′-GCCCTGTCCTCAGTGGGGATA-3′; actin, 5′-TGGAATCCTGTGGCATCCATGAAAC-3′ and 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′. For knockdown of MRCKα expression, the siRNA Mm_Cdc42bpa_3 (Qiagen, SI02771769) was transfected into BM-derived macrophages in accordance with the instructions of the manufacturer. The irrelevant oligonucleotide SI03650318 was used as a control. 36 h after transfection, cells were harvested for immunoblotting. The knockdown efficiency was checked by immunoblotting using anti-MRCKα antibody (sc-374568, 1:1000, Santa Cruz Biotechnology).
In Vitro GEF Assay
The genes encoding the DOCK8 DHR2 domain and its mutant were expressed in E. coli Arctic Express (Stratagene) as fusion proteins with the His-SUMO tag at their N termini for purification with nickel-nitrilotriacetic acid-agarose chromatography (Qiagen). The GST fusion Cdc42 and GST alone were expressed in BL21 (Agilent Technologies) and purified on glutathione-SepharoseTM 4B (GE Healthcare) immediately before use. The assays consisted of GST fusion Cdc42 (10 μm), BODIPY-FL GTP (2.4 μm, Invitrogen), and the His-SUMO tagged recombinant WT DOCK8 DHR2 domain (residues 1633–2071) or its VA mutant (specified concentrations) in reaction buffer (20 mm MES-NaOH, 150 mm NaCl, 10 mm MgCl2, and 20 μm GDP (pH 7.0)). For this purpose, GST-fusion Cdc42 (15.56 μm) was loaded with GDP by incubation in reaction buffer in a total volume of 96.4 μl on ice for 30 min and then mixed with BODIPY-FL GTP (0.1 mm, 3.6 μl) and allowed to equilibrate at 30 °C for 3 min. Recombinant DOCK8 DHR2 protein was equilibrated in reaction buffer in a total volume of 50 μl for 30 min at room temperature. The reaction was initiated by mixing GDP-loaded Cdc42/BODIPY-FL GTP (100 μl) and the recombinant DOCK8 DHR2 domain (50 μl) in a final volume of 150 μl and incubating at 30 °C. The change in the BODIPY-FL-GTP fluorescence (excitation, 488 nm, emission, 514 nm) was monitored for 20 min using an EnSpire multimode plate reader (PerkinElmer Life Sciences). Data were fitted using the curve fitting function in the GraphPad Prism 5 software program (GraphPad Software), and the initial slope during the first 10 s (in relative fluorescent unit [RFU] per second) was calculated and used for comparison of the GEF activity.
Cdc42 Activation Assays
BM-derived macrophages suspended in Hanks' balanced salt solution were stimulated with CCL2 (100 ng/ml). Aliquots of the cell extracts were kept for total lysate controls, and the remaining extracts were incubated with the GST fusion Cdc42-binding domain of PAK1 (Merck Millipore) at 4 °C for 60 min. The bound proteins and the total lysate control were analyzed by SDS-PAGE, and the blots were probed with anti-Cdc42 antibody (1:1000, 610928, BD Biosciences).
Statistical Analyses
Statistical analyses were performed using the GraphPad Prism software program. We first calculated the Gaussian distribution of the data using the Kolmogorov-Smirnov test. When two groups were compared, two-tailed Student's t test (Gaussian distribution) or Mann-Whitney test (no Gaussian distribution) was used. p < 0.05 was considered significant.
Author Contributions
A. S., F. S., and D. S. performed functional and immunohistochemical analyses and analyzed the data. T. U. and M. U. performed biochemical analyses. A. S., T. U., and T. H. helped write the manuscript. Y. F. conceived the project, interpreted the data, and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank T. Kitamura for providing the pMX vector and platinum-E packaging cells, A. Inayoshi and A. Aosaka for technical assistance, and Brian Quinn for English editing.
This work was supported by a grant-in-aid for scientific research on innovative areas from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; grants-in-aid for scientific research from the Japan Society for the Promotion of Science; the Leading Advanced Projects for Medical Innovation; and the Research on Development of New Drugs from Japan Agency for Medical Research and Development. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Movie 1.
- GEF
- guanine nucleotide exchange factor
- MRCK
- myotonic dystrophy kinase-related Cdc42-binding kinase
- DC
- dendritic cells
- BM
- bone marrow
- TG
- thioglycollate
- NMII
- nonmuscle myosin II.
References
- 1. Etienne-Manneville S., and Hall A. (2002) Rho GTPases in cell biology. Nature 420, 629–635 [DOI] [PubMed] [Google Scholar]
- 2. Leung T., Chen X. Q., Tan I., Manser E., and Lim L. (1998) Myotonic dystrophy kinase-related Cdc42-binding kinase acts as a Cdc42 effector in promoting cytoskeletal reorganization. Mol. Cell. Biol. 18, 130–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wilkinson S., Paterson H. F., and Marshall C. J. (2005) Cdc42-MRCK and Rho-ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nat. Cell Biol. 7, 255–261 [DOI] [PubMed] [Google Scholar]
- 4. Tan I., Yong J., Dong J. M., Lim L., and Leung T. (2008) A tripartite complex containing MRCK modulates lamellar actomyosin retrograde flow. Cell 135, 123–136 [DOI] [PubMed] [Google Scholar]
- 5. Gomes E. R., Jani S., and Gundersen G. G. (2005) Nuclear movement regulated by Cdc42, MRCK, myosin, and actin flow establishes MTOC polarization in migrating cells. Cell 121, 451–463 [DOI] [PubMed] [Google Scholar]
- 6. Lämmermann T., Renkawitz J., Wu X., Hirsch K., Brakebusch C., and Sixt M. (2009) Cdc42-dependent leading edge coordination is essential for interstitial dendritic cell migration. Blood 113, 5703–5710 [DOI] [PubMed] [Google Scholar]
- 7. Côté J. F., and Vuori K. (2002) Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J. Cell Sci. 115, 4901–4913 [DOI] [PubMed] [Google Scholar]
- 8. Ruusala A., and Aspenström P. (2004) Isolation and characterisation of DOCK8, a member of the DOCK180-related regulators of cell morphology. FEBS Lett. 572, 159–166 [DOI] [PubMed] [Google Scholar]
- 9. Harada Y., Tanaka Y., Terasawa M., Pieczyk M., Habiro K., Katakai T., Hanawa-Suetsugu K., Kukimoto-Niino M., Nishizaki T., Shirouzu M., Duan X., Uruno T., Nishikimi A., Sanematsu F., Yokoyama S., et al. (2012) DOCK8 is a Cdc42 activator critical for interstitial dendritic cell migration during immune responses. Blood 119, 4451–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Q., Davis J. C., Lamborn I. T., Freeman A. F., Jing H., Favreau A. J., Matthews H. F., Davis J., Turner M. L., Uzel G., Holland S. M., and Su H. C. (2009) Combined immunodeficiency associated with DOCK8 mutations. N. Engl. J. Med. 361, 2046–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Engelhardt K. R., McGhee S., Winkler S., Sassi A., Woellner C., Lopez-Herrera G., Chen A., Kim H. S., Lloret M. G., Schulze I., Ehl S., Thiel J., Pfeifer D., Veelken H., Niehues T., et al. (2009) Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J. Allergy Clin. Immunol. 124, 1289–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mizesko M. C., Banerjee P. P., Monaco-Shawver L., Mace E. M., Bernal W. E., Sawalle-Belohradsky J., Belohradsky B. H., Heinz V., Freeman A. F., Sullivan K. E., Holland S. M., Torgerson T. R., Al-Herz W., Chou J., Hanson I. C., et al. (2013) Defective actin accumulation impairs human natural killer cell function in patients with dedicator of cytokinesis 8 deficiency. J. Allergy Clin. Immunol. 131, 840–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Crawford G., Enders A., Gileadi U., Stankovic S., Zhang Q., Lambe T., Crockford T. L., Lockstone H. E., Freeman A., Arkwright P. D., Smart J. M., Ma C. S., Tangye S. G., Goodnow C. C., Cerundolo V., et al. (2013) DOCK8 is critical for the survival and function of NKT cells. Blood 122, 2052–2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Q., Dove C. G., Hor J. L., Murdock H. M., Strauss-Albee D. M., Garcia J. A., Mandl J. N., Grodick R. A., Jing H., Chandler-Brown D. B., Lenardo T. E., Crawford G., Matthews H. F., Freeman A. F., Cornall R. J., et al. (2014) DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. J. Exp. Med. 211, 2549–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Randall K. L., Chan S. S., Ma C. S., Fung I., Mei Y., Yabas M., Tan A., Arkwright P. D., Al Suwairi W., Lugo Reyes S. O., Yamazaki-Nakashimada M. A., Garcia-Cruz Mde L., Smart J. M., Picard C., Okada S., et al. . J. Exp. Med. 208, 2305–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Randall K. L., Lambe T., Johnson A., Treanor B., Kucharska E., Domaschenz H., Whittle B., Tze L. E., Enders A., Crockford T. L., Bouriez-Jones T., Alston D., Cyster J. G., Lenardo M. J., Mackay F., et al. (2009) DOCK8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nat. Immunol. 10, 1283–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lambe T., Crawford G., Johnson A. L., Crockford T. L., Bouriez-Jones T., Smyth A. M., Pham T. H., Zhang Q., Freeman A. F., Cyster J. G., Su H. C., and Cornall R. J. (2011) DOCK8 is essential for T-cell survival and the maintenance of CD8+ T-cell memory. Eur. J. Immunol. 41, 3423–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krishnaswamy J. K., Singh A., Gowthaman U., Wu R., Gorrepati P., Sales Nascimento M., Gallman A., Liu D., Rhebergen A. M., Calabro S., Xu L., Ranney P., Srivastava A., Ranson M., Gorham J. D., et al. (2015) Coincidental loss of DOCK8 function in NLRP10-deficient and C3H/HeJ mice results in defective dendritic cell migration. Proc. Natl. Acad. Sci. U.S.A. 112, 3056–3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murray P. J., and Wynn T. A. (2011) Protective and pathogenic function of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maus U. A., Wellmann S., Hampl C., Kuziel W. A., Srivastava M., Mack M., Everhart M. B., Blackwell T. S., Christman J. W., Schlöndorff D., Bohle R. M., Seeger W., and Lohmeyer J. (2005) CCR2-positive monocytes recruited to inflamed lungs downregulate local CCL2 chemokine levels. Am. J. Physiol. Lung Cell Mol. Physiol. 288, L350–L358 [DOI] [PubMed] [Google Scholar]
- 21. Yang J., Zhang Z., Roe S. M., Marshall C. J., and Barford D. (2009) Activation of Rho GTPases by DOCK exchange factors is mediated by a nucleotide sensor. Science 325, 1398–1402 [DOI] [PubMed] [Google Scholar]
- 22. Vicente-Manzanares M., Ma X., Adelstein R. S., and Horwitz A. R. (2009) Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 10, 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., and Horwitz A. R. (2003) Cell migration: integrating signals from front to back. Science 302, 1704–1709 [DOI] [PubMed] [Google Scholar]
- 24. Giannone G., Dubin-Thaler B. J., Rossier O., Cai Y., Chaga O., Jiang G., Beaver W., Döbereiner H. G., Freund Y., Borisy G., and Sheetz M. P. (2007) Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell 128, 561–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anderson T. W., Vaughan A. N., and Cramer L. P. (2008) Retrograde flow and myosin II activity within the leading cell edge deliver F-actin to the lamella to seed the formation of graded polarity actomyosin II filament bundles in migration fibroblasts. Mol. Biol. Cell 19, 5006–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nemethova M., Auinger S., and Small J. V. (2008) Building the actin cytoskeleton: filopodia contribute to the construction of contractile bundles in the lamella. J. Cell Biol. 180, 1233–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burnette D. T., Manley S., Sengupta P., Sougrat R., Davidson M. W., Kachar B., and Lippincott-Schwartz J. (2011) A role for actin arcs in the leading-edge advance of migrating cells. Nat. Cell Biol. 13, 371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Osmani N., Vitale N., Borg J. P., and Etienne-Manneville S. (2006) Scrib controls Cdc42 localization and activity to promote cell polarization during astrocyte migration. Curr. Biol. 16, 2395–2405 [DOI] [PubMed] [Google Scholar]
- 29. Zhou Y., Johnson J. L., Cerione R. A., and Erickson J. W. (2013) Prenylation and membrane localization of Cdc42 are essential for activation by DOCK7. Biochemistry 52, 4354–4363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lämmermann T., Bader B. L., Monkley S. J., Worbs T., Wedlich-Söldner R., Hirsch K., Keller M., Förster R., Critchley D. R., Fässler R., and Sixt M. (2008) Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 453, 51–55 [DOI] [PubMed] [Google Scholar]
- 31. Jacobelli J., Estin Matthews M., Chen S., and Krummel M. F. (2013) Activated T cell trans-endothelial migration relies on myosin-IIA contractility for squeezing the cell nucleus through endothelial cell barriers. PLoS ONE 8, e75151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maus U. A., Janzen S., Wall G., Srivastava M., Blackwell T. S., Christman J. W., Seeger W., Welte T., and Lohmeyer J. (2006) Resident alveolar macrophages are replaced by recruited monocytes in response to endotoxin-induced lung inflammation. Am. J. Respir. Cell Mol. Biol. 35, 227–235 [DOI] [PubMed] [Google Scholar]
- 33. Gotoh K., Tanaka Y., Nishikimi A., Inayoshi A., Enjoji M., Takayanagi R., Sasazuki T., and Fukui Y. (2008) Differential requirement for DOCK2 in migration of plasmacytoid dendritic cells versus myeloid dendritic cells. Blood 111, 2973–2976 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.