

Abstract
Recent genome-wide studies found that patients with hypotonia, developmental delay, intellectual disability, congenital anomalies, characteristic facial dysmorphic features, and low cholesterol levels suffer from Kaufman oculocerebrofacial syndrome (KOS, also reported as blepharophimosis-ptosis-intellectual disability syndrome). The primary cause of KOS is autosomal recessive mutations in the gene UBE3B. However, to date, there are no studies that have determined the cellular or enzymatic function of UBE3B. Here, we report that UBE3B is a mitochondrion-associated protein with homologous to the E6-AP C terminus (HECT) E3 ubiquitin ligase activity. Mutating the catalytic cysteine (C1036A) or deleting the entire HECT domain (amino acids 758–1068) results in loss of UBE3B's ubiquitylation activity. Knockdown of UBE3B in human cells induces changes in mitochondrial morphology and physiology, a decrease in mitochondrial volume, and a severe suppression of cellular proliferation. We also discovered that UBE3B interacts with calmodulin via its N-terminal isoleucine-glutamine (IQ) motif. Deletion of the IQ motif (amino acids 29–58) results in loss of calmodulin binding and a significant increase in the in vitro ubiquitylation activity of UBE3B. In addition, we found that changes in calcium levels in vitro disrupt the calmodulin-UBE3B interaction. These studies demonstrate that UBE3B is an E3 ubiquitin ligase and reveal that the enzyme is regulated by calmodulin. Furthermore, the modulation of UBE3B via calmodulin and calcium implicates a role for calcium signaling in mitochondrial protein ubiquitylation, protein turnover, and disease.
Keywords: calcium, mitochondria, oxidative stress, protein degradation, reactive oxygen species (ROS), ubiquitylation (ubiquitination), HECT, Kaufman oculocerebrofacial syndrome, super-resolution microscopy, ubiquitin/proteosome system
Introduction
Ubiquitylation is a reversible post-translational modification that impacts most cellular processes. However, it is best known for targeting proteins for degradation by the 26S proteasome, and the ubiquitin/proteasome system (UPS)2 is a major part of the cellular protein quality control network (1–5). Substrate modification can occur either by the attachment of a single ubiquitin molecule or a polyubiquitin chain, where one ubiquitin is conjugated to the next via an isopeptide bond (6). Conjugation of ubiquitin to one or more lysine residues on the substrate protein is mediated by the sequential action of a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3). Although there is limited diversity in E1 and E2 enzymes, numerous cellular E3 ligases specify the timing and substrate selection of ubiquitylation reactions.
E3 ligases are mainly composed of three major families, the really interesting new gene (RING) family, the homologous to the E6-AP C terminus (HECT) family, and the RING-between-RING (RBR) family. The RING ligases catalyze the direct transfer of ubiquitin from the E2 to the substrate (7, 8). Conversely, the HECT and RBR E3 ligases covalently transfer the ubiquitin from the E2 to a resident catalytic cysteine. This catalytic cysteine in the E3 ligase active site can then serve as the site of attachment for a thioester-linked polyubiquitin chain, which is then transferred to the substrate (9–12). Overall, E3 ubiquitin ligases are key regulatory determinants in the ubiquitylation reaction.
In eukaryotic cells the UPS mediates the turnover of outer mitochondrial membrane (OMM) proteins (13–19). For example, under mitochondrial stress, UPS-dependent degradation of OMM proteins is mediated by the E3 ubiquitin ligase, PARKIN (20–23). However, cells deficient in PARKIN are still able to turn over OMM proteins via the UPS (20, 21). Therefore, it is likely that, in addition to PARKIN, other E3 ubiquitin ligase(s) mediate the ubiquitylation of OMM proteins.
Kaufman oculocerebrofacial syndrome (KOS), also referred to as blepharophimosis-ptosis-intellectual-disability syndrome, is an autosomal recessively inherited disorder that is caused by biallelic mutations in the gene encoding a putative ubiquitin protein ligase (UBE3B). The disease is characterized by intellectual disabilities and developmental delay, hypotonia, microcephaly, structural eye anomalies, and other organ malformations, as well as distinctive facial dysmorphic features (24). Following the identification of the genetic variants that cause KOS, 14 patients from 11 unrelated families were reported (24–27). In cattle, a mutation that results in an altered Ube3b protein that lacks 40 amino acids (of which 20 are located in the conserved HECT domain) leads to a bovine phenotype that resembles human KOS and is known as ptosis, intellectual disability, retarded growth and mortality syndrome (28). However, none of these studies have defined a functional role for UBE3B in cellular protein homeostasis nor have these or any studies demonstrated E3 ubiquitin ligase activity for the UBE3B protein.
In a recent synthetic lethal screen done by our laboratory using an siRNA library, the chemotherapy agent temozolomide (TMZ), and a glioblastoma multiforme cell line, we found that knockdown (KD) of UBE3B sensitized cells to TMZ treatment (29). These findings are consistent with a role for UBE3B in the cellular response to mitochondrial oxidative stress, because it was shown that TMZ treatment triggers a rapid onset of ROS (29). Similarly, it was shown that the Caenorhabditis elegans ortholog of UBE3B, oxi-1, is required for protein degradation via the UPS, especially under oxidative stress conditions and that deletion of the mouse Ube3b gene led to reduced viability and brain size (25). Overall, UBE3B is highly expressed in the central nervous system, digestive tract, respiratory system, as well as in multiple cell lineages of skin and other soft tissues (25). However, despite a considerable body of evidence suggesting that UBE3B plays a significant role in neuronal cell capacity (24, 25, 28, 30), the function(s) and regulation of UBE3B remain uncharacterized.
In this study, we show that UBE3B is a HECT E3 ligase, with the catalytic cysteine at amino acid 1036 (Cys-1036). Mutation of this cysteine to alanine (C1036A) abolishes the ubiquitylation activity of UBE3B as determined using in vitro assays. We also show that UBE3B plays a role in maintaining mitochondrial morphology, as depletion of the protein results in more punctate mitochondria and altered mitochondrial physiology. Furthermore, we show that loss of UBE3B significantly reduces cell proliferation. Finally, we show that UBE3B interacts with calmodulin through its isoleucine-glutamine (IQ) motif, and deletion of this motif (UBE3BΔIQ) abolishes interaction. The UBE3BΔIQ protein also has increased ubiquitylation activity in vitro. Interestingly, expression of the UBE3BΔIQ protein in human cells induces apoptosis. Based on these data, we conclude that UBE3B is a calmodulin-regulated E3 ubiquitin ligase.
Results
UBE3B's Predicted Structure Resembles HECT E3 Ligases
From a previous report, it was proposed that UBE3B belongs to the HECT subfamily of E3 ubiquitin ligases (31). However, there are no reports on the predicted structure or function of UBE3B. Therefore, we used the software packages Protein Homology/analogY Recognition Engine (Phyre2) (32) and ClustalW2 (33, 34) to identify and align homologous proteins to UBE3B and to predict the three-dimensional structure of the different domains of UBE3B. As shown in Fig. 1A, the N terminus of UBE3B is predicted to contain the substrate binding domain and a calmodulin-binding (IQ) motif (35). The C terminus of UBE3B is the HECT domain, which is composed of two lobes as follows: the N-lobe that is predicted to bind to the ubiquitin-conjugating enzyme (E2), and the C-lobe that is predicted to contain the catalytic cysteine, which forms a covalent thioester bond with ubiquitin. The N- and C-lobes are connected via a flexible linker. To evaluate the conserved IQ motif and HECT domain of UBE3B, we aligned each with other homologous motifs/domains (Fig. 1, B and C, respectively). The top seven sequences that aligned with either the IQ motif or the HECT domain as ranked by Phyre2 are detailed in Tables 1 and 2, respectively.
FIGURE 1.

Alignment of UBE3B with select IQ motif proteins and HECT E3 ubiquitin ligases. A, schematic of UBE3B showing the IQ domain (amino acids 29–58) and the HECT domain (amino acids 757–1068). The proposed 3D structures of the IQ and HECT domains using Phyre2 are shown above the schematic. The N terminus of HECT domains are known to bind to substrate. The HECT domain is composed of two lobes as follows: the N-lobe binds the E2(s), and the C-lobe contains the catalytic cysteine that binds ubiquitin. B, alignment of UBE3B with calmodulin binding domains as predicted by Phyre2 and using ClustalW2. C, alignment of UBE3B with HECT E3 ligase domains as predicted by Phyre2 and using ClustalW2. The conserved catalytic cysteine is highlighted in red. * denotes a single fully conserved residue; : denotes conservation between groups of strongly similar properties, . denotes conservation between groups of weakly similar properties.
TABLE 1.
Details of the top seven sequence alignments with the IQ motif of UBE3B using Phyre2
The alignment coverage, confidence, and % ID for the IQ motif are shown. The alignment coverage gives the percentage of alignment between the query sequence and the template; the confidence represents the probability (from 0 to 100) that the match between the query sequence and the template is a true homology, and the % ID is the likelihood the model is accurate. For extremely high accuracy models, the % ID should be above 30–40%; however, if the confidence is high, even very low % ID (<15%) can be very useful.
| Rank | Template | Alignment coverage | Confidence | % ID |
|---|---|---|---|---|
| % | ||||
| 1 | Myosin-5a | 90 | 86.7 | 32 |
| 2 | Myosin Va | 83 | 79.0 | 31 |
| 3 | Unconventional myosin-1C | 73 | 77.4 | 39 |
| 4 | Myosin | 66 | 77.0 | 24 |
| 5 | Smooth muscle myosin heavy chain | 86 | 74.9 | 19 |
| 6 | Myosin heavy chain, muscle | 66 | 74.3 | 30 |
| 7 | Myosin heavy chain | 86 | 74.3 | 30 |
TABLE 2.
Details of the top seven sequence alignments with the HECT domain of UBE3B using Phyre2
The alignment coverage, confidence, and % ID for the HECT domain are shown. The alignment coverage gives the percentage of alignment between the query sequence and the template; the confidence represents the probability (from 0 to 100) that the match between the query sequence and the template is a true homology, and the % ID is the likelihood the model is accurate. For extremely high accuracy models, the % ID should be above 30–40%; however, if the confidence is high, even very low % ID (<15%) can be very useful.
| Rank | Template | Alignment coverage | Confidence | % ID |
|---|---|---|---|---|
| % | ||||
| 1 | Rsp5 | 97 | 100.0 | 34 |
| 2 | Huwe1 | 97 | 100.0 | 37 |
| 3 | HECT, E3 ligase | 97 | 100.0 | 34 |
| 4 | Nedd4-like | 96 | 100.0 | 36 |
| 5 | Smad | 96 | 100.0 | 33 |
| 6 | HECT, E3 ligase | 97 | 100.0 | 30 |
| 7 | Ubr5 | 38 | 100.0 | 30 |
UBE3B Associates with Mitochondria
Next, we examined the subcellular localization of UBE3B and asked whether the HECT domain affected localization. Stable cell lines were developed that express the following fusions of copGFP and UBE3B or its mutants: wild type UBE3B (UBE3B-copGFP); UBE3B missing the HECT domain (referred to as UBE3BΔ HECT-copGFP); and UBE3B with an inactive catalytic cysteine (UBE3B(C1036A)-copGFP), all with C-terminal copGFP tags. Localization of UBE3B-copGFP fusions was assessed through a combination of fluorescence microscopy and immunoblot analysis of subcellular fractions, using LN428 cell lines stably expressing copGFP or UBE3B-copGFP fusions. For fluorescence microscopy, mitochondria were first stained with MitoTracker Far Red and then fixed and stained with an antibody against the ER marker PDI. As shown in Fig. 2A, both UBE3B-copGFP and UBE3B(C1036A)-copGFP show strong co-localization with the mitochondria. Some overlap with the ER, and faint cytoplasmic staining was also evident, whereas UBE3BΔ HECT-copGFP shows a diffuse GFP signal throughout the cell. To assess the non-mitochondrial component of the GFP signal, cells were fractionated into mitochondrial, ER, and cytoplasmic fractions and probed with an antibody against copGFP (Fig. 2B). Full-length copGFP fusions were observed in the mitochondrial fraction, but not in the ER fraction. This suggests that full-length UBE3B is associated primarily, if not exclusively, with the mitochondria. The non-mitochondrial component of the observed GFP signal does not represent full-length UBE3B and is likely partly constituted of fragmented copGFP (25 kDa) species in the ER and cytoplasm. The purity of subcellular fractionation was assessed by immunoblotting. Mitochondrial fractions lack the cytoplasmic marker α-tubulin and show enrichment of the mitochondrial marker Tom20. The ER marker PDI was only observed in the ER fraction (Fig. 2, C and D). To confirm that UBE3B's subcellular recruitment to the mitochondria was not an artifact of expression of the tagged proteins, we isolated mitochondria from the control LN428 cells and identified endogenous UBE3B that was exclusively associated with the mitochondrial fraction (Fig. 2, C and E).
FIGURE 2.

UBE3B is associated with mitochondria. A, LN428 cells were transduced with lentivirus to stably express UBE3B, UBE3BΔHECT, or UBE3B(C1036A), all with C-terminal copGFP tags, and then were fixed and imaged with a Nikon A1rsi confocal microscope. MitoTracker DeepRed (excitation wavelength, 647 nm; emission wavelength, 665 nm) was used to stain mitochondria before fixation; cells were then immunostained for PDI, a marker for the endoplasmic reticulum (excitation wavelength, 568 nm; emission wavelength, 602 nm). DAPI (excitation wavelength, 360 nm; emission wavelength, 460 nm) was used to counterstain nuclei, as seen in the merged images. B, to confirm the immunofluorescence results, subcellular fractionation of the stable cell lines was performed, resulting in isolation of mitochondrial, ER, and cytoplasmic fractions, which were then probed by immunoblot (IB). An antibody against TurboGFP was used to detect UBE3B-copGFP proteins. Full-length UBE3B-copGFP proteins were detected only in mitochondrial fractions and in whole cell lysate. To confirm the quality of fractionation, fractions were probed with markers for different cellular compartments. C, mitochondrial fractions lack the cytoplasmic marker α-tubulin and show enrichment of the mitochondrial marker Tom20. D, purity of the ER fraction was assessed by immunoblot probe for the ER marker PDI, showing no cross-contamination with the mitochondrial fraction. E, to show that endogenous UBE3B associates with mitochondria and the immunofluorescence and subcellular fractionation results in A–D are not artifacts of overexpression or of the copGFP tag, we performed subcellular fractionation and immunoblot analysis for endogenous UBE3B in LN428 cells, using the cytoplasmic marker α-tubulin and the mitochondrial marker Tom40 to confirm fractionation.
Knockdown of UBE3B Changes Mitochondrial Morphology and Physiology and Suppresses Cellular Proliferation
To identify whether changes in UBE3B protein expression levels affected mitochondrial morphology and function, UBE3B was depleted (knocked down; KD) using siRNA (Fig. 3A). Using an MTS assay, it was determined that loss of UBE3B resulted in decreased NAD(P)H-dependent cellular oxidoreductase enzyme activity, an indicator of decreased cellular survival (Fig. 3B). The mitochondria from UBE3B-KD cells, as determined from fixed cell confocal microscopy, have more punctate mitochondria, instead of the more linear and reticular network observed in the scrambled (SCR) siRNA (negative control) cells (Fig. 3C). To confirm that the fragmented phenotype of the mitochondria in the UBE3B-KD cells was due to the loss of mitochondrial volume, we measured the total number of mitochondrial voxels and found that it was significantly decreased in the UBE3B-KD cells as compared with the healthy control cells (Fig. 3D). We then employed a novel approach to measure in vivo mitochondrial stress and damage via the MitoTimer reporter gene (36–38). This reporter gene expresses a mitochondrially targeted green fluorescent protein whose emission spectrum shifts irreversibly toward the red when the protein is oxidized. Because this shift is irreversible, the likelihood of this occurring increases with protein lifetime. Seventy two hours after co-transfection of pMitoTimer and either UBE3B siRNA or scrambled siRNA, the cells were imaged using live cell confocal microscopy. We observed a significantly higher red to green ratio in the UBE3B-KD cells. These results likely indicate an increase in mitochondrial oxidative stress but could also be caused by gross changes in protein translation and/or degradation causing an accumulation of red-shifted GFP molecules (Fig. 3E). Either interpretation suggests a significant change in mitochondrial physiology in cells depleted of UBE3B.
FIGURE 3.

Knockdown of UBE3B compromises mitochondrial morphology and function and reduces cell proliferation and colony formation. LN428 cells were transfected with UBE3B siRNA or SCR siRNA (90 nm, final concentration) for 72 h before being used for subsequent experiments. A, qRT-PCR was performed to measure the siRNA-mediated knockdown of UBE3B mRNA expression. β-Actin was used as the endogenous control, and mRNA expression was normalized to SCR siRNA cells. RQ indicates the relative quantification. B, to determine cellular metabolic activity, as an indicator of decreased cellular survival, 2000 cells/well were plated 24 h after siRNA transfection. After 48 h of incubation, an MTS assay was performed. C, to determine whether there are changes in mitochondrial morphology after depletion of UBE3B protein, confocal imaging was performed on fixed cells 72 h after siRNA transfection. ATP synthase β is the mitochondrial marker detected by immunofluorescence (excitation wavelength, 647 nm; emission wavelength, 666 nm). D, Z-stacking using confocal microscopy on the fixed cells from C was performed to determine the changes in mitochondrial voxels in UBE3B-KD cells as compared with scrambled (negative control) treated cells. E, to determine whether UBE3B knockdown induced mitochondrial stress, cells were co-transfected with UBE3B siRNA, and the ratiometric reporter gene Mito-Timer. Confocal imaging was performed 72 h after transfection. Quantitation of the collected images reveal an increase in mitochondrial stress in the UBE3B knockdown cells. F, LN428 cells were individually transduced with lentivirus expressing a scrambled shRNA control or either of two lentiviral shRNAs (sh2 and sh3) targeting different sequences of UBE3B. 48 h later, the loss of UBE3B mRNA expression was measured by qRT-PCR and normalized to the SCR control. G, 48 h after lentiviral infection, the SCR, sh2, and sh3 cells were seeded at 5000 cells/well in 24-well plates in 500 μl/well growth medium. The cell numbers of each plate were counted at 2 h after seeding and then again on days 3, 5, 7, and 10. The relative proliferation for each cell line was calculated by normalizing the cell number each day to the cell number at 2 h after seeding. H, 48 h after lentiviral infection, the SCR, sh2, and sh3 cells were seeded at 500 cells/well in 6-well plates in 5 ml/well growth medium and incubated for 10 days. The colonies were stained with crystal violet, and images were generated. I, quantitation of the clonogenic assay from the results in H, using Celigo, showed a strong decrease of colony numbers in both shRNA knockdown cells. J, to validate changes in mitochondrial morphology, UBE3B-sh2 and UBE3B-sh3 cells were stained with MitoTracker CMX-ROS and fixed, and correlative microscopy was performed with confocal (top row) and SIM super-resolution microscopy (bottom row), showing changes induced by UBE3B knockdown.
To further validate our observation that KD of UBE3B impairs cell growth and mitochondrial morphology, we used two shRNAs targeting UBE3B. Both shRNAs significantly reduced the UBE3B mRNA level (Fig. 3F). The depletion of UBE3B strongly reduced the proliferation and colony formation capability of both KD cells as compared with the SCR control cells (Fig. 3, G–I). Consistent with observations in the siRNA-treated cells, shRNA-mediated KD also caused changes in mitochondrial morphology. As shown when analyzed using correlative confocal and Structured Illumination Microscopy (SIM), loss of UBE3B resulted in an increase in punctate mitochondria and a reduced filamentous mitochondrial network (Fig. 3J).
HECT Domain of UBE3B Catalyzes Auto-ubiquitylation via an Active Site Cysteine
To determine whether UBE3B functions as a ubiquitin E3 ligase, as predicted (see above), the N-terminal HA-tagged UBE3B (HA-UBE3B) was immunoprecipitated, and an in vitro ubiquitylation activity assay was performed over time using His-tagged wild type ubiquitin (His-Ub-WT). We show here that UBE3B exhibits time-dependent auto-ubiquitylation activity, as evidenced by the laddering of the protein when probed using the anti-HA Ab (Fig. 4A).
FIGURE 4.
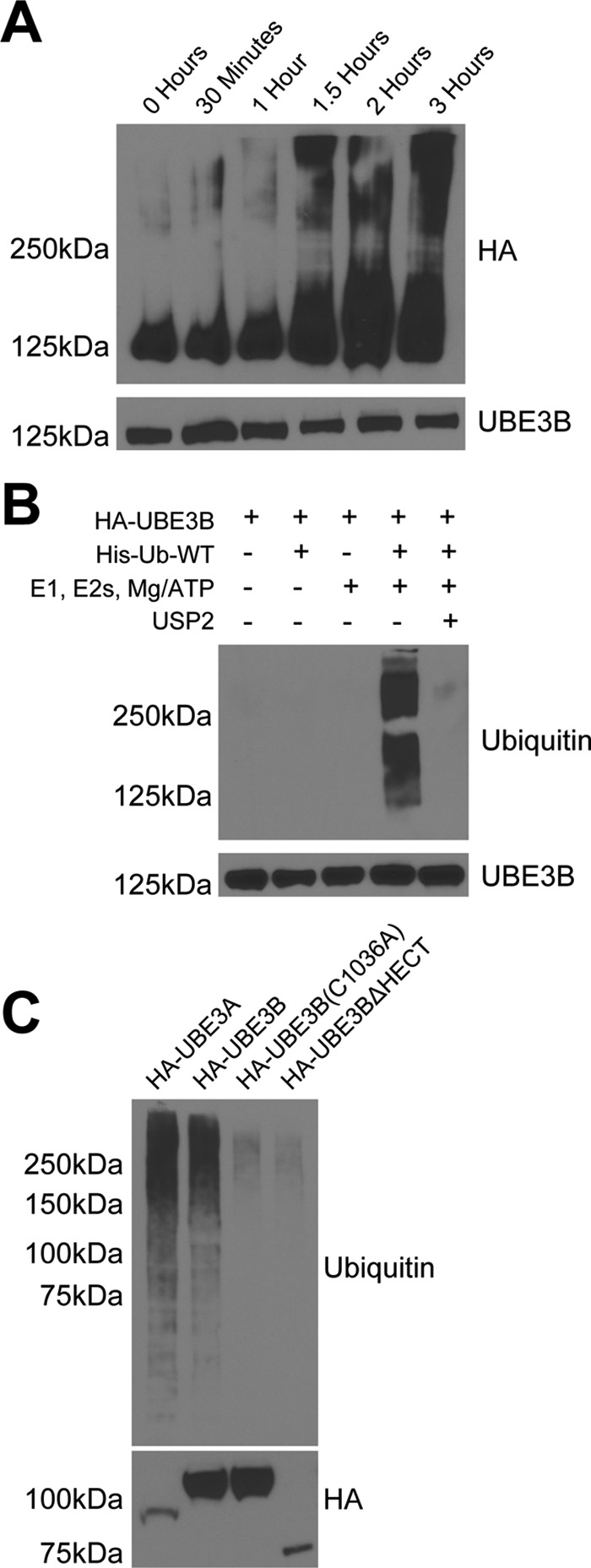
UBE3B exhibits ubiquitin ligase activity. Cell lysates from LN428 cells that stably express UBE3A, UBE3B, UBE3BΔHECT, or UBE3B(C1036A), all with N terminus HA tags, were immunoprecipitated using anti-HA affinity matrix and subjected to ubiquitylation activity assays. The ubiquitylation assays were carried out in 30-μl reactions with 0.1 μm E1, 0.25 μm of an E2 mixture, 1 μm ubiquitin aldehyde, 0.75 μg/μl His-ubiquitin, and 1× magnesium/ATP mixture, unless otherwise noted. A, ubiquitylation activity assay was performed in vitro using His-tagged wild type ubiquitin (His-Ub-WT). The expected size of HA-UBE3B is 125 kDa. Note the time-dependent increase in the high molecular mass signal. B, polyubiquitin chain formation was lost when ubiquitin or E1, E2s, or ATP were removed from the reaction mixture, or if 20 μg of the catalytic core of the deubiquitinating enzyme USP2 was added to a completed reaction for 30 min at 37 °C (63). C, polyubiquitin chains were lacking in reactions containing the catalytically inactive (HA-UBE3B(C1036A)) or HECT deleted (HA-UBE3BΔHECT) mutants of UBE3B. Completed reactions were applied to immunoblot for analysis of ubiquitylation with anti-hemagglutinin (HA) or ubiquitin (Ub) antibodies. Bottom panel shows comparable loading of the immunoprecipitated proteins. Representative immunoblots from three independent experiments are shown. Antibodies used for each of the immunoblots are listed on the side of the panels.
To show that this ubiquitylation activity was specific to UBE3B and not a reaction with a component in the mixture of E1, E2s, ubiquitin, and Mg2+/ATP, a dropout ubiquitylation assay was performed, where one enzyme/set of enzymes was missing from the reaction. We found that the polyubiquitylation activity of UBE3B required all the components, including the E1 and E2 mixture, ATP, and ubiquitin (Fig. 4B). Furthermore, the catalytic core of the deubiquitylating enzyme USP2 was added to the in vitro ubiquitylation assay and eliminated the signal (see last lane, Fig. 4B). This result demonstrates that the putative ubiquitin signal was due to protein ubiquitylation. Finally, to show that the HECT domain of UBE3B, specifically the proposed catalytic cysteine in the HECT domain, catalyzes the formation of the polyubiquitylation chains observed, the ubiquitylation activity assay was performed using UBE3B with an inactive catalytic cysteine (HA-UBE3B(C1036A)) and using UBE3B that lacked the HECT domain (HA-UBE3BΔHECT) (Fig. 4C). As shown, the mutation of Cys-1036 or the deletion of the HECT domain eliminated the ubiquitylation activity. These results demonstrate that UBE3B is a bona fide HECT domain E3 ubiquitin ligase.
Calmodulin Interacts with the IQ Motif of UBE3B
The presence of the IQ motif in the N terminus of UBE3B suggests that UBE3B and calmodulin interact. This would be unique as there are no reported E3 ubiquitin ligases that are calmodulin-regulated, with the exception of FBXL2, an F-box protein that is a subunit of the SCF ubiquitin protein ligase complex (39). Because HA-tagged UBE3B (HA-UBE3B) is readily immunoprecipitated from stable LN428/HA-UBE3B cells, we analyzed the immunoprecipitated proteins by mass spectrometry. Quantification results for two proteotypic calmodulin (CALM, Uniprot no. P62158) peptides showed a significant increase in signal for both peptides bound to either HA-UBE3B or HA-UBE3B (C1036A), with only a background-level signal in the control cells (Fig. 5A). The interaction between UBE3B and calmodulin was also confirmed by immunoprecipitation of HA-UBE3B followed by immunoblot for endogenous calmodulin (Fig. 5B).
FIGURE 5.

UBE3B associates with calmodulin through the IQ motif in a calcium-dependent manner. A, quantification results of HA-Ab affinity purification and high resolution LC-MS analysis for two proteotypic calmodulin peptides, VFDKDGNGYISAAELR and DTDSEEEIREAFR. Bar graphs represent the standardized mean values ± S.E. following affinity purification and differential mass spectrometry analysis of LN428 cells containing an empty vector control or expressing wild type HA-tagged UBE3B (HA-UBE3B) or the catalytic inactive UBE3B with an HA tag (HA-UBE3B(C1036A)). B, cells, indicated in the figure, were lysed, and the HA-tagged proteins were immunoprecipitated, as described under “Experimental Procedures,” to identify the proteins that interact with UBE3B, UBE3B(C1036A), or UBE3BΔHECT. Shown is an immunoblot to confirm the interaction of calmodulin with UBE3B as seen by LC-MS using anti-UBE3B and calmodulin antibodies. C, BioID analysis was performed using BirA-UBE3B and UBE3B-BirA with streptavidin immunoprecipitated to confirm that HA-UBE3B and calmodulin interact. Cells were treated with biotin (5 μm) for 24 h before harvest, as indicated. Streptavidin-purified proteins were then probed by immunoblot using streptavidin-HRP. Bottom right panel indicates the identification of calmodulin after streptavidin capture. D, 293FT cells were transiently transfected (48 h) with a plasmid expressing WT UBE3B-HA or UBE3BΔIQ-HA followed by immunoprecipitation with HA-affinity matrix beads. Deletion of the IQ motif demolished the interaction between UBE3B and calmodulin. E, 239FT cells were transiently transfected (48 h) with a plasmid expressing WT UBE3B-HA or UBE3BΔIQ-HA followed by immunoprecipitation with HA-affinity matrix beads. A ubiquitylation assay was then performed as described under “Experimental Procedures.” The results were analyzed by immunoblot using anti-ubiquitin, anti-His, and anti-HA antibodies to detect UBE3B and anti-CaM antibody to detect calmodulin. Ubiquitylation is increased in UBE3BΔIQ due to the loss of loss of the interaction with calmodulin. F, 239FT cells were transiently transfected (48 h) with a plasmid expressing WT UBE3B-HA or UBE3BΔIQ-HA. The expression of UBE3BΔIQ-HA triggered cell apoptosis as indicated by an elevation of the cleaved form of caspase3. G, calmodulin interaction with UBE3B is lost when immunoprecipitation beads are washed with CaCl2, and ubiquitylation is increased when calmodulin is not bound to UBE3B. Immunoprecipitations and the bead-based in vitro assay were performed as described above.
To more elegantly determine whether the UBE3B-calmodulin interaction is observed in cells and is not limited to a protein-protein interaction in cell lysates, we developed the BioID system to validate UBE3B-interacting proteins by fusing the promiscuous Escherichia coli biotin ligase BirA-R118G to either the N or C terminus of UBE3B. Stable cell lines were developed and were then pulsed with the addition of exogenous biotin (50 μm; 24 h), and the resulting biotinylated proteins represent those within 10 nm of UBE3B (40, 41). In support of the mass spectrometry and IP/immunoblot results above, we find that calmodulin is strongly biotinylated in cells expressing UBE3B-BirA but not in control cells, and only when biotin is added to the cells 24 h prior to lysate preparation (Fig. 5C). This supports our contention that UBE3B and calmodulin interact. As anticipated, the interaction with calmodulin is lost when we deleted the IQ motif of UBE3B (UBE3BΔIQ-HA) (Fig. 5D). Interestingly, the ubiquitylation activity of UBE3B is increased when calmodulin is no longer bound (Fig. 5E). We also found that expression of the UBE3BΔIQ-HA deletion mutant resulted in an increase in apoptosis, as indicated by an increase in cleaved caspase 3 levels (Fig. 5F). Most importantly, we see that calmodulin is released from UBE3B when the concentration of calcium is increased in the lysate (5 mm), and this results in an increase in ubiquitylation activity (Fig. 5G). These combined results suggest that UBE3B is a calmodulin/calcium-regulated E3 ubiquitin ligase and that calmodulin appears to act as a repressor of the E3 ligase activity of UBE3B.
Discussion
Our results demonstrate that UBE3B is a HECT domain containing E3 ubiquitin ligase that uses its catalytic cysteine to bind to ubiquitin and form polyubiquitin chains. We have also shown that the structure of UBE3B's HECT domain, specifically the region surrounding the catalytic cysteine, is highly conserved and homologous to other well studied E3 ligases, such as UBE3C, Huwe1, Nedd4, Itch, and Hace1. HECT E3 ligases are involved in numerous cellular processes, from UBE3C promoting growth and metastasis of renal cell carcinoma, to Nedd4 enhancing cell proliferation and autophagy, to Itch inhibiting skin inflammation (42–44). Reported disease-linked mutations in UBE3B associated with the most severe forms of KOS include the seven different nonsense, frameshift, and splice site mutations that likely cause protein truncation before or at the beginning of the HECT domain and therefore are predicted to abolish UBE3B's ligase activity (24–27). Consequently, our results, defining the HECT domain of UBE3B as the active E3 ubiquitin ligase domain, are consistent with these clinical findings.
The N terminus of UBE3B contains an isoleucine-glutamine (IQ) motif. The IQ motif is known to interact with calmodulin (CaM), the universal calcium sensor in eukaryotes (45–48). CaM can bind to a target in one of two ways, either in a calcium-bound (holoCaM) or calcium-free (apoCaM) form, thereby making its interaction with partners either Ca2+-dependent or Ca2+-independent (35). Calcium binding to calmodulin has been shown to induce major conformational changes in the IQ motifs and increase the flexibility of the myosin-1c tail (49). Our data show that calmodulin binds to UBE3B and modulates UBE3B's ubiquitylation activity in a calcium-dependent manner. Therefore, we propose that calmodulin might be a regulatory factor, rather than a UBE3B substrate. In contrast, one study performed by Wang et al. (50) showed that calcium binds to and activates the E3 ligase Nedd4 by releasing the C2 domain auto-inhibition via a calmodulin-independent mechanism. We propose that an increase in intracellular calcium levels after mitochondrial stress results in a decrease in the interaction between calmodulin and UBE3B, enhancing UBE3B's ubiquitylation activity (Fig. 6).
FIGURE 6.

Model for UBE3B activation in response to calcium signaling/mitochondrial stress. We propose that the E3 ligase UBE3B associates with the mitochondria and that in the absence of stress UBE3B is bound to calmodulin through its N-terminal IQ motif in a calcium-dependent manner. However, in response to mitochondrial stress the intracellular calcium levels increase, thereby activating calmodulin and releasing it from UBE3B. Calmodulin-free UBE3B can now bind to its substrates (potentially damaged mitochondrial proteins) and ubiquitylate substrates for degradation by the UPS.
As mentioned above, knockdown of UBE3B increases the sensitivity of glioblastoma cells (LN428) to TMZ treatment, and the C. elegans ortholog of UBE3B, oxi-1, was required for proteasomal degradation after oxidative stress (25, 29). In this study, we show the involvement of UBE3B in maintaining mitochondrial morphology since depletion of UBE3B in our human glioblastoma cells resulted in mitochondrial fragmentation, accompanied changes in mitochondrial physiology, and subsequent cell death. This raises the possibility that UBE3B may modulate mitochondrial fission/fusion dynamics and/or play a role in stress-induced mitophagy or apoptosis, as proposed for PARKIN and PINK1 (51), MARCH5 (52), and others (53).
In summary, we propose a model for the regulation of mitochondrial structure and ubiquitylation that includes the secondary messenger calmodulin, the HECT E3 ligase UBE3B, and the potentially damaged mitochondrial proteins (Fig. 6). We hypothesize that an increase in calcium signaling after mitochondrial stress activates calmodulin. Activated calmodulin in turn is released from UBE3B, allowing for its increased ubiquitylation activity to tag damaged mitochondrial proteins for proteasomal degradation. Identifying the substrates of UBE3B will be an important future direction in determining the clinical significance of UBE3B mutations and developing therapeutic strategies for patients who suffer from KOS.
Experimental Procedures
Materials
TransIT®-2020 transfection reagent (catalog no. MIR5400) was from Mirus, and the siPORTTM NeoFXTM transfection reagent (catalog no. AM4511) was from Life Technologies, Inc. Cell proliferation was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)-based CellTiter 96 Aqueous One Solution cell proliferation assay (catalog no. G3581) from Promega. The siRNA UBE3B-s40200 (catalog no. 4390824) was from Ambion. The Silencer® negative control siRNA#2 (catalog no. AM4613), TaqMan primer probe set for UBE3B (catalog no. 4331182), and β-actin were from Applied Biosystems. OptiMEM, α-Eagle's minimal essential medium, and phosphate-buffered saline (PBS) were from Invitrogen. Puromycin was from Clontech, and gentamycin was from Irvine Scientific. All the plastic tissue culture supplies were from Corning and Thermo Fisher Scientific. All PCR primers (Table 3) were from Eurofins MWG Operon. DreamTaq DNA polymerase (catalog no. FEREP0703) was from Thermo Fisher Scientific, and dNTPs (catalog no. 4303442) were from Life Technologies, Inc. The Pfu turbo DNA polymerase (catalog no. 600250) and QuikChange II XL site-directed mutagenesis kit (catalog no. 200521) were from Agilent Technologies. The TOPO® TA Cloning® kit for sequencing, with One Shot® TOP10 chemically competent E. coli (catalog no. K4575-40), Gateway® LR Clonase® II enzyme mix (catalog no. 11791-100), and T4 DNA ligase (catalog no. EL0011) were from Life Technologies, Inc. The plasmid p4054 HA-E6AP isoform II (catalog no. 8658) was purchased from Addgene. Restriction enzymes XbaI and BamHI were from New England Biolabs. The QIAquick PCR purification kit (catalog no. 28106) and QIAquick gel extraction kit (catalog no. 29706) were from Qiagen. The TALON® metal affinity resin (catalog no. 635501) and HisTALONTM Buffer set (catalog no. 635651) were from Clontech. The Amicon Ultra-15 centrifugal filter units (catalog no. UFC900324) were from EMD Millipore, and the DC protein assay kit II (catalog no. 500-0112EDU) was from Bio-Rad. The anti-His6 epitope tag Ab, Clone HIS.H8 (catalog no. MA121315), was from Thermo Fisher Scientific. FLOAT-A-LYZER G2 (catalog no. G235051) was from Spectrum Laboratories. The anti-HA affinity matrix (catalog no. 11815016001) was from Roche Applied Science. The mitochondria isolation kit for cultured cells (catalog no. 89874) was from Life Technologies, Inc. The endoplasmic reticulum isolation kit (catalog no.ER0100) was from Sigma. The ubiquitin-activating enzyme (UBE1) (catalog no. E-305) and ubiquitin aldehyde (catalog no. U-201) were from Boston Biochem. The ubiquitin-conjugating enzyme (E2) selection panels (catalog no. UB200) were from Life Sensors. The magnesium/ATP mixture (catalog no. 20-113) was from EMD Millipore. The protease inhibitor mini-tablets (catalog no. 88665) and IP lysis buffer (catalog no. 87788) were from Thermo Fisher Scientific. Biotin (catalog no. BP232-1) was from Thermo Fisher Scientific, and Dynabeads MyOne Streptavidin C (catalog no. 65001) was from Life Technologies, Inc. MitoTracker Deep Red was from Life Technologies, Inc. (catalog no. M22426).
TABLE 3.
Oligodeoxynucleotides used in this study
F is forward; R is reverse.
| Oligonucleotide name | Sequence (5′–3′)a |
|---|---|
| UBE3B-GFP-F | ATTCTAGACACCATGTTCACCCTGTCTCAGACCTCG |
| UBE3B-GFP-R | TAGCTCTAGAATGGAGAGTTCAAAGCCCGTGTTCATGC |
| UBE3BΔHECT-GFP-R | ATCGGATCCAATCCCCACTGGTTGTCTTGAACAGATT |
| UBE3B(C1036A)-F | TGCCCACCTCCTCCACCGCCTTCAACCTGCTCAAGC |
| UBE3B(C1036A)-R | GCTTGAGCAGGTTGAAGGCGGTGGAGGAGGTGGGCA |
| HA-UBE3A-F | CACCATGTACCCATACGATGTTCCAGATTACGCTGAGAAGCTG |
| HA-UBE3A-R | TTACAGCATGCCAAATCCTTTGGCATACGTGATGG |
| HA-UBE3B-F | CACCATGTACCCATACGATGTTCCAGATTACGCTTTCACCCTGTCTCAGACC |
| UBE3B-R | CTAGGAGAGTTCAAAGCCCGTGTTCATGCTGATGGCG |
| UBE3B-F | CACCATGTTCACCCTGTCTCAGACCTCGAGAGCATGG |
| UBE3BΔHECT-R | CTAGGAGAGTTCAAAGCCCGTGTTCATGCTGATGGCG |
| UBE3B-HA-R | CTAAGCGTAATCTGGAACATCGTATGGGTAGGAGAGTTCAAAGCCCGTG |
| Ubiquitin-F | CACCCAGATTTTCGTGAAAACCCTTACG |
| Ubiquitin-R | TTAACCACCACGAAGTCTCAACACAAGATG |
| UBE3B(ΔIQ)-F | CACCATGGCAGATGACCCTGAGTCCACTAAAAGAAGTGC |
a The bold bases indicate the mutated bases.
Primary Antibodies
UBE3B antibody (catalog no. SAB4503523) was from Sigma; ubiquitin (Ub) antibody (catalog no. AUB01) was from Cytoskeleton; α-tubulin antibody (catalog no. CP06) was from EMD Millipore; proliferating cell nuclear antigen antibody (catalog no. sc-56) was from Santa Cruz Biotechnology; cytochrome c oxidase IV (COX IV) antibody (catalog no. A21348) was from Life Technologies, Inc.; Turbo-green fluorescent protein (TurboGFP) antibody (catalog no. AB513) was from Evrogen; adenosine triphosphate synthase β (ATP synthase β) antibody (catalog no. MA1-930) was from Thermo Fisher Scientific; calmodulin antibody (catalog no. ab45689) was from Abcam; PDI antibody was from Life Technologies, Inc. (catalog no. RL77); and cleaved caspase 3 antibody was from Cell Signaling (catalog no. 9661s). The hybridoma cell clone 12CA5, expressing the anti-HA monoclonal antibody, was a generous gift from Kara Bernstein (University of Pittsburgh). The 12CA5 hybridoma cells were used to generate ascites, and the resulting Ab was purified using protein-A-agarose and isotyped (IgG2b/κ).
Secondary Antibodies
Goat anti-mouse horseradish peroxidase (GAM-HRP) conjugates and goat anti-rabbit horseradish peroxidase (GAR-HRP) conjugates were from Bio-Rad; goat anti-mouse Cy5 (catalog no. ab150115) was from Abcam, and goat anti-rabbit Cy3 (catalog no. A-11011) was from Life Technologies, Inc. The Novex® NuPAGE® SDS-polyacrylamide gel systems were from Life Technologies, Inc. Signal generation substrates were from Bio-Rad and Thermo Fisher Scientific. All electrophoresis reagents were from Bio-Rad.
Alignment of UBE3B with HECT E3 Ligases
Several different programs were used to align the sequences of known proteins with that of UBE3B and to predict the three-dimensional structure of the different domains of UBE3B, including Protein Homology/AnalogY Recognition Engine Version 2.0 (Phyre2) and ClustalW2. Phyre2 generates protein models using the principles and techniques of homology modeling, as described (32). The top seven and top six highest confidence homologous sequences for the IQ and HECT domains, respectively, of UBE3B were chosen for sequence alignment using ClustalW2. ClustalW2 is a sequence alignment program for three or more sequences (34).
Cell Culture
LN428 glioblastoma cells and culture conditions were as described previously (54). Cells were maintained at 37 °C in 5% CO2 and grown in α-Eagle's minimal essential medium containing 10% heat-inactivated fetal bovine serum (FBS), antibiotic/antimycotic, l-glutamine, and gentamycin. 293FT cells are a sub-clone of the human embryonic kidney cells 293, obtained from Thermo Fisher Scientific (catalog no. R70007). These were cultured in DMEM with 10% heat-inactivated FBS, 80 μg of penicillin/80 μg of streptomycin at 37 °C in 5% CO2.
Lentiviral Vectors for Expression of UBE3B and Mutants
Human UBE3B complementary DNA was PCR-amplified (using primers purchased from Eurofins MWG Operon) and cloned into the pENTR/D-TOPO plasmid to create the pENTR-UBE3B vector as per standard Topo-cloning methodology (55). With this plasmid as the template, UBE3B-tagged proteins or mutants were made either via standard PCR or with the QuikChange XL site-directed mutagenesis kit. The sequence of each primer is listed in Table 3. Briefly, we made HA-UBE3B, UBE3B-HA, HA-UBE3BΔHECT, and UBE3B(ΔIQ)-HA using primer pairs HA-UBE3B-F/UBE3B-R, UBE3B-F/UBE3B-HA-R, HA-UBE3B-F/UBE3BΔHECT-R, and UBE3B(ΔIQ)-F/UBE3B-R, respectively. Using the pENTR-HA-UBE3B plasmid as template, we made the HA-UBE3B(C1036A) mutation using primers UBE3B(C1036A)-F/UBE3B(C1036A)-R. Using p4054 HA-E6AP isoform II plasmid (Addgene) as template, we made HA-UBE3A using primer pair HA-UBE3A-F/HA-UBE3A-R. Once sequence-verified, the open reading frames from each of the plasmids pENTR-HA-UBE3B, pENTR-UBE3B-HA, pENTR-HA-UBE3BΔHECT, pENTR-HA-UBE3B(C1036A), pENTR-UBE3B(ΔIQ)-HA, and pENTR-HA-UBE3A were transferred into a Gateway-modified pLVX-IRES-Puro vector (Clontech), by the LR reaction using the Gateway LR Clonase II Enzyme Mix (Life Technologies, Inc.) as per the manufacturer's instruction. Positive clones were selected, and plasmids were extracted with the QIAprep spin miniprep kit (Qiagen). For the UBE3B-copGFP, UBE3BΔHECT-copGFP, and UBE3B(C1036A)-copGFP fusion plasmids, we used pENTR-UBE3B and pCT-CMV-MCS-copGFP-EF1-Puro (System Biosciences). We made UBE3B-copGFP and UBE3BΔ HECT-copGFP using primer pairs UBE3B-GFP-F/UBE3B-GFP-R and UBE3B-GFP-F/UBE3BΔHECT-GFP-R, respectively. Using pENTR-UBE3B(C1036A) plasmid as template, we made the UBE3B(C1036A)-copGFP mutation using primers UBE3B-GFP-F/UBE3B-GFP-R. The UBE3B and UBE3B(C1036A) open-reading frames were PCR-amplified to engineer the restriction enzyme site XbaI on both ends, whereas the UBE3BΔHECT-copGFP open reading frame was PCR-amplified to engineer the restriction enzyme sites XbaI and BamHI for cloning in-frame with copGFP using standard protocols. After PCR, products were digested with the respective enzymes; purified fragments were ligated into the pCT-CMV-copGFP-MCS-EF1-puro lentiviral vector hydrolyzed by XbaI or XbaI and BamHI. Positive colonies were selected and sequenced. Sequence-verified plasmids were transiently transfected, and lentivirus production was as described below. The pLPC-MYC-BirA plasmid was previously described (41). To create the BirA-UBE3B and UBE3B-BirA fusion plasmids, we had pENTR-BirA-UBE3B and pENTR-UBE3B-BirA plasmids synthesized by GenScript. Once sequence-verified, we transferred the open reading frames into the Gateway-modified pLVX-IRES-Puro vector as described above. All the vectors used and developed in this study are listed in Table 4.
TABLE 4.
Vectors developed for and used in this study
| Plasmid name | Insert description | Parental vector |
|---|---|---|
| pENTR-UBE3B | UBE3B | pENTR/d-TOPO |
| pENTR-HA-UBE3B | HA-UBE3B | pENTR/d-TOPO |
| pENTR-UBE3B-HA | UBE3B-HA | pENTR/d-TOPO |
| pENTR-HA-UBE3BΔHECT | HA-UBE3BΔHECT | pENTR/d-TOPO |
| pENTR-HA-UBE3B(C1036A) | HA-UBE3B (C1036A) | pENTR/d-TOPO |
| pENTR-HA-UBE3A | HA-UBE3A | pENTR/d-TOPO |
| pENTR-UBE3BΔIQ-HA | UBE3BΔIQ-HA | pENTR/d-TOPO |
| pCT-CMV-UBE3B-copGFP-Puro | UBE3B-copGFP | pCYTO-CMV-MCS-copGFP-EF1-Puro |
| pCT-CMV-UBE3BΔHECT-copGFP-Puro | UBE3BΔHECT-copGFP | pCYTO-CMV-MCS-copGFP-EF1-Puro |
| pCT-CMV-UBE3BΔHECT-copGFP-Puro | UBE3B (C1036A)-copGFP | pCYTO-CMV-MCS-copGFP-EF1-Puro |
| pLVX-HA-UBE3B-IRES-Puro | HA-UBE3B | pLVX-GW-IRES-Puro |
| pLVX-UBE3B-HA-IRES-Puro | UBE3B-HA | pLVX-GW-IRES-Puro |
| pLVX-HA-UBE3BΔHECT-IRES-Puro | HA-UBE3BΔHECT | pLVX-GW-IRES-Puro |
| pLVX-HA-UBE3B(C1036A)-IRES-Puro | HA-UBE3B (C1036A) | pLVX-GW-IRES-Puro |
| pLVX-HA-UBE3A-IRES-Puro | HA-UBE3A | pLVX-GW-IRES-Puro |
| pLVX-UBE3BΔIQ-HA-IRES-Puro | UBE3BΔIQ-HA | pLVX-GW-IRES-Puro |
| pLVX-BirA-UBE3B-Puro | BirA-UBE3B | pLVX-GW-IRES-Puro |
| pLVX-UBE3B-BirA-Puro | UBE3B-BirA | pLVX-GW-IRES-Puro |
Lentiviral Expression of UBE3B in LN428 Cells
Lentiviruses were prepared in collaboration with the University of Pittsburgh Cancer Institute Lentiviral Facility or the MCI Gene Expression, Editing, and Discovery Facility. Lentiviral particles were generated by co-transfection of four plasmids (control plasmid (pLVX-IRES-Puro) or one of the plasmids expressing UBE3B or UBE3B mutants, together with pMD2.g (VSVG), pVSV-REV, and pMDLg/pRRE) into 293FT cells using TransIT®-2020 transfection reagent. The collection and isolation of lentiviral particles and transduction of LN428 cells were performed as described previously (56, 57). Stable cell lines were developed by selection in puromycin (2 μg/ml) for 2 weeks. Expression of HA-UBE3B (or the indicated mutants or fusions) was confirmed by immunoblot. All the cell lines developed and used in this study are listed in Table 5.
TABLE 5.
Cell lines developed and used in this study
| Cell line name | Cell line description | Growth mediaa |
|---|---|---|
| LN428 | Human glioblastoma tumor cell line | Media 1 |
| LN428/UBE3B-copGFP | LN428 cells expressing UBE3B-copGFP | Media 2 |
| LN428/UBE3BΔHECT-copGFP | LN428 cells expressing UBE3BΔHECT-copGFP | Media 2 |
| LN428/UBE3B(C1036A)-copGFP | LN428 cells expressing UBE3B (C1036A)-copGFP | Media 2 |
| LN428/HA-UBE3B | LN428 cells expressing HA-UBE3B | Media 2 |
| LN428/UBE3B-HA | LN428 cells expressing UBE3B-HA | Media 2 |
| LN428/HA-UBE3BΔHECT | LN428 cells expressing HA-UBE3BΔHECT | Media 2 |
| LN428/HA-UBE3B(C1036A) | LN428 cells expressing HA-UBE3B (C1036A) | Media 2 |
| LN428/HA-UBE3A | LN428 cells expressing HA-UBE3A | Media 2 |
| LN428/BirA-UBE3B | LN428 cells expressing BirA-UBE3B | Media 2 |
| LN428/UBE3B-BirA | LN428 cells expressing UBE3B-BirA | Media 2 |
| 293FT | A sub-clone of human embryonic kidney cells 293 | Media 3 |
a Media 1 is α-minimal essential medium with 10% heat-inactivated FBS, 5 μg/ml gentamicin, 80 μg of penicillin, 80 μg of streptomycin, 0.32 μg of amphotericin/ml, 2 mm l-glutamine. Media 2 is Media 1 supplemented with puromycin (1.0 μg/ml). Media 3 is DMEM with 10% heat-inactivated FBS, 80 μg of penicillin, 80 μg of streptomycin.
Lentiviral Expression of shRNA to Mediate UBE3B Knockdown in LN428 Cells
Lentiviruses were prepared in collaboration with the MCI Gene Expression, Editing, and Discovery Facility. Lentiviral particles were generated as indicated above. The target sequences of sh2 and sh3 are CCGTGATGTATGTGAAAGTTT and CCCAGTGAAGAGTCTCCTAAA, respectively. The scrambled shRNA (SCR) was used as a negative control. The collection and isolation of lentiviral particles and transduction of LN428 cells were performed as described above.
Transient Expression of UBE3B-HA and UBE3B(ΔIQ)-HA in 293FT Cells
293FT cells (6 × 106) were seeded in a 150-mm dish and incubated overnight. Each transfection mixture was prepared by mixing 35 μg of plasmid (for expression of GFP, UBE3B-HA, or UBE3B(ΔIQ)-HA) with 70 μl of TransIT-X2 (catalog no. MIR6005, Mirus) in 2.7 ml of DMEM without FBS or antibiotics. The mixture was kept at room temperature for 20 min and then added to each 293FT pre-seeded 150-mm dish dropwise. Then, the cells were incubated for 48 h. All cells from each dish were collected and washed once with PBS. The cells were equally transferred into two 1.5-ml Eppendorf tubes and kept at −80 °C until used for immunoprecipitation and in vitro ubiquitylation analysis. One tube of cells was used for immunoprecipitation, and the second tube was for the in vitro ubiquitylation assay.
UBE3B Transient siRNA Knockdown in LN428 Cells
UBE3B was depleted in LN428 cells by transient siRNA transfection, as described previously (29). Briefly, 4 × 105 LN428 cells were wet reverse-transfected with either 90 nmol/liter siRNA UBE3B-s40200 (Ambion) or 90 nmol/liter Silencer® negative control siRNA#2 (Applied Biosystems). The siRNA was prepared with siPORTTM NeoFXTM and OptiMEM, and the transfections were incubated for 24 h at 37 °C before replacement with fresh media.
Quantitative RT-PCR Analysis
Expression of UBE3B mRNA after siRNA-mediated KD or shRNA-mediated KD was measured by quantitative RT-PCR (qRT-PCR) using an Applied Biosystems StepOnePlus system as described previously (58). We used the Applied Biosystems TaqMan gene expression assay human UBE3B:Hs00296200_m1, and we normalized gene expression to the expression of human β-actin (catalog no. 4333762T).
Cell Metabolic Activity Assay (MTS Assay)
Twenty four hours after siRNA-mediated UBE3B KD, cells were seeded into 96-well plates at a density of 2000 cells per well and allowed to grow for 48 h at 37 °C before determining the relative amount of metabolically active cells by an MTS assay (Promega). Results are the average of three separate experiments and normalized to the Silencer® negative control siRNA#2 cells, with error bars representing the mean ± S.E.
Cell Proliferation Assay
Forty eight hours after lentiviral transduction, the SCR, sh2, and sh3 cells were trypsinized and seeded at 5000 cells/well in 24-well plates with 500 μl/well growth medium. Cells were stained with 1:4000 dilution of Hoechst (catalog no. 62249, Thermo Fisher Scientific) in 500 μl of PBS, and the cell numbers in each plate were counted 2 h after seeding and again on days 3, 5, 7, and 10 using the Celigo image cytometry system (Nexcelom). Relative proliferation for each cell line was calculated by normalizing the cell number each day to the initial cell number (at 2 h).
Clonogenic Assay
Forty eight hours after lentiviral transduction, the SCR, sh2, and sh3 cells were trypsinized and seeded at 500 cells/well in 6-well plates with 5 ml/well growth medium and incubated for 10 days. Colonies were then stained with crystal violet dye, and the colonies with more than 50 cells in each well were counted using the Celigo image cytometry system (Nexcelom).
Cell Extract Preparation and Immunoblot
Mitochondrial and cytoplasmic fractions were isolated using the mitochondria isolation kit for cultured cells (Life Technologies, Inc.). Endoplasmic reticulum fractions were isolated using the endoplasmic reticulum isolation kit (Sigma). Collected subcellular fractions and whole cell extracts were prepared using 2× clear Laemmli buffer (2% SDS, 20% glycerol, and 63 mm Tris-HCl, pH 6.8). Protein concentrations were determined using the Bio-Rad protein assay kit II (catalog no. 500-0002), according to the manufacturer's instruction. Equal amounts of protein were loaded on precast 4–12% Tris-Bis Novex® NuPAGE® SDS-polyacrylamide gels (Life Technologies, Inc.).
Immunoprecipitation and in Vitro Ubiquitylation Assay
LN428 cells expressing HA-tagged UBE3B WT or mutant proteins (as well as HA-UBE3A as a positive control) were grown to 90% confluence in 100-mm dishes and harvested using IP Lysis Buffer supplemented with protease inhibitor (both from Thermo Fisher Scientific). The lysates were rotated for 1 h at 4 °C before pelleting the cell debris. The supernatant was incubated by rotating overnight at 4 °C with the anti-HA affinity matrix (Roche Applied Science). The beads were washed with Binding Buffer (10 mm Na2H2PO2, 10 mm Na2HPO4, and 150 μm NaCl, pH 7.0), followed by washes with Ubiquitylation Assay Buffer (10 mm Tris-HCl, pH 7.5, 100 mm NaCl, and 0.5 mm DTT). The ubiquitylation assay was performed on the beads for 2 h at 30 °C in a volume of 30 μl containing 0.1 μm E1 (Boston Biochem), 0.25 μm E2s (UBE2A/UBE2D1/UBE2D3/UBE2G2/UBE2L3/UBE2N/UBE2S/UBE2Z) (Life Sensors), 1 μm ubiquitin aldehyde (Boston Biochem), 0.75 μg/μl purified His-ubiquitin (see below), and 1× magnesium/ATP mixture (EMD Millipore). The ubiquitylation assays were stopped by adding 2× blue Laemmli (plus β-mercaptoethanol) and heating the samples at 70 °C for 15 min. Ubiquitylation activity was measured by immunoblot. The immunoprecipitation andin vitro ubiquitylation assay for the 293FT cells transiently expressing GFP, UBE3B(WT)-HA, or UBE3B(ΔIQ)-HA was the same as described above for the LN428 cells.
Affinity Capture of Biotinylated Proteins
LN428 cells that stably express BirA-UBE3B and UBE3B-BirA proteins were grown to 80% confluence in 150-mm dishes and incubated in complete media supplemented with 50 μm biotin to allow for biotin labeling of proteins as described previously (41). Briefly, 24 h after biotin addition, the cells were washed three times with PBS and lysed at 25 °C in 1 ml of Lysis Buffer (50 mm Tris, pH 7.4, 500 mm NaCl, 0.4% SDS, 5 mm EDTA, 1 mm DTT, and protease inhibitor; Thermo Fisher Scientific) and sonicated. Triton X-100 was added to 2% final concentration. After further sonication, an equal volume of 50 mm Tris, pH 7.4 (4 °C), was added before additional sonication (subsequent steps also at 4 °C) and centrifugation at 16,000 relative centrifugal force. Supernatants were incubated with 100 μl of Dynabeads MyOne Streptavidin C1 (Life Technologies, Inc.) overnight. Beads were collected and washed twice for 8 min at 25 °C (all subsequent steps at 25 °C) in 1 ml of Wash Buffer 1 (2% SDS in distilled H2O). This was repeated once with Wash Buffer 2 (0.1% deoxycholate, 1% Triton X-100, 500 mm NaCl, 1 mm EDTA, and 50 mm Hepes, pH 7.5), once with Wash Buffer 3 (250 mm LiCl, 0.5% Nonidet P-40, 0.5% deoxycholate, 1 mm EDTA, and 10 mm Tris, pH 8.1), and twice with Wash Buffer 4 (50 mm Tris, pH 7.4, and 50 mm NaCl). Bound proteins were removed from the magnetic beads with 50 μl of Laemmli SDS/sample buffer at 95 °C.
Expression and Purification of His-Ubiquitin
FLAG-tagged human wild type ubiquitin (pcDNA3.1-FLAG-humanUb) was a generous gift from Dr. Noriyuki Matsuda at Tokyo Metropolitan Institute of Medical Science. Complementary DNA was PCR-amplified (using primers purchased from Eurofins MWG Operon) with N-terminal polyhistidine tag (His) and cloned into pENTR/D-TOPO plasmid to create pENTR-His-Ub-WT via a standard Topo-cloning methodology (55). Once sequence-verified, the open reading frame from the plasmid pENTR-His-Ub-WT was transferred into a pDEST-17 vector (Invitrogen) by an LR reaction using the Gateway LR Clonase II enzyme mix (Life Technologies, Inc.) as per the manufacturer's instruction. Positive clones were selected, and plasmids were extracted with the QIAprep spin miniprep kit (Qiagen). The pDEST-17-Ub-WT plasmid was transformed into One-Shot BL21(DE3)pLysS chemically competent cells (Life Technologies, Inc.). To induce the expression of the His-Ub-WT protein, bacterial cultures were allowed to grow at 37 °C and 250 rpm to an A600 = 0.5–0.7, before the addition of isopropyl β-d-1-thiogalactopyranoside, for a final concentration of 1 μm isopropyl β-d-1-thiogalactopyranoside. The culture was allowed to grow for another 3.5 h. The bacteria were pelleted at 6000 × g for 15 min at 4 °C, followed by resuspension of the pellet in Buffer A (50 mm Tris-HCl, pH 7.6, 100 mm NaCl, 0.1 mm PMSF, and 1 tablet protease inhibitor/20 ml of Buffer A). The bacterial suspensions were pelleted at 6000 × g at 4 °C for 15 min, supernatants discarded, and the pellets frozen overnight at −80 °C. The recombinant His-Ub-WT proteins were purified using the TALON® metal affinity resin and HisTALONTM buffer set (Clontech) according to the manufacturer's protocol. The purified protein was dialyzed with Dialysis Buffer (50 mm Tris-HCl, pH 7.6, 100 mm NaCl, 0.5 mm EDTA, and 5 mm DTT), changing the buffer four times, every 6–8 h. The dialyzed proteins were concentrated using Amicon ultracentrifugal filter devices (EMD Millipore), and the protein concentrations were measured using the DCTM protein assay (Bio-Rad) with BSA as the standard.
Immunofluorescence and Mitochondrial Imaging
For siRNA experiments, cells were cultured on glass coverslips for 24 h before being fixed with 2% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 for 15 min, and blocked with 2% bovine serum albumin for 45 min, all at room temperature. Anti-ATP synthase β antibody (Thermo Fisher Scientific) at 1:250 dilution was incubated for 1 h, followed by goat anti-mouse Alexa Fluor 647 (Abcam) at 1:1000 for 1 h and DAPI stain for 30 s, all at room temperature. Slides were mounted and imaged on the Nikon A1 confocal microscope in the University of Pittsburgh Center for Biologic Imaging.
For subcellular localization experiments, cells were cultured in Fluorodish glass bottom dishes (World Precision Instruments) for 24 h before being stained with MitoTracker Deep Red (Life Technologies, Inc.), then fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100. Cells were blocked with 10% normal goat serum in PBS, then incubated with anti-PDI antibody (Life Technologies, Inc.) at 1:400 dilution for 2 h, and followed by secondary goat anti-mouse antibody conjugated to Alexa Fluor 488 (Life Technologies, Inc.). Dishes were then DAPI-stained and mounted. For shRNA studies, transduced cells were plated in Nunc Lab-Tek II (Thermo Scientific) chambered coverglass slides, for 24 h, then stained with MitoTracker CMX-ROS (Life Technologies, Inc.), and fixed. Cells were permeabilized as above and stained with NucBlue DNA dye (Thermo Fisher Scientific). Cells were then imaged with a Nikon A1rsi confocal microscope. Image-based registration was used to perform correlative microscopy on the same image fields using a Nikon N-SIM super-resolution microscope. Confocal and SIM imaging were performed at the Mitchell Cancer Institute Cellular and Biomolecular Imaging Facility.
Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) Analysis
Immunoprecipitated samples were separated on SDS-polyacrylamide gels (Bolt® 4–12% BisTris Plus Gel, Life Technologies, Inc.) to about 1 cm (150 V for 10 min) and stained with Simply BlueTM SafeStain (Life Technologies, Inc.). After washing with Milli-Q water, the stained regions were excised, washed with HPLC water, and destained with 50% acetonitrile, 25 mm ammonium bicarbonate until no visible blue staining. Gel sections were dehydrated with 100% acetonitrile, reduced with 10 mm dithiothreitol (DTT) at 56 °C for 1 h, followed by alkylation with 55 mm iodoacetamide at room temperature for 45 min in the dark. Gel sections were then again dehydrated with 100% acetonitrile to remove excess DTT and iodoacetamide and rehydrated with 20 ng/μl trypsin in 25 mm ammonium bicarbonate and digested overnight at 37 °C. The resultant tryptic peptides were extracted with 70% acetonitrile, 5% formic acid, speed-vacuum dried, and reconstituted in 18 μl of 0.1% formic acid.
Tryptic digests were analyzed using a high resolution liquid chromatography tandem mass spectrometer as described previously (57, 59). In brief, samples were loaded with a nanoAcquity autosampler (Waters, Waltham, MA) onto a capillary sample trap column, separated using a reversed phase gradient on a 0.075 × 100-mm PicoChipTMC18 column, and electrospray ionization source (New Objective, Inc., Woburn, MA). Mass analysis was performed on a hybrid LTQ/Orbitrap Velos mass spectrometer (Thermo Fisher, Waltham, MA). Data-dependent acquisition was used to acquire one high resolution full scan mass spectrum followed by 13 low resolution tandem mass spectra in the linear ion trap, with dynamic exclusion setting enabled to minimize redundant selection of precursor ions.
Cloud-based differential mass spectrometry (dMS) software developed by the University of Pittsburgh and Infoclinika (dMS 1.0, Seattle, WA) was used to quantify and identify proteins that exhibit a statistically significant difference in abundance between experimental conditions (60, 61). Briefly, the dMS software was used to detect and align mass spectral features present in the high resolution LC-MS data. The m/z, retention time, and intensity of each feature were combined into isotope groups for subsequent statistical and visual analysis. The COMET search algorithm (precursor mass tolerance = 20 ppm, fragment mass tolerance = 0.8 Da) and an indexed non-redundant human database (NCBI human, static modification of cysteine = 57.02 Da, variable modification of methionine = 15.99 Da) was used to identify the peptide sequence and protein identity of selected features present in the data set (62).
pMito-Timer Transfection and Analysis
Cells were simultaneously transfected with siRNA specific to UBE3B and the plasmid encoding for the Mito-Timer reporter (Addgene). Transfection was performed as described above with the addition of pMito-Timer DNA to the transfection mixture. Cells were then plated on Fluorodish glass-bottom dishes (World Precision Instruments) and imaged (live) 72 h after transfection with the Nikon A1rsi confocal microscope at the Mitchell Cancer Institute Cellular and Biomolecular Imaging Facility.
Author Contributions
A. B., N. Y., C. S. C., J. L. B., and R. W. S. designed the research; A. B., J. L., X.-H. W., N. B. D., J. A., L. Z., J. C., and X. Z. performed the research; A. B., J. L., J. A., X. Z., N. A. Y., and R. W. S. analyzed the data; and A. B. and R. W. S. wrote the paper.
Acknowledgments
We thank Dr. David Svilar for valuable comments during the preparation of this manuscript and Dr. Qingming Fang, Dr. Elise Fouquerrel, Zhongxun Yu, Ashley Brown, Sandy Schamus, and Morgan Jessup for technical help. The recombinant human ubiquitin expression vector (wild type) was a generous gift from Drs. Fumika Koyano and Noriyuki Matsuda (Tokyo Metropolitan Institute of Medical Science). Support for the University of Pittsburgh Cancer Institute Lentiviral Facility, the Biomedical Mass Spectrometry Center, and the University of Pittsburgh Cancer Institute Cancer Proteomics Facility was provided by the National Institutes of Health Cancer Center Support Grant CA047904. Support for the MCI Gene Expression, Editing, and Discovery Facility was provided by funds from the Abraham A. Mitchell Distinguished Investigator award.
This work was supported by National Institutes of Health Grants CA148629 and GM087798 (to R. W. S.) and Grant GM75061 and by Pittsburgh Center for Kidney Research Grant DK79307 from National Institutes of Health (to J. L. B.). R. W. S. is a scientific consultant for Trevigen, Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- UPS
- ubiquitin/proteasome system
- KOS
- Kaufman oculocerebrofacial syndrome
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- Ub
- ubiquitin
- PDI
- protein-disulfide isomerase
- ROS
- reactive oxygen species
- dMS
- differential mass spectrometry
- RBR
- RING-between-RING
- α-MEM
- α-minimal essential medium
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- OMM
- outer mitochondrial membrane
- TMZ
- temozolomide
- ER
- endoplasmic reticulum
- dMS
- differential mass spectrometry
- SCR
- scrambled
- CaM
- calmodulin
- qRT
- quantitative RT
- Ab
- antibody
- IP
- immunoprecipitation
- SIM
- Structured Illumination Microscopy.
References
- 1. Komander D. (2009) The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 37, 937–953 [DOI] [PubMed] [Google Scholar]
- 2. Tai H. C., and Schuman E. M. (2008) Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 9, 826–838 [DOI] [PubMed] [Google Scholar]
- 3. Kawabe H., and Brose N. (2011) The role of ubiquitylation in nerve cell development. Nat. Rev. Neurosci. 12, 251–268 [DOI] [PubMed] [Google Scholar]
- 4. Schmidt M., and Finley D. (2014) Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 1843, 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Varshavsky A. (2012) The ubiquitin system, an immense realm. Annu. Rev. Biochem. 81, 167–176 [DOI] [PubMed] [Google Scholar]
- 6. Komander D., and Rape M. (2012) The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 [DOI] [PubMed] [Google Scholar]
- 7. Budhidarmo R., Nakatani Y., and Day C. L. (2012) RINGs hold the key to ubiquitin transfer. Trends Biochem. Sci. 37, 58–65 [DOI] [PubMed] [Google Scholar]
- 8. Deshaies R. J., and Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 9. Hochstrasser M. (2006) Lingering mysteries of ubiquitin-chain assembly. Cell 124, 27–34 [DOI] [PubMed] [Google Scholar]
- 10. Wenzel D. M., and Klevit R. E. (2012) Following Ariadne's thread: a new perspective on RBR ubiquitin ligases. BMC Biol. 10, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huibregtse J. M., Scheffner M., Beaudenon S., and Howley P. M. (1995) A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. U.S.A. 92, 5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang M., and Pickart C. M. (2005) Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. EMBO J. 24, 4324–4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Benard G., and Karbowski M. (2009) Mitochondrial fusion and division: regulation and role in cell viability. Semin. Cell Dev. Biol. 20, 365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chan D. C. (2006) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125, 1241–1252 [DOI] [PubMed] [Google Scholar]
- 15. Neutzner A., Youle R. J., and Karbowski M. (2007) Outer mitochondrial membrane protein degradation by the proteasome. Novartis Found. Symp. 287, 4–14 [PubMed] [Google Scholar]
- 16. Neutzner A., Benard G., Youle R. J., and Karbowski M. (2008) Role of the ubiquitin conjugation system in the maintenance of mitochondrial homeostasis. Ann. N. Y. Acad. Sci. 1147, 242–253 [DOI] [PubMed] [Google Scholar]
- 17. Escobar-Henriques M., Westermann B., and Langer T. (2006) Regulation of mitochondrial fusion by the F-box protein Mdm30 involves proteasome-independent turnover of Fzo1. J. Cell Biol. 173, 645–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cohen M. M., Leboucher G. P., Livnat-Levanon N., Glickman M. H., and Weissman A. M. (2008) Ubiquitin-proteasome-dependent degradation of a mitofusin, a critical regulator of mitochondrial fusion. Mol. Biol. Cell 19, 2457–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Neutzner A., and Youle R. J. (2005) Instability of the mitofusin Fzo1 regulates mitochondrial morphology during the mating response of the yeast Saccharomyces cerevisiae. J. Biol. Chem. 280, 18598–18603 [DOI] [PubMed] [Google Scholar]
- 20. Chan N. C., Salazar A. M., Pham A. H., Sweredoski M. J., Kolawa N. J., Graham R. L., Hess S., and Chan D. C. (2011) Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tanaka A., Cleland M. M., Xu S., Narendra D. P., Suen D. F., Karbowski M., and Youle R. J. (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu S., Peng G., Wang Y., Fang S., and Karbowski M. (2011) The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell 22, 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gegg M. E., Cooper J. M., Chau K. Y., Rojo M., Schapira A. H., and Taanman J. W. (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Basel-Vanagaite L., Yilmaz R., Tang S., Reuter M. S., Rahner N., Grange D. K., Mortenson M., Koty P., Feenstra H., Farwell Gonzalez K. D., Sticht H., Boddaert N., Désir J., Anyane-Yeboa K., et al. (2014) Expanding the clinical and mutational spectrum of Kaufman oculocerebrofacial syndrome with biallelic UBE3B mutations. Hum. Genet. 133, 939–949 [DOI] [PubMed] [Google Scholar]
- 25. Basel-Vanagaite L., Dallapiccola B., Ramirez-Solis R., Segref A., Thiele H., Edwards A., Arends M. J., Miró X., White J. K., Désir J., Abramowicz M., Dentici M. L., Lepri F., Hofmann K., Har-Zahav A., et al. (2012) Deficiency for the ubiquitin ligase UBE3B in a blepharophimosis-ptosis-intellectual-disability syndrome. Am. J. Hum. Genet. 91, 998–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flex E., Ciolfi A., Caputo V., Fodale V., Leoni C., Melis D., Bedeschi M. F., Mazzanti L., Pizzuti A., Tartaglia M., and Zampino G. (2013) Loss of function of the E3 ubiquitin-protein ligase UBE3B causes Kaufman oculocerebrofacial syndrome. J. Med. Genet. 50, 493–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pedurupillay C. R., Baroy T., Holmgren A., Blomhoff A., Vigeland M. D., Sheng Y., Frengen E., Stromme P., and Misceo D. (2015) Kaufman oculocerebrofacial syndrome in sisters with novel compound heterozygous mutation in UBE3B. Am. J. Med. Genet. 167A, 657–663 [DOI] [PubMed] [Google Scholar]
- 28. Venhoranta H., Pausch H., Flisikowski K., Wurmser C., Taponen J., Rautala H., Kind A., Schnieke A., Fries R., Lohi H., and Andersson M. (2014) In frame exon skipping in UBE3B is associated with developmental disorders and increased mortality in cattle. BMC Genomics 15, 890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Svilar D., Dyavaiah M., Brown A. R., Tang J. B., Li J., McDonald P. R., Shun T. Y., Braganza A., Wang X. H., Maniar S., St Croix C. M., Lazo J. S., Pollack I. F., Begley T. J., and Sobol R. W. (2012) Alkylation sensitivity screens reveal a conserved cross-species functionome. Mol. Cancer Res. 10, 1580–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chahrour M. H., Yu T. W., Lim E. T., Ataman B., Coulter M. E., Hill R. S., Stevens C. R., Schubert C. R., ARRA Autism Sequencing Collaboration, Greenberg M. E., Gabriel S. B., and Walsh C. A. (2012) Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 8, e1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gong T. W., Huang L., Warner S. J., and Lomax M. I. (2003) Characterization of the human UBE3B gene: structure, expression, evolution, and alternative splicing. Genomics 82, 143–152 [DOI] [PubMed] [Google Scholar]
- 32. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goujon M., McWilliam H., Li W., Valentin F., Squizzato S., Paern J., and Lopez R. (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 35. Rhoads A. R., and Friedberg F. (1997) Sequence motifs for calmodulin recognition. FASEB J. 11, 331–340 [DOI] [PubMed] [Google Scholar]
- 36. Laker R. C., Xu P., Ryall K. A., Sujkowski A., Kenwood B. M., Chain K. H., Zhang M., Royal M. A., Hoehn K. L., Driscoll M., Adler P. N., Wessells R. J., Saucerman J. J., and Yan Z. (2014) A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J. Biol. Chem. 289, 12005–12015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ferree A. W., Trudeau K., Zik E., Benador I. Y., Twig G., Gottlieb R. A., and Shirihai O. S. (2013) MitoTimer probe reveals the impact of autophagy, fusion, and motility on subcellular distribution of young and old mitochondrial protein and on relative mitochondrial protein age. Autophagy 9, 1887–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hernandez G., Thornton C., Stotland A., Lui D., Sin J., Ramil J., Magee N., Andres A., Quarato G., Carreira R. S., Sayen M. R., Wolkowicz R., and Gottlieb R. A. (2013) MitoTimer: a novel tool for monitoring mitochondrial turnover. Autophagy 9, 1852–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen B. B., Coon T. A., Glasser J. R., and Mallampalli R. K. (2011) Calmodulin antagonizes a calcium-activated SCF ubiquitin E3 ligase subunit, FBXL2, to regulate surfactant homeostasis. Mol. Cell. Biol. 31, 1905–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim D. I., Birendra K. C., Zhu W., Motamedchaboki K., Doye V., and Roux K. J. (2014) Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. U.S.A. 111, E2453–E2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roux K. J., Kim D. I., Raida M., and Burke B. (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wen J. L., Wen X. F., Li R. B., Jin Y. C., Wang X. L., Zhou L., and Chen H. X. (2015) UBE3C promotes growth and metastasis of renal cell carcinoma via activating Wnt/β-catenin pathway. PLoS One 10, e0115622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li Y., Zhang L., Zhou J., Luo S., Huang R., Zhao C., and Diao A. (2015) Nedd4 E3 ubiquitin ligase promotes cell proliferation and autophagy. Cell. Prolif. 48, 338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Theivanthiran B., Kathania M., Zeng M., Anguiano E., Basrur V., Vandergriff T., Pascual V., Wei W. Z., Massoumi R., and Venuprasad K. (2015) The E3 ubiquitin ligase Itch inhibits p38α signaling and skin inflammation through the ubiquitylation of Tab1. Sci. Signal. 8, ra22. [DOI] [PubMed] [Google Scholar]
- 45. Jang D. J., Ban B., and Lee J. A. (2011) Characterization of novel calmodulin binding domains within IQ motifs of IQGAP1. Mol. Cells 32, 511–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huang H., Tan B. Z., Shen Y., Tao J., Jiang F., Sung Y. Y., Ng C. K., Raida M., Köhr G., Higuchi M., Fatemi-Shariatpanahi H., Harden B., Yue D. T., and Soong T. W. (2012) RNA editing of the IQ domain in Ca(v)1.3 channels modulates their Ca2+-dependent inactivation. Neuron 73, 304–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fischer C., Kugler A., Hoth S., and Dietrich P. (2013) An IQ domain mediates the interaction with calmodulin in a plant cyclic nucleotide-gated channel. Plant Cell Physiol. 54, 573–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Findeisen F., Rumpf C. H., and Minor D. L. Jr. (2013) Apo states of calmodulin and CaBP1 control CaV1 voltage-gated calcium channel function through direct competition for the IQ domain. J. Mol. Biol. 425, 3217–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lu Q., Li J., Ye F., and Zhang M. (2015) Structure of myosin-1c tail bound to calmodulin provides insights into calcium-mediated conformational coupling. Nat. Struct. Mol. Biol. 22, 81–88 [DOI] [PubMed] [Google Scholar]
- 50. Wang J., Peng Q., Lin Q., Childress C., Carey D., and Yang W. (2010) Calcium activates Nedd4 E3 ubiquitin ligases by releasing the C2 domain-mediated auto-inhibition. J. Biol. Chem. 285, 12279–12288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shirihai O. S., Song M., and Dorn G. W. 2nd (2015) How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 116, 1835–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu S., Cherok E., Das S., Li S., Roelofs B. A., Ge S. X., Polster B. M., Boyman L., Lederer W. J., Wang C., and Karbowski M. (2016) Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol. Biol. Cell 27, 349–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Franz A., Kevei É., and Hoppe T. (2015) Double-edged alliance: mitochondrial surveillance by the UPS and autophagy. Curr. Opin. Cell Biol. 37, 18–27 [DOI] [PubMed] [Google Scholar]
- 54. Goellner E. M., Grimme B., Brown A. R., Lin Y. C., Wang X. H., Sugrue K. F., Mitchell L., Trivedi R. N., Tang J. B., and Sobol R. W. (2011) Overcoming temozolomide resistance in glioblastoma via dual inhibition of NAD+ biosynthesis and base excision repair. Cancer Res. 71, 2308–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Trivedi R. N., Wang X. H., Jelezcova E., Goellner E. M., Tang J. B., and Sobol R. W. (2008) Human methyl purine DNA glycosylase and DNA polymerase β expression collectively predict sensitivity to temozolomide. Mol. Pharmacol. 74, 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fouquerel E., Goellner E. M., Yu Z., Gagné J. P., Barbi de Moura M., Feinstein T., Wheeler D., Redpath P., Li J., Romero G., Migaud M., Van Houten B., Poirier G. G., and Sobol R. W. (2014) ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 8, 1819–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fang Q., Inanc B., Schamus S., Wang X. H., Wei L., Brown A. R., Svilar D., Sugrue K. F., Goellner E. M., Zeng X., Yates N. A., Lan L., Vens C., and Sobol R. W. (2014) HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase β. Nat. Commun. 5, 5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tang J. B., Goellner E. M., Wang X. H., Trivedi R. N., St Croix C. M., Jelezcova E., Svilar D., Brown A. R., and Sobol R. W. (2010) Bioenergetic metabolites regulate base excision repair-dependent cell death in response to DNA damage. Mol. Cancer Res. 8, 67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Miedel M. T., Zeng X., Yates N. A., Silverman G. A., and Luke C. J. (2014) Isolation of serpin-interacting proteins in C. elegans using protein affinity purification. Methods 68, 536–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wiener M. C., Sachs J. R., Deyanova E. G., and Yates N. A. (2004) Differential mass spectrometry: a label-free LC-MS method for finding significant differences in complex peptide and protein mixtures. Anal. Chem. 76, 6085–6096 [DOI] [PubMed] [Google Scholar]
- 61. Paweletz C. P., Wiener M. C., Bondarenko A. Y., Yates N. A., Song Q., Liaw A., Lee A. Y., Hunt B. T., Henle E. S., Meng F., Sleph H. F., Holahan M., Sankaranarayanan S., Simon A. J., Settlage R. E., et al. (2010) Application of an end-to-end biomarker discovery platform to identify target engagement markers in cerebrospinal fluid by high resolution differential mass spectrometry. J. Proteome Res. 9, 1392–1401 [DOI] [PubMed] [Google Scholar]
- 62. Eng J. K., Jahan T. A., and Hoopmann M. R. (2013) Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24 [DOI] [PubMed] [Google Scholar]
- 63. Catanzariti A. M., Soboleva T. A., Jans D. A., Board P. G., and Baker R. T. (2004) An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 13, 1331–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]