

Abstract
Prions or PrPSc are proteinaceous infectious agents that consist of misfolded, self-replicating states of a sialoglycoprotein called the prion protein or PrPC. The current work tests a new hypothesis that sialylation determines the fate of prions in an organism. To begin, we produced control PrPSc from PrPC using protein misfolding cyclic amplification with beads (PMCAb), and also generated PrPSc with reduced sialylation levels using the same method but with partially desialylated PrPC as a substrate (dsPMCAb). Syrian hamsters were inoculated intraperitoneally with brain-derived PrPSc or PrPSc produced in PMCAb or dsPMCAb and then monitored for disease. Animals inoculated with brain- or PMCAb-derived PrPSc developed prion disease, whereas administration of dsPMCAb-derived PrPSc with reduced sialylation did not cause prion disease. Animals inoculated with dsPMCAb-derived material were not subclinical carriers of scrapie, as no PrPSc was detected in brains or spleen of these animals by either Western blotting or after amplification by serial PMCAb. In subsequent experiments, trafficking of brain-, PMCAb-, and dsPMCAb-derived PrPSc to secondary lymphoid organs was monitored in wild type mice. PrPSc sialylation was found to be critical for effective trafficking of PrPSc to secondary lymphoid organs. By 6 hours after inoculation, brain- and PMCAb-derived PrPSc were found in spleen and lymph nodes, whereas dsPMCAb-derived PrPSc was found predominantly in liver. This study demonstrates that the outcome of prion transmission to a wild type host is determined by the sialylation status of the inoculated PrPSc. Furthermore, this work suggests that the sialylation status of PrPSc plays an important role in prion lymphotropism.
Keywords: Fourier transform IR (FTIR), N-linked glycosylation, prion, prion disease, sialic acid, protein misfolding, secondary lymphoid organs, sialylation, spleen, N-linked glycans, cyclic amplification
Introduction
Prions or disease-associated infectious form(s) of the prion protein (PrPSc)3 are proteinaceous infectious agents that consist of misfolded, aggregated, self-replicating states of a sialoglycoprotein called the prion protein or PrPC (1, 2). Prions replicate by recruiting and converting PrPC molecules expressed by a host into misfolded PrPSc states (3, 4). Although other proteins or peptides linked to neurodegenerative diseases display certain characteristics of prion-like replication (5, 6), PrPSc is unique in several important aspects. First, among proteins capable of forming self-replicating states in a cell or organism, only PrPSc were found to be highly transmissible between organisms or species via natural routes (7, 8). Second, unlike non-prion protein amyloids, PrPSc can be titrated and show incredibly high infectivity titers similar to those of viral or microbial pathogens (9, 10). Third, PrPSc can successfully escape the immune system of a host (11, 12). In fact, PrPSc colonizes cells responsible for host defense including cells of the lymphoreticular system and replicates in SLOs autonomously from CNS (13–16). More surprising, despite low expression levels of PrPC in SLOs, SLOs are more permissive than CNS to low doses of PrPSc or foreign PrPSc acquired via cross-species transmission (17, 18). What makes mammalian prions unique among other self-replicating amyloidogenic proteins? Why are prions so incredibly transmissible, including transmission via peripheral routes? How do prions escape the host immune system?
PrPC is posttranslationally modified with two terminally sialylated N-linked glycans (19–21). Upon conversion, the N-linked glycans are carried over, giving rise to sialylated PrPSc (22, 23). Sialylated glycans play an essential role in a broad range of cellular functions, but are especially important in immunity (24). Terminal sialic acid residues on the surface of mammalian cells act as a part of a “self-associated molecular pattern,” providing molecular cues to the immune system for discriminating between “self,” “altered self,” or “non-self” (25, 26). Glycoproteins or glycolipids that lack sialic acids at terminal positions serve as a “pathogen-associated molecular pattern” used by mammalian immune systems to recognize pathogens or asialoglycoproteins that need to be removed (25). Bearing in mind that sialylation serves as a molecular marker of self versus non-self, we proposed a hypothesis that sialylation determines the fate of prions in an organism (27). In support of this hypothesis, here we show that PrPSc produced from partially desialylated PrPC does not induce prion disease in animals upon peripheral exposure. In contrast to sialylated PrPSc, PrPSc with reduced sialylation status is not efficiently transported to SLOs and, instead, appears to undergo faster clearance. This study suggests that sialylation of PrPSc is responsible for the highly transmissible nature of mammalian prions.
Results
Producing PrPSc with Reduced Sialylation Status
To test whether sialylation controls infectivity of PrPSc, PrPSc with reduced sialylation levels was produced using protein misfolding cyclic amplification with beads (PMCAb) that used partially desialylated PrPC as a substrate. Normal brain homogenate (NBH) was treated with bacterial sialidases that cleave both α2-3-linked and α2-6-linked sialic acid residues (sialidase-treated NBH will be referred to as dsNBH) and supplied as a substrate to PMCAb reactions seeded with 263K (hamster-adapted scrapie strain) brain material (PMCAb conducted in dsNBH will be referred to as dsPMCAb) (28). PMCAb reactions conducted in NBH and seeded with brain material were used as controls. To analyze changes in sialylation status, brain-, PMCAb-, and dsPMCAb-derived materials were treated with PK to remove any remaining PrPC, then denatured into PrP monomers, and the sialylation status of individual PrP molecules was analyzed by 2D Western blots. In agreement with previous studies (28, 29), 263K brain material showed a broad distribution of charge isoforms on 2D Western blots (Fig. 1A). In fact, the sialylation level of individual PrP molecules varied from heavily sialylated (at acidic pH) to lacking sialylation (at basic pH). However, the distribution of charge isoforms of dsPMCAb-derived material was shifted significantly toward basic pH relative to that of brain-derived 263K and lacked heavily sialylated isoforms (Fig. 1, A and B). As a reference, 263K brain material that was first denatured into monomers and then treated with sialidases showed a similar shift in distribution of charge isoforms, as did the dsPMCAb-derived material (Fig. 1A). The distribution of PMCAb-derived material showed a minor shift toward basic pH relative to that of brain-derived 263K (Fig. 1, A and B). Such a shift was consistent with the previous study and illustrates that selective exclusion of heavily sialylated PrPC from conversion into PrPSc takes place in the course of replication in PMCAb (28). In summary, 2D analysis established the following rank order with respect to PrPSc sialylation status: brain 263K > PMCAb-derived 263K ≫ dsPMCAb-derived 263K.
FIGURE 1.
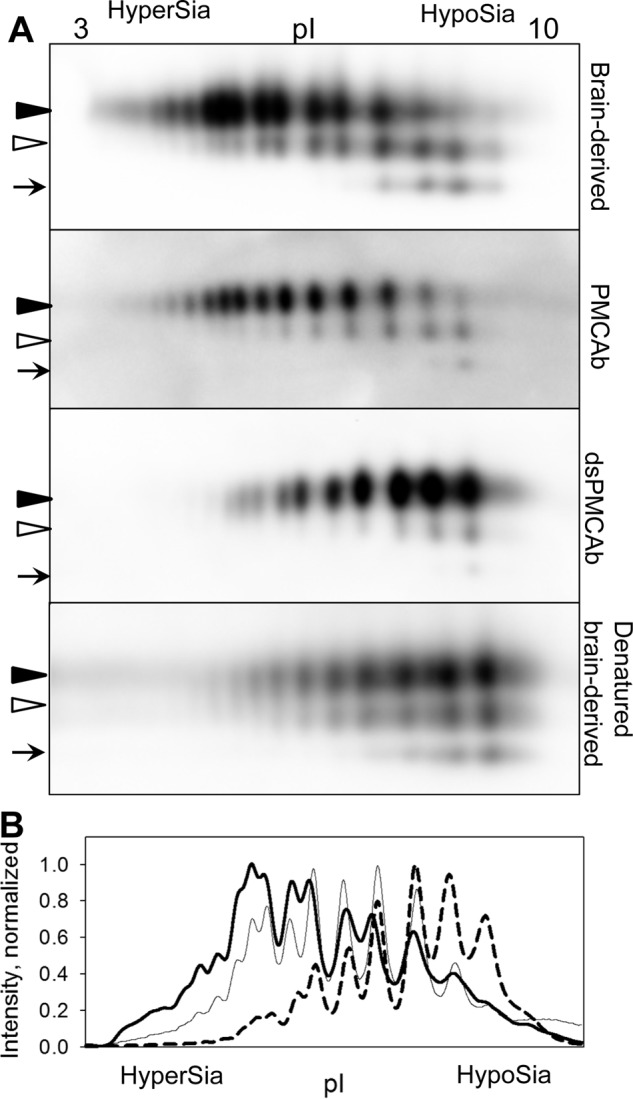
2D Western analysis of sialylation status of brain-, PMCAb-, and dsPMCAb-derived PrPSc. A, 2D Western blotting analysis of 263K brain-, PMCAb-, and dsPMCAb-derived material. 2D Western analysis of 263K brain-derived material denatured and then treated with sialidase is provided as a reference. Black and white triangles mark diglycosylated and monoglycosylated glycoforms, respectively, whereas arrows mark the unglycosylated form. All blots were stained with 3F4 antibody. HyperSia, hyper-sialylated; HypoSia, hypo-sialylated; pI, isoelectric point. B, sialylation profiles of diglycosylated isoforms of 263K brain- (solid thick line), PMCAb- (solid thin line), or dsPMCAb-derived material (dashed line). Profiles were built as described under “Experimental Procedures” using results of 2D Western blots.
PrPSc with Reduced Sialylation Status Does Not Cause Disease upon Peripheral Exposure
Syrian hamsters were inoculated i.p. with brain-, PMCAb-, or dsPMCAb-derived 263K. All animals inoculated with brain-derived 263K developed clinical signs of prion disease specific for 263K (Table 1). Their brain showed substantial accumulation of PrPSc, and smaller amounts of PrPSc were found in spleen (Fig. 2A). Five out of six animals inoculated with PMCAb-derived material developed clinical prion disease with clinical symptoms specific for 263K (Table 1). The five clinically positive animals showed considerable amounts of PrPSc in their brains and variable amounts in their spleens (Fig. 2A). No PrPSc was found in the brain or spleen from one clinically negative animal from this group. None of the animals inoculated with dsPMCAb-derived material showed any clinical signs of prion disease and were sacrificed at 352 days after inoculation (Table 1). No PrPSc was detected in brains or spleens of animals from this group by Western blotting (Fig. 2A). In summary, this experiment illustrated that the length of incubation time to disease and percentage of animals that succumb to the disease correlated well with the sialylation status of inoculated PrPSc.
TABLE 1.
Bioassay of 263K brain-, PMCAb-, and dsPMCAb-derived materials
| Inoculum | Incubation period (days after inoculation)a | n/nob |
|---|---|---|
| Brain-derived 263Kc | 155 ± 38 | 6/6 |
| PMCAb-derived 263K | 175 ± 57 | 5/6 |
| dsPMCAb-derived 263K | >352 | 0/6 |
a Average ± standard deviation for clinically sick animals.
b Number of clinically ill hamsters over the total number of inoculated hamsters.
c 104-fold diluted 263K brain material was used as an inoculum as a reference.
FIGURE 2.
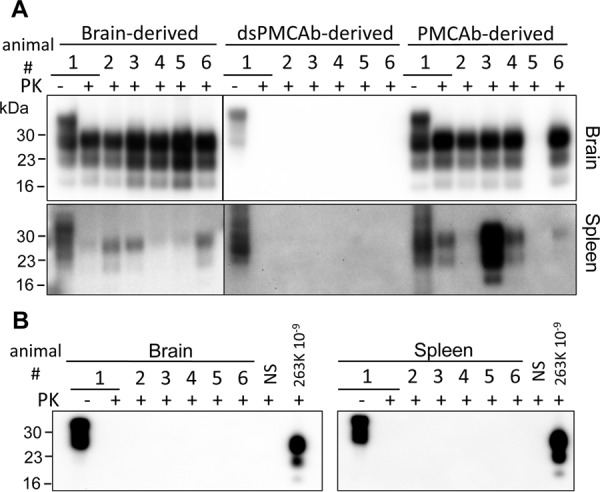
Western blotting and sPMCAb analysis of brains and spleens from animals inoculated with 263K brain-, PMCAb-, or dsPMCAb-derived material. Syrian hamsters were inoculated i.p. with 104-diluted 263K brain material, or 10-fold diluted PMCAb- or dsPMCAb-derived material (n = 6). The scrapie brain material was diluted 104-fold to match the amount of PK-resistant material to that in the 10-fold diluted PMCAb- and dsPMCAb-derived samples. Animals were euthanized at the terminal stage of the disease, or at 352 days after inoculation if no symptoms were evident. A, 10% brain or spleen homogenates were treated with PK and analyzed by Western blotting. B, sPMCAb reactions were seeded with 10% brain or spleen homogenates from hamsters inoculated with dsPMCAb-derived material and then subjected to four serial rounds, and reaction products were analyzed by Western blotting. As positive controls, sPMCAb reactions were seeded with 109-fold diluted 263K brain material. As negative controls for cross-contamination, non-seeded (NS) sPMCAb reactions were conducted in parallel. 3F4 antibody was used for staining in all Western blots.
Animals Inoculated with PrPSc with Reduced Sialylation Status Are Free of PrPSc
To test whether animals inoculated with dsPMCAb-derived material were subclinical carriers of prion infection, brain and spleen materials were analyzed using serial PMCAb (sPMCAb). In previous studies, three sPMCAb rounds were found to be sufficient for detecting a single infectious particle of 263K in animal tissues (9). Moreover, sPMCAb was shown to be 2 orders of magnitude more sensitive for detecting miniscule amounts of 263K PrPSc than animal bioassay (9). In the current study, sPMCAb reactions were seeded with 10% brain or spleen homogenates from animals infected with dsPMCAb-derived material and subjected to four serial rounds. As a positive control, sPMCAb reactions were seeded with 109-fold diluted 263K brain material. All sPMCAb reactions seeded with brain or spleen materials were negative, whereas reactions seeded with 109-fold diluted 263K produced positive signal on Western blot (Fig. 2B). These results argue that animals inoculated with dsPMCAb-derived material contain less than one prion infectious unit if any.
Infrared Microspectroscopy (IR-MSP) Did Not Detect Substantial Structural Differences That Would Explain the Loss of Infectivity of dsPMCAb-derived PrPSc
Although the reduced sialylation status of PrPSc correlates well with the loss of ability to infect a host, changes of the PrPSc 3D (or spatial) structure during PMCAb/dsPMCAb could also account for the drop in infectivity (30–32). In fact, previous studies that employed infrared microscopy revealed conformational changes in 263K brain material subjected to PMCA (32). To compare the conformation of PrPSc in three inoculums, PrPSc was purified from 263K brain-, PMCAb-, and dsPMCAb-derived materials and analyzed by IR-MSP. Brain-derived 263K showed a double peak at 1626/1634 cm−1 that corresponds to β-sheet-rich conformation, a small peak at 1695 cm−1 that also reports on β-sheet structures, and a peak at 1659 cm−1, which is conventionally assigned to a α-helical conformation (Fig. 3). In the context of the peak at 1659 cm−1, however, it has to be noted that the association between specific IR absorption bands and α-helical structure elements in PrP is currently under discussion (33). Analysis of PMCAb- and dsPMCAb-derived materials confirmed that structural changes occurred in PrPSc during serial amplification. Specifically, in PMCAb- and dsPMCAb-derived materials, only a single peak for β-sheet structures was detected at 1626 cm−1 instead of a double peak within the same region in brain-derived PrPSc (Fig. 3). Changes were also noticeable within the 1657–1659-cm−1 amide I region and between 1681 and 1695 cm−1. Remarkably, the spectra of PMCAb- and dsPMCAb-derived materials were very similar if not identical, yet PMCAb-derived 263K induced the disease, whereas infectivity was lost for dsPMCAb-derived material. To determine whether minor structural differences between PMCAb and dsPMCAb groups exist, hierarchical cluster analysis was performed (Fig. 3B). Previously, hierarchical cluster analysis was found to be very helpful in detecting differences in the conformation-sensitive amide I region of IR spectra of prion strains or isolates or their PMCA derivatives that were not obvious by visual inspection of spectra (32, 34, 35). The analysis revealed two main clusters: spectra of three 263K brains belong to one cluster, whereas all spectra of PMCAb- and dsPMCAb-derived materials formed another cluster. Although the structural differences between the two clusters were clearly noticeable, pronounced differences between PMCAb- and dsPMCAb-derived products could not be observed. In summary, although changes in PrPSc structure might account for some loss of infectivity in PMCAb and dsPMCAb materials, the structural differences between PMCAb and dsPMCAb materials did not appear substantial for explaining the loss of infectivity of dsPMCAb as compared with PMCAb.
FIGURE 3.
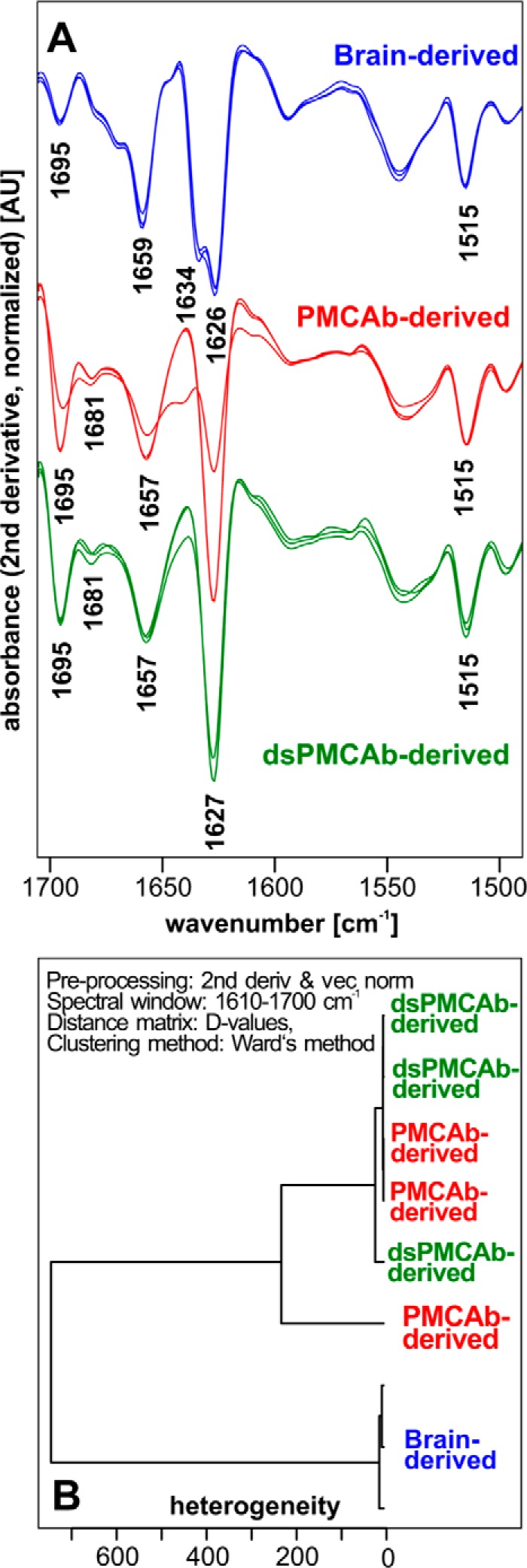
Assessing secondary structure by infrared microspectroscopy. A, IR spectra obtained from PrPSc materials purified from brains of three Syrian Hamster infected with 263K (blue), three independent PMCAb reactions (red), or three independent dsPMCAb reactions (green), each seeded with brain-derived 263K. Each spectrum represents a minimum/maximum normalized (tyrosine band at 1515 cm−1) second derivative spectrum obtained by averaging 10 individual point spectra. AU, arbitrary units. B, analysis of the conformational heterogeneity of 263K PrPSc material purified from brains of animals infected with 263K or PMCAb or dsPMCAb reactions (n = 3 independent brains or reactions). Dendrogram was obtained by hierarchical cluster analysis of the mean IR microspectra using the information content in the amide I region (1610–1670 cm−1), D-values as the inter-spectral distance measure, and Ward's algorithm as the clustering method. 2nd deriv, second derivative; vec norm, vector normalized.
Sialylation Status of PrPSc Controls Trafficking/Colonization of SLOs
For prions acquired via peripheral routes, colonization of SLOs typically precedes invasion of the nervous system and is considered to be an obligatory step before neuroinvasion (11, 36–38). Arrival of prions to SLOs could be detected as early as 2 h after intraperitoneal administration (39). To test whether PrPSc sialylation status controls prion trafficking to SLOs and other peripheral organs, wild type C57Bl mice were inoculated i.p. with 263K brain-, PMCAb-, and dsPMCAb-derived materials, and the amounts of PrPSc sequestered in spleen, lymph nodes, liver, and kidney were assessed 2, 6, and 18 h after inoculation (Fig. 4). Brains from the same animal groups were used as negative controls. Wild type mice were used instead of hamsters for inoculating hamster 263K scrapie materials to avoid interference due to immediate replication of 263K in hamster SLOs (40, 41). The amounts of PrPSc in three samples were carefully calibrated using the PK digestion assay to make sure that brain-, PMCAb-, and dsPMCAb-derived inoculums contain the same amounts of PrPSc.
FIGURE 4.

Analysis of PrPSc trafficking to SLOs, liver and kidney upon intraperitoneal inoculation. A, 263K brain-, PMCAb-, or dsPMCAb-derived materials were administered via intraperitoneal injection to C57Bl mice, and then the amounts of scrapie material in spleen, lymph nodes, liver, kidney, or brain were analyzed 2, 6, and 18 h after inoculation by Western blotting (n = 3 per animal group per each time point). 0.05% 263K scrapie brain homogenate (ScBH) treated with PK served as a reference in all Western blots. Western blots were stained with Ab3531 antibody. B and C, dynamics of PrPSc accumulation in spleen (B) and lymph nodes (C) for brain- (black circles), PMCAb- (white circles), or dsPMCAb-derived material (triangles) are presented on the top plots, while statistical analysis of PrPSc amounts in spleen (B) or lymph nodes (C) at 18 h after inoculation are shown on the bottom plots. For each animal group/tissue, the signal intensities were normalized per the intensities of 0.05% scrapie brain homogenate on the same Western blots. Statistical analysis was performed as described under “Experimental Procedures.” Data are presented as means ± S.D., * indicates significant differences (p < 0.05), whereas # indicates lack of significant differences (p > 0.05) (n = 3).
In groups inoculated with brain-derived 263K, PrPSc was detected in spleen and lymph nodes in one out of three animals at 2 h and in all animals at 6 and 18 h after inoculation (Fig. 4). In contrast, spleens of animals injected with dsPMCAb materials showed only a barely detectible PK-resistant signal at 6 and 18 h after inoculation. Lymph nodes also showed very weak signals in one out of three animals at each time point. These results indicate that dsPMCAb material is not transported to SLOs as efficiently as brain-derived 263K and/or is removed very quickly from circulation. PMCAb-derived material was found in spleen and lymph nodes, but the signal was not as strong as in corresponding groups injected with brain-derived 263K. In liver, the brain-derived 263K was detected only at 6 h, and PMCAb material was not detected at any time points, whereas dsPMCAb material was found in one out of three animals at 2 and 6 h and in all three animals at 18 h. These results suggest that partially desialylated PrPSc is targeted to the liver more effectively than normal PrPSc. In kidney, only animals inoculated with brain-derived 263K showed a transient signal at 6 h. As expected, PrPSc was not detected in brains in any animal groups at any time point. Although variations within animal groups/time points were observed, the following conclusions could be made. The efficiency of trafficking of PrPSc administered via peripheral route to SLOs correlated well with its sialylation status. dsPMCAb-derived material appeared to be targeted to liver, while showing the lowest signal across all organs tested at any specific time point in comparison with 263K or PMCAb-derived material. These results suggest that dsPMCAb-derived material is cleared faster than brain- or PMCAb-derived materials.
Discussion
Peripheral infection is the natural route of transmission for most prion diseases. The current study established that sialylation of PrPSc administered via peripheral route controls its ability to infect a host. 263K scrapie brain material amplified using partially desialylated PrPC failed to induce diseases in hamsters, whereas the material generated with normally sialylated PrPC induced clinical disease. Partially desialylated material did not cause clinical disease, and PrPSc was not detected in the brains or spleens of the animals at 352 days after inoculation by Western blotting. Moreover, no signal was detected after amplification of brain- or spleen-derived material in sPMCAb. Spleen was previously shown to be more permissive than brain to very low doses of prion infection, thus reducing the time considerably for detecting low-dose prion infections (17, 18). Therefore, considering lack of detectible PrPSc in spleen, our results strongly suggest that animals inoculated with dsPMCAb-derived material were not subclinical carriers of scrapie.
Slightly longer incubation times to disease and incomplete attack rate were found in animals inoculated with PMCAb-derived material in comparison with the group inoculated with brain-derived 263K. Previous studies revealed that subjecting hamster prion strains, including 263K, to serial PMCA or PMCAb alters the physical properties of PrPSc (31, 32). Bearing this in mind, the decline or loss of infectivity of PMCAb- and dsPMCAb-derived material, respectively, could be due to structural changes in PrPSc rather than changes in sialylation status. To address this question, the secondary structure of inoculated material was assessed using IR-MSP. IR-MSP measurements revealed that structural changes in PrPSc indeed occurred during PMCAb and dsPMCAb procedures. However, although very similar if not identical structural changes were observed in material produced in PMCAb and dsPMCAb, dsPMCAb material failed to induce clinical or subclinical disease in 100% of animals until 352 days of incubation (at which time point the animal bioassay was terminated). Although some decline in infectivity could be attributable to structural changes, we did not detect substantial secondary structure differences between dsPMCAb and PMCAb material by IR-MSP that would explain the loss of infectivity of the dsPMCAb material as compared with PMCAb material. In fact, sialylation status of the inoculum showed a better correlation with disease outcome. Previous studies showed a similar relationship between PrPSc sialylation status and disease outcome upon intracranial administration of 263K or SSLOW scrapie strains (28, 35). Moreover, our recent study revealed that in animals inoculated with PrPSc with reduced sialylation status via intracranial route, PrPSc was not detectible in spleens (35). This finding that PrPSc with reduced sialylation status does not spread from CNS to SLOs or spreads to SLOs but is effectively neutralized by the immune system is consistent with the results of the current work. Together, these data suggest that sialylation of PrPSc controls its ability to infect a host regardless of entry route.
Prions acquired via peripheral routes are captured by several types of cells, including macrophages, monocytes, B lymphocytes, and dendritic cells, and transported to SLOs (39, 41–43). Accumulation and replication of scrapie in SLOs are considered to be obligatory steps prior to neuroinvasion (11). Although a fraction of PrPSc sequestered by macrophages is degraded (44–47), the portion that survives degradation settles down in SLOs and seeds peripheral replication of prions by follicular dendritic cells of germinal centers (13–16). Prions that reach SLOs or that are produced via peripheral replication are subject to sialylation enhancement, which appears to be accomplished by extracellular sialyltransferases (48). It is likely that enhanced sialylation of PrPSc in SLOs prolongs the life time of PrPSc by protecting it from clearance by the innate immune system (48). Enhanced sialylation of PrPSc provides one of the possible explanations as to why SLOs are more permissive to prion infection than the CNS to cross-species transmission or small doses of prions, despite the much lower expression levels of PrPC in SLOs than in CNS (17, 18). The current study suggests that sialylation is responsible for the trafficking of PrPSc to SLOs. Scrapie material with reduced sialylation levels was not transported to spleen and lymph nodes as efficiently as the scrapie material with normal sialylation status. As such, partially desialylated material did not have a chance of getting extra-sialylated in SLOs. In PrPSc, the majority of sialic acid residues are attached to galactose via an α2-6 linkage (20). Recent work showed that sialic acid residues linked to N-glycans via α2-6 are responsible for the selective spread and adhesion of hepatocarcinoma cells to lymph nodes, in a process that involves Siglec-2 (49). It would be interesting to determine whether a similar mechanism accounts for trafficking of PrPSc to lymph nodes.
The total amounts of PrPSc deposited in all organs tested were lower for groups inoculated with PrPSc with reduced sialylation relative to the groups inoculated with PrPSc with normal sialylation. This result suggests that PrPSc sialylation might contribute to its stability in the periphery or its clearance rate. Several mechanisms might be responsible for clearing partially desialylated PrPSc. Lack of terminal sialylation of N-linked glycans exposes galactose residues that generate “eat me” signals for professional and non-professional macrophages. For example, erythrocytes or platelets with reduced sialylation are cleared in liver by Kupffer cells (50, 51). The clearance is mediated by asialoglycoprotein receptors that recognize asialoglycans on the cell surface (52). Likewise, the microbial pathogens that lack sialic acids are targeted by the innate immune system as foreign through recognition of their non-sialylated glycans that contribute to pathogen-associated molecular patterns (25). Several classes of host molecules are responsible for recognizing non-sialylated glycans on the surface of microbial pathogens or glycoproteins in circulation. Among them are galectins, a family of secreted proteins that recognize and bind to galactose and its derivatives (53). A similar mechanism could also play a role in clearing PrPSc with deficient sialylation status (54, 55). Alternative mechanisms might involve inhibitory Siglecs that recognize sialic acid patterns and suppress innate immune cells (56, 57), and/or factor H that dampens activation of alternative complement pathways by recognizing molecular patterns containing sialic acids on surfaces of host cells or pathogens (58, 59).
It has been shown that the life time of glycosylated molecules in blood circulation is controlled by their sialylation status (60, 61). Recent studies revealed that the density of sialic acid residues on a surface of PrPSc particles is strain-specific (29), suggesting that the strain-specific life time of circulating prions may be similarly controlled. Although PrPC molecules are highly heterogeneous with respect to their sialylation status (28, 62), subsets of PrPC sialoglycoforms are selected in a strain-specific manner to fit into strain-specific structures (29, 63). It remains to be established whether strain-specific differences in density of sialylation play a role in determining strain-specific rate of trafficking to SLOs and/or their degradation rates. Nevertheless, it is possible that the specific subpopulation of cells of innate immune system and the strength of their interaction with PrPSc are determined by strain-specific density of PrPSc sialylation.
In summary, the current study supports the new hypothesis that sialylation of PrPSc controls its fate in an organism (27). Sialylation of PrPSc was found to be important for effective trafficking of PrPSc to SLOs. Although sialylation of N-linked glycans of PrPSc was described more than 30 years ago (64), its role in prion pathogenesis has not previously been explored. Recent years have witnessed extraordinary growth of interest in sialylation and, specifically, in its role in host-pathogen interactions and communication between cells of the immune system (25, 65). Together with previous work (48), the current work proposes that sialylation camouflages PrPSc in peripheral sites and circulation, protecting prions from the innate immune system. As such, sialylation makes PrPSc unique among amyloidogenic proteins or peptides that propagate in a prion-like manner and are responsible for a number of neurodegenerative diseases.
Experimental Procedures
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal protocol was approved by the Institutional Animal Care and Use Committee of the University of Maryland, Baltimore, MD (Assurance Number A32000-01; Permit Number 0215002).
Animal Bioassay
Weanling Golden Syrian hamsters were inoculated i.p. under 2% O2/4 minimum alveolar concentration isoflurane anesthesia with 50 μl of 104-fold diluted 263K scrapie brain homogenates and 10-fold diluted PMCAb or dsPMCAb materials. After inoculation, animals were observed daily for disease using a “blind” scoring protocol and euthanized at the terminal stage of the disease. Animals that did not develop disease were euthanized at 352 days after inoculation.
PMCAb, dsPMCAb, and sPMCAb
10% NBH from healthy hamsters was prepared as described previously (66). To produce desialylated substrates for dsPMCAb, 10% NBH was treated with Arthrobacter ureafaciens (catalog number P0722L, New England Biolabs, Ipswich, MA) or Clostridium perfringens (catalog number P0720L, New England Biolabs, Ipswich, MA) sialidases that have broad substrate specificity as follows. After preclearance of NBH at 500 × g for 2 min and the addition of the enzyme buffer supplied by the manufacturer to supernatant, 200 units/ml sialidase were added to the supernatant and incubated on a rotator at 37 °C for 5 h, and the resulting material referred to as dsNBH substrate was used in dsPMCAb. PMCAb and dsPMCAb reactions were conducted as described previously (28, 67), using a Misonix S-4000 microplate horn (Qsonica LLC, Newtown, CT) in the presence of two 2/32 inch Teflon beads in each tube (McMaster-Carr, Elmhurst, IL). Both types of reactions were seeded with 105-fold diluted 263K scrapie brain homogenates. To produce PMCAb- or dsPMCAb-derived material, three serial rounds with 10-fold dilution between rounds were conducted, and then PMCAb and dsPMCAb products were diluted an additional 10-fold prior to inoculation. To test for the presence of prions in the brains or spleens of hamsters inoculated with dsPMCAb-derived materials, 90 μl of substrate was supplied with two 2/32 inch Teflon beads and seeded with 10 μl of 10% brain or spleen homogenates. Four serial rounds were conducted with 10-fold dilution of reaction products into a fresh substrate between rounds. 109-fold diluted 10% brain homogenate from a hamster terminally ill with 263K strain was used as positive control, and non-seeded sPMCAb reactions were used as negative controls. In all PMCAb, dsPMCAb, or sPMCAb reactions, one round consisted of 20-s sonications delivered at 170-watt energy output applied every 20 min during a 24 h period.
2D Western Blotting
Samples of 25 μl prepared in loading buffer as described above were solubilized for 1 h at room temperature in 200 μl of solubilization buffer (8 m urea, 2% (w/v) CHAPS, 5 mm tributyl phosphate, 20 mm Tris pH 8.0), alkylated by adding 7 μl of 0.5 m iodoacetamide, and incubated for 1 h at room temperature. Then, 1150 μl of ice-cold methanol was added, and samples were incubated for 2 h at −20 °C. After centrifugation at 16,000 × g at 4 °C, supernatant was discarded and the pellet was resolubilized in 160 μl of rehydration buffer (7 m urea, 2 m thiourea, 1% (w/v) DTT, 1% (w/v) CHAPS, 1% (w/v) Triton X-100, 1% (v/v) ampholyte, trace amount of bromphenol blue). Fixed pre-cast immobilized pH gradient (IPG) strips (catalog number ZM0018, Life Technologies) with a linear pH gradient 3–10 were rehydrated in 155 μl of the resulting mixture overnight at room temperature inside IPGRunner cassettes (catalog number ZM0003, Life Technologies). Isoelectrofocusing (first dimension separation) was performed at room temperature with rising voltage (175 V for 15 min, then 175–2000-V linear gradient for 45 min, then 2000 V for 30 min) with a Life Technologies Zoom Dual Power Supply using the XCell SureLock Mini-Cell Electrophoresis System (catalog number EI0001, Life Technologies). The IPG strips were then equilibrated for 15 min consecutively in (i) 6 m urea, 20% (v/v) glycerol, 2% SDS, 375 mm Tris-HCl, pH 8.8, 130 mm DTT and (ii) 6 m urea, 20% (v/v) glycerol, 2% SDS, 37 5 mm Tris-HCl, pH 8.8, 135 mm iodoacetamide and loaded on 4–12% Bis-Tris ZOOM SDS-PAGE pre-cast gels (catalog number NP0330BOX, Life Technologies). For the second dimension, SDS-PAGE in MES-SDS buffer (catalog number B0002, Life Technologies) was performed for 1 h at 170 V. Western blotting was performed as described below and stained using 3F4 antibody (catalog number SIG-39600, Covance).
2D Western blotting signal intensity was digitized for densitometry analysis using AlphaView software (ProteinSimple, San Jose, CA). For generating sialylation profiles for diglycosylated isoforms, densitometry analysis of 2D Western blots was performed using the “Lane profile” function in the AlphaView program; the highest curve signal value was taken as 100%.
Procedure for Purification of Scrapie Material
Extraction of PrPSc from brain tissue and PMCAb and dsPMCAb reactions for FTIR microspectroscopic analysis were performed as described by Daus et al. (32) with the following modifications for brain tissue. Hemispheres of mid-sagittally split hamster brains (∼0.5 g) were homogenized each in 10 ml of homogenization buffer. Brain tissue homogenates were subjected to the extraction procedure, and five final pellets of highly purified PrP27-30 were obtained from one hemisphere. For infrared spectroscopic analysis, final PrP27-30 pellets were washed as described (32) and resuspended in 10 μl of double-distilled H2O. 1-μl aliquots of these PrP27-30 suspensions were transferred for drying onto a CaF2 window of 1-mm thickness (Korth Kristalle GmbH, Altenholz, Germany).
Infrared Microspectroscopy and Cluster Analysis
For each type of scrapie material, three independent preparations were obtained and characterized by infrared microspectroscopy (IR-MSP). IR-MSP measurements were carried out as described previously (32). Briefly, mid-IR spectra were acquired in transmission mode using an IFS 28/B FTIR spectrometer from Bruker (Bruker Optics GmbH, Ettlingen, Germany) that was linked to an IRscope II infrared microscope (Bruker). IR microspectra were recorded with a spatial resolution of ∼80 μm. Nominal spectral resolution was 4 cm−1, and the zero filling factor was 4. For each background and for each sample spectrum, 512 individual interferograms were averaged and apodized using a Blackman-Harris three-term apodization function. To attain an improved signal-to-noise ratio and to address the aspect of within-sample heterogeneity, 10 point spectra were acquired and averaged per purified PrPSc sample. Data acquisition and spectral preprocessing were carried out by utilizing Bruker's instrument software OPUS v. 5.5. Second derivative spectra were obtained by means of a 9-smoothing point Savitzky–Golay derivative filter. All derivative spectra were minimum/maximum normalized to the tyrosine band at 1515 cm−1.
Unsupervised hierarchical cluster analysis was used to reveal groupings in the IR microspectra. The analysis was performed using the cluster analysis function of the OPUS data acquisition software with mean spectra from three independent preparations for each group as inputs. Pre-processing of the mean spectra involved application of a Savitzky–Golay second derivative filter with 13 smoothing points (68). Inter-spectral distances were calculated using the information from the secondary structure-sensitive amide I region (1610 and 1700 cm−1) as so-called D-values (scaled Pearson's correlation coefficients (69)), while Ward's algorithm (70) was used for hierarchical clustering.
Inoculation of Mice via Intraperitoneal Administration
Triton was extracted from PMCAb- and dsPMCAb-derived material using a detergent removal kit (catalog number 88305, Thermo Scientific), and the amounts of PrPSc were quantified using Western blotting. 10% (w/v) 263K brain homogenate was diluted using sterile PBS to a final concentration of 0.75% (w/v) for the amount of PrPSc to be equivalent in PMCAb- and dsPMCAb-derived materials. Mice (C57BL/6J, 6 weeks old) were injected i.p. with 800 μl of 0.75% brain-derived or PMCAb- or dsPMCAb-derived materials per mouse, using 1-ml syringes with a 26-gauge needle (catalog number 309625, BD Biosciences). Different animal groups were housed in different cages. In each animal group, three mice were sacrificed at each time point (2, 6, or 18 h after inoculations), and their brains, spleens, kidneys, mediastinal lymph nodes, and livers were collected for analysis.
Preparation of Tissue Homogenates and Analysis by Western Blotting
Harvested tissues were cut into pieces and crushed using the plunger of a 5-ml syringe. The tissue homogenates of spleen (5% w/v), mediastinal lymph nodes (2.5% w/v), brain (20% w/v), liver (20% w/v), or kidney (10% w/v) were prepared in M-Per lysis buffer (Mammalian Protein Extraction Reagent, catalog number 78501, Thermo Scientific) in the presence of protease inhibitors (cOmplete, Mini, EDTA-free, catalog number 04693159001, Roche Diagnostics), using a bead beater (Model 2412PS-12W-B30, BioSpec Products,) and 0.1-mm glass beads (catalog number 11079101, BioSpec products). The resulting homogenates were centrifuged for 30 s at 500 × g, and then supernatants (180 μl for spleen and lymph nodes, 270 μl for brain and kidneys, and 90 μl for liver) were collected and digested with PK (20 μg/ml, 37 °C, 30 min) in the presence of 0.5% Sarkosyl (N-dodecanoyl-N-methylglycine sodium salt, catalog number L5125, Sigma). PK-digested samples were concentrated by precipitation in 4 volumes of ice-cold acetone, incubated overnight at −20 °C, and then centrifuged for 30 min at 16,000 × g. The resulting pellets were suspended in 1× SDS loading buffer (50 μl for spleen, lymph nodes, brain, and kidneys or 35 μl for liver), heated for 10 min in a boiling water bath, and loaded into NuPAGE Novex 12% Bis-Tris Protein Gels (catalog number NP0343BOX, Thermo Fisher). Gel electrophoresis was performed at 170 V for 1 h at room temperature in MES-SDS buffer (catalog number NP0002, Thermo Fisher) in XCell SureLock Mini-Cell (catalog number EI0001, Thermo Fisher). After that, Western blotting was performed in XCell SureLock Mini-Cell Blot Module (catalog number EI0002, Thermo Fisher) for 1 h on ice (transfer buffer composition: 25 mm Tris, 190 mm glycine, 20% v/v methanol) at 33 V. The proteins were transferred to Immobilon membrane (catalog number IPVH00010, Millipore, Billerica, MA). After the completion of the transfer, the membrane was blocked in 5% fat-free milk in PBST (1× PBS, catalog number AB11072-01000, AmericanBio, 1% Tween 20, catalog number P749, Sigma-Aldrich) on a rotator for 45 min at room temperature. Then, the membrane was subsequently incubated on a rotator at room temperature in (i) primary antibody for 1 h; (ii) PBST for 10 min (three times); (iii) secondary antibody for 1 h; (iv) PBST for 10 min (three times). After that, the membrane was developed with Luminata Forte Western HRP Substrate (catalog number WBLUF0500, Millipore) for 10–30 s and visualized using the FluorChem E system (ProteinSimple). 0.05% 263K brain homogenate treated with PK (20 μg/ml, 37 °C, 30 min) served as internal reference for all gels.
Western blotting signal intensity was digitized for densitometry analysis using AlphaView software (ProteinSimple). For each animal group/tissue, the signal intensities were normalized per the intensities of 0.05% 263K brain homogenate on the same Western blots. Results are presented as the means ± S.D. (n = 3). Statistical significance (p) between groups was calculated by Student's t test, and indicated as * for significant (p < 0.05) and # for insignificant (p > 0.05).
Author Contributions
I. V. B. conceived and supervised the study. S. S, E. K., and M. L. D. performed the experiments. S. S., E. K., M. L. D., P. L., M. B., and I. V. B. analyzed the data. P. L., M. B., and I. V. B. wrote the manuscript.
Acknowledgment
We thank Pamela Wright for editing the manuscript.
This work was supported by National Institutes of Health Grant R01 NS045585 (to I. V. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PrPSc
- disease-associated infectious form(s) of the prion protein
- PrPC
- cellular form(s) of the prion protein
- NBH
- normal brain homogenate
- dsNBH
- sialidase-treated normal brain homogenate
- PMCAb
- protein misfolding cyclic amplification with beads
- dsPMCAb
- protein misfolding cyclic amplification with beads conducted using sialidase-treated normal brain homogenate
- sPMCAb
- serial PMCAb
- IR-MSP
- infrared microspectroscopy
- PK
- proteinase K
- SLO
- secondary lymphoid organ
- IPG
- immobilized pH gradient
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Prusiner S. B. (1982) Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144 [DOI] [PubMed] [Google Scholar]
- 2. Legname G., Baskakov I. V., Nguyen H. O. B., Riesner D., Cohen F. E., DeArmond S. J., and Prusiner S. B. (2004) Synthetic mammalian prions. Science 305, 673–676 [DOI] [PubMed] [Google Scholar]
- 3. Cohen F. E., and Prusiner S. B. (1998) Pathologic conformations of prion proteins. Annu. Rev. Biochem. 67, 793–819 [DOI] [PubMed] [Google Scholar]
- 4. Baskakov I. V., and Breydo L. (2007) Converting the prion protein: what makes the protein infectious. Biochim. Biophys. Acta 1772, 692–703 [DOI] [PubMed] [Google Scholar]
- 5. Jucker M., and Walker L. C. (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walker L. C., and Jucker M. (2015) Neurodegenerative diseases: expanding the prion concept. Annu. Rev. Neurosci. 38, 87–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miller M. W., and Williams E. S. (2004) Chronic wasting disease of cervids. Curr. Top. Microbiol. Immunol. 284, 193–214 [DOI] [PubMed] [Google Scholar]
- 8. Brown P., and Gajdusek D. C. (1991) The human spongiform encephalopathies: kuru, Creutzfeldt-Jakob disease, and the Gerstmann-Sträussler-Scheinker syndrome. Curr. Top. Microbiol. Immunol. 172, 1–20 [DOI] [PubMed] [Google Scholar]
- 9. Makarava N., Savtchenko R., Alexeeva I., Rohwer R. G., and Baskakov I. V. (2012) Fast and ultrasensitive method for quantitating prion infectivity titer. Nat. Commun. 3, 741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boerner S., Wagenführ K., Daus M. L., Thomzig A., and Beekes M. (2013) Towards further reduction and replacement of animal bioassays in prion research by cell and protein misfolding cyclic amplification assays. Lab. Anim. 47, 106–115 [DOI] [PubMed] [Google Scholar]
- 11. Aguzzi A., Nuvolone M., and Zhu C. (2013) The immunology of prion diseases. Nat. Rev. Immunol. 13, 888–902 [DOI] [PubMed] [Google Scholar]
- 12. Mabbott N. A. (2012) Prion pathogenesis and secondary lymphoid organs. Prion 6, 322–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McCulloch L., Brown K. L., Bradford B. M., Hopkins J., Bailey M., Rajewsky K., Manson J. C., and Mabbott N. A. (2011) Follicular dendritic cell-specific prion protein (PrP) expression alone is sufficient to sustain prion infection in the spleen. PLOS Pathog. 7, e1002402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kujala P., Raymond C. R., Romeijn M., Godsave S. F., van Kasteren S. I., Wille H., Prusiner S. B., Mabbott N. A., and Peters P. J. (2011) Prion uptake in the gut: identification of the first uptake and replication sites. PLOS Pathog. 7, e1002449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brown K. L., Stewart K., Ritchie D. L., Mabbott N. A., Williams A., Fraser H., Morrison W. I., and Bruce M. E. (1999) Scrapie replication in lyphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat. Med. 5, 1308–1312 [DOI] [PubMed] [Google Scholar]
- 16. Montrasio F., Frigg R., Glatzel M., Klein M. A., Mackay F., Aguzzi A., and Weissmann C. (2000) Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science 288, 1257–1259 [DOI] [PubMed] [Google Scholar]
- 17. Béringue V., Herzog L., Jaumain E., Reine F., Sibille P., Le Dur A., Vilotte J. L., and Laude H. (2012) Facilitated cross-species transmission of prions in extraneural tissue. Science 335, 472–475 [DOI] [PubMed] [Google Scholar]
- 18. Halliez S., Reine F., Herzog L., Jaumain E., Haïk S., Rezaei H., Vilotte J. L., Laude H., and Béringue V. (2014) Accelerated, spleen-based titration of variant Creutzfeldt-Jakob disease infectivity in transgenic mice expressing human prion protein with sensitivity comparable to that of survival time bioassay. J. Virol. 88, 8678–8686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Turk E., Teplow D. B., Hood L. E., and Prusiner S. B. (1988) Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem. 176, 21–30 [DOI] [PubMed] [Google Scholar]
- 20. Endo T., Groth D., Prusiner S. B., and Kobata A. (1989) Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry 28, 8380–8388 [DOI] [PubMed] [Google Scholar]
- 21. Stimson E., Hope J., Chong A., and Burlingame A. L. (1999) Site-specific characterization of the N-linked glycans of murine prion protein by high-performance liquid chromatography/electrospray mass spectrometry and exoglycosidase digestions. Biochemistry 38, 4885–4895 [DOI] [PubMed] [Google Scholar]
- 22. Stahl N., Baldwin M. A., Teplow D. B., Hood L., Gibson B. W., Burlingame A. L., and Prusiner S. B. (1993) Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry 32, 1991–2002 [DOI] [PubMed] [Google Scholar]
- 23. Rudd P. M., Endo T., Colominas C., Groth D., Wheeler S. F., Harvey D. J., Wormald M. R., Serban H., Prusiner S. B., Kobata A., and Dwek R. A. (1999) Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc. Natl. Acad. Sci. U.S.A. 96, 13044–13049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Varki A., and Gagneux P. (2012) Multifarious roles of sialic acid in immunity. Ann. N.Y. Acad. Sci. 1253, 16–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Varki A. (2008) Sialic acids in human health and disease. Trends Mol. Med. 14, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brown G. C., and Neher J. J. (2014) Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 15, 209–216 [DOI] [PubMed] [Google Scholar]
- 27. Baskakov I. V., and Katorcha E. (2016) Multifaceted role of sialylation in prion diseases. Front. Neurosci. 10, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Katorcha E., Makarava N., Savtchenko R., D'Azzo A., and Baskakov I. V. (2014) Sialylation of prion protein controls the rate of prion amplification, the cross-species barrier, the ratio of PrPSc glycoform and prion infectivity. PLoS Pathog. 10, e1004366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Katorcha E., Makarava N., Savtchenko R., and Baskakov I. V. (2015) Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci. Rep. 5, 16912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klingeborn M., Race B., Meade-White K. D., and Chesebro B. (2011) Lower specific infectivity of protease-resistant prion protein generated in cell-free reactions. Proc. Natl. Acad. Sci. U.S.A. 108, E1244–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gonzalez-Montalban N., and Baskakov I. V. (2012) Assessment of strain-specific PrPSc elongation rates revealed a transformation of PrPSc properties during protein misfolding cyclic amplification. PLoS ONE 7, e41210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Daus M. L., Wagenführ K., Thomzig A., Boerner S., Hermann P., Hermelink A., Beekes M., and Lasch P. (2013) Infrared microspectroscopy detects protein misfolding cyclic amplification (PMCA)-induced conformational alterations in hamster scrapie progeny seeds. J. Biol. Chem. 288, 35068–35080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smirnovas V., Baron G. S., Offerdahl D. K., Raymond G. J., Caughey B., and Surewicz W. K. (2011) Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 18, 504–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spassov S., Beekes M., and Naumann D. (2006) Structural differences between TSEs strains investigated by FT-IR spectroscopy. Biochim. Biophys. Acta 1760, 1138–1149 [DOI] [PubMed] [Google Scholar]
- 35. Katorcha E., Daus M. L., Gonzalez-Montalban N., Makarava N., Lasch P., Beekes M., and Baskakov I. V. (2016) Reversible off and on switching of prion infectivity via removing and reinstalling prion sialylation. Sci. Rep. 6, 33119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Glatzel M., Heppner F. L., Albers K. M., and Aguzzi A. (2001) Sympathetic innervation of lymphoreticular organs is rate limiting for prion neuroinvasion. Neuron 31, 25–34 [DOI] [PubMed] [Google Scholar]
- 37. Mabbott N. A., Mackay F., Minns F., and Bruce M. E. (2000) Temporal inactivation of follicular dendritic cells delays neuroinvasion of scrapie. Nat. Med. 6, 719–720 [DOI] [PubMed] [Google Scholar]
- 38. Prinz M., Heikenwalder M., Junt T., Schwarz P., Glatzel M., Heppner F. L., Fu Y. X., Lipp M., and Aguzzi A. (2003) Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 425, 957–962 [DOI] [PubMed] [Google Scholar]
- 39. Michel B., Meyerett-Reid C., Johnson T., Ferguson A., Wyckoff C., Pulford B., Bender H., Avery A., Telling G., Dow S., and Zabel M. D. (2012) Incunabular immunological events in prion trafficking. Sci. Rep. 2, 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kimberlin R. H., and Walker C. A. (1986) Pathogenesis of scrapie (strain 263K) in hamsters infected intracerebrally, intraperitoneally or intraocularly. J. Gen. Virol. 67, 255–263 [DOI] [PubMed] [Google Scholar]
- 41. Castro-Seoane R., Hummerich H., Sweeting T., Tattum M. H., Linehan J. M., Fernandez de Marco M., Brandner S., Collinge J., and Klöhn P. C. (2012) Plasmacytoid dendritic cells sequester high prion titres at early stages of prion infection. PLOS Pathog. 8, e1002538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huang F. P., Farquhar C. F., Mabbott N. A., Bruce M. E., and MacPherson G. G. (2002) Migrating intestinal dendritic cells transport PrPSc from the gut. J. Gen. Virol. 83, 267–271 [DOI] [PubMed] [Google Scholar]
- 43. Takakura I., Miyazawa K., Kanaya T., Itani W., Watanabe K., Ohwada S., Watanabe H., Hondo T., Rose M. T., Mori T., Sakaguchi S., Nishida N., Katamine S., Yamaguchi T., and Aso H. (2011) Orally administered prion protein is incorporated by M cells and spreads into lymphoid tissues with macrophages in prion protein knockout mice. Am. J .Pathol. 179, 1301–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beringue V., Demoy M., Lasmézas C. I., Gouritin B., Weingarten C., Deslys J. P., Andreux J. P., Couvreur P., and Dormont D. (2000) Role of spleen macrophages in the clearance of scrapie agent early in pathogenesis. J. Pathol. 190, 495–502 [DOI] [PubMed] [Google Scholar]
- 45. Carp R. I., and Callahan S. M. (1981) In vitro interaction of scrapie agent and mouse peritoneal macrophages. Intervirology 16, 8–13 [DOI] [PubMed] [Google Scholar]
- 46. Sassa Y., Inoshima Y., and Ishiguro N. (2010) Bovine macrophage degradation of scrapie and BSE PrPSc. Vet. Immunol. Immunopathol. 133, 33–39 [DOI] [PubMed] [Google Scholar]
- 47. Sassa Y., Yamasaki T., Horiuchi M., Inoshima Y., and Ishiguro N. (2010) The effect of lysosomal and proteasomal inhibitors on abnormal forms of prion protein degradation in murine macrophages. Microbiol. Immunol. 54, 763–768 [DOI] [PubMed] [Google Scholar]
- 48. Srivastava S., Makarava N., Katorcha E., Savtchenko R., Brossmer R., and Baskakov I. V. (2015) Post-conversion sialylation of prions in lymphoid tissues. Proc. Natl. Acad. Sci. U.S.A. 112, E6654–6662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang S., Chen X., Wei A., Yu X., Niang B., and Zhang J. (2015) α2,6-Linked sialic acids on N-glycans modulate the adhesion of hepatocarcinoma cells to lymph nodes. Tumour Biol. 36, 885–892 [DOI] [PubMed] [Google Scholar]
- 50. Aminoff D., Bruegge W. F., Bell W. C., Sarpolis K., and Williams R. (1977) Role of sialic acid in survival of erythrocytes in the circulation: interaction of neuraminidase-treated and untreated erythrocytes with spleen and liver at the cellular level. Proc. Natl. Acad. Sci. U.S.A. 74, 1521–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jansen A. J. G., Josefsson E. C., Rumjantseva V., Liu Q. P., Falet H., Bergmeier W., Cifuni S. M., Sackstein R., von Andrian U. H., Wagner D. D., Hartwig J. H., and Hoffmeister K. M. (2012) Desialylation accelerates platelet clearance after refrigeration and initiates GPIbα metalloproteinase-mediated cleavage in mice. Blood 119, 1263–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dini L., Autuori F., Lentini A., Oliverio S., and Piacentini M. (1992) The clearance of apoptotic cells in the liver is mediated by the asialoglycoprotein receptor. FEBS Lett. 296, 174–178 [DOI] [PubMed] [Google Scholar]
- 53. Vasta G. R. (2009) Roles of galectins in infection. Nat. Rev. Microbiol. 7, 424–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jin J.-K., Na Y.-J., Song J.-H., Joo H.-G., Kim S., Kim J.-I., Choi E.-K., Carp R. I., Kim Y.-S., and Shin T. (2007) Galectin-3 expression is correlated with abnormal prion protein accumulation in murine scrapie. Neurosci Lett. 420, 138–143 [DOI] [PubMed] [Google Scholar]
- 55. Mok S. W. F., Riemer C., Madela K., Hsu D. K., Liu F.-T., Gultner S., Heise I., and Baier M. (2007) Role of galectin-3 in prion infections of the CNS. Biochem. Biophys. Res. Commun. 359, 672–678 [DOI] [PubMed] [Google Scholar]
- 56. Carlin A. F., Uchiyama S., Chang Y. C., Lewis A. L., Nizet V., and Varki A. (2009) Molecular mimicry of host sialylated glycans allows a bacterial pathogen to engage neutrophil Siglec-9 and dampen the innate immune response. Blood 113, 3333–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cao H., and Crocker P. R. (2011) Evolution of CD33-related siglecs: regulating host immune functions and escaping pathogen exploitation? Immunology 132, 18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pangburn M. K., Pangburn K. L., Koistinen V., Meri S., and Sharma A. K. (2000) Molecular mechanisms of target recognition in an innate immune system: interactions among factor H, C3b, and target in the alternative pathway of human complement. J. Immunol. 164, 4742–4751 [DOI] [PubMed] [Google Scholar]
- 59. Kajander T., Lehtinen M. J., Hyvärinen S., Bhattacharjee A., Leung E., Isenman D. E., Meri S., Goldman A., and Jokiranta T. S. (2011) Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc. Natl. Acad. Sci. U.S.A. 108, 2897–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tanaka K., Siwu E. R. O., Minami K., Hasegawa K., Nozaki S., Kanayama Y., Koyama K., Chen W. C., Paulson J. C., Watanabe Y., and Fukase K. (2010) Noninvasive imaging of dendrimer-type N-glycan clusters: in vivo dynamics dependence on oligosaccharide structure. Angew. Chem. Int. Ed. Engl. 49, 8195–8200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. O'Sullivan J. M., Aguila S., McRae E., Ward S. E., Rawley O., Fallon P. G., Brophy T. M., Preston R. J. S., Brady L., Sheils O., Chion A., and O'Donnell J. S. (2016) N-Glycan truncation causes enhanced clearance of plasma-derived von Willebrand factor. J. Thromb. Haemost. 14, 2446–2457 [DOI] [PubMed] [Google Scholar]
- 62. Katorcha E., Klimova N., Makarava N., Savtchenko R., Pan X., Annunziata I., Takahashi K., Miyagi T., Pshezhetsky A. V., d'Azzo A., and Baskakov I. V. (2015) Knocking out of cellular neuraminidases Neu1, Neu3 or Neu4 does not affect sialylation status of the prion protein. PLoS ONE 10, e0143218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Makarava N., Savtchenko R., and Baskakov I. V. (2015) Two alternative pathways for generating transmissible prion disease de novo. Acta Neuropathol. Commun. 3, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bolton D. C., Meyer R. K., and Prusiner S. B. (1985) Scrapie PrP 27–30 is a sialoglycoprotein. J. Virol. 53, 596–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Varki A. (2010) Uniquely human evolution of sialic acid genetics and biology. Proc. Natl. Acad. Sci. U.S.A. 107, 8939–8946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Makarava N., Kovacs G. G., Savtchenko R., Alexeeva I., Budka H., Rohwer R. G., and Baskakov I. V. (2011) Genesis of mammalian prions: from non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathog. 7, e1002419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gonzalez-Montalban N., Makarava N., Ostapchenko V. G., Savtchenk R., Alexeeva I., Rohwer R. G., and Baskakov I. V. (2011) Highly efficient protein misfolding cyclic amplification. PLoS Pathog. 7, e1001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Savitzky A., and Golay M. J. E. (1964) Smoothing and differentiation of data by simplified least squares procedures. Anal. Chem. 36, 1627–1639 [Google Scholar]
- 69. Lasch P., and Naumann D. (2015) Infrared spectroscopy in microbiology. in Encyclopedia of Analytical Chemistry, pp. 1–32, John Wiley & Sons, Inc., New York [Google Scholar]
- 70. Ward J. H. (1963) Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 58, 236–244 [Google Scholar]