

Abstract
Selectin interactions with fucosylated glycan ligands mediate leukocyte rolling in the vasculature under shear forces. Crystal structures of P- and E-selectin suggest a two-state model in which ligand binding to the lectin domain closes loop 83–89 around the Ca2+ coordination site, enabling Glu-88 to engage Ca2+ and fucose. This triggers further allostery that opens the lectin/EGF domain hinge. The model posits that force accelerates transition from the bent (low affinity) to the extended (high affinity) state. However, transition intermediates have not been described, and the role of Glu-88 in force-assisted allostery has not been examined. Here we report the structure of the lectin and EGF domains of L-selectin bound to a fucose mimetic; that is, a terminal mannose on an N-glycan attached to a symmetry-related molecule. The structure is a transition intermediate where loop 83–89 closes to engage Ca2+ and mannose without triggering allostery that opens the lectin/EGF domain hinge. We used three complementary assays to compare ligand binding to WT selectins and to E88D selectins that replaced Glu-88 with Asp. Soluble P-selectinE88D bound with an ∼9-fold lower affinity to PSGL-1, a physiological ligand, due to faster dissociation. Adhesion frequency experiments with a biomembrane force probe could not detect interactions of P-selectinE88D with PSGL-1. Cells expressing transmembrane P-selectinE88D or L-selectinE88D detached from immobilized ligands immediately after initiating flow. Cells expressing E-selectinE88D rolled but detached faster. Our data support a two-state model for selectins in which Glu-88 must engage ligand to trigger allostery that stabilizes the high affinity state under force.
Keywords: carbohydrate-binding protein, cell adhesion, endothelial cell, glycobiology, inflammation, leukocyte
Introduction
Selectins are adhesion receptors that mediate the initial tethering and rolling of leukocytes on vascular surfaces during inflammation, hemostasis, and immune surveillance (1, 2). L-selectin is expressed on leukocytes, E-selectin is expressed on activated endothelial cells, and P-selectin is expressed on activated platelets and endothelial cells. Selectins mediate rolling through reversible interactions (bonds) with cell-surface ligands that are subjected to forces exerted by blood flow. Lower forces prolong bond lifetimes by decreasing off-rates (catch bonds), whereas higher forces shorten bond lifetimes by increasing off-rates (slip bonds).
Each of the selectins has an N-terminal C-type lectin domain followed by an EGF domain, a series of consensus repeats, a transmembrane domain, and a cytoplasmic tail (1, 2). The minimal ligand structure for selectins is sialyl Lewis x (sLex)2; NeuAcα2–3Galβ1–4[Fucα1–3]GlcNAcβ1-R), a terminal component of some N- and Ο-glycans on hematopoietic and endothelial cells. The lectin domain forms Ca2+-dependent interactions with fucose and additional interactions with sialic acid and galactose. L-selectin prefers sLex modified with sulfate on N-acetylglucosamine, which occurs on N- and O-glycans of mucins on endothelial cells of lymph nodes. In addition, P- and L-selectin bind cooperatively to an sLex-capped O-glycan, sulfated tyrosines, and other residues at the N terminus of P-selectin glycoprotein ligand-1 (PSGL-1), a mucin on leukocytes.
Crystal structures of soluble human P- and E-selectin proteins comprising the lectin and EGF domains and of some E-selectin proteins, also the first two consensus repeats, reveal two conformational states termed bent and extended (3–5). These differ in the orientations between the lectin and EGF domains and in the architecture of the ligand binding site. P- and E-selectin crystallized without ligand are in the bent state, and sLex soaked into these crystals binds to the bent conformation. In contrast, P-selectin cocrystallized with an N-terminal PSGL-1 fragment and E-selectin cocrystallized with sLex or an sLex mimetic are in the extended state. These structures are consistent with a two-state model in which ligand binding favors allosteric conversion to the extended state (4–6). The model also predicts that force applied to ligand bound to a membrane-anchored selectin will facilitate extension. Allosteric conversion to the extended state could represent a mechanism for catch bonds (7). Mutating an L-selectin residue that increases the flexibility of the lectin/EGF domain hinge reduces the force required to elicit catch bonds, consistent with this hypothesis (8). Mutating P-selectin residues predicted to lock the lectin domain in the conformation of the extended state increases the force-free affinity for PSGL-1 (9, 10). However, mutations that lock selectins in the putative low affinity conformations of the bent states have not been described.
A key feature of the two-state model is movement of a loop comprising residues 83–89 near the Ca2+ coordination site of the lectin domain (4, 5). In both bent and extended states of P- and E-selectin, conserved residues Glu-80, Asn-82, Asn-105, and Asn-106 simultaneously coordinate a Ca2+ ion and the 3- and 4-hydroxyl groups of fucose. In the bent state the 83–89 loop is open. In the extended state the loop closes, allowing Glu-88 to interact with Ca2+ and the 2-hydroxyl group of fucose. The closed loop also enables Arg-85 in P-selectin to interact with a tyrosine sulfate in PSGL-1 and Gln-85 in E-selectin to dock with fucose (4, 5). Glu-88 is conserved in L-selectin, but no structure has documented its orientation in the presence or absence of bound ligand. It is not known whether the Glu-88 interactions are essential for high affinity binding of selectins to their ligands, particularly when bonds are subjected to force. The two-state model further predicts that ligand-induced closure of the 83–89 loop precedes the large scale movements of interdomain extension (5, 6). However, no crystal structure has captured this intermediate conformation.
Here we report the structure of the lectin and EGF domains of human L-selectin bound to a fucose mimetic; that is, a terminal mannose on an N-glycan attached to the lectin domain of a neighboring molecule. The structure has an intermediate conformation in which the 83–89 loop closes to allow Glu-88 to interact with Ca2+ and mannose but without the large scale movements required for extension. Using site-directed mutagenesis, we demonstrate that all three selectins require Glu-88 to sustain bonds with sLex-containing ligands under force. Mutating Glu-88 to Asp locks selectins in their functionally inactive states and markedly impairs selectin-mediated cell rolling under flow.
Results
Crystal Structure of L-selectin Bound to Terminal Mannose on an N-Glycan from a Neighboring Molecule
We expressed the lectin and EGF domains of human L-selectin in Lec1 CHO cells that permit synthesis of Man5GlcNAc2 but not complex N-glycans (11). We crystallized the glycosylated L-selectin protein, which is unlike previous P- and E-selectin proteins in which N-glycans were enzymatically removed before crystallization (3–5). The L-selectin structure was solved at 2.2 Å resolution using molecular replacement (Table 1). Clear electron density was observed throughout the structure except for residues 125–131 and 145–156 in the distal regions of the EGF domain. Fig. 1A depicts a ribbon diagram of the structure. L-selectin has potential N-glycosylation sites at Asn-22 and Asn-66 in the lectin domain and at Asn-139 in the EGF domain. A single GlcNAc, termed N-glycan 1, is attached to Asn-22. A Man5GlcNAc2 pentasaccharide, termed N-glycan 2, is attached to Asn-66. No N-glycan is attached to Asn-139, although its side chain is visible.
TABLE 1.
Data collection and refinement statistics for L-selectin
| PDB entry | 3CFW |
|---|---|
| Data collection | |
| Space group | I 23 (197) |
| Cell dimensions | |
| a, b, c (Å) | 118.477 |
| α, β, γ (o) | 90 |
| Molecules in asymmetric unit | 1 |
| Resolution range (Å) | 25-2.2 |
| Rsym (%)a | 12.6 (64.4) |
| Completeness %a | 99.8 (98.9) |
| Total observations | 197,554 |
| Unique reflections | 14,192 |
| 〈I/s(I)〉a | 12.8 (2.3) |
| Model refinement | |
| Resolution (Å) | 25-2.2 |
| Rfactor (%) | 20.1 |
| Rfree (%) | 24.9 |
| 〈B value〉 (Å) | 30 |
| Root mean square deviations | |
| Bonds (Å) | 0.013 |
| Angles (o) | 1.52 |
a Values in parentheses are for the highest resolution shell.
FIGURE 1.
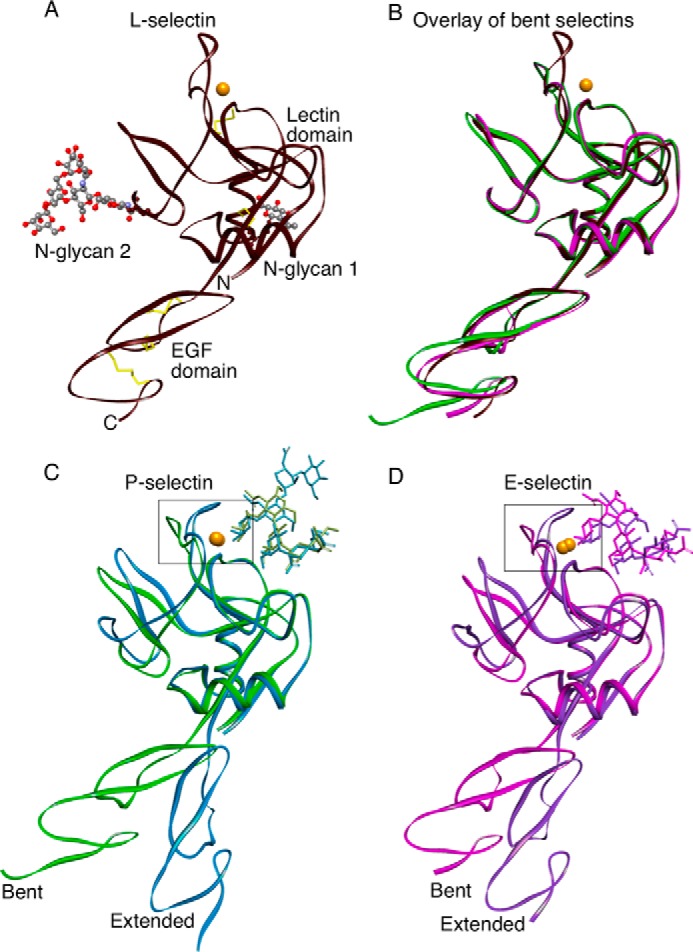
Ribbon representations of selectin structures. A, the lectin and EGF domains of L-selectin are in the bent orientation. Two N-glycans attached to the lectin domain are visible. Yellow sticks depict two disulfide bonds in the lectin domain and three disulfide bonds in the EGF domain. N and C show the amino and carboxyl ends of the protein. Coordinates for this structure have been deposited in the Protein Data Bank as 3CFW. B, overlay of the bent structures of L-selectin (brown, PDB code 3CFW), E-selectin (magenta, PDB code 1ESL), and P-selectin (green, PDB code 1G1Q). C and D, the bent structures of P-selectin with soaked sLex (green, PDB code 1G1R) or E-selectin with soaked sLex (magenta, PDB code 1G1T) are superimposed on the extended cocrystal structures P-selectin·PSGL-1 (blue, PDB code 1G1S) or E-selectin·sLex (purple, PDB code 4CSY). Gold spheres represent Ca2+ ions in all structures except for P-selectin·PSGL-1, where the sphere represents a Sr2+ ion. The boxed regions surrounding the Ca2+-coordination sites are enlarged in Fig. 3.
The L-selectin structure has a bent orientation between the lectin and EGF domains that closely resembles the bent states of P- and E-selectin (Fig. 1B). The singular difference is the loop comprising residues 83–89 near the Ca2+ coordination site in the lectin domain. The loop in L-selectin is closed, much like the closed loop in the extended structure of P-selectin cocrystallized with an sLex-containing PSGL-1 fragment and of E-selectin cocrystallized with sLex or an sLex mimetic (Fig. 1, C and D). Closer examination of L-selectin revealed that the Ca2+ coordination site is occupied by the terminal mannose of N-glycan 2 on a symmetry-related molecule in the crystal lattice (Fig. 2, A and B). The Ca2+ coordination sites of all three selectins have a Glu-Pro-Asn motif at residues 80–82, in common with a larger group of C-type lectins, including the prototypical mannose-binding protein, which binds both fucose and mannose (12). Thus, we obtained a cocrystal structure of L-selectin bound to an endogenous ligand. Binding of mannose stabilizes the 83–89 loop in the closed conformation (see below). This orientation is further stabilized by lattice contacts of Lys-85 with residues in a neighboring molecule (Fig. 2, C and D).
FIGURE 2.

L-selectin cocrystallizes with an endogenous ligand. A, lectin and EGF domains of L-selectin (brown) are rotated ∼30° clockwise from the orientation in Fig. 1A. N-Glycan 2 attached to Asn-66 of an adjacent symmetry-related molecule (bronze) occupies the ligand binding surface of L-selectin. The boxed region is magnified in panel B. B, Ball-and-stick representation of N-glycan 2 attached to Asn-66 on the symmetry-related molecule. The O3 and O4 hydroxyls of the terminal mannose (Man5) interact with the Ca2+ ion of L-selectin. C, contacts between Lys-85 on L-selectin (brown) and a symmetry-related molecule (bronze). The boxed region is magnified in D. The molecular backbone is displayed as a ribbon. Side chains of interacting residues are depicted with ball and stick. Side chains are colored like the parent molecules, except that interacting terminal oxygens are red and nitrogens are blue. Dashed lines represent hydrogen bonds. Gold spheres represent Ca2+ ions.
Ligand binding that closes the 83–89 loop is thought to trigger allosteric conversion from the bent to extended states of selectins (4–6). The L-selectin structure captures a ligand-bound conformation with a closed 83–89 loop that retains the overall bent orientation between the lectin and EGF domains. N-Glycan 2 is attached to Asn-66 on a surface loop that is normally part of a larger rigid-body movement in the lectin domain during extension. The simultaneous linkage of N-glycan 2 to the ligand binding region of one molecule and to Asn-66 on a neighboring molecule restrains the Asn-66-containing loop, decoupling linked allostery after ligand binding.
Closing Loop 83–89 Engages Glu-88 with Ca2+ and Glycan in All Selectin Structures
Binding of the terminal mannose of N-glycan 2 to L-selectin closely resembles binding of fucose to P- and E-selectin cocrystallized with sLex-containing ligands (Fig. 3, A–C). L-selectin engages the equatorial 3- and 4-hydroxyl groups of mannose, whereas P- and E-selectin engage the equatorial 3-hydroxyl and axial 4-hydroxyl groups of fucose. In all structures, Glu-80, Asn-82, Asn-105, and Asp-106 form hydrogen bonds with the hexose and ionic interactions with the Ca2+ ion. The closed 83–89 loop of L-selectin positions Glu-88 to simultaneously ligate Ca2+ and the 4-hydroxyl of mannose, whereas the closed loops of P- and E-selectin position Glu-88 to ligate Ca2+ and the 2-hydroxyl group of fucose. In E-selectin, unique loop residue Gln-85 also engages the 2-hydroxyl of fucose. Equivalent residues in mannose-binding protein (except for Gln-85) interact with Ca2+ and mannose, including Glu-193, which corresponds to Glu-88 in selectins (Fig. 3D). The loop containing Glu-193 remains closed in mannose-binding protein crystallized without ligand (13). In sharp contrast, the loop is open in P- and E-selectin crystallized without ligand and does not move when sLex is soaked into these crystals, leaving Glu-88 unengaged (Fig. 3, E and F).
FIGURE 3.

Closing loop 83–89 engages Glu-88 with Ca2+ and glycan in all selectin structures. The terminal mannose or fucose of each glycan ligand is shown. Lectin-domain residues that coordinate Ca2+ or form hydrogen bonds with mannose or fucose are represented as balls and sticks in L-selectin (PDB code 3CFW) (A), P-selectin·PSGL-1 (PDB code 1G1S) (B), E-selectin·sLex (PDB code 4CSY) (C), mannose-binding protein:Man9 (PDB code 2MSB) (D), P-selectin with soaked sLex (PDB code 1G1R) (E), and E-selectin with soaked sLex (PDB code 1G1T) (F). Gold spheres represent Ca2+ ions in all structures except for P-selectin·PSGL-1, where the sphere represents a Sr2+ ion. Lectin-domain residues Glu-80, Asn-82, Glu-88, Asn-105, and Asp-106 labeled in panel A are identical in panels B, C, E, and F. Topologically equivalent residues labeled in panel D have different numbers. Gln-85 in E-selectin·sLex is labeled. Also shown are the O2, O3, and O4 hydroxyls on the terminal fucose and the O3 and O4 hydroxyls on the terminal mannose. Regions of the lectin domain that obscure the ligand binding site have been omitted.
Replacing Glu-88 with Asp in Loop 83–89 of P-selectin Reduces Binding Affinity for PSGL-1 under Force-free Conditions
To assess the role of Glu-88 in high affinity binding to ligands, we expressed two soluble human P-selectin monomers comprising the lectin and EGF domains: the WT protein and an E88D mutant that replaced Glu-88 with Asp, thus shortening the side chain by 1.5 Å but retaining the core structure and charge. We used surface plasmon resonance to measure binding of each protein to 2-GSP-6, a glycosulfopeptide that recapitulates the N-terminal binding site on PSGL-1 (8). The 2-GSP-6 was biotinylated and captured on a streptavidin sensor at low density to avoid rebinding of P-selectin after dissociation. Sensor plots demonstrated rapid association and dissociation kinetics for both proteins, but dissociation was clearly more rapid for P-selectinE88D (Fig. 4, A and B). Data for both proteins best fit a one-state binding model. The measured koff for WT P-selectin was 3.1 ± 0.3 s−1, similar to previous results (14), whereas the koff for P-selectinE88D was greater than 10 s−1, the upper limits for detection on the instrument. Non-linear curve-fitting of the equilibrium binding isotherms indicated that P-selectinE88D bound with an ∼9-fold lower affinity than WT P-selectin (Fig. 4, C and D), at least in part from faster dissociation.
FIGURE 4.

Replacing Glu-88 with Asp in loop 83–89 of P-selectin reduces binding affinity for PSGL-1 under force-free conditions. A and B, overlays of increasing concentrations of P-selectin or P-selectinE88D binding to 2-GSP-6, a surrogate for PSGL-1, on a sensor surface. The horizontal line indicates when P-selectin or P-selectinE88D was injected. C and D, non-linear curve fits of the specific binding data generated from A and B. The data represent the mean ± S.D. from three separate experiments. RU, resonance units.
Replacing Glu-88 with Asp Prevents Binding of P-selectin to PSGL-1 Measured with a Biomembrane Force Probe
As a complementary assay, we used a biomembrane force probe to measure two-dimensional interactions of WT or E88D forms of P-selectin with 2-GSP-6 at high temporal and spatial resolution (Fig. 5A). Two micropipettes were placed on opposite sides of a chamber visualized with an inverted microscope. One micropipette held a biotinylated red blood cell used as a force transducer. Biotinylated 2-GSP-6 was attached to a streptavidin-coated glass bead, which was then attached to the biotinylated red blood cell, forming a force probe. The other micropipette held a glass bead (target) to which soluble P-selectin or P-selectinE88D was captured with an immobilized non-blocking mAb. Alternatively, the micropipette held as a target a transfected murine L1–2 pre-B cell expressing full-length P-selectin or P-selectinE88D. The micropipette bearing the target was brought in contact with the 2-GSP-6-coated probe for 0.1 s and then retracted. Adhesion was measured by deflection of the red blood cell. The adhesion frequency for 50 contacts was measured. Beads bearing P-selectin bound specifically to 2-GSP-6. However, beads bearing P-selectinE88D did not bind (Fig. 5B). Similarly, cells expressing P-selectin bound to 2-GSP-6, whereas cells expressing P-selectinE88D did not bind (Fig. 5C).
FIGURE 5.
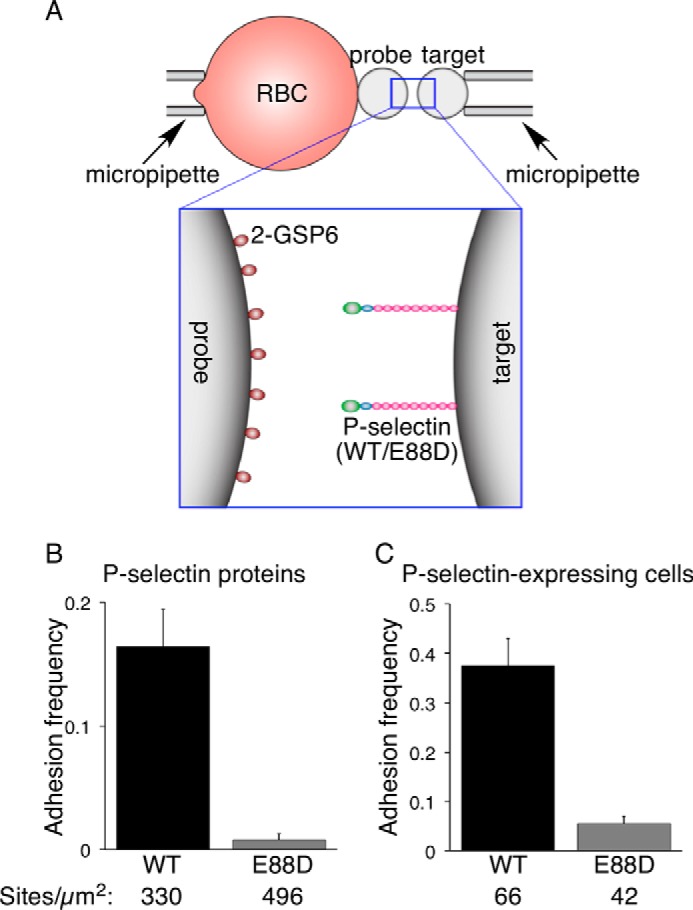
Replacing Glu-88 with Asp prevents binding of P-selectin to PSGL-1 measured with a biomembrane force probe. A, schematic of the biomembrane force probe. B and C, adhesion frequency data. A glass bead coated with the indicated density of P-selectin or P-selectinE88D (B) or a transfected cell expressing the indicated density of P-selectin or P-selectinE88D (C) (target) was brought in contact for 0.1 s with a 2-GSP-6-coated glass bead (probe) attached to a force-transducing red blood cell. The adhesion frequency for 50 contacts was measured for each probe/target pair. The data represent the mean ± S.E. of >10 probe/target pairs per bar.
Replacing Glu-88 with Asp Eliminates Rolling of Cells Expressing P- and L-selectin and Diminishes Rolling of Cells Expressing E-selectin on Glycan Ligands
To assess the role of Glu-88 in selectin binding to ligands under force, we expressed full-length WT and E88D forms of each human selectin in transfected murine L1–2 pre-B cells that do not express endogenous L-selectin. Cell sorting was used to obtain populations with matched densities of WT and E88D for each human selectin (Fig. 6, A–C). Selectin-expressing cells were allowed to settle on monolayers of CHO cells expressing PSGL-1 and the glycosyltransferases required to express sLex on N-glycans and core 2 branched O-glycans (CHO-C2/FTVII/PSGL-1 cells). Flow was then initiated at various wall shear stresses, and the number of rolling cells was quantified. Cells expressing each of the WT selectins rolled on the monolayers (Fig. 6, D, F, and G). Cells expressing L-selectin also rolled on immobilized 6-sulfo-sLex (Fig. 6E). In marked contrast, cells expressing P-selectinE88D or L-selectinE88D failed to roll on these ligands. Instead, they detached immediately after flow was initiated. Significantly fewer cells expressing E-selectinE88D rolled on the monolayers, particularly at higher shear stress (Fig. 6G). However, the adhesion defect was less pronounced than for cells expressing P-selectinE88D or L-selectinE88D.
FIGURE 6.

Replacing Glu-88 with Asp impairs rolling of selectin-expressing cells on glycan ligands. A–C, flow cytometry of transfected L1–2 cells expressing WT or E88D forms of L-selectin, P-selectin, or E-selectin labeled with FITC-conjugated anti-selectin mAb. D–G, cells expressing WT or E88D forms of L-selectin (D and E), P-selectin (F), or E-selectin (G) were introduced on a confluent monolayer of CHO-C2/FTVII/PSGL-1 cells (D, F, and G) or on immobilized 6-sulfo-sLex (E). Cells were allowed to settle for 5 min before flow was initiated at the indicated wall shear stress. Adherent cells were quantified in 3–5 frames of 20 s each. The data in A–C are representative of at least three experiments. The data in D–G represent the mean ± S.D. from three separate experiments.
Discussion
Our studies provide strong experimental support for the two-state model of selectin allostery. In particular, they demonstrate that the selectin 83–89 loop must precisely align Glu-88 to strengthen binding to glycan ligands.
Our structure of L-selectin binding to terminal mannose on a neighboring N-glycan validates the placement of selectins within a subset of C-type lectins that can interact with mannose or fucose (12). It is also consistent with an early report that mannose and mannose phosphate inhibit L-selectin-dependent adhesion of lymphocytes to high endothelial venules (15). L-selectin uses the same conserved residues around the Ca2+ coordination site to bind to mannose as P- and E-selectin use to bind to fucose. Thus, the structure mimics L-selectin binding to fucose in a physiological selectin ligand, but it has the advantage of freezing allosteric movement after binding. The 83–89 loop closes to dock Glu-88 with mannose and the Ca2+ ion, as in cocrystal structures of extended P- and E-selectin binding to sLex-containing ligands. However, L-selectin remains bent, because binding of the mannose terminating N-glycan 2 to another molecule restricts movement of the loop containing Asn-66, to which the glycan is anchored. This prevents downstream rearrangements that open the angle between the lectin and EGF domains (4–6). Therefore, our structure reveals how ligand binding initiates allostery by first closing the 83–89 loop. It also shows that binding of a single monosaccharide is sufficient to trigger allostery.
The engagement of Glu-88 in putative high affinity complexes of each selectin with glycan ligands suggested its importance. Glu-88 is conserved in E- and L-selectin of all species sequenced. In primates, P-selectin has Glu-88; in other species, Glu-88 is substituted with Gln, which has a side chain of identical length that could also interact with Ca2+ and with mannose or fucose. In our studies, shortening the side chain by only 1.5 Å markedly reduced the force-free affinity of P-selectinE88D for PSGL-1. Thus, the 83–89 loop has limited flexibility and requires a relatively long side chain at residue 88 to reach Ca2+ and sugar. The low affinity binding of P-selectinE88D to PSGL-1 is consistent with the hypothesized low affinity binding of sLex to the bent, open-loop crystal structures of P- and E-selectin, where Glu-88 does not engage Ca2+ or fucose (4). Moreover, our results demonstrate that ligand binding without Glu-88 is exquisitely sensitive to force. Cells expressing E88D mutants of P- or L-selectin immediately detached from immobilized ligands at all shear stresses tested. Binding of E-selectinE88D to sLex was less force-sensitive. This may be due to a unique, additional interaction of Gln-85 with fucose when loop 83–89 closes. In the P-selectin-PSGL-1 cocrystal structure, Arg-85 in P-selectin engages sulfated Tyr-10 (4). This interaction was not sufficient to prevent flow-induced detachment of cells expressing P-selectinE88D from immobilized PSGL-1. Conversely, if Glu-88 remains, mutating Tyr-10 does not prevent rolling of cells expressing P-selectin on PSGL-1 (16).
Why is Glu-88 so critical for selectin bonds? Fucose in sLex-containing ligands forms a ternary complex with Ca2+ and with Glu-80, Asn-82, Asn-105, and Asp-106. This region is part of a rigid surface that includes a loop from 77–82 with Glu-80 and Asn-82, the β5 strand from 103–108 with Asn-105 and Asp-106, and the β4 strand from 90–94 with Glu-92 and Tyr-94 (Fig. 7A). A disulfide bond between Cys-109 and Cys-90 connects the two β strands, limiting their movement. The broad surface surrounding the Ca2+ coordination site permits sLex-containing ligands to bind rapidly. However, they dissociate rapidly even without applied force unless closure of the 83–89 loop permits Glu-88 to interact with Ca2+ and fucose, strengthening the ternary complex. The fitting of data to a one-state binding model in our surface-plasmon-resonance experiments suggests that ligand binding to the open-loop form of P-selectin immediately triggers loop closure. Fucose binding moves Glu-80 and Asn-82 closer to Ca2+ and creates strain on the 83–89 loop. This induces loop closure by disruption of a hydrogen bond between the main-chain nitrogen of Cys-109 and the main-chain carbonyl oxygen of residue 87 and of interactions of the Asp-89 side chain with Arg-54 and Lys-55 (Fig. 7, B and C). Loop closure moves the Cα of residue 87 up 4.8 Å. The Cα of Glu-88 then moves 2.2 Å toward Ca2+ and fucose, just enough for the carboxyl group to dock.
FIGURE 7.
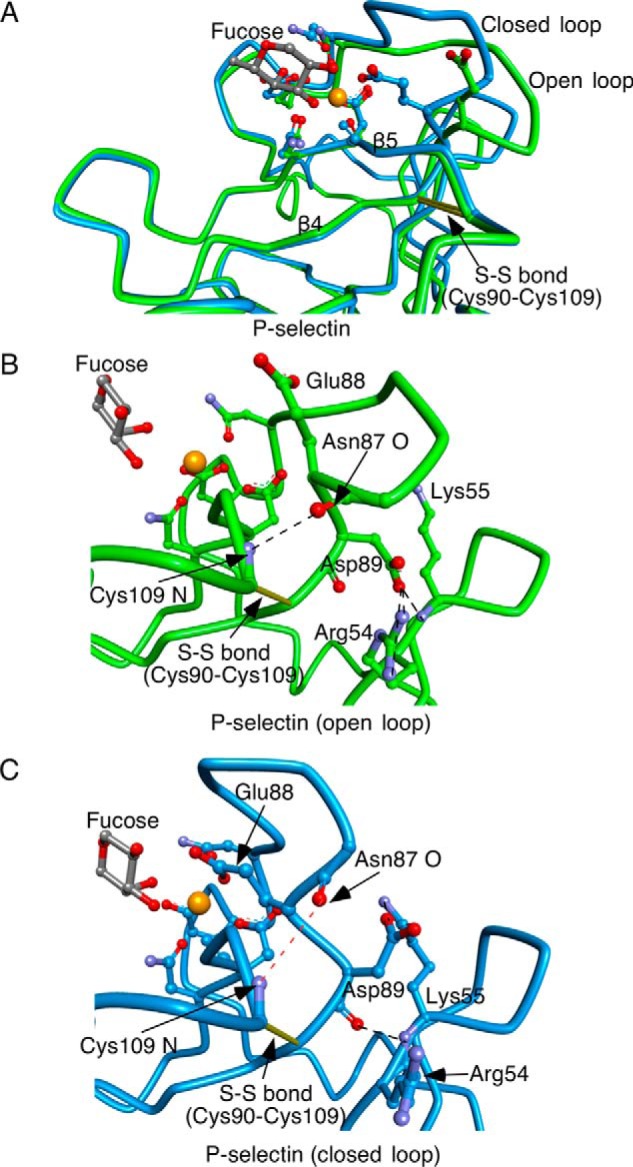
Ligand binding triggers closure of loop 83–89 in selectins. A, superimposition of P-selectin with soaked sLex (green, open loop 83–89) and P-selectin·PSGL-1 (blue, closed loop 83–89). B and C, enlarged views of region surrounding loop 83–89 in open (B) and closed (C) conformations. Yellow sticks depict disulfide bonds. Ball-and-stick representations depict side chains of residues that coordinate Ca2+ and form hydrogen bonds with fucose and of side chains and main chains of residues that restrict the open conformation of the loop. Dashed lines in black represent hydrogen bonds. A dashed red line in C indicates disruption of a hydrogen bond as Asn-87 moves away from Cys-109. Gold spheres represent Ca2+ or Sr2+ ions.
Force applied to sLex bound to the rigid surface creates further strain that accelerates loop closure. If Glu-88 does not immediately stabilize the interaction, even very low levels of force cause ligand to dissociate virtually instantaneously. Docking of Glu-88 closes a “lid” over the ligand and distributes force over a wider area. Force-facilitated extension of the lectin/EGF-domain interface shifts the equilibrium to the closed-loop configuration, locking catch bonds in place. Our data support a “kinetic-selection” model in which ligand binds rapidly to an open “inactive” (low affinity) receptor. Force then accelerates conversion to a closed “active” (high affinity) receptor that slows ligand dissociation (17). In the case of selectins, the 83–89 loop must close and engage Glu-88 to convert to the active receptor. Rapid conversion between the inactive and active conformations stabilizes rolling of cells under flow (9, 10). The E88D mutants lock selectins in their functionally inactive states, thus markedly impairing rolling.
Experimental Procedures
Proteins and Other Reagents
We prepared a cDNA construct encoding the signal peptide, lectin domain, and EGF domain of human L-selectin followed by an eight-residue epitope for the Ca2+-dependent mAb HPC4 (18) in the pEE14.1 expression vector (Lonza Biologics). We prepared a similar cDNA construct encoding the same domains of human P-selectin (19). Using the QuikChange mutagenesis kit (Stratagene), we also created a mutant P-selectin cDNA that substituted the codon encoding Glu-88 with a codon encoding Asp (E88D). The fidelity of both constructs was verified by DNA sequencing. The constructs were transfected into Lec1 CHO cells, which lack UDP-N-acetylglucosamine:α-3-d-mannoside β-1–2-N-acetylglucosaminyltransferase I (11). These cells express Man5GlcNAc2 but not complex N-glycans. Transfected cells expressing high levels of recombinant protein were selected and expanded. Recombinant soluble L-selectin, P-selectin, or P-selectinE88D in conditioned medium were isolated by affinity chromatography on immobilized HPC4 (19) and dialyzed into 20 mm MOPS, pH 7.4, 100 mm NaCl.
We prepared cDNAs encoding full-length forms of human P-selectin, L-selectin, and E-selectin in the pBK-EF vector (20). Using the QuikChange mutagenesis kit, we also created cDNAs encoding E88D mutants of each full-length selectin in the same vector. The fidelity of all constructs was verified by DNA sequencing. The constructs were transfected into murine L1–2 pre-B cells, which do not express endogenous selectins (21). Transfected cells were selected in G418. Clones expressing matched surface densities of each recombinant human selectin were selected by fluorescence-activated cell sorting.
The following reagents have been described: biotinylated 6-sulfo-sLex and 2-GSP-6 (8); CHO cells expressing core 2-N-acetylglucosaminyltransferase I, fucosyltransferase VII, and human PSGL-1 (CHO-C2/FTVII/PSGL-1) (16); anti-human P-selectin mAb G1 (22), anti-human E-selectin mAb ES1 (23), and anti-human L-selectin mAb DREG-56 (24).
Crystallization and Data Collection
Before crystallization trials, solution containing soluble L-selectin was adjusted to include final concentrations of 25 mm CaCl2 and 0.02% NaN3. Crystals of L-selectin were grown at 20 °C using the hanging drop vapor diffusion method. Drops containing equal volumes of protein (at 7.8 mg/ml) and precipitant (0.17 m sodium acetate trihydrate, 0.085 m Tris hydrochloride, pH 8.5, 25.5% w/v polyethylene glycol 4000, and 15% v/v glycerol anhydrous solution) were equilibrated against 0.5 ml of precipitant solution. Microcrystals were obtained within 2 days. The conditions were then optimized to yield diffraction-quality crystals. Some of these crystals were flash-cooled in liquid propane held at liquid nitrogen temperature. X-ray data were collected from a single crystal at 100 K at beam line X12B of the National Synchrotron Light Source at Brookhaven National Laboratory using a wavelength of 0.98 Å. Data were indexed, processed, scaled, and merged using the HKL2000 package (25) and programs of the CCP4 suite (26). The crystal diffracted to 2.2 Å and belonged to the cubic space group I23 with unit cell parameters of a = b = c = 118.48 Å. The asymmetric unit contained one protein molecule with 64.2% solvent content.
Structure Determination and Refinement
The three-dimensional structure of the lectin and EGF domains of human L-selectin was solved by molecular replacement. The search model was based on the human E-selectin structure (PDB code 1ESL). Using AmoRe (27), we located one copy of L-selectin in the asymmetric unit of the crystal. CNS (28) was used in all stages of structure refinement with 5% of the total reflections randomly selected before refinement to monitor the free R factor. At the beginning a model of L-selectin was refined using the simulated annealing slow-cooling protocol from 5000 K in the full resolution range of 25–2.2 Å. A bulk solvent correction and an anisotropic overall B-factor refinement were applied throughout the refinement. Cycles of refinement were alternated with manual correction of the model using the graphics program MAIN (29) and 2Fo − Fc and Fo − Fc electron density maps on a Silicon Graphics work station. The crystallographic statistics are reported in Table 1. Quality of the final model was assessed by Ramachandran plots, and model geometry was validated using PROCHECK (30) and WHAT IF (31). The final model for L-selectin comprises 1133 non-hydrogen protein atoms, 149 water molecules, 74 carbohydrate atoms, and one Ca2+ ion (PDB code 3CFW).
Molecular Superimpositions
Molecular overlays and figures were made using Discovery Studio 3.5 software (BIOVIA).
Surface Plasmon Resonance
Surface plasmon resonance experiments were performed on a Biacore 3000 instrument. The running buffer was 20 mm MOPS, pH 7.5, 150 mm NaCl, 1.5 mm CaCl2, 0.005% Tween 20. Biotinylated 2-GSP-6 was captured on a research grade, streptavidin-coated sensor chip pretreated according to the manufacturer's instructions. A control surface was mock-treated but was not injected with biotinylated 2-GSP-6. Increasing concentrations of soluble P-selectin or P-selectinE88D were injected over the surfaces at a high flow rate (60 μl/min) for a brief period (15 s) (14). The brief injection period was sufficient to reach equilibrium binding. Specific binding was measured by subtracting nonspecific binding to the control surface. Equilibrium binding data were analyzed by nonlinear curve fitting of the Langmuir binding isotherms using the BiaEvaluation software.
Biomembrane Force Probe Assay
Measurement of adhesion frequencies with a biomembrane force probe was performed as previously described (32). Briefly, two micropipettes were placed on opposite sides of a chamber visualized with an inverted microscope. One micropipette held a biotinylated red blood cell used as a force transducer. Biotinylated 2-GSP-6 was attached to a streptavidin-coated glass bead, which was then attached to the biotinylated red blood cell, forming a force probe. The other micropipette held a glass bead as a target to which soluble P-selectin or P-selectinE88D was captured using immobilized mAb HPC4. Alternatively, the micropipette held as the target a transfected L1–2 pre-B cell expressing full-length P-selectin or P-selectinE88D. Site densities of P-selectin or P-selectinE88D on beads or cells were measured by flow cytometry (33, 34). The target was driven by a computer-controlled piezo translator attached to its holding pipette to contact the stationary force probe for a predetermined area and duration to allow bond formation. After target retraction, it either immediately separated from the probe (no adhesion, scored 0) or brought with it the probe that remained bound by stretching the flexible red blood cell (adhesion, scored 1) for a short time until detachment. The image analysis software tracked deflection with 0.6-ns temporal resolution and 5-nm spatial resolution, which for a spring constant of 0.3 piconewton (pN)/nm, translated to 1.5-pN force resolution. The likelihood of adhesion was estimated from the frequency of adhesions (Pa) observed in 50 repeated contact cycles using a single pair of beads/cells.
Flow Chamber Assay
Adhesive interactions under laminar flow were measured by video microscopy as described previously (35). Transfected L1–2 pre-B cells expressing matched surface densities of WT or E88D forms of human L-selectin, E-selectin, or P-selectin (106 cells/ml in Hanks' balanced salt solution containing 0.5% human serum albumin) were allowed to settle for 5 min on confluent monolayers of CHO-C2/FTVII/PSGL-1 cells. In some experiments cells expressing WT or E88D forms of L-selectin were settled on biotinylated 6-sulfo-sLex captured on immobilized streptavidin. Flow was then initiated at 0.5, 1.0, or 1.5 dynes/cm2. After 15 s, the number of rolling cells in 3–5 separate fields was counted over a 20-s interval. Rolling was selectin-dependent because inclusion of 10 mm EDTA or 20 μg/ml of the respective blocking anti-selectin mAb (DREG-56, G1, or ES1) eliminated all rolling cells.
Author Contributions
P. M.-D., A. G. K., V. O., S. T., T. M., Z. L., and S. R. P. performed the experiments. P. M.-D., C. Z., and R. P. M. designed the research. All authors analyzed the data. P.M.-D. and R. P. M. wrote the paper, with input from all authors.
Acknowledgment
We thank Cindy Carter for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants HL034363, AI077343, and HL085607. R. P. M. is a co-founder of Selexys Pharmaceutical Corp., now part of Novartis, and of Tetherex Pharmaceutical Corp. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 3CFW) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- sLex
- sialyl Lewis x
- PSGL-1
- P-selectin glycoprotein ligand-1.
References
- 1. McEver R. P., and Zhu C. (2010) Rolling cell adhesion. Annu. Rev. Cell Dev. Biol. 26, 363–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McEver R. P. (2015) Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 107, 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Graves B. J., Crowther R. L., Chandran C., Rumberger J. M., Li S., Huang K.-S., Presky D. H., Familletti P. C., Wolitzky B. A., and Burns D. K. (1994) Insight into E-selectin/ligand interaction from the crystal structure and mutagenesis of the lec/EGF domains. Nature 367, 532–538 [DOI] [PubMed] [Google Scholar]
- 4. Somers W. S., Tang J., Shaw G. D., and Camphausen R. T. (2000) Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell 103, 467–479 [DOI] [PubMed] [Google Scholar]
- 5. Preston R. C., Jakob R. P., Binder F. P., Sager C. P., Ernst B., and Maier T. (2016) E-selectin ligand complexes adopt an extended high-affinity conformation. J. Mol. Cell Biol. 8, 62–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Springer T. A. (2009) Structural basis for selectin mechanochemistry. Proc. Natl. Acad. Sci. U.S.A. 106, 91–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lou J., and Zhu C. (2007) A structure-based sliding-rebinding mechanism for catch bonds. Biophys. J. 92, 1471–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lou J., Yago T., Klopocki A. G., Mehta P., Chen W., Zarnitsyna V. I., Bovin N. V., Zhu C., and McEver R. P. (2006) Flow-enhanced adhesion regulated by a selectin interdomain hinge. J. Cell Biol. 174, 1107–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Phan U. T., Waldron T. T., and Springer T. A. (2006) Remodeling of the lectin-EGF-like domain interface in P- and L-selectin increases adhesiveness and shear resistance under hydrodynamic force. Nat. Immunol. 7, 883–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Waldron T. T., and Springer T. A. (2009) Transmission of allostery through the lectin domain in selectin-mediated cell adhesion. Proc. Natl. Acad. Sci. U.S.A. 106, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Patnaik S. K., and Stanley P. (2006) Lectin-resistant CHO glycosylation mutants. Methods Enzymol. 416, 159–182 [DOI] [PubMed] [Google Scholar]
- 12. Weis W. I., Taylor M. E., and Drickamer K. (1998) The C-type lectin superfamily in the immune system. Immunol. Rev. 163, 19–34 [DOI] [PubMed] [Google Scholar]
- 13. Weis W. I., Kahn R., Fourme R., Drickamer K., and Hendrickson W. A. (1991) Structure of the calcium-dependent lectin domain from a rat mannose-binding protein determined by MAD phasing. Science 254, 1608–1615 [DOI] [PubMed] [Google Scholar]
- 14. Mehta P., Cummings R. D., and McEver R. P. (1998) Affinity and kinetic analysis of P-selectin binding to P-selectin glycoprotein ligand-1. J. Biol. Chem. 273, 32506–32513 [DOI] [PubMed] [Google Scholar]
- 15. Stoolman L. M., Tenforde T. S., and Rosen S. D. (1984) Phosphomannosyl receptors may participate in the adhesive interaction between lymphocytes and high endothelial venules. J. Cell Biol. 99, 1535–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ramachandran V., Nollert M. U., Qiu H., Liu W-J., Cummings R. D., Zhu C., and McEver R. P. (1999) Tyrosine replacement in P-selectin glycoprotein ligand-1 affects distinct kinetic and mechanical properties of bonds with P- and L-selectin. Proc. Natl. Acad. Sci. U.S.A. 96, 13771–13776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yakovenko O., Tchesnokova V., Sokurenko E. V., and Thomas W. E. (2015) Inactive conformation enhances binding function in physiological conditions. Proc. Natl. Acad. Sci. U.S.A. 112, 9884–9889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stearns D. J., Kurosawa S., Sims P. J., Esmon N. L., and Esmon C. T. (1988) The interaction of a Ca2+-dependent monoclonal antibody with the protein C activation peptide region. Evidence for obligatory Ca2+ binding to both antigen and antibody. J. Biol. Chem. 263, 826–832 [PubMed] [Google Scholar]
- 19. Mehta P., Patel K. D., Laue T. M., Erickson H. P., and McEver R. P. (1997) Soluble monomeric P-selectin containing only the lectin and epidermal growth factor domains binds to P-selectin glycoprotein ligand-1 on leukocytes. Blood 90, 2381–2389 [PubMed] [Google Scholar]
- 20. Liu W.-J., Ramachandran V., Kang J., Kishimoto T. K., Cummings R. D., and McEver R. P. (1998) Identification of N-terminal residues on P-selectin glycoprotein ligand-1 required for binding to P-selectin. J. Biol. Chem. 273, 7078–7087 [DOI] [PubMed] [Google Scholar]
- 21. Diacovo T. G., Puri K. D., Warnock R. A., Springer T. A., and von Andrian U. H. (1996) Platelet-mediated lymphocyte delivery to high endothelial venules. Science 273, 252–255 [DOI] [PubMed] [Google Scholar]
- 22. Geng J.-G., Bevilacqua M. P., Moore K. L., McIntyre T. M., Prescott S. M., Kim J. M., Bliss G. A., Zimmerman G. A., and McEver R. P. (1990) Rapid neutrophil adhesion to activated endothelium mediated by GMP-140. Nature 343, 757–760 [DOI] [PubMed] [Google Scholar]
- 23. Patel K. D., Moore K. L., Nollert M. U., and McEver R. P. (1995) Neutrophils use both shared and distinct mechanisms to adhere to selectins under static and flow conditions. J. Clin. Invest. 96, 1887–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kishimoto T. K., Warnock R. A., Jutila M. A., Butcher E. C., Lane C., Anderson D. C., and Smith C. W. (1991) Antibodies against human neutrophil LECAM-1 (LAM-1/Leu-8/DREG-56 antigen) and endothelial cell ELAM-1 inhibit a common CD18-independent adhesion pathway in vitro. Blood 78, 805–811 [PubMed] [Google Scholar]
- 25. Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 26. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 27. Navaza J. (2001) Implementation of molecular replacement in AMoRe. Acta Crystallogr. D Biol. Crystallogr. 57, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 28. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., and Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 29. Turk D. (2013) MAIN software for density averaging, model building, structure refinement and validation. Acta Crystallogr. D Biol. Crystallogr. 69, 1342–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., and Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 31. Vriend G. (1990) WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 8, 52–56 [DOI] [PubMed] [Google Scholar]
- 32. Chen W., Zarnitsyna V. I., Sarangapani K. K., Huang J., and Zhu C. (2008) Measuring receptor-ligand binding kinetics on cell surfaces: from adhesion frequency to thermal fluctuation methods. Cell. Mol. Bioeng. 1, 276–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chesla S. E., Selvaraj P., and Zhu C. (1998) Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys. J. 75, 1553–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang J., Zarnitsyna V. I., Liu B., Edwards L. J., Jiang N., Evavold B. D., and Zhu C. (2010) The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature 464, 932–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yago T., Fu J., McDaniel J. M., Miner J. J., McEver R. P., and Xia L. (2010) Core 1-derived O-glycans are essential E-selectin ligands on neutrophils. Proc. Natl. Acad. Sci. U.S.A. 107, 9204–9209 [DOI] [PMC free article] [PubMed] [Google Scholar]