

Abstract
Mutations in hemoglobin can cause a wide range of phenotypic outcomes, including anemia due to protein instability and red cell lysis. Uncovering the biochemical basis for these phenotypes can provide new insights into hemoglobin structure and function as well as identify new therapeutic opportunities. We report here a new hemoglobin α chain variant in a female patient with mild anemia, whose father also carries the trait and is from the Turkish city of Kirklareli. Both the patient and her father had a His-58(E7) → Leu mutation in α1. Surprisingly, the patient's father is not anemic, but he is a smoker with high levels of HbCO (∼16%). To understand these phenotypes, we examined recombinant human Hb (rHb) Kirklareli containing the α H58L replacement. Mutant α subunits containing Leu-58(E7) autoxidize ∼8 times and lose hemin ∼200 times more rapidly than native α subunits, causing the oxygenated form of rHb Kirklareli to denature very rapidly under physiological conditions. The crystal structure of rHb Kirklareli shows that the α H58L replacement creates a completely apolar active site, which prevents electrostatic stabilization of bound O2, promotes autoxidation, and enhances hemin dissociation by inhibiting water coordination to the Fe(III) atom. At the same time, the mutant α subunit has an ∼80,000-fold higher affinity for CO than O2, causing it to rapidly take up and retain carbon monoxide, which prevents denaturation both in vitro and in vivo and explains the phenotypic differences between the father, who is a smoker, and his daughter.
Keywords: carbon monoxide, heme, hemoglobin, mutant, oxidative stress
Introduction
Most human hemoglobin variants are caused by point mutations, and to date (September, 2016), more than 1000 types have been listed in the HbVar database (1). Of those, nearly 75 and 95% are located in the coding regions of the α and β genes, respectively. The vast majority of them are rare and clinically silent. However, a small number of the mutations are associated with symptoms of hemolytic anemia, methemoglobinemia, cyanosis, and polycythemia due to altered oxygen affinity, resistance to autoxidation, and globin stability.
This study describes a new α-globin chain variant found during Hb investigations performed on a 23-year-old woman of Turkish descent, because routine hematological and biochemical data led to suspicion of iron deficiency anemia. After ruling out more common causes of anemia, cDNA sequencing of her α- and β-hemoglobin genes and mass spectral analyses of her hemoglobin polypeptides were performed and showed that the patient had an α1 His-58 → Leu mutation. Subsequent analysis of the patient's father showed that he also had the same α1 trait, which was named Hb Kirklareli after his home city in Turkey. The father, who was a smoker, had no anemia but showed abnormally high HbCO levels of ∼16%. The α1 H58L mutation appears to be the underlying cause of the clinical phenotype, but additional structural and biophysical analyses were required to verify this conclusion and provide mechanistic interpretations.
Birukou et al. (2) had already constructed and characterized a series of distal histidine mutants in both the α and β subunits of recombinant human hemoglobin using the expression systems originally developed by Somatogen, Inc. (3), and Shen et al. (4). Thus, Birukou et al. (2) had already reported the O2, CO, and NO binding properties of the mutant α (H58L) subunit. Crystal structures of the deoxyHb and HbCO forms of rHb α(H58L)β(WT) and α(WT)β(H63L) had been determined using tetramers with the native N-terminal valines but had not been reported in a peer-reviewed journal article (PDB3 access codes 3QJD, 3QJB, 3QJE, and 3QJC, respectively (5)). As a result, we were in a position to examine in structural detail the cause of the inherent instability of Hb Kirklareli and how CO stabilizes this mutant.
We previously characterized in detail the effects of the His-64(E7) → Leu mutation in mammalian Mb on ligand binding, autoxidation, hemin dissociation, and apoglobin unfolding (6–11). In this work, we measured for the first time the rates of autoxidation, hemin loss, and precipitation of rHb Kirklareli, and we have shown how CO binding to the mutant α subunit stabilizes the protein and inhibits its oxidative degradation both in vitro and in vivo.
Results
Hematological Analyses for the Patient
The propositus was a 21-year-old Turkish female diagnosed with iron deficiency anemia (IDA). Secondary IDA due to blood loss, gastritis, and common forms of congenital dyserythropoietic anemia was excluded. On physical examination, her liver, spleen, and pancreas were not enlarged. Blood samples were sent to our laboratory for additional investigations of the causes of her IDA. Hypochromic microcytic anemia was confirmed (Table 1) with a low serum iron (188 μg/liter), low transferrin saturation (4%), low ferritin level (4.5 μg/liter), high soluble transferrin receptor (9.2 mg/dl), and high total iron-binding capacity (536 μg/dl). Serum concentrations of haptoglobin and bilirubin were within normal ranges (data not shown). The determination of the erythrocyte enzyme levels showed normal results. Peripheral blood smears stained with brilliant cresol blue demonstrated normal erythrocytic inclusion bodies (< 10%). The level of carbon monoxide hemoglobin was 4.0%, and the patient is a non-smoker.
TABLE 1.
Hematological parameters for propositus and her family
Abbreviations used are as follows: TC, tobacco consumption; NS, non-smoker; S, smoker; Hb, total blood hemoglobin concentration; PVC (or hematocrit), packed volume of red cells; RBC, red blood cell count; WBC, white blood cell count; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin (weight); MCHC, mean corpuscular hemoglobin concentration; PLT, platelet count; RETI, reticulocyte percentage of red cells; HbCO, percentage of hemoglobin with bound carbon monoxide; MetHb, percentage of hemoglobin that is oxidized.
| Subject | Sex/Age | TC | Hb | PVC (l/l %) | RBC (1012/l) | WBC (109/l) | MCV | MCH | MCHC | PLT (109/l) | RETI | HbCO | MetHb |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g/dl | fl | pg | g/dl | % | % | % | |||||||
| Fathera | M/51 | S | 18.3 | 63.2 | 6.33 | 6.90 | 99.8 | 28.9 | 29.0 | 191.0 | 1.36 | 15.9 | 2.7 |
| Mother | F/50 | S | 11.0 | 36.1 | 4.23 | 10.17 | 85.4 | 26.0 | 30.7 | 417.7 | 0.60 | 4.0 | 1.2 |
| Propositusa | F/29 | NS | 9.9 | 31.2 | 4.13 | 5.22 | 75.6 | 24.2 | 31.8 | 256.5 | 0.90 | 4.5 | 0.4 |
| Brother | M/28 | S | 17.3 | 54.9 | 5.82 | 9.16 | 94.3 | 29.7 | 31.5 | 310 | 0.96 | 2.1 | 1.3 |
a Carrier of Hb Kirklareli trait (α1 H58L).
Hb Analyses
Electrophoretic analysis at pH 8.6 and 6.2 failed to show the presence of any Hb variant. The heat stability and the isopropyl alcohol tests of the crude hemolysate seemed to show no significant deviation from the normal. However, it is possible that any highly unstable hemoglobins could have precipitated during lysis and been lost in the stroma pellet without stabilization by CO binding. As a result, only native HbA would have been present in the crude lysate. Analyses of the HbCO-stabilized samples by cation-exchange HPLC revealed an abnormal fraction, HbX, which eluted 1.6 min after HbA0 and accounted for ∼22% of the total Hb (Fig. 1A). Globin chain analysis by reverse phase HPLC showed a mutant globin (αX) eluting 6.7 min behind the normal αA-globin chain (Fig. 1B). Of the three other members of the family examined, only the father was a carrier of the same αX variant.
FIGURE 1.

Observation of variant Hb tetramers and α chains by anion-exchange and reverse phase HPLC. A, elution profile from a 21 × 250-mm PolyCAT A column using the chromatographic method described by Divoky et al. (51). Labels used are as follows: HbA0 (αA2βA2), the major component of adult hemoglobin Hb A; and HbX, Hb Kirklareli (α2(H58L)β2(WT)). B, reverse phase HPLC for hemoglobin isolated from the patient (both peaks in A) using the method described by Bisse et al. (48). Labels used are as follows: βA, β(WT); αA, α(WT); and αX, α(H58L).
Elucidation of the Amino Acid Replacement
The variant αX chain was purified by reverse phase HPLC and then submitted for a series of mass spectrometry analyses. LC-MS analysis of this purified fraction revealed the presence of an αX-globin chain variant (15,102.58 ± 0.28 Da), which was 24 Da lower than the molecular mass of the normal αA-globin chain (15,126.44 ± 0.24 Da) (Fig. 2). Peptide mapping was performed using specific enzymatic digestion (AspN), followed by nanoliquid chromatography-tandem mass spectrometry (nanoLC-MS/MS). The mass corresponding to the α AspN6 peptide (amino acids 47–63) was searched for in the LC-MS/MS data. A molecular ion was present and its MS/MS spectrum submitted to de novo sequencing. The MS/MS spectrum of its triple charge ion at m/z 565.66 yielded an amino acid sequence of a normal αA6 peptide except that the histidine at position 58 was replaced by a leucine or isoleucine (Fig. 3). This result was also confirmed by the collision-induced dissociation (CID) mass spectrum of the double charged peptide αA6 at m/z 847.98 (Fig. 3). Thus, the primary sequence of the peptide generated by AspN digestion (amino acids 47–63) was DLSHGSAQVKG(L/I)GKKVA.
FIGURE 2.

MS spectra of the purified α chains. Transformed data revealed molecular masses of 15,126.6 ± 0.3 and 15,102.58 ± 0.2 Da corresponding to the WT (A) and mutant (B) α chains, respectively. The mutant α chain shows a mass 24 Da lower than that of the WT.
FIGURE 3.

Fragment ions from the MS/MS spectrum obtained from the 3+ (m/z 565. 66) and 2+ (m/z 847.98) molecular ions from the 17-amino acid mutated AspN peptide αA6 (47DLSHGSAQVKG(I/L)58GKKVA63) showing the His to Leu or Ile substitution in αX.
DNA Analysis
Direct DNA sequencing of the α1-globin gene revealed a sequence change c.177A→T in the propositus and her father, confirming that codon 58 (E7 helical position) of the α1-globin gene was heterozygous for a His → Leu substitution. DNA analysis using multiplex PCRs showed that the father was also heterozygous for the −α3.7 deletion, which is the most common type of α-thalassemia worldwide. In this case, only one α gene is defective, and this condition is not normally associated with hematological and clinical abnormalities and seems not to interact with Hb Kirklareli. Sequencing of the TMPRSS6 gene showed no mutation, ruling out anemia due to alterations in hepcidin expression.
O2 and CO Binding to Hb Kirklareli
Birukou et al. (2) measured rates and equilibrium constants for O2 and CO binding to a series of His(E7) mutants in the α and β chains of recombinant human HbA in the R or high affinity conformation. Laser photolysis techniques and ligand replacement experiments were used to measure bimolecular rebinding and unimolecular dissociation for the last step in ligand binding to hemoglobin tetramers (i.e. Hb4X3 + X ⇌ Hb4X4, where X is either O2 or CO). The latter reactions can be analyzed to obtain association and dissociation rate constants for the α and β chains without requiring complex models for cooperative ligand binding and are associated with the high affinity or R-state quaternary structure of hemoglobin (12). These R-state parameters can be checked by examining the same rate constants for isolated α and β subunits as described in Birukou et al. (2), Mathews and Olson (13), and Olson et al. (12).
A summary of these rate and equilibrium constants for O2 and CO binding to native and WT (recombinant) α and β subunits and to α(H58L) subunits in recombinant Hb Kirklareli is given in Table 2. Within experimental error, the parameters for native subunits are identical to those for subunits in the recombinant proteins with either the normal N-terminal valine or a V1M mutation. In these proteins, the ligand affinities of the native α and β subunits are, within experimental error, identical to each other. The P50 values for O2 and CO binding to the high affinity forms of human Hb tetramers are ∼0.38 ± 0.10 and 0.0016 ± 0.0005 μm, respectively (2). The ratio of CO to O2 affinity (M-value) is equal to 240 ± 100 for both native subunits (Table 2). In contrast to WT α subunits, the P50 values for O2 and CO binding to the α H58L subunit are 7.1 ± 5.1 and 0.00009 ± 0.00003 μm, respectively, with an M-value of 79,000 ± 60,000. Thus, the α subunit in Hb Kirklareli has an ∼19-fold lower affinity for O2 and an ∼18-fold higher affinity for CO. The net result is that the mutant α subunit will avidly bind and retain CO even in the presence of large amounts of O2.
TABLE 2.
O2 and CO binding parameters for subunits in the R-state form of tetrameric rHb Kirklareli, WT rHbA, and native HbA in 0.1 m sodium phosphate at pH 7.0, 20 °C, taken from Birukou et al. (2)
The symbols k′, k, and K represent association rate, dissociation rate, and association equilibrium constants, respectively, for either CO or O2 binding to the subunits of hemoglobin. P50 is defined as 1/K and represents an equilibrium dissociation constant. M is the ratio of CO to O2 affinities.
| Subunit | kO2′ | kO2 | 1/KO2 (P50 R state) | kCO′ | kCO | 1/KCO (P50 R state) | M (KCO/KO2) |
|---|---|---|---|---|---|---|---|
| μm−1s−1 | s−1 | μm | μm−1s−1 | s−1 | μm | ||
| β native | 82 ± 15 | 28 ± 6 | 0.32 ± 0.10 | 7.1 ± 2.4 | 0.007 ± 0.003 | 0.0010 ± 0.0006 | 320 ± 140 |
| β WT | 60 ± 12 | 31 ± 13 | 0.26 ± 0.04 | 7.1 ± 2.0 | 0.008 ± 0.001 | 0.0011 ± 0.0006 | 230 ± 84 |
| α native | 32 ± 4 | 13 ± 2.9 | 0.39 ± 0.06 | 2.9 ± 0.5 | 0.005 ± 0.002 | 0.0017 ± 0.0007 | 230 ± 75 |
| α WT | 29 ± 11 | 14 ± 8 | 0.53 ± 0.14 | 4.0 ± 1.1 | 0.011 ± 0.004 | 0.0028 ± 0.0015 | 190 ± 82 |
| α H58L | 100 ± 60 | 680 ± 180 | 7.1 ± 5.1 | 22 (20)a,b | 0.0019a | 0.00009a | 80,000a |
a The CO binding experiments for tetrameric rHb α(H58L)β(WT) were only done two times independently in Birukou et al. (2). However, experiments were also done with isolated α(H58L) monomers, and similar rate parameters were observed so the estimated errors for the CO binding parameters for the mutant α subunits are assumed to be ≤30%.
b A sample of purified HbCO Kirklareli from the patient was examined in microsecond flash photolysis experiments, and a fast phase representing bimolecular rebinding to the mutant α subunit was observed with a rate constant equal to ∼20 μm−1 s−1 for CO binding to the R state, as was observed for α(H58L) subunits in both recombinant Hb tetramers and monomers (2).
We also tried to examine O2 equilibrium binding to rHb Kirklareli and a small amount of native Hb Kirklareli purified from the patient's blood (see Fig. 4). These experiments proved to be very difficult, because the HbCO Kirklareli samples require oxidation to remove all CO bound to the mutant α subunits and then a reduction in air to generate an unstable HbO2 form. To slow and reverse autoxidation, we had to use buffer conditions leading to relatively low P50 values for the native HbA control. As shown in Fig. 4, both the patient-derived and recombinant Hb Kirklareli show little or no cooperativity, with a P50 value similar to that for the native HbA control, which did show an nHill value of ≥2.4. The lack of cooperativity for the mutant Hb is probably due to an ordered addition of ligand. O2 binds first to β subunits in the low affinity T-state followed by binding to the R-state mutant α subunits, which have an intrinsically 20-fold lower affinity for O2 due to the H58L replacement (Table 2). As a result, there is little net increase in ligand affinity even after the switch to the R-state. Lower cooperativity could also be due to dimerization, but, as described below, rHb Kirklareli has a smaller tetramer to dimer dissociation equilibrium constant than HbA.
FIGURE 4.

Oxygen equilibrium curves for native HbA, purified Hb Kirklareli from the patient, and recombinant Hb α(H58L)β(WT) in the absence of phosphates at pH 7. 4, 20 °C. Both the patient's and recombinant Hb Kirklareli showed no cooperativity. Although the P50 of all three globins were similar, the Hb Kirklareli samples had a low affinity component that was hard to saturate and presumably represented O2 binding to the α H58L subunits. However, the samples were very unstable and hard to keep reduced.
Structure of Recombinant Hb Kirklareli
The crystal structures of α(H58L)β(WT) (i.e. Hb Kirklareli) in both deoxygenated and CO-bound forms were determined as described under “Experimental Procedures.” The crystal parameters, statistics of X-ray data collection, and refinement characteristics are provided in Table 3 along with the PDB accession codes. The structures of the mutant tetramers were almost identical to those of native HbA, except in the distal portions of the heme pocket. Electron density maps for the distal pocket of the mutated α subunit in the presence and absence of ligand are shown in Fig. 5. A comparison of the α subunit active sites of native HbA (A) and rHb Kirklareli (B) without and with bound CO are shown in Fig. 6. In deoxygenated native α subunits, a distal pocket water molecule is present in the active site, stabilized by hydrogen bonding to the distal histidine, and must be displaced before ligands can bind. The net result is a 5–10-fold inhibition of the rate of binding of all ligands due to the requirement to displace this distal pocket water molecule (2, 14).
TABLE 3.
Crystallization and structure determination parameters of Leu(E7) Hb mutants
| Protein | DeoxyHb α(Leu(E7))β(WT) | HbCO α(Leu(E7))β(WT) |
|---|---|---|
| PDB entry code | 3QJD | 3QJB |
| Crystal data | ||
| Resolution range (Å) | 61.79–1.45 | 99.0–1.8 |
| Space group | P21 | P41212 |
| Unit cell parameters (Å, °) | a = 62.7, b = 82.1, c = 53.5, β = 99.5 | a = b = 53.6, c = 191.2 |
| Reflections (measured/unique) | 215,542/76,521 | 111,460/26,524 |
| Completeness (%) | 80.9 (14.2)a | 98.1 (96.9) |
| 〈I〉/s〈I〉 | 13.0 (1.9) | 32.1 (12.5) |
| Redundancy | 2.8 (1.5) | 4.1 (4.0) |
| Rmerge (%) | 4.4 (16.1) | 5.1 (14.8) |
| Refinement: | ||
| Resolution range (Å) | 20.60–1.56 | 37.18–1.80 |
| R-factor (%) | 16.9 | 17.0 |
| Rfree (%) | 19.1 | 20.5 |
| Root mean square deviations from ideal values | ||
| Bond length (Å) | 0.006 | 0.027 |
| Bond angles (°) | 1.193 | 0.989 |
| Ramachandran plot | ||
| Residues in most favorable region (%) | 98.3 | 98.6 |
| Residues in additional allowed region (%) | 1.7 | 1.4 |
a Parameters in parentheses are for the outer resolution shell.
FIGURE 5.
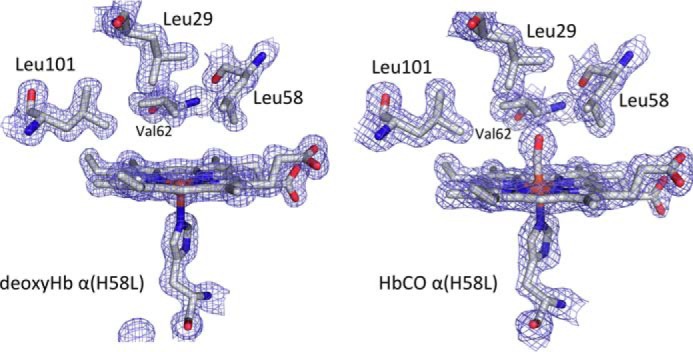
Electron density (2Fo − Fc) maps of the distal pockets of rHb Kirklareli in the reduced deoxygenated (3QJD) and CO bound forms (3QJB).
FIGURE 6.
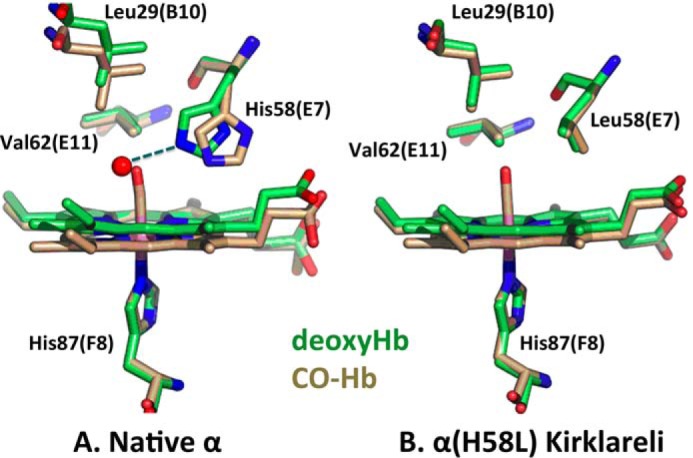
Stick models of deoxy- (green) and CO-forms (beige) of α native and α(H58L) distal pockets in adult human hemoglobin. A, native α structures were created from the PDB entries 2DN2 for the CO-form and 2DN3 for deoxy-form (63); and B, α(H58L) structures were created from 3QJB for the CO-form and 3QJD for the deoxy-form. Color code is as follows: carbon, green (deoxyHb) or beige (HbCO); red, oxygen; blue, nitrogen; pink, iron. The red sphere in A represents a distal pocket water molecule found ∼2.7 Å away from the His-58 Nϵ atom with an occupancy of ∼80% (5).
In contrast, electron density associated with water is not present in the distal pocket of the deoxygenated α(H58L) subunit, which appears completely apolar (Figs. 5 and 6). Thus, the H58L mutation enhances the binding of all ligands by removing the steric water barrier to entry into the distal pocket. However, in the case of O2 binding, the net result of the α H58L mutation is a marked decrease in affinity because the bound dioxygen is no longer stabilized by hydrogen bonding. The distal E7 histidine (His-58 in α subunits) electrostatically stabilizes the highly polar FeO2 complex almost 1000-fold in both Hb subunits and myoglobin (6, 14). In contrast, polar interactions with the neutral FeCO complex are very weak. Thus, replacement of αHis-58 with Leu results in a significant enhancement of the affinities of all ligands, including CO, due to loss of steric hindrance by the internal water molecule in the native deoxygenated subunit. However, at the same time, there is a large highly selective decrease in O2 affinity due to loss of hydrogen bonding to the bound ligand (Table 2). The net result is a >300-fold increase in the ratio of CO to O2 affinity (M-value in Table 2).
The ultra-high affinity of α(H58L) subunits for CO accounts for the high levels of HbCO found in the father who is a heterozygote for the Hb Kirklareli trait and a smoker. The amount of HbCO in his blood is probably very similar to the percentage of mutant α subunits present in his red cells. The high M-value also accounts for why CO cannot be displaced even in the presence 1 atm of pure O2. To completely remove CO from the α H58L subunit, we had to oxidize rHb Kirklareli with ferricyanide. Similar effects are seen for His(E7) → Leu mutations in human β subunits (2) and mammalian Mbs (6, 15). The affinity and rates of CO binding to H64L sperm whale Mb are so high that this mutant protein has been used to assay for carbon monoxide production during heme oxygenase reactions (16).
As described below, the oxygenated form of Hb Kirklareli autoxidizes very rapidly. Although the structure of the CO form of rHb α(H58L)β(WT) shows no differences in the α1β2 interface in the tetramer, an increase in the tetramer to dimer equilibrium dissociation constant, K4,2, could account for the large autoxidation rate of the mutant. Adult human HbO2 dimers autoxidize roughly 20–30 times more rapidly than tetramers and lose hemin at much higher rates as well (17, 18). As controls, we examined the gel filtration elution profiles of the reduced CO forms of HbA and rHb Kirklareli as a function of total protein concentration using the methods described by Manning et al. (19). The K4,2 for HbCO Kirklareli was estimated to be ∼0.3 μm, which is ∼4-fold smaller than the value estimated for HbA in our experiments, 1.1 μm. The latter value is very similar to previously reported K4,2 values (20, 21). Thus, the rapid rates of degradation of Hb Kirklareli are not due to increased dissociation into dimers.
Autoxidation, Hemin Dissociation, and Denaturation of rHb Kirklareli
Although we noted anecdotally that rHbs with the His(E7) → Leu replacements autoxidize rapidly (2, 5, 22), we had not measured the rates of this process quantitatively. The same His(E7) → Leu mutation in sperm whale MbO2 causes an ∼100-fold increase in the rate of autoxidation (kautox), from ∼0.1 (WT) to 10 h−1 (mutant) at pH 7, 37 °C (8). Thus, a large increase in kautox was expected for α(H58L) subunits.
Spectral changes and time courses for the autoxidation of HbA and rHb Kirklareli at pH 7, 37 °C, are shown in Fig. 7, and the observed autoxidation rates are given in Table 4. In all cases, catalase, superoxide dismutase, and EDTA were present to prevent additional oxidative reactions due to the production of H2O2 and to simulate the conditions in red cells. In the case of native HbA, the spectral changes show clear isosbestic points, indicating a smooth transition from HbO2 to metHb, with little or no hemichrome intermediate formation or appearance of turbidity due to precipitation. The observed time courses for HbA at high concentration show some acceleration and an overall rate constant equal to ∼0.04 h−1.
FIGURE 7.

Autoxidation of HbA, recombinant Hb Kirklareli, and rHb Kirklareli containing α(H58L)CO subunits at pH 7, 37 °C. A–C, spectra were deconvoluted into components for HbO2, metHb, hemichrome species, and turbidity (base line increases and dependence on 1/λ4 for light scattering). Then the concentrations of HbO2, metHb, and hemichrome as a function of time were computed. An estimate of the amount of aggregated apoglobin was computed as the loss of total hemoglobin, i.e. [Hb]total, initial − ([HbO2] + [metHb] + [hemichrome]). A, spectral changes for autoxidation of HbA: red lines at 0 and 100 min; blue lines at 200 and 300 min; green lines at 400 and 600 min; black lines at 900 min and every 300 min afterward. In this case, only HbO2 and metHb are present, and the time course for the decay of HbO2 is shown in the inset. B, spectral changes for the autoxidation of fully oxygenated rHb Kirklareli. In this case, significant amounts of turbidity begins to occur after an hour or two, and large precipitates occur at long times. Inset, times courses for HbO2 decay, metHb appearance, turbidity, and hemichrome are shown. No HbCO was present. C, autoxidation of rHb Kirklareli with CO bound to the mutant α subunits. In this case, little precipitation occurs, and again the inset shows time courses for HbO2, metHb, hemichrome, and turbidity. Note that the concentration of HbCO stays constant. D, time courses for the decay of the HbO2 forms of all three hemoglobin samples with the inset showing the initial rates of autoxidation.
TABLE 4.
Autoxidation and hemin loss parameters for rHb Kirklareli in 0.1 m sodium phosphate at pH 7.0, 37 °C
Observed time courses for these processes are shown in Figs. 2 and 3. The parameters kautox and k−H represent first-order rate constants for autoxidation and hemin (H) dissociation, respectively.
| Hemoglobin 100 μm | kautox | Hemoglobin | k−H (α subunit) | k−H (β subunit) |
|---|---|---|---|---|
| h−1 | 10 μm | h−1 | h−1 | |
| Native HbA | 0.043 ± 0.01 | native metHbA | 0.57 ± 0.10 | 5.8 ± 0.4 |
| (0.027, 0.077)a | ||||
| rHb Kirklareli (αO2βO2) | 0.30 ± 0.01 | met-rHb Kirklareli | 19 ± 1.5 | (≥19) |
| rHb Kirklareli (αCOβO2) | 0.17 ± 0.01 |
a The deconvoluted autoxidation time courses for HbA-O2 show acceleration with an initial rate of ∼0.03 h−1 and final rate of ∼ 0.08 h−1.
In contrast, the spectral changes for the oxidation of rHb Kirklareli are highly complex, showing no isosbestic points. Autoxidation is clearly very rapid, with large initial decreases in absorbance at 578 nm, the α band peak for HbO2. However, in the first 4 h after oxidation occurs, protein aggregates begin to form, causing turbidity that results in marked increases in absorbance at 700 nm. These aggregates then begin to coalesce, precipitate over the next 10 h, and sink to the bottom of the cuvette, leading to decreases in absorbance at 700 nm. The inset to Fig. 7B shows time courses for the following: A, the decay of completely oxygenated rHb Kirklareli; B, the formation of the met-Hb; C, the appearance of hemichrome intermediates; and D, appearance and decay of turbidity. A large fraction of the initial protein precipitates along with its associated heme, as judged by the grayish green color of the solid material at the bottom of the cuvettes. This autoxidation experiment is highly reproducible, including the formation of the insoluble Heinz body-like material at the bottom of the solution.
The cause of rapid precipitation of the newly formed met-rHb Kirklareli is due to rapid hemin dissociation. As shown in Fig. 8 and Table 4, when native metHbA is mixed with excess H64Y/V68F apoMb as a hemin-scavenging reagent, the observed time course at pH 7, 37 °C, is biphasic with an overall half-time of roughly 40 min. Hargrove et al. (23) have shown that the fast phase is due to hemin dissociation from β subunits (∼6 h−1) and the slow phase to dissociation from α subunits (∼0.6 h−1). The observed rates depend on total hemoglobin concentration, with dimers showing significantly higher rates of hemin dissociation than tetramers. In the case of met-rHb Kirklareli, hemin dissociation is very rapid and almost monophasic with an observed rate ≥20 h−1, and complete hemin loss occurs in ≤10 min. These results show that the H58L mutation causes an ∼40-fold increase in the rate of hemin loss from the mutant met-α subunits.
FIGURE 8.
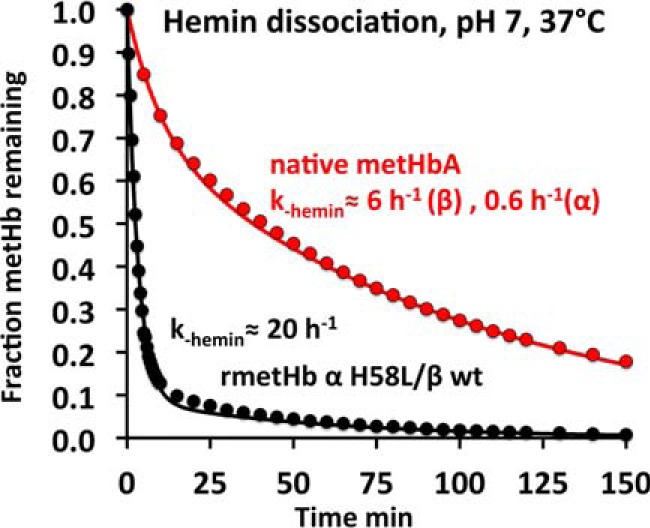
Time courses for hemin dissociation from native metHbA and met-rHb Kirklareli in 0. 1 m phosphate buffer at pH 7, 37 °C. The “brown” metHb sample at 10 μm was mixed with 100 μm H64Y/V68F apoMb, which turns “green” when it scavenges hemin, and with a 10-fold excess of the scavenging agent, the observed rates represent the rate of hemin dissociation from the Hb sample (23, 72). As shown, the rate of hemin dissociation from the mutant α subunits in rHb Kirklareli is roughly 30 times faster than from WT α subunits.
A similar ∼20-fold increase in k−H was observed for the His-64(E7) → Leu mutant of sperm whale metMb (9). The biochemical cause of the increase in k−H is due to loss of direct stabilization of coordinated water by hydrogen bonding to the distal histidine. In effect, His(E7) helps fix the six-coordinate hemin in the distal pocket by this highly favorable electrostatic interaction with covalently bound water. When His(E7) is replaced with apolar amino acids, no water is coordinated to the iron atom creating an unstable pentacoordinate hemin complex, which readily dissociates from both metMb and metHb (7, 9).
The high rate of autoxidation and hemin loss from the α(H58L) subunit in Hb Kirklareli accounts for the instability of the mutant shown in Fig. 7B and in red cells. Although no hemin-scavenging agent was present in the autoxidation experiments, dissociated hemin can react with itself to form dimers and higher order aggregates that lead to dark green precipitates (24). As a result, rapid loss of hemin leads to irreversible formation of apohemoglobin species, which are themselves highly unstable at 37 °C and rapidly precipitate. In the case of Hb Kirklareli, the situation is complex with the mutant α subunit autoxidizing and losing hemin first. The resultant semi-hemoglobin (apo-α/holo-β) is itself unstable and causes the WT β subunit to be more susceptible to oxidation, hemin loss, and precipitation.
Most of these deleterious effects are mitigated when rHb Kirklareli is pretreated with carbon monoxide and then exposed to oxygen. In the experiment shown in Fig. 7C, carbon monoxide on the WT β subunits of rHbCO Kirklareli was replaced with O2 by equilibration of the sample with 1 atm of O2 in the presence of a strong light. The affinity of α(H58L) for CO is so high that this treatment does not result in loss of carbon monoxide from the mutant subunit, and a mixed ligand hybrid is formed, α(H58L)CO/βO2. This same species forms in vivo in response to smoking, as judged by the father's high levels of HbCO (approaching ∼1 CO bound per mutant α subunit or ∼25% of the total heme groups for the heterozygote phenotype). During incubation in air at 37 °C, the CO remains on the high affinity mutant α subunit preventing any oxidation of the α iron atom or loss of heme. Autoxidation of the partner WT βO2 subunit does occur and is ∼4 times faster than in native HbA (kautox ≈0.17 h−1, see Table 4 and Fig. 7D), but little or no precipitation occurs (Fig. 7C). As result, the partially oxidized α(H58L)CO/metβ is stable and can be reversibly re-reduced both in vitro and in red cells, accounting for why the father, who smokes, does not have anemia.
Discussion
Shortly after the publication of the first high resolution crystallographic structure of human hemoglobin, Perutz and Lehmann (25) wrote a remarkable paper entitled “The Molecular Pathology of Human Hemoglobin.” The clinical phenotypes of over 125 different hemoglobinopathies were interpreted in terms of structural alterations in globin structure. From the structure and several well characterized hemoglobinopathies, it was clear that both the proximal (F8 helical position) and distal (E7 helical position) histidines are indispensable for preventing autoxidation and for regulating the ligand affinity. Their initial ideas have been verified by additional studies of Hb variants and site-directed mutagenesis of recombinant Mbs (26) and the α and β chains of recombinant human hemoglobin (2, 27). Hb M-Boston (α2 H58Y (28, 29)), Hb Flurlingen (α2 H58Q, c.177C→G (30)), and Hb Boghé (α2 H58Q, c.177C→A (30)) are so far the only mutant hemoglobins in which the distal histidine at the E7 helical position in α subunits is replaced by another amino acid. Patients with Hb M Boston (α H58Y) and Hb M Saskatoon (β H63Y(E7)) show a history of cyanosis with low oxygen saturation and markedly increased levels of methemoglobinemia (31), which are quite different from the clinical phenotype of Hb Kirklareli (see Table 1). Hb Flurlingen exhibited a mild phenotype resembling α-thalassemia, although Hb Boghé showed normal clinical features even though the amino acid substitutions were the same (30).
Hb Zürich in which the distal histidine in β subunits is replaced with an arginine does have a clinical phenotype that is similar to but more severe than that of Hb Kirklareli (32, 33). The Hb Zurich β H63R mutation is associated with acute hemolytic episodes that are affected by whether or not the patient smokes (34–36). For both Hb Zurich and Hb Kirklareli, markedly increased levels of HbCO are observed in patients who smoke. Although the propositus with the Hb Kirklareli trait showed a slightly elevated HbCO level (4%), her father, who is a smoker, showed a markedly higher level (∼16%).
We considered other causes of the propositus' anemia. First, we looked for TMPRSS6 gene mutations, which have also been shown to cause iron deficiency (37). However, no mutations were identified in the TMPRSS6 gene from either the propositus or her father. In addition, the propositus had no history of gastritis, another leading cause of iron deficiency. Thus, the Hb Kirklareli trait does appear to be the major cause of her iron deficiency, even though Heinz bodies were not seen. When it occurs, autoxidation and denaturation of Hb Kirklareli could be severe enough to cause either lysis or rapid removal by the spleen without the accumulation of the intermediate Heinz body-like precipitates (Fig. 7B). However, if CO is bound to the mutant α subunits, then no precipitation or hemolysis occurs (Fig. 7C).
The similar clinical phenotypes of Hb Kirklareli and Hb Zurich seem incongruous because in one case the distal histidine is replaced with an apolar amino acid, and in the other case, it is replaced with a positively charged basic amino acid. However, a comparison of the active sites of α(H58L) and β(H63R) subunits shows why both mutations lead to poor O2 binding, high relative CO affinity, and rapid rates of autoxidation and hemin dissociation (Fig. 9).
FIGURE 9.

Active site structures of native HbA αO2, rHb Kirklareli, α(H58L)O2, and Hb Zurich β(H63R)O2. The structure of native HbA-O2 was taken from Park et al. ((63), PDB code 2DN1). Superposition of the α subunits of native HbA in 2DN1 (oxy), 2DN3 (CO), and rHb Kirklareli-CO (3QJB) shows that their heme pockets are identical except for the orientations of the distal E7 residues. The distal His residues in the two native α structures occupy slightly altered positions due to differences in the strength of hydrogen bonding to bound CO versus O2. The α Leu(E7) residue in rHbCO Kirklareli fits nicely in the space occupied by the distal His residues in HbA. Because the Leu(E7) residue does not form a hydrogen bond with bound ligands, we assumed that its orientation in the hypothetical HbO2 Kirklareli structure would be identical to that in the HbCO and deoxy forms, and bound O2 was simply coordinated to heme iron using the coordinates in the HbAO2 structure. The structure of Hb Zurich-O2 was based on the Hb Zurich-CO structure published by Tucker et al. (38). Because the coordinates were not available in the Protein Data Bank, the oxy model was generated from oxy HbA ((63), PDB code 2DN1) by mutating the distal E7 residue His-63 to Arg in the program COOT (65). The Arg side chain was then manipulated so that it swings out of the heme pocket toward the CD corner and forms a hydrogen bond with one of the heme propionate oxygens, following the conformation published in Tucker et al. (38). A similar procedure was used by D. Gell to construct the active site of β subunits in Hb Zurich in Thom et al. (73).
In both cases an unhindered apolar active site is created. In Hb Kirklareli, the mutant α subunit has a completely apolar site with the bound O2 surrounded by Leu-29(B10), Phe-43(CD1), Leu-58(E7), Val-62(E11), and Leu-101(G8). There are no favorable electrostatic interactions to increase O2 affinity, inhibit autoxidation, stabilize coordinated water, and prevent hemin dissociation. In both the CO and deoxy crystal structures of Hb Zurich, the Arg-63(E7) side chain is pointing outward, adjacent to the heme 6-propionate (38, 39). Perutz and co-workers (38) argued that the large Arg side chain will not fit easily into the distal pocket and that the highly polar guanidinium group is stabilized by interaction with solvent water molecules and the negatively charged carboxyl group of the heme 6-propionate.
In the 1980s, Caughey and co-workers (40, 41) characterized the spectral and functional properties of Hb Zurich and observed the following: 1) bound CO in the mutant β subunit is in a more apolar environment than WT β subunits, with a C–O stretching frequency about 10 cm−1 higher than the native subunits; 2) Hb Zurich autoxidizes, forms hemichromes, and precipitates 3–10 times more rapidly than HbA at most pH values (42); and 3) O2 binds more poorly, and CO binds much more tightly to the β (H63R) subunits in Hb Zurich (43). Springer et al. (15) and Rohlfs et al. (44) showed that the same His(E7)-Arg mutation in sperm whale Mb causes a 50-fold increase in the rate of autoxidation, a 14-fold increase in CO affinity, and a 13-fold decrease in O2 affinity, resulting in a 200-fold increase in the ratio of CO to O2 affinity.
All of these functional effects of the β H63R replacement in Hb Zurich are similar to those observed for the α H58L mutation in Hb Kirklareli. Both hemoglobinopathies result in anemia for non-smokers due to rapid oxidative degradation of the mutant hemoglobins, marked elevation of HbCO levels in smokers (15–20%) due to preferential CO binding, and no anemia in smokers due to CO stabilization of the mutant subunits.
Zinkman et al. (36) looked more carefully at the relationship between the rates and extents of red cell lysis and HbCO levels in 15 patients with the Hb Zurich trait. The correlation between smokers with high HbCO percentages and low rates and extents of lysis were striking. A similar correlation occurs between the father (smoker) and daughter (non-smoker) with the Hb Kirklareli trait. These correlations suggest that inhalation of small amounts of CO may be therapeutic to inhibit oxidative degradation of the mutant subunits. Our biochemical results also suggest that anti-oxidant therapy might be beneficial to reduce the stress caused by autoxidation, hemin loss, globin precipitation, and red cell lysis. Free hemin is itself quite toxic, generating free radicals, promoting lipid oxidation, and causing inflammation by binding to TLR4-like receptors on macrophages (45–47).
Experimental Procedures
Hematological Analyses of Blood Samples
Samples from the propositus and her parents were collected into tubes containing EDTA but not any anticoagulant. Informed consent was given prior to collection. Hematological evaluations were made with automated cell counters and with standard procedures. The red cell lysates were analyzed by electrophoresis on agarose at pH 8.6 and 6.0 (48) and by different tests, such as Heinz body formation, isopropyl alcohol, and heat stability at pH 7.0 and 60 °C (49). The activities of different erythrocyte enzymes were assayed as described previously by Beutler (50). Hb analysis was performed using cation-exchange HPLC, which was also used to purify the abnormal Hb tetramer, and reverse phase HPLC (48) was used to separate and analyze the denatured α and β apoglobin chains.
Isolation of a significant amount of the variant was difficult because of the lack of a charge difference between Hb Kirklareli and HbA and involved a HPLC procedure with a 21 × 250 mm PolyCAT A column. This chromatographic method was described previously by Divoky et al. (51). To avoid autoxidation, hemoglobin in the freshly prepared hemolysate was converted into the more stable HbCO form, by saturating with 1 atm of pure carbon monoxide (CO) prior to purification. The Hb fractions were collected, re-equilibrated with CO, and then concentrated with a 10-kDa membrane filter cartridge. Because of the difficulty of isolating the mutant tetramer, which co-elutes with HbA, the amount of material obtained from the patient was small and used only to verify the properties observed for the recombinant Hb Kirklareli that could be expressed and purified in large amounts without any HbA contamination.
Protein Sequence Analysis
The techniques used to obtain globin chains and determine sequence variations were identical to those described previously (52). The analyses of crude hemolysates and purified α chains were performed by liquid chromatography coupled to liquid mass spectrometry (LC-MS) (see Figs. 1 and 2 and “Results”). The molecular masses of the intact globin chains were measured by LC-MS-liquid chromatography (Agilent Technologies, Palo Alto, CA) coupled with mass spectrometry (micrOTOF-Q, Bruker Daltonics, Bremen, Germany). The chromatography was performed on a reversed phase column (Nucleosil C18 column 4 × 125 mm, 5 μm, 300 Å, Macherey-Nagel, Dürren, Germany) using water/trifluoroacetic acid 0.1% (solvent A) and acetonitrile/trifluoroacetic acid 0.1% (solvent B) as the mobile phase (profile gradient, from 40 to 55% B in 35 min with a flow rate of 0.3 ml/min).
Purified α chains were digested with endopeptidase AspN, and the peptides were analyzed using nano-LC-MS/MS (see under “Results” and Fig. 3). Digestion with AspN was done with a 1:50 (w/w) enzyme to protein ratio in darkness for 6 h, and the resulting digested peptides were analyzed by nanoLC-MS/MS.
NanoLC-MS/MS analyses were performed on a nano ACQUITY Ultra-Performance-LC system (UPLC) coupled to a Q-TOF mass spectrometer equipped with a nano-electrospray source (maXis, Bruker Daltonics, Bremen, Germany). The UPLC system was equipped with a Symmetry C18 pre-column (20 × 0.18 mm, 5 μm particle size, Waters) and an ACQUITY UPLC® BEH130 C18 separation column (75 μm × 200 mm, 1.7 μm particle size, Waters). The solvent system consisted of 0.1% v/v formic acid in water (solvent A) and 0.1% v/v formic acid in acetonitrile (solvent B). Peptides were trapped on the column by 3 min of flow (5 μl/min) of 99% A and 1% B. Elution was performed at 45 °C with a flow rate of 400 nl/min, using a linear gradient of 1 to 40% B over 35 min.
The mass spectrometer was operated in positive mode, with the following settings. The source temperature was set to 200 °C and dry gas flowing at 4 liters/min. The nano-electrospray voltage was optimized to −4500 V. For tandem MS experiments, the system was operated with automatic switching between MS and MS/MS modes in the range of 50–2200 m/z. The four most abundant peptides (absolute intensity threshold of 1500, preferably with doubly, triply, and quadruply charged ions) were selected from each MS spectrum for further isolation and CID fragmentation using argon as the collision gas. The complete system was fully controlled by Hystar 3.2 (Bruker Daltonics).
Mascot 2.3.02 (Matrix Science, London, UK) software was used to search the MS/MS data for comparison with the SwissProt database using the following parameters. Asp-N was selected as the enzyme. Variable modifications (carbamidomethyl (C) and oxidation (M)) with mass tolerances on precursor and fragment ions of 20 ppm and 0.07 Da were used. For peptides not corresponding to expected masses for normal hemoglobin chains, the corresponding MS/MS spectra were submitted to manual de novo sequencing.
DNA Analysis
Molecular analysis of the globin genes and the TMPRSS6 gene were investigated. Finberg et al. (37) and others have shown that TMPRSS6 codes for a type II transmembrane serine protease that regulates expression of hepicidin and that mutations in this gene can lead to iron-refractory iron deficiency anemia. The cDNA for TMPRSS6 was isolated from white blood cells as described previously (53). Multiplex PCRs were performed to investigate α-thalassemia deletions and α-globin gene triplication using previously reported primers (54). Multiplex ligation-dependent probe amplification techniques (P140 and P102, MRC Holland Amsterdam, The Netherlands) were performed to screen for deletions of both α- and β-globin genes as described previously (55). Direct sequencing of amplified α1- and α2-globin genes was performed (56, 57) and compared with the reference sequences NM_000558.3 (HBA1) and NM_000517.3 (HBA2). To gain insight into the iron deficiency of the patient, the TPMPRSS6 gene was analyzed by sequencing coding regions, intro-exon junctions, and the proximal promoter (58).
Preparation and Crystallization of rHb Kirklareli
The α(H58L)β(WT) rHb mutant was expressed from the pHE2 plasmid in Escherichia coli JM109 strains (4, 59) and purified as described in the supplemental material in Birukou et al. (2, 5).
Recombinant HbCO Kirklareli was crystallized using the batch method initially designed by Perutz (60). Concentrated HbCO and sodium/potassium phosphate, pH 6.7, solutions were mixed together to yield final concentrations of ∼15 mg/ml HbCO in 2.3–2.5 m sodium/potassium phosphate. 200 μl of the mixture followed by a drop of toluene was placed in a sealed glass vial that had been purged with CO. Seeding with finely crushed WT HbCO crystals was used to initiate nucleation. Crystals of 50–100 μm grew over 2–3 days at room temperature.
DeoxyHb Kirklareli was obtained from the CO-bound protein by successive oxidation and reduction because the mutated α(H58L) subunit has such a high affinity for CO that it cannot be converted directly to the O2 form. First, the met-form of the protein was generated by incubating concentrated HbCO with excess potassium ferricyanide in a round bottom flask attached to a Roto-evaporator apparatus. The flask was rotated in an ice bath under constant illumination with strong light to promote CO photodissociation. The gas space above the sample was continuously purged with O2. Conversion to the met-form was verified by color changes and confirmed by recording spectra of aliquots of the sample and monitoring the appearance of the 409-nm Soret peak. The oxidized protein was first passed through a Sephadex G-25 column to remove excess ferricyanide and CO, then reduced with excess dithionite, and passed quickly through another G-25 column at 4 °C to generate the HbO2 form.
Crystallization of recombinant deoxyHb Kirklareli was performed according to the procedure described by Brucker (61), which was adapted from Perutz (60). All precipitant solutions were brought into a N2-filled anaerobic glovebox (Vacuum Atmospheres, Hawthorne, CA) and degassed for 10–20 min. The HbO2 samples were reduced with a 10-fold excess of sodium dithionite in the glove box to remove any residual O2 and metHb. Crystals grew over 2–4 weeks from mixtures of deoxyHb and ammonium phosphate/sulfate at final concentrations of 10 mg/ml Hb and 2.26–2.80 m precipitant.
Determination of Crystal Structures
15% glycerol was added as a cryoprotectant to the appropriate mother liquor just before data collection. For CO complexes, the mounting solution was saturated with 1 atm of pure carbon monoxide; for deoxyHb, the mounting solution was saturated with 1 atm of ultrapure N2, and a few grains of sodium dithionite were added to remove any oxygen contamination that occurred during transfer.
Complete X-ray diffraction data sets for each of the globins were collected at 100 K using CuKα radiation from a Rigaku RUH3R rotating anode X-ray generator and a R-AXIS IV++ image plate detector (Rigaku Americas Co., TX). Data were collected, scaled, and reduced using d*TREK software (62).
The structures were solved in PHENIX by direct Fourier synthesis using the structures of native human HbCO and deoxyHb (63), respectively, as starting models. The structures were refined using PHENIX (64), with manual map inspection and model building performed in COOT (65). Refinement included initial rounds of simulated annealing to calculate unbiased maps to confirm the placement of the mutated residue and ligand. The quality of the model was regularly checked using MolProbity (66). The accession codes for the models, crystal parameters, and statistics of X-ray data collection and refinement are provided in Table 3, and electron density maps are given in Fig. 5. Crystallographic figures were prepared using PyMOL Molecular Graphics System, version 1.2r3pre (Schrödinger, LLC).
Measurements of Ligand Binding and Autoxidation
Birukou et al. (2) measured rates of O2 and CO binding to both intact rHb α(H58L)β(WT) tetramers and the isolated α(H58L) chains with the native N-terminal valine and with a V1M mutation. CO association rate constants were measured in laser photolysis experiments; CO dissociation rate constants were measured by mixing HbCO solutions anaerobically with high [NO] as a replacing ligand; and O2 association and dissociation rate constants were measured in photolysis experiments in which the Hb was equilibrated with mixtures of CO and O2 (2, 12). A summary of the rate constants for WT α, β, and mutant α(H58L) subunits is given in Table 2 for the high affinity or R-state forms of native HbA and rHb Kirklareli.
To measure O2 equilibrium binding and autoxidation, the CO forms of native HbA were converted directly to HbO2, which was verified by recording spectra of aliquots of the sample until the peaks in the Soret and α visible bands stabilized at 415 and 578 nm, respectively. In the case of rHb Kirklareli, the metHb form was made initially by oxidation with ferricyanide as described previously. Then the met-rHb Kirklareli sample was quickly reduced with excess dithionite and passed quickly through another G-25 column using buffer equilibrated with 1 atm of pure O2 at 4 °C to generate the HbO2 form, which was used immediately.
For autoxidation experiments, the HbO2 samples were rapidly diluted to an appropriate concentration in warm buffer (37 °C) in a 1-cm cuvette and placed in a Cary 50 UV-visible spectrophotometer (Agilent Technologies) to begin recording the spectral changes accompanying autoxidation at 37 °C. Complete sets of spectra from 480 to 700 nm were recorded every 5 min. Samples were examined in triplicate, using a cuvette sample changer. In almost all cases a native HbA control was also run with the mutant sample for direct comparisons. The resultant spectra were deconvoluted into fractional amounts of HbO2, metHb, hemichrome, and turbidity using standard basis spectra for the oxygenated, ferric, and hemichrome forms of the hemoglobin sample. The method of least squares and multiple regression for non-linear functions described by Bevington (67) was used to fit the observed spectra. The algorithm was written in the R programming language with the minpack.lm and signal packages installed. The function nls.LM in the minpack.lm package was used and applies the Levenberg-Marquardt algorithm for non-linear least squares curve fitting. The sgolayfilt function in the R signal package was used for smoothing the spectral data and uses the Savitzky-Golay smoothing algorithm. Turbidity effects were estimated from baseline elevation at 700 nm and a spectrum showing a 1/λ4 dependence for light scattering. For native HbA-O2, little or no turbidity was observed, whereas for rHbO2 Kirklareli the fractional amount of light scattering was very large (compare A and B, Fig. 7).
Oxygen equilibrium binding experiments were performed according to the method described by Imai et al. (68) in an apparatus constructed and programmed by Unzai et al. (69) and Maillett (70) at Rice University. Approximately 6 ml of 60 μm oxyhemoglobin diluted into 50 mm HEPES, 0.1 m NaCl, 0.1 mm EDTA, pH 7.4, was placed into the sample chamber and was continuously stirred to ensure rapid equilibration with the gas space. Humidified ultrapure N2 gas was blown into the chamber at a flow rate of 10–20 ml/min, and the solution was deoxygenated for roughly 1 h. Then the gas was switched to compressed air, and re-oxygenation was measured. PO2 was directly monitored with a Clark-type oxygen electrode. Changes in saturation were monitored by recording absorbance at 560 nm using a Shimadzu UV-2100 spectrophotometer (Tokyo, Japan). Because the mutant α subunit in rHb Kirklareli readily autoxidizes, we added the enzymatic reducing system described by Hayashi et al. (71), kept the temperature at 20 °C, and used phosphate-free buffer to keep the O2 affinity relatively high to inhibit autoxidation even further.
Measurements of Hemin Dissociation
Hemin dissociation time courses for met-HbA and met-rHb Kirklareli were measured using the method of Hargrove et al. (72), as applied to hemoglobin (23). In these experiments, cold (4 °C), freshly prepared metHb was rapidly diluted to ∼10 μm in 0.1 m sodium phosphate buffer containing 40 μm H64Y/V68F apoMb as a heme-scavenging agent. Sucrose (600 mm) was present to inhibit precipitation of the apohemoglobin product. The cuvette containing the reaction solution was then rapidly heated to 37 °C in a water bath and placed into a heated cell holder in a Cary 50 spectrophotometer. Spectra were recorded from 400 to 700 nm every minute for ∼4 h. In this case, the spectra were deconvoluted into fractional decreases in holo-metHb (brownish color), increases in holo-H64Y/V68F Mb (greenish color), and increases in turbidity, again using basis spectra for completely oxidized metHb, holo-H64Y/V68F metMb, and light scattering. The algorithm and program were the same as that used to analyze the autoxidation data (67).
Measurement of Tetramer to Dimer Dissociation Constants
The high rates of autoxidation and hemin loss for Hb Kirklareli could also be due to dissociation into dimers, which autoxidize roughly 20 times faster than tetramers and lose hemin roughly 10-fold more rapidly (17). To compare the tetramer-dimer disassociation constant (K4,2) for rHb α(H58L)β(WT) and HbA, we used the analytical gel filtration analysis developed by Manning et al. (19–21), (Fig. 10). HbCO samples in 200 mm potassium phosphate, pH 7, were loaded onto a 24-ml Superose-12 HR 10/30 GL column using an 100-μl loop. The concentration of the eluted protein was determined to be the initial concentration of the loaded sample multiplied by the ratio of the elution peak width at the half-height to the sample volume loaded (19). The elution position of fully tetrameric Hb (Vt) was approximated to be 13.65 ml by loading concentrated Hb, and the elution position of dimeric Hb (Vd) was approximated to be 14.39 ml by loading Hb at very dilute concentrations. The percentage of tetramer (%T) in solution was determined initially at a specific elution position (V) corresponding to a specific [Hb] using Equation 1 (19),
| (Eq. 1) |
FIGURE 10.

A, analytical gel filtration chromatograms of CO-bound HbA and rHb αH58L β(WT). Final concentrations of 0.55 μm HbA and 0.54 μm rHb αH58L β(WT) were eluted off the 24-ml Superose-12 HR 10/30 GL column equilibrated with 200 mm potassium phosphate, pH 7. B, K4,2 determination from analytical gel filtration analysis. The percentage of tetrameric Hb (%T) was determined based on elution peak positions and linear least square fitting to Equation 2 (19).
We then determined K4,2 for the Hbs using Equation 2 through simple linear least square fitting, utilizing the Solver package in MS Excel 2011.
| (Eq. 2) |
Interestingly, at almost all the concentrations loaded, the elution positions of rHb α(H58L)β(WT) always preceded that of HbA (Fig. 10A), indicating that the mutant had a higher propensity to stay tetrameric. Accordingly, from our fittings, we determined the K4,2 of HbA to be 1.1 μm, and the K4,2 of rHb α(H58L)β(WT) to be ∼4-fold lower at 0.3 μm (Fig. 10B). Thus, the rapid degradation of HbO2 Kirklareli is not due to a greater tendency to dissociate into dimers.
Author Contributions
E. B., T. E., and K. W. treated the patient and performed the initial blood hemoglobin and DNA analyses. C. S.-R., A. V. D., and T. D. A. performed the mass spectral analyses to identify the mutated peptide and the genome sequencing to identify the base substitution. A. S. B. C. performed the autoxidation and hemin loss experiments. J. S. and I. B. determined the crystal structures of the α and β Leu(E7) recombinant hemoglobins and made graphics drawings. J. S. performed the O2 equilibrium curve measurements. P. P. S. measured the tetramer to dimer dissociation constants of native HbA and recombinant Hb Kirklareli. J. S. O. helped to design the various ligand binding and denaturation experiments and organized the writing of the manuscript to which all authors contributed.
Acknowledgments
We are indebted to Dr. Sabine Preisler-Adams, from the Institut für Humangenetik Westfälische Wilhelms-Universität, D-48149 Münster, Germany, for performing the DNA analysis before her death. We also greatly appreciate the cooperation of the members of the family in this study.
This work was supported by Grant C-0612 from Robert A. Welch Foundation (to J. S. O.), National Institutes of Health Grant P01 HL110900 (to J. S. O.), the Ministry of Science and Arts, Baden-Württemberg, Germany (to University Medical Center, Freiburg, Germany (to E. B., T. E., and K. W.)), the Fondation pour la Recherche Médicale, and the Proteomic French Infrastructure (ProFI) Grant ANR-10-INSB-08-03 (to C. S.-R., A. D., and T. D. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PDB
- Protein Data Bank
- rHb
- recombinant human Hb
- IDA
- iron deficiency anemia
- CID
- collision-induced dissociation.
References
- 1. Hardison R. C., Chui D. H. K., Riemer C. R., Miller W., Carver M. F., Molchanova T. P., Efremov G. D., and Huisman T. H. J. (1998) Access to “A Syllabus of Human Hemoglobin Variants” (1996) via the World Wide Web. Hemoglobin 22, 113–127 [DOI] [PubMed] [Google Scholar]
- 2. Birukou I., Schweers R. L., and Olson J. S. (2010) Distal histidine stabilizes bound O2 and acts as a gate for ligand entry in both subunits of adult human hemoglobin. J. Biol. Chem. 285, 8840–8854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Looker D., Mathews A. J., Neway J. O., and Stetler G. L. (1994) Expression of recombinant human hemoglobin in Escherichia coli. Methods Enzymol. 231, 364–374 [DOI] [PubMed] [Google Scholar]
- 4. Shen T. J., Ho N. T., Zou M., Sun D. P., Cottam P. F., Simplaceanu V., Tam M. F., Bell D. A. Jr., and Ho C. (1997) Production of human normal adult and fetal hemoglobins in Escherichia coli. Protein Eng. 10, 1085–1097 [DOI] [PubMed] [Google Scholar]
- 5. Birukou I. (2011) Determination of Pathways for Oxygen Binding to Human Hemoglobin A. in Biochemistry and Cell Biology. Ph.D. thesis, Rice University, Houston, TX [Google Scholar]
- 6. Phillips G. N. Jr., Teodoro M., Li T., Smith B., Gilson M. M., and Olson J. S. (1999) Bound CO is a molecular probe of electrostatic potential in the distal pocket of myoglobin. J. Phys. Chem. B. 103, 8817–8829 [Google Scholar]
- 7. Quillin M. L., Arduini R. M., Olson J. S., and Phillips G. N. Jr. (1993) High-resolution crystal structures of distal histidine mutants of sperm whale myoglobin. J. Mol. Biol. 234, 140–155 [DOI] [PubMed] [Google Scholar]
- 8. Brantley R. E. Jr., Smerdon S. J., Wilkinson A. J., Singleton E. W., and Olson J. S. (1993) The mechanism of autoxidation of myoglobin. J. Biol. Chem. 268, 6995–7010 [PubMed] [Google Scholar]
- 9. Hargrove M. S., Wilkinson A. J., and Olson J. S. (1996) Structural factors governing hemin dissociation from metmyoglobin. Biochemistry 35, 11300–11309 [DOI] [PubMed] [Google Scholar]
- 10. Hargrove M. S., Krzywda S., Wilkinson A. J., Dou Y., Ikeda-Saito M., and Olson J. S. (1994) Stability of myoglobin: a model for the folding of heme proteins. Biochemistry 33, 11767–11775 [DOI] [PubMed] [Google Scholar]
- 11. Samuel P. P., Smith L. P., Phillips G. N. Jr, and Olson J. S. (2015) Apoglobin stability is the major factor governing both cell-free and in vivo expression of holomyoglobin. J. Biol. Chem. 290, 23479–23495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Olson J. S., Foley E. W., Maillett D. H., and Paster E. V. (2003) Measurement of rate constants for reactions of O2, CO, and NO with hemoglobin. Methods Mol. Med. 82, 65–91 [DOI] [PubMed] [Google Scholar]
- 13. Mathews A. J., and Olson J. S. (1994) Assignment of rate constants for O2 and CO binding to α and β subunits within R- and T-state human hemoglobin. Methods Enzymol. 232, 363–386 [DOI] [PubMed] [Google Scholar]
- 14. Olson J. S., and Phillips G. N. J. (1997) Myoglobin discriminates between O2, NO and CO by electrostatic interactions with the bound ligand. J. Biol. Inorg. Chem. 2, 544–552 [Google Scholar]
- 15. Springer B. A., Egeberg K. D., Sligar S. G., Rohlfs R. J., Mathews A. J., and Olson J. S. (1989) Discrimination between oxygen and carbon monoxide and inhibition of autoxidation by myoglobin. Site-directed mutagenesis of the distal histidine. J. Biol. Chem. 264, 3057–3060 [PubMed] [Google Scholar]
- 16. Nambu S., Matsui T., Goulding C. W., Takahashi S., and Ikeda-Saito M. (2013) A new way to degrade heme: the Mycobacterium tuberculosis enzyme MhuD catalyzes heme degradation without generating CO. J. Biol. Chem. 288, 10101–10109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mollan T. L., Jia Y., Banerjee S., Wu G., Kreulen R. T., Tsai A. L., Olson J. S., Crumbliss A. L., and Alayash A. I. (2014) Redox properties of human hemoglobin in complex with fractionated dimeric and polymeric human haptoglobin. Free Radic. Biol. Med. 69, 265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang L., Levy A., and Rifkind J. M. (1991) Autoxidation of hemoglobin enhanced by dissociation into dimers. J. Biol. Chem. 266, 24698–24701 [PubMed] [Google Scholar]
- 19. Manning L. R., Dumoulin A., Jenkins W. T., Winslow R. M., and Manning J. M. (1999) Determining subunit dissociation constants in natural and recombinant proteins. Methods Enzymol. 306, 113–129 [DOI] [PubMed] [Google Scholar]
- 20. Manning L. R., Jenkins W. T., Hess J. R., Vandegriff K., Winslow R. M., and Manning J. M. (1996) Subunit dissociations in natural and recombinant hemoglobins. Protein Sci. 5, 775–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manning L. R., Russell J. E., Padovan J. C., Chait B. T., Popowicz A., Manning R. S., and Manning J. M. (2007) Human embryonic, fetal, and adult hemoglobins have different subunit interface strengths. Correlation with lifespan in the red cell. Protein Sci. 16, 1641–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schweers R. (2003) Electrostatic Regulation of O2 and CO Binding in the α and β Subunits of Recombinant Human Hemoglobin. Ph.D. thesis, Rice University, Houston, TX [Google Scholar]
- 23. Hargrove M. S., Whitaker T., Olson J. S., Vali R. J., and Mathews A. J. (1997) Quaternary structure regulates hemin dissociation from human hemoglobin. J. Biol. Chem. 272, 17385–17389 [DOI] [PubMed] [Google Scholar]
- 24. de Villiers K. A., Kaschula C. H., Egan T. J., and Marques H. M. (2007) Speciation and structure of ferriprotoporphyrin IX in aqueous solution: spectroscopic and diffusion measurements demonstrate dimerization, but not mu-oxo dimer formation. J. Biol. Inorg. Chem. 12, 101–117 [DOI] [PubMed] [Google Scholar]
- 25. Perutz M. F., and Lehmann H. (1968) Molecular pathology of human haemoglobin. Nature 219, 902–909 [DOI] [PubMed] [Google Scholar]
- 26. Springer B. A., Sligar S. G., Olson J. S., and Phillips G. N. (1994) Mechanisms of ligand recognition in myoglobin. Chem. Rev. 94, 699–714 [Google Scholar]
- 27. Mathews A. J., Rohlfs R. J., Olson J. S., Tame J., Renaud J. P., and Nagai K. (1989) The effects of E7 and E11 mutations on the kinetics of ligand binding to R state human hemoglobin. J. Biol. Chem. 264, 16573–16583 [PubMed] [Google Scholar]
- 28. Nagatomo S., Jin Y., Nagai M., Hori H., and Kitagawa T. (2002) Changes in the abnormal α-subunit upon CO-binding to the normal β-subunit of Hb M Boston: resonance Raman, EPR and CD study. Biophys. Chem. 98, 217–232 [DOI] [PubMed] [Google Scholar]
- 29. Pik C., and Raine D. N. (1964) The chemical identification of two cases of Hb M (Hb M Boston and Hb M Saskatoon) occurring in England. Clin. Chim. Acta 10, 90–92 [DOI] [PubMed] [Google Scholar]
- 30. Qadah T., Finlayson J., Dennis M., Newbound C., and Ghassemifar R. (2015) Experimental characterization of Hb Flurlingen (HBA2: c.177 C > G, p.His > Gln) and Hb Boghe (HBA2: c.177 C > A, p.His > Gln) reveals contradictory HBA2 expression and translation patterns despite identical amino acid substitutions. Hemoglobin 39, 340–345 [DOI] [PubMed] [Google Scholar]
- 31. Pulsinelli P. D., Perutz M. F., and Nagel R. L. (1973) Structure of hemoglobin M Boston, a variant with a five-coordinated ferric heme. Proc. Natl. Acad. Sci. U.S.A. 70, 3870–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huisman T. H., Horton B., Bridges M. T., Betke K., and Hitzig W. H. (1961) A new abnormal human hemoglobin-Hb: Zurich. Clin. Chim. Acta 6, 347–355 [DOI] [PubMed] [Google Scholar]
- 33. Virshup D. M., Zinkham W. H., Sirota R. L., and Caughey W. S. (1983) Unique sensitivity of Hb Zurich to oxidative injury by phenazopyridine: reversal of the effects by elevating carboxyhemoglobin levels in vivo and in vitro. Am. J. Hematol 14, 315–324 [DOI] [PubMed] [Google Scholar]
- 34. Wallace W. J., Volpe J. A., Maxwell J. C., and Caughey W. S. (1976) Properties of hemoglobin A and hemoglobin Zurich (β63 histidine replaced by arginine): quantitative evaluation of functional abnormalities in hemoglobins. Biochem. Biophys. Res. Commun. 68, 1379–1386 [DOI] [PubMed] [Google Scholar]
- 35. Zinkham W. H., Houtchens R. A., and Caughey W. S. (1980) Carboxyhemoglobin levels in an unstable hemoglobin disorder (Hb Zurich): effect on phenotypic expression. Science 209, 406–408 [DOI] [PubMed] [Google Scholar]
- 36. Zinkham W. H., Houtchens R. A., and Caughey W. S. (1983) Relation between variations in the phenotypic expression of an unstable hemoglobin disorder (hemoglobin Zurich) and carboxyhemoglobin levels. Am. J. Med. 74, 23–29 [DOI] [PubMed] [Google Scholar]
- 37. Finberg K. E., Heeney M. M., Campagna D. R., Aydinok Y., Pearson H. A., Hartman K. R., Mayo M. M., Samuel S. M., Strouse J. J., Markianos K., Andrews N. C., and Fleming M. D. (2008) Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet. 40, 569–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tucker P. W., Phillips S. E., Perutz M. F., Houtchens R., and Caughey W. S. (1978) Structure of hemoglobins Zurich [His-E7(63)β replaced by Arg] and Sydney [Val E11(67)β replaced by Ala] and role of the distal residues in ligand binding. Proc. Natl. Acad. Sci. U.S.A. 75, 1076–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Phillips S. E., Hall D., and Perutz M. F. (1981) Structure of deoxyhaemoglobin Zurich (HisE7(63 β) - greater than Arg). J. Mol. Biol. 150, 137–141 [DOI] [PubMed] [Google Scholar]
- 40. Choc M. G., and Caughey W. S. (1981) Evidence from infrared and 13C NMR spectra for discrete rapidly interconverting conformers at the carbon monoxide binding sites of hemoglobins A and Zurich. J. Biol. Chem. 256, 1831–1838 [PubMed] [Google Scholar]
- 41. Shimada H., Dong A., Matsushima-Hibiya Y., Ishimura Y., and Caughey W. S. (1989) Distal His–Arg mutation in bovine myoglobin results in a ligand binding site similar to the abnormal β site of hemoglobin Zurich (β 63 His–Arg). Biochem. Biophys. Res. Commun. 158, 110–114 [DOI] [PubMed] [Google Scholar]
- 42. Di Iorio E. E., Winterhalter K. H., Mansouri A., Blumberg W. E., and Peisach J. (1984) Studies on the oxidation of hemoglobin Zurich (β 63 E7 Arg). Eur. J. Biochem. 145, 549–554 [DOI] [PubMed] [Google Scholar]
- 43. Giacometti G. M., Brunori M., Antonini E., Di Iorio E. E., and Winterhalter K. H. (1980) The reaction of hemoglobin Zurich with oxygen and carbon monoxide. J. Biol. Chem. 255, 6160–6165 [PubMed] [Google Scholar]
- 44. Rohlfs R. J., Mathews A. J., Carver T. E., Olson J. S., Springer B. A., Egeberg K. D., and Sligar S. G. (1990) The effects of amino acid substitution at position E7 (residue 64) on the kinetics of ligand binding to sperm whale myoglobin. J. Biol. Chem. 265, 3168–3176 [PubMed] [Google Scholar]
- 45. Belcher J. D., Chen C., Nguyen J., Milbauer L., Abdulla F., Alayash A. I., Smith A., Nath K. A., Hebbel R. P., and Vercellotti G. M. (2014) Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 123, 377–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schaer D. J., Buehler P. W., Alayash A. I., Belcher J. D., and Vercellotti G. M. (2013) Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 121, 1276–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schaer D. J., and Alayash A. I. (2010) Clearance and control mechanisms of hemoglobin from cradle to grave. Antioxid. Redox Signal. 12, 181–184 [DOI] [PubMed] [Google Scholar]
- 48. Bissé E., Zorn N., Heinrichs I., Eigel A., Van Dorsselaer A., Wieland H., Kister J., and Marden M. C. (2000) Characterization of a new electrophoretically silent hemoglobin variant. Hb saale OR α2β 2 84(EF8)Thr → Ala. J. Biol. Chem. 275, 21380–21384 [DOI] [PubMed] [Google Scholar]
- 49. Huisman T. H., and Jonxsis J. H. (1977) The Hemoglobinopathies:Techniques of Identification. Clinical and Biochemical Analysis, pp. 70–400, Marcel Dekker, New York [Google Scholar]
- 50. Beutler E. (1975) Red Cell Metabolism. A Manual of Biochemical Methods, 2nd Ed., pp. 3–178, Grune & Stratton, New York [Google Scholar]
- 51. Divoky V., Bissé E., Wilson J. B., Gu L. H., Wieland H., Heinrichs I., Prior J. F., and Huisman T. H. (1992) Heterozygosity for the IVS-I-5 (G→C) mutation with a G→A change at codon 18 (Val→Met; Hb Baden) in cis and a T→G mutation at codon 126 (Val→Gly; Hb Dhonburi) in trans resulting in a thalassemia intermedia. Biochim. Biophys. Acta 1180, 173–179 [DOI] [PubMed] [Google Scholar]
- 52. Alayi T. D., Van Dorsselaer A., Epting T., Bissé E., and Schaeffer-Reiss C. (2014) Hb A2-Konz [δ50(D1)Ser → Thr; HBD: c.151T > A]: a new δ chain hemoglobin variant characterized by mass spectrometry and high performance liquid chromatography. Hemoglobin 38, 133–136 [DOI] [PubMed] [Google Scholar]
- 53. Bissé E., Hovasse A., Preisler-Adams S., Epting T., Wagner O., Kögel G., Van Dorsselaer A., and Schaeffer-Reiss C. (2011) Hb Riesa or β93 (F9) Cys→Ser, a new electrophoretically silent haemoglobin variant interfering with haemoglobin A1c measurement. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 879, 2952–2956 [DOI] [PubMed] [Google Scholar]
- 54. Liu Y. T., Old J. M., Miles K., Fisher C. A., Weatherall D. J., and Clegg J. B. (2000) Rapid detection of α-thalassaemia deletions and α-globin gene triplication by multiplex polymerase chain reactions. Br. J. Haematol. 108, 295–299 [DOI] [PubMed] [Google Scholar]
- 55. So C. C., So A. C., Chan A. Y., Tsang S. T., Ma E. S., and Chan L. C. (2009) Detection and characterisation of β-globin gene cluster deletions in Chinese using multiplex ligation-dependent probe amplification. J. Clin. Pathol. 62, 1107–1111 [DOI] [PubMed] [Google Scholar]
- 56. Sanguansermsri T., Matragoon S., Changloah L., and Flatz G. (1979) Hemoglobin Suan-Dok (α 2 109 (G16) Leu replaced by Arg β 2): an unstable variant associated with α-thalassemia. Hemoglobin 3, 161–174 [DOI] [PubMed] [Google Scholar]
- 57. Eng L. I., Baer A., Lewis A. N., and Welch Q. B. (1973) Hemoglobin constant spring (slow-moving hemoglobin X components) and hemoglobin in Malayan aborigines. Am. J. Hum. Genet. 25, 382–387 [PMC free article] [PubMed] [Google Scholar]
- 58. Guillem F., Lawson S., Kannengiesser C., Westerman M., Beaumont C., and Grandchamp B. (2008) Two nonsense mutations in the TMPRSS6 gene in a patient with microcytic anemia and iron deficiency. Blood 112, 2089–2091 [DOI] [PubMed] [Google Scholar]
- 59. Shen T. J., Ho N. T., Simplaceanu V., Zou M., Green B. N., Tam M. F., and Ho C. (1993) Production of unmodified human adult hemoglobin in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 90, 8108–8112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Perutz M. F. (1968) Preparation of haemoglobin crystals. J. Cryst. Growth 2, 54–55 [Google Scholar]
- 61. Brucker E. A. (2000) Genetically crosslinked hemoglobin: a structural study. Acta Crystallogr. D Biol. Crystallogr. 56, 812–816 [DOI] [PubMed] [Google Scholar]
- 62. Pflugrath J. W. (1999) The finer things in X-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 55, 1718–1725 [DOI] [PubMed] [Google Scholar]
- 63. Park S. Y., Yokoyama T., Shibayama N., Shiro Y., and Tame J. R. (2006) 1.25 A resolution crystal structures of human haemoglobin in the oxy, deoxy and carbonmonoxy forms. J. Mol. Biol. 360, 690–701 [DOI] [PubMed] [Google Scholar]
- 64. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 66. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bevington P. R. (1969) Data Reduction and Error Analysis for The Physical Sciences, pp. 204–246, McGraw-Hill Book Co., New York [Google Scholar]
- 68. Imai K., Morimoto H., Kotani M., Watari H., and Hirata W. (1970) Studies on the function of abnormal hemoglobins. I. An improved method for automatic measurement of the oxygen equilibrium curve of hemoglobin. Biochim. Biophys. Acta 200, 189–196 [DOI] [PubMed] [Google Scholar]
- 69. Unzai S., Eich R., Shibayama N., Olson J. S., and Morimoto H. (1998) Rate constants for O2 and CO binding to the α and β subunits within the R and T states of human hemoglobin. J. Biol. Chem. 273, 23150–23159 [DOI] [PubMed] [Google Scholar]
- 70. Maillett D. H. (2003) Engineering Hemoglobin and Myoglobins for Efficient O2 Transport. Ph.D. thesis, Rice University, Houston, TX [Google Scholar]
- 71. Hayashi A., Suzuki T., and Shin M. (1973) An enzymic reduction system for metmyoglobin and methemoglobin, and its application to functional studies of oxygen carriers. Biochim. Biophys. Acta 310, 309–316 [DOI] [PubMed] [Google Scholar]
- 72. Hargrove M. S., Singleton E. W., Quillin M. L., Ortiz L. A., Phillips G. N. Jr., Olson J. S., and Mathews A. J. (1994) His64(E7→Tyr apomyoglobin as a reagent for measuring rates of hemin dissociation. J. Biol. Chem. 269, 4207–4214 [DOI] [PubMed] [Google Scholar]
- 73. Thom C. S., Dickson C. F., Olson J. S., Gell D. A., and Weiss M. J. (2015) in NATHAN and OSKI's Hematology of Infancy and Childhood (Orkin S. H., Nathan D. G., Ginsburg D., Look A. T., Fisher D. E., and Lux S. E., eds) pp. 630–672, Saunders Elsevier, Philadelphia, PA [Google Scholar]