

Abstract
Stromal interaction molecule 1 (STIM1) regulates store-operated Ca2+ entry (SOCE) and other ion channels either as an endoplasmic reticulum Ca2+-sensing protein or when present in the plasma membrane. However, the role of STIM1 in insulin-secreting β-cells is unresolved. We report that lowering expression of STIM1, the gene that encodes STIM1, in insulin-secreting MIN6 β-cells with RNA interference inhibits SOCE and ATP-sensitive K+ (KATP) channel activation. The effects of STIM1 knockdown were reversed by transduction of MIN6 cells with an adenovirus gene shuttle vector that expressed human STIM1. Immunoprecipitation studies revealed that STIM1 binds to nucleotide binding fold-1 (NBF1) of the sulfonylurea receptor 1 (SUR1) subunit of the KATP channel. Binding of STIM1 to SUR1 was enhanced by poly-lysine. Our data indicate that SOCE and KATP channel activity are regulated by STIM1. This suggests that STIM1 is a multifunctional signaling effector that participates in the control of membrane excitability and Ca2+ signaling events in β-cells.
Keywords: beta cell (B-cell), calcium imaging, cell signaling, electrophysiology, endoplasmic reticulum (ER), Ca2+ signaling, KATP channels, STIM1, SUR1, store-operated ion channels
Introduction
The primary physiological role of pancreatic β-cells is to regulate metabolism by sensing changes in blood glucose concentration and secreting insulin accordingly. Interplay between glucose metabolism, closure of ATP-sensitive K+ (KATP) channels, and Ca2+ signaling evoked by activation of voltage-dependent Ca2+ channels (VDCCs)2 form the molecular basis of the so-called consensus model of glucose-stimulated insulin secretion. Nevertheless, this simple model does not address the temporal and spatial diversity of Ca2+-dependent signal transduction events generated in β-cells consequent to stimulation with secretagogues (1).
Ca2+ homeostasis and signal transduction are controlled by multiple ion transport mechanisms and organelles. Store-operated Ca2+ entry (SOCE) plays a critical role regulating spatial and temporal changes in cytoplasmic Ca2+ concentration ([Ca2+]c), endoplasmic reticulum (ER) Ca2+ homeostasis and protein biosynthesis, mitochondrial function, secretion, and cell viability (2, 3). The ER regulates Ca2+ signaling by acting as a subcellular store for Ca2+ that can be rapidly and transiently released into the cytoplasm, and the ER luminal Ca2+ concentration ([Ca2+]ER) regulates gating of store-operated Ca2+ (SOC) channels located in the plasma membrane (PM).
The presence of SOCE in β-cells was first reported in 1994 (4–6). Patch clamp electrophysiology has been used to characterize some of the electrical properties of SOC current (ISOC) in β-cell lines and primary β-cells isolated from rodent islets of Langerhans (5–7). The store-operated conductance in insulin-secreting cells was found to be an inwardly rectifying current carried by a non-selective cation (CRAN) channel rather than a highly Ca2+-selective Ca2+-release activated Ca2+ (CRAC) channel (5, 8). The electrical characteristics of ISOC in insulin-secreting cells are similar to mammalian transient receptor potential (trp)-related channels, a gene family that includes Ca2+ store-operated channels (9, 10), and several trp genes are expressed in β-cells (11, 12). Members of the Orai family of genes (Orai1, Orai2, and Orai3) and some TRPC channels (TRPC1, TRPC2, and TRPC4) form store-operated channels either as homomeric or heteromeric complexes (13). Although Affymetrix GeneChip microarrays show that rodent β-cell lines as well as human islets express Orai genes (T1Dbase), the roles of the proteins encoded by these genes in β-cell store-operated Ca2+ entry and β-cell stimulus-secretion coupling remain to be clarified.
Stromal interaction molecule-1 (STIM1), a single transmembrane-spanning Ca2+ binding phosphoprotein located in both the ER and PM, functions as a luminal Ca2+ sensor of the ER and couples changes in [Ca2+]ER with activation of SOCE (14). STIM1 is activated by cellular stimuli and stress conditions that lower [Ca2+]ER. After a decrease in [Ca2+]ER, STIM1 translocates to plasma membrane-associated membrane (PAM) complexes, specialized regions where the ER and PM are closely apposed. At the PAM, STIM1 directly interacts with plasmalemmal store-operated ion channels and other protein targets that control a wide range of cellular signaling events (15, 16).
STIM1 is expressed in β-cells, but its role in β-cell Ca2+ signaling has not been fully resolved (17). Confocal microscopy demonstrated that subsequent to decreasing [Ca2+]ER with thapsigargin, an inhibitor of sarco(endo)plasmic reticulum calcium ATPase (SERCA), fluorescently-tagged STIM1 expressed in MIN6 β-cells translocates to regions near the plasma membrane (18) consistent with STIM1 regulating β-cell SOC channels. STIM1 translocation and formation of sub-PM puncta or clusters of STIM1 molecules can also occur in response to cAMP elevation but without activation of SOCE (19).
To better understand the role of STIM1 in β-cells, we utilized patch clamp electrophysiology, Ca2+ imaging, and RNA interference. We demonstrate for the first time that in insulin-secreting cells STIM1 participates in the activation of store-operated Ca2+ current but also directly interacts with KATP channels. Our data suggest that STIM1 regulates β-cell excitability by coupling multiple PM signaling mechanisms with changes in [Ca2+]ER.
Results
Store-operated Ca2+ Entry in MIN6 Cells
Application of carbachol, a muscarinic receptor agonist, in the absence of external Ca2+ evoked a transient increase in [Ca2+]c (Fig. 1A). After discharge of the intracellular Ca2+ stores, the addition of extracellular Ca2+ caused a biphasic increase in [Ca2+]c characteristic of SOCE found in a wide range of cell types (Fig. 1A). The response of MIN6 cells with depleted stores to Ca2+ reintroduction was significantly reduced (peak amplitude reduced by 11% and area under the Ca2+ response curve (area-under-curve (AUC)) by 24%) by nimodipine (5 μm), an inhibitor of voltage-gated Ca2+ channels (Fig. 1, B and C). These data indicate that Ca2+ influx through L-type voltage-gated Ca2+ channels makes a small, but significant, contribution to the increase in [Ca2+]c under these conditions. This nimodipine-sensitive influx could result from spontaneous activity of L-type channels at the resting potential (20) or from a small SOCE-induced depolarization and weak activation of VDCCs. The broad spectrum channel inhibitor, 1-[2-(4-methoxyphenyl)-2-[3-(4-methoxyphenyl) propoxy]ethyl-1H-imidazole hydrochloride (SKF 96365), is reported to inhibit SOCE in β cells (8) and was also tested. MIN6 cells were plated into 96-well plates and Fura 2-loaded in Ca2+-free solution containing 1 μm thapsigargin (Tg) to deplete ER stores and activate SOCE. The Fura 2 loading solution was replaced with Ca2+-free bath solution containing Tg ± SKF96365. Ca2+ addition to the bath solution induced SOCE that was dose-dependently inhibited by SKF96365 (Fig. 1D).
FIGURE 1.
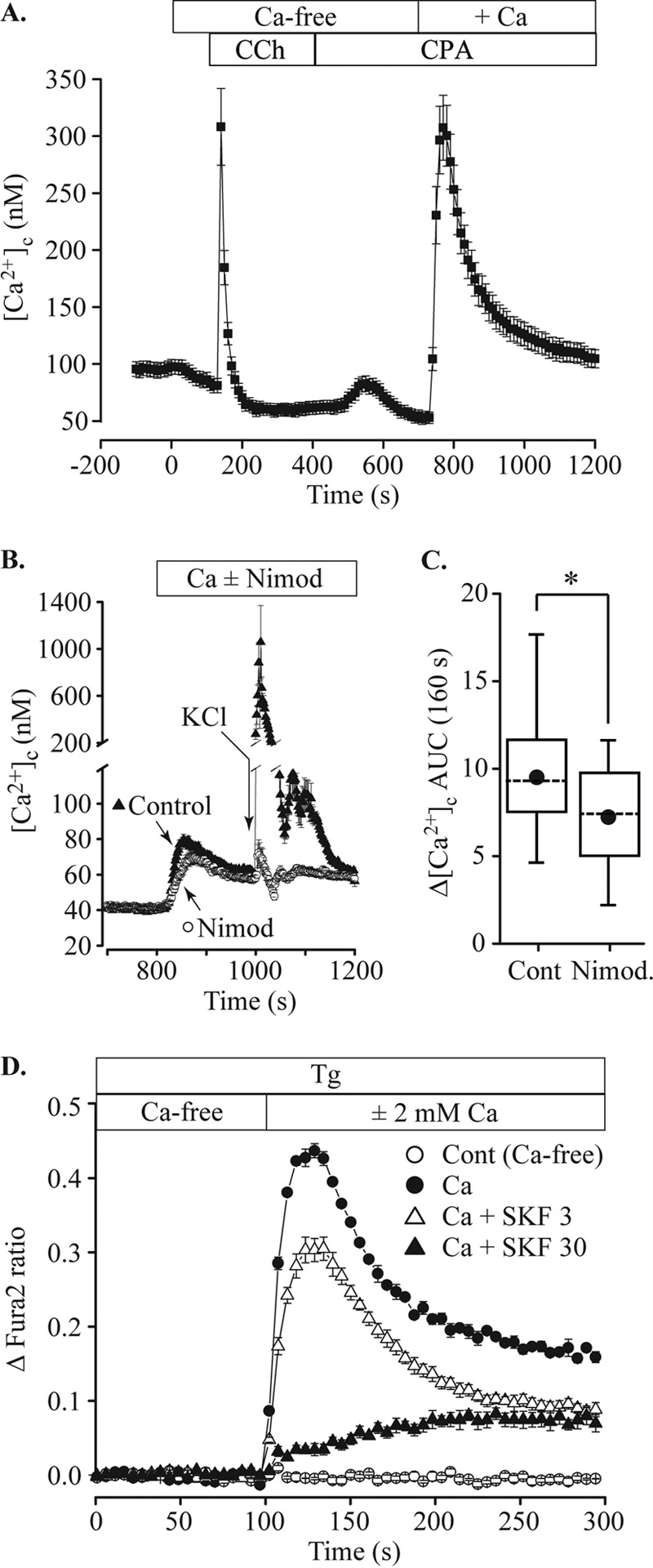
Store-operated Ca2+ entry in MIN6 cells. A, MIN6 cells were loaded with Fura 2 and perifused with test solutions containing 2 mm glucose at 37 °C. Cells were initially bathed in normal, 2.5 mm Ca2+ extracellular solution to establish a baseline. The bath solution was then exchanged for Ca2+-free solution (10 μm EGTA), Ca2+-free with 250 μm carbachol (CCh) followed by Ca2+-free with 10 μm cyclopiazonic acid (CPA) to discharge and prevent refilling of intracellular Ca2+ stores. Reintroduction of 2.5 mm Ca2+ caused a rise of cytosolic [Ca2+] ([Ca2+]c) representing SOCE. B, the addition of 5 μm nimodipine (Nimod) before the reintroduction of extracellular Ca2+ caused a small decrease in the peak amplitude. The efficacy of nimodipine to block voltage-gated Ca2+ channels was confirmed by applying a pulse of 56 mm KCl solution (indicated by arrow). Whereas under control conditions KCl elicited a large rise in [Ca2+]c, this rise was inhibited by nimodipine. C, the AUC of the SOCE response shown in B was measured for the 160-s period after Ca2+ addition and was significantly reduced by nimodipine (52 control cells, 24 nimodipine-treated; *, p = 0.002, ANOVA). D, MIN6 cells in a 96-well plate were loaded with Fura 2 in Ca2+-free solution containing 2 mm glucose and 1 μm Tg. The cells were then washed with fresh bath solution containing Tg. Ca2+-free bath solution (Cont) or Ca2+-containing solution with Tg and SKF96365 (final, 2 mm [Ca2+], SKF96365 concentration indicated in μm) was injected at 100 s. SOCE was dose-dependently blocked by SKF96365.Data are plotted as the mean ± S.E., error bars shown are larger than symbols, averaged from 12 wells for each solution and are representative of three independent experiments.
Knockdown of STIM1 Reduces SOCE in MIN6 Cells
STIM1 functions as a sensor of [Ca2+]ER, coupling ER Ca2+ store content with generation of SOCE and cytosolic Ca2+ oscillations, but knowledge of the role of STIM1 in β-cell Ca2+ signaling is limited. We employed a knockdown strategy using viral transduction with short-hairpin RNAs (shRNA) directed against STIM1. Cells expressing a scrambled sequence with no known target (shRNA-scr, ss) were used as controls. By qPCR screening of RNA isolated from cells expressing different shRNA constructs, we identified a construct (shRNA-STIM1, sh) that significantly reduced STIM1 mRNA expression and protein levels by 84 and 70%, respectively (Fig. 2A). In shRNA-STIM1 cells, SOCE peak amplitude and AUC were 67 and 61%, respectively, lower than shRNA-scr control cells (Fig. 2, B–D). The specificity of shRNA construct targeting and absence of off-target effects were confirmed by transducing shRNA-STIM1 cells with an adenovirus encoding the human isoform of STIM1 (AdV-hSTIM1, av). Reconstituting STIM1 protein expression (Fig. 2A) in this way partially restored SOCE (Fig. 2, B–D). We also used PCR with primers spanning exon 11 to test for the expression of long and short isoforms of STIM1 (21, 22) and found no evidence for expression of the long isoform in MIN6 cells (data not shown).
FIGURE 2.
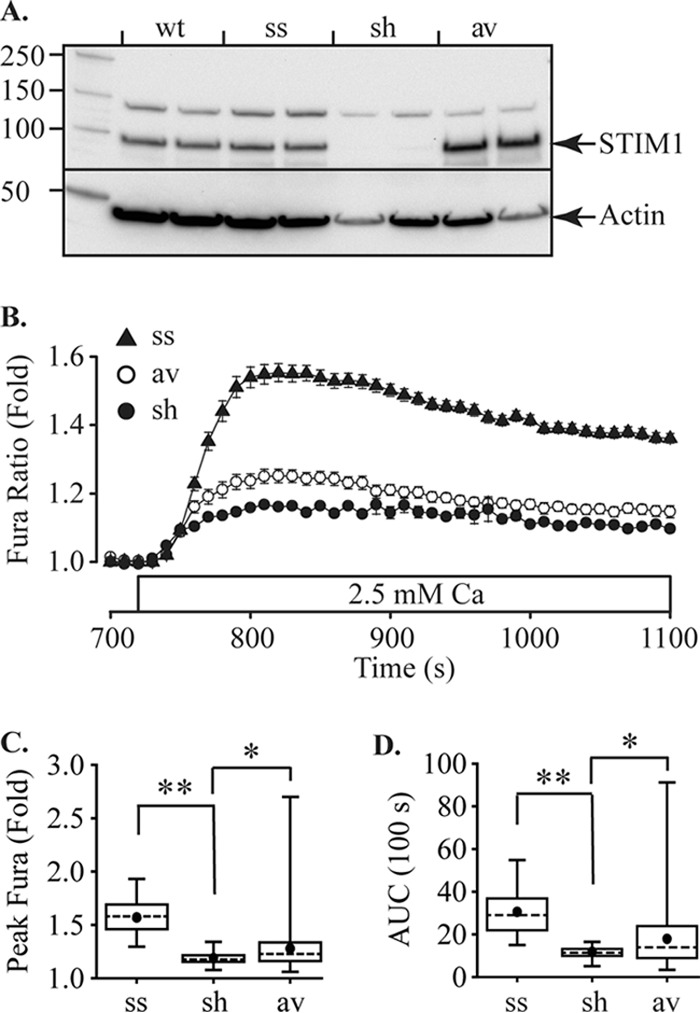
Store-operated Ca2+ entry in MIN6 cells is regulated by STIM1. A, Western blotting shows expression of STIM1 in MIN6 cells and knockdown of STIM1 protein expression by shRNA-STIM1 (sh) relative to either wild-type MIN6 cells (wt) or cells with shRNA-scr (ss). Expression of STIM1 was reconstituted in the shRNA-STIM1 cells by transduction with an adenovirus expression vector encoding the human isoform of STIM1 (av). The left lane shows Mr markers and then duplicate lanes for each cell line. Anti-STIM1 is shown in the top part of the gel and anti-actin as a loading control in the lower part. B, stable MIN6 cell lines were treated using the protocol shown in Fig. 1A. Traces show only the records immediately before and after reintroduction of Ca2+ (indicated by open bar). SOCE in shRNA-STIM1 cells (sh) is inhibited relative to shRNA-scr (ss) and was partially rescued by adenoviral transduction with human STIM1 (av). Traces show the mean ± S.E. The peak SOCE amplitude (C) and AUC for the first 100 s after Ca2+ addition (D) were quantified for ss, sh, and av cells (28, 26 and 139 cells, respectively). Both peak and AUC were significantly reduced for sh cells relative to ss cells and were significantly recovered by av (*, p = 0.05; **, p < 0.01, ANOVA).
Characterization of Inward Currents Activated by STIM1 in β-Cells
After confirming that SOCE occurs in MIN6 cells by Ca2+ imaging, experiments to investigate ion currents were performed in the whole cell configuration using the standard bath solution and Cs+ pipette solution. Cells were transduced with an ER-targeted Ca2+ reporter to confirm that ER stores were depleted by dialysis with the base pipette solution. Under these conditions, an inward current was activated (Fig. 3A, upper trace) that followed depletion of ER Ca2+ stores (Fig. 3A, lower trace). This current reached peak amplitude within 5 min after break-in and showed slow inactivation. The inward current displayed a linear current-voltage relation that reversed at −12.7 ± 2.3 mV (n = 27 cells, Fig. 3B). This property is consistent with previous reports of a store-operated non-selective cation current in β-cells (5). A non-selective blocker of SOCE, SKF96365 (2, 8), reversibly inhibited this inward current (Fig. 3C) by 92.3 ± 2.6% (100 μm, n = 6 cells).
FIGURE 3.
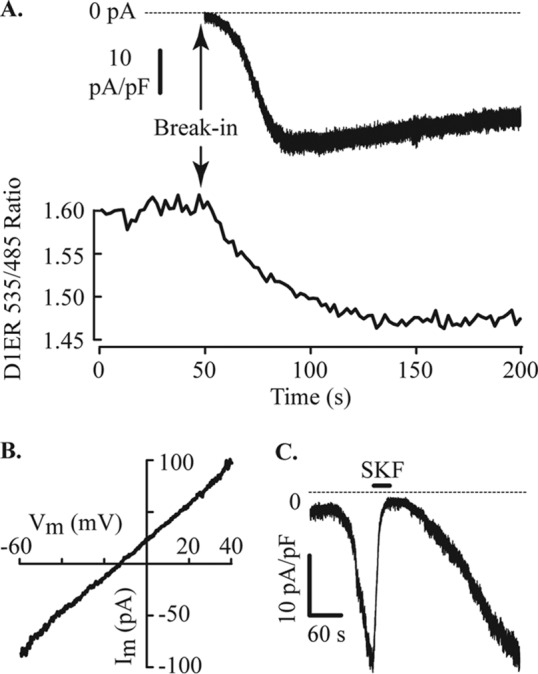
Store depletion and activation of an inward current in MIN6 cells. A, membrane current and [Ca2+]ER were measured simultaneously in cells expressing D1ER, a FRET-based genetically encoded indicator of ER Ca2+. Whole cell recording of membrane current with normal bath solution and Cs+ pipette solution designed to passively deplete ER stores is shown. The FRET ratio decreased, proportional to [Ca2+]ER, immediately after break-in to the whole cell configuration. An inward membrane current started to develop within a few seconds and reached a peak after ∼1 min in this cell. The whole cell capacitance (Cm) of the cell illustrated was 4.2 pF. B, the current-voltage relation of the current after a voltage ramp from the holding potential of −60 mV to +40 mV at 1 V/s. This inward current has a linear current-voltage relation, and the mean reversal potential was −12.7 ± 2.3 mV (n = 27, −13.3 mV in the cell illustrated). C, extracellular application of the channel blocker SKF96365 (100 μm) reversibly inhibits the inward current. Break-in for this whole cell record occurred at the start of the trace. Shown is a representative example of six cells with a mean current inhibition of 92.3 ± 2.6% induced by SKF96365.
We determined ion selectivity of the inward current by substituting cations present in the extracellular solutions. Replacement of extracellular Na+ with N-methyl-d-glucamine (NMDG+) had no effect on the inward current amplitude (Fig. 4A), suggesting that the current is not carried through non-selective cation channels. Given that store depletion activates Ca2+ influx, we applied a Ca2+-free extracellular solution. This Ca2+-free solution increased, rather than inhibited, this current (Fig. 4B), suggesting that the current activated under these conditions is not a pure Ca2+ current but is consistent with extracellular Ca2+ inhibiting influx through K+ channels (23, 24).
FIGURE 4.
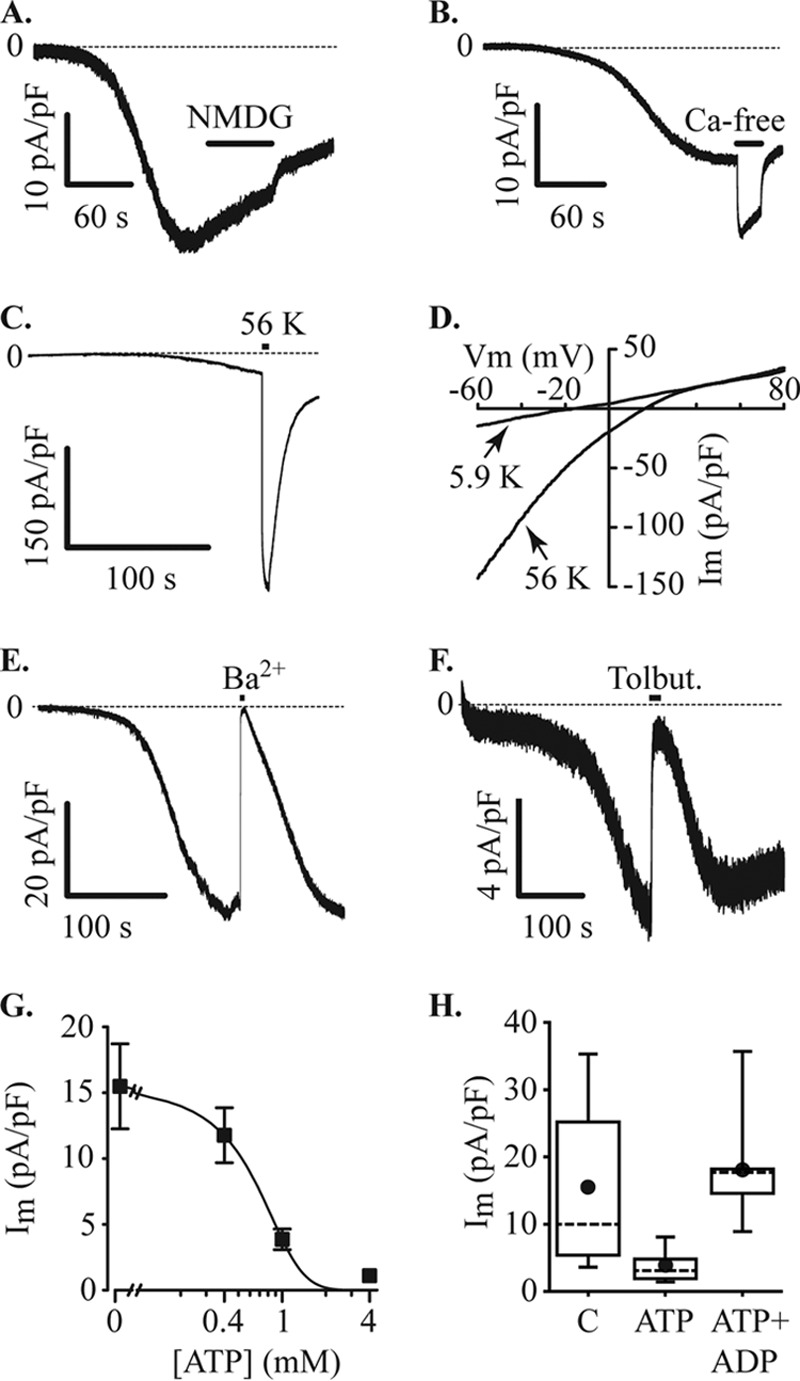
Inward currents through KATP channels. MIN6 cells were voltage-clamped at −60 mV in the whole cell configuration with a Cs+ pipette solution containing 200 μm EGTA. Under these recording conditions, with normal bath solution, an inward current developed, and the effects of ion substitution were tested. A, the current was not inhibited by substituting extracellular Na+ with NMDG+ (NMG, application indicated by filled bar). B, the inward current amplitude increased upon removal of extracellular Ca2+ (Ca-free, filled bar). C, raising extracellular K+ from 5.9 to 56 mm (56 K, filled bar) produced an increase in current amplitude (−30.9 ± 9.1 pA/pF at normal extracellular K+ and −247.2 ± 75.0 pA/pF at 56 mm K+, n = 7). D, increasing K+ produced a positive shift of the reversal potential for the current. In the cell illustrated, the reversal potential shifted from −16.1 mV to +15.9 mV and recovered to −14.3 mV, the mean change was from −17.6 ± 2.5 mV at normal (5.9 mm K+) to +13.0 ± 3.1 mV at 56 mm K+ and −15.2 ± 2.2 after returning to normal K+. Im, inward current. E, substitution of extracellular Ca2+ for Ba2+ also inhibited the inward current. Current inhibition caused by a 10-s pulse of Ba2+ was 97.1 ± 1.2% (n = 7). F, the inward current was reversibly inhibited by 100 μm tolbutamide (Tolbut). G, increasing concentrations of ATP in the pipette solution inhibited the inward current (Im). Data points were curve-fitted indicating an IC50 of 0.6 mm (data from 9–13 cells at each concentration). H, inhibition of inward current (Im) by 1 mm ATP was reversed by 1 mm ADP (C, control (no ATP), n = 13 cells; ATP, n = 9 cells; ATP+ADP, n = 9 cells).
The base pipette solution used in these studies is K+-free, and the extracellular K+ is 5.9 mm. Under these conditions the predicted K+ reversal potential is expected to approximate the observed reversal potential of the inward current. To determine whether the observed inward current could be carried by K+, we tested the effects of raising extracellular K+ to 56 mm. This increase of extracellular K+ induced a large increase in current amplitude (Fig. 4C) accompanied by a positive shift in the reversal potential of the current (Fig. 4D). The reversal potential shifted from −17.6 ± 2.5 mV to +13.0 ± 3.1 mV and recovered to −15.2 ± 2.2 mV (n = 7, measured using voltage ramps). This inward current was reversibly inhibited by substitution of extracellular Ca2+ with Ba2+ (Fig. 4E) and by 100 μm tolbutamide (Fig. 4F), suggesting involvement of KATP channels. This hypothesis was tested by adding different concentrations of ATP to the patch pipette solutions. The addition of ATP dose-dependently reduced inward current amplitudes with an IC50 of 0.6 mm (Fig. 4G). The inhibitory effect of ATP (1 mm) was reversed by simultaneous addition of an equal concentration of ADP (Fig. 4H). These data suggest that this current is an inward current passing through KATP channels. The inhibitory effect of SKF96365 (Fig. 3C) is consistent with this suggestion as this compound was recently shown to reversibly inhibit KATP channels in smooth muscle cells (25).
Several studies have described SOCE in β-cells (17), suggesting that a SOC current is activated in MIN6 cells after ER Ca2+ store depletion. To unmask the potential contribution of a SOC current, we used a Cs+ pipette solution in combination with bath solutions containing either glyburide (1 μm), a sulfonylurea that blocks KATP channels, or with tetraethylammonium chloride (TEA; 140 mm, substituted for Na+) to block inward currents through K+ channels. In the presence of extracellular glyburide, an inward current was observed (Fig. 5A) with a reversal potential similar to that measured in the absence of glyburide (Fig. 5B). These data are inconsistent with a highly Ca2+-selective current conducted by Orai channels and suggest that if a SOC current is present, it was not adequately isolated by adding extracellular glyburide. However, in the presence of extracellular TEA, dialysis of wild-type MIN6 cells to deplete ER stores generated a small inward current with a mean current density of 0.5 ± 0.1 pA/pF (n = 7 cells); this current was inhibited by exposing the cells to a Ca2+-free bath solution (Fig. 5C). This small Ca2+ current is likely to represent activation of SOC channels in β-cells. Under these recording conditions, activation of the store-independent arachidonic acid-regulated Ca2+ (ARC)-channel current present in β-cells (26) is not expected to occur.
FIGURE 5.
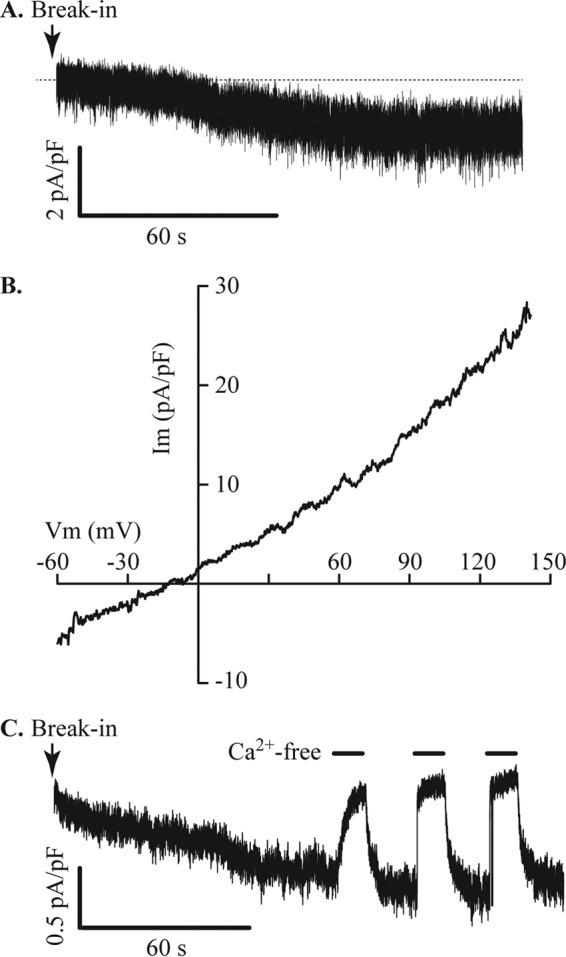
Store-operated Ca2+ current in MIN6 cells. A, glyburide (1 μm) and nimodipine (10 μm) were added to the bath solution to block KATP and L-type VDCCs, respectively, in cells dialyzed with Cs+ pipette solution containing 5 mm EGTA. Under these conditions a small inward current developed. B, measurement of the reversal potential of the current shown in A indicates that this current is not Ca2+-selective. Im, membrane current; Vm, membrane potential. C, using an extracellular solution containing TEA (substituted for Na+) to block inward K+ currents through KATP channels, a small inward current carried by Ca2+ was revealed. This current was inhibited by transient puffer applications of Ca2+-free bath solution, indicated by filled bars. The current illustrated is representative of seven cells with a mean current density of 0.5 ± 0.1 pA/pF.
The store-operated Ca2+ current (Fig. 5C) in shRNA-scr MIN6 cells was 0.2 ± 0.02 pA/pF (n = 11 cells). Knockdown of STIM1 expression in shRNA-STIM1 cells suppressed this current below the limit for reliable measurement. These data are consistent with the expression of a STIM1-regulated store-operated Ca2+-influx pathway in MIN6 cells similar to that described in other types of cells (27).
STIM1 Regulates KATP Currents in MIN6 Cells
Ca2+ imaging and electrophysiology data shows that knockdown of STIM1 using shRNA results in the predicted reduction of SOCE in MIN6 cells. However, STIM1 has previously been shown to interact with other target proteins including an inhibitory effect on CaV1.2 L-type Ca2+ channels (28–30) that is important for triggering insulin secretion (31, 32). To test for the potential effects of STIM1 on alternative targets important for regulating insulin secretion from β-cells, we measured membrane currents in cells stably expressing shRNA-STIM1 or control shRNA-scr. Knockdown of STIM1 reduced the amplitude of the KATP current by 54% (11.1 ± 1.6 pA/pF, n = 18 cells and 5.1 ± 1.1 pA/pF, n = 30 cells) for shRNA-scr (ss) and shRNA-STIM1 cells (sh), respectively (Fig. 6A). Reconstitution of STIM1 expression by AdV-hSTIM1 transduction restored this current, and current densities in shRNA-STIM1 cells and AdV-hSTIM1/shRNA-STIM1 (av) cells were 2.8 ± 1.3 pA/pF (n = 6 cells) and 18.8 ± 2.4 pA/pF (n = 8 cells), respectively (Fig. 6A). We detected no effect of shRNA-Stim1 on voltage-dependent Ca2+ currents (Fig. 6B) or Ca2+-dependent exocytosis (Fig. 6C).
FIGURE 6.
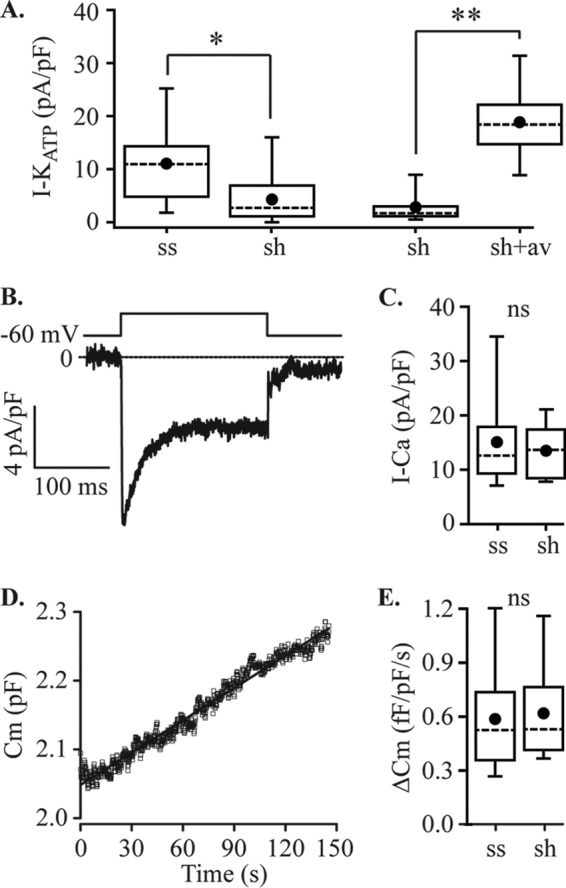
Effects of STIM1 knockdown on MIN6 cell membrane currents and Ca2+-dependent exocytosis. A, ShRNA-STIM1 (sh) reduced the amplitude of the inward current recorded with normal bath and Cs+ pipette solutions, as shown in Fig. 3, by ∼50% relative to shRNA-scr (ss) controls (*, p = 0.014; ss, n = 18 cells; sh, n = 24 cells), and this effect was reversed by adenoviral transduction of sh cells with human STIM1 (sh, n = 6 cells; sh + av, n = 8 cells; **, p < 0.001, ANOVA). B and C, STIM1 knockdown had no effect on the amplitude of voltage-dependent Ca2+ currents measured using the same solutions with voltage steps from −60 mV to 0 mV (ss, n = 7 cells; sh, n = 8 cells, not significantly different, ANOVA). A representative example of a record is shown in B, and quantification of peak current data for ss and sh cells is shown in C. D and E, exocytosis measured by capacitance tracking in cells dialyzed with 1.5 μm Ca2+ was also unaffected by STIM1 knockdown. A representative example of a capacitance record is shown in D, and quantification of the data for ss and sh cells shown in E (ss, n = 10 cells; sh, n = 8 cells; ns, not significantly different, ANOVA). The rate of exocytosis was determined by linear fitting of the data using Origin software. For the cell shown in D, the linear fit is illustrated and gave a y intercept of 2.05 pF, used to normalize data to cell size, and the slope was 0.76 fF/pF/s. Cm, whole cell capacitance.
STIM1 Protein Interacts with the Sulfonylurea Receptor 1 (SUR1) Subunit of KATP Channels
To further investigate the potential role of STIM1 in regulating KATP channels, we performed immunoprecipitation studies to determine whether there was a physical interaction between STIM1 and the KATP channel subunits SUR1 and Kir6.2. For these studies mouse islet protein extracts or lysates from HEK293T cells transfected with human STIM1(N131Q/N171Q) and FLAG-tagged SUR1 with or without Kir6.2 were used. The Asn-131/171 mutations in STIM1 prevent glycosylation and cell surface expression (33). Cell lysates were subject to immunoprecipitation (IP) using antibodies against SUR1, FLAG, or STIM1 with pulldown using protein A-agarose beads followed by Western immunoblotting (IB). All three lysates showed pulldown of STIM1 by the anti-SUR1 antibody (Fig. 7A). These data suggest an interaction between STIM1 and SUR1 as pulldown occurred in the absence of Kir6.2 in the HEK cell lysates (Fig. 7, C and D). This experiment was reversed with IP by anti-STIM1 and IB using anti-SUR1. Pulldown of SUR1 was observed in all three samples and was enhanced by poly-lysine (50 μg/ml) addition during the IP step (Fig. 7B). Incubation of cell lysates with anti-FLAG antibody is shown to pull down STIM1 in cells expressing FLAG-SUR1 but not in cells that did not express FLAG-SUR1 (Fig. 7C). Similarly, cell lysates treated with protein A-agarose beads alone did not pull down STIM1. These data demonstrate that the pulldown of STIM1 is not due to direct interaction of the FLAG antibody or of the beads with STIM1. Pulldown of STIM1 using anti-FLAG IP was observed in either the presence or absence of Kir6.2 (Figs. 7, A–D), suggesting that STIM1 interacts with SUR1.
FIGURE 7.

Interaction of STIM1 and KATP. A, IP studies using mouse islet (mIslet) extracts or HEK293T cell extracts expressing STIM1 and FLAG-SUR1 in the presence or absence of Kir6.2. IP was performed using anti-SUR1 antibody followed by IB with anti-STIM1.The SUR1 antibody pulled down STIM1 in all three samples. The input protein (IB only with anti-STIM1) is shown in the right three lanes. The band indicated as HC represents the heavy chain of the IP antibody, detected by the secondary antibody, and is only seen in the three IP samples. Co-immunoprecipitation was observed in two independent experiments using mouse islet or HEK cell proteins performing IP with anti-STIM1 and IB with anti-SUR1. B, IP was performed using anti-STIM1 and IB with anti-SUR1 using the same protein samples as in A, representative of two independent experiments with this protocol. The STIM1 antibody pulled down SUR1 in all three samples, and pulldown was enhanced by adding 50 μg/ml poly-lysine to the samples during IP. The SUR1 antibody exhibited a secondary band of slightly higher molecular weight than the heavy chain band, and subsequent experiments were performed using anti-FLAG to avoid any confounding effects of this band. LC represents the light chain of the IP antibody, and In is the mouse islet input protein. C, control IP studies using HEK293T cells expressing STIM1 in the presence or absence of FLAG-SUR1. IP performed with protein A-agarose beads (Beads) alone with samples expressing STIM1 and FLAG-SUR1 failed to pull down STIM1. IP with the FLAG antibody in cells expressing STIM1 but not FLAG-SUR1 also failed to pull down STIM1. IP using the FLAG antibody did, however, pull down STIM1 in cells expressing both STIM1 and FLAG-SUR1. STIM1 was detected by IB with anti-STIM1 along with nonspecific bands for the heavy-chain (HC) and light-chain (LC) of the FLAG antibody in the two FLAG-IP lanes but not the beads-only lane. IP of STIM1 using the FLAG antibody was observed in six experiments using four different protein samples. D, IP with the FLAG antibody pulled down STIM1 in the presence or absence of Kir6.2 under control conditions, and this pulldown was enhanced by the addition of 50 μg/ml poly-lysine (PolyLys). The image shown is representative of three independent experiments, and the increase in band density with poly-lysine was 27 ± 3% (n = 9, range 17 −44%) relative to control for cells expressing STIM1 and FLAG-SUR1 and 11 ± 1% (n = 6, range 8–16%) for cells expressing STIM1, FLAG-SUR1, and Kir6.2. Densities of STIM1 IB bands were significantly higher for poly-lysine-treated extracts relative to controls for these samples with STIM1 and FLAG-SUR1 (paired t test, p = 0.003) and also for samples with STIM1, FLAG-SUR1, and Kir6.2 (paired t test, p < 0.001). E, the STIM1 band density was 97 ± 2% (n = 6, range 92–102%, not significantly different, paired t test) relative to control for cells expressing STIM1 and FLAG-SUR1 and 94 ± 5% (n = 6, range 83–105%, not significantly different, paired t test) for cells expressing STIM1, FLAG-SUR1, and Kir6.2. F, the FLAG band density was significantly lower with poly-lysine (91 ± 6%, n = 10, range 75–133%, p = 0.04, three independent experiments, paired t test) relative to control for cells expressing STIM1 and FLAG-SUR1 and 83 ± 5% (n = 7, range 74–99%, p = 0.03, two independent experiments, paired t test) for cells expressing STIM1, FLAG-SUR1, and Kir6.2.
Poly-lysine is known to reverse the stimulatory action of phosphatidylinositol 4,5-bisphosphate (PIP2) on KATP channels (34) and enhance sulfonylurea binding to SUR1 (35). PIP2 also inhibits the binding of syntaxin-1A to SUR1, reversing syntaxin-1A mediated KATP channel inhibition (36). These studies led us to investigate whether poly-lysine might influence the interaction of STIM1 with SUR1 either through the effects on PIP2 or by competing with the lysine-rich C terminus of STIM1 for binding. The interaction of STIM1 and FLAG-SUR1 was enhanced by the addition of 50 μg/ml poly-lysine to the IP reaction (Fig. 7D). This effect was more pronounced in studies with STIM1 and FLAG-SUR1 without Kir6.2 than in studies with Kir6.2 (3 independent experiments, mean band density 1.27 ± 0.03 higher with poly-lysine for samples expressing STIM1 and FLAG-SUR1 and 1.11 ± 0.01 higher for samples with STIM1, FLAG-SUR1, and Kir6.2). This enhancement is similar to the reported effect of poly-lysine to increase sulfonylurea binding to SUR1 by ∼30% (35). To determine whether the effect of poly-lysine was due to enhanced binding of the IB antibody to its target, we performed control Western blots using anti-STIM1 (Fig. 7E) or anti-FLAG (Fig. 7F) in the presence or absence of poly-lysine. Poly-lysine had no significant effect on anti-STIM1 binding and slightly, but significantly, decreased anti-FLAG binding. These data suggest that the enhanced IP signal in the presence of poly-lysine is due to a change in binding of STIM1 and SUR1.
To further investigate the interaction of STIM1 with SUR1, we transfected cells with STIM1 and a plasmid encoding the FLAG-tagged nucleotide binding fold-1 (FLAG-NBF1) region of SUR1. NBF1 has previously been shown to play a role in binding EPAC2 (37) and syntaxin-1A (38, 39). IP studies revealed that STIM1 and FLAG-NBF1 co-immunoprecipitated, suggesting that NBF1 forms at least one interaction site between STIM1 and SUR1 proteins (Fig. 8).
FIGURE 8.
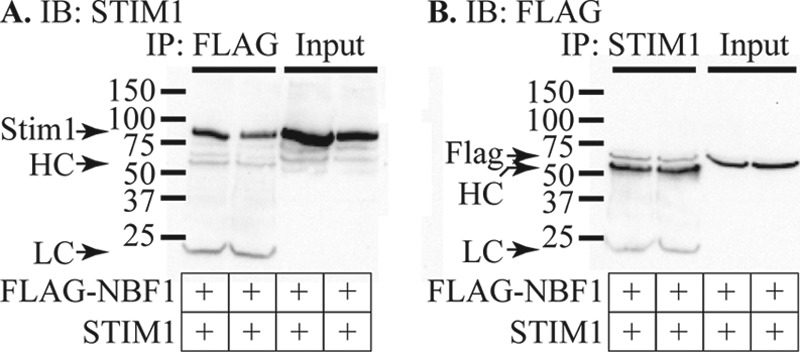
STIM1 interacts with NBF-1 of SUR1. HEK293T cells were transfected with STIM1 and FLAG-NBF1 and IP performed using anti-FLAG (A) or anti-STIM1 (B). The data shown are representative of three independent IP experiments using four protein samples from different transfections. A, two protein samples from different transfections were collected and subject to IP using anti-FLAG followed by IB using anti-STIM1. The two left lanes show the results of IP, whereas the two right lanes show the corresponding input protein. The bands marked HC and LC that appear only in the IP lanes represent the heavy and light chains of the IP antibody. B, the same two protein samples as used in A were subjected to IP using anti-STIM1 followed by IB with anti-FLAG. IP lanes are shown on the left side of the panel, whereas the input protein is shown on the right. IP with anti-STIM1 pulled down FLAG-NBF1 and additional bands for the heavy and light chains of the IP antibody can be seen only in the IP lanes.
To determine whether shRNA-STIM1 has an off-target effect on expression of the KATP channel subunits SUR1 and inwardly rectifying K+ channel 6.2 (Kir6.2), we performed qPCR. SUR1 and Kir6.2 mRNA levels were not different in cells expressing shRNA-scr or shRNA-STIM1. In three independent experiments performed with triplicate samples in each, Kir6.2 expression in shRNA-STIM1 cells ranged from 92 to 117% of shRNA-scr controls, and SUR1 expression was 104–157% of controls. These data indicate that the reduction in inward current amplitude in shRNA-STIM1 cells was not due to decreased expression of KATP channel subunits and are consistent with an unexpected regulatory role for STIM1 in the control of KATP channel activity.
Discussion
Previous work in MIN6 cells as well as mouse and human primary β-cells demonstrated translocation and accumulation of fluorescently labeled STIM1 to punctae in subplasmalemmal ER after ER Ca2+ store depletion with thapsigargin or cyclopiazonic acid, inhibitors of SERCA (sarco(endo)plasmic reticulum calcium ATPase) pumps (18, 19). The role of endogenous STIM1 in β-cell signaling was not established in those studies. Here, we demonstrate that STIM1 regulates SOCE in MIN6 cells. We also discovered that STIM1 interacts with SUR1, a subunit of KATP channels, and functions in KATP channel activation. Binding of STIM1 to SUR1 was detected in transfected HEK293T cells as well as mouse islets of Langerhans. Our data indicate that STIM1 interacts with multiple proteins in β-cells that contribute to the regulation of β-cell Ca2+ signaling dynamics and membrane excitability. These findings suggest that STIM1 is a regulator of multiple signaling events in insulin-secreting cells.
It is known that SOCE in non-β-cells is regulated by direct interaction of STIM1 with the Ca2+ channel protein Orai1 and other regulatory proteins (27). Whether endogenously expressed Orai1 and STIM1 functionally interact and form the molecular basis of SOCE in β-cells has not been established. In view of previous reports that store depletion activates a non-selective cation current in β-cells (8), it seems unlikely that Orai1 would be the sole channel-forming subunit as these channels are highly Ca2+ selective when activated by STIM1 (40). Non-selective store-operated currents can be formed by an Orai1-TRPC1 complex regulated by STIM1 (41–45). An alternative possibility is that STIM1-Orai1 forms the SOCE mechanism and that the previously reported non-selective current represents the secondary activation of Ca2+-sensitive Trp channels. Ca2+-activated TRPM4 and TRPM5 channels have been shown to play a role in regulating insulin secretion (46–49). Expression of a Ca2+-selective SOCE mechanism distinct from previously reported non-selective cation currents would be consistent with the small currents we observe using whole cell recording.
An unexpected discovery from our experiments was that knockdown of STIM1 using shRNA reduced KATP channel activation. We considered the possibility that this represented an “off target” effect of the shRNA. However, reconstitution of KATP channel activation in shRNA-STIM1-expressing cells transduced with an adenovirus construct that encoded human STIM1 excludes a nonspecific effect of shRNA-STIM1. This suggests that STIM1 plays a role in regulating KATP channel activity. Quantitative PCR assays indicated that shRNA-STIM1 did not affect SUR1 or Kir6.2 expression, therefore, indicating it unlikely that STIM1 participates in KATP channel subunit biosynthesis. Nevertheless, our studies raise the prospect that loss of STIM1 expression either directly impacts KATP channel gating or indirectly modifies KATP channel activation via interaction with KATP channel regulatory proteins. A physical interaction between STIM1 and SUR1 mediated through NBF1 is suggested by our immunoprecipitation data, but it is not yet clear whether this is a direct interaction or indirect through an intermediary protein.
At least two mechanisms for direct interaction of STIM1 and SUR1 are possible based on the distinct mechanisms by which STIM1 regulates Orai and TRPC1. Orai1 is activated by interaction of the STIM-Orai activation region (SOAR) of STIM1 with cytosolic N- and C-terminal domains of Orai1 (50). In contrast, STIM1 activates TRPC1 through an electrostatic interaction of a KK amino acid pair in STIM1 with a DD pair in TRPC1 (51). In silico analysis of human SUR1 reveals a region with homology to the extended transmembrane Orai1 N-terminal (ETON) domain of Orai1 that interacts with STIM1 and also four DD pairs, including two pairs within NBF1. NBF1 is known to be important for MgADP activation of KATP (52). Additional experiments will be necessary to establish whether either of these sites plays a role in the molecular mechanism for STIM1-regulated KATP activity.
A number of STIM1 binding partners have been identified in addition to Orai and TRP channels (53, 54). Whether any of these alternative binding partners might contribute to an indirect effect on KATP channel regulation by STIM1 is unknown. One possibility is that actin binds STIM1 (53) and the actin cytoskeleton regulates KATP channels in β-cells (55, 56). Leptin is a hormone that activates KATP channels (55, 57), an effect mediated by disruption of actin filaments (55). Recent work has shown that Ca2+ influx through store-operated channels promotes actin depolymerization (58), raising the possibility that co-localization of STIM1 with KATP channels might promote local actin depolymerization and channel activation. Indeed, studies of STIM1 translocation in β-cells indicate aggregation of STIM1 punctae in actin-poor regions of the membrane (18). Thus, the effect of STIM1 on KATP might be an indirect effect of localized Ca2+ influx through Orai channels causing cytoskeletal actin disruption.
Whether the regulation of KATP activity is mediated by STIM1 in the ER, similar to the regulation of Orai1 and SOCE, or by plasma membrane-resident STIM1, similar to the ARC (arachidonic acid-regulated Ca2+) channel, is unknown at present. However, our immunoprecipitation studies indicate that a mutated STIM1, which is not glycosylated and inserted in the PM (33), and SUR1, expressed in the absence of Kir6.2, interact. These data suggest that STIM1 and SUR1 interact within the ER but do not exclude interplay of PM STIM1 with SUR1.
It is interesting to note that whereas reconstitution of the expression of STIM1 in MIN6 cells using human STIM1 restored KATP activity to above control levels, SOCE was only partially restored. Western blot analysis revealed that our rescue studies increased STIM1 protein levels above that seen in control cells (Fig. 6). A recent report indicates that high STIM1 expression can trap Orai1 intracellularly and prevent its trafficking into the plasma membrane (59). Our observation of partial rescue of SOCE may be attributable to an unfavorable ratio of STIM1 to Orai1 given that Adv-hSTIM1 raised expression of STIM1 protein to 7–12-fold higher than endogenous levels. KATP channels also undergo dynamic trafficking to the PM (60–63). Our data indicating that overexpression of STIM1 fully restored KATP activity but only partially restored SOCE might indicate that STIM1 does not affect KATP translocation in the same way as Orai1, if the trafficking trap mechanism applies in our studies. However, whether STIM1 influences KATP trafficking remains to be determined.
The KATP channel is well established as a key regulator of β-cell electrical excitability in response to nutritional status and hormonal inputs (57, 64, 65). Our data indicate that STIM1, through either direct or indirect interaction, regulates the activity of KATP channels in addition to regulating SOCE and plays a role in regulating β-cell excitability through these two processes. We propose a working model that incorporates a physiological role for STIM1 in regulating KATP channel activity and thereby β-cell membrane potential and Ca2+ oscillations. After β-cells are exposed to a high concentration of extracellular glucose, an initial fall in cytosolic Ca2+ concentration occurs (termed Phase 0) (66, 67). This phase of the Ca2+ response to glucose stimulation precedes β-cell depolarization and is caused by glucose-induced Ca2+ uptake into ER Ca2+ stores (67). Estimates of the resting [Ca2+]ER in β-cells are 250–500 μm (68–70), a similar range to estimates for the KD of Ca2+ binding to the EF-SAM domain of STIM1 (71, 72). This suggests that store filling induced by glucose elevation might decrease STIM1 activity and potentially play a role in regulating KATP. An increase in [Ca2+]ER will induce translocation of STIM1 away from the plasma membrane to inactivate SOCE. A second possibility is that the rise in intracellular ATP will cause displacement of STIM1 from binding to SUR1, analogous to the effects of ATP on syntaxin binding (39). Displacement of STIM1 from SUR1 by ATP would contribute to the inhibition of KATP channels after a rise in blood glucose.
Our new observation that STIM1 knockdown inhibits KATP activation suggests that STIM1 has a positive effect on channel activity. Therefore, the translocation of STIM1 away from the plasma membrane or displacement of STIM1 from SUR1 might be predicted to reduce KATP activity and could contribute to channel inhibition after a rise in blood glucose. The importance of this mechanism relative to inhibition caused by the increase in the ATP/ADP ratio is unknown. However, STIM1-mediated changes in SOCE and KATP activity could contribute to the initiation of cell depolarization and [Ca2+]c oscillations. In contrast, release of intracellular stores will cause translocation of STIM1 to PAM regions where it can interact with and activate Orai1 to refill stores and perhaps also interact with and activate KATP.
It has been demonstrated that SOCE activates type 8 adenylyl cyclase in both HEK 293 cells and MIN6 cells (73). This effect could lead to elevation of intracellular cAMP that is an important regulator of β-cell electrical activity and secretion mediated by protein kinase A and Epac2 (64, 74). A role for SOCE in regulating [Ca2+]c oscillations in β-cells has been proposed as a consequence of ATP consumption and regulation of KATP activity (75). Metabolic oscillations and slow changes in KATP activity have been proposed to be important for glucose-induced oscillations (76). It is possible that slow changes in KATP activity as a consequence of store depletion and refilling and trafficking of STIM1 might contribute to normal bursting activity in islets.
In summary, we report that STIM1 is a multifunctional regulator of signaling dynamics in β-cells. In addition to defining the role of STIM1 as a regulator of SOCE in β-cells, our data revealed that STIM1 participates in KATP channel activation through an interaction with SUR1. This suggests a novel pathway for the control of β-cell excitability and function by interplay between ER Ca2+ stores and KATP channels.
Experimental Procedures
Cell Culture
MIN6 cells (passages 19–32) and HEK293T cells were cultured at 37 °C with 5% CO2 in Dulbecco's modified Eagle's medium with 25 mm d-glucose (DMEM-H, Invitrogen) supplemented with 10% fetal bovine serum (Hyclone), 100 units/ml penicillin, and 10 μg/ml streptomycin (Gibco).
Cytoplasmic Ca2+ Imaging
[Ca2+]c was measured using Fura 2 as previously described (77). MIN6 cells were grown on glass coverslips, and the cells were incubated for 15 min at 37 °C with 1 μm Fura 2 acetoxymethyl ester (Invitrogen), 0.02% (v/v) Pluronic acid in Krebs-Ringer buffer (KRBH). KRBH contained 119 mm NaCl, 25 mm NaHCO3, 4.7 mm KCl, 1.2 mm MgSO4, 2.5 mm CaCl2, 1.2 mm KH2PO4, 10 mm HEPES-NaOH (pH 7.4), and 2 mm d-glucose. All solutions were made using ultrapure water. After loading, cells were transferred onto the temperature-controlled stage of an inverted microscope (Nikon 2000u, Nikon Ti or Olympus IX81) and visualized with a 40× superfluor oil immersion objective. Cells were superfused continuously at 37 °C with KRBH. Fura 2 excitation wavelengths were 340 and 380 nm, and the fluorescence emission was detected at 510 nm. The ratio of the emission intensities of Fura 2 excited at 340 and 380 nm (R340/380) were recorded using MetaFluor software (Molecular Devices) for image acquisition and analysis (78).
[Ca2+]c measurement in some studies was performed using a FlexStation 3 plate reader in 96-well plate format using solutions and wavelengths for recording as described above. Test solutions were injected into individual wells containing MIN6 cells at 60–80% confluence. Experimental parameters and data acquisition were controlled using SoftMax Pro 5.4.6 software (Molecular Devices).
Electrophysiology
Whole cell recordings of membrane currents were made at room temperature (21–24 °C) using an EPC-9 amplifier controlled by PatchMaster software (HEKA Electronik, Lambrecht/Pfalz, Germany). Electrodes were pulled from patch clamp glass capillaries (catalog no. G85150T-4, Warner Instruments, Hamden, CT) using a Sutter P97 puller. MIN6 cells were bathed in KRBH. Isosmotic substitution of NaCl (NMDG-Cl or KCl) was used in some experiments, or CaCl2 was omitted (Ca+-free, 10 μm EGTA) or substituted with BaCl2 (in this Ba2+ solution, MgSO4 was replaced with MgCl2). For some experiments a TEA bath solution comprised 140 mm TEA-Cl, 10 mm CaCl2, 1.2 mm MgCl2, and 10 mm HEPES (pH 7.4). Glucose was added to these solutions as indicated. Patch pipettes were filled with a base solution containing 90 mm Cs2SO4, 10 mm NaCl, 1 mm MgCl2, 5 mm HEPES, and 0.2 mm EGTA, pH adjusted to 7.4 with CsOH. ATP, ADP, and GTP were added to the pipette solution as indicated. Cells were held at −60 mV for current recording, and voltage ramps were applied to measure reversal potentials. PatchMaster software compensated junction potentials and was used to generate voltage ramps at 1 V/s, the software was also used to measure cell capacitance to normalize current amplitudes to cell size. Whole cell capacitance measurements of Ca2+-dependent exocytosis were made using the LockIn module of PatchMaster with an applied sine wave of 10 mV amplitude at 1 kHz and a pipette solution containing (in mm) 125 cesium glutamate, 10 NaCl, 10 KCl, 1 MgCl2, 10 EGTA, 11 CaCl2, 3 MgATP, 0.2 LiGTP, and 5 HEPES (pH 7.4). Extracellular test solutions were applied by pressure ejection from glass micro-capillaries using a PicoSpritzer III (Parker Instrumentation). Experiments with fluorescence imaging used a PTI DeltaRam monochromator light source controlled with MetaFluor software. For FRET experiments, emission images at 485 and 535 nm were captured simultaneously using a DualView beam splitter and MetaFluor software.
Quantitative Real-time PCR
Total RNA was isolated and purified from two to four replicate plates of each cell type using an RNeasy Mini kit (Qiagen) according to the manufacturer's instructions. RNA (1 μg) was reverse-transcribed to cDNA using oligo(dT15) primers according to the manufacturer's instructions (Promega). Primer sequences for S18 were 5′-GCCATCACTGCCATTAAGGG and 5′-CCAGTCTGGGATCTTGTACTG and for STIM1 were 5′-GAAGCAAAATGCAGAGAGGC and 5′-CATCATCCAGGGAAGAGCTG. Primers for Kir6.2 were 5′-GTGTTCACCACGCTGGTGGAC and 5′-CAGGTCACCGTGGGCGAAGG. Primers for SUR1 were 5′-GGACAGAAGATCGGGATCTGC and 5′-GAAGGAGGACTTCCCACTGCC. Each primer was used at a concentration of 500 nm in a 25-μl reaction volume containing Brilliant II SYBR Green qPCR master mix (Agilent Technologies). Quantitative real-time PCR was completed in a Stratagene MX3000P real-time PCR thermal cycler under the following conditions: initial hold at 95 °C for 10 min followed by 40 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s. Fluorescence intensity data were collected during each cycle at 72 °C. Amplicon dissociation was initiated immediately after the final PCR extension, with a 30-s hold at 72 °C followed by increasing the temperature to 95 °C with 0.25 °C steps. One cDNA was produced from each RNA sample, and each sample was tested in duplicate by qPCR. Cq values were averaged for duplicates, and relative quantification calculations were completed as described by Pfaffl (79).
Inhibition of STIM1 Expression Using shRNA
Cells in which STIM1 expression was transiently or stably deleted were used. For transient suppression of STIM1, MIN6 cells were transfected with a plasmid vector that expressed small interfering RNA (siRNA) targeted to STIM1. MIN6 cells stably expressing short hairpin RNA targeted to STIM1 (shRNA-STIM1) or shRNA containing a scrambled sequence that targeted no known genes (shRNA-scr) were produced by transduction with lentiviral particles (Santa Cruz Biotechnology). Stably transduced cells were maintained in DMEM-H medium containing 2 μg/ml puromycin. In some experiments, STIM1 expression was reconstituted by transducing cells with an adenoviral vector to express the human isoform of STIM1 (AdV-shRNA-hSTIM1) (ViraQuest Inc.).
Western Immunoblotting of Endogenous MIN6 Protein
Western blotting was used to supplement qPCR to assess the effectiveness of STIM1 knockdown and restoration in cells stably expressing shRNA-STIM1. Whole-cell protein lysates were collected from two replicate plates of MIN6 shRNA-STIM1, shRNA-STIM1/AdV-hSTIM1, shRNA-scr, and control, wild-type cells. Lysis buffer contained 69 mm Tris-HCl, 1% SDS, 10% glycerol, and 0.04% protease inhibitor mixture (Roche Applied Science) (pH 6.8). Lysates were immediately frozen and stored at −80 °C until use. Samples were prepared for SDS-PAGE by adding β-mercaptoethanol (10% v/v) to 50 μg of protein and then heated to 95 °C. SDS-PAGE was performed on a 4–20% gradient gel (Bio-Rad) at 130 V. Proteins were transferred to PVDF membranes (Immobilon-P, Millipore) and blocked in Tris-buffered saline with Tween 20 (TBST, 20 mm Tris-Base, 136 mm NaCl, 0.1% Tween 20, pH 7.4) buffer containing 5% nonfat dry milk for 45 min. Membranes were incubated overnight with gentle rocking at 4 °C in anti-STIM1 antibody (1:250; BD Biosciences catalog no. 610954, lot #5199779) or anti-β-actin antibody (1:1000; Sigma catalog no. A5316, lot # 119K4765) in nonfat dry milk. The blots were incubated for 1 h at room temperature in horseradish peroxidase (HRP)-conjugated anti-mouse IgG secondary antibody (detailed below) diluted 1:1000 plus Streptactin-HRP (Bio-Rad) diluted 1:6600. Blots were developed using the SuperSignal West Pico Chemiluminescence Substrate kit (Thermo Scientific) for 5 min, and protein bands were detected using a Bio-Rad Gel Documentation system with QuantityOne software. Band intensities were quantified to determine protein knockdown efficiency using Quantity One software (Bio-Rad Laboratories).
Co-immunoprecipitation and Western Blotting of HEK293T Cell Lysates
HEK293T cells were transfected with plasmids (1 μg/10 cm plate) for human STIM1(N131Q/N171Q), FLAG-SUR1, FLAG-NBF1, and Kir6.2 using Lipofectamine 2000 according to manufacturer's instructions. After 2 days cells were lysed with 0.5 ml ice-cold radioimmune precipitation assay buffer (50 mm Tris-Base, 150 mm NaCl, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS (pH 8.0)) with 1 mm dithiothreitol, 25 μm pepstatin A, 0.2 μm soybean trypsin inhibitor, and 1 mm Perfabloc SC added fresh before use. The lysate was collected and centrifuged at 13,000 rpm (Eppendorf 5415D microcentrifuge) for 10 min at 4 °C. The supernatant was transferred into a clean tube, and protein concentration was determined using a Bio-Rad DC protein assay kit. Protein samples were stored at −80 °C until use.
For immunoprecipitation, 100 μg of protein lysate was incubated for 16 h at 4 °C with antibodies (1–5 μg) as indicated. Then the antigen-antibody complex was immobilized on protein A-agarose beads (20 μl of 50% beads aqueous slurry, Thermo Scientific) for 3 h at 4 °C. The beads were washed 5 times with 250 μl of CHAPS lysis buffer (50 mm Tris-Base, 150 mm NaCl, 1 mm EDTA, 1% CHAPS (pH 8.0)) containing 10% β-mercaptoethanol and pelleted by centrifugation at 13,000 rpm for 30 s at 4 °C. The beads were resuspended with 20 μl of 2× SDS sample buffer (final concentrations: 25 mm Tris, 192 mm glycine, 1% SDS (pH 8.3)), and 20 μl of radioimmune precipitation lysis buffer. The samples were then centrifuged for 60 s at 13000 rpm, and the supernatant was used for Western blotting. Primary antibodies used were anti-SUR1 (Abcam catalog no. S289-16, Lot # GR-103710-18), anti-FLAG, clone M2 (Sigma catalog no. F1804, lot # SLBN5629V), and anti-STIM1 (as described for the Western blots above). Antibody specificity was assessed in HEK293T cells by comparing Western blots of control cells with cells transfected with the appropriate plasmid to induce protein expression.
The immunoprecipitated protein samples (∼30 μg of protein) and molecular weight markers (Precision Plus All Blue Protein Standard, Bio-Rad catalog no. 161–0373) were then resolved by SDS-PAGE (4–20% gradient protean GTX mini gels, Bio-Rad) and wet-transferred to Immobilon-P PVDF membranes (Millipore) using a buffer containing 25 mm Tris-base, 192 mm glycine, and 20% methanol (pH 8.3). The blots were washed once with TBST and incubated for 45 min at room temperature with blocking buffer (TBST with 5% blotting grade nonfat skim milk (Bio-Rad); membranes were incubated with specific antibodies (1:1,000 dilution in blocking buffer) for 16 h at 4 °C followed by 3 washes with TBST and incubation with HRP-coupled anti-mouse secondary antibody (Abcam catalog no. ab6728, lot # GR152005-0; 1:10,000 dilution) for 1 h at room temperature. After 3 washes with TBST, membranes were incubated with SuperSignal West Pico Chemiluminescence Substrate kit (Thermo Scientific) for 5 min, and protein bands were detected using a Bio-Rad Gel Documentation system with QuantityOne software. For quantification of band density, background-corrected measurements were made on images with no saturated pixels from three different exposure times to test for linearity.
Statistical Analysis
Student's t test or ANOVA was used for intergroup comparisons (p < 0.05 was considered statistically significant) as detailed in the figure legends. Data traces are plotted as the mean ± S.E. Box and whisker plots show mean (solid circle), median (dashed line), 25 and 75% values (box), and minimum/maximum values (whiskers).
Author Contributions
C. A. L., R. F. K., H. A. N., J. N., and M. W. R. conducted the experiments, analyzed the results, and edited the manuscript. C. A. L. conducted the patch clamp electrophysiology experiments. C. A. L., R. F. K., and M. W. R. conducted the calcium imaging experiments. R. F. K. performed qPCR. H. A. N. and J. N. performed immunoprecipitation and Western blotting. M. W. R. conceived the idea for the project and wrote the paper with C. A. L.
Acknowledgments
We thank Dr. Xiangquan Li for excellent technical assistance with the shRNA experiments during the early stages of this work. We thank the late Dr. R. Y. Tsien for the generous gift of D1ER and Dr. T. J. Shuttleworth for providing the STIM1(N131Q/N171Q) plasmid. We also thank Dr. Louis H. Philipson, Dr. Natalia Tamarina, and Dr. George G. Holz for many helpful discussions during the course of our studies.
This work was supported, in whole or in part, by National Institutes of Health Grants R01DK074966 (to M. W. R.) and R01DK092616 (to M. W. R.). This work was also supported by American Diabetes Association Research Grant 1-12-BS-109 (to C. A. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health National Institutes of Health/NIDDK or the American Diabetes Association.
- VDCC
- voltage-dependent Ca2+ channel
- SOCE
- store-operated Ca2+ entry
- SOC
- store-operated Ca2+ channel
- STIM1
- stromal interaction molecule 1
- STIM1
- gene encoding STIM1
- KATP
- ATP-sensitive potassium channel
- ER
- endoplasmic reticulum
- PM
- plasma membrane
- [Ca2+]c
- cytoplasmic calcium concentration
- [Ca2+]ER
- endoplasmic reticulum Ca2+ concentration
- SUR1
- sulfonylurea receptor 1
- NBF1
- nucleotide binding fold-1
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- PAM
- plasma membrane-associated membrane
- AUC
- area-under-curve
- Tg
- thapsigargin
- qPCR
- quantitative polymerase chain reaction
- NMDG
- N-methyl-d-glucamine
- TEA
- tetraethylammonium chloride
- pF
- picofarads
- IP
- immunoprecipitation
- IB
- Western immunoblotting
- KRBH
- Krebs-Ringer buffer
- TBST
- Tris-buffered saline with Tween 20
- ANOVA
- analysis of variance
- ss
- shRNA-scr cells
- sh
- shRNA-STIM1 cells
- av
- AdV-hSTIM1 cells.
References
- 1. Henquin J. C., Nenquin M., Ravier M. A., and Szollosi A. (2009) Shortcomings of current models of glucose-induced insulin secretion. Diabetes Obes. Metab. 11, 168–179 [DOI] [PubMed] [Google Scholar]
- 2. Jairaman A., and Prakriya M. (2013) Molecular pharmacology of store-operated CRAC channels. Channels 7, 402–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moccia F., Zuccolo E., Soda T., Tanzi F., Guerra G., Mapelli L., Lodola F., and D'Angelo E. (2015) Stim and Orai proteins in neuronal Ca2+ signaling and excitability. Front Cell Neurosci. 9, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Leech C. A., Holz G. G. 4th, and Habener J. F. (1994) Voltage-independent calcium channels mediate slow oscillations of cytosolic calcium that are glucose-dependent in pancreatic beta cells. Endocrinology 135, 365–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Worley J. F. 3rd, McIntyre M. S., Spencer B., and Dukes I. D. (1994) Depletion of intracellular Ca2+ stores activates a maitotoxin-sensitive nonselective cationic current in beta cells. J. Biol. Chem. 269, 32055–32058 [PubMed] [Google Scholar]
- 6. Worley J. F. 3rd, McIntyre M. S., Spencer B., Mertz R. J., Roe M. W., and Dukes I. D. (1994) Endoplasmic reticulum calcium store regulates membrane potential in mouse islet beta cells. J. Biol. Chem. 269, 14359–14362 [PubMed] [Google Scholar]
- 7. Mears D., and Zimliki C. L. (2004) Muscarinic agonists activate Ca2+ store-operated and -independent ionic currents in insulin-secreting HIT-T15 cells and mouse pancreatic beta cells. J. Membr. Biol. 197, 59–70 [DOI] [PubMed] [Google Scholar]
- 8. Roe M. W., Worley J. F. 3rd, Qian F., Tamarina N., Mittal A. A., Dralyuk F., Blair N. T., Mertz R. J., Philipson L. H., and Dukes I. D. (1998) Characterization of a Ca2+ release-activated nonselective cation current regulating membrane potential and [Ca2+]i oscillations in transgenically derived beta cells. J. Biol. Chem. 273, 10402–10410 [DOI] [PubMed] [Google Scholar]
- 9. Ambudkar I. S., and Ong H. L. (2007) Organization and function of TRPC channelosomes. Pflugers Arch. 455, 187–200 [DOI] [PubMed] [Google Scholar]
- 10. Berna-Erro A., Redondo P. C., and Rosado J. A. (2012) Store-operated Ca2+ entry. Adv. Exp. Med. Biol. 740, 349–382 [DOI] [PubMed] [Google Scholar]
- 11. Islam M. S. (2011) TRP channels of islets. Adv. Exp. Med. Biol. 704, 811–830 [DOI] [PubMed] [Google Scholar]
- 12. Jacobson D. A., and Philipson L. H. (2007) TRP channels of the pancreatic beta cell. Handb. Exp. Pharmacol. 179, 409–424 [DOI] [PubMed] [Google Scholar]
- 13. Redondo P. C., and Rosado J. A. (2015) Store-operated calcium entry: unveiling the calcium handling signalplex. Int. Rev. Cell Mol. Biol. 316, 183–226 [DOI] [PubMed] [Google Scholar]
- 14. Zhang S. L., Yu Y., Roos J., Kozak J. A., Deerinck T. J., Ellisman M. H., Stauderman K. A., and Cahalan M. D. (2005) STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hogan P. G. (2015) The STIM1-ORAI1 microdomain. Cell Calcium 58, 357–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hooper R., Samakai E., Kedra J., and Soboloff J. (2013) Multifaceted roles of STIM proteins. Pflugers Arch 465, 1383–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leech C. A., Kopp R. F., Philipson L. H., and Roe M. W. (2014) Beta cell store-operated ion channels. In Islets of Langerhans (Islam M. S., ed.) 2nd Ed., pp. 337–368, Springer, New York [Google Scholar]
- 18. Tamarina N. A., Kuznetsov A., and Philipson L. H. (2008) Reversible translocation of EYFP-tagged STIM1 is coupled to calcium influx in insulin secreting beta cells. Cell Calcium 44, 533–544 [DOI] [PubMed] [Google Scholar]
- 19. Tian G., Tepikin A. V., Tengholm A., and Gylfe E. (2012) cAMP induces stromal interaction molecule 1 (STIM1) puncta but neither Orai1 protein clustering nor store-operated Ca2+ entry (SOCE) in islet cells. J. Biol. Chem. 287, 9862–9872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smith P. A., Aschroft F. M., and Fewtrell C. M. (1993) Permeation and gating properties of the L-type calcium channel in mouse pancreatic beta cells. J. Gen. Physiol. 101, 767–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Darbellay B., Arnaudeau S., Bader C. R., Konig S., and Bernheim L. (2011) STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. 194, 335–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horinouchi T., Higashi T., Higa T., Terada K., Mai Y., Aoyagi H., Hatate C., Nepal P., Horiguchi M., Harada T., and Miwa S. (2012) Different binding property of STIM1 and its novel splice variant STIM1L to Orai1, TRPC3, and TRPC6 channels. Biochem. Biophys. Res. Commun. 428, 252–258 [DOI] [PubMed] [Google Scholar]
- 23. Carrillo E., Pacheco L., Balleza D., and Gomez-Lagunas F. (2015) K+-dependent selectivity and external Ca2+ block of Shab K+ channels. PLoS ONE 10, e0120431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gomez-Lagunas F., Melishchuk A., and Armstrong C. M. (2003) Block of Shaker potassium channels by external calcium ions. Proc. Natl. Acad. Sci. U.S.A. 100, 347–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tanahashi Y., Wang B., Murakami Y., Unno T., Matsuyama H., Nagano H., and Komori S. (2016) Inhibitory effects of SKF96365 on the activities of K+ channels in mouse small intestinal smooth muscle cells. J. Vet. Med. Sci. 78, 203–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yeung-Yam-Wah V., Lee A. K., Tse F. W., and Tse A. (2010) Arachidonic acid stimulates extracellular Ca2+ entry in rat pancreatic beta cells via activation of the noncapacitative arachidonate-regulated Ca2+ (ARC) channels. Cell Calcium 47, 77–83 [DOI] [PubMed] [Google Scholar]
- 27. Lopez J. J., Albarran L., Gómez L. J., Smani T., Salido G. M., and Rosado J. A. (2016) Molecular modulators of store-operated calcium entry. Biochim. Biophys. Acta 1863, 2037–2043 [DOI] [PubMed] [Google Scholar]
- 28. Dionisio N., Smani T., Woodard G. E., Castellano A., Salido G. M., and Rosado J. A. (2015) Homer proteins mediate the interaction between STIM1 and CaV1.2 channels. Biochim. Biophys. Acta 1853, 1145–1153 [DOI] [PubMed] [Google Scholar]
- 29. Park C. Y., Shcheglovitov A., and Dolmetsch R. (2010) The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330, 101–105 [DOI] [PubMed] [Google Scholar]
- 30. Wang Y., Deng X., Mancarella S., Hendron E., Eguchi S., Soboloff J., Tang X. D., and Gill D. L. (2010) The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330, 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Braun M., Ramracheya R., Bengtsson M., Zhang Q., Karanauskaite J., Partridge C., Johnson P. R., and Rorsman P. (2008) Voltage-gated ion channels in human pancreatic beta cells: electrophysiological characterization and role in insulin secretion. Diabetes 57, 1618–1628 [DOI] [PubMed] [Google Scholar]
- 32. Drews G., Krippeit-Drews P., and Düfer M. (2010) Electrophysiology of islet cells. Adv. Exp. Med. Biol. 654, 115–163 [DOI] [PubMed] [Google Scholar]
- 33. Mignen O., Thompson J. L., and Shuttleworth T. J. (2007) STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store depletion or translocation to the plasma membrane. J. Physiol. 579, 703–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shyng S. L., and Nichols C. G. (1998) Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 282, 1138–1141 [DOI] [PubMed] [Google Scholar]
- 35. Klein A., Lichtenberg J., Stephan D., and Quast U. (2005) Lipids modulate ligand binding to sulphonylurea receptors. Br. J. Pharmacol. 145, 907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liang T., Xie L., Chao C., Kang Y., Lin X., Qin T., Xie H., Feng Z. P., and Gaisano H. Y. (2014) Phosphatidylinositol 4,5-biphosphate (PIP2) modulates interaction of syntaxin-1A with sulfonylurea receptor 1 to regulate pancreatic beta-cell ATP-sensitive potassium channels. J. Biol. Chem. 289, 6028–6040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shibasaki T., Sunaga Y., Fujimoto K., Kashima Y., and Seino S. (2004) Interaction of ATP sensor, cAMP sensor, Ca2+ sensor, and voltage-dependent Ca2+ channel in insulin granule exocytosis. J. Biol. Chem. 279, 7956–7961 [DOI] [PubMed] [Google Scholar]
- 38. Chang N., Liang T., Lin X., Kang Y., Xie H., Feng Z. P., and Gaisano H. Y. (2011) Syntaxin-1A interacts with distinct domains within nucleotide-binding folds of sulfonylurea receptor 1 to inhibit beta-cell ATP-sensitive potassium channels. J. Biol. Chem. 286, 23308–23318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kang Y., Zhang Y., Liang T., Leung Y. M., Ng B., Xie H., Chang N., Chan J., Shyng S. L., Tsushima R. G., and Gaisano H. Y. (2011) ATP modulates interaction of syntaxin-1A with sulfonylurea receptor 1 to regulate pancreatic beta-cell KATP channels. J. Biol. Chem. 286, 5876–5883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McNally B. A., and Prakriya M. (2012) Permeation, selectivity and gating in store-operated CRAC channels. J. Physiol. 590, 4179–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liao Y., Plummer N. W., George M. D., Abramowitz J., Zhu M. X., and Birnbaumer L. (2009) A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc. Natl. Acad. Sci. U.S.A. 106, 3202–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jardin I., Salido G. M., and Rosado J. A. (2008) Role of lipid rafts in the interaction between hTRPC1, Orai1, and STIM1. Channels 2, 401–403 [DOI] [PubMed] [Google Scholar]
- 43. Huang G. N., Zeng W., Kim J. Y., Yuan J. P., Han L., Muallem S., and Worley P. F. (2006) STIM1 carboxyl terminus activates native SOC, I-CRAC and TRPC1 channels. Nat. Cell Biol. 8, 1003–1010 [DOI] [PubMed] [Google Scholar]
- 44. Yuan J. P., Kim M. S., Zeng W., Shin D. M., Huang G., Worley P. F., and Muallem S. (2009) TRPC channels as STIM1-regulated SOCs. Channels 3, 221–225 [DOI] [PubMed] [Google Scholar]
- 45. Salido G. M., Jardín I., and Rosado J. A. (2011) The TRPC ion channels: association with Orai1 and STIM1 proteins and participation in capacitative and non-capacitative calcium entry. Adv. Exp. Med. Biol. 704, 413–433 [DOI] [PubMed] [Google Scholar]
- 46. Cheng H., Beck A., Launay P., Gross S. A., Stokes A. J., Kinet J. P., Fleig A., and Penner R. (2007) TRPM4 controls insulin secretion in pancreatic beta cells. Cell Calcium. 41, 51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Colsoul B., Schraenen A., Lemaire K., Quintens R., Van Lommel L., Segal A., Owsianik G., Talavera K., Voets T., Margolskee R. F., Kokrashvili Z., Gilon P., Nilius B., Schuit F. C., and Vennekens R. (2010) Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in TRPM5−/− mice. Proc. Natl. Acad. Sci. U.S.A. 107, 5208–5213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Enklaar T., Brixel L. R., Zabel B. U., and Prawitt D. (2010) Adding efficiency: The role of the CAN ion channels TRPM4 and TRPM5 in pancreatic islets. Islets 2, 337–338 [DOI] [PubMed] [Google Scholar]
- 49. Marigo V., Courville K., Hsu W. H., Feng J. M., and Cheng H. (2009) TRPM4 impacts on Ca2+ signals during agonist-induced insulin secretion in pancreatic beta cells. Mol. Cell Endocrinol. 299, 194–203 [DOI] [PubMed] [Google Scholar]
- 50. Wang X., Wang Y., Zhou Y., Hendron E., Mancarella S., Andrake M. D., Rothberg B. S., Soboloff J., and Gill D. L. (2014) Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun. 5, 3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zeng W., Yuan J. P., Kim M. S., Choi Y. J., Huang G. N., Worley P. F., and Muallem S. (2008) STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol. Cell 32, 439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Masia R., Caputa G., and Nichols C. G. (2007) Regulation of KATP channel expression and activity by the SUR1 nucleotide binding fold 1. Channels 1, 315–323 [DOI] [PubMed] [Google Scholar]
- 53. Ambily A., Kaiser W. J., Pierro C., Chamberlain E. V., Li Z., Jones C. I., Kassouf N., Gibbins J. M., and Authi K. S. (2014) The role of plasma membrane STIM1 and Ca2+entry in platelet aggregation: STIM1 binds to novel proteins in human platelets. Cell Signal. 26, 502–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Saitoh N., Oritani K., Saito K., Yokota T., Ichii M., Sudo T., Fujita N., Nakajima K., Okada M., and Kanakura Y. (2011) Identification of functional domains and novel binding partners of STIM proteins. J. Cell Biochem. 112, 147–156 [DOI] [PubMed] [Google Scholar]
- 55. Harvey J., Hardy S. C., Irving A. J., and Ashford M. L. (2000) Leptin activation of ATP-sensitive K+ (KATP) channels in rat CRI-G1 insulinoma cells involves disruption of the actin cytoskeleton. J. Physiol. 527, 95–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Koriyama N., Kakei M., Nakazaki M., Yaekura K., Ichinari K., Gong Q., Morimitsu S., Yada T., and Tei C. (2000) PIP2 and ATP cooperatively prevent cytosolic Ca2+-induced modification of ATP-sensitive K+ channels in rat pancreatic beta cells. Diabetes 49, 1830–1839 [DOI] [PubMed] [Google Scholar]
- 57. Kieffer T. J., Heller R. S., Leech C. A., Holz G. G., and Habener J. F. (1997) Leptin suppression of insulin secretion by the activation of ATP-sensitive K+ channels in pancreatic beta cells. Diabetes 46, 1087–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hartzell C. A., Jankowska K. I., Burkhardt J. K., and Lewis R. S. (2016) Calcium influx through CRAC channels controls actin organization and dynamics at the immune synapse. Elife 5, e14850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hodeify R., Selvaraj S., Wen J., Arredouani A., Hubrack S., Dib M., Al-Thani S. N., McGraw T., and Machaca K. (2015) A STIM1-dependent “trafficking trap” mechanism regulates Orai1 plasma membrane residence and Ca2+ influx levels. J. Cell Sci. 128, 3143–3154 [DOI] [PubMed] [Google Scholar]
- 60. Manna P. T., Smith A. J., Taneja T. K., Howell G. J., Lippiat J. D., and Sivaprasadarao A. (2010) Constitutive endocytic recycling and protein kinase C-mediated lysosomal degradation control KATP channel surface density. J. Biol. Chem. 285, 5963–5973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Park S. H., Ryu S. Y., Yu W. J., Han Y. E., Ji Y. S., Oh K., Sohn J. W., Lim A., Jeon J. P., Lee H., Lee K. H., Lee S. H., Berggren P. O., Jeon J. H., and Ho W. K. (2013) Leptin promotes KATP channel trafficking by AMPK signaling in pancreatic beta cells. Proc. Natl. Acad. Sci. U.S.A. 110, 12673–12678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sivaprasadarao A., Taneja T. K., Mankouri J., and Smith A. J. (2007) Trafficking of ATP-sensitive potassium channels in health and disease. Biochem. Soc. Trans. 35, 1055–1059 [DOI] [PubMed] [Google Scholar]
- 63. Zerangue N., Schwappach B., Jan Y. N., and Jan L. Y. (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron 22, 537–548 [DOI] [PubMed] [Google Scholar]
- 64. Leech C. A., Dzhura I., Chepurny O. G., Kang G., Schwede F., Genieser H. G., and Holz G. G. (2011) Molecular physiology of glucagon-like peptide-1 insulin secretagogue action in pancreatic beta cells. Prog. Biophys. Mol. Biol. 107, 236–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang S. N., Shi Y., Yang G., Li Y., Yu J., and Berggren P. O. (2014) Ionic mechanisms in pancreatic beta cell signaling. Cell Mol. Life Sci. 71, 4149–4177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chow R. H., Lund P. E., Löser S., Panten U., and Gylfe E. (1995) Coincidence of early glucose-induced depolarization with lowering of cytoplasmic Ca2+ in mouse pancreatic beta cells. J. Physiol. 485, 607–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Roe M. W., Mertz R. J., Lancaster M. E., Worley J. F. 3rd, and Dukes I. D. (1994) Thapsigargin inhibits the glucose-induced decrease of intracellular Ca2+ in mouse islets of Langerhans. Am. J. Physiol. 266, E852–E862 [DOI] [PubMed] [Google Scholar]
- 68. Maechler P., Kennedy E. D., Sebö E., Valeva A., Pozzan T., and Wollheim C. B. (1999) Secretagogues modulate the calcium concentration in the endoplasmic reticulum of insulin-secreting cells: studies in aequorin-expressing intact and permeabilized INS-1 cells. J. Biol. Chem. 274, 12583–12592 [DOI] [PubMed] [Google Scholar]
- 69. Tengholm A., Hellman B., and Gylfe E. (1999) Glucose regulation of free Ca2+ in the endoplasmic reticulum of mouse pancreatic beta cells. J. Biol. Chem. 274, 36883–36890 [DOI] [PubMed] [Google Scholar]
- 70. Varadi A., and Rutter G. A. (2002) Dynamic imaging of endoplasmic reticulum Ca2+ concentration in insulin-secreting MIN6 Cells using recombinant targeted cameleons: roles of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA)-2 and ryanodine receptors. Diabetes 51, S190–S201 [DOI] [PubMed] [Google Scholar]
- 71. Huang Y., Zhou Y., Wong H. C., Chen Y., Chen Y., Wang S., Castiblanco A., Liu A., and Yang J. J. (2009) A single EF-hand isolated from STIM1 forms dimer in the absence and presence of Ca2+. FEBS J. 276, 5589–5597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Stathopulos P. B., Li G. Y., Plevin M. J., Ames J. B., and Ikura M. (2006) Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: an initiation mechanism for capacitive Ca2+ entry. J. Biol. Chem. 281, 35855–35862 [DOI] [PubMed] [Google Scholar]
- 73. Martin A. C., Willoughby D., Ciruela A., Ayling L. J., Pagano M., Wachten S., Tengholm A., and Cooper D. M. (2009) Capacitative Ca2+ entry via Orai1 and stromal interacting molecule 1 (STIM1) regulates adenylyl cyclase type 8. Mol. Pharmacol. 75, 830–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Furman B., Ong W. K., and Pyne N. J. (2010) Cyclic AMP signaling in pancreatic islets. Adv. Exp. Med. Biol. 654, 281–304 [DOI] [PubMed] [Google Scholar]
- 75. Liu W. (2013) Store-operated calcium entry could prevent continuous spiking of membrane potential to sustain normal intracellular calcium oscillations and normal potential bursting in pancreatic beta cells. Math Biosci. 243, 240–250 [DOI] [PubMed] [Google Scholar]
- 76. Ren J., Sherman A., Bertram R., Goforth P. B., Nunemaker C. S., Waters C. D., and Satin L. S. (2013) Slow oscillations of KATP conductance in mouse pancreatic islets provide support for electrical bursting driven by metabolic oscillations. Am. J. Physiol. Endocrinol Metab 305, E805–E817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Landa L. R. Jr, Harbeck M., Kaihara K., Chepurny O., Kitiphongspattana K., Graf O., Nikolaev V. O., Lohse M. J., Holz G. G., and Roe M. W. (2005) Interplay of Ca2+ and cAMP signaling in the insulin-secreting MIN6 beta-cell line. J. Biol. Chem. 280, 31294–31302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Harbeck M. C., Chepurny O., Nikolaev V. O., Lohse M. J., Holz G. G., and Roe M. W. (2006) Simultaneous optical measurements of cytosolic Ca2+ and cAMP in single cells. Sci. STKE 2006, pl6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]