

Abstract
A small library of well defined heparan sulfate (HS) polysaccharides was chemoenzymatically synthesized and used for a detailed structure-activity study of fibroblast growth factor (FGF) 1 and FGF2 signaling through FGF receptor (FGFR) 1c. The HS polysaccharide tested contained both undersulfated (NA) domains and highly sulfated (NS) domains as well as very well defined non-reducing termini. This study examines differences in the HS selectivity of the positive canyons of the FGF12-FGFR1c2 and FGF22-FGFR1c2 HS binding sites of the symmetric FGF2-FGFR2-HS2 signal transduction complex. The results suggest that FGF12-FGFR1c2 binding site prefers a longer NS domain at the non-reducing terminus than FGF22-FGFR1c2. In addition, FGF22-FGFR1c2 can tolerate an HS chain having an N-acetylglucosamine residue at its non-reducing end. These results clearly demonstrate the different specificity of FGF12-FGFR1c2 and FGF22-FGFR1c2 for well defined HS structures and suggest that it is now possible to chemoenzymatically synthesize precise HS polysaccharides that can selectively mediate growth factor signaling. These HS polysaccharides might be useful in both understanding and controlling the growth, proliferation, and differentiation of cells in stem cell therapies, wound healing, and the treatment of cancer.
Keywords: fibroblast growth factor (FGF), fibroblast growth factor receptor (FGFR), heparan sulfate, signaling, structure-function, block copolymers, domain structures
Introduction
Heparan sulfate is a glycosaminoglycan (GAG),2 a linear sulfated polysaccharide of 1→4-linked α-d-glucosamine (GlcN) and uronic acid, β-d-glucuronic acid (GlcA), or α-l-iduronic acid (IdoA) (1). The mammalian GAG heparan sulfate contains a domain structure comprising undersulfated sequences rich in GlcNAc (where Ac is acetyl) residues (called “NA domains”) and highly sulfated sequences rich in GlcNS (where S is sulfo) residues (called “NS domains”) (Fig. 1) (2–4). These domains structurally vary based on the species and tissues from which an HS is obtained (5), and the NS domain is of particular importance in cellular behavior and disease processes (6). HS is biosynthesized in the endoplasmic reticulum and Golgi as a constituent of proteoglycans (PGs) attached to one of a number of core proteins (7, 8). The biosynthesis of the GAG chain within the Golgi controls the placement of domains within HS (8, 9). These PGs are primarily localized to the cell membrane or in the extracellular matrix (7, 8) where they play a critical biological role as a co-receptor in growth factor signaling (10, 11). HS domain structure and position on the GAG chain can also be catabolically modified through the action of extracellular sulfatases (7, 12) and heparanase (7, 13). A plethora of HS structures are possible, and many are observed in animals.
FIGURE 1.

Structures of natural and synthetic heparan sulfates. A generalized chemical structure of animal-sourced HS is shown, and below it the same structure is drawn using conventional symbols (59). Synthetic heparan sulfates 1-8 are shown in their symbolic representations. The presence of substituents is indicated above and below the symbols with the carbon position number (or “N” for nitrogen substituent) and “OH” for unsubstituted or “S” for sulfo group substituted. An “OH/S” indicates that the position can be either unsubstituted or sulfated. The “/” at the non-reducing terminus indicates an ambiguous monosaccharide that was not controlled during the final step of synthesis.
In the past, heparan sulfate used in biochemical studies was generally extracted from animal tissues following proteolysis. The resulting HS GAG is large (Mr >10,000), polydisperse (for Mr 10,000–30,000, weight average molecular weight/number average molecular weight values of 1.2–1.6), and microheterogeneous (e.g. possessing multiple structural domains and variable saccharide sequences) (5, 14, 15, 17). The structural complexity of heparan sulfate GAG complicates the study of its structure-activity relationship (SAR) with regard to its protein-mediated signaling activities. Related less heterogeneous and more highly sulfated heparin, chemically modified heparins, or heparin-derived oligosaccharides have been applied to simplify these SAR studies (18–21). Unfortunately, natural heparan sulfate has highly variable compositions and sequences, and only a very limited number of chemically modified heparin structures can be reliably prepared from natural polysaccharides and in quite small quantities (e.g. micrograms to milligrams). Structurally defined heparan sulfate-derived or heparin-derived oligosaccharides from natural GAGs or organic synthesis are often too small to exhibit many important biological activities in comparison with the “full-length” native polysaccharides (18, 19, 21).
Recently, it has become possible to chemoenzymatically synthesize larger heparan sulfate chains having domain structures (22). Recombinant heparan sulfate-polymerizing and -modifying enzymes have been utilized for reactions in vitro (23–25). The GAG chain backbones can be efficiently and controllably synthesized in vitro using GAG synthases to add the monosaccharide units from UDP-sugar donors onto an acceptor (Fig. 2). When building GAGs from natural UDP-sugars (UDP-GlcNAc and UDP-GlcA) or non-natural UDP-sugars (UDP-GlcNTFA where TFA is trifluoroacetyl) (26), in vitro GAG chain synthesis can be performed in one of two preferred formats: stepwise elongation (i.e. one sugar unit at a time) or in a synchronized polymerization reaction (i.e. block addition via multiple sugar units). Both of these formats yield well defined products with narrow size distributions (monodisperse or nearly so) and potentially much more controllable compositions than the GAG produced in vivo (21, 27–29).
FIGURE 2.

Chemoenzymatic synthesis of heparan sulfates. The first step involving the iterative synthesis of the trisaccharide acceptor from GlcA-pNP and UDP-donor sugars is not shown. a, the second step is polysaccharide backbone synthesis from the trisaccharide acceptor and UDP-donor sugars (shown at the top) under stoichiometric control. b, the chemical conversion of GlcNTFA to GlcN (for a GlcNAc-containing domain, this part of the chain remains basically untransformed at this step). c, the chemical conversion of GlcN to GlcNS. d, enzymatic treatment with C5-epimerase and 2-O-sufotransferase. e, enzymatic treatment with 6-O-sufotransferase 1 and 6-O-sufotransferase 3.
In our approach, the polysaccharide backbone is assembled with blocks of either (→4)-β-d-GlcA (1→4)-α-d-GlcNAc(1→) or (→4)-β-d-GlcA (1→4)-α-d-GlcNTFA(1→). Mild chemical treatments remove the TFA moieties from the glucosamine and replace it with N-sulfo groups to generate a precursor structure with blocks of (→4)-β-d-GlcA (1→4)-α-d-GlcNAc(1→) or (→4)-β-d-GlcA (1→4)-α-d-GlcNS(1→) that are enzymatically modified with C5-epimerase, which converts GlcA into IdoA, and O-sulfotransferases that transfer sulfo groups from the donor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to the various hydroxyl groups of the GAG (30–36). Such resulting synthetic heparan sulfate chains have a controlled domain structure useful for SAR studies (22).
Heparan sulfate regulates the activity of the 22-member family of extracellular fibroblast growth factors (FGFs) involved in critically important cellular activities including angiogenesis, cellular proliferation, cellular motility, differentiation (37–39), and adhesion (40, 41). The FGFs signal through their cognate membrane-bound fibroblast growth factor receptors (FGFRs), a group of seven distinct protein receptors (42–44), and an HS co-receptor. Kinetic experiments using surface plasmon resonance suggest that two extracellular FGFs first bind to the HS chain(s) of membrane-anchored HSPGs and then recruit two FGFRs to assemble into a signaling complex (45). Assembly of an FGF-HS-FGFR ternary complex (46) then activates signaling across the transmembrane helix, which then activates the intracellular tyrosine kinase domain (47). The individual binding affinities of the HS chains for FGFs and FGFRs have been determined, but the actual structure of the ternary complex remains unclear (45, 48–52). The dimeric protein complex, FGF2-FGFR2, forms a positively charged cleft or “canyon” lined with basic amino acid residues that interact with one or two complementary negatively charged HS chains with high (nm range) affinity (43, 53, 54). The FGF-HS-FGFR ternary complex is believed to be a symmetric structure of 2:2:2 stoichiometry (55). Previous studies show that highly sulfated NS domains at the non-reducing terminus of heparan sulfate bind with higher affinity and promote FGF2-FGFR1 signaling (22, 56).
The current study examines the impact of the domain structure and the structure at the non-reducing terminus of chemoenzymatically synthesized heparan sulfate GAG chains on FGF-1 and FGF-2 signaling through FGFR-1c. Heparan sulfate-mediated FGF-FGFR signaling was determined using a murine immortalized bone marrow (BaF3) cell line developed by Ornitz and co-workers (43, 54) that expresses FGFR type 1c without expressing either HSPGs or FGF. The ternary complex signaling process is determined by measuring heparan sulfate-mediated cellular proliferation.
Results
Design of Synthetic Heparan Sulfate Targets for Testing
The domains present within natural heparan sulfate consist of sequences of high sulfation (NS domains) and low sulfation (NA domains) (2–4). Although these domains vary in size and number of sulfo groups among species and tissues (5), the regions do share some common features. First, their placement and sizes are believed to be controlled through the placement of N-sulfo groups by the action of the different N-deacetylase N-sulfotransferase isoforms (3, 4, 8, 9, 11, 30). Second, the domain closest to the core protein at the reducing end of the heparan sulfate chain is generally an NA domain, whereas the domains at the non-reducing end of non-signaling and signaling heparan sulfate chains are typically NA and NS domains, respectively (2, 4). Moreover, the conversion of a non-signaling heparan sulfate chain to a signaling heparan sulfate chain in disease processes associated with rapid cell proliferation, such as cancer, can also take place through the action of heparanase, which cleaves between a high and low sulfate domain, thus exposing a GlcN residue associated with an NS domain at the non-reducing end of the chain (7, 13).
Eight heparan sulfate chains were designed to begin to study the contribution of domain structures in HS for FGF-FGFR signaling (Fig. 1, 1-8). These chains consist of one or two long domains assembled at the non-reducing end of a GlcA-pNP acceptor. One chain, 2, contained a long (∼28-disaccharide repeat) NS domain assembled at the non-reducing end of a GlcA-pNP acceptor, resembling heparin, serving as a positive control in signaling assays. A second chain, 1, contained a long (∼15-repeat) NA domain assembled at the non-reducing end of the acceptor (Fig. 3), resembling a single domain heparan sulfate chain, serving as a negative control in signaling assays. A third chain, 3, contained two domains comprising a long (∼13-repeat) NS domain assembled at the non-reducing end of a long (∼15-repeat) NA domain assembled at the reducing end of the GlcA-pNP acceptor. On the remaining five synthetic heparan sulfate chains (4-8), this long (∼16-repeat) reducing end NA domain was terminated at the non-reducing end with NS domains of variable lengths capped with a single GlcN residue substituted with either an N-sulfo or N-acetyl group.
FIGURE 3.

PAGE analysis of various synthetic heparosan and TFA-protected heparosan precursors and HPLC-MS analysis of the precursor to synthetic heparan sulfate 1. The heparosan polysaccharide and TFA-protected heparosan precursors were assembled on the GlcA-GlcNAc-GlcA-pNP or GlcA-GlcNTFA-GlcA-pNP acceptors analyzed by HPLC-MS. A, 8% PAGE was used to analyze these samples together with individual hyaluronan and a mixture of hyaluronan (HA) standards (Hyalose, LLC; note that hyaluronan and heparosan migrate similarly but not identically on PAGE). Samples (∼1 μg) were loaded onto the gel and run at 250 V applied for 20 min. The gel was stained overnight with 0.05% Alcian Blue stain. Lane 1, hyaluronan LoLadder (fastest migrating band is 27 kDa); lanes 2 and 13, 10-kDa hyaluronan; lanes 3 and 12, 6.5-kDa hyaluronan; lane 4, NAc precursor of 1; lanes 5 and 11, NAc precursor of 1 (SAX-polished); lane 6, NTFA precursor of 4 (five sugars added); lane 7, NAc precursor of 7 (nine sugars added); lane 8, NTFA precursor of 5 (nine sugars added); lane 9, NAc precursor of 8 (11 sugars added); lane 10, NTFA precursor of 6 (11 sugars added). HPLC-MS analysis of the precursor to synthetic heparan sulfate 1 (purified; same as shown in A, lanes 5 and 11) was performed. B, the HPLC chromatogram with the broad peak at 6.94 min corresponding to the polysaccharide chain. C, the mass spectrum of polysaccharide chains in the HPLC peak eluting at 6.94 min. D, the deconvoluted mass spectrum of polysaccharide chains in the 6.94-min HPLC peak with a degree of polymerization (dp; the number of monosaccharides in the chain) and molecular mass distribution of precursor to synthetic heparan sulfate 1 showing a molecular mass consistent with the 6.4-kDa target.
Block Copolymer Elongation and Formation
Chain synthesis began on a commercially available GlcA-pNP acceptor. This acceptor was iteratively extended through the alternative addition of UDP-GlcNAc and UDP-GlcA donors to prepare the precursor for targets 1 and 3-8 or UDP-GlcNTFA and UDP-GlcA donors to prepare precursor for target 2, resulting in trisaccharides GlcA-GlcNAc-GlcA-pNP and GlcA-GlcNTFA-GlcA-pNP. These trisaccharides were characterized by electrospray ionization mass spectrometry (MS) and anion exchange high performance liquid chromatography (HPLC).
The synthesized trisaccharides are excellent acceptors for stoichiometrically controlled extension. The length of the long domain was controlled by the ratio of the trisaccharide acceptor and the UDP-sugar donors. The molecules are quasimonodisperse due to the synchronization of the synthase-catalyzed polymerization reaction with acceptor as described (27). The size of the resulting chains, (GlcA-GlcNAc)15-GlcA-pNP (as well as all the other synthetic HS chains), was determined by HPLC-MS. The (GlcA-GlcNTFA)28-GlcA-pNP chain corresponds to the first precursor to synthetic heparan sulfate 2 (Figs. 1 and 4, top).
FIGURE 4.

NMR analyses of HS intermediates. Proton NMR (600 MHz) of the first and second intermediate products of synthetic heparan sulfate 2 was carried out. Top, the NTFA precursor, corresponding to compound 2. Bottom, the NSulfo precursor is formed through de-N-trifluoracetylation and N-sulfonation of the NTFA precursor.
The (GlcA-GlcNAc)16-GlcA-pNP chains were further modified by iterative transfer of UDP-GlcNTFA and UDP-GlcA adding five, 9, or 11 additional monosaccharide units. In each case, these chains were capped with either a final GlcNTFA or a GlcNAc residue (Fig. 1) to afford the first precursor of the designed synthetic heparan sulfates 4-8. It is important to note that these non-reducing terminal extensions were completely defined structures. Although there are multiple species (∼8 major) observed for the block portion of the synthetic chain, the stepwise synthesis results in precise additions. This control is needed to assess the SAR of the end of HS chains.
Glucosamine De-N-trifluoracetylation and N-Sulfonation
Once the precursors to synthetic heparan sulfates 2-8 were synthesized with the desired GlcNAc- and GlcNTFA-containing domains, the NTFA groups were deprotected and subsequently N-sulfonated. Each TFA-containing block copolymer was dissolved in a mildly basic solution of Et3N, MeOH, and H2O and stirred overnight. Under these conditions, the GlcNTFA residues were completely deprotected, exposing free amino groups, which were chemically N-sulfonated using NMe3·SO3 to afford GlcNAc and GlcNS domains containing block copolymers, the NSulfo precursors to synthetic heparan sulfates 2-8. Conversely, the acetyl groups of NA domains remain virtually intact throughout this process.
The sizes of the resulting dicopolymer intermediates, as examined using polyacrylamide gel electrophoresis (PAGE) and LC-MS, were consistent with those expected based on their synthesis. The disaccharide composition of the NSulfo precursors to synthetic heparan sulfates 4, 5, 6, and 7 were examined by disaccharide compositional analysis using HPLC-MS analysis to confirm that the expected extensions had taken place (Table 1).
TABLE 1.
Disaccharide composition of the precursors and the oligosaccharides after O-sulfation
Disaccharide composition was calculated from HPLC-MS peak integration (Fig. 4) using appropriate response factors. The theoretical percentage of dp2(NS) (where dp is the degree of polymerization) was calculated as NS/(NS + NAc − 1), and the percentage of dp2(NAc) was calculated as (NAc − 1)/(NS + NAc − 1) (see structure in Fig. 1). TriS, NS6S, NS2S, and NS disaccharides arise from the NS domains, and 2S6S, 6S, 2S, and 0S disaccharides arise from the NS domains. —, not detected.
| Disaccharide composition |
Experimental |
Theoretical |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TriS | NS6S | NS2S | NS | 2S6S | 6S | 2S | 0S | Total NS | Total NAc | Total NS | Total NAc | |
| % | % | % | ||||||||||
| NSulfo precursor 4 | — | — | — | 17 | — | — | — | 83 | 17 | 83 | 12 | 88 |
| NSulfo precursor 5 | — | — | — | 26 | — | — | — | 74 | 26 | 74 | 21 | 79 |
| NSulfo precursor 6 | — | — | — | 30 | — | — | — | 70 | 30 | 70 | 25 | 75 |
| NSulfo precursor 7 | — | — | — | 28 | — | — | — | 72 | 28 | 72 | 21 | 79 |
| Heparan sulfate 1 | — | 16 | — | — | — | 7 | — | 77 | 16 | 85 | 0 | 100 |
| Heparan sulfate 2 | 65 | 23 | — | — | — | 1 | 7 | 3 | 88 | 11 | 100 | 0 |
| Heparan sulfate 3 | 37 | 15 | — | — | — | 10 | 3 | 36 | 52 | 49 | 48 | 52 |
| Heparan sulfate 4 | 2 | 6 | — | — | — | 15 | 32 | 44 | 8 | 91 | 12 | 88 |
| Heparan sulfate 5 | 6 | 14 | — | — | — | 21 | 17 | 41 | 20 | 79 | 21 | 79 |
| Heparan sulfate 6 | 5 | 14 | — | — | — | 19 | 29 | 31 | 19 | 79 | 25 | 75 |
| Heparan sulfate 7 | 5 | 12 | — | — | — | 21 | 23 | 39 | 17 | 83 | 21 | 79 |
| Heparan sulfate 8 | — | 20 | — | — | — | 17 | — | 63 | 20 | 80 | 25 | 75 |
The structure of the NTFA and NSulfo precursors for each synthetic heparan sulfate was evaluated by one-dimensional (1D) 1H nuclear magnetic resonance (NMR). The 1H spectra for the NTFA and NSulfo precursors of synthetic heparan sulfate 2 are shown in Fig. 4. The conversion of the GlcNTFA residue to a GlcNS residue was confirmed by the change in chemical shift of the anomeric proton (H1) signal. NMR spectra of all block copolymers show two signals in the anomeric region of ∼4–6 ppm. The signals at ∼5.5 and ∼4.4 ppm correspond to the anomeric proton of GlcNTFA and GlcA, respectively. Incomplete N-sulfonation would be indicated by a peak at ∼5.6 ppm, but such a signal cannot be seen in the spectra. Taken together, the NMR data indicate that the block copolymers were successfully N-sulfonated.
Disaccharide Composition of Heparan Sulfate Precursors
The disaccharide composition of the heparan sulfate NSulfo was next determined by exhaustively treating each with heparin lyases, labeling with 2-aminoacridone (AMAC), and performing HPLC-MS. The resulting total ion chromatogram obtained by HPLC-MS analysis showed only 0S and NS disaccharides associated with each of the HS block copolymers. The HPLC-MS analysis of the NSulfo precursor of heparan sulfate 4 shown in Fig. 5, for example, is consistent with its structure after compensating for the different response factors for the 0S and NS disaccharides. The experimentally observed disaccharide compositions of the NSulfo precursor of heparan sulfates 4-7, which have a complex block structure, correspond quite well to their theoretical compositions (Table 1). The data suggest that these polysaccharide compounds have the anticipated structures.
FIGURE 5.

Disaccharide compositional analysis of synthetic HS. Disaccharides afforded through treatment with heparin lyases I, II, and III were analyzed using HPLC-MS. Extracted ion chromatograms (EIC) are shown on the left, and mass spectra are shown on the right (0S, before any modification; NS, after N-sulfonation; and TriS, after O-sulfation.) A, HPLC analysis of heparan sulfate disaccharide standards with detection by an ion trap mass spectrometer. B, disaccharide analysis of the NSulfo precursor intermediate for synthetic heparan sulfate 4. C, disaccharide analysis of synthetic heparan sulfate 4 (complete with O-sulfotransferase modifications).
C5-Epimerization and O-Sulfonation
The HS block copolymers were treated exhaustively with C5-epimerase in the presence of 2-O-sulfotransferase followed by 6-O-sulfotransferase 1 and 6-O-sulfotransferase 3. These enzymes all act in the N-sulfo domains to form fully modified high sulfo S-domains comprising →GlcNS6S→IdoA2S→ repeating units. The 6-O-sulfotransferases could also modify the N-acetyl N-domains to a limited extent.
Characterization of Heparan Sulfate Products
The disaccharide composition of the synthetic heparan sulfates obtained through C5-epimerization and O-sulfonation were next analyzed by LC-MS (Fig. 2 and Table 1). The disaccharide analysis of synthetic heparan sulfate 4 is shown in Fig. 5. Upon treatment with heparin lyases, all the synthetic heparan sulfates afforded complex mixtures containing different amounts of the eight possible disaccharides (Table 1). The NS domains comprised TriS and NS6S, consistent with expectation. The NA domains primarily comprised 0S, 2S, and 6S. High levels of 0S and 6S were anticipated. The exhaustive treatment of NA domains with C5-epimerase in the presence of 2-O-sulfotransferase affording surprising amounts of 2S-containing sequences is most likely the result of the presence of unexpectedly large amounts of GlcA2S.
Bioactivity/Cellular Proliferation of Synthetic Heparan Sulfates
The synthetic heparan sulfates were tested for cellular proliferation with the FGFR1c-expressing cells in the presence of FGF1 or FGF2 in a 96-well plate (22). The assay was performed in a single 96-well plate in triplicate, giving a small standard deviation, confirming the precision of this measurement. First, a standard curve of the optical density (at 590 nm) as a function of y was constructed to demonstrate the linearity of the cellular proliferation assay (Fig. 6A). The negative control, no added heparan sulfate, gave a baseline cell count of 4,000 ± 750 and 33,000 ± 1,100 for FGF1 and FGF2 signaling through FGFR1c, respectively (Fig. 6B). The difference in baseline may be an indication that FGF2 may promote some signaling in the absence of GAG, although signaling is greatly augmented by addition of these polysaccharides. Porcine intestinal mucosal heparin served as a positive control, showing a cell count of 26,000 ± 1,900 through FGF1 but a significantly higher cell count of 49,000 ± 990 through FGF2. Next, porcine intestinal heparan sulfate was tested, and although it showed a very low cell count of 7,000 ± 460 through FGF1, it unexpectedly showed a slightly higher signaling cell count of 52,000 ± 1,400 than observed for heparin through FGF2. This observation implies potential for the variability for naturally sourced GAGs.
FIGURE 6.
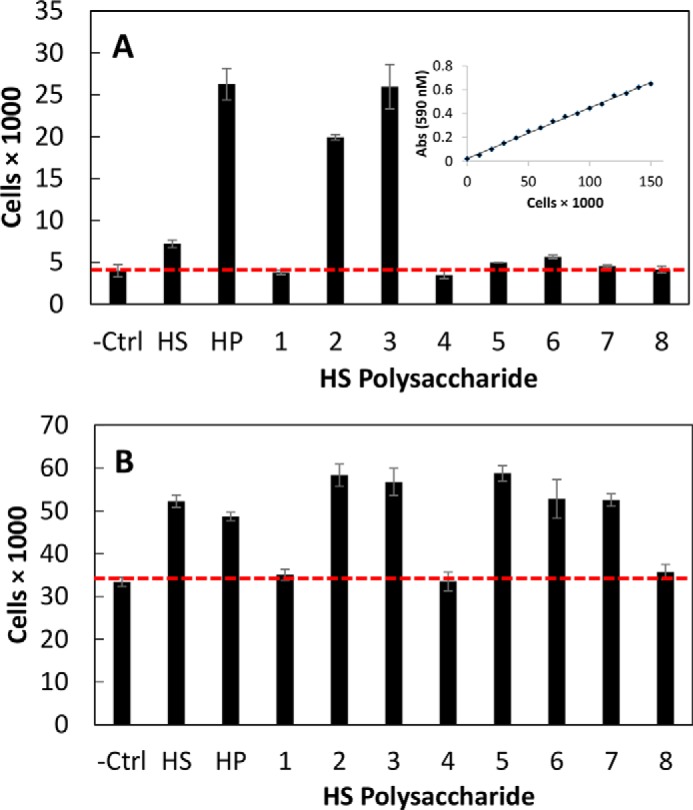
Fibroblast growth factor receptor bioactivity of natural and synthetic HS. Heparan sulfate-mediated FGF-FGFR signaling was analyzed using a BaF3 cellular proliferation assay in a 96-well plate (22). A, FGF1 signaling through FGFR1c. The inset shows a standard curve of cell proliferation assay. B, FGF2 signaling through FGFR1c. Abs, absorbance; Ctrl, control; HP, heparin. Error bars represent S.D.
As expected, synthetic heparan sulfate 2 containing only a single NS domain with ∼28 repeating disaccharides and terminated with a GlcNS residue at the non-reducing end showed high FGF1 and FGF2 signaling activities (26,000 ± 330 and 49,000 ± 2,600, respectively) similar to that observed for heparin. Similarly, the two-domain synthetic heparan sulfate 3 with an NA domain of ∼15 repeating disaccharides at the reducing end and an NS domain of ∼15 repeating disaccharides at the non-reducing end also showed potent FGF1 and FGF2 signaling activities (26,000 ± 2,600 and 57,000 ± 3,200, respectively) comparable with the positive control heparin. Synthetic heparan sulfate 1 having a single NA domain of ∼15 repeating disaccharides and terminated with a GlcA residue showed no FGF1 and FGF2 signaling activities (i.e. buffer alone baseline values), suggesting that a non-reducing terminal NS domain is critical for robust signaling.
Of particular interest were the signaling activities of synthetic heparan sulfates 4-8 having an NA domain of ∼16 repeating disaccharides (derived from 1, the inactive HS) at the reducing end with defined short variable length NS domains at their non-reducing ends. Synthetic heparan sulfates 7 and 8 with a reducing end NA domain of ∼16 repeating disaccharides and non-reducing end NS domains of four and five repeating disaccharides were terminated with a GlcNAc residue. Synthetic heparan sulfates 7 and 8 behave similarly to synthetic heparan sulfate 1, showing no FGF1 signaling activities. Interestingly, synthetic heparan sulfates 7 and 8 showed some FGF2 signaling activity. Synthetic heparan sulfates 4, 5, and 6 with non-reducing end NS domains of two, four, and five disaccharides and terminated with a GlcNS residue showed differential FGF1 and FGF2 signaling activities. Synthetic heparan sulfates 5 and 6 with longer four- and five-disaccharide non-reducing end NS domains showed low FGF1 and high FGF2 signaling activities. In contrast, synthetic heparan sulfate 4 with the shortest non-reducing end NS domain showed no FGF1 and FGF2 signaling activities, suggesting that a non-reducing end terminal NS domain of at least four to five disaccharides is necessary for signaling. The BaF3 cell-based assay was performed a second time and confirmed the relative similar levels of signaling for the various synthetic heparan sulfates.
Discussion
Using our chemoenzymatic approach for the synthesis of heparan sulfate polysaccharides on simple aglycone acceptors, we had previously synthesized block polysaccharides having different arrangements of uniform size NS and NA domains (22). The symmetric FGF2-HS2-FGFR2 ternary complex model (55) was best supported by the results of our prior study suggesting that NS domains at the non-reducing end were required for FGF signaling (22). Because of the topological constraints of the two heparan sulfate chains being attached to the core protein through their reducing ends (in the native FGF2-HSPG2-FGFR2 complex the two heparan sulfate chains are attached to the core protein through their reducing ends), the interacting NS domains must be located on the non-reducing end of each heparan sulfate chain (Fig. 7). These two heparan sulfate chains are docked through their non-reducing ends into the basic canyon located on the top face of the FGF2-FGFR2 protein complex called the heparin binding site. Furthermore, the electrostatic and topological characteristics of the basic canyon are different for each protein signaling complex; i.e. the canyon of FGF12-FGFR1c2 should be distinct from that of FGF22-FGFR1c2 (57).
FIGURE 7.

Proposed model and structure/activity relationship of heparan sulfate-mediated FGF-FGFR signaling through an FGF2-HSPG2-FGFR1c2 complex. Heparan sulfate structural characteristics required to facilitate signaling complex formation differ between FGF1 and FGF2. FGF1 (left) requires a terminal NS domain of 10–11 disaccharides and a terminal GlcNS for signaling. In contrast, FGF2 (right) utilizes a shorter non-reducing NS domain (∼5 disaccharides) and is tolerant of a non-reducing end GlcNAc.
Most SAR studies in the past focused on the structural characteristics of heparin or HS required for binding to various FGFs in the absence of receptor (58). These studies provide only limited information because they do not consider the FGFR component of the interaction (57). Furthermore, without a cellular component, such as used in the BaF3 assay, it is always unclear whether binding is sufficient for signaling (49). Biochemical studies directly assessing FGF-FGFR signaling clearly support the symmetric FGF2-HS2-FGFR2 ternary complex model in which the non-reducing ends of two HS chains are involved in the interaction (22, 52, 60). Studies on chemically modified heparins and various chondroitin sulfates generally show heparin giving the greatest signaling among the heparin derivatives and dermatan sulfate (chondroitin sulfate B) giving the greatest signaling for the various chondroitin sulfates (61). Although these previous studies provide a better understanding of the SAR, they provide no information about the precise structural features directly at the non-reducing ends of the chains involved in FGF signaling.
FGF2 signaling through FGFR1c has been shown to interact via the NS domains of HS chains (62) and FGF2 binding to FGFR1c (57). The current study is focused on developing a more precise SAR for heparan sulfate-mediated signaling through one pair of homologous growth factor-receptor complexes, FGF12-FGFR1c2 and FGF22-FGFR1c2. A new paradigm for the chemoenzymatic synthesis of a variety of larger heparan sulfate chains having between 16 and 28 repeating units was required to carry out this study.
Our synthetic approach utilized three enzymatic steps using UDP-sugar donors to build the polysaccharide backbone. In the first step, a commercially available glycoside acceptor, GlcA-pNP, was iteratively extended to the heparosan trisaccharide acceptor GlcA-GlcNAc-GlcA-pNP or GlcA-GlcNTFA-GlcA-pNP. In the second step, the trisaccharide acceptors were efficiently elongated through stoichiometrically controlled extension to afford either an NA block or the NTFA precursor of an NS block. In the third step, either a second block was added under stoichiometric control, or a short oligosaccharide domain was added through iterative synthesis. For the end-capped polymers, the last residue added at the non-reducing end of the chain was controlled to be either GlcNAc or GlcNTFA.
Once the suitable polysaccharide precursors were assembled, the GlcNTFA residues were quantitatively converted to GlcNS as could be demonstrated by NMR and through the use of HPLC-MS-based disaccharide analysis. It is important to note that, in comparison with the natural N-deacetylase N-sulfotransferase-based processes, our strategy for chemical installation of the NSulfo groups (i) is virtually complete and (ii) allows strict placement and segregation of the NA and NS domains.
Final enzymatic treatment with C5-epimerase and 2-OST followed by 6-OST-1 and 6-OST-3 afforded synthetic heparan sulfates 1-8. After confirming their structures, a 96-well plate BaF3 cell proliferation assay was conducted to afford a more detailed SAR of heparan sulfate signaling through FGF12-FGFR1c2 or FGF22-FGFR1c2.
The heparan sulfate binding site canyon in FGF12-FGFR1c2 appears to prefer a longer non-reducing terminal NS domain than that of FGF22-FGFR1c2 (Fig. 7). Porcine intestinal heparin strongly signals through FGF12-FGFR1c2 as do synthetic heparan sulfates 2 and 3, but porcine intestinal heparan sulfate and synthetic heparan sulfate 6 only weakly signal. This suggests the minimum NS binding domain to be four to five disaccharide repeats terminated with a GlcNS residue for weak signaling and an NS binding domain of ∼15 disaccharide sequences terminated with a GlcNS for robust signaling. In contrast, the heparan sulfate binding site canyon in FGF22-FGFR1c2 appears to utilize a shorter non-reducing terminal NS domain. Robust signaling was observed for synthetic heparan sulfates 5 and 6, but no signaling was observed for 4, suggesting that the optimal length of the non-reducing terminal NS domain is >4–5 disaccharides with a terminal GlcNS residue. Furthermore, in contrast to FGF12-FGFR1c2, signaling was observed for synthetic heparan sulfates 7 and 8 with a non-reducing terminal NS domains of four and five disaccharides, respectively, with a terminal GlcNAc residue, demonstrating flexibility in the requirement for terminal NS domains.
The results of this study demonstrate that, at least for the FGF12-FGFR1c2 and FGF22-FGFR1c2 signal transduction complexes, it is possible to design and chemoenzymatically synthesize heparan sulfates that can selectively mediate signaling. Basically, in this case, our HS species with shorter NS terminal domains allow differential targeting of one FGF signaling complex over another homologous complex. The application of this approach to the other 20 members of the FGF family and the other six FGFRs needs to be explored. Ultimately, the use of synthetic heparan sulfates to selectively control FGF-FGFR signaling might play an important role in the control of stem cell differentiation or developmental biology and might suggest new therapeutic approaches for the treatment of cancer and enhancement of wound healing.
Experimental Procedures
Materials
Triethylamine, trimethylamine-sulfur trioxide complex, endotoxin-free water, carbazole, MES, HEPES, AMAC, dimethyl sulfoxide (DMSO), sodium cyanoborohydride, and DEAE-Sepharose were from Sigma-Aldrich. D2O (99.9%) and Norell Select Series 5-mm NMR tubes were purchased from Sigma-Aldrich and used directly from packaging. Ammonium acetate (NH4Ac), calcium chloride (CaCl2), acetic acid (HAc), Whatman 3MM paper, n-butyl alcohol, and HPLC grade acetonitrile were from Fisher Scientific. Hyaluronan standards for PAGE were obtained from Hyalose, LLC (Oklahoma City, OK). All other chemical reagents were purchased from Sigma-Aldrich and used without further purification.
The Chimera G bifunctional heparosan polymerizing catalyst was a fusion of maltose-binding protein (MBP) to PmHS2(1–167)-PmHS1(134–318)-PmHS2(353–651) prepared in recombinant Escherichia coli as described previously (63). Two mutant monofunctional derivatives of MBP-Chimera G that transfer either UDP-GlcA (D479N,D481N in PmHS2 sequence) or UDP-GlcNAc (D181N,D183N in PmHS1 sequence) were constructed by site-directed mutagenesis in an analogous process to PmHS1 (64). UDP-GlcA, UDP-GlcNAc, and GlcA-pNP were purchased from Sigma unless otherwise noted. UDP-GlcNTFA was chemoenzymatically synthesized as described previously (26). The GAG polysaccharides were quantified by the carbazole assay with a glucuronic acid standard (65).
A group of recombinant HS-modifying enzymes, including human C5-epimerase (NCBI accession number NM_015554.1), hamster 2-O-sulfotransferase (GenBankTM accession number D88811.1), murine 6-O-sulfotransferase 1 (NCBI accession number NM_015818.2), and murine 6-O-sulfotransferase 3 (NCBI accession number NM_015820.3), were used to prepare HS polymers. The enzymes were each expressed as a form of MBP fusions. The expression of these enzymes was carried out in E. coli, and enzymes were purified by an amylose-agarose column (New England Biolabs) as described previously (5). A sulfo donor, PAPS, was prepared in house using a method published previously (66).
Syringe filters (0.2 μm), Microcon YM-3 centrifugal filter units with 3,000 molecular weight cutoff (MWCO), and Amicon Ultra-15 3,000 MWCO spin columns were purchased from Millipore (Billerica, MA); the latter were washed three times with deionized water before use. Vivapure Q mini H spin columns were purchased from Sartorious Stedim Biotech (Bohemia, NY). Dialysis membranes (1,000 MWCO) were from Spectrum Laboratories, Inc. (Houston, TX).
Unsaturated disaccharide standards of HS (0S, ΔUA-GlcNAc; NS, ΔUA-GlcNS; 6S, ΔUA-GlcNAc6S; 2S, ΔUA2S-GlcNAc; NS2S, ΔUA2S-GlcNS; NS6S, ΔUA-GlcNS6S; 2S6S, ΔUA2S-GlcNAc6S; TriS, ΔUA2S-GlcNS6S where ΔUA is 4-deoxy-α-l-threo-hex-4-enopyranosyluronic acid) were purchased from Iduron (Cheshire, UK). E. coli expression and purification of the recombinant Flavobacterium heparinum heparin lyases I (heparin lyase), II, and III (heparin-sulfate lyase) (Enzyme Commission (EC) numbers 4.2.2.7, 4.2.2.-, and 4.2.2.8, respectively) were performed in our laboratory as described previously (67).
BaF3 cells expressing FGFR1c were generously provided by Dr. David M. Ornitz of Washington University, St. Louis, MO. FGF1 and FGF2, newborn bovine calf serum, penicillin/streptomycin solution, sterile phosphate-buffered saline (PBS), and Geneticin (G418) were purchased from Invitrogen. Interleukin 3 (IL3) was purchased from Peprotech (Rocky Hill, NJ). RPMI 1640 medium was purchased from Sigma. Sterile polystyrene 75-cm2 tissue culture treated flasks were purchased from VWR (Radnor, PA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and Breathe EZ breathable membrane were from Sigma.
Defined Polysaccharide Synthesis
Polysaccharides containing either (i) alternating NA blocks of GlcA-GlcNAc or NS precursor blocks of GlcA-GlcNTFA repeating disaccharide units or (ii) an NA block with various specific sugar extensions at the non-reducing termini were synthesized.
Initially, in all syntheses, a heparosan trisaccharide was prepared from the successive transfer of UDP-GlcNAc (or GlcNTFA) and UDP-GlcA, donors to GlcA-p-nitrophenyl glycoside, by a series of addition reactions catalyzed by PmHS2. Here, the recombinant pmHS2 was constructed as an N-terminal fusion to His6 using a PET-15b vector (Novagen) expressed in BL21 star (DE3) cells (Invitrogen).
In subsequent block reactions, 0.5–2 mm heparosan trisaccharide (GlcA-GlcNAc (or TFA)-GlcA-pNP) was used as an acceptor to maintain a continuous block as desired. All block reactions received 12–28 mm UDP-GlcA and, depending on the desired block, either 25 mm UDP-GlcNAc or 12–20 mm UDP-GlcNTFA (for preparing the N- or S-domain, respectively) (20, 21). Reaction buffer contained 50–130 mm HEPES, pH 7.2, and 1 mm MnCl2. Each reaction received 1 μg/μl purified MBP-Chimera G enzyme. This enzyme, PmHS2(1–167)-PmHS1 (134–318)-PmHS2(353–651), was selected as the catalyst for NS block polysaccharide synthesis because it exhibits at least a 10- and 2-fold higher specific activity using UDP-GlcNAc and UDP-GlcNTFA when compared with PmHS2, respectively. Also, Chimera G is roughly twice as acceptor-dependent as PmHS2 due to a lower level of de novo synthesis (i.e. the initiation with UDP-sugars only, not an exogenously supplied oligosaccharide). Each step was incubated at 30 °C for 16 h. After polymerization was complete, the bulk of the enzyme was removed by extraction with an equal volume of n-butyl alcohol and vortexing followed by phase separation via centrifugation at 14,000 × g for 5 min. The various HS precursors in the lower aqueous phase were then purified as described later.
The molecular masses of each block section and the total polymer were determined by a combination of polyacrylamide gel/Alcian Blue staining analyses with size-defined standards (22, 68), size exclusion chromatography coupled to multiangle light scattering (29), and/or by LC-MS (68).
The precursor for HS 2 was isolated from the extracted reaction mixture using strong anion exchange (SAX) chromatography on Sepharose Q resin (GE Healthcare) with an ammonium formate step gradient (0.2 m wash for 10 column volumes and then an elution with 0.7, 0.8, and 0.9 m for 1.5 column volumes each). The pooled 0.7–0.9 m eluates were then frozen, and the volatile salt was removed by repeated lyophilization from water. It is important to note that the SAX step was used here instead of paper chromatography (described later) for this long TFA-containing block because polymers composed solely of this more hydrophobic heparosan derivative migrated away from the origin, thus complicating facile and efficient recovery.
The reaction mixture containing the precursor for HS polysaccharide 1 was used as the starting material for targets 3-8. To create a more homogenous polymer with an equivalent and defined non-reducing terminus, the repeating GlcNAc-GlcA-pNP polysaccharide 1 product was further treated with a synthase and one UDP-sugar and then purified to reduce its size heterogeneity. First, the HS precursor was end-capped to ensure all chains terminated with a GlcA residue via reaction with ∼5 molar eq of UDP-GlcA and 0.5 μg/μl wild-type MBP-PmHS1 enzyme (27) to create a uniform population of NA polysaccharide acceptor. Second, a SAX chromatography step on Sepharose Q resin with a linear gradient (0.15 m wash for 5 column volumes and then a gradient to 1 m over 10 column volumes in 25 min) followed by repeated lyophilization of the target fractions from water was used. The end capping and SAX steps were then repeated in tandem for another cycle. The final central SAX peak was harvested to reduce the sample complexity from the original ∼12 major species to ∼8 major species (where “major” is defined as any species that is present at an intensity level of at least 30% or more of the most abundant peak ion) as observed by LC-MS (69). The molecular weight of the central peak polymer of resulting precursor 1 was 6,400 by LC-MS.
The precursor for HS 3 (made in a reaction of the pre-SAX-purified precursor for HS 1 as the acceptor for reactions with UDP-GlcNTFA and UDP-GlcA donors) was isolated by preparative descending paper chromatography (65:35 ethanol, 1 m ammonium acetate, pH 5.5, development solvent with Whatman 3MM); the polysaccharide remains at the origin of the paper strip, whereas any excess UDP-sugars and the UDP by-product migrate substantially down the paper. The origin with the target was cut out, air-dried, washed with ethanol, dried again, and then eluted with water. The samples were then frozen, and the volatile salt was removed by three cycles of lyophilization from water (typical overall yield, >95%).
To create precursors for HS 4-8, extensions ranging from five to 11 monosaccharide units were sequentially added to the non-reducing terminus of the SAX-polished, GlcA end-capped precursor to HS 1 acceptor. These stepwise addition reactions (e.g. first adding UDP-hexosamine, purification, and then adding UDP-GlcA, etc.) used 1.2 molar eq of donor and a 1 μg/μl concentration of the appropriate monofunctional Chimera G catalyst (e.g. first the hexosamine transferase and then the GlcA-transferase, etc.) under the same general reaction conditions as described for the block polymers. The choice of hexosamine in any position of the chain was controlled by the UDP-sugar, either UDP-GlcNAc or UDP-GlcNTFA, used in a given step. At some steps, an intermediate polymer was split into two parallel reactions that received either one or the other hexosamine precursor, thus affording control of the N-sulfation status of the terminal non-reducing sugar.
After each stepwise sugar addition, the target molecule was isolated by batch mode paper chromatography; in this high capacity method, the polysaccharide was spotted onto squares of Whatman 3MM paper at ∼0.3 mg/cm2, air-dried, and washed with eight changes of 65:35 ethanol, 1 m ammonium acetate, pH 5.5 (15–45 min each) to remove excess UDP-sugars and UDP. The squares were then washed with ethanol twice (10 min each), air-dried, and then eluted with water. The volatile salt was removed by repeated lyophilization from water as described before.
Detrifluoroacetylation and N-Sulfonation
Briefly, the N-trifluoroacetylated block copolymer was dissolved in a solution of 1:1:0.5 MeOH:H2O:Et3N at a concentration of 1 mg/ml and stirred overnight at room temperature to expose the amine functionality protected by the TFA group. The de-N-trifluoroacetylated block copolymer was then loaded onto a 3,000 MWCO Amicon Ultra spin unit and washed with distilled H2O three times at 14,000 × g for 10 min. The retentate containing the de-N-trifluoroacetylated block copolymer was recovered and lyophilized.
N-Sulfonation of block copolymers was then performed according to a modified procedure from Maruyama et al. (70) using NMe3·SO3 as the sulfonating agent. The lyophilized de-N-trifluoroacetylated polysaccharide was then dissolved in distilled H2O (1 mg/ml) at pH 7. Na2CO3 and NMe3·SO3 were then added in a 3:1 (w/w) ratio to polysaccharide (e.g. 3 mg each of Na2CO3 and NMe3·SO3 for 1 mg of starting polymer) and stirred for 12 h at 45 °C. A second equivalent portion of Na2CO3 and NMe3·SO3 was then added to the reaction and stirred for an additional 12 h at 45 °C. The reaction mixture was loaded into a 3,000 MWCO Amicon Ultra spin unit and washed with distilled H2O three times at 14,000 × g for 10 min. The retentate containing the N-sulfonated polysaccharide was recovered and lyophilized to afford a white fluffy powder.
Characterization of HS Block Copolymer Intermediates by NMR
Analysis of the resulting block copolymers using PAGE (56) confirmed that the polymer backbone remained intact following the de-N-trifluoroacetylation and N-sulfonation reactions. The four block copolymers were also characterized by 1D 1H NMR spectroscopy after the chemical N-sulfonation step. All samples were dissolved in 400 μl of D2O (99.9%; Sigma-Aldrich) and lyophilized three times to remove the exchangeable protons. The samples were redissolved in 400 μl of 99.9% D2O and transferred to NMR microtubes. All NMR experiments were performed at 298 K on a Bruker Advance II 600 MHz instrument with Topspin 2.1.6 software. 1Dl 1H spectra were recorded for 32 scans.
Enzymatic O-Sulfonation
The N-sulfonated polysaccharide backbones were subjected to modifications by C5-epimerase, 2-O-sulfotransferase, and 6-O-sulfotransferase 1 and 6-O-sulfotransferase 3 to introduce IdoA residues, 2-O-sulfo groups, and 6-O-sulfo groups, respectively. For the C5-epimerase (30) and 2-O-sulfotransferase modifications, the reaction was carried out in one-pot format to drive the reversible epimerization reaction forward. Briefly, the backbone polysaccharides (0.1 mg/ml) were incubated with C5-epimerase (0.2 mg/ml) in a buffer containing 1 mm CaCl2 and 50 mm MES, pH 7.0, at 37 °C. After 30-min incubation, 2-O-sulfotransferase (0.1 mg/ml) and PAPS (100 μm) were added to the reaction mixture. The reaction mixture was then incubated at 37 °C overnight. The product was purified using DEAE-Sepharose column chromatography as described previously (15). After 2-O-sulfonation, the product (0.1 mg/ml) was further modified with 6-O-sulfotransferase 1 (0.1 mg/ml) and 6-O-sulfotransferase 3 (0.1 mg/ml) in 50 mm MES, pH 7.0, PAPS (100 μm) at 37 °C overnight. The 6-O-sulfonated products were then also purified through a DEAE-Sepharose column.
Polysaccharide Purification
Each chemoenzymatically synthesized HS sample was freeze-dried and then dissolved in endotoxin-free water at a concentration of 2–8 mg/ml. Each HS sample (0.8–3.1 mg) was bound on the Vivapure® Q Maxi H mini SAX columns that had been pre-equilibrated with water by centrifugation at 2,000 × g for 10 min. Columns were washed thrice with 400 μl of water and thrice with 400 μl of 0.2 m aqueous NaCl. The HS was then eluted by washing thrice with 400 μl of 1.0 m and thrice with 2.0 m aqueous NaCl. The combined eluted samples were desalted by 1,000 MWCO dialysis membranes and filtered through 0.2-μm syringe filters. The recovered samples were subjected to carbazole assay (65) to measure each HS used to prepare stock solutions for chemical and biological analyses.
Disaccharide Compositional Analysis of Precursor and Block Copolymer Products by Using HPLC-MS
The disaccharide compositions of block copolymers were analyzed by high performance liquid chromatography-mass spectrometry (71). Heparosan oligosaccharides (1–10 μg) were digested by adding 100 μl of digestion buffer (50 mm ammonium acetate containing 2 mm calcium chloride adjusted to pH 7.0) and recombinant heparin lyases I, II, and III (10 milliunits each in Tris-HCl buffer, pH 7.0). The samples were digested at 37 °C overnight. The reaction was terminated by eliminating the enzyme via trapping in the retentate of 3,000 MWCO spin columns. The filter units were washed twice with 300 μl of distilled water, and the filtrates with disaccharides were collected, combined, and lyophilized. The dried digested HS samples were AMAC-labeled by adding 20 μl of 0.1 m AMAC in DMSO:acetic acid (17:3, v/v) and incubating at room temperature for 10 min followed by adding 20 μl of 1 m aqueous sodium cyanoborohydride and incubating for 1 h at 45 °C. A mixture containing all eight disaccharide standards prepared at 12.5 ng/μl was similarly AMAC-labeled and used for each run as an external standard. After the AMAC labeling reaction, the samples were centrifuged, and each supernatant was recovered.
For the disaccharide analysis of intermediate samples before extensive purification, LC-MS analyses were performed on an Agilent 1200 LC/MSD Instrument (Agilent Technologies, Inc., Wilmington, DE) equipped with a 6300 ion trap and a binary pump. LC was performed at 45 °C using an Agilent Poroshell 120 ECC18 (2.7-μm, 3.0 × 50-mm) column. Mobile phase A was 50 mm ammonium acetate aqueous solution, and mobile phase B was methanol. The flow rate was 300 μl/min. The concentration of mobile phase A increased from 5 to 45% during 10 min and then rose to 100% mobile phase B in the following 0.2 min, and a 4-min hold at 100% mobile phase B was applied to elute all compounds. The mass spectrometer was operated in negative ionization mode with a skimmer potential of −40.0 V, a capillary exit of −40.0 V, and a source temperature of 350 °C. Mass range of the spectrum was 300–900 m/z. Nitrogen (8 liters/min; 40 p.s.i.) was used as drying and nebulizing gas.
For disaccharide analysis of samples after purification, a triple quadrupole mass spectrometry system equipped with an electrospray ionization source (Thermo Fisher Scientific, San Jose, CA) was used as a detector. The online MS analysis was in the multiple reaction monitoring mode. The conditions and collision energies for the all of the disaccharide multiple reaction monitoring transitions were as described before (16).
Cell Culture
BaF3 cells (a murine immortalized bone marrow cell line) were grown in RPMI 1640 medium supplemented with 10% newborn calf serum, 5 ml of penicillin/streptomycin solution, 50 μm β-mercaptoethanol, and 400 μg/ml G418 as described previously (43, 54). In the absence of the FGF and GAG combination, BaF3 cells can be grown with the addition of 5 ng/ml murine IL3 (Life Technologies) to the medium. Cells were grown at 37 °C with 5% CO2 in T75 culture flasks. Cells were passaged every 3 days with initial seeding densities of 2 × 105 cells/ml.
Prior to use in the signaling assay, cells were centrifuged at 200 × g for 5 min, spent medium was removed through vacuum aspiration, and the cell pellet washed with 5 ml of IL3-free RPMI 1640 medium. The centrifugation and washing step were repeated four times to remove any residual IL3. The resulting cell pellet was then used in the signaling assay below.
BaF3 Signaling Assay
RPMI 1640 medium with FGF1 or FGF2 (5 μm final) or no growth factor was added to the washed BaF3 cell pellet to form a suspension (5 × 106 cells/ml) that was dispensed into a 96-well plate at 50,000 cells/well. Solutions of the individual HS block copolymers or heparin (positive control) in PBS were added to obtain a final concentration of 2 μg/ml. The negative control was PBS vehicle only. The plate was incubated at 37 °C in 5% CO2 for 24 h. Each sample was tested in triplicate wells.
Cell proliferation was measured with an MTT assay. Briefly, 40 μl of a 2.5 mg/ml MTT solution was added to each well, and incubated for 3 h. Then 100 μl of 10% sodium dodecyl sulfate (SDS) in 0.01 n HCl was added to each well and gently shaken overnight to dissolve the formazan crystals. Optical density was measured at 590 nm and at 690 nm as a control, the value of which was subtracted from the absorbance at 590 nm to give a background-corrected optical density. Optical density was correlated to cell count using a standard curve.
Author Contributions
V. S. did chemical synthesis and polysaccharide analysis and coordinated the study. M. S. did cell activity assays. X. L. did MS analysis. X. Z. did NMR analysis. Y. Y. did polysaccharide purification. L. L. helped in experimental design and MS analysis. D. E. G. did polysaccharide chain synthesis. Y. X. did enzymatic sulfation. F. Z. coordinated the study and assisted in writing. P. L. D. contributed to conception, design, and writing the manuscript. J. L. helped in conception and design. R. J. L. contributed to conception, design, and writing the manuscript.
Acknowledgment
We thank Dr. Anais Chavaroche-Wickham for the monofunctional MBP-Chimera G mutagenesis work.
This work was supported by National Institutes of Health Grants HL094463 and HL62244. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GAG
- glycosaminoglycan
- GlcN
- α-d-glucosamine
- PG
- proteoglycan
- HS
- heparan sulfate
- GlcA
- β-d-glucuronic acid
- IdoA
- α-l-iduronic acid
- GlcNAc
- N-acetylglucosamine
- SAR
- structure-activity relationship
- PAPS
- 3′-phosphoadenosine 5′-phosphosulfate
- FGFR
- fibroblast growth factor receptor
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- MWCO
- molecular weight cutoff
- AMAC
- 2-aminoacridone
- NS
- block containing GlcNS in the disaccharide repeats and a high content of O-sulfo groups
- NA
- domain containing only GlcNAc in the disaccharide repeating units and low level O-sulfo groups
- SAX
- strong anion exchange
- MBP
- maltose-binding protein
- TFA
- trifluoroacetyl
- pNP
- p-nitrophenyl
- OST
- O-sulfotransferase
- TriS
- trisulfated.
References
- 1. Griffin C. C., Linhardt R. J., Van Gorp C. L., Toida T., Hileman R. E., Schubert R. L. 2nd, and Brown S. E. (1995) Isolation and characterization of heparan sulfate from crude porcine intestinal mucosa peptidoglycan heparin. Carbohydrate Research 276, 183–197 [DOI] [PubMed] [Google Scholar]
- 2. Gallagher J. T. (1997) Structure-activity relationship of heparan sulfate. Biochem. Soc. Trans. 25, 1206–1209 [DOI] [PubMed] [Google Scholar]
- 3. Safaiyan F., Lindahl U., and Salmivirta M. (2000) Structural diversity of N-sulfated heparan sulfate domains: distinct modes of glucuronyl C5 epimerization, iduronic acid 2-O-sulfation, and glucosamine 6-O-sulfation. Biochemistry 39, 10823–10830 [DOI] [PubMed] [Google Scholar]
- 4. Jastrebova N., Vanwildemeersch M., Lindahl U., and Spillmann D. (2010) Heparan sulfate domain organization and sulfation modulate FGF-induced cell signaling. J. Biol. Chem. 285, 26842–26851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kusche-Gullberg M., Nybakken K., Perrimon N., and Lindahl U. (2012) Drosophila heparan sulfate, a novel design. J. Biol. Chem. 287, 21950–21956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smits N. C., Kurup S., Rops A. L., ten Dam G. B., Massuger L. F., Hafmans T., Turnbull J. E., Spillmann D., Li J. P., Kennel S. J., Wall J. S., Shworak N. W., Dekhuijzen P. N., van der Vlag J., and van Kuppevelt T. H. (2010) The heparan sulfate motif (GlcNS6S-IdoA2S)3, common in heparin, has a strict topography and is involved in cell behavior and disease. J. Biol. Chem. 285, 41143–41151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lambaerts K., Wilcox-Adelman S. A., and Zimmermann P. (2009) The signaling mechanisms of syndecan heparan sulfate proteoglycans. Curr. Opin. Cell Biol. 21, 662–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lindahl U., and Li J. P. (2009) Interactions between heparan sulfate and proteins—design and functional implications. Int. Rev. Cell Mol. Biol. 276, 105–159 [DOI] [PubMed] [Google Scholar]
- 9. Salmivirta M., Lidholt K., and Lindahl U. (1996) Heparan sulfate: a piece of information. FASEB J. 10, 1270–1279 [DOI] [PubMed] [Google Scholar]
- 10. Capila I., and Linhardt R. J. (2002) Heparin-protein interactions. Angew. Chem. Int. Ed. Engl. 41, 391–412 [DOI] [PubMed] [Google Scholar]
- 11. Kreuger J., Spillmann D., Li J. P., and Lindahl U. (2006) Interactions between heparan sulfate and proteins: the concept of specificity. J. Cell Biol. 174, 323–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Staples G. O., Shi X., and Zaia J. (2011) Glycomics analysis of mammalian heparan sulfates modified by the human extracellular sulfatase HSulf2. PLoS One 6, e16689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramani V. C., Purushothaman A., Stewart M. D., Thompson C. A., Vlodavsky I., Au J. L., and Sanderson R. D. (2013) The heparanase/syndecan-1 axis in cancer: mechanisms and therapies. FEBS J. 280, 2294–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rosenberg R. D., Armand G., and Lam L. (1978) Structure-function relationship of heparin species. Proc. Natl. Acad. Sci. U.S.A. 75, 3065–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gallagher J. T., Turnbull J. E., and Lyon M. (1992) Patterns of sulphation in heparan sulphate: polymorphism based on a common structural theme. Int. J. Biochem. 24, 553–660 [DOI] [PubMed] [Google Scholar]
- 16. Sun X., Li L., Overdier K. H., Ammons L. A., Douglas I. S., Burlew C. C., Zhang F., Schmidt E. P., Chi L., and Linhardt R. J. (2015) Analysis of total human urinary glycosaminoglycan disaccharides by liquid chromatography-tandem mass spectrometry. Anal. Chem. 87, 6220–6227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Puvirajesinghe T. M., Ahmed Y. A., Powell A. K., Fernig D. G., Guimond S. E., and Turnbull J. E. (2012) Array-based functional screening of heparin glycans. Chem. Biol. 19, 553–558 [DOI] [PubMed] [Google Scholar]
- 18. Pervin A., Gallo C., Jandik K. A., Han X.-J., and Linhardt R. J. (1995) Preparation and structural characterization of large heparin-derived oligosaccharides. Glycobiology 5, 83–95 [DOI] [PubMed] [Google Scholar]
- 19. Hileman R. E., Smith A. E., Toida T., and Linhardt R. J. (1997) Preparation and structure of heparin lyase-derived heparan sulfate oligosaccharides. Glycobiology 7, 231–239 [DOI] [PubMed] [Google Scholar]
- 20. Liu R., Xu Y., Chen M., Weïwer M., Zhou X., Bridges A. S., DeAngelis P. L., Zhang Q., Linhardt R. J., and Liu J. (2010) Chemoenzymatic design of heparan sulfate oligosaccharides. J. Biol. Chem. 285, 34240–34249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu Y., Masuko S., Takieddin M., Xu H., Liu R., Jing J., Mousa S. A., Linhardt R. J., and Liu J. (2011) Chemoenzymatic synthesis of structurally homogeneous ultra-low molecular weight heparins. Science 334, 498–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sterner E., Masuko S., Li G., Li L., Green D. E., Otto N. J., Xu Y., DeAngelis P. L., Liu J., Dordick J. S., and Linhardt R. J. (2014) Fibroblast growth factor-based signaling through synthetic heparan sulfate block copolymers studied using high-cell density three-dimensional cell printing. J. Biol. Chem. 289, 9754–9765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. DeAngelis P. L. (2013) Methods for the Pasteurella glycosaminoglycan synthases: enzymes that polymerize hyaluronan, chondroitin, or heparosan chains. Methods Mol. Biol. 1022, 215–227 [DOI] [PubMed] [Google Scholar]
- 24. Chen J., Avci F. Y., Muñoz E. M., McDowell L. M., Chen M., Pedersen L. C., Zhang L., Linhardt R. J., and Liu J. (2005) Enzymatic redesigning of biologically active heparan sulfate. J. Biol. Chem. 280, 42817–42825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. DeAngelis P. L., Liu J., and Linhardt R. J. (2013) Chemoenzymatic synthesis of glycosaminoglycans: re-creating, re-modeling, and re-designing nature's longest or most complex carbohydrate chains. Glycobiology 23, 764–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Masuko S., Bera S., Green D. E., Weïwer M., Liu J., DeAngelis P. L., and Linhardt R. J. (2012) Chemoenzymatic synthesis of UDP-GlcNAc and UDP-GalNAc analogs for the preparation of unnatural glycosaminoglycans. J. Org. Chem. 77, 1449–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sismey-Ragatz A. E., Green D. E., Otto N. J., Rejzek M., Field R. A., and DeAngelis P. L. (2007) Chemoenzymatic synthesis with distinct Pasteurella heparosan synthases. J. Biol. Chem. 282, 28321–28327 [DOI] [PubMed] [Google Scholar]
- 28. Williams K. J., Halkes K. M., Kamerling J. P., and DeAngelis P. L. (2006) Critical elements of oligosaccharide acceptor substrates for the Pasteurella multocida hyaluronan synthase. J. Biol. Chem. 281, 5391–5397 [DOI] [PubMed] [Google Scholar]
- 29. Jing W., and DeAngelis P. L. (2004) Synchronized chemoenzymatic synthesis of monodisperse hyaluronan polymers. J. Biol. Chem. 279, 42345–42349 [DOI] [PubMed] [Google Scholar]
- 30. Li J., Hagner-McWhirter A., Kjellén L., Palgi J., Jalkanen M., and Lindahl U. (1997) Biosynthesis of heparin/heparan sulfate. cDNA cloning and expression of d-glucuronyl C5-epimerase from bovine lung. J. Biol. Chem. 272, 28158–28163 [DOI] [PubMed] [Google Scholar]
- 31. Habuchi H., Kobayashi M., and Kimata K. (1998) Molecular characterization and expression of heparan-sulfate 6-sulfotransferase. Complete cDNA cloning in human and partial cloning in Chinese hamster ovary cells. J. Biol. Chem. 273, 9208–9213 [DOI] [PubMed] [Google Scholar]
- 32. Shworak N. W., Liu J., Fritze L. M., Schwartz J. J., Zhang L., Logeart D., and Rosenberg R. D. (1997) Molecular cloning and expression of mouse and human cDNAs encoding heparan sulfate d-glucosaminyl 3-O-sulfotransferase. J. Biol. Chem. 272, 28008–28019 [DOI] [PubMed] [Google Scholar]
- 33. Esko J. D., and Selleck S. B. (2002) Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 71, 435–471 [DOI] [PubMed] [Google Scholar]
- 34. Peterson S., Frick A., and Liu J. (2009) Design of biologically active heparan sulfate and heparin using an enzyme-based approach. Nat. Prod. Rep. 26, 610–627 [DOI] [PubMed] [Google Scholar]
- 35. Liu J., Moon A. F., Sheng J., and Pedersen L. C. (2012) Understanding substrate specificity of the heparan sulfate sulfotransferases by an integrated biosynthesis and crystallographic approach. Curr. Opin. Struct. Biol. 22, 550–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sheng J., Xu Y., Dulaney S. B., Huang X., and Liu J. (2012) Uncovering biphasic catalytic mode of C5-epimerase in heparan sulfate biosynthesis. J. Biol. Chem. 287, 20996–21002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Montesano R., Vassalli J.-D., Baird A., Guillemin R., and Orci L. (1986) Basic fibroblast growth factor induces angiogenesis in vitro. Proc. Natl. Acad. Sci. U.S.A. 83, 7297–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Iwabu A., Smith K., Allen F. D., Lauffenburger D. A., and Wells A. (2004) Epidermal growth factor induces fibroblast contractility and motility via a protein kinase C δ-dependent pathway. J. Biol. Chem. 279, 14551–14560 [DOI] [PubMed] [Google Scholar]
- 39. Marie P. J. (2003) Fibroblast growth factor signaling controlling osteoblast differentiation. Gene 316, 23–32 [DOI] [PubMed] [Google Scholar]
- 40. Melder R. J., Koenig G. C., Witwer B. P., Safabakhsh N., Munn L. L., and Jain R. K. (1996) During angiogenesis, vascular endothelial growth factor and basic fibroblast growth factor regulate natural killer cell adhesion to tumor endothelium. Nat. Med. 2, 992–997 [DOI] [PubMed] [Google Scholar]
- 41. Sanchez-Heras E., Howell F. V., Williams G., and Doherty P. (2006) The fibroblast growth factor receptor acid box is essential of interactions with N-cadherin and all of the major isoforms of neural cell adhesion molecule. J. Biol. Chem. 281, 35208–35216 [DOI] [PubMed] [Google Scholar]
- 42. Ornitz D. M., and Itoh N. (2001) Fibroblast growth actors. Genome Biol. 2, reviews3005.1–reviews3005.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ornitz D. M., Xu J., Colvin J. S., McEwen D. G., MacArthur C. A., Coulier F., Gao G., and Goldfarb M. (1996) Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 271, 15292–15297 [DOI] [PubMed] [Google Scholar]
- 44. Mohammadi M., Olsen S. K., and Ibrahimi O. A. (2005) Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 16, 107–137 [DOI] [PubMed] [Google Scholar]
- 45. Ibrahimi O. A., Zhang F., Hrstka S. C., Mohammadi M., and Linhardt R. J. (2004) Kinetic model for FGF, FGFR, and proteoglycan signal transduction complex assembly. Biochemistry 43, 4724–4730 [DOI] [PubMed] [Google Scholar]
- 46. Brown A., Robinson C. J., Gallagher J. T., and Blundell T. L. (2013) Cooperative heparin-mediated oligomerization of fibroblast growth factor-1 (FGF1) precedes recruitment of FGFR2 to ternary complexes. Biophys. J. 104, 1720–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jaye M., Schlessinger J., and Dionne C. A. (1992) Fibroblast growth factor receptor tyrosine kinases: molecular analysis and signal transduction. Biochim. Biophys. Acta 1135, 185–199 [DOI] [PubMed] [Google Scholar]
- 48. Mulloy B., and Linhardt R. J. (2001) Order out of complexity: protein structures that interact with heparin. Curr. Opin. Struct. Biol. 11, 623–628 [DOI] [PubMed] [Google Scholar]
- 49. Powell A. K., Fernig D. G., and Turnbull J. E. (2002) Fibroblast growth factors 1 and 2 interact differently with heparin/heparan sulfate: implications for dynamic assembly of a ternary signaling complex. J. Biol. Chem. 277, 28554–28563 [DOI] [PubMed] [Google Scholar]
- 50. Rusnati M., Coltrini D., Caccia P., Dell'Era P., Zoppetti G., Oreste P., Valsasina B., and Presta M. (1994) Distinct role of 2-O-, N-, and 6-O-sulfate groups of heparin in the formation of the ternary complex with basic fibroblast growth factor and soluble FGF receptor-1. Biochem. Biophys. Res. Commun. 203, 450–458 [DOI] [PubMed] [Google Scholar]
- 51. Robinson C. J., Harmer N. J., Goodger S. J., Blundell T. L., and Gallagher J. T. (2005) Cooperative dimerization of fibroblast growth factor 1 (FGF1) upon a single heparin saccharide may drive the formation of 2:2:1 FGF1·FGFR2c·heparin ternary complexes. J. Biol. Chem. 280, 42274–42282 [DOI] [PubMed] [Google Scholar]
- 52. Wu Z. L., Zhang L., Yabe T., Kuberan B., Beeler D. L., Love A., and Rosenberg R. D. (2003) The involvement of heparan sulfate (HS) in FGF1/HS/FGFR1 signaling complex. J. Biol. Chem. 278, 17121–17129 [DOI] [PubMed] [Google Scholar]
- 53. Zhang X., Ibrahimi O. A., Olsen S. K., Umemori H., Mohammadi M., and Ornitz D. M. (2006) Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 281, 15694–16700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ornitz D. M., and Leder P. (1992) Ligand specificity and heparin dependence of fibroblast growth factor receptors 1 and 3. J. Biol. Chem. 267, 16305–16311 [PubMed] [Google Scholar]
- 55. Schlessinger J., Plotnikov A. N., Ibrahimi O. A., Eliseenkova A. V., Yeh B. K., Yayon A., Linhardt R. J., and Mohammadi M. (2000) Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 6, 743–750 [DOI] [PubMed] [Google Scholar]
- 56. Naimy H., Buczek-Thomas J. A., Nugent M. A., Leymarie N., and Zaia J. (2011) Highly sulfated nonreducing end-derived heparan sulfate domains bind fibroblast growth factor-2 with high affinity and are enriched in biologically active fractions. J. Biol. Chem. 286, 19311–19319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ibrahimi O. A., Yeh B. K., Eliseenkova A. V., Zhang F., Olsen S. K., Igarashi M., Aaronson S. A., Linhardt R. J., and Mohammadi M. (2005) Analysis of mutations in FGF and a pathogenic mutation in FGFR provide direct evidence in support of the two-end model for FGFR dimerization. Mol. Cell. Biol. 25, 671–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Faham S., Linhardt R. J., and Rees D. C. (1998) Diversity does make a difference: fibroblast growth factor-heparin interactions. Curr. Opin. Struct. Biol. 8, 578–586 [DOI] [PubMed] [Google Scholar]
- 59. Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., and Etzler M. E. (eds) (2009) Essentials of Glycobiology, 2nd Ed., pp. 1–29, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 60. Zhang Z., Coomans C., and David G. (2001) Membrane heparan sulfate proteoglycan-supported FGF2-FGFR1 signaling: evidence in support of the “cooperative end structures” model. J. Biol. Chem. 276, 41921–41929 [DOI] [PubMed] [Google Scholar]
- 61. Sterner E., Meli L., Kwon S. J., Dordick J. S., and Linhardt R. J. (2013) FGF-FGFR signaling mediated through glycosaminoglycans in microtiter plate and cell-based microarray platforms. Biochemistry 52, 9009–9019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nguyen T. K., Raman K., Tran V. M., and Kuberan B. (2011) Investigating the mechanism of the assembly of FGF1-binding heparan sulfate motifs. FEBS Lett. 585, 2698–2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Otto N. J., Green D. E., Masuko S., Mayer A., Tanner M. E., Linhardt R. J., and DeAngelis P. L. (2012) Structure/function analysis of Pasteurella multocida heparosan synthases: toward defining enzyme specificity and engineering novel catalysts. J. Biol. Chem. 287, 7203–7212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kane T. A., White C. L., and DeAngelis P. L. (2006) Functional characterization of PmHS1, a Pasteurella multocida heparosan synthase. J. Biol. Chem. 281, 33192–33197 [DOI] [PubMed] [Google Scholar]
- 65. Baik J. Y., Dahodwala H., Oduah E., Talman L., Gemmill T. R., Gasimli L., Datta P., Yang B., Li G., Zhang F., Li L., Linhardt R. J., Campbell A. M., Gorfien S. F., and Sharfstein S. T. (2015) Optimization of bioprocess conditions improves production of a CHO cell-derived, bioengineered heparin. Biotechnol. J., 10, 1067–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhou X., Chandarajoti K., Pham T. Q., Liu R., and Liu J. (2011) Expression of secreted forms of sulfotransferases in Kluyveromyces lactis to synthesize heparin and heparan sulfate. Glycobiology 21, 771–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Su H., Blain F., Musil R. A., Zimmermann J. J., Gu K., and Bennett D. C. (1996) Isolation and expression in Escherichia coli of hepB and hepC, genes coding for the glycosaminoglycan-degrading enzymes heparinase II and heparinase III, respectively, from Flavobacterium heparinum. Appl. Environ. Microbiol. 62, 2723–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li G., Masuko S., Green D. E., Xu Y., Li L., Zhang F., Xue C., Liu J., DeAngelis P. L., and Linhardt R. J. (2013) N-Sulfotestosteronan, a novel substrate for heparan sulfate 6-O-sulfotransferases and its analysis by oxidative degradation. Biopolymers 99, 675–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li L., Zhang F., Zaia J., and Linhardt R. J. (2012) Top-down approach for the direct characterization of low molecular weight heparins using LC-FT-MS. Anal. Chem. 84, 8822–8829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Maruyama T., Toida T., Imanari T., Yu G., and Linhardt R. J. (1998) Conformational changes and anticoagulant activity of chondroitin sulfate following its O-sulfonation. Carbohydr. Res. 306, 35–43 [DOI] [PubMed] [Google Scholar]
- 71. Yang B., Chang Y., Weyers A. M., Sterner E., and Linhardt R. J. (2012) Disaccharide analysis of glycosaminoglycan mixtures by ultra-high-performance liquid-chromatography-mass spectrometry. J. Chromatogr. A. 1224, 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]