

Abstract
The hormone prolactin (PRL) contributes to breast cancer pathogenesis through various signaling pathways, one of the most notable being the JAK2/signal transducer and activator of transcription 5 (STAT5) pathway. PRL-induced activation of the transcription factor STAT5 results in the up-regulation of numerous genes implicated in breast cancer pathogenesis. However, the molecular mechanisms that enable STAT5 to access the promoters of these genes are not well understood. Here, we show that PRL signaling induces chromatin decompaction at promoter DNA, corresponding with STAT5 binding. The chromatin-modifying protein high mobility group nucleosomal binding domain 2 (HMGN2) specifically promotes STAT5 accessibility at promoter DNA by facilitating the dissociation of the linker histone H1 in response to PRL. Knockdown of H1 rescues the decrease in PRL-induced transcription following HMGN2 knockdown, and it does so by allowing increased STAT5 recruitment. Moreover, H1 and STAT5 are shown to function antagonistically in regulating PRL-induced transcription as well as breast cancer cell biology. While reduced STAT5 activation results in decreased PRL-induced transcription and cell proliferation, knockdown of H1 rescues both of these effects. Taken together, we elucidate a novel mechanism whereby the linker histone H1 prevents STAT5 binding at promoter DNA, and the PRL-induced dissociation of H1 mediated by HMGN2 is necessary to allow full STAT5 recruitment and promote the biological effects of PRL signaling.
Keywords: breast cancer, chromatin remodeling, prolactin, STAT transcription factor, transcription regulation, linker histone H1, non-histone chromosomal protein HMG-17 (HMGN2), prolactin receptor, signal transducers and activators of transcription 5 (STAT5)
Introduction
In the mammary gland, signals from the polypeptide hormone prolactin (PRL) are essential for normal development (1–4), and these physiological effects are mediated through the homologous transcription factors signal transducer and activator of transcription 5a and 5b (referred to as STAT5) (5). PRL signals by binding to the PRL receptor (PRLR),2 a transmembrane receptor of the cytokine receptor superfamily. Binding of PRL to the PRLR results in the activation of STAT5 via phosphorylation by the tyrosine kinase Janus kinase 2 (JAK2) (6–8). Phosphorylated STAT5 dimerizes, and the active STAT5 dimers translocate to the nucleus. In the nucleus, STAT5 recognizes and binds to consensus elements in the DNA, which induces the expression of STAT5 target genes (9).
PRL-induced STAT5 activation results in the up-regulation of many pro-proliferative genes (10–12). Although essential for proper mammary gland development, aberrant PRL signaling and STAT5 activation also contribute to breast cancer pathogenesis. Transgenic mice that overexpress PRL in the mammary epithelium develop mammary tumors, which can be either estrogen receptor-positive or -negative (13). In epidemiologic studies, women with elevated serum PRL are at an increased risk of developing breast cancer (14–17). PRL also enhances the proliferation, motility, and survival of breast cancer cells (18–20). Similarly, overexpression of STAT5 in the mammary gland of transgenic mice results in mammary tumor development (21), whereas hemizygous loss of Stat5a delays cancer progression driven by simian virus 40 T antigen in the mouse mammary gland (22). In human breast cancer cell lines, STAT5 promotes cell survival and anchorage-independent growth (23). Given the role of PRL and STAT5 in breast cancer pathogenesis, the factors regulating PRL-induced, STAT5-mediated gene expression merit close investigation.
Although STAT5 activation is critical in mediating the effects of PRL, transcription factor activation alone is not enough to translate cellular signals into the proper gene expression patterns. Upon activation, transcription factors only bind to a small fraction of their consensus elements in the genome, typically occupying <1% of their potential binding sites based on DNA sequence alone (24). Therefore, a significant question remaining in the study of transcriptional regulation is how certain consensus elements become accessible for transcription factor binding, whereas others remain unbound. Chromatin structure appears to be a major determinant of transcription factor binding patterns. In genome-wide studies, transcription factor consensus elements that are located within accessible chromatin, marked by DNase I hypersensitivity or active histone modifications, are preferentially bound by their respective transcription factors following activation (24–26). In the mammary epithelium, chromatin accessibility plays a distinct role in determining cell type-specific and context-specific patterns of transcription factor binding and gene expression. STAT5-regulated genes that are expressed mainly in the mammary epithelium are not recognized by STAT5 in cell types where these genes are not expressed (27). These findings indicate that STAT5 loci exhibit cell type-specific patterns of accessibility, enabling STAT5 binding only in the proper context and cell type. Correspondingly, mammary-specific genes have been shown to exhibit an open chromatin organization that is specific not just to mammary tissue but also to the appropriate developmental stage (28). One study has shown that DNA methylation in particular is associated with impaired STAT5 recruitment (29). However, additional chromatin-remodeling events that enable or prevent STAT5 recruitment have not been well characterized.
Our laboratory has previously shown that the chromatin-modifying protein high mobility group nucleosomal binding domain 2 (HMGN2) promotes PRL-induced, STAT5-mediated transcription (30). Following PRL stimulation, HMGN2 is recruited to the promoter of the cytokine-inducible SH2-containing protein (CISH) gene, where HMGN2 facilitates STAT5 recruitment (30). However, the mechanism by which HMGN2 facilitates STAT5 recruitment had not been identified. HMGN proteins are non-histone chromosomal proteins that bind to nucleosomes and induce structural changes in chromatin (31). In particular, HMGN proteins have been shown to compete with the linker histone H1 for binding sites on chromatin, antagonizing the chromatin-condensing activity of H1 (32). In this study, we hypothesized that PRL treatment induces chromatin remodeling at the promoters of PRL-responsive genes via HMGN2, thus enabling STAT5 binding and transcriptional activation. Here, we show that PRL treatment results in the displacement of both a well positioned nucleosome and the linker histone H1 at STAT5 consensus elements in the CISH promoter. HMGN2 facilitates the displacement of H1, and H1 loss is necessary to allow full STAT5 binding and transcriptional activation. Finally, H1 was found to antagonize STAT5 in promoting the proliferation of breast cancer cells. This is the first report to identify H1 occupancy as a critical regulator of STAT5-mediated transcription, specifically linking chromatin remodeling to STAT5 recruitment and biological function.
Results
STAT5 Inhibition Results in Decreased Proliferation of Breast Cancer Cells
Because the transcription factor STAT5 is a critical mediator of PRL-induced signaling, we first assessed the role of STAT5 in mediating the biological effects of PRL in breast cancer cells. T47D breast cancer cells were chosen based on their high endogenous levels of PRLR expression (18). To assess the role of STAT5, a chemical inhibitor of STAT5 (CAS 285986-31-4) was utilized (33). This compound prevents STAT5 phosphorylation and DNA binding by selectively targeting the Src homology 2 domain of STAT5, which mediates protein-protein interactions of STAT5 both with activated receptors and in dimer formation (33). In the absence of STAT5 inhibition, PRL treatment resulted in phosphorylation of STAT5 in the cytoplasmic fraction and increased levels of both phosphorylated and total STAT5 in the nuclear fraction, indicative of nuclear translocation of the activated protein (Fig. 1A). Treatment with the STAT5 inhibitor diminished these effects, resulting in reduced phosphorylation and reduced nuclear translocation of STAT5 in response to PRL treatment (Fig. 1A). The effect of STAT5 inhibition on cell proliferation was next assessed by BrdU incorporation. In the absence of STAT5 inhibition, PRL treatment resulted in increased cell proliferation, as has been reported previously (18). Treatment with the STAT5 inhibitor resulted in decreased cell proliferation in a dose-dependent manner and reduced the ability of PRL to stimulate proliferation (Fig. 1B). These results confirm that STAT5 plays a key role in breast cancer cell proliferation and further implicate STAT5 as a critical mediator of the biological effects of PRL.
FIGURE 1.

STAT5 inhibition results in decreased proliferation of breast cancer cells. A, effectiveness of chemical inhibition of STAT5 in preventing PRL-induced STAT5 phosphorylation and nuclear translocation. T47D cells were pretreated with the STAT5 inhibitor CAS 285986-31-4 or DMSO as the vehicle control (VC) for 1 h before stimulation with PRL. Nuclear and cytoplasmic lysates were isolated, and activation of STAT5 was analyzed by Western blotting using an antibody against phosphorylated (p-) STAT5. The membranes were then stripped and reprobed with an antibody against total STAT5. Histone H4 and tubulin were used as loading controls for the nuclear and cytoplasmic fractions, respectively. B, STAT5 inhibition reduces cell proliferation and prevents the PRL-induced increase in proliferation. T47D cells were treated with the indicated concentrations of STAT5 inhibitor or vehicle control, with or without PRL, for 3 days. BrdU incorporation was measured by absorbance as an indication of cell proliferation. Results are presented as the mean ± S.E. (error bars) of three independent experiments. Within each individual experiment, each set of treatment conditions was carried out in triplicate. Statistical significance was determined by two-sided t test assuming equal sample variance, comparing without versus with PRL (No PRL versus +PRL) at each concentration of STAT5 inhibitor. *, p ≤ 0.05; **, p ≤ 0.01.
PRL Treatment Induces Chromatin Decompaction and Promotes Binding of the Transcriptional Machinery
Because STAT5 is an important mediator of the biological effects of PRL, we sought to elucidate the molecular mechanisms regulating STAT5 recruitment and PRL-induced gene expression. We first assessed whether PRL signaling induces chromatin remodeling at the promoters of immediate early genes or whether PRL-induced genes are poised with an open chromatin structure and bound RNA polymerase II (RNAPII). To begin the analyses, we focused on the PRL-induced immediate early gene CISH. CISH has been shown to be overexpressed in breast cancer and stimulates proliferation by activating the ERK pathway (34). STAT5-induced CISH transcription has been well characterized (10, 30, 35), making it a robust model for initial analyses. To assess chromatin compaction at the CISH promoter, various histone proteins were assessed by ChIP over a time course of PRL treatment. The region surrounding STAT5 consensus elements, termed γ-interferon-activating sequence motifs, in the CISH proximal promoter was assayed (see Fig. 3B for a map of the CISH promoter). PRL treatment resulted in a loss of histone H3 by 30 min of treatment with a maximum loss at 45 min (Fig. 2A). To corroborate these results, the presence of histone H4 was also determined by ChIP and was likewise decreased after 45 min of PRL treatment (Fig. 2B). To further assess the effect of PRL signaling on chromatin compaction, the linker histone H1 was analyzed. The H1 subtype H1.2 was selected for these studies because H1.2 has been shown to play a specific role in regulating the cell cycle progression of breast cancer cells, in particular by promoting gene repression (36–41). Concordantly, PRL treatment resulted in a loss of histone H1 from the CISH promoter (Fig. 2C). These results indicate that PRL signaling induces chromatin decompaction at target promoter DNA through the dissociation of both nucleosome core particles and linker histones.
FIGURE 3.

PRL treatment results in increased chromatin accessibility at STAT5-binding sites. A, schematic of the qPCR amplicons used for the MNase protection assay at the CISH promoter. Amplification indicates that the DNA region was protected from MNase digestion, suggesting that it was nucleosome-bound. B, PRL treatment results in nucleosome remodeling at the CISH promoter. Top, T47D cells were treated with or without PRL for 1 h. Nuclei were permeabilized, and the chromatin was digested with MNase. Mononucleosomal DNA was purified, and MNase protection was determined by qPCR using amplicons tiling across the CISH promoter (A). Enrichment of mononucleosomal DNA was calculated relative to amplification of undigested genomic DNA. Values are plotted at the midpoint of each amplicon. Results are presented as the mean ± S.E. (error bars) of three independent experiments. Statistical significance was determined by two-way repeated measures ANOVA. The shaded region indicates statistical significance between the PRL-treated and untreated conditions, p ≤ 0.05. Bottom, diagram of the presumed nucleosome core particle positions at the CISH promoter indicated by the MNase protection assay above, assuming ∼150 bp of protection per nucleosome core particle. STAT5 consensus elements within the CISH promoter are indicated, corresponding to the axis positions shown above. C, diagram of CISH luciferase reporter constructs with sequential truncation of the distal promoter. Distances are relative to the CISH TSS. D, T47D cells were transfected with the CISH luciferase reporter constructs in C along with a Renilla luciferase control vector. Luciferase readings were normalized to the Renilla luciferase internal control, and -fold change was calculated relative to the −975 bp construct without PRL treatment. Results are presented as the mean ± S.E. of three independent experiments. Within each individual experiment, each transfection was carried out in triplicate, and each sample was read in duplicate. Statistical significance was determined by two-way repeated measures ANOVA. RLU, relative luciferase units. **, p ≤ 0.01 for −975 bp versus −84 bp, with PRL treatment. n.s., p > 0.05 for −975 bp versus all other constructs, with PRL treatment.
FIGURE 2.

PRL treatment induces chromatin decompaction and promotes binding of the transcriptional machinery. A–F, ChIP-qPCR analysis of the CISH promoter following a time course of PRL treatment. Nuclear lysates were precipitated with antibodies against H3 (A), H4 (B), H1 (C), RNAPII (D), phosphorylated (p-) RNAPII (E), or STAT5 (F). Normal IgG served as a control for nonspecific binding. For all histones and STAT5, primers amplify the region of the CISH promoter from −81 to −9 bp relative to the TSS, which includes STAT5 consensus elements. For RNAPII and phospho-RNAPII, primers amplify the region from +46 to +132 bp of the CISH TSS. See Fig. 3B for a map of the CISH promoter. The amount of DNA recovered was calculated relative to the input control and is graphed as a percentage of input. Results are presented as the mean ± S.E. (error bars), n ≥ 3 independent experiments. Statistical significance was determined by a two-sided ratio paired t test, comparing PRL treatment time points with the untreated sample. STAT5 ChIP was analyzed by two-sided paired differences t test. p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
We next assessed the recruitment of components of the basal transcription machinery in response to PRL. Consistent with the time course of chromatin decompaction identified above, PRL treatment resulted in increased binding of RNAPII, with a maximum response at 45 min of treatment (Fig. 2D). RNAPII was present in the active phosphorylated form (Fig. 2E). Moreover, these responses correlated with the time course of STAT5 binding at CISH (Fig. 2F). The open chromatin architecture identified in response to PRL signaling is thus associated with increased binding of the basal transcription machinery. These results imply a role for chromatin remodeling in regulating PRL-induced transcriptional activation of CISH.
PRL Treatment Results in Increased Chromatin Accessibility at STAT5-binding Sites
Given the observed chromatin decompaction at the CISH promoter, we next evaluated how PRL signaling affects the nucleosome landscape by mapping the CISH promoter using a nucleosome-scanning assay. Chromatin was digested with micrococcal nuclease (MNase), mononucleosomal DNA was isolated, and protected regions were amplified by quantitative real-time PCR (qPCR) using overlapping amplicons spanning across the CISH promoter (about −500 to +500 bp; Fig. 3A). This region was chosen to allow for detection of at least one full nucleosome both upstream and downstream of the STAT5 binding sites analyzed by ChIP (Fig. 2). Before PRL treatment, the analyzed region of the CISH promoter exhibited three regions of protection, indicative of a −2, −1, and +1 nucleosome (Fig. 3B). The −1 nucleosome, located upstream of the transcription start site (TSS), was positioned overlying four STAT5 consensus elements (Fig. 3B, bottom). PRL treatment resulted in decreased protection of this region, indicative of eviction of the −1 nucleosome in a substantial fraction of the treated cell population (Fig. 3B; also see Fig. 6D). The −2 nucleosome also exhibited chromatin remodeling following PRL treatment, whereas the +1 nucleosome was insensitive to PRL (Fig. 3B). These results suggest that PRL-induced chromatin remodeling at the CISH promoter results in increased accessibility specifically at the STAT5 consensus elements, presumably allowing increased STAT5 binding following activation.
FIGURE 6.

HMGN2 facilitates the loss of histone H1 but does not affect nucleosome core particle remodeling. A–C, HMGN2 knockdown impairs H1 dissociation and RNAPII loading at CISH but does not affect the dissociation of H3. T47D cells expressing shCTL or shHMGN2 were treated with or without PRL for 45 min and were analyzed by ChIP-qPCR at the CISH promoter. Nuclear lysates were precipitated with antibodies against H3 (A), H1 (B), or RNAPII (C). Normal IgG served as a nonspecific control. CISH primers are described in the legends to Figs. 2 and 5A. The amount of DNA recovered was calculated relative to the input control and is graphed as a percentage of input. Results are presented as the mean ± S.E. (error bars), n ≥ 3 independent experiments. Statistical significance was determined by two-way repeated measures ANOVA. D, HMGN2 knockdown does not affect nucleosome core particle positioning by MNase accessibility. T47D cells expressing shCTL or shHMGN2 were treated with or without PRL for 1 h. Nuclei were permeabilized, and the chromatin was digested with MNase. Mononucleosomal DNA was purified, and MNase protection was determined by qPCR using amplicons tiling across the CISH promoter (Fig. 3A). Enrichment of mononucleosomal DNA was calculated relative to amplification of undigested genomic DNA. Values are plotted at the midpoint of each amplicon. Results are presented as the mean ± S.E. of three independent experiments. Statistical significance was determined by two-way repeated measures ANOVA. The shaded region indicates statistical significance between the PRL-treated and untreated conditions, p ≤ 0.05. The shCTL and shHMGN2 conditions were not significantly different. E, HMGN2 overexpression in T47D cells. Shown is Western blotting analysis of cells stably infected to express exogenous HMGN2 with either a C-terminal (HMGN2-C) or N-terminal FLAG tag (HMGN2-N) or an empty vector (EV) control. Nuclear lysates were probed with an antibody against HMGN2, and histone H4 was used as a loading control. FLAG-tagged HMGN2 runs slightly higher than endogenous HMGN2. F and G, HMGN2 overexpression does not further promote STAT5 binding or H1 dissociation at CISH. Cells from E were analyzed by ChIP-qPCR as in A–C, using antibodies against STAT5 (F) or H1 (G). Results are presented as the mean ± S.E. of three independent experiments. Statistical significance was determined by two-way repeated measures ANOVA. n.s., p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
Because PRL treatment resulted in chromatin remodeling upstream of the STAT5 binding sites as well, we sought to determine whether there are other important regulatory elements in this region that enhance or repress PRL-induced CISH transcription. To this end, a Dual-Luciferase reporter assay was utilized. Our laboratory has previously shown that PRL induces transcription from a luciferase reporter containing ∼1 kb of the CISH proximal promoter sequence (10). Sequential truncations of this CISH luciferase reporter were created to identify regions critical for CISH transcription (Fig. 3C). The reporter was first truncated to −503 bp to correlate with the region analyzed in the MNase protection assay, followed by smaller successive truncations. PRL treatment induced high expression from the full-length reporter construct (−975 bp; Fig. 3D). With increasing truncation of the distal promoter sequence, PRL-induced reporter expression remained at a similarly high level, up to and including the −226 bp construct (Fig. 3D). These data suggest that the transcription factor binding sites critical for PRL-induced transcription are contained within the 226 bp upstream of the CISH TSS because truncation of the upstream sequence had no significant effect on reporter expression. This critical region contains all four of the STAT5 consensus elements discussed above. The smallest reporter sequence tested (−84 bp), which only contains two of the STAT5 consensus elements, displayed high baseline activity but no further induction by PRL treatment (Fig. 3D). From these data, the more distal STAT5 consensus elements appear to be necessary for the proper coordination of PRL-induced CISH transcription, which may well involve cooperation between all four STAT5 consensus elements.
HMGN2 Promotes the Transcription of STAT5 Target Genes
Despite the importance of STAT5 as a transcription factor, the chromatin remodeling events that enable STAT5 binding have not been well characterized. Our laboratory has shown previously that the chromatin-modifying protein HMGN2 promotes PRL-induced STAT5 binding at CISH (30). To determine the mechanism by which HMGN2 promotes STAT5 binding, T47D cells stably expressing short hairpin RNA targeting HMGN2 (shHMGN2) or a nonspecific control (shCTL) were utilized (Fig. 4A). In shHMGN2-expressing cells, HMGN2 was depleted by nearly 90% on average by Western blotting analysis. Moreover, HMGN2 knockdown resulted in decreased binding of HMGN2 at the CISH promoter (Fig. 4B). The effect of HMGN2 knockdown on expression from the CISH luciferase reporter truncations was assessed. HMGN2 knockdown resulted in decreased expression from the −975 bp reporter as well as the reporters containing truncations of the CISH promoter, up to and including the −226 bp construct (Fig. 4C). These data suggest that HMGN2 mediates CISH transcription specifically through the proximal 226 bp of the promoter, containing four STAT5 binding sites.
FIGURE 4.

HMGN2 promotes the transcription of STAT5 target genes. A, HMGN2 knockdown in T47D cells. Shown is Western blotting analysis of cells stably infected with shRNA vectors targeting HMGN2 (shHMGN2) or a nonspecific control (shCTL). Whole cell lysates were probed with an antibody against HMGN2, and tubulin was used as a loading control. B, HMGN2 knockdown results in decreased binding at CISH. T47D shCTL and shHMGN2 cells from A were analyzed by ChIP-qPCR. Nuclear lysates were precipitated with an antibody against HMGN2, and normal IgG served as a control for nonspecific binding. Primers amplify the region of the CISH promoter from −81 to −9 bp. Recovered DNA is graphed as a percentage of input. Results are presented as the mean ± S.E. (error bars), n ≥ 3 independent experiments. Statistical significance was determined by a two-sided ratio paired t test. C, T47D shCTL and shHMGN2 cells were transfected with the CISH luciferase reporter constructs in Fig. 3C along with a Renilla luciferase control vector. Luciferase readings were normalized to the Renilla luciferase internal control, and -fold change was calculated relative to the −975 bp construct with PRL treatment. Results are presented as the mean ± S.E. of three independent experiments. Within each individual experiment, each transfection was carried out in triplicate, and each sample was read in duplicate. Statistical significance was determined by two-way repeated measures ANOVA. RLU, relative luciferase units. D, STAT5 mediates the PRL-induced expression of CISH, IER3, and PHLDA2. T47D cells were pretreated with the STAT5 inhibitor CAS 285986-31-4 or DMSO as the VC for 1 h before treatment with PRL for 1 h. RNA was isolated and analyzed by qRT-PCR for the genes indicated. -Fold change was calculated relative to VC with no PRL treatment. Results are presented as the mean ± S.E., n ≥ 3 independent experiments. Statistical significance was determined by a two-sided ratio paired t test, comparing STAT5 inhibition versus VC with PRL treatment. E, HMGN2 promotes the expression of CISH, IER3, and PHLDA2. T47D shCTL and shHMGN2 cells were treated with PRL for the indicated times. RNA was isolated and analyzed by qRT-PCR for the genes indicated. -Fold change was calculated relative to shCTL with no PRL treatment. Results are presented as the mean ± S.E., n ≥ 3 independent experiments. Statistical significance was determined by a two-sided ratio paired t test, comparing shCTL versus shHMGN2 at each time point. *, p ≤ 0.05; **, p ≤ 0.01; ****, p ≤ 0.0001.
We next asked whether the function of HMGN2 generalizes to other PRL-induced STAT5 target genes in addition to CISH. PRL-induced genes were previously identified by our laboratory through global RNA profiling (42, 43), and the genes immediate early response 3 (IER3) and pleckstrin homology-like domain, family A, member 2 (PHLDA2) were chosen for further analysis based on their breast cancer relevance (see “Discussion”). PRL treatment resulted in increased expression of CISH, IER3, and PHLDA2 here (Fig. 4, D and E). Furthermore, treatment with the STAT5 inhibitor prevented the PRL-induced expression of CISH, IER3, and PHLDA2 in a dose-dependent manner (Fig. 4D), confirming that these genes are regulated by STAT5. We next assessed whether HMGN2 facilitates the STAT5-mediated expression of these genes. HMGN2 knockdown resulted in decreased expression of CISH, IER3, and PHLDA2 in response to PRL treatment (Fig. 4E). These data indicate that HMGN2 plays a key role in facilitating PRL-induced, STAT5-mediated gene expression.
HMGN2 Affects STAT5 Chromatin Accessibility
We next sought to address whether HMGN2 promotes STAT5 binding at the IER3 and PHLDA2 gene promoters as it does at CISH (Fig. 5A) (30). To this end, we searched the promoter regions of IER3 and PHLDA2 using the Sequence Manipulation Suite (44) to identify STAT5 consensus elements (TTCnnnGAA). The region from 5 kb upstream to 1 kb downstream of the TSS was chosen for analysis because it has been shown that in mouse mammary tissue, over half of the genes whose expression is functionally regulated by STAT5 are bound by STAT5 within this region (27). The analysis of these promoters identified one STAT5 consensus element for IER3 and two for PHLDA2; the analyzed region for IER3 was then extended to 10 kb upstream of the TSS, which yielded a second site. PRL-induced STAT5 recruitment to these sites was assessed by ChIP-qPCR (Fig. 5B). Whereas strong PRL-induced STAT5 recruitment was identified at CISH, none of the analyzed sites within the IER3 or PHLDA2 promoters exhibited significant STAT5 recruitment (Fig. 5B). In examining the ENCODE ChIP sequencing database, STAT5 binding has been observed within the ∼600 bp surrounding the TSS of IER3 in hematopoietic cell lines (45, 46). Based on this observation, additional sites within this proximal region of IER3 as well as PHLDA2 were assessed for STAT5 enrichment by ChIP-qPCR. Again, no notable recruitment of STAT5 was observed at these sites (Fig. 5B). These results lead us to conclude that, although STAT5 regulates the expression of IER3 and PHLDA2, the location of STAT5 recruitment probably occurs either at a non-canonical binding site or outside of the proximal 5–10 kb of the promoter sequence. Because HMGN2 knockdown resulted in decreased IER3 and PHLDA2 expression, HMGN2 may likewise promote STAT5 binding at such sites.
FIGURE 5.

HMGN2 affects STAT5 chromatin accessibility. A, HMGN2 knockdown results in decreased STAT5 binding at CISH. T47D cells expressing shCTL or shHMGN2 were treated with or without PRL for 45 min and analyzed by ChIP-qPCR. Nuclear lysates were precipitated with an antibody against STAT5, and normal IgG served as a control for nonspecific binding. Primers amplify the region of the CISH promoter from −81 to −9 bp, which includes STAT5 consensus elements. Recovered DNA is graphed as a percentage of input. Results are presented as the mean ± S.E. (error bars) of three independent experiments. Statistical significance was determined by two-way repeated measures ANOVA. *, p ≤ 0.05. B, STAT5 is probably not recruited to the promoter region of IER3 or PHLDA2. T47D cells were analyzed by ChIP-qPCR for STAT5 recruitment following 45 min of PRL treatment. The x axis labels indicate the location of each qPCR amplicon relative to the TSS of the indicated gene. All amplicons target STAT5 consensus elements, except IER3 −0.2 kb, IER3 TSS, and PHLDA2 +0.1 kb. CISH primers are described in A. Results are presented as the mean ± S.E. of three independent experiments. Statistical significance was determined by a two-sided t test assuming equal sample variance. ****, p ≤ 0.0001 comparing without and with PRL (No PRL and +PRL) at CISH. No other regions exhibited significant STAT5 enrichment. C, HMGN2 knockdown does not affect STAT5 activation, as indicated by STAT5 phosphorylation and nuclear translocation. T47D cells expressing shCTL or shHMGN2 were treated with or without PRL for 45 min. Nuclear lysates were isolated and analyzed by Western blotting using an antibody against phosphorylated (p-) STAT5. The membrane was stripped and reprobed with an antibody against total STAT5. Knockdown of HMGN2 was verified, and histone H4 was used as a loading control. D, HMGN2 knockdown does not affect global chromatin accessibility. Nuclei from T47D shCTL and shHMGN2 cells were permeabilized, and the chromatin was digested with MNase for the indicated times. Purified DNA was analyzed by agarose gel electrophoresis.
Next, we tested the hypothesis that HMGN2 promotes STAT5 recruitment by regulating the access of STAT5 to the DNA. First, the effect of HMGN2 on STAT5 activation was assessed. Notably, HMGN2 knockdown did not affect PRL-induced STAT5 activation, because nuclear localization and phosphorylation of STAT5 were not affected by HMGN2 knockdown (Fig. 5C). We next assessed whether HMGN2 knockdown had a global effect on chromatin accessibility. Global chromatin accessibility to MNase was not affected by HMGN2 knockdown, because cells with or without HMGN2 knockdown released mononucleosomal DNA to a similar extent throughout a time course of digestion (Fig. 5D). These results suggest that HMGN2 affects STAT5 accessibility to the chromatin through local, gene-specific effects.
HMGN2 Facilitates the Loss of Histone H1 but Does Not Affect Nucleosome Core Particle Remodeling
The contribution of HMGN2 to the PRL-induced chromatin decompaction identified in Fig. 2 was next assessed. HMGN2 knockdown did not affect the PRL-induced dissociation of nucleosome core particles, as determined by ChIP for H3 at CISH (Fig. 6A). Because PRL treatment also resulted in dissociation of the linker histone H1 from the CISH promoter (Fig. 2C), we determined whether HMGN2 was involved in this displacement. HMGN2 knockdown did impair the dissociation of H1, because H1 occupancy was maintained following PRL treatment (Fig. 6B). HMGN2 knockdown also resulted in decreased loading of RNAPII at CISH (Fig. 6C). Together, these data suggest that HMGN2 promotes STAT5 binding and RNAPII loading by facilitating the eviction of H1 from the CISH promoter.
To address the possibility that HMGN2 may affect nucleosome positioning without affecting total levels of H3, the MNase protection assay was carried out following HMGN2 knockdown, covering the region upstream of the CISH TSS, which had previously exhibited PRL-induced chromatin remodeling (Fig. 3B). However, HMGN2 knockdown did not affect chromatin remodeling by MNase protection (Fig. 6D). Of note, this assay would not be expected to identify changes in H1 occupancy, because the additional protection imparted by linker histones when bound to the core particle (∼20 bp) is below the level of resolution of this amplicon-based assay. In addition, because mononucleosomal DNA is isolated before the amplification step, only DNA regions bound to a core particle are included in the analysis. Taken together, these data imply that HMGN2 most likely acts to facilitate STAT5 binding without affecting nucleosome core particle positioning or eviction.
To complement the results for HMGN2 knockdown, the effect of HMGN2 overexpression on STAT5 and H1 binding was investigated. Experiments were carried out using T47D cells stably overexpressing one of two different HMGN2 constructs, with either a C-terminal or N-terminal FLAG tag, to minimize potential structural effects from adding a tag (Fig. 6E). HMGN2 was overexpressed within the nuclear lysate by an average of 80 and 50%, respectively, by Western blotting analysis. However, HMGN2 overexpression had no further effect on PRL-induced STAT5 binding (Fig. 6F) or H1 dissociation at CISH (Fig. 6G). These results indicate that the endogenous protein levels of HMGN2 in T47D cells are not rate-limiting in these processes.
HMGN2 Does Not Affect Post-translational Histone Modifications
Because HMGN2 can affect histone acetylation (47), ChIP for acetylated histone H3K9 and H3K14 at the CISH promoter was performed upon HMGN2 knockdown. Increased levels of H3K9 and H3K14 acetylation were observed at the CISH promoter with PRL treatment (Fig. 7A). However, HMGN2 knockdown had no significant effect on H3K9 or H3K14 acetylation (Fig. 7A).
FIGURE 7.

HMGN2 does not affect post-translational histone modifications. A, HMGN2 does not affect histone acetylation at H3K9 or H3K14. T47D cells expressing shCTL or shHMGN2 were treated with or without PRL for 45 min and were analyzed by ChIP-qPCR at the CISH promoter. Nuclear lysates were precipitated with antibodies against acetylated H3K9 (H3K9Ac), H3K14 (H3K14Ac), or total H3. CISH primers are described in the legends to Figs. 2 and 5A. The percentage of input DNA recovered with the acetyl-specific antibodies was normalized to the percentage of input recovered for total H3 in a parallel sample. Results are presented as the mean ± S.E. (error bars), n ≥ 3 independent experiments. Statistical significance was determined by two-way repeated measures ANOVA. B, the CISH promoter is not enriched for histone trimethylation at H3K9 or H3K27. ChIP-qPCR analysis was carried out as in A using antibodies against H3K9me3 or H3K27me3. Positive (+) controls for enrichment are ZNF554 for H3K9me3 and α-Satellite for H3K27me3 (EMD Millipore). The amount of DNA recovered was calculated relative to the input control and is graphed as a percentage of input. Results are presented as the mean ± S.E. of three independent experiments. Statistical significance was determined by two-way ANOVA. n.s., p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01.
Binding of H1.2 has been associated with the repressive histone marks H3K9 trimethylation (H3K9me3) and H3K27me3 in certain contexts (40, 48, 49). Therefore, ChIP for H3K9me3 and H3K27me3 at the CISH promoter was performed upon HMGN2 knockdown. Neither histone methylation mark was significantly enriched at CISH under the conditions tested, despite significant pull-down of positive control regions (Fig. 7B). These data imply that HMGN2 most likely acts to facilitate CISH transcription without affecting post-translational histone modifications.
HMGN2 Promotes Histone H1 Loss to Regulate STAT5 Binding and CISH Transcription
Given these results, we hypothesized that the decrease in PRL-induced gene expression following HMGN2 knockdown is mediated by and specific to the inability to remove H1 in the absence of HMGN2. To test this hypothesis, we examined whether H1 knockdown would rescue gene expression following HMGN2 knockdown. For these experiments, the control and HMGN2 knockdown stable cells were transiently transfected with siRNA targeting H1, subtype H1.2 (siH1; Fig. 8A). siH1 sequences 1 and 2 resulted in an average of 66 and 60% knockdown, respectively, by Western blotting analysis. Cells were then assessed for PRL-induced transcription of CISH, IER3, and PHLDA2. For all three genes, the decrease in expression induced by HMGN2 knockdown was rescued by the additional knockdown of histone H1, and these results were confirmed using two different siRNA sequences (Fig. 8B). These data suggest that the decrease in PRL-induced expression following HMGN2 knockdown is due to the continued presence of H1, presumably preventing STAT5 from accessing its binding sites. Of note, H1 knockdown did not appreciably affect the baseline mRNA expression in cells without PRL treatment, indicating that the activation of STAT5 is necessary for transcriptional activation even after H1 knockdown (Fig. 8B).
FIGURE 8.

HMGN2 promotes histone H1 loss to regulate STAT5 binding and gene transcription. A, efficiency of H1.2 knockdown. T47D cells were transiently transfected with siRNA targeting H1.2 using two different sequences (siH1.2 #1 and #2) or a nonspecific control (siCTL). Whole cell lysates were analyzed by Western blotting and probed with antibodies against H1.2 or tubulin (loading control). B, H1 knockdown rescues gene expression following HMGN2 knockdown. T47D shCTL and shHMGN2 cells were transiently transfected with the siRNA constructs in A and treated with or without PRL for 1 h. RNA was isolated, and cDNA was synthesized by RT-PCR and analyzed by qPCR. -Fold change was calculated relative to shCTL and siCTL with no PRL treatment. Results are presented as the mean ± S.E. (error bars), n ≥ 3 independent experiments. Statistical significance was determined by a two-sided ratio paired t test. C, H1 knockdown rescues STAT5 binding following HMGN2 knockdown. Cells were treated as in (B) and were analyzed by ChIP-qPCR for STAT5 at CISH following 45 min of PRL treatment. Recovered DNA was normalized to input and calculated as -fold enrichment compared with shHMGN2 and siCTL. Results are presented as the mean ± S.E. of three independent experiments. Statistical significance was determined by a two-sided t test assuming equal sample variance. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001.
These results led us to hypothesize that H1 dissociation is an important event in allowing STAT5 to access the DNA. Given this, we sought to elucidate whether the rescued CISH expression was indeed mediated by increased STAT5 binding. As above, control and HMGN2 knockdown stable cells were transiently transfected with siH1.2 and were assayed for STAT5 binding by ChIP. Indeed, H1 knockdown resulted in increased STAT5 binding at the CISH promoter in shHMGN2 cells (Fig. 8C). These data suggest that H1 loss is an important step in allowing full access of STAT5 to the promoter DNA to drive transcription.
H1 Knockdown Enhances Gene Expression and Breast Cancer Cell Proliferation in Response to Reduced STAT5 Activation
Because H1 knockdown rescued PRL-induced gene expression following HMGN2 knockdown, we next asked whether H1 knockdown also rescues PRL-induced gene expression following intermediate levels of STAT5 inhibition. We hypothesized that, with reduced levels of active STAT5, H1 knockdown would improve chromatin accessibility to facilitate binding of the remaining pool of active STAT5, thus rescuing transcriptional activation. Cells were treated with 200 μm STAT5 inhibitor, which resulted in decreased PRL-induced STAT5 activation but, importantly, did not cause complete inhibition (Fig. 1A). As observed with HMGN2 knockdown, the decreased gene expression caused by partial STAT5 inhibition was rescued by H1 knockdown (Fig. 9A). Under the conditions assessed, PHLDA2 did not exhibit PRL-induced expression, although STAT5 inhibition and H1 knockdown did affect expression to a small but significant degree (Fig. 9A). Longer PRL stimulation (>2 h) may be necessary to elicit PHLDA2 expression under these treatment conditions. In sum, these results further suggest that H1 and STAT5 function antagonistically to regulate gene expression.
FIGURE 9.

H1 knockdown enhances gene expression and breast cancer cell proliferation in response to reduced STAT5 activation. A, H1 knockdown rescues gene expression following partial STAT5 inhibition. T47D cells were transfected with siCTL, siH1-1, or siH1-2 (Fig. 8A). Transfectants were pretreated with the STAT5 inhibitor (200 μm) or DMSO VC for 1 h, followed by PRL treatment for 2 h. RNA was isolated, and cDNA was synthesized by RT-PCR and analyzed by qPCR. -Fold change was calculated relative to VC, siCTL with no PRL treatment. Results are presented as the mean ± S.E. (error bars), n ≥ 3 independent experiments. Statistical significance was determined by a two-sided ratio paired t test. B, H1 knockdown rescues cell proliferation in response to intermediate levels of STAT5 inhibition. T47D cells were transfected with siCTL or siH1 (pooled 1 and 2). Transfectants were treated with the indicated concentrations of STAT5 inhibitor, with or without PRL, for 3 days. BrdU incorporation was measured by absorbance as an indication of cell proliferation. Results are presented as the mean ± S.E. of three independent experiments. Within each individual experiment, each set of treatment conditions was carried out in triplicate. Statistical significance was determined by two-sided t test assuming equal sample variance. Statistical significance shown in the figure is comparing siCTL + PRL versus siH1 + PRL at the indicated concentration of STAT5 inhibitor. Other statistically significant comparisons are as follows: siCTL No PRL versus siH1 No PRL (p ≤ 0.01 at 0 inhibitor; p ≤ 0.05 at 25 μm); siH1 No PRL versus siH1 + PRL (p ≤ 0.05 at 25 μm; p = 0.052 at 50 μm). *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001.
Given the influence of H1 on STAT5-mediated transcription, we next determined whether H1 knockdown also mitigates the decrease in PRL-induced cell proliferation induced by STAT5 inhibition (Fig. 1B). At low and intermediate levels of STAT5 inhibition, H1 knockdown resulted in increased PRL-induced proliferation and protected against the effects of STAT5 inhibition compared with control cells (Fig. 9B). Of note, with high levels of STAT5 inhibition, H1 knockdown was not able to rescue proliferation (Fig. 9B). This result implies that, as expected, H1 knockdown facilitates STAT5 binding when there are intermediate levels of active STAT5 but cannot rescue STAT5 function following high levels of inhibition. These results indicate that H1 and STAT5 functionally regulate the biological effects of PRL on breast cancer cells.
Genes Regulated by H1 Are Enriched for STAT Signaling Pathways
These studies identify H1 as an important factor in regulating STAT5 binding to the DNA in response to PRL. Because little is known about the mechanisms regulating STAT accessibility, we asked whether the mechanism identified here might generalize to other contexts of STAT activation as well. A recent publication identified genes up-regulated by H1.2 knockdown in MCF7 breast cancer cells (40). Using the Enrichr enrichment analysis tool (50, 51), we discovered that the data set of genes up-regulated by H1 knockdown was significantly enriched for the Jak/STAT signaling pathway from the KEGG pathway database (Fig. 10A). The prolactin signaling pathway was also highly enriched, with a p value of 0.0026 and adjusted p value of 0.059 (Fig. 10A). In addition, the genes up-regulated by H1 knockdown were significantly enriched for genes bound by STAT5 in ChIP sequencing of the mouse mammary epithelium (Fig. 10B). Genes bound by other STAT factors were also significantly enriched (Fig. 10B). These findings further validate our model (Fig. 11) and suggest that linker histone H1 occupancy may serve as a general mechanism regulating STAT transcription factor binding.
FIGURE 10.

Genes regulated by H1 are enriched for STAT signaling pathways. A and B, genes found to be up-regulated by H1.2 knockdown in MCF7 cells by Kim et al. (40) were analyzed by the Enrichr enrichment analysis tool (50, 51). Significantly enriched gene sets of interest are plotted with their adjusted p values (q values), which were calculated using the Benjamini-Hochberg method for correction for multiple-hypothesis testing. KEGG pathways are shown in A along with the associated KEGG identifiers. ChIP enrichment analysis through ChEA 2015 is shown in B, with numbers indicating the publication PMID for each study.
FIGURE 11.
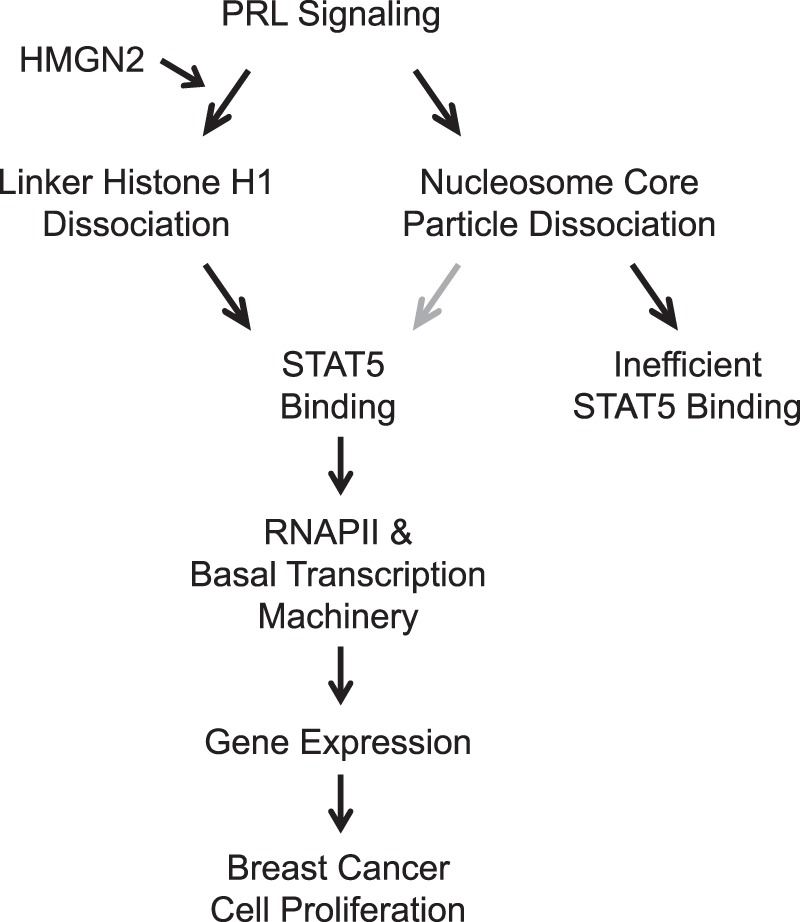
Diagram of PRL-induced chromatin remodeling and regulation of STAT5 binding. Gray arrow, possible contribution (see “Discussion”).
Discussion
Transcription factors only bind to a small fraction of their respective consensus elements throughout the genome; however, the mechanisms that regulate transcription factor accessibility are not well understood. Here, we show that PRL treatment induces chromatin decompaction at the promoter of a STAT5 target gene through the loss of both the linker histone H1 and nucleosome core particles. The loss of H1, executed here by HMGN2, is a necessary step in allowing full access of STAT5 and the transcriptional machinery to the promoter DNA, driving PRL-induced transcription and ultimately breast cancer cell proliferation (Fig. 11). Although DNA methylation has been previously implicated in preventing STAT5 accessibility (29), additional chromatin factors regulating STAT5 binding have not been well characterized. This is the first report identifying H1 eviction as a key PRL-induced event, directly regulating the access of STAT transcription factor binding at functional gene targets. Studies of other transcription factors have shown that consensus elements that are marked by active histone modifications or exhibit DNase I hypersensitivity in unstimulated cells are preferentially bound by transcription factors upon activation (24–26), indicating that the underlying chromatin state of unstimulated cells predetermines transcription factor binding patterns. Whereas our present study supports chromatin accessibility regulating transcription factor binding, our study is significant in that we have identified signaling-induced chromatin remodeling that is necessary for transcription factor binding. We show here that STAT5 accessibility is dynamically regulated by PRL-induced chromatin remodeling, suggesting that transcription factor accessibility is not just a predetermined state but is also actively regulated by signaling pathways.
We have shown previously that HMGN2 is recruited to the CISH promoter following PRL treatment, where HMGN2 promotes STAT5 binding and CISH transcription (30). Here, we sought to determine the specific mechanism of HMGN2 in promoting STAT5 binding. HMGN proteins have previously been shown to compete with the linker histone H1 for binding to chromatin, antagonizing the chromatin-condensing activity of H1 (32). The HMGN2 binding sites on chromatin overlap with the proposed binding sites for H1 (52), and competition has been observed in living cells (32). Moreover, competition between HMGN1 and H1 increases the transcriptional activity of chromatin (53). Like HMGN2, the chromatin-associated protein poly(ADP-ribose) polymerase 1 (PARP1) has also been shown to compete with H1 for binding sites on chromatin, displacing H1 to promote gene transcription (36, 54). Similar to HMGN2 and STAT5 in our system, a previous study has shown that PARP1 cooperates with the breast cancer-relevant transcription factor GATA3 to activate transcription of the cyclin D1 (CCND1) gene to promote cell proliferation (36). Another study has also shown that knockdown of PARP1 results in increased incorporation of H1 and reduced RNAPII loading at target gene promoters (54). It is notable that, in the context of PRL-induced transcription, HMGN2 appears to stimulate transcription specifically through modulating H1 incorporation without affecting H3 displacement or modification. Similarly, in both studies above, PARP1 knockdown resulted in increased incorporation of H1 and decreased target gene expression without affecting H3 occupancy (36, 54). Therefore, H1 can have significant effects on gene transcription independent of H3 and the nucleosome core particle organization.
Although our results here establish that H1 loss is necessary for full STAT5 binding at CISH, it is not yet clear whether the observed nucleosome core particle loss is a requirement for STAT5 binding. It is possible that nucleosome core particle positioning before PRL treatment masks the STAT5-binding sites and plays an important role in preventing STAT5 and RNAPII from accessing the DNA. Nucleosome core particle loss may be necessary for STAT5 binding, because STAT5 is not a recognized pioneer factor. However, nucleosome core particle loss does not appear to be sufficient for full STAT5 recruitment, because cells with HMGN2 knockdown exhibited decreased levels of PRL-induced STAT5 binding despite exhibiting the same extent of nucleosome core particle loss as control cells. Although not sufficient for full STAT5 recruitment, core particle loss may nonetheless contribute to STAT5 binding, because cells with HMGN2 knockdown still exhibit PRL-induced STAT5 binding, albeit significantly less than in control cells with proper displacement of H1. The residual STAT5 binding following HMGN2 knockdown may be a result of residual HMGN2 expression, or it may indicate that H3/nucleosome core particle loss is sufficient to allow lower levels of STAT5 engagement, with full STAT5 binding achieved only when H1 is removed from the chromatin as well. Further studies are necessary to fully elucidate the role of nucleosome positioning in regulating STAT5 accessibility to consensus elements.
Like CISH, the genes IER3 and PHLDA2 have also been implicated in breast cancer pathogenesis. IER3 has been shown to be overexpressed in invasive tumors compared with preinvasive tumors in both transgenic mouse models and human breast cancers (55). IER3 also promotes anchorage-independent growth and survival in breast cancer cells (55). PHLDA2 expression is associated with successful tumor engraftment in breast cancer patient-derived xenograft models, and PHLDA2 knockdown in breast cancer cells results in decreased cell invasion and proliferation (56).
To the best of our knowledge, this report provides the first functional evidence of PHLDA2 expression being driven by STAT5. IER3 expression has previously been implicated as being STAT5-driven (57); however, the functional binding site was not identified. From our studies here, we conclude that the STAT5 binding sites regulating IER3 and PHDLA2 expression are located outside of the proximal 5–10 kb of the promoter sequence and/or occur at non-canonical consensus elements. This finding is not unexpected, because one study has shown that close to 30% of genes that are at least partially dependent on STAT5 are not bound by STAT5 within −50 to +1 kb of the TSS (27). Therefore, STAT5 may regulate IER3 and PHLDA2 expression through enhancer elements or other more distant or intronic regulatory regions. The importance of STAT factors at enhancers is well recognized (58–60). In particular, STAT5 has been shown to bind to a mammary-specific autoregulatory enhancer in the intergenic region between Stat5a and Stat5b, and this feed-forward loop contributes to the high levels of STAT5 in mammary tissue (61). Moreover, the high levels of STAT5 drive the formation of additional mammary-specific enhancers and superenhancers (62). Notably, mutation of a STAT5 consensus element within a constituent enhancer of the STAT5-driven mammary Wap superenhancer by genome editing technologies prevents enhancer formation (62). Likewise, HMGN2 and H1 have also been implicated in enhancer function. Knock-out of Hmgn1 and Hmgn2 in mouse embryo fibroblasts results in the loss of DNase I-hypersensitive sites, particularly at enhancer regions (63). In breast cancer cells, the depletion of certain H1 subtypes, including H1.2, is associated with histone marks indicative of strong enhancers (37). A recent publication has also shown that the pioneer factor forkhead box A (FOXA) displaces H1 at enhancers in mouse hepatocytes, and the displacement of H1 promotes accessibility of the underlying nucleosomes and binding of liver-specific transcription factors (64). Therefore, HMGN2 may likewise function to displace H1 at enhancers, allowing STAT5 binding and strong enhancer formation.
The data reported here indicate that H1 is not only an important regulator of STAT5 binding, but this function also contributes to breast cancer cell biology. We have shown that, whereas STAT5 inhibition results in decreased cell proliferation, H1 knockdown significantly increases the ability of PRL to drive proliferation in the face of reduced STAT5 activation. These data highlight the relevance of this pathway in breast cancer biology. A potential weakness of this study is the possibility of off-target effects from the STAT5 inhibitor, particularly at higher concentrations, and this should be considered when interpreting the observed results. The concentrations of STAT5 inhibitor used in this study, however, are consistent with other studies in the literature (65, 66). These results are also intriguing in the context of previous reports regarding the effect of H1 on proliferation. Another group has shown that inducible shRNA-mediated knockdown of H1.2 causes G1 arrest in T47D cells and induces an apoptotic phenotype in MCF7 breast cancer cells, whereas other cell types were unaffected (39). However, H1.2 knockdown in MCF7 cells has also been shown to rescue G1 arrest, as induced by PARP1 knockdown (36). These varying results probably indicate the significant context-dependent effects of H1. It is becoming increasingly well recognized that H1 does not simply condense chromatin but rather plays a very nuanced and gene-specific role in regulating gene expression. The proliferation data presented here suggest that these effects of H1 knockdown are specific to the PRL/STAT5 signaling pathway. A limitation of this study is that all experiments have been carried out in T47D cells. Additional experiments will be necessary to establish the generalizability of the presented model. However, gene enrichment analysis suggests that the effect of H1 in regulating STAT5 binding may generalize to other STAT family transcription factors in other cell types as well (Fig. 10).
This work furthers our understanding of how PRL signaling alters the biology of breast cancer cells by influencing chromatin dynamics and highlights the relevance of the linker histone H1 in regulating transcription factor binding and gene expression. Taken together, these results suggest that the regulatory axis of HMGN2/H1 may serve as a target for future breast cancer therapeutics.
Experimental Procedures
Cell Culture and Treatment
T47D human breast cancer cells were obtained from the ATCC (Manassas, VA) and cultured in DMEM (Life Technologies, Inc.) supplemented with 10% FBS (Sigma-Aldrich or Atlanta Biologicals (Flowery Branch, GA)) and 100 units/ml penicillin/streptomycin (Life Technologies). Phoenix-AM-PHO retroviral packaging cells were provided by Dr. Jonathan Licht (University of Florida, Gainesville, FL) and were cultured in DMEM with 10% FBS and penicillin/streptomycin. Cells were cultured on tissue culture-treated polystyrene from Falcon (Corning, Inc., Corning, NY) and grown in a humidified 37 °C incubator containing 5% CO2. Cell lines were tested to be free from mycoplasma contamination using the Myco-Alert Mycoplasma detection kit (Lonza Inc., Walkersville, MD). For PRL treatment, cells were arrested in serum-free medium containing phenol red-free medium, 0.1% BSA, and 100 units/ml penicillin/streptomycin for 48 h before treatment. PRL was a gift from Dr. Anthony Kossiakoff (University of Chicago). PRL was added to yield a final concentration of 250 ng/ml for the indicated time points. For STAT5 inhibition, cells were treated with the indicated concentrations of the nonpeptidic nicotinoyl hydrazine compound STAT5 inhibitor CAS 285986-31-4 (EMD Millipore, Billerica, MA) or DMSO (EMD Millipore) as the vehicle control for 1 h before PRL treatment. For experiments comparing multiple treatment concentrations, DMSO was added to the intermediate concentrations as well so that all conditions contained the same total amount of vehicle.
Cell Lysis and Western Blotting
Nuclear extracts were prepared using the Nuclear Complex Co-IP kit (Active Motif, Carlsbad, CA) according to the manufacturer's protocol. For detection of phosphorylated proteins, 1× phosphatase inhibitor mixture (EMD Millipore, catalogue no. 524624) was added to lysis buffers. Whole cell lysates were prepared using radioimmune precipitation buffer: 25 mm HEPES, 150 mm NaCl, 5 mm MgCl2, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS, 1× protease inhibitor mixture (EMD Millipore, catalogue no. 539134). Lysates were resolved by SDS-PAGE and transferred to PVDF membranes (EMD Millipore). Membranes were blocked with either casein blocker (Thermo Scientific, catalogue no. 37532) or 5% (w/v) BSA prepared in Tris-buffered saline (TBS) containing 0.1% (v/v) Tween 20 for phospho-specific antibodies. Blots were subsequently incubated with primary antibodies diluted in blocking buffer overnight at 4 °C. Bound antibodies were visualized with HRP-conjugated secondary antibodies against mouse or rabbit IgG (GE Healthcare) using Luminata Crescendo Western HRP substrate (EMD Millipore). Blots were developed using the FujiFILM LAS-3000 imaging system (Fujifilm Medical Systems, Stamford, CT). The following antibodies were used for Western blotting analysis: phospho-STAT5 (Invitrogen, 71-6900), STAT5 (Santa Cruz Biotechnology (Dallas, TX), sc-835), α-tubulin (Invitrogen, 32-2500), H4 (Abcam (Cambridge, MA), ab7311), HMGN2 (Cell Signaling Technology (Danvers, MA), 9437S), H1.2 (ab4086 (Abcam)).
Cell Proliferation
T47D cells were plated at an initial density of 1 × 104 cells/well in a 96-well plate. Cells were arrested on the following day. After 24 h of arrest, cells were treated with the indicated concentrations of STAT5 inhibitor, vehicle control (DMSO), and/or PRL for 3 days. Cell proliferation was assessed using the BrdU Cell Proliferation Assay kit (Cell Signaling Technology) according to the manufacturer's protocol, using a 4-h incubation time for BrdU incorporation.
Chromatin Immunoprecipitation
ChIP was performed as described previously (67) until DNA extraction. Immunoprecipitated DNA was extracted using the QIAquick PCR purification kit (Qiagen, Valencia, CA). DNA was subjected to qPCR analysis. Primer sequences can be found in Table 1. Enrichment was calculated as a percentage of total input DNA. The following antibodies were used for ChIP: RNAPII (Santa Cruz Biotechnology, sc-899), phospho-RNAPII (Abcam, ab5408), H3 (Abcam, ab1791), H4 (Abcam, ab7311), H1.2 (Abcam, ab4086), STAT5 (Cell Signaling Technology, 9363S (for Figs. 5B and 6F); Santa Cruz Biotechnology, sc-1081 X (for all other experiments)), HMGN2 (Cell Signaling Technology, 9437S), acetyl-H3K9 (Abcam, ab10812), acetyl-H3K14 (EMD Millipore, 07-353), H3K9me3 (EMD Millipore, 17-625), H3K27me3 (EMD Millipore, 17-622).
TABLE 1.
List of primers for ChIP
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| CISH, −45 bp (STAT5) | AGCCCGCGGTTCTAGGAA | AGCTGCTGCCTAATCCTTTGTC |
| CISH, +89 bp (RNAPII) | CACTGCCTCTCAGTCCCTGT | CGGCTGGAGGGAACCAGT |
| IER3, −8.0 kb | GTGTCAGTACTGGGGTTTGGT | TGGTTCTGAGTCACACATGGC |
| IER3, −2.9 kb | AGAGCTAGTGTTCACATCTGTCC | CACCCACTGTTCAAGGTCAC |
| IER3, −0.2 kb | TTGTGAGTGTGTGAGTCGTG | GGATCCTGTGGCTAAAGTGAG |
| IER3, TSS | CGAGAGTGACACATGGTGAG | AAGGACCCGCCCAATTT |
| PHLDA2, −1.4 kb | GCCCTTCAGGGTCACTGTAAA | GCTGCTACCTGCATGCCATT |
| PHLDA2, −0.7 kb | TCTTAGCCTCGGTTTTCGCT | TGCTGGTCTAGCCCTTTCCA |
| PHLDA2, +0.1 kb | CCGCGCTTCTTCTTCCATAG | TCGGCACGACATGAAATCC |
| ZNF554 (H3K9me3) | CGGGGAAAAGCCCTATAAAT | TCCACATTCACTGCATTCGT |
| α-Satellite (H3K27me3) | CTGCACTACCTGAAGAGGAC | GATGGTTCAACACTCTTACA |
MNase Protection Assay
Cells were permeabilized in 35 mm HEPES, pH 7.4, 150 mm sucrose, 80 mm KCl, 5 mm K2HPO4, 5 mm MgCl2, 0.5 mm CaCl2, 250 ng/ml 1-oleoyl-sn-glycero-3-phosphocholine for 3 min at room temperature. Nuclei were washed and resuspended in MNase buffer: 10 mm Tris, pH 7.4, 15 mm NaCl, 60 mm KCl, 0.15 mm spermine, 0.5 mm spermidine, 1 mm CaCl2. Chromatin was digested with MNase (10 Worthington units/ml; Sigma-Aldrich) for 20 or 30 min at room temperature. Reactions were stopped by adding an equal amount of 5% SDS, 250 mm EDTA, pH 8.0. Digestions were incubated with 3 mg/ml Proteinase K overnight at 65 °C. Digested DNA was purified by phenol/chloroform extraction followed by ethanol precipitation, and RNA was removed by treatment with 0.1 mg/ml RNase A at 37 °C. Mononucleosomal DNA fragments (∼150 bp) were isolated by agarose gel electrophoresis followed by gel extraction using the QIAquick gel extraction kit (Qiagen). Genomic DNA was isolated using the Wizard genomic DNA purification kit (Promega, Madison, WI). qPCR was performed on a LightCycler 480 II (Roche Applied Science) using the primers in Table 2 and SYBR Advantage GC Premix (Takara Bio, Mountain View, CA). Primers were designed using PCRTiler version 1.42 (68) and PrimerQuest (Integrated DNA Technologies, Inc., Coralville, IA). Relative enrichment of mononucleosomal DNA was calculated relative to amplification of undigested genomic DNA using the comparative Ct method.
TABLE 2.
List of primers for the MNase protection assay
M, amplicon midpoint, relative to CISH TSS (bp).
| M | Forward primer (5′–3′) | Reverse Primer (5′-3′) |
|---|---|---|
| −453 | CACACGCCGACAGACCTC | GGGAAGGGCCCTCTTATCTC |
| −438 | GCTCGCAGGGAGGACAA | GGGAAGGGCCCTCTTATCT |
| −376 | TTCCACCGCGAGATAAGAGG | GGGTCGGGGAAGTTAAGGAG |
| −344 | GACGCAGAATGCCAGAAGG | GGCAGGTCGGAGAGATCAGT |
| −322 | GGGCCCCTCCTTAACTTCC | GGCAGGTCGGAGAGATCAGT |
| −284 | CCACTGATCTCTCCGACCT | CAATAGCAGCGCGTGGA |
| −256 | GACCTGCCCCTCTGGGTA | AATCGCGACGCTGAAGGT |
| −214 | GTCCACGCGCTGCTATTG | GACAGATTTCCAAGAACTTTCCA |
| −174 | GTCGCGATTGGTCAGCTC | GGGCCCTGAGCAGTGAAA |
| −157 | TTCCTGGAAAGTTCTTGGAAATC | GGCCCTGAGCAGTGAAA |
| −54 | TCAGCCCGCGGTTCTA | TTTGTCTGCCGCGTTCC |
| −42 | CCCGCGGTTCTAGGAAGA | GGCGAGCTGCTGCCTAAT |
| −29 | TCCGGGAAGGGCTGGAA | GGCGAGCTGCTGCCTAATC |
| +20 | AAAGGATTAGGCAGCAGCTC | GACAGGGACTGAGAGGCAGT |
| +76 | CACTGCCTCTCAGTCCCT | AGCGCGTGCTGGGTA |
| +90 | TGCCTCTCAGTCCCTGTCC | GGCTGGAGGGAACCAGTG |
| +164 | CCCACTGGTTCCCTCCAG | AGAGAGCCGCGCTTACCC |
| +208 | GACATGGTCCTCTGCGTTCAG | GTGGTGGCCGGGAAGG |
| +225 | ACATGGTCCTCTGCGTTCAG | TAGCACGAAGCCCCTGTTCT |
| +249 | GTAAGCGCGGCTCTCTG | TCAAGCCCTCCCAATGC |
| +295 | AGAACAGGGGCTTCGTGCTA | CTTGCTCCTGAGGTCCGTCT |
| +324 | GCATTGGGAGGGCTTGA | GGAGTGGGAGTGTCACAG |
| +384 | GCTGTGACACTCCCACTC | CCGGTTTCCCAATCCACA |
| +412 | CCCCCACACACTTACCTCAA | CCACCCTGTGAGTTCCTCCT |
Luciferase Reporter Construction
The CISH promoter region (−975 to +60 bp) was previously PCR-amplified and cloned into the promoterless pGL4.10[luc2] luciferase reporter vector (Promega), termed pGL4-CISH (10). Truncations of the pGL4-CISH luciferase reporter vector were created here by inverse PCR. The pGL4-CISH plasmid was PCR-amplified using Phusion High Fidelity DNA polymerase (New England BioLabs, Ipswich, MA) using primers with phosphorylated ends (Integrated DNA Technologies), followed by self-ligation. The primer sequences (5′–3′) are as follows: −503 bp Forward, CGCCGACAGACCTCCTT; −375 bp Forward, AATCTGGGCGCGGGTTG; −226 bp Forward, TTCAGCGTCGCGATTGG; −84 bp Forward, CTCAGCCCGCGGTTCTA; Reverse (for all constructs), AGGCTAGCGAGCTCAGG. All constructs were confirmed by traditional DNA sequencing at the Genomics Core Facility (Northwestern University, Chicago, IL).
Dual-Luciferase Reporter Assay
T47D cells were plated in a 24-well plate 24 h before transfection. Transfection was carried out using Lipofectamine 3000 (Life Technologies) according to the manufacturer's protocol. Cells were transfected with the described CISH luciferase reporter truncation plasmids along with the constitutive Renilla luciferase control vector pGL4.73[hRluc/SV40] (Promega), which was used at a ratio of 1:100 to pGL4-CISH. Equal molar ratios of the CISH luciferase reporter truncations were transfected, and pUC19 DNA (New England Biolabs) was used as filler to equalize the total amount of DNA transfected per well. Cells were arrested 24 h after transfection. Following 24 h of arrest, cells were treated with PRL for 24 h. Reporter expression was then assessed using the Dual-Luciferase reporter assay system (Promega) following the manufacturer's protocol. Luminescence was read on a Synergy HT microplate reader (Bio-Tek, Winooski, VT). Luciferase readings were normalized to expression of the internal Renilla control.
RNA Interference
Stable shRNA-mediated knockdown of HMGN2 via retroviral infection has been described previously (30, 42), using the retroviral vector pRFP-C-RS expressing shRNA targeting HMGN2 (5′-GTGTCAGGCAATCTGGACTTTCCAGTGAT-3′; catalogue no. TF319505, Origene, Rockville, MD). siRNA targeting H1.2 (catalogue no. SR302039B (siH1.2-1) and SR302039A (siH1.2-2)) and non-targeting control (catalogue no. SR30004 (siCTL)) were purchased from Origene. T47D cells were transfected with siRNA at a final concentration of 2–10 nm. Reverse transfections were performed using RNAiMAX (Life Technologies, Inc.) following the manufacturer's protocol.
RNA Extraction and Quantitative RT-PCR (qRT-PCR)
Total RNA was isolated using the RNeasy Plus minikit (Qiagen) according to the manufacturer's protocol. cDNA was synthesized with the iScript cDNA synthesis kit (Bio-Rad) using 1 μg of RNA as template. cDNA was amplified using gene-specific primers (Table 3) using Power SYBR Green PCR master mix (Applied Biosystems) on a QuantStudio 12K Flex real-time PCR system (Applied Biosystems). -Fold change values were calculated using the comparative Ct method using GAPDH or 18S rRNA for normalization.
TABLE 3.
List of primers for qRT-PCR
| Gene (cDNA) | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| CISH | AGAGGAGGATCTGCTGTGCAT | GGAACCCCAATACCAGCCAG |
| IER3 | CTCGAGTGGTCCGGCG | ACGATGGTGAGCAGCAGAAA |
| PHLDA2 | CCGCCGCGGGCCATA | GCACGGGAAGTTCTTCTGCT |
| 18S rRNA | CCCCATGAACGAGGGAATT | GGGACTTAATCAACGCAAGCTT |
| GAPDH | CATGAGAAGTATGACAACAGCCT | AGTCCTTCCACGATACCAAAGT |
Retroviral Production
HMGN2 cDNA constructs, fused to either a C-terminal or N-terminal FLAG tag and flanked by a 5′ EcoRI site and a 3′ SalI site, were ordered as custom gBlock gene fragments (Integrated DNA Technologies, Inc.). The constructs were digested with EcoRI and SalI restriction enzymes (New England Biolabs) and ligated into the retroviral pBabe-GFP vector. The constructs were confirmed by traditional DNA sequencing at the Genomics Core Facility (Northwestern University). pBabe-GFP HMGN2-FLAG plasmids were transfected into Phoenix packaging cells using FuGene 6 (Promega). Retroviral supernatant was collected 48 h post-transfection and filtered through a 0.45-μm filter (EMD Millipore). T47D cells were infected with viral supernatant supplemented with fresh growth medium containing 8 μg/ml Polybrene (EMD Millipore) by spin infection at 500 × g at 32 °C for 2 h. Infected cells were enriched by fluorescence-activated cell sorting for GFP.
Chemicals
All chemicals were purchased from Sigma-Aldrich unless otherwise specified.
Statistical Analysis
All statistical analysis was performed using GraphPad Prism version 6.0 (GraphPad Software, La Jolla, CA). All data are presented as the mean ± S.E. from at least three independent experiments. Statistical analysis was performed as described in the figure legends. Statistical significance was considered at a value of p ≤ 0.05.
Author Contributions
S. M. S. designed and performed all of the experiments and analyzed the data. C. V. C. conceived and coordinated the study. S. M. S., C. V. C., J. D. L., and J. J. K. designed the study and interpreted the data. S. M. S. drafted the manuscript, and C. V. C., J. D. L., and J. J. K. critically revised the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Katherine Harrington from the Clevenger laboratory for insightful discussions and Dr. Eliza Small from the Licht laboratory for technical expertise in the MNase protection assay.
This work was supported by NCI, National Institutes of Health, Grant R01CA173305 (to C. V. C.) and National Research Service Award F30CA171858 (to S. M. S.), by grants from the Lynn Sage Foundation (to C. V. C.), and by a Malkin Scholar Award from the Robert H. Lurie Comprehensive Cancer Center of Northwestern University (to S. M. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PRLR
- PRL receptor
- RNAPII
- RNA polymerase II
- MNase
- micrococcal nuclease
- TSS
- transcription start site
- H3K9
- H3K14, and H3K27, histone H3 lysine 9, 14, and 27, respectively
- H3K9me3 and H3K27me3
- H3K9 and H3K27 trimethylation, respectively
- VC
- vehicle control
- ANOVA
- analysis of variance
- qPCR
- quantitative PCR
- qRT-PCR
- quantitative RT-PCR.
References
- 1. Vomachka A. J., Pratt S. L., Lockefeer J. A., and Horseman N. D. (2000) Prolactin gene-disruption arrests mammary gland development and retards T-antigen-induced tumor growth. Oncogene 19, 1077–1084 [DOI] [PubMed] [Google Scholar]
- 2. Horseman N. D., Zhao W., Montecino-Rodriguez E., Tanaka M., Nakashima K., Engle S. J., Smith F., Markoff E., and Dorshkind K. (1997) Defective mammopoiesis, but normal hematopoiesis, in mice with a targeted disruption of the prolactin gene. EMBO J. 16, 6926–6935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ormandy C. J., Camus A., Barra J., Damotte D., Lucas B., Buteau H., Edery M., Brousse N., Babinet C., Binart N., and Kelly P. A. (1997) Null mutation of the prolactin receptor gene produces multiple reproductive defects in the mouse. Genes Dev. 11, 167–178 [DOI] [PubMed] [Google Scholar]
- 4. Ormandy C. J., Binart N., and Kelly P. A. (1997) Mammary gland development in prolactin receptor knockout mice. J. Mammary Gland Biol. Neoplasia 2, 355–364 [DOI] [PubMed] [Google Scholar]
- 5. Wakao H., Gouilleux F., and Groner B. (1995) Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J. 14, 854–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lebrun J. J., Ali S., Sofer L., Ullrich A., and Kelly P. A. (1994) Prolactin-induced proliferation of Nb2 cells involves tyrosine phosphorylation of the prolactin receptor and its associated tyrosine kinase JAK2. J. Biol. Chem. 269, 14021–14026 [PubMed] [Google Scholar]
- 7. Rui H., Kirken R. A., and Farrar W. L. (1994) Activation of receptor-associated tyrosine kinase JAK2 by prolactin. J. Biol. Chem. 269, 5364–5368 [PubMed] [Google Scholar]
- 8. Pezet A., Ferrag F., Kelly P. A., and Edery M. (1997) Tyrosine docking sites of the rat prolactin receptor required for association and activation of stat5. J. Biol. Chem. 272, 25043–25050 [DOI] [PubMed] [Google Scholar]
- 9. Schindler C., Shuai K., Prezioso V. R., and Darnell J. E. Jr. (1992) Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science 257, 809–813 [DOI] [PubMed] [Google Scholar]
- 10. Fang F., Antico G., Zheng J., and Clevenger C. V. (2008) Quantification of PRL/Stat5 signaling with a novel pGL4-CISH reporter. BMC Biotechnol. 8, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fang F., Zheng J., Galbaugh T. L., Fiorillo A. A., Hjort E. E., Zeng X., and Clevenger C. V. (2010) Cyclophilin B as a co-regulator of prolactin-induced gene expression and function in breast cancer cells. J. Mol. Endocrinol. 44, 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brockman J. L., Schroeder M. D., and Schuler L. A. (2002) PRL activates the cyclin D1 promoter via the Jak2/Stat pathway. Mol. Endocrinol. 16, 774–784 [DOI] [PubMed] [Google Scholar]
- 13. Rose-Hellekant T. A., Arendt L. M., Schroeder M. D., Gilchrist K., Sandgren E. P., and Schuler L. A. (2003) Prolactin induces ERα-positive and ERα-negative mammary cancer in transgenic mice. Oncogene 22, 4664–4674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eliassen A. H., Tworoger S. S., and Hankinson S. E. (2007) Reproductive factors and family history of breast cancer in relation to plasma prolactin levels in premenopausal and postmenopausal women. Int. J. Cancer 120, 1536–1541 [DOI] [PubMed] [Google Scholar]
- 15. Hankinson S. E., Willett W. C., Michaud D. S., Manson J. E., Colditz G. A., Longcope C., Rosner B., and Speizer F. E. (1999) Plasma prolactin levels and subsequent risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 91, 629–634 [DOI] [PubMed] [Google Scholar]
- 16. Tworoger S. S., Eliassen A. H., Sluss P., and Hankinson S. E. (2007) A prospective study of plasma prolactin concentrations and risk of premenopausal and postmenopausal breast cancer. J. Clin. Oncol. 25, 1482–1488 [DOI] [PubMed] [Google Scholar]
- 17. Tworoger S. S., Eliassen A. H., Zhang X., Qian J., Sluss P. M., Rosner B. A., and Hankinson S. E. (2013) A 20-year prospective study of plasma prolactin as a risk marker of breast cancer development. Cancer Res. 73, 4810–4819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rasmussen L. M., Frederiksen K. S., Din N., Galsgaard E., Christensen L., Berchtold M. W., and Panina S. (2010) Prolactin and oestrogen synergistically regulate gene expression and proliferation of breast cancer cells. Endocr. Relat. Cancer 17, 809–822 [DOI] [PubMed] [Google Scholar]
- 19. Maus M. V., Reilly S. C., and Clevenger C. V. (1999) Prolactin as a chemoattractant for human breast carcinoma. Endocrinology 140, 5447–5450 [DOI] [PubMed] [Google Scholar]
- 20. Perks C. M., Keith A. J., Goodhew K. L., Savage P. B., Winters Z. E., and Holly J. M. (2004) Prolactin acts as a potent survival factor for human breast cancer cell lines. Br. J. Cancer 91, 305–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iavnilovitch E., Cardiff R. D., Groner B., and Barash I. (2004) Deregulation of Stat5 expression and activation causes mammary tumors in transgenic mice. Int. J. Cancer 112, 607–619 [DOI] [PubMed] [Google Scholar]
- 22. Ren S., Cai H. R., Li M., and Furth P. A. (2002) Loss of Stat5a delays mammary cancer progression in a mouse model. Oncogene 21, 4335–4339 [DOI] [PubMed] [Google Scholar]
- 23. Tang J. Z., Zuo Z. H., Kong X. J., Steiner M., Yin Z., Perry J. K., Zhu T., Liu D. X., and Lobie P. E. (2010) Signal transducer and activator of transcription (STAT)-5A and STAT5B differentially regulate human mammary carcinoma cell behavior. Endocrinology 151, 43–55 [DOI] [PubMed] [Google Scholar]
- 24. Ballaré C., Castellano G., Gaveglia L., Althammer S., González-Vallinas J., Eyras E., Le Dily F., Zaurin R., Soronellas D., Vicent G. P., and Beato M. (2013) Nucleosome-driven transcription factor binding and gene regulation. Mol. Cell 49, 67–79 [DOI] [PubMed] [Google Scholar]
- 25. John S., Sabo P. J., Thurman R. E., Sung M. H., Biddie S. C., Johnson T. A., Hager G. L., and Stamatoyannopoulos J. A. (2011) Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 43, 264–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guccione E., Martinato F., Finocchiaro G., Luzi L., Tizzoni L., Dall' Olio V., Zardo G., Nervi C., Bernard L., and Amati B. (2006) Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell Biol. 8, 764–770 [DOI] [PubMed] [Google Scholar]
- 27. Yamaji D., Kang K., Robinson G. W., and Hennighausen L. (2013) Sequential activation of genetic programs in mouse mammary epithelium during pregnancy depends on STAT5A/B concentration. Nucleic Acids Res. 41, 1622–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rijnkels M., Freeman-Zadrowski C., Hernandez J., Potluri V., Wang L., Li W., and Lemay D. G. (2013) Epigenetic modifications unlock the milk protein gene loci during mouse mammary gland development and differentiation. PLoS One 8, e53270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hedrich C. M., Rauen T., Apostolidis S. A., Grammatikos A. P., Rodriguez Rodriguez N., Ioannidis C., Kyttaris V. C., Crispin J. C., and Tsokos G. C. (2014) Stat3 promotes IL-10 expression in lupus T cells through trans-activation and chromatin remodeling. Proc. Natl. Acad. Sci. U.S.A. 111, 13457–13462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fiorillo A. A., Medler T. R., Feeney Y. B., Liu Y., Tommerdahl K. L., and Clevenger C. V. (2011) HMGN2 inducibly binds a novel transactivation domain in nuclear PRLr to coordinate Stat5a-mediated transcription. Mol. Endocrinol. 25, 1550–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bustin M. (1999) Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol. Cell. Biol. 19, 5237–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Catez F., Yang H., Tracey K. J., Reeves R., Misteli T., and Bustin M. (2004) Network of dynamic interactions between histone H1 and high-mobility-group proteins in chromatin. Mol. Cell. Biol. 24, 4321–4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Müller J., Sperl B., Reindl W., Kiessling A., and Berg T. (2008) Discovery of chromone-based inhibitors of the transcription factor STAT5. ChemBioChem 9, 723–727 [DOI] [PubMed] [Google Scholar]
- 34. Raccurt M., Tam S. P., Lau P., Mertani H. C., Lambert A., Garcia-Caballero T., Li H., Brown R. J., McGuckin M. A., Morel G., and Waters M. J. (2003) Suppressor of cytokine signalling gene expression is elevated in breast carcinoma. Br. J. Cancer 89, 524–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pinz S., Unser S., Buob D., Fischer P., Jobst B., and Rascle A. (2015) Deacetylase inhibitors repress STAT5-mediated transcription by interfering with bromodomain and extra-terminal (BET) protein function. Nucleic Acids Res. 43, 3524–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shan L., Li X., Liu L., Ding X., Wang Q., Zheng Y., Duan Y., Xuan C., Wang Y., Yang F., Shang Y., and Shi L. (2014) GATA3 cooperates with PARP1 to regulate CCND1 transcription through modulating histone H1 incorporation. Oncogene 33, 3205–3216 [DOI] [PubMed] [Google Scholar]
- 37. Millán-Ariño L., Islam A. B., Izquierdo-Bouldstridge A., Mayor R., Terme J. M., Luque N., Sancho M., López-Bigas N., and Jordan A. (2014) Mapping of six somatic linker histone H1 variants in human breast cancer cells uncovers specific features of H1.2. Nucleic Acids Res. 42, 4474–4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Harshman S. W., Hoover M. E., Huang C., Branson O. E., Chaney S. B., Cheney C. M., Rosol T. J., Shapiro C. L., Wysocki V. H., Huebner K., and Freitas M. A. (2014) Histone H1 phosphorylation in breast cancer. J. Proteome Res. 13, 2453–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sancho M., Diani E., Beato M., and Jordan A. (2008) Depletion of human histone H1 variants uncovers specific roles in gene expression and cell growth. PLoS Genet. 4, e1000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim J. M., Kim K., Punj V., Liang G., Ulmer T. S., Lu W., and An W. (2015) Linker histone H1.2 establishes chromatin compaction and gene silencing through recognition of H3K27me3. Sci. Rep. 5, 16714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nacht A. S., Pohl A., Zaurin R., Soronellas D., Quilez J., Sharma P., Wright R. H., Beato M., and Vicent G. P. (2016) Hormone-induced repression of genes requires BRG1-mediated H1.2 deposition at target promoters. EMBO J. 35, 1822–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fiorillo A. A., Medler T. R., Feeney Y. B., Wetz S. M., Tommerdahl K. L., and Clevenger C. V. (2013) The prolactin receptor transactivation domain is associated with steroid hormone receptor expression and malignant progression of breast cancer. Am. J. Pathol. 182, 217–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Medler T. R., Craig J. M., Fiorillo A. A., Feeney Y. B., Harrell J. C., and Clevenger C. V. (2016) HDAC6 deacetylates HMGN2 to regulate Stat5a activity and breast cancer growth. Mol. Cancer Res. 14, 994–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stothard P. (2000) The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques 28, 1102–1104 [DOI] [PubMed] [Google Scholar]
- 45. ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gerstein M. B., Kundaje A., Hariharan M., Landt S. G., Yan K. K., Cheng C., Mu X. J., Khurana E., Rozowsky J., Alexander R., Min R., Alves P., Abyzov A., Addleman N., Bhardwaj N., et al. (2012) Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ueda T., Postnikov Y. V., and Bustin M. (2006) Distinct domains in high mobility group N variants modulate specific chromatin modifications. J. Biol. Chem. 281, 10182–10187 [DOI] [PubMed] [Google Scholar]
- 48. Cao K., Lailler N., Zhang Y., Kumar A., Uppal K., Liu Z., Lee E. K., Wu H., Medrzycki M., Pan C., Ho P. Y., Cooper G. P. Jr., Dong X., Bock C., Bouhassira E. E., and Fan Y. (2013) High-resolution mapping of h1 linker histone variants in embryonic stem cells. PLoS Genet. 9, e1003417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Deng T., Postnikov Y., Zhang S., Garrett L., Becker L., Racz I., Holter S. M., Wurst W., Fuchs H., Gailus-Durner V., de Angelis M. H., and Bustin M. (2016) Interplay between H1 and HMGN epigenetically regulates OLIG1&2 expression and oligodendrocyte differentiation. Nucleic Acids Res. 10.1093/nar/gkw1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen E. Y., Tan C. M., Kou Y., Duan Q., Wang Z., Meirelles G. V., Clark N. R., and Ma'ayan A. (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuleshov M. V., Jones M. R., Rouillard A. D., Fernandez N. F., Duan Q., Wang Z., Koplev S., Jenkins S. L., Jagodnik K. M., Lachmann A., McDermott M. G., Monteiro C. D., Gundersen G. W., and Ma'ayan A. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Alfonso P. J., Crippa M. P., Hayes J. J., and Bustin M. (1994) The footprint of chromosomal proteins HMG-14 and HMG-17 on chromatin subunits. J. Mol. Biol. 236, 189–198 [DOI] [PubMed] [Google Scholar]
- 53. Ding H. F., Bustin M., and Hansen U. (1997) Alleviation of histone H1-mediated transcriptional repression and chromatin compaction by the acidic activation region in chromosomal protein HMG-14. Mol. Cell. Biol. 17, 5843–5855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krishnakumar R., and Kraus W. L. (2010) PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol. Cell 39, 736–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang C., Trent S., Ionescu-Tiba V., Lan L., Shioda T., Sgroi D., and Schmidt E. V. (2006) Identification of cyclin D1- and estrogen-regulated genes contributing to breast carcinogenesis and progression. Cancer Res. 66, 11649–11658 [DOI] [PubMed] [Google Scholar]
- 56. Moon H. G., Oh K., Lee J., Lee M., Kim J. Y., Yoo T. K., Seo M. W., Park A. K., Ryu H. S., Jung E. J., Kim N., Jeong S., Han W., Lee D. S., and Noh D. Y. (2015) Prognostic and functional importance of the engraftment-associated genes in the patient-derived xenograft models of triple-negative breast cancers. Breast Cancer Res. Treat. 154, 13–22 [DOI] [PubMed] [Google Scholar]
- 57. Sato T., Tran T. H., Peck A. R., Liu C., Ertel A., Lin J., Neilson L. M., and Rui H. (2013) Global profiling of prolactin-modulated transcripts in breast cancer in vivo. Mol. Cancer 12, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kang K., Yamaji D., Yoo K. H., Robinson G. W., and Hennighausen L. (2014) Mammary-specific gene activation is defined by progressive recruitment of STAT5 during pregnancy and the establishment of H3K4me3 marks. Mol. Cell. Biol. 34, 464–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vahedi G., Takahashi H., Nakayamada S., Sun H. W., Sartorelli V., Kanno Y., and O'Shea J. J. (2012) STATs shape the active enhancer landscape of T cell populations. Cell 151, 981–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sugathan A., and Waxman D. J. (2013) Genome-wide analysis of chromatin states reveals distinct mechanisms of sex-dependent gene regulation in male and female mouse liver. Mol. Cell. Biol. 33, 3594–3610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Metser G., Shin H. Y., Wang C., Yoo K. H., Oh S., Villarino A. V., O'Shea J. J., Kang K., and Hennighausen L. (2016) An autoregulatory enhancer controls mammary-specific STAT5 functions. Nucleic Acids Res. 44, 1052–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shin H. Y., Willi M., Yoo K. H., Zeng X., Wang C., Metser G., and Hennighausen L. (2016) Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat. Genet. 48, 904–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Deng T., Zhu Z. I., Zhang S., Postnikov Y., Huang D., Horsch M., Furusawa T., Beckers J., Rozman J., Klingenspor M., Amarie O., Graw J., Rathkolb B., Wolf E., Adler T., et al. (2015) Functional compensation among HMGN variants modulates the DNase I hypersensitive sites at enhancers. Genome Res. 25, 1295–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Iwafuchi-Doi M., Donahue G., Kakumanu A., Watts J. A., Mahony S., Pugh B. F., Lee D., Kaestner K. H., and Zaret K. S. (2016) The pioneer transcription factor FoxA maintains an accessible nucleosome configuration at enhancers for tissue-specific gene activation. Mol. Cell 62, 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aksamitiene E., Achanta S., Kolch W., Kholodenko B. N., Hoek J. B., and Kiyatkin A. (2011) Prolactin-stimulated activation of ERK1/2 mitogen-activated protein kinases is controlled by PI3-kinase/Rac/PAK signaling pathway in breast cancer cells. Cell. Signal. 23, 1794–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hodge L. S., Ziesmer S. C., Yang Z. Z., Secreto F. J., Novak A. J., and Ansell S. M. (2014) Constitutive activation of STAT5A and STAT5B regulates IgM secretion in Waldenstrom's macroglobulinemia. Blood 123, 1055–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee T. I., Johnstone S. E., and Young R. A. (2006) Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc. 1, 729–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gervais A. L., Marques M., and Gaudreau L. (2010) PCRTiler: automated design of tiled and specific PCR primer pairs. Nucleic Acids Res. 38, W308–W312 [DOI] [PMC free article] [PubMed] [Google Scholar]