

Abstract
Re-establishing blood supply is the primary goal for reducing myocardial injury in subjects with ischemic heart disease. Paradoxically, reperfusion results in nitroxidative stress and a marked inflammatory response in the heart. TRAF3IP2 (TRAF3 Interacting Protein 2; previously known as CIKS or Act1) is an oxidative stress-responsive cytoplasmic adapter molecule that is an upstream regulator of both IκB kinase (IKK) and c-Jun N-terminal kinase (JNK), and an important mediator of autoimmune and inflammatory responses. Here we investigated the role of TRAF3IP2 in ischemia/reperfusion (I/R)-induced nitroxidative stress, inflammation, myocardial dysfunction, injury, and adverse remodeling. Our data show that I/R up-regulates TRAF3IP2 expression in the heart, and its gene deletion, in a conditional cardiomyocyte-specific manner, significantly attenuates I/R-induced nitroxidative stress, IKK/NF-κB and JNK/AP-1 activation, inflammatory cytokine, chemokine, and adhesion molecule expression, immune cell infiltration, myocardial injury, and contractile dysfunction. Furthermore, Traf3ip2 gene deletion blunts adverse remodeling 12 weeks post-I/R, as evidenced by reduced hypertrophy, fibrosis, and contractile dysfunction. Supporting the genetic approach, an interventional approach using ultrasound-targeted microbubble destruction-mediated delivery of phosphorothioated TRAF3IP2 antisense oligonucleotides into the LV in a clinically relevant time frame significantly inhibits TRAF3IP2 expression and myocardial injury in wild type mice post-I/R. Furthermore, ameliorating myocardial damage by targeting TRAF3IP2 appears to be more effective to inhibiting its downstream signaling intermediates NF-κB and JNK. Therefore, TRAF3IP2 could be a potential therapeutic target in ischemic heart disease.
Keywords: cytokine, inflammation, ischemia, myocardial infarction, oxidative stress, transcription factor
Introduction
Ischemic heart disease is a leading cause of morbidity and mortality worldwide (1). Although reperfusion of the ischemic myocardium is the primary goal of reducing myocardial injury and dysfunction, paradoxically it can also produce pathological effects by the (i) excessive generation of reactive oxygen and nitrogen species, (ii) activation of the redox-sensitive dimeric transcription factors NF-κB and AP-1, (iii) up-regulation of inflammatory mediators, (iv) recruitment and activation of immune and inflammatory cells, and (v) induction of cardiomyocyte apoptosis, which ultimately results in myocardial injury, dysfunction (ischemia/reperfusion injury or I/R injury), and adverse remodeling (2, 3). Despite progress in our understanding of its underlying molecular mechanisms, myocardial injury post-I/R remains an important clinical problem.
A number of studies have firmly established causal roles for the nuclear transcription factors NF-κB and AP-1 in myocardial I/R injury (4–12). Various stimuli, including oxidative stress, cytokines, and growth factors, activate NF-κB by inducing the hyperphosphorylation and degradation of its inhibitory subunit IκB. A multiprotein complex comprised of IKKα, IKKβ, and a regulatory subunit IKKγ/NEMO was shown to mediate phosphorylation of IκB, followed by ubiquitination and degradation, resulting in NF-κB activation, nuclear translocation, and gene induction (4, 13, 14).
Like NF-κB, AP-1 is a highly inducible redox-sensitive nuclear transcription factor. It is composed mainly of Jun and Fos subunits. Phosphorylation of c-Jun by JNK results in nuclear translocation and transcriptional up-regulation of AP-1-dependent inflammatory and apoptotic gene expression. Of note, ischemia/reperfusion (I/R),5 but not ischemia alone, activates JNK (10). Similar to inhibition of IKK and NF-κB, jnk gene deletion or inhibition attenuates I/R-induced cardiomyocyte death (11, 12). These reports demonstrate that activation of NF-κB or AP-1 is detrimental, and suggest that targeting their expression or activation is cardioprotective in myocardial I/R injury.
The cytoplasmic adapter molecule TRAF3IP2 mediates the activation of both NF-κB and AP-1 (15, 16). Although TRAF3IP2 physically associates with IKK and activates NF-κB (15), we have previously demonstrated that its physical interaction with IKKγ results in JNK activation in cardiomyocytes (17), suggesting that induction of TRAF3IP2 could result in activation of proinflammatory signal transduction pathways regulated by both IKK/NF-κB and JNK/AP-1.
Similar to NF-κB and AP-1, TRAF3IP2 is a highly inducible redox-sensitive adapter molecule. Its promoter region contains potential binding sites for AP-1, NF-κB, C/EBPβ, CREB, and IRF1 (18, 19), indicating that oxidative stress induces TRAF3IP2 expression, and upon induction, it may regulate its own expression and that of multiple inflammatory mediators, ultimately resulting in tissue injury and dysfunction. In fact, we have previously demonstrated that advanced oxidation protein products, which serve as markers of oxidative stress and mediators of inflammation, induce cardiomyocyte death in part via Nox2/Rac1/superoxide-dependent TRAF3IP2/JNK signaling (20). Its pathological role is also demonstrated in various autoimmune and inflammatory diseases (21, 22). However, it is not known whether TRAF3IP2 plays a causal role in myocardial I/R injury. Using both genetic (conditional cardiomyocyte-specific Traf3ip2 gene deletion) and interventional (ultrasound-targeted microbubble destruction (UTMD)-mediated delivery of phosphorothioated TRAF3IP2 antisense oligonucleotides into the LV chamber) approaches, we report for the first time that TRAF3IP2 plays a causal role in myocardial I/R injury, dysfunction, and adverse remodeling. We also determined that TRAF3IP2 might be a better therapeutic target than either NF-κB or JNK alone in reducing myocardial injury post-I/R.
Results
I/R Induces TRAF3IP2 Expression in the Heart
Previously, we demonstrated that 30-min ischemia/2-h reperfusion induces oxidative stress and activation of the oxidative stress-responsive transcription factors NF-κB and AP-1 in the heart (23–26). Because TRAF3IP2 is an oxidative stress-responsive cytoplasmic adapter molecule and upstream regulator of NF-κB and AP-1 (15, 16), we hypothesized that I/R will induce TRAF3IP2 expression in the heart. Wild type mice underwent 30-min ischemia/2-h reperfusion (Fig. 1A), and TRAF3IP2 expression was analyzed by immunoblotting. The level of TRAF3IP2 expressed in the myocardium of wild type mice was not significantly altered by either sham surgery (Fig. 1A), or treatment with ischemia alone (Fig. 1B). However, TRAF3IP2 expression was markedly up-regulated in a time-dependent manner in the heart post-I/R (Fig. 1B). Although increased expression was observed as early as 30 min after reperfusion, its levels increased further at 2 h reperfusion, and remained elevated even at 6 h reperfusion (Fig. 1B). Immunohistochemistry revealed that TRAF3IP2 is present in cardiomyocytes localized to cytoplasm and membranes, and I/R increased its expression specifically in the ischemic (Is) zone (Fig. 1C). Confirming our hypothesis, these results indicate that I/R up-regulates TRAF3IP2 expression in the heart (Fig. 1).
FIGURE 1.
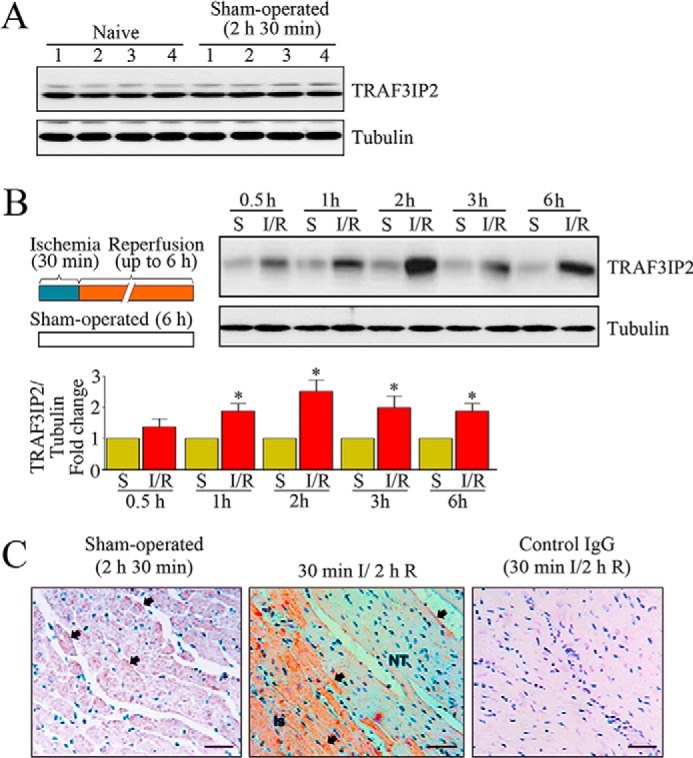
Reperfusion (R) following 30 min of ischemia (I) induces TRAF3IP2 expression in the heart. A, sham operation fails to induce TRAF3IP2 expression. Control mice underwent sham operation. TRAF3IP2 expression was analyzed in LV tissue by immunoblotting. LV tissue from naive animals served as controls. Numbers at the top denote individual animals. B, I/R up-regulates TRAF3IP2 expression. Control mice underwent 30 min ischemia and reperfusion for up to 6 h. Details of the I/R protocol are shown on the left. TRAF3IP2 expression in the ischemic zones was analyzed by immunoblotting, and a representative immunoblot is shown. Sham-operated (S) and ischemia (I) alone groups served as controls. Intensity of immunoreactive bands from 5 to 6 animals was quantified by densitometry and summarized at the bottom. *, p < 0.01 versus Sham or Ischemia alone. C, localization of TRAF3IP2 in the heart following 30-min ischemia/2-h reperfusion. TRAF3IP2 expression was localized by immunohistochemistry (Is, ischemic zone; NT, normal tissue). Control IgG: normal mouse IgG2a. Arrows denote positive immunoreactivity.
Conditional Cardiomyocyte-specific Traf3ip2 Gene Deletion Inhibits I/R-induced Nitroxidative Stress, Inflammatory Response, Myocardial Dysfunction, Injury, and Adverse Remodeling
After having demonstrated that I/R up-regulates TRAF3IP2 expression in the heart (Fig. 1B), and is localized predominantly in cardiomyocytes (Fig. 1C), we next determined whether TRAF3IP2 plays a causal role in I/R-induced myocardial injury, dysfunction, and adverse remodeling. We generated tamoxifen-regulated, cardiomyocyte (CM)-specific Traf3ip2 knock-out mice (Cre+/−;TRAF3IP2fl/+; CM-KO). At 2 months of age, both CM-KO and littermate controls (Cre+/−;TRAF3IP2+/+) received tamoxifen, and after 1 month, TRAF3IP2 levels were analyzed in the heart. Prior to tamoxifen administration, TRAF3IP2 expression was similar in both CM-KO and control mice (Fig. 2A, upper panel). However, 1 month after tamoxifen administration, TRAF3IP2 expression was markedly reduced only in the CM-KO mice (Fig. 2A, left lower panel). Furthermore, whereas TRAF3IP2 expression was readily detectable in both cardiomyocytes (CM) and cardiac fibroblasts isolated from the CM-KO mice without tamoxifen administration, its expression was markedly and selectively attenuated in CM following tamoxifen administration (Fig. 2B, upper panel). However, tamoxifen administration failed to affect TRAF3IP2 expression in non-target organs like lung, liver, and kidneys from the CM-KO mice (Fig. 2C). These results indicate that Traf3ip2 gene deletion was specific to cardiomyocytes.
FIGURE 2.

Conditional, cardiomyocyte-specific TRAF3IP2 gene deletion attenuates I/R-induced contractile dysfunction and myocardial injury. A and B, administration of tamoxifen attenuated myocardial TRAF3IP2 expression in cardiomyocyte-specific TRAF3IP2 gene knock-out (CM-KO), but not control mice. One month after last tamoxifen (Tam.) administration, TRAF3IP2 expression in control and CM-KO mice was analyzed by immunoblotting (A). CM, but not cardiac fibroblasts (CF), from tamoxifen administered CM-KO expressed almost no TRAF3IP2 expression (B, upper panel). *, p < 0.01 versus respective control. C, tamoxifen fails to affect TRAF3IP2 expression in non-cardiac tissue from CM-KO mice. Lung, liver, and kidneys from animals described in A were analyzed for TRAF3IP2 expression by immunoblotting. D, I/R fails to up-regulate TRAF3IP2 expression in the CM-KO hearts. Control and CM-KO mice underwent 30 min ischemia/2 h reperfusion (I/R protocol is detailed on the left). TRAF3IP2 expression in the ischemic zones was analyzed by immunoblotting, and densitometric analysis from 5 to 6 animals is summarized on the right. *, p < 0.01 versus control-Sham; †, p < 0.05 versus control-Sham; §, p < 0.01 versus control-I/R. E and F, TRAF3IP2 gene deletion attenuates I/R-induced myocardial dysfunction (E) and cardiac injury (F). Details of the I/R protocol are shown in the respective upper panels. Myocardial function was analyzed by echocardiography, and FS is shown as a change from baseline (E). A representative image of infarct is shown in the inset (F), and infarct size as % area at risk from 6 to 8 animals is summarized in the bottom. *, p < 0.001 versus baseline or respective control; †, p < 0.05 versus control (Con.).
One month after tamoxifen administration, both control and CM-KO mice underwent 30 min I and 2 or 24 h reperfusion. Sham-operated and ischemia alone groups served as controls. Immunoblotting revealed that whereas I/R up-regulated TRAF3IP2 expression in control mice, its levels were significantly attenuated in the CM-KO group (Fig. 2D). Furthermore, echocardiography revealed that at baseline, the fractional shortening (FS) was similar in both control and CM-KO mice (∼47 ± 2.64%; Fig. 2E). However, FS was reduced to a larger extent after I/R in control (26 ± 3.46%; 55% reduction; Fig. 2E) compared with CM-KO mice (36 ± 4.2%; 21% reduction, Fig. 2E). Infarct size was measured after 24 h by TTC staining and planimetry (26). Animals with an area at risk of less than 18% were excluded from the study as these animals had little or no infarct. The mean I/R-induced infarct size of control mice was 46.7% (Fig. 2F). Notably, the I/R-induced infarct size was markedly attenuated in the CM-KO mice (a 71% reduction; Fig. 2F). These results indicate that Traf3ip2 gene deletion in a conditional cardiomyocyte-specific manner blunts contractile dysfunction and injury post-I/R (Fig. 2).
Sustained nitroxidative stress contributes to contractile dysfunction and myocardial injury post-I/R (1, 27–29). Therefore we next determined whether I/R increases nitroxidative stress in the heart, and whether Traf3ip2 gene deletion blunts this response. Both control and CM-KO mice underwent I/R (Fig. 3A). The results show that whereas I/R increases H2O2 production in control mice within 30 min of reperfusion, its levels increased further at 1 h and declined gradually thereafter (Fig. 3A). Traf3ip2 gene deletion significantly reduced this effect (Fig. 3A). Similarly, I/R increased O2˙̄ generation, and this effect was also reduced by Traf3ip2 gene deletion (Fig. 3B). Supporting these data, tissue levels of lipid peroxidation products (malondialdehyde (MDA)/4-hydroxyalkenals (4-HNE)) were also markedly increased in hearts from control, but not CM-KO mice post-I/R (Fig. 3C). I/R also significantly increased the inducible form of nitric-oxide synthase (iNOS) in control mice, but to a significantly lesser degree in the CM-KO mice (Fig. 3D). Similarly, I/R-induced nitrotyrosine levels were also markedly reduced in the CM-KO mice (Fig. 3E). These results indicate that Traf3ip2 gene deletion blunts nitroxidative stress in the heart post-I/R (Fig. 3).
FIGURE 3.

Conditional, cardiomyocyte-specific Traf3ip2 gene deletion attenuates I/R-induced oxidative and nitrative stress in the heart post-I/R. A and B, Traf3ip2 gene deletion significantly attenuated I/R-induced H2O2 (A) and superoxide (O2˙̄; B) generation. Control (C) and CM-KO mice underwent 30 min ischemia and reperfusion for up to 3 h. Details of the I/R protocol are shown at the top. H2O2 production in ischemic zones was analyzed by Amplex Red assay. O2˙̄ generation was quantified by cytochrome c assay. *, p < 0.01 versus control-Sham or I alone; §, p < 0.01 versus control-I/R (n = 5–6/group). C, Traf3ip2 gene deletion blunts lipid peroxidation. Control and CM-KO mice underwent 30 min ischemia/2 h reperfusion. The ischemic zones were analyzed for lipid peroxidation by quantifying MDA and 4-HNE levels using a commercially available colorimetric assay kit. *, p < 0.001 versus respective Sham or ischemia group; †, p < 0.01 versus control-I/R (n = 5–6/group). D, Traf3ip2 gene deletion inhibits I/R-induced iNOS expression. Following 30 min ischemia/2 h reperfusion, iNOS expression in the ischemic zones was analyzed by immunoblotting. Densitometric analysis from 5 to 6 animals/group is shown on the right. *, p < 0.01 versus the respective ischemia group; †, p < 0.05 versus control-I/R. E, Traf3ip2 gene deletion attenuates nitrotyrosine levels. Following 30 min ischemia/2 h reperfusion, tissue nitrotyrosine levels were analyzed by ELISA. *, p < 0.001 versus the respective ischemia group; †, p < 0.05 versus control-I/R (n = 5–6/group).
Increased oxidative stress plays a key role in the activation of NF-κB and AP-1, both of which regulate the expression of various inflammatory mediators. Because I/R-induced oxidative stress and TRAF3IP2 expression in the heart post-I/R, we next investigated whether Traf3ip2 gene deletion blunts activation of IKK/NF-κB and JNK/AP-1, and induction of inflammatory mediators in the heart post-I/R. Results in Fig. 4A show that although I/R induced activation of IKKβ, p65, JNK, and c-Jun (Fig. 4A), and expression of IL-18, LIX, and ICAM1 (Fig. 4B), these changes were markedly attenuated in the CM-KO mice. Furthermore, RT2 ProfilerTM PCR Arrays revealed a marked increase in the mRNA expression of chemokines (Ccl2), chemokine receptors (Ccr2 and Ccr3), inflammatory cytokines (IL-1β, IL-6, IL-17A, IL-17F, IL-18, IL-33, and TNF-α), adhesion molecules (ICAM1 and LFA-1), growth factors (TGFβ and CTGF), and matrix metalloproteinases (2 and 9), and fibrillar collagens (Ia1 and IIIa1) in control mice, and these effects were markedly attenuated in the CM-KO mice (Table 1).
FIGURE 4.

Conditional, cardiomyocyte-specific Traf3ip2 gene deletion attenuates I/R-induced, IKK, p65, JNK, and c-Jun activation, and induction of myocardial and systemic inflammatory mediators. A–C, Traf3ip2 gene deletion inhibits I/R-induced IKK, p65, JNK, and AP-1 activation (A), and expression of inflammatory cytokines, chemokines, and adhesion molecules (B and C). Post-ischemic myocardium was analyzed for activation of IKK, p65, JNK, and AP-1 (A), and expression of IL-18, LIX, and ICAM1 expression by immunoblotting (B). Although representative immunoblots are shown on the left, densitometric analysis of immunoreactive bands from 5 to 6 mice is shown on the right or bottom. *, p < at least 0.05 versus control-I/R. Following 24 h reperfusion, systemic IL-18, IL-1β, IL-6, IL-17A, IL-17F, IL-33, and TNF-α levels were analyzed by corresponding ELISA (C). *, p < 0.01 versus respective sham or control-I/R. †, p < 0.05 versus control-I/R. D, Traf3ip2 gene deletion attenuates I/R-induced neutrophil infiltration. MPO, a biochemical marker for neutrophil infiltration into tissues, was measured in post-ischemic reperfused myocardium using a colorimetric assay. *, p < 0.01 versus control-sham; †, p < 0.05 versus control-I/R.
TABLE 1.
Conditional cardiomyocyte-specific Traf3ip2 gene deletion significantly attenuated I/R-induced inflammatory and extracellular matrix genes
Following 30 min I/2 h R, gene expression was analyzed by Qiagen pathway-focused RT2 Profiler™ PCR Arrays. hprt was used as an internal control. The results are shown as fold-change after considering gene expression in LV tissue of corresponding sham-operated animals as 1. The following abbreviations were used: Ccl, C-C motif chemokine ligand; Ccr, C-C motif chemokine receptor; Il, interleukin; Itgb2, integrin subunit β2; Mif, macrophage migration inhibitory factor; Pf4, platelet factor 4; Tgfb, transforming growth factor β; Tnfrsf1a, tumor necrosis factor receptor superfamily member 1A; Cdh3, cadherin 3; Ctgf, connetive tissue growth factor; Ctnn, catenin α; Ecm1, extracellular matrix protein 1; Emilin1, elastin microfibril interfacer 1; Fn1, fibronectin 1; Itga1, integrin subunit α1; Itgb1, integrin subunit β1; Thbs2, thrombospondin 2; TNC, tenascin C; IL6ST, interleukin 6 signal transducer; Itgal, integrin subunit αL; LFA-1, lymphocyte function-associated antigen 1; Icam1, intercellular adhesion molecule 1.
| Inflammation | I/R vs sham; fold-change |
|
|---|---|---|
| Control | CM-KO | |
| Ccl2 (MCP-1) | 2.5 ± 0.80 | 1.20 ± 0.12a |
| Ccl6 | 2.2 ± 0.32 | 0.81 ± 0.09a |
| Ccl9 (MIP1g) | 3.9 ± 0.98 | 1.22 ± 0.14a |
| Ccl17 | 6.8 ± 1.12 | 2.43 ± 0.32a |
| Ccr2 (MCP-1-R) | 3.8 ± 0.92 | 0.80 ± 0.08+ |
| Ccr3 (Eotaxin receptor) | 2.9 ± 0.78 | 1.10 ± 0.01a |
| Ccr5 | 3.13 ± 1.12 | 0.78 ± 0.03a |
| Cxcl5 (LIX) | 3.98 ± 0.83 | 1.12 ± 0.12a |
| Il1b | 2.31 ± 0.71 | 0.81 ± 0.04a |
| Il2rg | 0.93 ± 0.32 | 0.78 ± 0.41 |
| Il10ra | 2.11 ± 0.89 | 3.18 ± 1.12 |
| Il17a | 2.1 ± 0.78 | 0.89 ± 0.09a |
| Il17f | 1.98 ± 0.31 | 0.43 ± 0.24 |
| Il33 | 2.34 ± 0.22 | 1.18 ± 0.32 |
| Itgb2 | 2.9 ± 0.71 | 1.89 ± 0.51 |
| Mif | 1.9 ± 0.18 | 0.62 ± 0.12a |
| Pf4 | 1.2 ± 0.08 | 0.81 ± 0.02 |
| Tgfb1 | 3.1 ± 0.24 | 1.78 ± 0.11a |
| Tnfa | 2.98 ± 0.23 | 1.12 ± 0.32a |
| Tnfrsf1a | 2.0 ± 0.18 | 0.32 ± 0.01a |
| Extracellular matrix | ||
| Cdh3 | 8.3 ± 0.32 | 3.10 ± 0.56a |
| Col1a1 | 2.9 ± 0.21 | 0.78 ± 0.09a |
| Col3a1 | 1.8 ± 0.21 | 0.41 ± 0.07a |
| Col5a1 | 8.1 ± 1.12 | 2.31 ± 0.02a |
| CTGF | 5.1 ± 0.91 | 2.14 ± 0.91a |
| Ctnna1 | 0.12 ± 0.01 | 0.08 ± 0.23 |
| Ecm1 | 2.3 ± 0.26 | 1.12 ± 0.02a |
| Emilin1 | 2.1 ± 0.31 | 1.08 ± 0.32 |
| Fn1 | 7.3 ± 1.11 | 2.31 ± 0.54a |
| Itga4 | 1.2 ± 0.32 | 0.07 ± 0.01a |
| Itga5 | 2.8 ± 0.78 | 1.12 ± 0.49a |
| Itgal | 0.2 ± 0.01 | 0.12 ± 0.01 |
| Itgb1 | 0.8 ± 0.12 | 0.04 ± 0.01a |
| Itgb2 | 2.1 ± 0.42 | 1.11 ± 0.03 |
| Mmp2 | 2.3 ± 0.01 | 0.71 ± 0.02a |
| Mmp9 | 2.1 ± 0.26 | 0.32 ± 0.02a |
| THBS2 | 3.2 ± 0.21 | 1.32 ± 0.02a |
| TNC | 21 ± 1.19 | 13.8 ± 1.21a |
| RT-qPCR | ||
| Il6 | 3.1 ± 0.39 | 1.2 ± 0.32a |
| Il6R (IL-6RA) | 4.2 ± 0.78 | 2.3 ± 0.21a |
| IL6ST (gp130) | 3.7 ± 0.91 | 1.9 ± 0.13a |
| Il18 | 3.4 ± 0.32 | 0.78 ± 0.21a |
| Cxcl2 (MIP-2a) | 2.39 ± 0.18 | 1.12 ± 0.31a |
| Itgal (LFA-1) | 3.0 ± 0.71 | 1.19 ± 0.12a |
| Icam1 | 2.9 ± 0.32 | 1.23 ± 0.21a |
a p < at least 0.05 versus control.
Having demonstrated that the gene expression of various inflammatory mediators is markedly reduced in hearts from the CM-KO mice following 30-min ischemia/2-h reperfusion (Fig. 4 and Table 1), we next analyzed changes in protein levels of inflammatory mediators at 24-h reperfusion using a Quantibody® Mouse Cytokine Antibody Array that simultaneously detects changes in 200 proteins. Again, the results showed a marked inhibition in the protein levels of various inflammatory mediators (cytokines, chemokines, and adhesion molecules), and MMPs in the CM-KO hearts (Table 2). Furthermore, systemic levels of IL-1β, IL-6, IL-17A, IL-17F, IL-18, IL-33, and TNF-α were also significantly reduced in the CM-KO mice (Fig. 4C). Traf3ip2 gene deletion also attenuated tissue myeloperoxidase (MPO) activity, a surrogate marker for neutrophils, in the heart post-I/R (Fig. 4D).
TABLE 2.
Conditional cardiomyocyte-specific Traf3ip2 gene deletion markedly attenuates various pro-inflammatory and remodeling-associated factors in post-ischemic heart
Following 30 min ischemia/24 h reperfusion, changes in protein expression were analyzed by RayBiotech Quantibody® Mouse Cytokine Antibody Array. The results are shown as fold-change after considering protein expression in corresponding sham-operated animals as 1. Results represent mean ± S.E. of n = 4/group, and are considered statistically significant if p < 0.05. Proteins that showed a 1.5-fold change or more are depicted. The following abbreviations were used: MCSF, macrophage-colony stimulating factor; TWEAK, TNF-related weak inducer of apoptosis; TWEAKR, TWEAK receptor; MAdCAM-1, mucosal vascular addressin cell adhesion molecule 1; VCAM-1, vascular cell adhesion molecule 1; β-FGF, basic fibroblast growth factor; ACE, angiotensin converting enzyme; Ang, angiotensin.
| Target | I/R vs sham; fold-change |
|
|---|---|---|
| Control | CM-KO | |
| Inflammation | ||
| Cytokines | ||
| G-CSF | 2.1 ± 0.04 | 0.98 ± 0.12a |
| gp130 | 2.32 ± 0.23 | 0.78 ± 0.12a |
| IL-1β | 3.12 ± 0.54 | 1.12 ± 0.21a |
| IL-6 | 2.3 ± 0.32 | 0.78 ± 0.07a |
| IL-10 | 0.21 ± 0.01 | 4.12 ± 0.48 |
| IL-17A | 1.98 ± 0.98 | 0.54 ± 0.04a |
| IL-17F | 2.1 ± 1.12 | 0.79 ± 0.32a |
| IFN-γ | 3.12 ± 0.92 | 0.64 ± 0.08 |
| Lipocalin-2 | 1.98 ± 0.78 | 0.32 ± 0.01a |
| MCSF | 2.13 ± 1.12 | 0.56 ± 0.01a |
| RAGE | 2.34 ± 0.21 | 0.89 ± 0.21a |
| TNF-α | 2.56 ± 0.34 | 0.73 ± 0.01a |
| TWEAK | 2.13 ± 0.45 | 1.78 ± 0.54 |
| TWEAKR | 3.08 ± 0.73 | 1.18 ± 0.12a |
| P-selectin | 1.98 ± 0.31 | 0.43 ± 0.08a |
| Chemokines, adhesion molecules | ||
| CCL5 | 2.29 ± 0.31 | 0.89 ± 0.08a |
| CXCL16 | 2.34±.21 | 0.78 ± 0.11a |
| Fractalkine | 3.4 ± 0.81 | 1.21 ± 0.32a |
| 6Ckine | 2.1 ± 0.21 | 0.58 ± 0.11a |
| E-selectin | 1.98 ± 0.03 | 0.71 ± 0.01 |
| ICAM1 | 3.23 ± 0.21 | 1.21 ± 0.10a |
| KC (CXCL1) | 2.98 ± 0.32 | 0.89 ± 0.12a |
| LIX (CXCL5) | 3.11 ± 0.19 | 0.54 ± 0.01a |
| MIP-2 | 2.21 ± 0.23 | 0.38 ± 0.01a |
| MAdCAM-1 | 3.1 ± 0.21 | 0.98 ± 0.31a |
| P-cadherin | 1.98 ± 0.11 | 0.41 ± 0.01a |
| VCAM-1 | 3.12 ± 0.73 | 1.18 ± 0.42a |
| Fibrosis and ECM modulating proteins | ||
| β-FGF | 5.32 ± 0.49 | 2.13 ± 0.32a |
| Periostin | 2.13 ± 0.43 | 0.89 ± 0.21a |
| Testican | 1.81 ± 0.23 | 1.32 ± 0.07 |
| MMP-2 | 7.8 ± 1.38 | 2.18 ± 0.12a |
| Pro-MMP-9 | 3.21 ± 0.54 | 1.12 ± 0.23a |
| MMP-10 | 4.9 ± 0.89 | 1.19 ± 0.42a |
| Hypertension | ||
| ACE | 1.12 ± 0.01 | 0.12 ± 0.01a |
| Ang-III | 2.1 ± 0.78 | 0.78 ± 0.01a |
| Renin-3 | 8.1 ± 1.24 | 2.31 ± 0.49a |
a p at least 0.05 versus control.
Traf3ip2 Gene Deletion Attenuates Adverse Myocardial Remodeling
Restoring blood flow in time to the ischemic region, whereas helping to regain function, also induces oxidative stress and intense inflammation, which can result in cardiomyocyte death (1). Oxidative stress and proinflammatory cytokines contribute to cardiac remodeling (30–32), characterized by increased fibrosis, hypertrophy of surviving cardiomyocytes, myocardial hypertrophy, and transition to heart failure. Because Traf3ip2 gene deletion significantly reduced myocardial oxidative stress, inflammation and injury post-I/R, we next determined whether its gene deletion also inhibits I/R-induced adverse myocardial remodeling and transition to failure. Because myocardial remodeling is a progressive process, which continues long after the initial cardiomyocyte injury and loss, and is an important predictor of heart failure development, we performed cross-sectional studies and analyzed myocardial function at 2, 4, 8, and 12 weeks post-I/R by echocardiography in both control and CM-KO mice. The data revealed a gradual deterioration in contractile function (increased LVIDs and LV Vols) with concomitant decrease in LVPWs and FS, suggesting possible onset of heart failure in control mice at 12 weeks post-I/R (Fig. 5A). Furthermore, increased cardiomyocyte cross-sectional area (Fig. 5B), MMP2 and -9 activities (Fig. 5C), and perivascular and interstitial fibrosis (Fig. 5D) were observed in the non-ischemic region of hearts at 12 weeks post-I/R in control mice. However, these changes were markedly attenuated in the CM-KO mice, demonstrating that targeting TRAF3IP2 is cardioprotective, and attenuates I/R-induced adverse myocardial remodeling (Fig. 5).
FIGURE 5.

Conditional, cardiomyocyte-specific Traf3ip2 gene deletion attenuates I/R-induced adverse myocardial remodeling. A, Traf3ip2 gene deletion blunts progression of I/R-contractile dysfunction and progression to heart failure. Following 30 min ischemia and reperfusion for up 12 weeks, myocardial function was analyzed by echocardiography. LVIDs, Left ventricular internal dimension at end-systole; LV Vol, LV volume; LVPWs, left ventricular posterior wall thickness at end-systole. *, p < at least 0.05 versus control-I/R (n = 5 = 6/group). B, Traf3ip2 gene deletion attenuates development of hypertrophy in the non-ischemic zone. Cardiomyocyte cross-sectional area after a 12-week reperfusion was analyzed in H&E-stained sections by ImageJ software. Although representative images are shown on the left, the mean area was quantified and summarized on the right. *, p < 0.001 versus control-I/R (n = 5–6 animals/group, 100 cells/animal). C, Traf3ip2 gene deletion attenuates I/R-induced MMP2 and MMP9 activation. After indicated reperfusion periods, non-ischemic zones were analyzed for MMP activation by immunoblotting using antibodies that detect both pro and active forms (n = 3/group). D, Traf3ip2 gene deletion attenuates I/R-induced fibrosis. After a 12-week reperfusion, fibrosis in non-ischemic zones was analyzed by Picrosirius Red staining. Although representative images are shown on the left, mean collagen positive area is summarized on the right (n = 5–6/group). *, p < at least 0.05 versus respective sham-operated group; †, p < 0.05 versus control-I/R.
UTMD-mediated Delivery of Phosphorothioated Antisense Deoxyoligonucleotides (AS-ODN) Targeting TRAF3IP2 Is Highly Effective in Attenuating I/R-induced Myocardial Injury
Because Traf3ip2 gene deletion is cardioprotective following I/R (Fig. 2F), we next determined whether targeting TRAF3IP2 using an interventional approach could inhibit I/R-induced myocardial injury. Because no pharmacological inhibitors specific for TRAF3IP2 were available, we used a phosphorothioated AS-ODN strategy. We had successfully used this strategy before, where we demonstrated that delivery of phosphorothioated AS-ODN against TNF-α encapsulated in PESDA (perfluorocarbon-exposed sonicated dextrose albumin) microbubbles into the LV chamber and their release by therapeutic ultrasound (ultrasound-targeted microbubble destruction or UTMD), significantly attenuated I/R-induced TNF-α expression in the heart (33). Using a similar approach, here we targeted TRAF3IP2 to determine whether inhibition of TRAF3IP2 reduces myocardial injury post-I/R, and whether TRAF3IP2 is a better therapeutic target than its downstream effectors NF-κBp65 and JNK in reducing myocardial injury post-I/R.
The AS-ODN against p65 and JNK1 have been described previously (34, 35). To target TRAF3IP2, we designed 4 individual AS-ODNs (Table 3; Integrated DNA Technologies), which we first tested for efficacy in vitro by transfection into neonatal mouse cardiomyocytes. Inhibition of target protein expression was confirmed by immunoblotting. Control or scrambled ODN served as non-targeting controls. Transfection with phosphorothioated AS-ODN against p65 markedly attenuated basal p65 levels (Fig. 6A, left panel) without affecting the expression of JNK1, used as off-target control. Similarly, JNK1 AS-ODN attenuated JNK1, but not p65 expression (Fig. 6A, right panel). All four TRAF3IP2 AS-ODN efficiently inhibited basal TRAF3IP2 expression; however, Oligo#4 appeared to be the most effective (Fig. 6B). The scrambled ODN containing the same nucleotides, but rearranged randomly, did not affect TRAF3IP2 expression (Fig. 6B, middle panel). ASK1, IKKβ, JNK1, and Akt served as off-targets, and TRAF3IP2 Oligo#4, as well as the scrambled ODN had no affect on their basal expression (Fig. 6C). Notably, TRAF3IP2 AS-ODN was highly effective in inhibiting hypoxia/reoxygenation (H/R)-induced cardiomyocyte injury, as evidenced by reduced lactate dehydrogenase (LDH) release (Fig. 6D), caspase-3 activation (Fig. 6E, left panel), and cleaved poly(ADP-ribose) polymerase levels (Fig. 6E, right panel).
TABLE 3.
Sequence of phosphorothioated AS-ODN against p65, JNK1, and TRAF3IP2
| Target | ODN | ODN sequencea | Length |
|---|---|---|---|
| p65 | AS | 5′-G*A*A*A*C*A*G*A*T*C*G*T*C*C*A*T*G*G*T-3′ | 19 |
| Scrambled | 5′-G*G*A*A*C*A*G*T*T*C*G*T*C*T*A*T*G*G*C-3′ | 19 | |
| JNK1 | AS | 5′-T*G*T*T*G*T*C*A*C*G*T*T*T*A*C*T*T*C*T*G-3 | 20 |
| Control | 5′-C*C*T*T*C*C*C*T*G*A*A*G*G*T*T*C*C*T*C*C-3′ | 20 | |
| TRAF3IP2 | Oligo#1-AS | 5′-G*C*C*A*C*A*T*A*C*T*C*T*T*C*C*T*C*C*C*T-3′ molecular mass: 6229.0 Da; 3′ end position: 2023 | 20 |
| Oligo#2-AS | 5′-G*C*C*A*C*A*T*A*C*T*C*T*T*C*C*T*C*C*C*T*G-3′ molecular mass: 6574.3 Da; 3′ end position: 2022 | 21 | |
| Oligo#3-AS | 5′-G*C*C*A*C*A*T*A*C*T*C*T*T*C*C*T*C*C*C-3′ molecular mass: 5908.8 Da; 3′ end position: 2024 | 19 | |
| Oligo#4-AS | 5′-A*G*C*C*A*C*A*T*A*C*T*C*T*T*C*C*T*C*C*C*T-3′ molecular mass: 6558.3 Da; 3′ end position: 2023 | 21 | |
| Oligo#4scrambled | 5′-A*C*T*C*C*A*T*C*C*G*T*C*C*T*A*C*T*C*A*T*C-3′ molecular mass 6558.3 Da | 21 |
a p65, the p65 subunit of NF-κB; JNK1, c-Jun N-terminal kinase 1; TRAF3IP2, TRAF3 interacting protein 2; asterisk represents phosphorothioated.
FIGURE 6.

TRAF3IP2 AS-ODN attenuated hypoxia/reoxygenation (H/R)-induced cardiomyocyte injury in vitro. A, phosphorothioated p65 AS-ODN (p65 AS) attenuated basal p65 expression in NMCM. A representative immunoblot showing reduced p65 expression in NMCM transduced with p65 AS-ODN (500 nm; left panel). Likewise, phosphorothioated JNK1 AS-ODN (JNK1 AS; 500 nm) attenuated basal JNK1 expression in NMCM (right panel). B, phosphorothioated TRAF3IP2 AS-ODN#4 (500 nm), but not its scrambled ODN, attenuated basal TRAF3IP2 expression in NMCM (right panel). NMCM were transfected with 1 of 4 different AS-ODN (500 nm), and analyzed for TRAF3IP2 expression by immunoblotting. Densitometric analysis from three independent experiments is summarized at the bottom. The scrambled ODN, however, failed to affect basal TRAF3IP2 expression (right-hand panel; densitometric analysis from three independent experiments is summarized at the bottom). c, untransfected controls. C, TRAF3IP2 AS-ODN#4 or the scrambled ODN had no off-target effects. Transfection with AS-ODN#4 or scrambled ODN affected ASK1, IKKβ, JNK, nor Akt at basal conditions. D and E, TRAF3IP2 (versus p65 or JNK1) AS-ODN is highly effective in attenuating H/R-induced neonatal cardiomyocyte injury. NMCM transfected with AS-ODN against TRAF3IP2, p65, or JNK1 were exposed to H/R. LDH release into culture supernatant (D), and cleaved caspase-3 and poly(ADP-ribose) polymerase (PARP) levels (E) were analyzed by a colorimetric assay (D) and immunoblotting (E). Normoxia (N) and hypoxia alone (H) served as controls. *, p < 0.01 versus respective scrambled or control AS-ODN; †, p < at least 0.05 versus H/R (n = 12/group).
Having demonstrated the efficacy of TRAF3IP2 AS-ODN#4 in vitro (Fig. 6D), we then investigated its ability in reducing myocardial I/R injury (infarct size) in vivo, and compared it with targeting p65 or JNK1 alone. PESDA alone or PESDA + scrambled ODN served as controls. Infarct size was quantified after 24 h as in Fig. 2F. The results showed that 30-min ischemia/24-h reperfusion (Fig. 7A) induced significant injury (increased infarct size; injured areas are indicated by white arrows), and administration of control AS-ODN had no detectable effect on this outcome (Fig. 7B). In contrast, p65 and JNK1 AS-ODN each attenuated infarct size between 40 and 44% (Fig. 7B); however, TRAF3IP2 AS-ODN appeared to be most effective at reducing infarct size (Fig. 7B). Neither PESDA alone nor PESDA + scrambled ODN modulated I/R-induced myocardial injury (Fig. 7B). Moreover, each of these AS-ODNs markedly suppressed their target protein expression (Fig. 7C-E). However, the control ODN failed to modulate I/R-induced TRAF3IP2, p65 or JNK1 expression (Fig. 7, C–E). Similar to control ODN, the scrambled ODN also failed to affect I/R-induced myocardial injury (Fig. 8A). Importantly, the TRAF3IP2 AS-ODN, whereas inhibiting TRAF3IP2 expression in the heart, failed to affect its expression in lung, liver, or kidney (Fig. 8B). Furthermore, no histological changes were observed in those non-cardiac organs (Fig. 8C). Liver function also remained unchanged as evidenced by similar levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in serum (Fig. 8D). Together, these results demonstrate that UTMD-mediated delivery of phosphorothioated AS-ODN against TRAF3IP2 into the LV in a clinically relevant time frame markedly inhibits I/R-induced TRAF3IP2 expression, myocardial injury (smaller infarct), and adverse remodeling.
FIGURE 7.

Ultrasound-targeted TRAF3IP2, p65, or JNK1 AS-ODN attenuated I/R-induced myocardial injury. A, details of the I/R protocol are shown. B, TRAF3IP2 AS-ODN is highly effective in reducing I/R-induced myocardial injury. C56BL/6 mice underwent 30 min ischemia/24 h reperfusion. At the initiation of reperfusion, PESDA-bound TRAF3IP2, p65, or JNK1 AS-ODN (AS) were delivered arterially into left ventricle for 10 min with simultaneous application of ultrasound. 24 h later, animals were sacrificed and infarct size quantified. Arrows indicate the infarcted region. Although representative images are shown at the top, results from 6 to 8 mice/group are summarized on the bottom. C–E, administration of TRAF3IP2, p65, or JNK1 AS-ODN inhibits their target protein expression in the heart. Following delivery of AS-ODN, expression of respective target proteins was analyzed by immunoblotting. Although a representative immunoblot is shown on the left, densitometric analysis of immunoreactive bands from 5 to 6 animals is summarized on the right. B–E, *, p < 0.01 versus sham; †, p < 0.05 versus I/R ± control; ††, p < 0.01 versus I/R + PESDA (n = 6–8/group).
FIGURE 8.

Ultrasound-targeted TRAF3IP2 AS-ODN attenuates I/R-induced myocardial injury-control studies. A, following 30 min ischemia, animals were administered with PESDA-encapsulated TRAF3IP2 AS-ODN with simultaneous application of therapeutic ultrasound. PESDA alone or PESDA-encapsulated scrambled ODN served as controls. Arrows indicate infarcted region. Although representative images are shown on the top, results from 6 to 8 mice/group are summarized in the bottom of panel A, †, p < 0.05 or ††, p < 0.01 versus I/R + PESDA alone (n = 6–8/group). B, TRAF3IP2 AS-ODN significantly inhibits TRAF3IP2 expression in heart, but not lung, liver, or kidney. A representative immunoblot (n = 2–4/group) showing TRAF3IP2 expression in various organs following 30 min ischemia/24 h reperfusion is shown. C and D, administration of scrambled AS-ODN or TRAF3IP2 AS-ODN had no effect on histology of non-cardiac organs or liver function. Lung, liver, and kidneys from animals administered with TRAF3IP2 AS-ODN were analyzed by H&E for histological changes. Scrambled ODN served as a control. Liver function (D) was analyzed by quantifying serum ALT and AST levels by ELISA.
Discussion
The major findings of this study are: (i) I/R up-regulates TRAF3IP2 expression in the heart, and (ii) Traf3ip2 gene deletion in a conditional cardiomyocyte-specific manner suppresses I/R-induced nitroxidative stress, inflammatory response, myocardial dysfunction, injury and adverse remodeling. (iii) Targeting TRAF3IP2 by UTMD-mediated delivery of phosphorothioated AS-ODN markedly attenuates myocardial I/R injury, and (iv) TRAF3IP2 appears to be a better target than its downstream signaling intermediates NF-κB and JNK in reducing I/R injury. Together, the results from these genetic and clinically relevant interventional studies demonstrate that TRAF3IP2 could be a potential therapeutic target in myocardial injury and adverse remodeling post-I/R.
Myocardial I/R injury is a multifactorial disease. Factors, including increased nitroxidative stress, inflammation, intracellular Ca2+ overload, shift in pH, opening of mitochondrial permeability transition pore, endothelial dysfunction, microvascular injury, and altered metabolism, all acting in concert contribute to its pathogenesis (1, 28). Because TRAF3IP2 is an oxidative stress responsive adapter molecule and a critical intermediate in inflammatory signaling (17, 19, 20), we focused mainly on oxidative stress and inflammation. Our results show that I/R markedly increased oxygen-free radical production (H2O2 and O2˙̄) in the heart, resulting in increased lipid peroxidation as evidenced by higher levels of MDA and 4-HNE levels, and Traf3ip2 gene deletion attenuated these deleterious effects. Although several oxidoreductases have been identified in the post-ischemic heart, activation of the NADPH oxidases Nox4 and Cybb (Nox2) contribute to H2O2 and O2˙̄ generation. Interestingly, Nox4 and Nox2 are NF-κB, AP-1, or IRF1 responsive genes (36–38). Because TRAF3IP2 is an upstream regulator of these transcription factors (18, 19), it is highly likely that Traf3ip2 gene deletion might have resulted in reduced expression and/or activation of these critical enzymes. We have also shown that Traf3ip2 gene deletion inhibited Nos2 expression, also a NF-κB-responsive gene (39), in the heart post-I/R. This was accompanied by reduced levels of nitrotyrosine, a surrogate marker of peroxynitrite. Peroxynitrite is a potent pro-oxidant formed by the reaction between O2˙̄ and NO, and is a known mediator of reperfusion injury (29, 40). It is thus possible that Traf3ip2 gene deletion might have resulted in reduced infarct size due to inhibition of nitroxidative stress in the heart post-I/R.
Of note, both oxidative stress and nitrative stress contribute to inflammation and injury, and Traf3ip2 gene deletion inhibited the expression of multiple inflammatory mediators, including IL-1β, IL-6, IL-17, IL-18, and TNF-α in the heart post-I/R. Traf3ip2 gene deletion also blunted I/R-induced LIX expression in the heart. LIX is an ELR+ CXC chemokine, and a potent neutrophil chemoattractant, and is induced by I/R (25, 41). In fact, Traf3ip2 gene deletion attenuated I/R-induced increases in MPO activity, a surrogate marker of neutrophil infiltration. Traf3ip2 gene deletion also attenuated the expression of adhesion molecule ICAM-1 and LFA-1, both of which play a role in neutrophil adhesion to endothelial cells and subsequent extravasation. Because several of these inflammatory mediators are known negative myocardial inotropes, it is highly likely that reduced expression of cytokines, chemokines, and adhesion molecules might have contributed to reduced inflammatory cell infiltration, improved function, and smaller infarct in the Traf3ip2 knockout mice.
Analogous to the results obtained in the CM-KO mice, UTMD-mediated delivery of phosphorothioated Traf3ip2 AS-ODN in a clinically relevant time frame (during reperfusion) markedly attenuated myocardial I/R injury. Although targeting p65 and JNK1 each attenuated the infarct size between 40 and 44%, targeting TRAF3IP2 was highly effective and reduced the infarct size by a significant 61%. Interestingly, whereas inhibiting TRAF3IP2 expression in the heart, administration of TRAF3IP2 AS-ODN did not affect TRAF3IP2 expression in non-target organs like lung, liver, and kidney. Furthermore, no histological changes were observed in these non-target organs, indicating that targeted delivery of AS-ODN to the heart can safely and effectively reduce myocardial injury post-I/R, and without adversely affecting other major organs. These results suggest that UTMD-mediated delivery of TRAF3IP2 AS-ODN in a clinically relevant time frame is a viable therapeutic approach in ischemic heart disease.
Although the exact mechanism of ultrasound-mediated transfection is not fully known, several possibilities have been discussed. For example, therapeutic ultrasound may enhance AS-ODN entry into cells by increasing vascular endothelial disruption in the target organ, facilitating passage between cells and across the microvascular barrier (42–45). It is also possible that destabilization and microporation of cell membranes might enhance AS-ODN uptake. Importantly, the membrane damage was shown to be reversible, with resealing of holes (46). Moreover, UTMD increases uptake of the therapeutic agent by 10–12-fold in the heart (45). Mukherjee et al. (47) have previously reported endothelial membrane disruption, pore formation, and increased intercellular gaps in rat hearts following ultrasound application with acoustic power in the range between 0.8 and 1.0 W/cm2. These authors also observed no structural changes with a power of 0.6 W/cm2, as used in this study (47). Of note, UTMD is now being used in larger animals, including pigs, to deliver viral vectors, drugs, and DNA (48). Therefore, UTMD-mediated delivery of AS-ODN may be a viable therapeutic approach in ischemic heart disease.
We have demonstrated that targeting TRAF3IP2 reduces activation of IKK, NF-κB, JNK, and AP-1 in the heart post-I/R. It is well established that activation of IKK and NF-κB is detrimental in I/R injury. For example, NfkB1 (the p50 subunit of the NF-κB dimer) gene deletion inhibited I/R-induced NF-κB activation and myocardial injury (49). Administration of Bay 65-1942, an ATP-competitive inhibitor that selectively targets IKKβ kinase activity significantly reduced NF-κB activation, inflammatory cytokine expression, and myocardial injury post-I/R (8). Another major downstream target of TRAF3IP2 is JNK. Interestingly, ischemia alone promotes JNK1 nuclear translocation, but not activation (50). However, reperfusion results in its activation (50), possibly due to increased reactive oxygen species generation during the initial stages of reperfusion. It has also been shown that I/R induces JNK activation, preferentially the 55-kDa isoform (JNK1) both in vitro and in vivo (10). Importantly, administration of a peptide inhibitor that specifically targets JNK reduced I/R injury ex vivo (12). Furthermore, genetic ablation of jnk1 or jnk2, or transgenic overexpression of a dominant-negative mutant form of JNK1/2 resulted in reduced basal JNK activity, I/R-induced cardiomyocyte death and myocardial injury (11). Here we have demonstrated that targeting NF-κBp65 or JNK1 by UTMD-mediated delivery of AS-ODN markedly reduces myocardial injury post-I/R, and TRAF3IP2 appears to be a better therapeutic target compared with p65 or JNK1 alone, possibly due to suppression of pro-inflammatory and/or pro-death pathways regulated by both IKK/NF-κB and JNK/AP-1 in the heart post-I/R. It is also possible that targeting TRAF3IP2 might have blocked amplification of inflammatory pathways by inhibiting the cross-talk between IKK and JNK, resulting ultimately in reduced myocardial injury.
In addition to activation of critical signaling intermediates like IKK/NF-κB and JNK/AP-1, induction of TRAF3IP2 is also known to activate the mitogen-activated protein kinase p38 (p38 MAPK). Activation of p38 MAPK induces the expression of various inflammatory mediators in part via NF-κB- and/or AP-1-dependent signaling. Importantly, inhibition of p38 MAPK suppresses cardiomyocyte death and improves myocardial function post-I/R (51). Targeting p38 MAPK also reduces I/R-induced endothelial adhesion molecule expression, neutrophil infiltration, and myocardial injury. Therefore, it is highly likely that suppression of p38 MAPK activation might have also contributed to reduced myocardial injury post-I/R in CM-KO mice and following UTMD-mediated delivery of TRAF3IP2 AS-ODN in wild type mice. Thus, TRAF3IP2 could be a potential therapeutic target in ischemic heart disease.
Future Perspectives
Unlike its causal role in the I/R injury (49), activation of NF-κB is cardioprotective in the MI (permanent ligation of coronary artery) model (52). However, JNK activation plays a deleterious role in both I/R (11, 12) and MI (53) models. Because TRAF3IP2 is an upstream regulator of both NF-κB and JNK, it will be critical to determine whether targeting TRAF3IP2 reduces myocardial injury and adverse remodeling post-MI.
Experimental Procedures
Animals and Generation of Conditional Cardiomyocyte-specific Traf3ip2 Knock-out Mice
All animal studies conformed to NIH guidelines, and were approved by the Institutional Animal Care and Use Committees at Tulane University, New Orleans, LA, and University of Texas Health Sciences Center, San Antonio, TX. Generation of TRAF3IP2-loxP targeted mice (exon 2 was flanked by loxP sites; floxed or fl) on a C57Bl/6 background was previously described (54). The floxed mice were crossed with αMHC-MerCreMer mice (stock number 005650; The Jackson Laboratory) to generate Cre+/−;TRAF3IP2fl/+ (CM-TRAF3IP2-KO or CM-KO) mice. Two month-old male CM-KO mice were injected with tamoxifen in corn oil (Sigma, 20 mg/kg body wt, IP) daily for 4 days to delete TRAF3IP2 specifically in cardiomyocytes. As experimental controls, we used two genotypes: Cre−/−;TRAF3IP2fl/fl and Cre+/−;TRAF3IP2+/+ mice. All the animals were injected with tamoxifen and Cre−/−;TRAF3IP2fl/fl mice were used to confirm that the observed phenotype was not due to loxP insertion into the TRAF3IP2 allele, and Cre+/−;TRAF3IP2+/+ mice were used to confirm that Cre expression was not the cause of the phenotype. One month after the last tamoxifen injection, TRAF3IP2 expression in LV tissue from control and CM-KO mice was analyzed by immunoblotting. Hearts from the two control groups and WT mice appeared normal by histology, and were similar to hearts from untreated naive mice (data not shown). Therefore, in all subsequent experiments, we used Cre+/−;TRAF3IP2+/+ mice as controls. One month after the last tamoxifen administration, both control and CM-KO mice underwent the I/R procedure as previously described (26) using a chronically instrumented closed chest mouse model. The animal numbers are detailed in Tables 4 and 5. A subgroup of animals underwent ischemia only (30 min ischemia and no reperfusion). Sham-operated animals were prepared identically, but without ligating the left anterior descending coronary artery. At the indicated time points, the hearts were rapidly excised and rinsed in ice-cold physiological saline. The right ventricle and atria were trimmed away, and the left ventricle was divided into ischemic and non-ischemic zones and snap frozen in liquid N2 and stored at −80 °C.
TABLE 4.
Treatment groups using a genetic approach
The following abbreviations were used: control, Cre+/−;TRAF3IP2+/+; CM-KO, conditional cardiomyocyte-specific TRAF3IP2 knockout mice (Cre+/−;TRAF3IP2fl/+).
| Group | Model | n |
|---|---|---|
| 1. | Control-sham-operated | 24 |
| 2. | Control-I/R | 36 |
| 3. | CM-KO-sham-operated | 24 |
| 4. | CM-KO-I/R | 36 |
TABLE 5.
Treatment groups using a interventional approach
| Group | Wild type C57Bl/6 mice treatment | n |
|---|---|---|
| 1. | Sham-operated + ultrasound | 8 |
| 2. | I/R + no treatment + ultrasound | 12 |
| 3. | I/R + PESDA alone + ultrasound | 12 |
| 4. | I/R + control AS-ODN + ultrasound | 12 |
| 5. | I/R + p65 AS-ODN + ultrasound | 12 |
| 6. | I/R + JNK1 AS-ODN + ultrasound | 12 |
| 7. | I/R + TRAF3IP2 AS-ODN + ultrasound | 12 |
| 8. | I/R + scrambled ODN + ultrasound | 12 |
UTMD-mediated AS-ODN Delivery
PESDA is an echocardiographic contrast agent, and retains the drug binding properties of albumin site 1 and the ability to bind AS-ODN with high affinity (55). The delivery system takes advantage of both the drug binding properties of PESDA and the fact that PESDA microbubbles can be destroyed with ultrasound (56). UTMD can release ODNs locally in high concentration, resulting in higher uptake to exert the therapeutic effect. Site-specific delivery by UTMD may also lower systemic concentrations of ODN, resulting in fewer side effects. The phosphorothioate internucleotide linkages increase endo- and exonuclease resistance in vivo. Preparation of PESDA microbubbles, encapsulation of phosphorothioated AS-ODN (100 μg), arterial delivery into LV, and application of ultrasound, have all been described in detail previously (33). PESDA microbubbles were prepared on the day of experiment and used within 4 h. AS-ODN mixed with PESDA was infused arterially into anesthetized wild type mice using a small silastic catheter (Micro-Renathane, Braintree Scientific, Inc., Braintree MA) advanced into the LV via the right carotid artery. The animals then underwent 30 min ischemia. During the reperfusion period, PESDA microbubbles with AS-ODN were delivered slowly over a period of 10 min with simultaneous application of therapeutic ultrasound to the anterior chest wall (1 MHz, 0.6 W/cm2, 500 mm2 applicator, 15 min; Sonicator® 730, Mettler Electronics Corp., Anaheim, CA) as described previously in rats (33). After treatment, the catheter was removed, the artery ligated, and the animals allowed to recover. No cell death was observed in WT mice that underwent UTMD (no changes in cardiac troponin I levels and TUNEL-positive cells).6
Echocardiography, Infarct Size Measurement, Cardiomyocyte Size, and Fibrosis
FS was calculated from M-mode images taken in short axis view at the level of the papillary muscles using a Vevo 770 high-resolution ultrasound system (FUJIFILM VisualSonics, Toronto, ON) with a 30-MHz frequency real-time microvisualization scan head (RMV707). FS was calculated using the equation %FS = LVEDD − LVESD/LVEDD × 100, where LVEDD is LV end diastolic diameter, and LVESD is LV end systolic diameter. Infarct size was quantified after 24 h reperfusion using TTC (26). TTC normally stains tissue dark red; infarcted tissue will be unstained and appear white. The LV area (LV), area at risk (AR), and area of necrosis (AN) for all slices were calculated by planimetry. The AR was expressed as a percentage of LV (percentage of AR = total AR/total LV × 100), and the AN was expressed as a percentage of the AR (infarct size = total AN/total AR × 100). Hearts used for infarct size measurement were not employed in biochemical or molecular analyses. Cardiomyocyte size was determined by measuring the cross-sectional area in H&E-stained sections and quantified using ImageJ software. Cardiac fibrosis was analyzed by picrosirius red staining. For quantitative morphometric analysis, five random sections were electronically scanned into RGB images, which were subsequently analyzed using the ImageJ software, and include both perivascular and interstitial fibrosis (57).
mRNA Expression and MMP Activation
Total RNA was isolated from frozen LV tissue using TRIzol reagent (Sigma) and 1 μg of RNA was reverse transcribed into cDNA using a reverse transcription kit (Agilent Technologies). mRNA expression was analyzed by RT2 ProfilerTM PCR Arrays from Qiagen (Inflammatory Cytokines & Receptors PCR Array, PAMM-011Z; and Extracellular Matrix & Adhesion Molecule PCR Array, PAMM-013Z) that simultaneously detect 84 genes each. Expressions of genes that were below detection level and those that did not show a significant difference were excluded from the results in Table 1. mRNA expression of genes that were not included in arrays was analyzed by RT-qPCR using Applied Biosystems TaqManTM probes. All data were normalized to corresponding hprt (hypoxanthine phosphoribosyltransferase) levels and analyzed using the 2−ΔΔCt method. Activation of MMP2 and -9 was analyzed by immunoblotting using antibodies that detect both pro and active forms (MMP2, NB200–114G, Novus; MMP9, M9570, Sigma) (58).
Protein Quantibody Array, Western Blot Analysis, ELISA, Immunohistochemistry, and MPO Activity
A Quantibody® Mouse Cytokine Antibody Array 4000 (QAM-CAA-4000; RayBiotech) that detects 200 proteins simultaneously was used to analyze changes in protein expression in LV homogenates (57). Expressions of proteins that were below detection level and those that did not show a significant difference were excluded from the results in Table 2. Preparation of protein homogenates, immunoblotting, and densitometry were performed as described previously (17, 19, 20, 26). The source and concentration of antibodies used in immunoblotting are previously described (17, 19, 20, 26, 57). Anti-iNOS antibodies were from Novus (#NB-300; 1:500). Serum levels of IL-1β (number BMS6002), IL-6 (number BMS603/2), IL-17A (number BMS6026), IL-17F (number BMS6020), IL-18 (number BMS618/3), IL-33 (number BMS6025), and TNF-α (number BMS607/3) were analyzed by ELISA (eBioscience). TRAF3IP2 expression was localized by immunohistochemistry using anti-TRAF3IP2 antibodies (1:50; number sc-100647, Santa Cruz Biotechnology, Inc.) (57). Normal mouse IgG2a (number sc-3878; Santa Cruz Biotechnology, Inc.) served as a control. MPO activity in ischemic LV tissue was analyzed by a Lactate Dehydrogenase Activity Assay Kit (Sigma) as described previously (26).
Hydrogen Peroxide (H2O2) Production, Superoxide Generation, Lipid Peroxidation, and Nitrotyrosine Levels
H2O2 production in ischemic zones was analyzed by Amplex Red assay as previously described (59). Superoxide generation was quantified by cytochrome c assay as previously described (59). Lipid peroxidation was analyzed using a Lipid Peroxidation Assay kit (Calbiochem) that quantifies both MDA/4-HNE in LV homogenates (26). Nitrotyrosine levels in cleared tissue homogenates were analyzed by ELISA (60).
Serum ALT and AST Activity
Serum ALT and AST activities were analyzed by the Mouse Alanine Aminotransferase ELISA kit (MAK052) and the Mouse Aspartate Aminotransferase kit (MAK055) from Sigma.
Isolation of Neonatal Cardiomyocytes (NMCM), Hypoxia/Reoxygenation and LDH Assay
Cardiomyocytes were isolated from 1 to 3-day-old neonatal C57BL/6 mice as previously described using enzymatic digestion (61). At 70% confluence, the cells were incubated with in DMEM/M199 medium without fetal bovine serum for 12 h. Hypoxia was induced by replacing the initial culture medium with DMEM without glucose and serum, and flushed with 95% N2 and 5% CO2 for 15 min. The cells were then placed in a modular incubator chamber (Billups Rothenberg, Inc., Del Mar, CA) flushed with 95% N2 and 5% CO2 for 60 min. The sealed chamber was then placed into a 37 °C incubator. After the 4-h hypoxia incubation, the medium was replaced with fresh medium containing 10% serum, and an atmosphere of 95% air and 5% CO2 for reoxygenation. After 8 h, LDH released into medium was quantified. To determine the role of p65, JNK, and TRAF3IP2 in H/R-induced cell injury, cells plated at a density of 2 × 105/cm2 in 30-mm Falcon plates were transfected with the respective AS-ODN (500 nm) using FuGENE 6. After 6 h, cells were washed with serum-free medium, and exposed to H/R as described above. LDH levels were analyzed by Lactate Dehydrogenase Assay Kit (MAK066–1KT) from Sigma.
Statistical Analysis
Data were analyzed using Microsoft Excel, Clampfit (Molecular Device, Sunnyvale, CA), and Origin 7 (OriginLab Corp., Northampton, MA) programs. Normality criteria were evaluated to select the correct parametric or non-parametric test using the Shapiro-Wilk estimator. Because the sample size was small (n = 4–8/group), the test determined that the non-parametric approach should be used to avoid type II errors. Because two groups were used (control and CM-KO), pairwise comparisons were made using the Tukey adjustment. The statistical tests were performed using the software SPSS 23.0.0.1 (Chicago, IL). The overall significance level was set at 0.05. The results are presented as the mean ± S.E.
Author Contributions
B. C. designed the experimental strategy and interpreted the data. J. M. E., A. J. V., S. M., H. K., V. D., S. B. B., and W. P. F. performed experiments. U. S. and H. K. generated TRAF3IP2 floxed mice and conditional cardiomyocyte-specific TRAF3IP2 knockout mice. J. M. E., A. J. V., and B. C. analyzed the data. J. M. E., A. J. V., and B. C. wrote the manuscript.
The work was supported in part by United States Department of Veterans Affairs, Office of Research and Development-Biomedical Laboratory Research and Development (ORD-BLRD) Service Award I01-BX002255. The authors declare that they have no conflicts of interest with the contents of this article. The contents of this report do not represent the views of the Department of Veterans Affairs or the United States government. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
J. M. Erikson, A. J. Valente, S. Mummidi, H. K. Kandikattu, V. G. DeMarco, S. B. Bender, W. P. Fay, U. Siebenlist, and B. Chandrasekar, unpublished observations.
- I/R
- ischemia/reperfusion
- ALT
- alanine transaminase
- AS-ODN
- antisense deoxynucleotides
- AST
- aspartate aminotransferase
- C/EBP
- CCAAT enhancer-binding protein
- CREB
- cAMP responsive element-binding protein 1
- IRF
- interferon regulatory factor
- LFA
- lymphocyte function-associated antigen
- LIX
- lipopolysaccharide-induced CXC chemokine
- NEMO
- NF-κB essential modulator
- TTC
- 2,3,5-triphenyltetrazolium chloride
- UTMD
- ultrasound-targeted microbubble destruction
- CM-KO
- cardiomyocyte knock-out
- FS
- fractional shortening
- PESDA
- perfluorocarbon-exposed sonicated dextrose albumin
- 4-HNE
- 4-hydroxyalkenals
- LV
- left ventricle
- MDA
- malondialdehyde
- NMCM
- neonatal cardiomyocytes
- LDH
- lactate dehydrogenase
- MPO
- myeloperoxidase
- AS-ODN
- antisense deoxyoligonucleotide
- MMP
- matrix metalloprotease
- qPCR
- quantitative PCR.
References
- 1. Hausenloy D. J., and Yellon D. M. (2013) Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J. Clin. Invest. 123, 92–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Turer A. T., and Hill J. A. (2010) Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am. J. Cardiol. 106, 360–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Entman M. L., Youker K. A., Frangogiannis N., Lakshminarayanan V., Nossuli T., Evans A., Kurrelmeyer K., Mann D. L., and Smith C. W. (2000) Is inflammation good for the ischemic heart: perspectives beyond the ordinary. Z. Kardiol 89, Suppl. 9, IX/82–87 [DOI] [PubMed] [Google Scholar]
- 4. Jones W. K., Brown M., Wilhide M., He S., and Ren X. (2005) NF-kappaB in cardiovascular disease: diverse and specific effects of a “general” transcription factor? Cardiovasc. Toxicol. 5, 183–202 [DOI] [PubMed] [Google Scholar]
- 5. Dawn B., Xuan Y. T., Marian M., Flaherty M. P., Murphree S. S., Smith T. L., Bolli R., and Jones W. K. (2001) Cardiac-specific abrogation of NF-κB activation in mice by transdominant expression of a mutant IκBα. J. Mol. Cell Cardiol. 33, 161–173 [DOI] [PubMed] [Google Scholar]
- 6. Kim Y. S., Kim J. S., Kwon J. S., Jeong M. H., Cho J. G., Park J. C., Kang J. C., and Ahn Y. (2010) BAY 11–7082, a nuclear factor-κB inhibitor, reduces inflammation and apoptosis in a rat cardiac ischemia-reperfusion injury model. Int. Heart. J 51, 348–353 [DOI] [PubMed] [Google Scholar]
- 7. Squadrito F., Deodato B., Squadrito G., Seminara P., Passaniti M., Venuti F. S., Giacca M., Minutoli L., Adamo E. B., Bellomo M., Marini R., Galeano M., Marini H., and Altavilla D. (2003) Gene transfer of IκBα limits infarct size in a mouse model of myocardial ischemia-reperfusion injury. Lab. Invest. 83, 1097–1104 [DOI] [PubMed] [Google Scholar]
- 8. Moss N. C., Stansfield W. E., Willis M. S., Tang R. H., and Selzman C. H. (2007) IKKβ inhibition attenuates myocardial injury and dysfunction following acute ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 293, H2248–H2253 [DOI] [PubMed] [Google Scholar]
- 9. Onai Y., Suzuki J., Kakuta T., Maejima Y., Haraguchi G., Fukasawa H., Muto S., Itai A., and Isobe M. (2004) Inhibition of IκB phosphorylation in cardiomyocytes attenuates myocardial ischemia/reperfusion injury. Cardiovasc. Res. 63, 51–59 [DOI] [PubMed] [Google Scholar]
- 10. Yin T., Sandhu G., Wolfgang C. D., Burrier A., Webb R. L., Rigel D. F., Hai T., and Whelan J. (1997) Tissue-specific pattern of stress kinase activation in ischemic/reperfused heart and kidney. J. Biol. Chem. 272, 19943–19950 [DOI] [PubMed] [Google Scholar]
- 11. Kaiser R. A., Liang Q., Bueno O., Huang Y., Lackey T., Klevitsky R., Hewett T. E., and Molkentin J. D. (2005) Genetic inhibition or activation of JNK1/2 protects the myocardium from ischemia-reperfusion-induced cell death in vivo. J. Biol. Chem. 280, 32602–32608 [DOI] [PubMed] [Google Scholar]
- 12. Milano G., Morel S., Bonny C., Samaja M., von Segesser L. K., Nicod P., and Vassalli G. (2007) A peptide inhibitor of c-Jun NH2-terminal kinase reduces myocardial ischemia-reperfusion injury and infarct size in vivo. Am. J. Physiol. Heart Circ. Physiol. 292, H1828–H1835 [DOI] [PubMed] [Google Scholar]
- 13. Harhaj E. W., and Dixit V. M. (2012) Regulation of NF-κB by deubiquitinases. Immunol. Rev. 246, 107–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Israel A. (2010) The IKK complex, a central regulator of NF-κB activation. Cold Spring Harb. Perspect. Biol. 2, a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leonardi A., Chariot A., Claudio E., Cunningham K., and Siebenlist U. (2000) CIKS, a connection to IκB kinase and stress-activated protein kinase. Proc. Natl. Acad. Sci. U.S.A. 97, 10494–10499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li X., Commane M., Nie H., Hua X., Chatterjee-Kishore M., Wald D., Haag M., and Stark G. R. (2000) Act1, an NF-κB-activating protein. Proc. Natl. Acad. Sci. U.S.A. 97, 10489–10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Valente A. J., Clark R. A., Siddesha J. M., Siebenlist U., and Chandrasekar B. (2012) CIKS (Act1 or TRAF3IP2) mediates angiotensin-II-induced Interleukin-18 expression, and Nox2-dependent cardiomyocyte hypertrophy. J. Mol. Cell Cardiol. 53, 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao Z., Qian Y., Wald D., Xia Y. F., Geng J. G., and Li X. (2003) IFN regulatory factor-1 is required for the up-regulation of the CD40-NF-κB activator 1 axis during airway inflammation. J. Immunol. 170, 5674–5680 [DOI] [PubMed] [Google Scholar]
- 19. Venkatesan B., Valente A. J., Das N. A., Carpenter A. J., Yoshida T., Delafontaine J. L., Siebenlist U., and Chandrasekar B. (2013) CIKS (Act1 or TRAF3IP2) mediates high glucose-induced endothelial dysfunction. Cell Signal. 25, 359–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Valente A. J., Yoshida T., Clark R. A., Delafontaine P., Siebenlist U., and Chandrasekar B. (2013) Advanced oxidation protein products induce cardiomyocyte death via Nox2/Rac1/superoxide-dependent TRAF3IP2/JNK signaling. Free Radic. Biol. Med. 60, 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qian Y., Liu C., Hartupee J., Altuntas C. Z., Gulen M. F., Jane-Wit D., Xiao J., Lu Y., Giltiay N., Liu J., Kordula T., Zhang Q. W., Vallance B., Swaidani S., Aronica M., Tuohy V. K., Hamilton T., and Li X. (2007) The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat. Immunol. 8, 247–256 [DOI] [PubMed] [Google Scholar]
- 22. Ha H. L., Wang H., Pisitkun P., Kim J. C., Tassi I., Tang W., Morasso M. I., Udey M. C., and Siebenlist U. (2014) IL-17 drives psoriatic inflammation via distinct, target cell-specific mechanisms. Proc. Natl. Acad. Sci. U.S.A. 111, E3422–E3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chandrasekar B., Colston J. T., Geimer J., Cortez D., and Freeman G. L. (2000) Induction of nuclear factor κB but not κB-responsive cytokine expression during myocardial reperfusion injury after neutropenia. Free Radic. Biol. Med. 28, 1579–1588 [DOI] [PubMed] [Google Scholar]
- 24. Chandrasekar B., and Freeman G. L. (1997) Induction of nuclear factor κB and activation protein 1 in postischemic myocardium. FEBS Lett. 401, 30–34 [DOI] [PubMed] [Google Scholar]
- 25. Chandrasekar B., Smith J. B., and Freeman G. L. (2001) Ischemia-reperfusion of rat myocardium activates nuclear factor-κB and induces neutrophil infiltration via lipopolysaccharide-induced CXC chemokine. Circulation 103, 2296–2302 [DOI] [PubMed] [Google Scholar]
- 26. Venkatachalam K., Prabhu S. D., Reddy V. S., Boylston W. H., Valente A. J., and Chandrasekar B. (2009) Neutralization of interleukin-18 ameliorates ischemia/reperfusion-induced myocardial injury. J. Biol. Chem. 284, 7853–7865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Granger D. N., and Kvietys P. R. (2015) Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 6, 524–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kalogeris T., Baines C. P., Krenz M., and Korthuis R. J. (2012) Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 298, 229–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ferdinandy P., and Schulz R. (2003) Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br. J. Pharmacol. 138, 532–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frangogiannis N. G. (2014) The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 11, 255–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pall M. L. (2013) The NO/ONOO-cycle as the central cause of heart failure. Int. J. Mol. Sci. 14, 22274–22330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kurian G. A., Rajagopal R., Vedantham S., and Rajesh M. (2016) The role of oxidative stress in myocardial ischemia and reperfusion injury and remodeling: revisited. Oxid. Med. Cell Longev. 2016, 1656450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Erikson J. M., Freeman G. L., and Chandrasekar B. (2003) Ultrasound-targeted antisense oligonucleotide attenuates ischemia/reperfusion-induced myocardial tumor necrosis factor-α. J. Mol. Cell Cardiol. 35, 119–130 [DOI] [PubMed] [Google Scholar]
- 34. Schlaak J. F., Barreiros A. P., Pettersson S., Schirmacher P., Meyer Zum Büschenfelde K. H., and Neurath M. F. (2001) Antisense phosphorothioate oligonucleotides to the p65 subunit of NF-κB abrogate fulminant septic shock induced by S. typhimurium in mice. Scand. J. Immunol. 54, 396–403 [DOI] [PubMed] [Google Scholar]
- 35. Gunawan B. K., Liu Z. X., Han D., Hanawa N., Gaarde W. A., and Kaplowitz N. (2006) c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 131, 165–178 [DOI] [PubMed] [Google Scholar]
- 36. Anrather J., Racchumi G., and Iadecola C. (2006) NF-κB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 281, 5657–5667 [DOI] [PubMed] [Google Scholar]
- 37. Williams C. R., Lu X., Sutliff R. L., and Hart C. M. (2012) Rosiglitazone attenuates NF-κB-mediated Nox4 upregulation in hyperglycemia-activated endothelial cells. Am. J. Physiol. Cell Physiol. 303, C213–C223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eklund E. A., Luo W., and Skalnik D. G. (1996) Characterization of three promoter elements and cognate DNA binding protein(s) necessary for IFN-γ induction of gp91-phox transcription. J. Immunol. 157, 2418–2429 [PubMed] [Google Scholar]
- 39. Xie Q. W., Kashiwabara Y., and Nathan C. (1994) Role of transcription factor NF-κB/Rel in induction of nitric-oxide synthase. J. Biol. Chem. 269, 4705–4708 [PubMed] [Google Scholar]
- 40. Levrand S., Vannay-Bouchiche C., Pesse B., Pacher P., Feihl F., Waeber B., and Liaudet L. (2006) Peroxynitrite is a major trigger of cardiomyocyte apoptosis in vitro and in vivo. Free Radic. Biol. Med. 41, 886–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rovai L. E., Herschman H. R., and Smith J. B. (1998) The murine neutrophil-chemoattractant chemokines LIX, KC, and MIP-2 have distinct induction kinetics, tissue distributions, and tissue-specific sensitivities to glucocorticoid regulation in endotoxemia. J. Leukoc. Biol. 64, 494–502 [DOI] [PubMed] [Google Scholar]
- 42. Chen S., and Grayburn P. A. (2017) Ultrasound-targeted microbubble destruction for cardiac gene delivery. Methods Mol. Biol. 1521, 205–218 [DOI] [PubMed] [Google Scholar]
- 43. Zen K., Okigaki M., Hosokawa Y., Adachi Y., Nozawa Y., Takamiya M., Tatsumi T., Urao N., Tateishi K., Takahashi T., and Matsubara H. (2006) Myocardium-targeted delivery of endothelial progenitor cells by ultrasound-mediated microbubble destruction improves cardiac function via an angiogenic response. J. Mol. Cell Cardiol. 40, 799–809 [DOI] [PubMed] [Google Scholar]
- 44. Wang J., Zhao Z., Shen S., Zhang C., Guo S., Lu Y., Chen Y., Liao W., Liao Y., and Bin J. (2015) Selective depletion of tumor neovasculature by microbubble destruction with appropriate ultrasound pressure. Int. J. Cancer 137, 2478–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shohet R. V., Chen S., Zhou Y. T., Wang Z., Meidell R. S., Unger R. H., and Grayburn P. A. (2000) Echocardiographic destruction of albumin microbubbles directs gene delivery to the myocardium. Circulation 101, 2554–2556 [DOI] [PubMed] [Google Scholar]
- 46. Wei K., Skyba D. M., Firschke C., Jayaweera A. R., Lindner J. R., and Kaul S. (1997) Interactions between microbubbles and ultrasound: in vitro and in vivo observations. J. Am. Coll. Cardiol. 29, 1081–1088 [DOI] [PubMed] [Google Scholar]
- 47. Mukherjee D., Wong J., Griffin B., Ellis S. G., Porter T., Sen S., and Thomas J. D. (2000) Ten-fold augmentation of endothelial uptake of vascular endothelial growth factor with ultrasound after systemic administration. J. Am. Coll. Cardiol. 35, 1678–1686 [DOI] [PubMed] [Google Scholar]
- 48. Schlegel P., Huditz R., Meinhardt E., Rapti K., Geis N., Most P., Katus H. A., Müller O. J., Bekeredjian R., and Raake P. W. (2016) Locally targeted cardiac gene delivery by AAV microbubble destruction in a large animal model. Hum. Gene Ther. Methods 27, 71–78 [DOI] [PubMed] [Google Scholar]
- 49. Frantz S., Tillmanns J., Kuhlencordt P. J., Schmidt I., Adamek A., Dienesch C., Thum T., Gerondakis S., Ertl G., and Bauersachs J. (2007) Tissue-specific effects of the nuclear factor κB subunit p50 on myocardial ischemia-reperfusion injury. Am. J. Pathol. 171, 507–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. He H., Li H. L., Lin A., and Gottlieb R. A. (1999) Activation of the JNK pathway is important for cardiomyocyte death in response to simulated ischemia. Cell Death Differ. 6, 987–991 [DOI] [PubMed] [Google Scholar]
- 51. Ma X. L., Kumar S., Gao F., Louden C. S., Lopez B. L., Christopher T. A., Wang C., Lee J. C., Feuerstein G. Z., and Yue T. L. (1999) Inhibition of p38 mitogen-activated protein kinase decreases cardiomyocyte apoptosis and improves cardiac function after myocardial ischemia and reperfusion. Circulation 99, 1685–1691 [DOI] [PubMed] [Google Scholar]
- 52. Misra A., Haudek S. B., Knuefermann P., Vallejo J. G., Chen Z. J., Michael L. H., Sivasubramanian N., Olson E. N., Entman M. L., and Mann D. L. (2003) Nuclear factor-κB protects the adult cardiac myocyte against ischemia-induced apoptosis in a murine model of acute myocardial infarction. Circulation 108, 3075–3078 [DOI] [PubMed] [Google Scholar]
- 53. Wei J., Wang W., Chopra I., Li H. F., Dougherty C. J., Adi J., Adi N., Wang H., and Webster K. A. (2011) c-Jun N-terminal kinase (JNK-1) confers protection against brief but not extended ischemia during acute myocardial infarction. J. Biol. Chem. 286, 13995–14006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pisitkun P., Ha H. L., Wang H., Claudio E., Tivy C. C., Zhou H., Mayadas T. N., Illei G. G., and Siebenlist U. (2012) Interleukin-17 cytokines are critical in development of fatal lupus glomerulonephritis. Immunity 37, 1104–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Srinivasan S. K., Tewary H. K., and Iversen P. L. (1995) Characterization of binding sites, extent of binding, and drug interactions of oligonucleotides with albumin. Antisense Res. Dev. 5, 131–139 [DOI] [PubMed] [Google Scholar]
- 56. Porter T. R., Iversen P. L., Li S., and Xie F. (1996) Interaction of diagnostic ultrasound with synthetic oligonucleotide-labeled perfluorocarbon-exposed sonicated dextrose albumin microbubbles. J. Ultrasound Med. 15, 577–584 [DOI] [PubMed] [Google Scholar]
- 57. Yariswamy M., Yoshida T., Valente A. J., Kandikattu H. K., Sakamuri S. S., Siddesha J. M., Sukhanov S., Saifudeen Z., Ma L., Siebenlist U., Gardner J. D., and Chandrasekar B. (2016) Cardiac-restricted overexpression of TRAF3 interacting protein 2 (TRAF3IP2) results in spontaneous development of myocardial hypertrophy, fibrosis, and dysfunction. J. Biol. Chem. 291, 19425–19436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Siddesha J. M., Valente A. J., Sakamuri S. S., Yoshida T., Gardner J. D., Somanna N., Takahashi C., Noda M., and Chandrasekar B. (2013) Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK. J. Mol. Cell Cardiol. 65, 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Frazziano G., Al Ghouleh I., Baust J., Shiva S., Champion H. C., and Pagano P. J. (2014) Nox-derived ROS are acutely activated in pressure overload pulmonary hypertension: indications for a seminal role for mitochondrial Nox4. Am. J. Physiol. Heart Circ. Physiol. 306, H197–H205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ma X. L., Gao F., Nelson A. H., Lopez B. L., Christopher T. A., Yue T. L., and Barone F. C. (2001) Oxidative inactivation of nitric oxide and endothelial dysfunction in stroke-prone spontaneous hypertensive rats. J. Pharmacol. Exp. Ther. 298, 879–885 [PubMed] [Google Scholar]
- 61. Colston J. T., de la Rosa S. D., Koehler M., Gonzales K., Mestril R., Freeman G. L., Bailey S. R., and Chandrasekar B. (2007) Wnt-induced secreted protein-1 is a prohypertrophic and profibrotic growth factor. Am. J. Physiol. Heart Circ. Physiol. 293, H1839–H1846 [DOI] [PubMed] [Google Scholar]