

Abstract
The antiviral myxovirus resistance protein 1 (MX1) is an interferon-induced GTPase that plays an important role in the defense of mammalian cells against influenza A viruses. Mouse MX1 interacts with the influenza ribonucleoprotein complexes (vRNPs) and can prevent the interaction between polymerase basic 2 (PB2) and the nucleoprotein (NP) of influenza A viruses. However, it is unclear whether mouse MX1 disrupts the PB2-NP interaction in the context of pre-existing vRNPs or prevents the assembly of new vRNP components. Here, we describe a conditionally active mouse MX1 variant that only exerts antiviral activity in the presence of a small molecule drug. Once activated, this MX1 construct phenocopies the antiviral and NP binding activity of wild type MX1. The interaction between PB2 and NP is disrupted within minutes after the addition of the small molecule activator. These findings support a model in which mouse MX1 interacts with the incoming influenza A vRNPs and inhibits their activity by disrupting the PB2-NP interaction.
Keywords: antiviral agent, GTPase, influenza virus, inhibition mechanism, molecular biology, negative-strand RNA virus, virology, MX1, influenza A, ribonucleoprotein, interaction, viral replication
Introduction
The myxovirus resistance (MX) genes are evolutionarily conserved in nearly all vertebrates. MX gene expression is induced by type I or III interferon, and the corresponding gene products can inhibit a wide range of viruses (1). Human MxA, for example, can suppress the replication of influenza and Thogoto viruses (both Orthomyxoviridae), vesicular stomatitis virus (a rhabdovirus), and hepatitis B virus (a hepadnavirus), and mouse MX1 inhibits influenza and Thogoto virus replication (2).
MX proteins are classified as large GTPases (3, 4). The crystal structure of MxA revealed how the GTPase domain, the bundle-signaling element (BSE),4 and the stalk domain are positioned relative to each other in space (5). These three domains each have specific functions in antiviral activity. The GTPase domain is the most conserved part in the family of large GTPases, and the capacity of MX to bind with GTP determines its antiviral activity (6). The BSE is connected to the GTPase domain via a hinge. Gao et al. (5, 7) suggested that this BSE is crucial for transmitting conformational changes, caused by GTPase activity, to the third domain of MX, i.e. the stalk. The stalk domain is important for oligomerization and target recognition. It contains three interfaces and a loop region (loop L4), which mediate oligomerization through a crisscross interaction pattern. This ultimately results in the formation of oligomeric rings with the stalk domains pointing inward and the GTPase domains located at the periphery of the ring. Loop L4, present at the tip of this stalk, and directed toward the center of the MxA oligomeric ring, is important for viral target recognition (8–10). A yet unproven model proposes that MX proteins, organized in rings, wrap around their viral targets (e.g. the vRNPs) and cooperatively inhibit or disturb the function of those viral targets. However, this model has recently been challenged by the results of Nigg and Pavlovic (11), who reported that oligomerization is not crucial for the antiviral activity of human MxA.
Substantial progress has been made in the last few years in our understanding of the molecular details of the antiviral mechanism of MX proteins. However, it remains unclear how MX proteins interact with influenza A vRNPs and what the molecular consequences are of such an interaction. There is clear evidence that human influenza A viruses are more resistant to human MxA than avian influenza viruses are (12). This difference in sensitivity is associated with amino acid differences in the nucleoprotein (NP) of human and avian influenza A viruses (13–15). This suggests that NP is a direct or indirect target of mammalian MX1 proteins. In line with this, we and others previously showed that mouse MX1 can interact with NP. There is also evidence that influenza A PB2 is a target of and binds with mouse MX1 (14, 16, 17).
PB2 and NP are part of the vRNPs, which are the minimal units required for influenza RNA transcription and replication. The vRNPs consist of the viral RNA genome, multiple NP molecules, and one RNA-dependent RNA polymerase complex containing PB1, PB2, and polymerase acidic protein (PA) (18). We showed that the interaction between NP and PB2 is strongly reduced in the presence of MX1 (10, 14, 19). An appealing model is therefore that mouse MX1 prevents or disrupts the PB2-NP interaction and thereby inhibits viral polymerase activity.
To elucidate whether mouse MX1 can disrupt pre-existing PB2-NP interactions or rather prevent de novo assembly of these interactions, we developed a dormant MX1 variant that could be activated post-translationally. We showed that the active form of this conditional MX1 variant behaves as the wild type protein based on its antiviral activity, nuclear localization, and interaction with NP. Finally, we used this activatable MX1 variant to show that MX1 can actively disrupt pre-existing PB2-NP interactions.
Results
Generation of a Conditionally Inactive MX1 Variant That Can Be Rapidly Activated
We previously reported that mouse MX1 can prevent the interaction between the influenza A virus vRNP components PB2 and NP, which could explain how this protein suppresses influenza A virus replication (14). However, it is unclear whether MX1 prevents vRNP assembly or (also) inactivates pre-existing vRNPs. To address this question, we first have to overcome the problem that mammalian cells that express mouse MX1 are highly resistant to influenza A virus infection. This means that de novo synthesis and assembly of vRNPs are strongly reduced in these cells. We therefore aimed to design a dormant MX1 variant that could be rapidly activated by a small compound stimulus. We took advantage of the FRB*-FKBP-based inducible heterodimerization system to accomplish this (20, 21). In this system, chimeric constructs are generated whereby one of the proteins of interest is fused to FRB* (i.e. FRB with a T2098L mutation) and a second protein is fused to FKBP. In the presence of a suitable rapamycin analog (rapalog), the FRB* and FKBP domains can rapidly and selectively heterodimerize.
Mouse MX1 normally resides in the nucleus, a location that is required for it to suppress influenza A virus replication (22). The nuclear localization signal (NLS) of mouse MX1 resides in the C-terminal α helix of the bundle signaling element, which is localized close to the N terminus in the three-dimensional structure of MX1 (7). Our initial plan was to make a cytoplasmic version of MX1 by mutating its NLS, which would abolish its capacity to inhibit influenza A virus replication. This MX1 variant with a non-functional NLS would be genetically fused with an FRB* domain. By co-expressing an NLS-containing FKBP protein, it then becomes possible to conditionally (depending on the presence of rapalog) provide the MX1 variant with an NLS in trans and thereby promote nuclear entry and the capacity to again exert anti-influenza virus activity. For this intended conditional NLS transcomplementation, we generated a construct in which FLAG-tagged FKBP was fused to a heterologous nuclear localization signal, derived from the SV40 large T antigen (Fig. 1A).
FIGURE 1.

Design and initial characterization of mouse MX1 constructs fused with FRB*. A, schematic representation of FRB*-mouse MX1 fusions (N- or C-terminal fusion) and of the FKBP construct with a N-terminal FLAG tag followed by the nuclear localization signal of SV40 large T antigen. B, influenza A minireplicon assay to evaluate MX1-inhibitory potential. HEK293T cells were transfected with pCAXL-PB1, -PB2, -PA, and -NP plasmids (25 ng each), pHW-NSLuc (100 ng), and pRL-CMV (25 ng). The cells were co-transfected with 75 ng of the respective MX1 constructs (FRB*-MX1, MX1-FRB*, MX1 WT, FRB*-MX1T69A, MX1T69A-FRB*, or MX1T69A) as indicated. In addition, 75 ng of PCAXL or pCAXL-F-NLS-FKBP was co-transfected. Six hours after transfection, cells were treated with 100 nm rapalog (in ethanol) or with the equivalent volume of ethanol (no rapalog). The normalized luciferase activity in the lysates was determined 24 h after transfection. Bars represent the average of the triplicates. This panel is representative of three independent experiments. Statistics was performed by pairwise comparison of the effect of the addition of rapalog with a Fisher's protected two way-analysis of variance test. The data are represented as mean ± S.E. **, p < 0.01; ****, p < 0.0001; ns, not significant. C, MX1, NP, and F-NLS-FKBP expression in cell lysates as prepared in B were determined by Western blotting with antibodies recognizing MX1, NP, and FLAG tag (F-NLS-FKBP). This panel is representative of three independent experiments. D, FRB*-MX1 reduces the antiviral effect of co-expressed MX1 in a minireplicon assay. HEK293T cells were (co)transfected with the indicated plasmids. In five settings, pCAXL-Mx1 (50 ng) was co-transfected with increasing amounts (50–400 ng) of pCAXL-FRB*-Mx1 and F-NLS-FKBP (ratios ranging from 2:1 to 1:8). Six hours after transfection, cells were treated or not with 100 nm rapalog, and lysates were made 24 h after transfection. The normalized relative luciferase activity in the lysates is shown in the graphs. Bars represent the average of triplicates. This graph is representative of two independent experiments. The data are represented as mean ± S.E.
As a control for our strategy, we generated FRB*-MX1 and MX1-FRB*, i.e. constructs in which MX1 carried its wild type NLS. These two constructs were expected to suppress influenza A virus RNA polymerase activity provided that the fused FRB* domain would not hinder this function. We first evaluated whether FRB*-MX1 and MX1-FRB* had anti-influenza virus activity using a minireplicon system that is based on A/Puerto Rico/8/34 (PR8) virus vRNP components. HEK293T cells were transfected with expression plasmids encoding PB1, PB2, PA, and NP as well as a firefly luciferase viral RNA-like reporter together with a wild type MX1, the inactive MX1T69A mutant, FRB*-MX1, MX1-FRB*, FRB*-MX1T69A, or MX1T69A-FRB*. In half of the settings, the F-NLS-FKBP expression plasmid was co-transfected as well. After transfection, cells were treated with rapalog or with vehicle. As expected, wild type MX1 strongly reduced the influenza A polymerase complex activity (Fig. 1B). However, in the absence of F-NLS-FKBP and rapalog, FRB*-MX1 and MX1-FRB* were inactive, suggesting that the N- and C-terminally fused FRB* domains interfered with the antiviral activity of MX1. Remarkably, in the presence of F-NLS-FKBP and rapalog, FRB*-MX1 regained antiviral activity in the PR8 minireplicon system to a degree comparable with that of wild type MX1 (Fig. 1B). This suggests that fusion of FRB* to the N terminus of MX1 interferes with the antiviral activity of mouse MX1 by a mechanism that can be reversed in the presence of F-NLS-FKBP by the addition of rapalog. Thus, coincidently, we obtained a dormant MX1 variant (FRB*-MX1) that could be activated by the addition of an exogenous small molecule (rapalog) without mutating the endogenous NLS sequence.
MX1-FRB* failed to suppress the influenza A RNA polymerase activity regardless of the presence or absence of F-NLS-FKBP and rapalog. It has been reported that fusion of FRB* to a protein can have a destabilizing effect and lead to degradation of the fusion protein (21, 22). To assess whether such a destabilizing effect could explain the lack of antiviral activity of FRB*-MX1 or MX1-FRB*, we determined protein expression levels in lysates from transfected cells by Western blotting analysis. When compared with the other MX1 constructs, MX1-FRB* and MX1T69A-FRB* expression levels in the detergent-extractable cell fraction were strongly reduced (Fig. 1C). Co-expression of F-NLS-FKBP and the addition of rapalog did not increase the expression levels of MX1-FRB* and MX1T69A-FRB*. Therefore, the low expression levels may explain the lack of antiviral activity of the MX1-FRB* variant. In contrast to the C-terminal FRB* fusions, the expression levels of FRB*-MX1 and FRB*-MX1T69A were only slightly reduced when compared with those of MX1 and MX1T69A (Fig. 1C). To gain some insight into how the FRB* domain fused to the N terminus of MX1 disturbed the antiviral activity in a reversible way, we co-expressed FRB*-MX1 (with or without F-NLS-FKBP) with wild type MX1 at different ratios. In the absence of rapalog, the activity of MX1 dropped dose-dependently when FRB*-MX1 was co-expressed, suggesting a dominant negative effect of FRB*-MX1 (Fig. 1D). As a control for dominant negative interference, MX1 was co-expressed with MX1T69A. However, when rapalog and F-NLS-FKBP were present, the dominant negative effect of FRB*-MX1 on MX1 was at least partially reversed (Fig. 1D). This reversible dominant negative effect on MX1 suggests that FRB*-MX1 can still form dimers or even higher order oligomers.
FRB*-MX1 and F-NLS-FKBP Dimerize in a Rapalog-dependent Way
To further document the dependence on rapalog addition to activate FRB*-MX1 in the presence of F-NLS-FKBP, we performed a dose range experiment in which different concentrations of this small molecule compound were added to transfected cells. Starting from a concentration of 25 nm rapalog, FRB*-MX1 inhibited the PR8 polymerase activity in a F-NLS-FKBP-dependent way and reached a maximum at 100 nm (Fig. 2). Taken together, this shows that N- or C-terminal fusion of an FRB* domain to mouse MX1 is detrimental for its antiviral activity. However, in the case of FRB*-MX1, the antiviral activity, based on an influenza A minireplicon reporter assay, can be restored in the presence of F-NLS-FKBP by the addition of rapalog in a dose-dependent way.
FIGURE 2.

Rapalog restores the antiviral activity of FRB*-MX1 in a dose-dependent way in the presence of F-NLS-FKBP. A, HEK293T cells were transfected with PB1, PB2, PA, and NP expression plasmids (25 ng each), pHW-NSLuc (100 ng), and pRL-CMV (25 ng). The cells were co-transfected with 150 ng of pCAXL, 75 ng of F-NLS-FKBP, 75 ng of FRB*-MX1, or 75 ng of F-NLS-FKBP and 75 ng of FRB*-MX1, as indicated. Six hours after transfection, different doses of rapalog were added to the cells. The normalized luciferase activity in the lysates was determined 24 h after transfection. Rel Luc activity, relative luciferase activity. The experiment was performed in technical triplicates, and the data are represented as mean ± S.E. The statistical probability that differences were significant between pairs of different constructs at a fixed rapalog concentration was assessed by a Fisher's protected least significant difference test. ****, p < 0.0001. B, representative Western blotting analysis of FRB*-MX1, NP, and F-NLS-FKBP in lysates of cell transfected as in A. The results shown in this figure are representative of two independent experiments.
Subcellular Localization of FRB*-MX1 and F-NLS-FKBP
The subcellular localization of MX1, FRB*-MX1, MX1-FRB*, MX1T69A, FRB*-MX1T69A, and F-NLS-FKBP in the absence or presence of rapalog was also visualized. In the absence of F-NLS-FKBP, FRB*-MX1 is expressed mainly in the nucleus and in a punctate pattern and, like MX1, it can also be detected in the cytoplasm (Fig. 3A). When F-NLS-FKBP was expressed alone, it was distributed evenly over the whole cell despite its added NLS, presumably because it is small enough to diffuse freely in and out of the nucleus (predicted molecular mass: 14.3 kDa) and because of a masking effect of the NLS by the FLAG tag. When FRB*-MX1 and F-NLS-FKBP were expressed together, F-NLS-FKBP was redistributed to the MX1 nuclear speckles in the presence of rapalog (Fig. 3). This suggests that rapalog induces the dimerization of F-NLS-FKBP and FRB*-MX1.
FIGURE 3.

Confocal microscopy images of transfected HEK293T cells. A, HEK293T cells were transfected with 75 ng of the respective Mx1 constructs (Mx1, FRB*-MX1, MX1-FRB*, MX1T69A, FRB*-MX1T69A) or with F-NLS-FKBP. After 24 h, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and stained for MX1 (green) or FLAG (F-NLS-FKBP) (red). Cell nuclei were visualized with Hoechst (blue). Images were recorded with a confocal microscope (Leica Sp5 AOBS confocal system). B, FRB*-MX1 and F-NLS-FKBP were co-localized. HEK293T cells were transfected with 75 ng of F-NLS-FKBP and 75 ng of the respective MX1 constructs (MX1, FRB*-MX1, MX1-FRB*, MX1T69A, or FRB*-MX1T69A). Six hours after transfection, cells were treated with 100 nm rapalog (in ethanol) or with the equivalent volume of ethanol (no rapalog). Twenty-four hours after transfection, the cells were fixed, permeabilized, and stained as described in A.
To verify that F-NLS-FKBP and FRB*-MX1 heterodimerize in the presence of rapalog, we performed a co-immunoprecipitation experiment. In addition, we estimated how fast this dimerization could occur after adding rapalog to the cells. F-NLS-FKBP was pulled down with an anti-FLAG antibody, and co-immunoprecipitation of FRB*-MX1 was evaluated by Western blotting. This experiment revealed that FRB*-MX1 interacts with F-NLS-FKBP only in the presence of rapalog (Fig. 4). Interestingly, FRB*-MX1 already co-immunoprecipitated with F-NLS-FKBP when rapalog was added just 5 min prior to cell lysis (Fig. 4).
FIGURE 4.
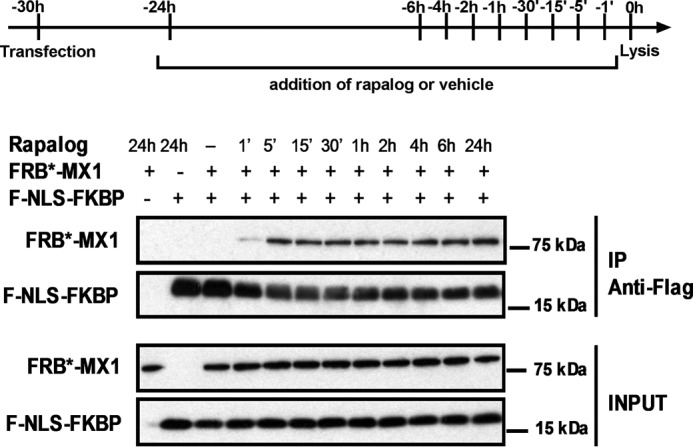
Rapid rapalog-dependent interaction between FRB*-MX1 and F-NLS-FKBP. HEK293T cells were co-transfected with 1 μg of FRB*-MX1 and 1 μg of F-NLS-FKBP expression vector. Rapalog (final concentration 100 nm) was added to the cells at different time points before lysis, as indicated. As control, cells treated with vehicle for 24 h were included. The cells were lysed 30 h after transfection. F-NLS-FKBP was immunoprecipitated from the cell lysates with anti-FLAG antibody, and immunoprecipitated (IP) proteins were visualized by Western blotting using rabbit anti-MX1 serum and anti-FLAG to detect FRB*-MX1 and F-NLS-FKBP, respectively. Part of the cell lysates prior to immunoprecipitation was analyzed in the same way (INPUT). The results shown are representative of three independent experiments.
Activated FRB*-MX1 Interacts with Influenza A NP
MX1 can interact with influenza A virus NP (14). Therefore, we tested whether FRB*-MX1 could also interact with NP and, if so, whether this interaction was dependent on the presence of F-NLS-FKBP and rapalog. HEK293T cells were transfected with expression plasmids encoding NP and MX1 or FRB*-MX1, and the latter setup was with or without co-transfected F-NLS-FKBP. The cells were treated with rapalog or vehicle starting at 16 h before cell lysis. Subsequently, NP was pulled down, and then the presence of co-immunoprecipitated MX1 or FRB*-MX1 protein was determined. As expected, wild type MX1 formed a complex with NP, irrespective of the presence or absence of rapalog (Fig. 5). FRB*-MX1, however, only interacted with NP when both F-NLS-FKBP and rapalog were present (Fig. 5). Therefore, interaction of FRB*-MX1 with influenza A virus NP correlates with its antiviral configuration.
FIGURE 5.

Interaction of FRB*-MX1 with influenza NP depends on rapalog and F-NLS-FKBP. HEK293T cells were transfected with 1 μg of expression vectors coding for MX1 WT, MX1T69A, FRB*-MX1 or FRB*-MX1T69A. In addition, 0.5 μg of pCAXL-NP and/or 1 μg of pCAXL-FNLS-FKBP was co-transfected as indicated. Six hours after transfection, cells were treated with 100 nm rapalog or with vehicle. Twenty-four hours after transfection, cells were lysed and NP was immunoprecipitated. FRB*-MX1 and NP in the immunoprecipitated (IP) and input fraction were visualized by Western blotting with antibodies recognizing MX1 and NP (anti-RNP antibody). The result shown is a representative of three independent experiments.
Activated FRB*-MX1 Protects Cells against Influenza A Virus Infection
FRB*-MX1 can inhibit influenza A polymerase complex activity in a minireplicon system in the presence of F-NLS-FKBP and rapalog. In a next step, we wanted to verify whether this conditionally activatable MX1 variant could also suppress influenza A virus replication in an infection setting. To investigate this, HEK293T cells were transfected with expression vectors for MX1 or FRB*-MX1 and F-NLS-FKBP. Cells transfected with GTPase-deficient MX1T69A were included as a negative control. Thirty hours after transfection, the cells were infected with PR8 virus at a multiplicity of infection (MOI) of 1. At different time points before and after infection, rapalog was added to the cells. Sixteen hours after infection, the cells were analyzed by flow cytometry to determine the percentage of infected (vRNP-positive) cells in the transfected (MX1-positive) population. As expected, the majority of wild type MX1-expressing cells had very low vRNP levels, whereas most of the MX1T69A-expressing cells were infected (Fig. 6). In the absence of rapalog, most of the FRB*-MX1-expressing cells were infected (Fig. 6). However, when rapalog was added 1 h before, at the same time as or 1 h after infection, up to 50% of FRB*-MX1-expressing cells displayed very low vRNP levels, suggesting that in these cells, the virus failed to replicate (Fig. 6). FRB*-MX1 loses its ability to suppress vRNP expression when rapalog is added 4 h after the start of infection (Fig. 6). Taken together, FRB*-MX1, which is activated by rapalog shortly before or after infection, can inhibit influenza A virus replication.
FIGURE 6.

FRB*-MX1 activated with rapalog after infection inhibits influenza A virus replication. A, HEK293T cells were transfected with 200 ng of MX1, 200 ng of MX1T69A, 200 ng of FRB*-MX1T69A, or 200 ng of FRB*-MX1 and 200 ng of F-NLS-FKBP expression vector as indicated. Thirty hours later, the cells were infected with PR8 virus at an MOI of 1. Rapalog (100 nm) was added 1 h before, at the same time of, 1 h after, or 4 h after viral infection. Cells transfected with MX1 or MX1T69A expression vectors were treated with rapalog (100 nm) starting from 6 h after transfection for a total of 38 h. Sixteen hours after infection, the cells were collected, fixed, permeabilized, and stained for MX1 (anti-MX1) and NP (anti-RNP) and analyzed on an LSR-II flow cytometer. The percentage of infected (RNP-positive) and uninfected (RNP-negative) cells in the transfected (MX1-positive) population was determined using FACSDiva and FlowJo software and is indicated in the top right and lower right corner of each plot. For each setup, one representative fluorescence-activated cell plot is shown. The experiment was repeated three times with similar results. B, graph showing the percentage of transfected (MX1-positive) cells that are PR8 virus-infected based on flow cytometry analysis as in A. Data points are from three independent experiments (averages of triplicates), the horizontal line indicates the mean, and the error bars depict the standard errors. In A, “FRB*-MX1” implies that F-NLS-FKBP was co-transfected. For statistical analysis, infected cell proportions were analyzed by logistic regression (logit was used as the link function). *, p < 0.05; ***, p < 0.001; ****, p < 0.0001; ns, not significant.
Activated FRB*-MX1 Disrupts the PB2-NP Interaction
We previously reported that mouse MX1 interferes with the interaction of PB2 and NP, which correlates with its antiviral activity (14). This reduced PB2-NP interaction could be the result of an active disruption of the PB2-NP interaction in pre-existing vRNPs or the result of inhibition of the formation of new vRNPs. It is very difficult to distinguish between these two possibilities, because either way, this implies that MX1 blocks infection at an early step. Either the very small amount of incoming vRNPs is inactivated by MX1, or the incoming vRNPs remain associated and newly produced vRNPs are prevented from being formed by MX1. The conditionally activatable FRB*-MX1 variant described above now allowed vRNP formation to proceed for a certain amount of time before the addition of rapalog, to initiate activation of its antiviral activity. In this way, it is possible to investigate the effect of MX1 on a large enough pool of pre-existing vRNPs.
HEK293T cells were transfected with the minireplicon system of PR8 virus, and we co-transfected these cells with MX1 WT, MX1T69A, FRB*-MX1, F-NLS-FKBP, and FRB*-MX1 together with F-NLS-FKBP or FRB*-MX1T69A together with F-NLS-FKBP (Fig. 7). At different time points before cell lysis, rapalog was added to activate FRB*-MX1 in cells that also expressed F-NLS-FKBP. Co-immunoprecipitations were performed in both directions, i.e. PB2V5 was pulled down followed by analysis of co-immunoprecipitated NP, and, vice versa, we pulled down NP and assessed whether PB2V5 was co-immunoprecipitated. As expected, the PB2V5-NP interaction was disrupted by wild type MX1, but not by MX1T69A (Fig. 7). In addition, FRB*-MX1, but not FRB*-MX1T69A, disrupted the NP-PB2V5 interaction, but only when both F-NLS-FKBP and rapalog were present. Interestingly, disruption of the NP-PB2V5 complex in the presence of FRB*-MX1 was apparent even when rapalog was added only 15 min before cell lysis. Taken together, this suggests that activated FRB*-MX1 can disrupt pre-existing viral PB2-NP interactions.
FIGURE 7.

Activated FRB*-MX1 disrupts the interaction between PB2V5 and NP. HEK293T cells were transfected with PB1, PB2V5, PA and NP expression plasmids and pHW-NSLuc (100 ng each). The cells were co-transfected with 1 μg of expression vector for MX1 WT, MX1T69A, FRB*-MX1, and F-NLS-FKBP and 0.5 μg of NP expression vector as indicated. The cells were treated with 100 nm rapalog at different time points after transfection as indicated. Vehicle was added as a negative control for the −24 h time point (−). Cells were lysed 30 h after transfection. PB2V5 and NP were immunoprecipitated (IP) with anti-V5 and anti-NP antibodies, respectively. Proteins in the NP (IP anti-NP) and PB2V5 (IP anti-V5) immunoprecipitates and total cell lysates (input) were visualized by Western blotting with antibodies specific for NP (anti-RNP antibody) and PB2V5 (anti-V5 antibody). The result shown is representative of three independent experiments.
Discussion
Mouse MX1 inhibits influenza virus replication by blocking primary transcription (23). However, until now the details of the antiviral mechanism of MX1 have remained elusive. We and others previously reported that the presence of mouse MX1 prevents the interaction between the influenza A virus vRNP components PB2 and NP, which could explain how this protein suppresses influenza A virus replication (14, 23). Nonetheless, it is unclear whether MX1 prevents vRNP assembly or (also) inactivates pre-existing vRNP complexes. Mammalian cells that express mouse MX1 suppress influenza A virus replication and are highly resistant to influenza A virus infection, which means that de novo synthesis and assembly of vRNPs are strongly reduced. This makes it technically very difficult to address whether MX1 prevents vRNP assembly or (also) inactivates pre-existing vRNP complexes. We therefore aimed to design an antivirally inactive MX1 variant that could be rapidly activated by a small compound stimulus.
To generate such a conditional MX1 variant, we made use of the inducible heterodimerization between FRB* and FKBP in the presence of the cell-permeable small compound rapalog. We explored two conditional MX1 variants, i.e. one with the FRB* domain fused at the N terminus (FRB*-MX1) and one with an FRB* domain at the C terminus (MX1-FRB*). As such, both variants were inactive in a PR8 minireplicon assay, suggesting that the addition of the FRB* domain hinders the antiviral activity of the MX1 protein. However, in the presence of F-NLS-FKBP and rapalog, the variant with the FRB* domain at the N terminus regained antiviral activity almost to a similar level as the wild type MX1 protein. The antiviral activity of FRB*-MX1 was dependent on the presence of both F-NLS-FKBP and rapalog, and positively correlated with the dose of rapalog. The activatable phenotype of the FRB*-MX1 variant was also confirmed in a PR8 infection experiment. Also, during infection, antiviral activity of FRB*-MX1 was dependent on the presence of both F-NLS-FKBP and rapalog. Nevertheless, the resulting antiviral activity appeared to be lower than that of wild type MX1 (Fig. 6). This is not unexpected because newly translated MX1 in pCAXL-Mx1-transfected cells is immediately active.
To further corroborate that the activated FRB*-MX1 variant functionally resembles the wild type MX1 protein, we investigated the binding of FRB*-MX1 with a viral target, i.e. the NP protein. For this, we performed a co-immunoprecipitation experiment in cells expressing NP and FRB*-MX1 with or without F-NLS-FKBP and in the absence or presence of rapalog. This experiment revealed that FRB*-MX1 could bind to NP, but only in its active form, i.e. in the presence of F-NLS-FKBP and rapalog. Finally, we also performed a co-immunoprecipitation experiment to investigate the effect of FRB*-MX1 on the PB2-NP interaction. This experiment showed that activated FRB*-MX1 was able to inhibit the PB2-NP interaction to a similar level as the wild type MX1 protein. Taken together, these results suggest that the active form of FRB*-MX1, i.e. in complex with F-NLS-FKBP and rapalog, functionally resembles the wild type MX1 protein.
As soon as 5 min after the addition of rapalog, we observed a clear interaction between F-NLS-FKBP and FRB*-MX1, and this interaction became more pronounced over time. This suggests that the heterodimerization of F-NLS-FKBP and FRB*-MX1 occurs very fast after the addition of rapalog. Such a fast activation of FRB*-MX1 is crucial to investigate the effect of FRB*-MX1 on the PB2-NP interaction in existing vRNPs independent of its effect on the formation of new vRNPs. In line with this, the interaction between PB2 and NP disappeared, even if rapalog was added for only 15 min to cells that expressed FRB*-MX1 and F-NLS-FKBP. This suggests that FRB*-MX1 is activated very rapidly, and actively disturbs pre-existing vRNPs. Taken together, given that activated FRB*-MX1 functionally resembles its wild type counterpart in terms of inhibiting influenza A virus replication, this suggests that wild type mouse MX1 can target the incoming influenza A vRNPs and actively disturb them.
The potency of activated FRB*-MX1 to inhibit influenza A virus in the context of an infection was also studied. This experiment showed that FRB*-MX1 could inhibit influenza infection if it was activated at the latest 1 h after infection. If rapalog was added 4 h after infection, FRB*-MX1 could no longer suppress the expression of newly produced vRNPs. This result is not unexpected, if one keeps in mind that mouse MX1 is only active in the nucleus. After virus entry, the vRNPs are transported to the cell nucleus where viral transcription and replication take place. After a few hours, these vRNPs are then transported to the cell cytoplasm and cell membrane, where they assemble into new virus particles. As MX1 is present in the nucleus, it can target the vRNPs only when these are also present in the nucleus. Thus, MX1 needs to be activated before or at the same time as the vRNPs are imported or are present in the nucleus, and before they are transported out of the nucleus. Therefore, our results indicate that mouse MX1 can actively disturb the PB2-NP interaction within existing vRNPs in the nucleus. To investigate the effect of MX1 later in infection, another read-out than NP expression is needed to measure antiviral activity. It could be interesting to investigate whether MX1, for example, could inhibit the export of vRNPs out of the nucleus.
We showed that the antiviral activity of FRB*-MX1 is activated in the presence of F-NLS-FKBP and rapalog. Exactly how FRB*-MX1 is activated by rapalog in the presence of F-NLS-FKBP remains elusive. FRB*-MX1 can still enter the nucleus, and its localization is not altered in the presence of F-NLS-FKBP and upon the addition of rapalog. This suggests that the activation mechanism is not based on an altered localization. An interesting observation is that FRB*-MX1 is not able to bind the influenza NP protein until it is activated. This could mean that the FRB* domain shields the NP interaction interface of MX1, or alters its conformation and thereby prevents its interaction with NP. This suggests that the presence of F-NLS-FKBP and rapalog alters or stabilizes the conformation of FRB*-MX1, allowing its interaction with NP. Another possibility, which is not mutually exclusive, is that the addition of the FRB* domain to MX1 alters its conformation, resulting in a loss of GTPase activity, leading to a loss in antiviral activity. The addition of F-NLS-FKBP and rapalog could then restore the natural conformation of MX1, leading to an active GTPase protein. Although we assume that the FRB* domain destabilizes or alters the conformation of MX1, this disturbance may be only partial. FRB*-MX1 still localizes to distinct spots in the nucleus. In addition, when FRB*-MX1 and the wild type MX1 are co-expressed in the minireplicon system, the FRB*-MX1 protein has a dominant negative effect on the antiviral activity of the wild type MX1 protein, suggesting that both proteins can still form dimers (Fig. 1D). In contrast to FRB*-MX1, the opposite variant MX1-FRB* could not be activated in the presence of F-NLS-FKBP and rapalog, which correlated with strongly reduced expression levels. This could suggest that the addition of the FRB* domain at the C terminus of MX1 has a more profound destabilizing effect, resulting in protein degradation. This loss of function could not be reverted by F-NLS-FKBP and rapalog. Taken together, we described the generation of an activatable MX1 protein, FRB*-MX1, which in its active form functionally resembles the wild type MX1 protein. We used this conditional MX1 variant to show that active MX1 protein can actively disrupt the PB2-NP interaction in existing vRNPs.
Experimental Procedures
Cells
HEK293T cells were maintained in DMEM supplemented with 10% fetal calf serum, 2 mm l-glutamine, 0.4 mm sodium pyruvate, non-essential amino acids, 100 units/ml penicillin, and 0.1 mg/ml streptomycin. Madin-Darby canine kidney cells were maintained in DMEM supplemented with 10% fetal calf serum, 2 mm l-glutamine, non-essential amino acids, 100 units/ml penicillin, and 0.1 mg/ml streptomycin.
Viruses
Mouse-adapted PR8 (H1N1 (14)) was propagated by multicycle grow on Madin-Darby canine kidney cell monolayers in the presence of trypsin (2 μg/ml) and concentrated from the culture supernatant by centrifugation at 25,000 × g for 16 h at 4 °C. Pelleted virions were resuspended in phosphate-buffered saline containing 20% glycerol, aliquoted, and stored at −80 °C until use.
Plasmids
The mammalian expression plasmids pCAXL-Mx1, -Mx1T69A, -PB1, -PB2, PB2V5, -PA, and -NP and the pHW-NSLuc influenza A polymerase reporter plasmid have been described (14). The pRL-CMV plasmid (Promega, catalog number E2261) contains a Renilla luciferase gene under the control of a CMV promoter and was used to normalize for transfection efficiency. FRB* carrying the T2098L mutation (20) and FKBP-coding plasmids were a kind gift from Dr. Bram van Raam (Ghent University and VIB). The FRB*-MX1 (WT or T69A) and MX1 (WT or T69A)-FRB* fusions were generated by fusion PCR, using the vectors pCAXL-Mx1 (WT or T69A) and an FRB*-containing plasmid as templates. The fusion proteins were cloned into pCAXL between XhoI and MluI restriction sites. F-NLS-FKBP was generated by adding the coding information for a FLAG tag (DYKDDDDK) and the nuclear localization signal of SV40 large T antigen (PKKKRKV) 5′ of the coding information for FKBP, and the fusion was subsequently cloned into pCAXL between XhoI and MluI restriction sites. The resulting plasmids were named pCAXL-FRB*-Mx1 (WT or T69A), pCAXL-Mx1 (WT or T69A)-FRB*, and pCAXL-F-NLS-FKBP (Fig. 1).
Influenza A Virus Minireplicon System
HEK293T cells were seeded at 5 × 104 cells per well in 24-well plates 24 h before transfection. Transfections were performed in triplicate using the calcium phosphate precipitation method. To determine the effect of different MX1 variants on the influenza A RNA-dependent RNA polymerase complex activity, as reported by the firefly luciferase levels, 75 ng of the respective MX1 expression vectors were co-transfected with plasmids pCAXL-PB1, -PB2, -PA, and -NP (25 ng each), firefly luciferase reporter pHW-NSLuc (100 ng), and pRL-CMV (25 ng). In addition, 75 ng of pCAXL or pCAXL-F-NLS-FKBP was co-transfected. At different time points after transfection, cells were treated or not with different concentrations of A/C Heterodimerizer (Clontech, catalog number 635057; hereafter named rapalog) or vehicle (ethanol) as indicated in the figure legends. Cells were lysed 48 h after transfection with luciferase lysis buffer (25 mm Tris-phosphate, 2 mm DTT, 2 mm CDTA, 10% glycerol, and 1% Triton X-100). Luciferase activity in these lysates was measured by using the Dual-Luciferase® Reporter Assay System (Promega, catalog number E-1960) and a GloMax® 96 Microplate Luminometer (Promega). The ratio of the firefly and Renilla luciferase activities (firefly/Renilla luciferase × 1000) was calculated to normalize for transfection efficiency. Expression of transfected MX1 or MX1 fusion constructs, NP and F-NLS-FKBP, in the cell lysates was confirmed by Western blotting analysis.
Antibodies and Chemicals
A rabbit polyclonal antiserum raised against the C-terminal part of mouse MX1 has been described (14). The monoclonal anti-V5 used in co-immunoprecipitation was from Pierce (catalog number MA5-15253), the monoclonal anti-V5-HRP antibody used for Western blots was from Invitrogen (catalog number R96125), and the monoclonal anti-FLAG® M2 was purchased from Sigma (catalog number F3165). Monoclonal anti-influenza A virus NP clones A1 and A3 (ascites blend, mouse, NR-4282) and polyclonal anti-influenza virus ribonucleoprotein (RNP) complexes, A/Scotland/840/74 (H3N2) (antiserum, goat, NR-3133), were obtained from the National Institutes of Health Biodefense and Emerging Infections Research Resources Repository (NIAID, National Institutes of Health). The fluorescently labeled antibodies Alexa Fluor® 488 donkey anti-rabbit IgG (catalog number A21206), Alexa Fluor® 594 donkey anti-mouse IgG (catalog number A21203), and Alexa Fluor® 647 donkey anti-goat IgG (catalog number 21447) were from Life Technologies Europe B.V. N-Ethylmaleimide (NEM) was obtained from Sigma (catalog number E-3876) and dissolved in ethanol at a concentration of 2 m. Protease inhibitor cocktail tablets were from Roche Applied Science (catalog number 11 873 580 001). Rapalog (also called A/C Heterodimerizer) dissolved in ethanol was purchased from Clontech (catalog number 635057).
Immunofluorescence Microscopy
HEK293T cells were seeded a 5 × 104 cells per well on glass coverslips in 24-well plates. Twenty-four hours later, the cells were transfected using the calcium phosphate precipitation method with 75 ng of pCAXL-Mx1, pCAXL-FRB*-Mx1, pCAXL-FRB*Mx1, pCAXL-MX1T69A, pCAXL-FRB*Mx1T69A, or pCAXL-F-NLS-FKBP. In another set of experiments, HEK293T cells were transfected with pCAXL-F-NLS-FKBP together with pCAXL-Mx1, pCAXL-FRB*Mx1, pCAXL-Mx1T69A, or pCAXL-FRB*Mx1T69A. Six hours later, the cells were treated with 100 nm rapalog or vehicle (as control). Twenty-four hours after transfection, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and stained with rabbit anti-MX1 serum (diluted 1/1000) followed by Alexa Fluor® 488-labeled donkey anti-rabbit IgG (diluted 1/600). F-NLS-FKBP was visualized with anti-FLAG monoclonal antibody (diluted 1/400) followed by Alexa Fluor® 594-labeled donkey anti-mouse IgG (diluted 1/600). Cell nuclei were visualized with Hoechst (Invitrogen, catalog number H21492; diluted 1/1000). Images were recorded with a confocal microscope (Leica Sp5 AOBS confocal system) using a 63× HCX PL Apo 1.4 oil immersion objective. A bandpass filter of 510–555 nm was positioned before the detector to measure the fluorescence of Alexa Fluor® 488 after excitation with an argon 488-nm laser.
Co-immunoprecipitation
To study the interaction of MX1 with NP, HEK293T cells (seeded at 1.2 × 106 cells per 9-cm dish) were transfected using the calcium phosphate precipitation method with 0.5 μg of pCAXL or pCAXL-NP, 1 μg of pCAXL-Mx1 or pCAXL-FRB*-Mx1, and 1 μg of pCAXL or pCAXL-F-NLS-FKBP. At different time points after transfection, the cells were treated with 100 nm rapalog (in ethanol) or vehicle (as control). Total lysates were prepared 18 h later in 600 μl of low salt lysis buffer, pH 7.2, containing 25 mm NEM (50 mm Tris-HCl, pH 7.2, 150 mm NaCl, 5 mm EDTA, 1% NP-40, 25 mm NEM, and a protease inhibitor cocktail). The cells were lysed for 20 min on ice and centrifuged for 3 min at 16,000 × g to remove insoluble material. NP was immunoprecipitated from 135 μl of the cleared lysates with monoclonal anti-NP for 3 h at 4 °C. Immune complexes were collected by incubation for 1 h at 4 °C in the presence of protein G-Sepharose beads (GE Healthcare, catalog number 17-0618-01) followed by centrifugation. Immunoprecipitates were washed six times with high-salt lysis buffer, pH 7.2 (50 mm Tris-HCl, pH 7.2, 500 mm NaCl, 5 mm EDTA, and 1% NP-40). Proteins were eluted from the beads by heating for 10 min at 95 °C in 2× Laemmli buffer. Finally, lysates and immunoprecipitated fractions were separated by SDS-PAGE (8% acrylamide), and MX1 proteins, NP, and F-NLS-FKBP were visualized by Western blotting with antibodies directed against MX1, NP (polyclonal goat anti-RNP), and FLAG tag, respectively.
To analyze the interaction between PB2 and NP, HEK293T cells (seeded at 1.2 × 106 cells per 9-cm dish) were transfected using calcium phosphate precipitation with expression plasmids pCAXL-PB1, -PB2V5, -PA, and -NP and pHW-NSLuc (100 ng each) together with 1 μg of pCAXL or pCAXL-Mx1 or with 1 μg of pCAXL-FRB*-Mx1 and 1 μg of pCAXL or pCAXL-F-NLS-FKBP. After 6 h or at specific times before lysis (as mentioned in the figure legend), the cells were treated with 100 nm rapalog or vehicle, and 18 h later, cell lysates were prepared as described above. The cleared lysates were split in two. Monoclonal anti-V5 antibody was added to one part of the lysate, and anti-NP monoclonal antibodies were added to the other part. After a 3-h incubation at 4 °C, immune complexes were isolated with protein G-Sepharose beads and visualized by Western blotting with antibodies directed against MX1, anti-V5-HRP, and NP (polyclonal goat anti-RNP).
Flow Cytometric Analysis of Influenza A Virus-infected Cells
HEK293T cells (5 × 105 cells per 6-cm dish) were transfected with 200 ng of pCAXL or with 200 ng of pCAXL-Mx1, -Mx1T69A, -FRB*-Mx1, or FRB*-MXT69A. In addition, 200 ng of pCAXL-F-NLS-FKBP or 200 ng of pCAXL was co-transfected. The FuGENE HD transfection reagent (Promega catalog number E-2311) was used for transfection according to the manufacturer's instructions. After 30 h, the cells were infected with mouse-adapted PR8 at an MOI of 1 for 16 h. At different time points before or after infection, the cells were treated with 100 nm rapalog or with vehicle as control. Next, the cells were collected, fixed with 4% paraformaldehyde, and permeabilized with 0.2% Triton X-100. The cells were stained for MX1 (with anti-MX1, diluted 1/400) and influenza virus NP (with anti-RNP, diluted 1/2,000). Alexa Fluor® 488-labeled donkey anti-rabbit IgG (diluted 1/600) and Alexa Fluor® 633-labeled donkey anti-goat IgG (diluted 1/1200) were used as secondary antibodies. The number of MX1- and RNP-positive cells was determined with an LSR-II flow cytometer (BD Biosciences), and the data were analyzed using FACSDiva (BD Biosciences) and FlowJo X (TreeStar) software.
Statistics
The data were analyzed with GraphPad Prism 7 (Figs. 1 and 2) or with GenStat software (Fig. 6). For the PR8 minireplicon, the relative luciferase activities were compared by using a two-way analysis of variance, with the replicates of experiments being set as blocks. Significance was assessed by an F test. For the infection assay (Fig. 6), a logistic regression was performed on the data of infected cell proportions, as implemented in GenStat. Infected cell proportion were logit-transformed to provide additivity and stabilize the variance. The significance of the activity of the different MX1 constructs was assessed by an F test.
Author Contributions
J. V. and D. D. V. conceived the FRB*-Mx1 constructs; J. V., L. V. H., J. S., D. D. V., and A. K. performed experiments. J. V., L. V. H., and X. S. wrote the manuscript.
Acknowledgment
We thank Dr. Bram van Raam for sharing FRB* and FKBP coding plasmids.
This work was supported by Ghent University Special Research Fund (Project BOF12/GOA/014) (to X. S. and J. V.) This work was also supported by grants from IWT-Vlaanderen (to J. S. and D. D. V.) and FP7 ITN UniVacFlu (to A. K.). The authors declare that they have no conflicts of interest with the contents of this article.
- BSE
- bundle-signaling element
- NP
- nucleoprotein
- RNP
- ribonucleoprotein
- vRNP
- viral RNP
- PA
- polymerase acidic protein
- FKBP
- FK506 binding protein
- FRB
- FKBP rapamycin-binding protein
- NLS
- nuclear localization signal
- MOI
- multiplicity of infection
- CDTA
- 1,2-cyclohexylenedinitrilotetraacetic acid
- NEM
- N-ethylmaleimide.
References
- 1. Haller O., and Kochs G. (2011) Human MxA protein: an interferon-induced dynamin-like GTPase with broad antiviral activity. J. Interferon Cytokine Res. 31, 79–87 [DOI] [PubMed] [Google Scholar]
- 2. Verhelst J., Hulpiau P., and Saelens X. (2013) Mx proteins: antiviral gatekeepers that restrain the uninvited. Microbiol. Mol. Biol. Rev. 77, 551–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haller O., and Kochs G. (2002) Interferon-induced Mx proteins: dynamin-like GTPases with antiviral activity. Traffic 3, 710–717 [DOI] [PubMed] [Google Scholar]
- 4. Praefcke G. J., and McMahon H. T. (2004) The dynamin superfamily: universal membrane tubulation and fission molecules? Nat. Rev. Mol. Cell Biol. 5, 133–147 [DOI] [PubMed] [Google Scholar]
- 5. Gao S., von der Malsburg A., Dick A., Faelber K., Schröder G. F., Haller O., Kochs G., and Daumke O. (2011) Structure of myxovirus resistance protein a reveals intra- and intermolecular domain interactions required for the antiviral function. Immunity 35, 514–525 [DOI] [PubMed] [Google Scholar]
- 6. Pitossi F., Blank A., Schröder A., Schwarz A., Hüssi P., Schwemmle M., Pavlovic J., and Staeheli P. (1993) A functional GTP-binding motif is necessary for antiviral activity of Mx proteins. J. Virol. 67, 6726–6732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao S., von der Malsburg A., Paeschke S., Behlke J., Haller O., Kochs G., and Daumke O. (2010) Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature 465, 502–506 [DOI] [PubMed] [Google Scholar]
- 8. Mitchell P. S., Patzina C., Emerman M., Haller O., Malik H. S., and Kochs G. (2012) Evolution-guided identification of antiviral specificity determinants in the broadly acting interferon-induced innate immunity factor MxA. Cell Host Microbe 12, 598–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patzina C., Haller O., and Kochs G. (2014) Structural requirements for the antiviral activity of the human MxA protein against Thogoto and influenza A virus. J. Biol. Chem. 289, 6020–6027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Verhelst J., De Vlieger D., and Saelens X. (2015) Co-immunoprecipitation of the mouse Mx1 protein with the influenza A virus nucleoprotein. J. Vis. Exp. 98, e52871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nigg P. E., and Pavlovic J. (2015) Oligomerization and GTP-binding requirements of MxA for viral target recognition and antiviral activity against influenza A virus. J. Biol. Chem. 290, 29893–29906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dittmann J., Stertz S., Grimm D., Steel J., García-Sastre A., Haller O., and Kochs G. (2008) Influenza A virus strains differ in sensitivity to the antiviral action of Mx-GTPase. J. Virol. 82, 3624–3631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mänz B., Schwemmle M., and Brunotte L. (2013) Adaptation of avian influenza A virus polymerase in mammals to overcome the host species barrier. J. Virol. 87, 7200–7209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Verhelst J., Parthoens E., Schepens B., Fiers W., and Saelens X. (2012) Interferon-inducible protein Mx1 inhibits influenza virus by interfering with functional viral ribonucleoprotein complex assembly. J. Virol. 86, 13445–13455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zimmermann P., Mänz B., Haller O., Schwemmle M., and Kochs G. (2011) The viral nucleoprotein determines Mx sensitivity of influenza A viruses. J. Virol. 85, 8133–8140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang T., Pavlovic J., Staeheli P., and Krystal M. (1992) Overexpression of the influenza virus polymerase can titrate out inhibition by the murine Mx1 protein. J. Virol. 66, 4154–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stranden A. M., Staeheli P., and Pavlovic J. (1993) Function of the mouse Mx1 protein is inhibited by overexpression of the PB2 protein of influenza virus. Virology 197, 642–651 [DOI] [PubMed] [Google Scholar]
- 18. Engelhardt O. G., and Fodor E. (2006) Functional association between viral and cellular transcription during influenza virus infection. Rev. Med. Virol. 16, 329–345 [DOI] [PubMed] [Google Scholar]
- 19. Verhelst J., Spitaels J., Nürnberger C., De Vlieger D., Ysenbaert T., Staeheli P., Fiers W., and Saelens X. (2015) Functional comparison of Mx1 from two different mouse species reveals the involvement of loop L4 in the antiviral activity against influenza A viruses. J. Virol. 89, 10879–10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bayle J. H., Grimley J. S., Stankunas K., Gestwicki J. E., Wandless T. J., and Crabtree G. R. (2006) Rapamycin analogs with differential binding specificity permit orthogonal control of protein activity. Chem. Biol. 13, 99–107 [DOI] [PubMed] [Google Scholar]
- 21. Stankunas K., Bayle J. H., Havranek J. J., Wandless T. J., Baker D., Crabtree G. R., and Gestwicki J. E. (2007) Rescue of degradation-prone mutants of the FK506-rapamycin binding (FRB) protein with chemical ligands. Chembiochem 8, 1162–1169 [DOI] [PubMed] [Google Scholar]
- 22. Edwards S. R., and Wandless T. J. (2007) The rapamycin-binding domain of the protein kinase mammalian target of rapamycin is a destabilizing domain. J. Biol. Chem. 282, 13395–13401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pavlovic J., Haller O., and Staeheli P. (1992) Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J. Virol. 66, 2564–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]