

Abstract
MsrPQ is a newly identified methionine sulfoxide reductase system found in bacteria, which appears to be specifically involved in the repair of periplasmic proteins oxidized by hypochlorous acid. It involves two proteins: a periplasmic one, MsrP, previously named YedY, carrying out the Msr activity, and MsrQ, an integral b-type heme membrane-spanning protein, which acts as the specific electron donor to MsrP. MsrQ, previously named YedZ, was mainly characterized by bioinformatics as a member of the FRD superfamily of heme-containing membrane proteins, which include the NADPH oxidase proteins (NOX/DUOX). Here we report a detailed biochemical characterization of the MsrQ protein from Escherichia coli. We optimized conditions for the overexpression and membrane solubilization of an MsrQ-GFP fusion and set up a purification scheme allowing the production of pure MsrQ. Combining UV-visible spectroscopy, heme quantification, and site-directed mutagenesis of histidine residues, we demonstrated that MsrQ is able to bind two b-type hemes through the histidine residues conserved between the MsrQ and NOX protein families. In addition, we identify the E. coli flavin reductase Fre, which is related to the dehydrogenase domain of eukaryotic NOX enzymes, as an efficient cytosolic electron donor to the MsrQ heme moieties. Cross-linking experiments as well as surface Plasmon resonance showed that Fre interacts with MsrQ to form a specific complex. Taken together, these data support the identification of the first prokaryotic two-component protein system related to the eukaryotic NOX family and involved in the reduction of periplasmic oxidized proteins.
Keywords: electron transfer, Escherichia coli (E. coli), flavin mononucleotide (FMN), heme, membrane protein, NADPH oxidase, YedZ, flavin reductase Fre, methionine sulfide reductase MsrPQ
Introduction
The repair of oxidative damage to proteins associated with the formation of methionine sulfoxide is an important issue for living organisms. This is possible thanks to the presence of the ubiquitous methionine sulfoxide reductase enzymes (Msr), which catalyze the reduction of the methionine sulfoxide into the original methionine (1). Very recently, a new type of Msr has been discovered in Gram-negative bacteria (2, 3). This system appears to be specifically involved in the repair of periplasmic proteins oxidized by hypochlorous acid HOCl. HOCl is generated in particular within phagocytic cells of the mammalian innate immune system to kill internalized pathogens. It is a powerful antimicrobial associated with its capability to oxidize methionine residues into methionine sulfoxide, leading to protein misfolding, loss of their cellular function, and aggregation (4).
This new methionine sulfoxide reductase system, named MsrPQ, involves two proteins encoded in the same operon (2, 3). MsrP, which carries out the Msr activity, is a periplasmic soluble protein with a molybdenum atom in its active site. It was previously named YedY (5) until its Msr activity was discovered. To be functional in vivo, MsrP has to be specifically associated with MsrQ, an integral b-type heme containing membrane-spanning protein, previously named YedZ (6). MsrQ was proposed to be the specific electron donor for MsrP, allowing catalysis of methionine sulfoxide reduction (3).
Detailed functional characterization of this new Msr system is an important issue to better understand its involvement in the bacterial defense against HOCl stress. This system could play an important role in some pathogenic bacteria to escape the mammalian innate immune system and to cope with the oxidative burst and HOCl killing. Whereas MsrP (YedY) has been structurally well characterized (5), showing similarities with the sulfite oxidase molybdenum-enzyme family (7–9), biochemical studies on MsrQ (YedZ) are much more scarce (10). Long before the identification of its physiological function, bioinformatic studies recognized MsrQ as an archetypal domain present in an important superfamily of heme-containing membrane proteins initially designated as the YedZ family (11). More recently, this domain, present in bacterial and eukaryotic representatives, has been renamed as the FRD domain (for ferric reductase domain) (12), based on its identification within STEAP (six-transmembrane epithelial antigen of the prostate) and FRE (yeast ferric reductases) proteins, which are both involved in iron metabolism (13–15). Furthermore, the FRD domain is also present within the NADPH oxidase (NOX/DUOX) family of transmembrane enzymes involved in the production of reactive oxygen species (ROS)3 (16).
The archetype of this FRD membrane domain, with structural similarity to cytochrome b of the mitochondrial bc1 complex, consists of six transmembrane segments (TM) with two bis-histidyl motifs in TM3 and TM5, allowing the chelation of two b-type hemes. Phylogenetic analyses suggested that among the FRD family, MsrQ proteins form a specific group together with the eukaryotic ferric reductases of the STEAP family on one side and the FRE and NOX family on the other side (16). In particular, differences with the NOX and FRE family can be seen within the number of predicted histidine residues involved in heme binding (Fig. 1). In the MsrQ/STEAP family, one of these histidines was replaced by an arginine residue. In addition, whereas the STEAP proteins possess two histidyl residues, MsrQ presents three histidines among the four (Fig. 1). These data suggested that the STEAP/MsrQ proteins might lack one of the two b-type hemes present in the NOX and FRE enzymes (17).
FIGURE 1.

Structural relationships between MsrQ (formerly YedZ), STEAP, and NOX/DUOX protein families. A, domain cluster prediction in the primary sequence of proteins belonging to the MsrQ, STEAP, and NOX/DUOX families (red, green, and blue bar on the left, respectively). The FRD/MsrQ domain is highlighted, and TM numbering refers to position of the TM within this domain; when applicable, an additional TM is not numbered. Histidine residues (H), localized in the third (TM III) and fifth (TM V) FRD transmembrane segments and conserved among the members of these protein families, are framed by an orange rectangle. The distance separating two histidine residues inside a transmembrane segment is indicated below. The N-terminal dehydrogenase domain in STEAP 2-3 is homologous to the FNO-like domain (Pfam accession number PF00420). The peroxidase-like and EF-hand domains in Duox correspond to the Pfam domains PF03098 (peroxidase-like) and PF0036 (EF-hands). The C-terminal dehydrogenase domain common to NOX, DUOX, and FRE proteins corresponds to the FNR-like domain (PF00175). B, partial sequence alignment of members of the MsrQ(YedZ) family (red line to the left of the alignment), STEAP family (green line), and NOX family (blue line), adapted from Ref. 16. The amino acids are colored according to average BLOSUM62 score (correlated to amino acid conservation) in each alignment column: bright blue, >3; purple, between 3 and 1.5; bright green, between 1.5 and 0.5. The transmembrane helix positions were predicted with the TMHMM prediction server.
MsrQ is also remarkable in that it contains only the transmembrane domain, without any additional cytosolic domains (Fig. 1). In fact, NOX, FRE, and STEAP proteins contain additional dehydrogenase cytosolic domain(s), allowing NADPH and flavin binding (12). The presence of such cofactor sites permits efficient electron transfer from NADPH to the membrane b-type hemes, which, for NOX, leads to the catalysis of oxygen reduction and ROS formation and, for ferric reductase, to the reduction of ferric complexes required for iron assimilation. In the case of MsrQ, the membrane reduced quinone pool was proposed to be the source of electrons for the MsrQ heme reduction (3). Nevertheless, taking into account the efficiency of the dehydrogenase systems to reduce the membrane b-type hemes in the FRD family, the existence of soluble cytosolic dehydrogenase protein functioning as MsrQ electron donor might be envisioned. However, for MsrQ, the dehydrogenase would not be an associated cytosolic domain but a separate soluble partner.
In this work, we have optimized overproduction, solubilization, and purification of MsrQ. From this new preparation, we have shown that MsrQ contains two b-type hemes, with UV-visible spectra identical to those of NOX proteins. The presence of the two b-type hemes was also supported by mutations of the conserved histidine heme ligands within TM3 and TM5, which led to the loss of one or two b-type hemes. In addition, we have identified Fre,4 a cytosolic NAD(P)H flavin reductase, as a potential soluble physiological dehydrogenase partner for MsrQ in Escherichia coli. Altogether, these data suggest that the Fre and MsrQ proteins form a prokaryotic two-component system for electrons transfer through membrane, which could be structurally related to eukaryotic NADPH oxidase enzymes of the NOX family.
Results
MsrQ Overexpression, Solubilization, and Purification
The optimization of MsrQ production in E. coli was performed following a standardized protocol, which has allowed the overproduction of many E. coli MG1655 membrane proteins fused to GFP (18). Using a MsrQ-GFP fusion gene cloned into the pET28 vector, 72 different expression conditions, involving the use of seven E. coli strains, three culture media, and three induction temperatures, were screened simultaneously. For each of these conditions, the GFP fluorescence was measured on the whole bacterial cell population to assess the MsrQ-GFP production level. As shown in Fig. 2A, the E. coli BL21 (DE3) strain exhibited the highest MsrQ-GFP production level after an overnight induction at 20 °C in a rich culture medium (Terrific Broth, TB). These conditions were selected for MsrQ-GFP production. Cytoplasmic membranes prepared after induction were subsequently solubilized using six different detergents. The soluble and insoluble fractions were recovered and analyzed by immunoblotting, using anti-His tag antibody, to detect the presence of MsrQ-GFP fusion protein (Fig. 2B). DDM was the only detergent able to fully solubilize MsrQ-GFP protein. Whereas detergents belonging to the Fos-choline family were totally ineffective, other detergents, such as LDAO or β-OG, only ensured a partial extraction of the protein from the membrane (Fig. 2B).
FIGURE 2.

Expression and purification of MsrQ-GFP and MsrQ proteins. A, optimized conditions for MsrQ-GFP expression were screened as indicated under “Experimental Procedures.” GFP fluorescence emission was measured at 510 nm with an excitation wavelength of 485 nm. B, identification of the optimal MsrQ-GFP solubilization conditions. Membranes expressing the MsrQ-GFP protein were solubilized with six different detergents (DDM, β-OG, LDAO, Fc12, Fc14, and Fc16) as indicated under “Experimental Procedures.” After solubilization, the presence of MsrQ-GFP in the soluble (S) or insoluble (P) fractions was analyzed by Western blotting using an antibody raised against the C-terminal histidine tag. C and D, MsrQ-GFP fusion construct (C) and SDS-PAGE analysis (D) of the MsrQ purification. Lane 1, MsrQ-GFP Ni-NTA elution; lane 2, MsrQ Ni-NTA flow-through after TEV cleavage; lane 3, MsrQ after Mono-Q purification step.
MsrQ-GFP was purified from membranes solubilized in DDM and loaded onto an Ni-NTA-agarose column (Fig. 2, C and D). 6.5 mg of pure MsrQ-GFP protein per liter of culture were obtained, a yield 2.5 times higher than that reported previously (18). Following an overnight incubation with TEV protease, the MsrQ protein without the GFP domain was recovered in the flow-through of a second Ni-NTA-agarose column. A subsequent purification through an anion exchange chromatography led to a yield of 1.5 mg of pure MsrQ protein/liter of culture (Fig. 2D).
UV-visible Characterization
UV-visible spectroscopy showed that the as-isolated oxidized MsrQ-GFP protein exhibited two intense absorption peaks at 412 and 490 nm, characteristics of oxidized b-type heme and a GFP chromophore, respectively (Fig. 3A). Accordingly, the purified oxidized MsrQ protein, obtained after removal of the GFP part of the fusion, still exhibited the absorption peak centered at 412 nm, and now peaks at 534 and 562 nm were clearly visible (Fig. 3A). Following dithionite reduction, reduced minus oxidized difference spectra of both MsrQ-GFP and MsrQ proteins were found to be identical, revealing three new absorption peaks at 558, 530, and 426 nm corresponding to the α, β, and Soret heme bands, respectively (Fig. 3B). The GFP contribution was abrogated in these difference spectra. This absorption spectrum, identical to that of reduced human neutrophil flavocytochrome b558 (19), is characteristic of b-type hemoproteins in their reduced states. Using the MsrQ heme content reported in Table 1, the extinction coefficient value for the oxidized MsrQ form at 412 nm was calculated to be 51,950 m−1 cm−1. For the reduced MsrQ form at 426 and 558 nm, the extinction coefficient values were found to be 69,015 m−1 cm−1 and 11,075 m−1 cm−1, respectively.
FIGURE 3.

UV-visible spectral characterization of WT and mutated MsrQ proteins in 50 mm Tris/HCl, pH 7.6, 0.016%. A, oxidized MsrQ-GFP (dashed line) and MsrQ (solid line) proteins at 6.1 μm. B, reduced minus oxidized difference spectra of the MsrQ protein (7 μm), measured between 380 and 600 nm. The protein was reduced by the addition of sodium dithionite. Arrows, absorption peaks and shoulders. C, oxidized MsrQ-GFP; wild type (■) and single mutant H151A (●), H164A (▾), and H91A (▴) proteins. Protein concentration was normalized using the GFP absorption peak at 490 nm. The total MsrQ heme content on each protein was determined from the absorbance at 412 nm, corrected from the residual absorption of GFP at this wavelength. Heme content was as follows: wild type, 100%; H151A, 57%; H164A, 37%; H91A, 7%.
TABLE 1.
Quantification of MsrQ heme/protein ratio
Heme content was determined by the pyridine hemochromogen assay. MsrQ concentration was calculated using amino acid analysis or infrared spectroscopy.
| MsrQ concentration | Heme concentration | [Heme]/[MsrQ] ratio | |
|---|---|---|---|
| μm | μm | ||
| Amino acid analysis | 7.9 ± 0.4 | 19.8 ± 1.4 | 2.5 ± 0.3 |
| Infrared spectroscopy | 9.5 ± 0.6 | 19.8 ± 1.4 | 2.1 ± 0.3 |
Heme Stoichiometry Determination
To quantify the MsrQ heme content, a purified MsrQ sample was divided into two parts. The first part of the sample was used for the quantification of heme using the pyridine hemochromogen assay (20). The heme concentration was found to be 19.8 ± 1.4 μm (Table 1). The second part of the sample was used to determine the MsrQ protein concentration by two different techniques. First, quantitative amino acid analysis was carried out. A protein concentration of 7.9 ± 0.4 μm was determined, leading to the calculation of a heme/protein ratio of 2.5 ± 0.3 (Table 1). Second, the protein concentration was determined by infrared spectroscopy using the amide I band corresponding to the carbonyl vibration of the peptide bonds (21). The heme/MsrQ protein ratio quantified by this approach was found to be 2.1 ± 0.3 (Table 1). These data show that MsrQ contains two b-type hemes per polypeptide chain.
Histidine Coordination
In the human neutrophil flavocytochrome b558, four histidine residues located in the third and fifth transmembrane domains are involved in the coordination of two b-type hemes (22). Among these histidines, three were remarkably conserved in the MsrQ primary sequence and localized in the third (His-91) and fifth (His-151 and His-164) transmembrane helices (Fig. 1B) (16). As suggested by the topological representation of MsrQ, histidines 91 and 164 could be considered as a first heme-binding site facing the periplasmic compartment, whereas the histidine 151 could be part of a second binding site on the cytoplasmic side.
To address the role of histidines 91, 151, and 164 in heme binding, histidine mutants of MsrQ-GFP protein were designed. The impact of these mutations on heme content was analyzed by UV-visible spectroscopy on the purified proteins (Fig. 3C). The protein concentration of each mutant was normalized using the GFP absorption peak at 490 nm, and the heme content was calculated from the absorbance value at 412 nm, corrected from the absorbance contribution of GFP at this wavelength. As illustrated in Fig. 3C, about half (57%) of the total MsrQ heme content remained present in the H151A mutant protein. These data are in agreement with the involvement of His-151 in the coordination of one of the two MsrQ hemes. Furthermore, whereas the H91A single mutation led to the almost total loss (93%) of the two hemes, the H164A single mutation induced the loss of 63% of the total MsrQ heme content (Fig. 3C). This suggests that His-164 is involved in the coordination of one of the two hemes of MsrQ. Together with the above mentioned topological representation of MsrQ (Fig. 1A), these results suggest that one heme-binding site involves His-164 and most likely His-91, similarly to that reported for NOX enzyme (Fig. 1A) (22). The loss of the two hemes in H91A mutant suggests that His-91 could play an additional role in the global stabilization of the second b-type heme cofactor present in MsrQ. The data also indicate that His-151 is involved in the coordination of the second heme. However, in this case, the second canonical heme binding histidine is missing in the third transmembrane helix (Fig. 1B), suggesting an atypical coordination for the second heme present in MsrQ.
Search for MsrQ Cytosolic Electron Donors
Previous bioinformatics analyses suggested an evolutionary relationship between MsrQ and the FRE-NOX protein families (11, 16). We hypothesized that an E. coli cytosolic NADPH-dependent flavoprotein related to the cytosolic dehydrogenase domain of FRE-NOX enzymes could be the source of the electrons for MsrQ heme reduction. This FRE-NOX cytosolic dehydrogenase domain is homologous to the ferredoxin-NADP+ reductase family (FNR), which bears NADPH and flavin binding sites (23). To identify such E. coli proteins, a 3D screening of the currently available protein structures in the PDB was performed with the DALI and PDBeFold servers using the spinach FNR structure as a template (the spinach FNR, first structure solved, was used because it historically defined this family). The flavin reductase Fre4 (24) and the flavodoxin NADPH reductase Fpr (25) were found to be the two E. coli flavin-dependent enzymes with the highest similarity with the spinach FNR (26). Fre and Fpr are both NAD(P)H flavin-dependent oxidoreductases, which catalyze the transfer of electrons between NADPH and their cellular targets via a flavin, tightly bound (Fpr) or not bound (Fre) to the polypeptide chain. Thus, Fpr is a flavoprotein (25), whereas Fre uses free flavins as substrates (24). Among these two, the physiological role of Fre has never been clearly stated or identified, whereas Fpr is well known for its involvement in glycyl radical generation within pyruvate formate lyase and anaerobic ribonucleotide reductase (25, 27). Thus, we focused our investigation on Fre to test whether it could be a potential dehydrogenase partner for MsrQ.
The ability of Fre to catalyze MsrQ heme reduction was investigated under anaerobiosis, in the presence of NADPH and free FMN. Reduced minus oxidized difference spectra recorded in anaerobic conditions showed that the three absorption peaks at 426, 530, and 558 nm characteristic of b-type heme in a reduced state appeared following incubation of MsrQ with the Fre system (Fig. 4A). No reduction of MsrQ was observed in the absence of Fre, with NADPH alone, or with NADPH and FMN (Fig. 4A). These data indicate that MsrQ is reduced thanks to the free reduced FMN generated by the Fre activity. Full MsrQ heme reduction was observed after a reaction time proportional to the amount of Fre added into the reaction cuvette (Fig. 4B). From the linear part of the MsrQ reduction kinetics recorded at 558 nm, a specific activity of 2,570 ± 110 nmol of MsrQ reduced by min and by mg of Fre was calculated (Fig. 4B). It should be noted that this value was obtained in the presence of 250 μm NADPH and 30 μm FMN, corresponding to the conditions for Fre maximum velocity (24, 28). In the absence of MsrQ, the Fre NADPH-FMN reductase specific activity, measured in the same conditions, was determined to be 2,250 ± 50 nmol of NADPH oxidized by min and by mg of Fre (not shown). These data show that Fre specific activity for MsrQ reduction is almost identical to that of FMN reduction. These results demonstrate that Fre is able to efficiently catalyze the electron transfer from NADPH to MsrQ.
FIGURE 4.

Reduction of MsrQ by the cytosolic flavin oxidoreductase Fre from E. coli. The experiments were carried out at 20 °C under anaerobiosis, in 50 mm Tris/HCl buffer, pH 7.8, 0.016% DDM, 120-μl final volume. A, full reduction of MsrQ (7.4 μm) after a 30-min incubation in the presence of 0.035 μg of Fre, 250 μm NADPH, and 30 μm FMN (▴). MsrQ reduction was evaluated from the appearance of the absorbance bands at 426 and 558 nm in the reduced minus oxidized difference spectra (absorbance spectrum of MsrQ recorded after incubation in the presence of NADPH, FMN, and Fre minus that recorded in the presence of MsrQ alone). Control experiments were recorded in the same conditions but in the absence of Fre (with NADPH and FMN) (●), or in the absence of Fre and FMN (NADPH alone) (■). The broad absorption band centered at 445 nm (●) corresponds to a spectrum of oxidized FMN at a concentration of 30 μm. Note that NADPH, NADP+, and reduced FMN do not exhibit notable absorption bands in the 400–600 nm region. B, selected kinetic traces of MsrQ reduction by Fre, followed by the increase of the absorbance at 558 nm, in the presence of 30 μm FMN, 250 μm NADPH, and various amounts of Fre. The spectrophotometric cuvette contained 7.4 μm oxidized MsrQ, and the reaction was initiated by adding, from the left to the right, 0.107, 0.067, and 0.026 μg of Fre. The activities were determined from the slope of the linear part of the kinetic traces (dashed lines). The activity measurements were realized in triplicate. A specific activity of 2,570 ± 110 nmol of MsrQ reduced by min and by mg of Fre was calculated.
Interaction between Fre and MsrQ
The formation of a specific complex between Fre and MsrQ, which could play some role in the heme reduction process, was studied. At first, this has been investigated by cross-linking reactions performed in vitro using ethylene glycol-bis-(succinimidylsuccinate) (EGS) cross-linking reagent (Fig. 5A). SDS-PAGE analysis shows that EGS treatment triggers a 45 kDa additional band, the intensity of which increases in a Fre concentration-dependent manner in the presence of MsrQ. The 45 kDa molecular mass perfectly fits the theoretical weight expected for Fre-MsrQ cross-linked complex, showing a specific interaction between Fre and MsrQ. In contrast, no cross-link was observed when Fre or MsrQ was incubated with a control protein, such as lysozyme (Fig. 5A) or BSA (not shown). The specificity of this interaction was further investigated by testing the effect of increasing NaCl concentration in the reaction medium (Fig. 5B). Under such conditions, the Fre-MsrQ complex is maintained fairly stable, suggesting hydrophobic interactions between the two partners. Interestingly, no cross-linked complex could be seen with Fre when using the fusion protein MsrQ-GFP (supplemental Fig. S1). Thus, GFP fused on the cytosolic side of MsrQ hampers complex formation with Fre. These data demonstrate that the observed Fre-MsrQ complex is oriented on the cytosolic side, as expected from a physiological point of view.
FIGURE 5.
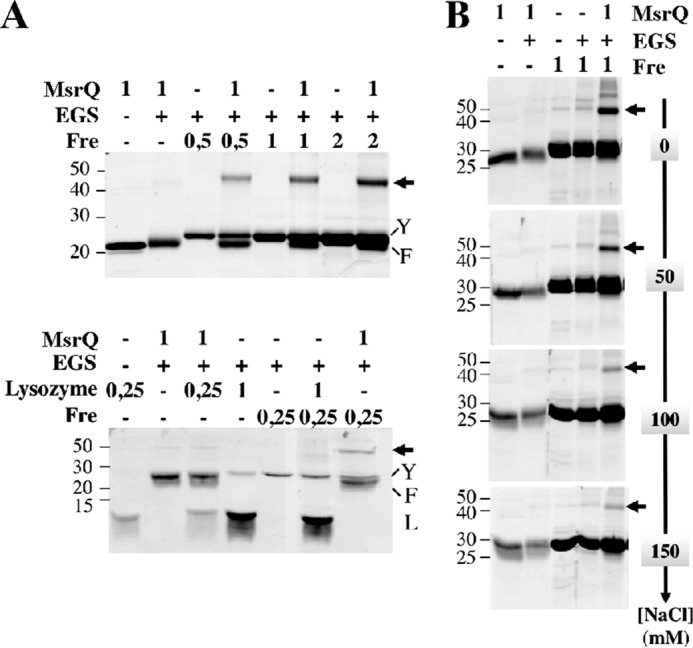
Interaction analysis between Fre and MsrQ. A, purified proteins MsrQ (10 μl at 1 mg/ml), lysozyme (10 μl at 0.25 or 1 mg/ml), and Fre (10 μl at 0.5, 1, or 2 mg/ml) were first preincubated for 1 h, alone or together, without the cross-linking reagent. As indicated above the gels, the cross-linking reactions were then started, by adding (+) or not (−) 0.5 mm EGS. After a 20-min incubation, the reactions were immediately subjected to SDS-PAGE analysis. B, effect of sodium chloride addition (0–150 mm) during the cross-linking reactions.
The Fre-MsrQ interaction was further analyzed using surface plasmon resonance (Fig. 6). The results showed a specific interaction between Fre and MsrQ proteins in a concentration-dependent manner. The specificity of the interaction between MsrQ and Fre functionalized surface was confirmed at 50 and 150 mm NaCl concentration (Fig. 6 and supplemental Fig. S2A) as well as the use of BSA-functionalized surface as a negative control (supplemental Fig. S2B). Using a steady state affinity model for analysis, apparent Kd values of MsrQ for Fre were determined to be 15 ± 4 and 18 ± 3 μm, in the presence of 150 and 50 mm NaCl, respectively. These data reflect the fact that Fre could be a physiological reducing system of MsrQ.
FIGURE 6.
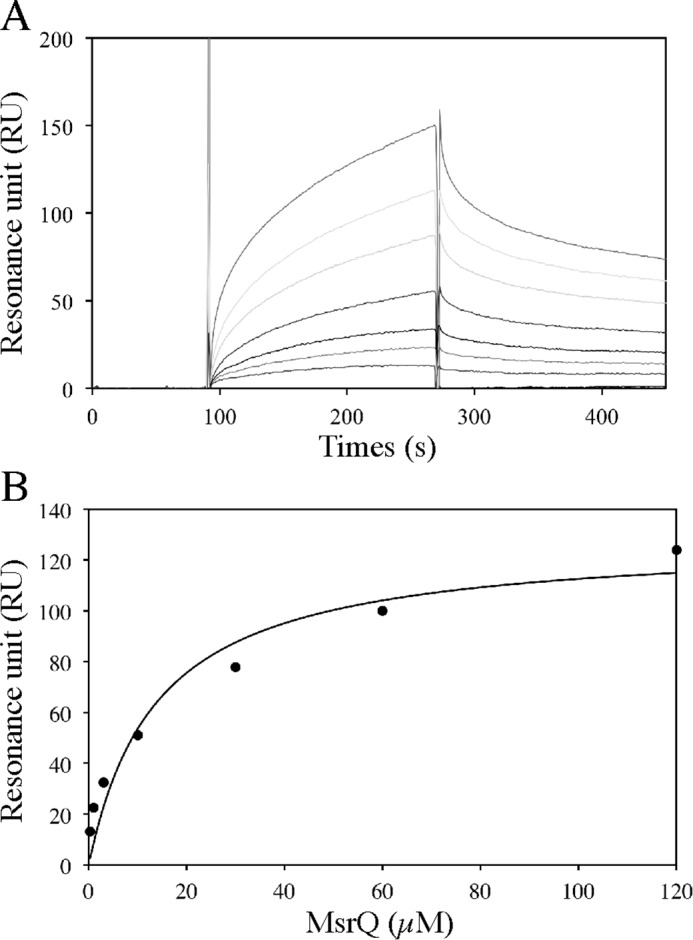
Surface plasmon resonance analysis of MsrQ binding to immobilized E. coli flavin reductase Fre. A, the purified MsrQ protein (60 μl) was injected over an Fre functionalized surface at a flow rate of 20 μl/min in the running buffer: 50 mm Tris-HCl, pH 8.0, 0.016% DDM, 150 mm NaCl. Additional correction for the bulk effect due to the excess of detergent associated with MsrQ was performed in the association phase. The sensorgram was obtained for a range of MsrQ concentration at 120, 60, 30, 10, 3, 1, and 0.3 μm (from top to bottom). This surface plasmon resonance analysis was done two times with two freshly functionalized surfaces. B, steady state analysis, from the data set presented above, of the MsrQ/Fre interaction, assuming a stoichiometric ratio of 1.
Discussion
Although MsrQ (YedZ) was identified many years ago as an integral transmembrane-spanning hemoprotein (6), its cellular function has been only very recently identified (2, 3). MsrQ belongs to an enzymatic system involving MsrP (YedY), a new type of methionine sulfoxide reductase, which repairs periplasmic methionine sulfoxide-containing proteins. In this system, MsrQ, thanks to its heminic cofactor(s), acts as the MsrP-specific electron donor for methionine sulfoxide reduction (3).
In this work, we have carried out a detailed characterization of MsrQ and reinvestigated its heme content. We also showed that the flavin reductase Fre, in addition to the previously characterized membrane-bound quinone pool (3), could act as an efficient cytosolic electron donor for MsrQ.
Bioinformatics studies showed that MsrQ is related to the FRE/NOX family, with a similar b-type heme transmembrane domain (11, 16). However, differences can be seen within the number of predicted histidine residues that could be involved in heme binding. Indeed, whereas FRE/NOXs possess two bis-histidyl motifs in TM3 and TM5 required to bind two b-type hemes, one histidyl in TM3 is missing in MsrQ, suggesting the presence of only one b-type heme in MsrQ.
Here, taking advantage of an MsrQ-GFP fusion strategy previously designed for a systematic study on E. coli membrane proteins (18), we have optimized conditions for MsrQ-GFP fusion overproduction and established a purification scheme allowing a production of MsrQ compatible with quantitative biochemical characterizations. Our present work, carried out on pure MsrQ preparations, which allowed an accurate determination of the protein concentration, demonstrated the presence of two b-type hemes per MsrQ polypeptide. This result clearly differs from previous studies, which have reported the presence of only one heme per MsrQ (YedZ) protein (18). This discrepancy regarding heme/protein ratio may come from a heme depletion during purification. Indeed, it was observed here that in the last purification step of MsrQ, some fractions of MsrQ were apo-forms (data not shown).
We further showed that single mutation of histidines at positions 164 and 151 led to the loss of only one of the two MsrQ hemes. Thus, in agreement with the topological representation of MsrQ (Fig. 1), our results suggest that His-91 and His-164 are directly involved in the coordination of the first b-type heme, whereas His-151 is involved in the coordination of the second b-type heme. However, because a second histidyl ligand associated with His-151 to coordinate this heme is missing and replaced by a well conserved arginine residue (Arg-77), the question of its involvement as the second heme ligand might be considered. In fact, because of the strong basicity of its guanidinium side chain, arginine has been not clearly observed as an iron ligand in metalloproteins. It could be possible that in addition to the His-151, a second histidine ligand may come from another neighboring helix, as reported previously in E. coli formate dehydrogenase, which contains two b-type hemes in a four-TM subunit (29). Further studies are needed to characterize this second heme ligand in MsrQ.
In any case, these data suggest that the two b-type hemes present in MsrQ can be at least differentiated based on MsrQ topology. As shown by the MsrQ (YedZ)-PhoA/GFP analysis (6), the first heme, with the canonical heme binding histidines (His-91 and His-164), is expected to face the periplasmic compartment, whereas the second heme, with the atypical coordination, would be located on the cytosolic side of MsrQ.
Recently, from genetic experiments, it has been proposed that MsrQ gets electrons from the membrane-bound quinones to reduce MsrP (3). Nevertheless, with such a two-heme topology spanning the membrane, MsrQ seems also well designed to receive electrons from redox-soluble cytosolic component(s) and transfer them across the membrane to the molybdenum cofactor of MsrP located in the periplasmic space. Indeed, we showed here that MsrQ can be efficiently reduced by the cytosolic NAD(P)H:flavin reductase Fre in the presence of free FMN and NADPH. The flavin reductase Fre uses flavins as substrate, which are further released from its active site after their reduction by NAD(P)H (24). We showed that the rate of MsrQ reduction by Fre in the presence of FMN and NADPH is identical to that of FMN reduction by Fre in the presence of NADPH. Because the oxidation of one molecule of NADPH involves two electrons and the reduction of MsrQ involves two electrons (two b-type hemes per MsrQ molecule), these data strongly suggest that all of the FMN molecules reduced by Fre are fully oxidized by MsrQ. Thus, the reduction of MsrQ by the Fre system appears to be solely limited by Fre turnover, and no free reduced FMN is likely to accumulate during the reaction, because it is promptly oxidized by MsrQ.
In vivo, such a property could be strongly beneficial, because the accumulation of free reduced flavins in the cell is deleterious (30). In fact, the instable and highly reactive free reduced flavins can reduce molecular oxygen to form toxic ROS species and could also carry out other unwanted reduction processes of cell components. Such an issue has been well described in the cases of the two-component monooxygenase systems, which involve, as distinct proteins, a flavin reductase that generates a reduced flavin and a monooxygenase that utilizes the reduced flavin as a substrate for monooxygenation (30). For these two-component enzymes, an optimal reduced flavin transfer between the flavin reductase and the monooxygenase has been shown to be one of the key points in their catalytic properties (30).
Furthermore, cross-linking experiments and surface plasmon resonance studies carried out in this work showed that Fre and MsrQ form a specific complex with a rather low Kd value of 15 μm. Such interaction could also contribute to the efficient and specific transfer of the reduced flavin between Fre and MsrQ, because this has been reported in the case of some two-component monooxygenase enzymes (30).
Altogether, these results suggest that Fre could be an efficient electron donor to MsrQ in the cell and could represent an alternative electron source to MsrQ compared with the membrane-bound quinones (3). In particular, it could be hypothesized that the Fre MsrQ-reductase activity might play an essential role in conditions where the formation of the reduced quinone pool is deficient (e.g. when the E. coli respiratory chain is inactivated due to stress conditions). These could include the presence of reactive chlorine species against which the MsrP/Q system is supposed to act. Further studies are clearly required to better specify the physiological role of Fre for the reduction of methionine sulfoxide associated with hypochlorite damage.
Finally, these data provide a new perspective on the previously underlined phylogenetic relationship between MsrQ, STEAP, and NOX/FRE proteins (12). As illustrated in Fig. 7, when compared with MsrQ, both the STEAP and NOX membrane-bound protein families display a soluble dehydrogenase domain on their cytosolic side, fused to their N or C terminus, respectively. The identification of Fre as a potential dehydrogenase partner for MsrQ suggests the existence of a two-component system with homologies to the NOX/FRE. Indeed, strong similarity between the known structural part of Nox2 dehydrogenase domain and Fre does exist (supplemental Fig. S3). Thus, the identification of the MsrQ/Fre two-component system is a strong support to Sumimoto's (31) hypothesis of a prokaryotic origin for the eukaryotic NOX enzyme, resulting from the fusion of a membrane cytochrome b and a FNR-like dehydrogenase domain. The MsrQ/Fre two-component system might be viewed as a prokaryotic intermediate prefiguring the fused NOX enzymes.
FIGURE 7.

Topological comparison between the MsrQ/Fre two-component system and the eukaryotic NOX/DUOX and STEAP one-component systems. The membrane-bound FRD domains are shown in dark blue. The soluble domains, FNR (yellow), FNO-like (green), EF-hand (purple), and peroxidase-like (dark green), are represented using the same colors as those used in Fig. 1A. The electron acceptors for each system are shown in the corresponding periplasmic/extracellular compartment.
Experimental Procedures
Chemicals and Reagents
Detergents were obtained from Anatrace. All other chemicals were from Sigma-Aldrich. The recombinant flavin reductase Fre from E. coli was overexpressed and purified as described previously (24).
Expression of Recombinant MsrQ(YedZ)-GFP Protein
The pET28-YedZ-GFP construct was kindly provided by Jan Willem de Gier (18). In this construct, the yedZ gene comes from the E. coli strain MG1655. Conditions for optimal expression of MsrQ-GFP in E. coli cells were screened using the RoBioMol platform (Institut de Biologie Structurale, Grenoble, France). Seven E. coli BL21 (DE3)-derived strains, transformed with the plasmid pET28-YedZ-GFP (BL21 (DE3), BL21 pLysS (DE3), C41 (DE3), C43 (DE3), RIL (DE3), Rosetta2 (DE3), and RP (DE3)) were tested. Three culture media (TB, LB, and autoinduction), two induction times (4 h and overnight), three induction temperatures (37, 25, and 20 °C), and one concentration of isopropyl 1-thio-β-d-galactopyranoside (0.5 mm) were analyzed concomitantly. Cultures were carried out in 96-well plates at 37 °C, with 200-rpm shaking. After 3 h of growth, cultures were induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside and grown further according to the desired conditions. The production level of MsrQ-GFP was analyzed on whole cells by measuring the fluorescence associated with GFP (excitation 485 nm, emission 510 nm).
Site-directed Mutagenesis
MsrQ-GFP site-directed mutagenesis experiments were performed with the QuikChange mutagenesis kit (Stratagene), using two sets of complementary mutant oligonucleotides. DNA sequences were verified on both strands (Beckman Coulter Genomics).
Membrane Preparation
A bacterial cell pellet (from a 6-liter culture) was resuspended in 50 mm Tris-HCl, pH 8.0, supplemented with one tablet of Complete EDTA-free protease inhibitors (Roche Applied Science). Cells were disrupted twice using a Microfluidizer M-110P (Microfluidics International) at 10,000 p.s.i., and the homogeneous lysate was centrifuged at 8,000 × g for 15 min at 4 °C. The supernatant was further centrifuged at 150,000 × g for 45 min at 4 °C. The membrane fraction, recovered in the pellet, was resuspended in 50 mm Tris-HCl, pH 8.0, homogenized with a Potter homogenizer, and stored at −80 °C until used.
Membrane Solubilization
Solubilization tests of MsrQ-GFP were performed with several detergents using the following concentrations: 19.5 mm DDM, 80 mm β-OG, 21.7 mm LDAO, 4.5 mm Fos-choline-12, 0.36 mm Fos-choline-14, and 0.06 mm Fos-choline-16. For each assay, the membranes containing MsrQ-GFP proteins (60 μl at 16 mg/ml) were solubilized with a detergent solution (60 μl) twice concentrated. After an incubation of 1 h on ice, this suspension was centrifuged for 20 min at 200,000 × g. The solubilized membrane proteins were recovered in the supernatant. The pellet, corresponding to the insoluble membrane fraction, was resuspended in 120 μl of 50 mm Tris-HCl, pH 8.0, buffer. The soluble and insoluble protein fractions were analyzed on SDS-PAGE and electrotransferred onto a nitrocellulose sheet. Immunodetection of MsrQ-GFP was performed using an anti-His-HRP-conjugated antibody and revealed by a colorimetric reaction using Sigma-Fast tablets (Sigma-Aldrich).
MsrQ-GFP Purification
Membrane fractions were diluted to 5 mg/ml in 50 mm Tris-HCl, pH 8.0. Membrane solubilization was carried out by the addition of 1.2 mg of DDM for 1.1 mg of membrane proteins, and the suspension was incubated for 1 h at 4 °C on a rotating wheel. After centrifugation for 30 min at 150,000 × g, the supernatant containing the solubilized proteins was recovered and mixed overnight with 10 ml of Ni-NTA-agarose resin (Qiagen) pre-equilibrated in buffer A (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.016% DDM, 15 mm imidazole). After two extensive washing steps with 50 ml of buffer A and then with 50 ml of buffer B (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.016% DDM, 25 mm imidazole), the MsrQ-GFP protein was eluted using 50 ml of buffer C (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.016% DDM, 250 mm imidazole). After desalting on a PD10 Sephadex G-25 M column (GE Healthcare), the C-terminal GFP domain of the recombinant protein was cleaved off after an overnight incubation at room temperature with the His-tagged TEV protease. Removal of the GFP moiety and the TEV was performed by purification on an Ni-NTA-agarose column. The unbound fraction that contained MsrQ protein was concentrated and run over a Mono-Q column (GE Healthcare) pre-equilibrated in buffer D (50 mm Tris-HCl, pH 8.0, 0.016% DDM). The purified MsrQ protein was recovered in the flow-through and stored at 4 °C until used.
Analytical Methods
Determination of protein concentrations was carried out by quantitative amino acid analysis on three different protein dilutions. The samples were dried and hydrolyzed for 24 h under reduced pressure at 110 °C in constant boiling 6 n HCl containing 1% (w/v) phenol. Amino acid contents were analyzed with a Biochrom 30 amino acid analyzer. An internal standard (norleucine) was added to each sample to determine the amount of the sample recovered during the assay. Alternatively, protein quantification was performed using infrared spectroscopy (Detect Direct, Merck Millipore). Measurements were performed in triplicate according to the manufacturer's instructions. The intensity of the amide I band (between 1,600 and 1,690 cm−1), corresponding to the C=O stretching vibration of the peptide bond, was measured. The sample concentration was determined in reference to a calibration curve of 10 concentration points (0.125–5 mg/ml) of bovine serum albumin prepared in PBS. When not specified, protein concentration was determined by the Bradford method (Bio-Rad).
Heme content was determined by the pyridine hemochromogen assay, as described by Rieske (20).
In Silico Screening
The spinach FNR structure (PDB code 1FNB) was used to carry out a 3D screening using DALI (32) and PDBeFold (33, 34) servers. Around 80,000 entries were examined, and only two structures, Fre (PDB code 1QFJ) and Fpr (PDB code 1FDR), were found to be close enough to FNR in E. coli. The validation of these first hits was assessed with Pfam (35). The sequence alignment of known FNR homologues was performed according to the presence of the NAD and FAD binding domains. In this large collection of protein domain families, 2,546 sequences were screened, and the first two E. coli proteins were Fpr and Fre.
Cross-linking Experiments
Protein cross-linking reactions were performed in 50 mm HEPES buffer, pH 8.0, using EGS as cross-linking reagent. Before starting the reaction, Tris buffer present in the purified protein samples was exchanged on a PD10 column against 50 mm Hepes buffer, pH 8.0, containing 0.016% DDM (MsrQ) or 10% glycerol (Fre, lysozyme, and BSA). Depending on the conditions, the purified proteins (0.25–2 mg/ml) were first incubated without EGS for 1 h on ice in a 30-μl reaction volume. Sodium chloride (50, 100, or 150 mm) was then added to the reaction mixture. The cross-linking reaction was started by the addition of 0.5 mm EGS prepared extemporaneously in dimethyl sulfoxide. After a 20-min incubation at room temperature, reactions were stopped with 1 μl of 1 m Tris-HCl buffer, pH 8.0, and the protein samples were immediately subjected to an SDS-PAGE analysis.
Enzymatic Activity Measurements
The activity measurements were performed in 50 mm Tris-HCl, pH 7.6, 0.016% DDM at 20 °C under anaerobic conditions, using a glove box (Jacomex B210) equipped with an oxymeter (Arelco Arc) and filled with a nitrogen atmosphere containing <2 ppm O2. The glove box was equipped with a UV-visible cell coupled to an Uvikon XL spectrophotometer by optical fibers (Photonetics). Flavin reductase activity was measured from the decrease of the absorbance at 450 nm due to the reduction of free FMN (12.3 mm−1 cm−1) in the presence of NADPH. The spectroscopic cuvette (120-μl final volume) contained 30 μm FMN and 250 μm NADPH. The reaction was initiated by adding 0.02–0.15 μg of Fre. MsrQ heme reduction assays were carried out in a spectrophotometric cuvette (120-μl final volume), containing 30 μm FMN, 250 NADPH, 11 μm MsrQ, and was initiated by adding 0.02–0.15 μg of Fre. Reduced minus oxidized difference spectra were performed to follow the redox state of MsrQ. The kinetics of MsrQ reduction were followed at 558 nm.
Surface Plasmon Resonance (SPR)
SPR experiments were conducted on a Biacore 3000 system using a CM4 sensor chip, functionalized using HBS-P buffer from the manufacturer at a 5-μl/min flow rate. Flow cells (Fc) 1 and 2 were activated as recommended. Fc1 was blocked with 1 m ethanolamine and served as a control surface. Fc2 was functionalized by injecting 40 μl of Fre (60 μg/ml) diluted in sodium acetate (10 mm, pH 5) at 5 μl/min. The remaining activated groups were blocked with 1 m ethanolamine (30 μl). All runs were carried out at 20 °C and at a flow rate of 20 μl/min with buffer D supplemented with 50 or 150 mm NaCl as running buffer. Increasing MsrQ concentrations were injected onto the Fre surface. After each injection, the surface was regenerated by a 10-μl injection of 10 mm NaOH.
Author Contributions
F. F. conceived and coordinated the study, analyzed and interpreted the data throughout the work, and wrote the paper. C. J.-B. and A. P. performed together the experiments throughout the work. A. P. contributed to the writing, and C. J.-B. contributed to the preparation of figures. J.-P. A. performed and analyzed experiments corresponding to Table 1. C. V. provided technical assistance, contributed to the preparation of figures, and improved the paper. I. P.-H. performed additional SPR experiments presented in the supplemental materials. C. C.-T. performed experiments related to Fig. 4. V. N. coordinated and analyzed experiments from Fig. 4 and contributed to the writing of the paper and global analysis of the data. J. D. contributed to the analysis presented in supplemental Fig. S3.
Supplementary Material
Acknowledgments
We thank Jan Willem de Gier for the gift of the pET28-YedZ-GFP plasmid. This work used the MP3, the Robiomol, and the SPR platforms of the Grenoble Instruct Center (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB).
This work was supported by the French Agence Nationale de la Recherche (ANR), under program ANR-2010-BLAN-1536-01 (Prototype NOX). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1–S3.
It should be noted that Fre, the E. coli flavin reductase, and FRE, the ferric reductases from Saccharomyces cerevisiae are different proteins, catalyzing different enzymatic reactions. We discriminate these two proteins by using capital letters, FRE, for ferric reductase and lowercase, Fre, for flavin reductase. This homonymy is even more uncomfortable when considering that Fre from E. coli is homologue to the C-terminal dehydrogenase domain of FRE from S. cerevisiae (as it is with respect to NOX proteins).
- ROS
- reactive oxygen species
- DDM
- dodecyl maltoside
- EGS
- ethylene glycol-bis-(succinimidylsuccinate)
- FNR
- ferredoxin NADP+ reductase
- Fpr
- E. coli ferredoxin NADP+ reductase
- FRE
- yeast ferric reductase
- Fre
- NAD(P)H:flavin reductase from Escherichia coli
- LDAO
- N,N-lauryl-(dimethyl)-amine oxide
- NOX
- NADPH oxidase
- β-OG
- octyl-β-d-glucoside
- TM
- transmembrane segment(s)
- Fc
- flow cell(s)
- Ni-NTA
- nickel-nitrilotriacetic acid
- SPR
- surface plasmon resonance
- PDB
- Protein Data Bank
- TEV
- tobacco etch virus.
References
- 1. Boschi-Muller S., and Branlant G. (2014) Methionine sulfoxide reductase: chemistry, substrate binding, recycling process and oxidase activity. Bioorg. Chem. 57, 222–230 [DOI] [PubMed] [Google Scholar]
- 2. Melnyk R. A., Youngblut M. D., Clark I. C., Carlson H. K., Wetmore K. M., Price M. N., Iavarone A. T., Deutschbauer A. M., Arkin A. P., and Coates J. D. (2015) Novel mechanism for scavenging of hypochlorite involving a periplasmic methionine-rich peptide and methionine sulfoxide reductase. MBio 6, e00233–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gennaris A., Ezraty B., Henry C., Agrebi R., Vergnes A., Oheix E., Bos J., Leverrier P., Espinosa L., Szewczyk J., Vertommen D., Iranzo O., Collet J.-F., and Barras F. (2015) Repairing oxidized proteins in the bacterial envelope using respiratory chain electrons. Nature 528, 409–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gray M. J., Wholey W.-Y., and Jakob U. (2013) Bacterial responses to reactive chlorine species. Annu. Rev. Microbiol. 67, 141–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Loschi L., Brokx S. J., Hills T. L., Zhang G., Bertero M. G., Lovering A. L., Weiner J. H., and Strynadka N. C. J. (2004) Structural and biochemical identification of a novel bacterial oxidoreductase. J. Biol. Chem. 279, 50391–50400 [DOI] [PubMed] [Google Scholar]
- 6. Drew D., Sjöstrand D., Nilsson J., Urbig T., Chin C.-N., de Gier J.-W., and von Heijne G. (2002) Rapid topology mapping of Escherichia coli inner-membrane proteins by prediction and PhoA/GFP fusion analysis. Proc. Natl. Acad. Sci. U.S.A. 99, 2690–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Workun G. J., Moquin K., Rothery R. A., and Weiner J. H. (2008) Evolutionary persistence of the molybdopyranopterin-containing sulfite oxidase protein fold. Microbiol. Mol. Biol. Rev. 72, 228–248, table of contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pushie M. J., Doonan C. J., Moquin K., Weiner J. H., Rothery R., and George G. N. (2011) Molybdenum site structure of Escherichia coli YedY, a novel bacterial oxidoreductase. Inorg. Chem. 50, 732–740 [DOI] [PubMed] [Google Scholar]
- 9. Havelius K. G. V., Reschke S., Horn S., Döring A., Niks D., Hille R., Schulzke C., Leimkühler S., and Haumann M. (2011) Structure of the molybdenum site in YedY, a sulfite oxidase homologue from Escherichia coli. Inorg. Chem. 50, 741–748 [DOI] [PubMed] [Google Scholar]
- 10. Brokx S. J., Rothery R. A., Zhang G., Ng D. P., and Weiner J. H. (2005) Characterization of an Escherichia coli sulfite oxidase homologue reveals the role of a conserved active site cysteine in assembly and function. Biochemistry 44, 10339–10348 [DOI] [PubMed] [Google Scholar]
- 11. von Rozycki T., Yen M.-R., Lende E. E., and Saier M. H. Jr. (2004) The YedZ family: possible heme binding proteins that can be fused to transporters and electron carriers. J. Mol. Microbiol. Biotechnol. 8, 129–140 [DOI] [PubMed] [Google Scholar]
- 12. Zhang X., Krause K.-H., Xenarios I., Soldati T., and Boeckmann B. (2013) Evolution of the ferric reductase domain (FRD) superfamily: modularity, functional diversification, and signature motifs. PLoS One 8, e58126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ohgami R. S., Campagna D. R., McDonald A., and Fleming M. D. (2006) The Steap proteins are metalloreductases. Blood 108, 1388–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ohgami R. S., Campagna D. R., Greer E. L., Antiochos B., McDonald A., Chen J., Sharp J. J., Fujiwara Y., Barker J. E., and Fleming M. D. (2005) Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 37, 1264–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dancis A., Roman D. G., Anderson G. J., Hinnebusch A. G., and Klausner R. D. (1992) Ferric reductase of Saccharomyces cerevisiae: molecular characterization, role in iron uptake, and transcriptional control by iron. Proc. Natl. Acad. Sci. U.S.A. 89, 3869–3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sanchez-Pulido L., Rojas A. M., Valencia A., Martinez-A C., and Andrade M. A. (2004) ACRATA: a novel electron transfer domain associated to apoptosis and cancer. BMC Cancer 4, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kleven M. D., Dlakić M., and Lawrence C. M. (2015) Characterization of a single b-type heme, FAD, and metal binding sites in the transmembrane domain of six-transmembrane epithelial antigen of the prostate (STEAP) family proteins. J. Biol. Chem. 290, 22558–22569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Drew D., Slotboom D.-J., Friso G., Reda T., Genevaux P., Rapp M., Meindl-Beinker N. M., Lambert W., Lerch M., Daley D. O., Van Wijk K.-J., Hirst J., Kunji E., and De Gier J.-W. (2005) A scalable, GFP-based pipeline for membrane protein overexpression screening and purification. Protein Sci. 14, 2011–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Segal A. W., and Jones O. T. (1978) Novel cytochrome b system in phagocytic vacuoles of human granulocytes. Nature 276, 515–517 [DOI] [PubMed] [Google Scholar]
- 20. Rieske J. S. (1967) [76] The quantitative determination of mitochondrial hemoproteins. Methods Enzymol. 10, 488–493 [Google Scholar]
- 21. Jackson M., and Mantsch H. H. (1995) The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 30, 95–120 [DOI] [PubMed] [Google Scholar]
- 22. Biberstine-Kinkade K. J., DeLeo F. R., Epstein R. I., LeRoy B. A., Nauseef W. M., and Dinauer M. C. (2001) Heme-ligating histidines in flavocytochrome b558: identification of specific histidines in gp91phox. J. Biol. Chem. 276, 31105–31112 [DOI] [PubMed] [Google Scholar]
- 23. Karplus P. A., Daniels M. J., and Herriott J. R. (1991) Atomic structure of ferredoxin-NADP+ reductase: prototype for a structurally novel flavoenzyme family. Science 251, 60–66 [PubMed] [Google Scholar]
- 24. Fieschi F., Nivière V., Frier C., Décout J. L., and Fontecave M. (1995) The mechanism and substrate specificity of the NADPH:flavin oxidoreductase from Escherichia coli. J. Biol. Chem. 270, 30392–30400 [DOI] [PubMed] [Google Scholar]
- 25. Bianchi V., Reichard P., Eliasson R., Pontis E., Krook M., Jörnvall H., and Haggård-Ljungquist E. (1993) Escherichia coli ferredoxin NADP+ reductase: activation of E. coli anaerobic ribonucleotide reduction, cloning of the gene (fpr), and overexpression of the protein. J. Bacteriol. 175, 1590–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nivierè V., Fieschi F., Décout J. L., and Fontecave M. (1996) Is the NAD(P)H:flavin oxidoreductase from Escherichia coli a member of the ferredoxin-NADP+ reductase family?: evidence for the catalytic role of serine 49 residue. J. Biol. Chem. 271, 16656–16661 [DOI] [PubMed] [Google Scholar]
- 27. Blaschkowski H. P., Neuer G., Ludwig-Festl M., and Knappe J. (1982) Routes of flavodoxin and ferredoxin reduction in Escherichia coli: CoA-acylating pyruvate: flavodoxin and NADPH: flavodoxin oxidoreductases participating in the activation of pyruvate formate-lyase. Eur. J. Biochem. 123, 563–569 [PubMed] [Google Scholar]
- 28. Nivière V., Fieschi F., Dećout J. L., and Fontecave M. (1999) The NAD(P)H:flavin oxidoreductase from Escherichia coli: evidence for a new mode of binding for reduced pyridine nucleotides. J. Biol. Chem. 274, 18252–18260 [DOI] [PubMed] [Google Scholar]
- 29. Jormakka M., Törnroth S., Byrne B., and Iwata S. (2002) Molecular basis of proton motive force generation: structure of formate dehydrogenase-N. Science 295, 1863–1868 [DOI] [PubMed] [Google Scholar]
- 30. Sucharitakul J., Tinikul R., and Chaiyen P. (2014) Mechanisms of reduced flavin transfer in the two-component flavin-dependent monooxygenases. Arch. Biochem. Biophys. 555, 33–46 [DOI] [PubMed] [Google Scholar]
- 31. Sumimoto H. (2008) Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 275, 3249–3277 [DOI] [PubMed] [Google Scholar]
- 32. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 34. Krissinel E. (2007) On the relationship between sequence and structure similarities in proteomics. Bioinformatics 23, 717–723 [DOI] [PubMed] [Google Scholar]
- 35. Finn R. D., Mistry J., Tate J., Coggill P., Heger A., Pollington J. E., Gavin O. L., Gunasekaran P., Ceric G., Forslund K., Holm L., Sonnhammer E. L. L., Eddy S. R., and Bateman A. (2010) The Pfam protein families database. Nucleic Acids Res. 38, D211–D222 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.