

Abstract
Grafting of neural stem cells (NSCs) or GABA-ergic progenitor cells (GPCs) into the hippocampus could offer an alternative therapy to hippocampal resection in patients with drug-resistant chronic epilepsy, which afflicts >30% of temporal lobe epilepsy (TLE) cases. Multipotent, self-renewing NSCs could be expanded from multiple regions of the developing and adult brain, human embryonic stem cells (hESCs), and human induced pluripotent stem cells (hiPSCs). On the other hand, GPCs could be generated from the medial and lateral ganglionic eminences of the embryonic brain and from hESCs and hiPSCs. To provide comprehensive methodologies involved in testing the efficacy of transplantation of NSCs and GPCs in a rat model of chronic TLE, NSCs derived from the rat medial ganglionic eminence (MGE) and MGE-like GPCs derived from hiPSCs are taken as examples in this unit. The topics comprise description of the required materials, reagents and equipment, methods for obtaining rat MGE-NSCs and hiPSC-derived MGE-like GPCs in culture, generation of chronically epileptic rats, intrahippocampal grafting procedure, post-grafting evaluation of the effects of grafts on spontaneous recurrent seizures and cognitive and mood impairments, analyses of the yield and the fate of graft-derived cells, and the effects of grafts on the host hippocampus.
Keywords: cell transplantation, chronic temporal lobe epilepsy, Cognitive and mood impairments, GABA-ergic progenitors, glial-cell line derived neurotrophic factor, hippocampal neurogenesis, human induced pluripotent stem cells, medial ganglionic eminence, phenotypic differentiation of graft-derived cells, spontaneous recurrent seizures
Introduction
Chronic temporal lobe epilepsy (TLE) is characterized by recurrent partial complex seizures, memory impairments, depression, and substantial decline in hippocampal neurogenesis (Astur et al., 2002; Hattiangady et al., 2004, 2011; Detour et al., 2005; Coras et al., 2010; Hattiangady and Shetty, 2010; Shetty, 2011). Antiepileptic drug therapy, though widely used for controlling seizures, has no effect on the course of the disease and fails to restrain seizures in >30% of TLE patients (Fisher et al., 1998; Strine et al., 2005). Intracerebral transplantation of NSCs or γ-amino butyric acid (GABA) positive progenitor cells (GPCs) is evolving as an attractive therapy for promoting regeneration and repair in various brain disorders including TLE (Shetty and Bates, 2015). Fascination for using NSCs are linked to their properties such as multipotency and ability for self-renewal and the ease by which they can be obtained from multiple regions of the developing and adult brain, human embryonic stem cells (hESCs), and human induced pluripotent stem cells (hiPSCs) (Shetty and Hattiangady, 2007; Hattiangady and Shetty, 2012; Shetty, 2014). On the other hand, interest in utilizing GPCs stems from the advent of novel directed differentiation methods to obtain them in large numbers from hESCs and hiPSCs (Liu et al., 2013).
Studies in neurological disease models have shown that NSCs can survive intracerebral grafting, engraft into the injured brain areas, release a multitude of neurotrophic factors, positively influence the survival of host cells and tissues, and promote functional recovery. Similarly, GPC grafting has shown considerable promise for alleviating deficits in prototypes of several neurological disorders (Shetty and Bates, 2015). Transplantation of apt NSCs in TLE may considerably restrain spontaneous recurrent seizures (SRS) because of their ability to give rise to significant numbers of neurons synthesizing the inhibitory neurotransmitter GABA and/or astrocytes synthesizing the anticonvulsant protein the glial cell line–derived neurotrophic factor (GDNF) ((Waldau et al., 2010; Hattiangady and Shetty, 2012; Hattiangady et al., 2015). Grafting of NSCs may also improve cognitive function in chronic TLE because of their ability to engraft into neurogenic regions of the dentate gyrus and thereby influence the extent of hippocampal neurogenesis. On the other hand, GPC grafting has been shown to reduce seizures through addition of new GABA-ergic interneurons and improved GABA-ergic neurotransmission in the hippocampus of brains afflicted with TLE (Hunt et al., 2013; Hattiangady et al., 2013; Henderson et al., 2014; Cunningham et al., 2014).
In this unit, to provide a detailed methodology involved in evaluating the usefulness of transplantation of NSCs and GPCs in a rat model of chronic TLE, we describe the protocol for grafting NSCs expanded from the MGE of the embryonic day-14 rat fetuses and MGE-like GPCs derived from hiPSCs into hippocampi of rats exhibiting chronic TLE. The protocols mainly include description of the required materials, reagents and equipment, methods for obtaining rat MGE-NSCs and hiPSC-derived MGE-like GPCs in culture, generation of chronically epileptic rats (CERs), intrahippocampal grafting procedure, post-grafting evaluation of the effects of grafts on spontaneous recurrent seizures and cognitive and mood impairments, analyses of the yield and the fate of graft-derived cells, and the effects of grafts on the host hippocampus.
Figure 1 shows a schematic representation of MGE-NSC grafting experiments described in this manuscript.
Figure 1.
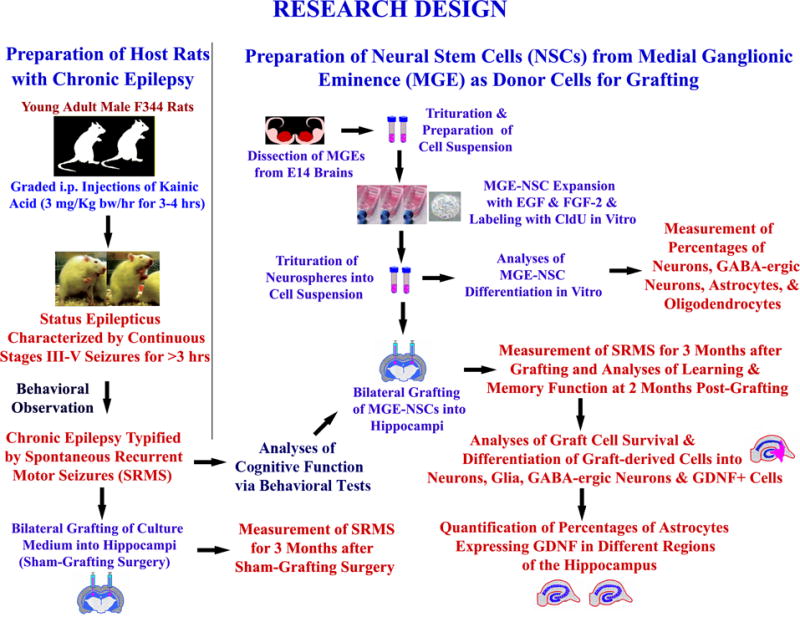
Experimental design of an MGE-NSC grafting study performed in chronically epileptic rats. It shows the creation of host rats with chronic epilepsy for grafting and sham-grafting surgery (on the left side), and the preparation of fresh neural stem cells (NSCs) expanded in vitro from embryonic medial ganglionic eminence (MGE) as donor cells (upper half on right). The various measurements performed after MGE-NSC grafting include frequency, duration, and severity of spontaneous seizures, learning and memory function, graft-cell survival and differentiation, and effects of grafts on expression of GDNF in hippocampal astrocytes (lower half on right). Reproduced from Waldau et al. (2010)
NOTE: All protocols using live animal studies must be first reviewed and approved by the Institutional Animal Care and Use Committee (IACUC). The experimenter must strictly follow all the guidelines recommended by the IACUC while performing the experiments in animal models.
BASIC PROTOCOL 1: GENERATION OF RATS EXHIBITING CHRONIC TLE: INDUCTION OF STATUS EPILEPTICUS (SE) IN ADULT MALE F344 RATS
In this protocol, we describe how to generate rats exhibiting chronic temporal lobe epilepsy characterized by SRS and cognitive and mood dysfunction using a chemoconvulsant chemical [i.e., kainic acid (KA)] to induce status epilepticus (SE). As generation of rats exhibiting chronic TLE requires a time frame of 3 to 5 months, the experiments to be performed on chronically epileptic rats need to be planned well in advance. Furthermore, as the extent of SRS varies between animals (Rao et al., 2006a, 2007; Waldau et al., 2010; Hattiangady et al., 2011), having a larger pool of rats exhibiting chronic TLE would help in choosing animals exhibiting a similar extent (frequency and intensity) of SRS for the transplantation study.
Materials
Experimental animals: 4- to 5-month-old male Fischer 344 (F344) rats
Kainic acid (KA; Milestone PharmTech)
Saline (0.9% NaCl)
Diazepam
Ringer’s lactate solution, sterile
Regular rat chow soaked in water (soft pellets) and transgel
Additional reagents and equipment for intraperitoneal and subcutaneous injections of drugs to rats (Donovan and Brown, 2006)
Establish the animal model
-
1Order 4- to 5-month-old male F344 rats and allow them to acclimatize to the new environment at the vivarium for at least a week.
- Other stains of rats such as Sprague-Dawley may also be used, but these appear to require higher or additional doses of KA for induction of SE (see Hellier et al., 1998 for details). Acute seizure behavior varies depending on the age and sex of the animal, and hence the protocol described here is good only for 4- to 5-month old male F344 rats. If induction of SE is planned for female, younger, or aged rats, it is important to standardize the required dose and injections of KA for eliciting SE in these models.
-
2Prepare a desired amount of the KA solution (e.g., 3.0 mg/ml in sterile saline).
- As KA can be obtained from multiple sources, it will be important to stick to a single source to avoid confounds in SE induction between different groups of rats. We currently use the KA sold by Milestone PharmTech, which has worked well in our experiments.
-
3Measure the weight of each rat and inject KA intraperitoneally (Donovan and Brown, 2006) at a dose of 3.0 mg/kg body weight at hourly intervals.
- Three to four injections of KA are typically sufficient for inducing SE in most rats for the age group mentioned above. It is possible that some rats may develop SE with just two injections of KA while some others may need additional (i.e., >4) injections at a full dose (3 mg/kg body weight) or at a half dose (1.5 mg/kg body weight) for inducing SE. Therefore, it is important to closely observe and score the type and intensity of acute seizures after two injections of KA and empirically determine whether or not additional KA injections would be required to induce SE on a rat-by-rat basis [See protocol below for Scoring of acute seizures].
- Furthermore, a small fraction of rats (5% to 10%) exhibit immobility and fail to show the features of SE even with four injections of KA. Such rats need to be excluded from the study, as additional KA injections would typically lead to mortality in these animals.
Scoring of acute seizures induced by KA
It will be difficult to score stage I to II seizures, which are characterized by salivation, excessive grooming behavior, mastication, wet dog shakes, etc. However, stages III to V seizures are much easier to follow and can be scored using a modified Racine scale (Racine, 1972; Ben-Ari, 1985; Hellier et al., 1998; Rao et al., 2006a).
-
4Score seizures as follows:
- Stage III seizure is characterized by the unilateral forelimb clonus (Fig. 2)
- Stage IV seizure is identified by the bilateral forelimb clonus (or the piano playing or praying posture; Fig. 2D.7.2)
- Stage V seizure is typified by the bilateral forelimb clonus coupled with rearing and falling.
- The definition of the onset of SE in experimental animals varies in different studies depending upon the behavior of the animal model employed in the study. Based on our experience in scoring acute seizures in a large number of male F344 rats injected with KA over the last 10 years, our definition of the onset of SE has also evolved. We now identify the onset of SE in male F344 rats as the occurrence of the first stage V seizure followed by continuous stages III to V seizures for over 10 min (Type 1 SE) or continuous stage IV seizures for over 10 min (Type 2 SE).
- Rats exhibiting Type 1 or Type 2 SE typically continue to have intermittent stage III to V seizures for over 3 hr at a rate of 5 to 10 seizures per hr. Therefore, once a rat exhibits either Type 1 or Type 2 SE, it is prudent to discontinue KA injections and to score the rat for seizures in the next 2–3 hrs to determine the extent of SE. Virtually all rats that exhibit Type 1 or Type 2 SE and display 5 to 10 seizures during the 1st and 2nd hr after SE would develop chronic epilepsy characterized by SRS within 2–5 months post-SE. However, it is possible that a small fraction (~5%) of rats exhibiting Type 1 or Type 2 SE may not display additional seizures in the 2nd and 3rd hour after SE. It will be important to exclude these rats from the study, as such rats rarely develop robust chronic TLE.
-
5Terminate acute seizures with diazepam injection at a dose of 5 mg/kg body weight once the animal completes 2 or 3 hrs of seizure activity.
- In our earlier studies, we left the rats after SE to recover without any antiepileptic medication (Rao et al., 2006a, 2007). While such rats developed robust chronic epilepsy (typified by increased frequencies and intensities of SRS), they were prone to increased mortality both immediately after SE and also in the chronic phase of epilepsy. Therefore, over the last five years, we have tested the effect of diazepam injection at a dose of 5 mg/kg body weight once the animal completes 2–3 hrs of seizure activity. Based on our experience, diazepam injection after 2–3 hrs of seizure activity reduces mortality of animals that undergo SE and also does not interfere with the development of chronic TLE.
- Animals treated with diazepam after SE display reduced frequency and intensity of SRS in the chronic phase of epilepsy, in comparison to animals treated with no diazepam after SE. Therefore, it is important to stick to one SE protocol. Examples include raising CERs with moderate frequencies of SRS through diazepam treatment at a specific time-point after SE onset (i.e. 2 or 3 hrs after SE onset) or generating CERs with very high frequencies of SRS by avoiding antiepileptic drug treatment after SE onset. This methodology facilitates selection of rats with similar SRS frequency for different animal groups in an experiment.
Figure 2.
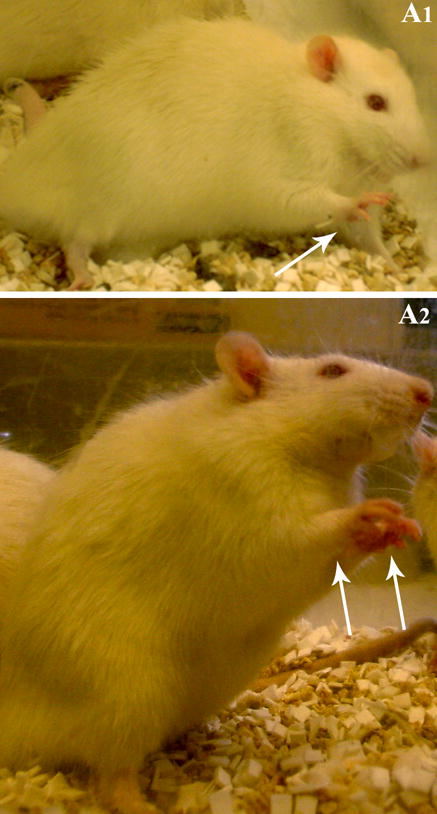
Examples of a Stage III seizure characterized by the unilateral forelimb clonus (A1) and a stage IV seizure typified by a bilateral forelimb clonus (A2).
Care for animals after SE
-
6Observe animals for lethargy and distress after SE. Provide moistened standard rat chow and transgel in a small dish placed inside the cage within a range that is easily accessible to the rat. Additionally, to prevent dehydration, give subcutaneous injections of Ringer’s lactate solution (5 to 10 ml/day) every day for 3 to 5 days following SE.
- Rats that undergo SE will exhibit hematuria for a day or two, and hence multiple red spots will be seen on cage bedding.
- Typically, hematuria stops on its own within two days after SE.
-
7
Select rats exhibiting chronic TLE (see Basic Protocol 2).
BASIC PROTOCOL 2: SELECTING RATS EXHIBITING CHRONIC TLE
Measure behavioral SRS in animals
Following SE, there will be a silent period of 1 to 2 months during which no or only occasional SRS are observed. Therefore, commencing the measurement of behavioral SRS in the 3rd month after SE is ideal. In most of our studies, we intermittently score the frequency and duration of stage III to V SRS at 3 to 6 months post-SE (i.e., 8 hr/week; 4 hr/session; 2 sessions/week, total 32 hr/month) to determine the extent/pattern of chronic epilepsy. From the recorded seizures, calculate the following parameters for every month of observation: the frequency of all (stages III to V) SRS, the frequency of stage V seizures (the most severe form of SRS), the average duration of individual SRS (i.e., the total amount of time spent in seizures/the total number of seizures), and the percentage of time spent in SRS (i.e., the total amount of time spent in seizures/the total duration of observation × 100).
Intermittent scoring of the frequency and intensity of SRS for several months (i.e., 8 hr/week; 4 hr/session; 2 sessions/week, total 32 hr/month) has been found to be sufficient for determining the extent/intensity of chronic epilepsy typified by SRS in male F344 rats (Rao et al., 2006a, 2007; Waldau et al., 2010; Hattiangady et al., 2011). However, measuring the frequency and intensity of SRS with additional hours of observation or continuous (24/7) video monitoring will be superior and especially important if animals mostly exhibit SRS in clusters.
Select CERs for grafting studies
Select groups of age-matched CERs exhibiting a similar extent of SRS (in terms of both frequency and intensity) from a larger pool of CERs for transplantation studies.
The extent of SRS can vary between epileptic animals (Rao et al., 2006a, 2007; Waldau et al., 2010). Choosing animals exhibiting a similar extent of SRS for different experimental groups would facilitate the comparison of changes in the seizure frequency and intensity with specific treatment such as grafting of NSCs or GPCs, sham-grafting surgery alone, epileptic rats receiving cyclosporine alone, or epileptic rats receiving neither surgery nor grafting (i.e. epilepsy alone rats).
BASIC PROTOCOL 3: ANALYZING COGNITIVE FUNCTION IN CERS CHOSEN FOR GRAFTING STUDIES
In order to facilitate the assessment of improvement in hippocampus-dependent cognitive or memory function with NSC grafting, it is important to examine the extent of cognitive or memory dysfunction prior to cell grafting in the chosen CERs. While one can assess hippocampal-dependent cognitive or memory function using quite a few tests, we have selected object location test (OLT, a hippocampus-dependent memory test) as an example for assessing object location memory function in this article. The different aspects of this test are described below.
Materials
Chronically epileptic rats (CERs; see Basic Protocols 1 and 2)
Open field apparatus, suitable for rats
Noldus-Ethovision or ANY-maze video tracking system (Stoelting; http://www.any-maze.com/)
Object Location Test (OLT)
This test is performed for assessing the cognitive ability of rats to detect subtle changes in the environment. Maintenance of this function depends upon the integrity of the hippocampus circuitry.
Apparatus
This test is performed in an open field box measuring 100 cm (L) × 100 cm (W) × 60 cm (H). Each animal is subjected to three successive trials in this test (Fig. 3).
Figure 3.
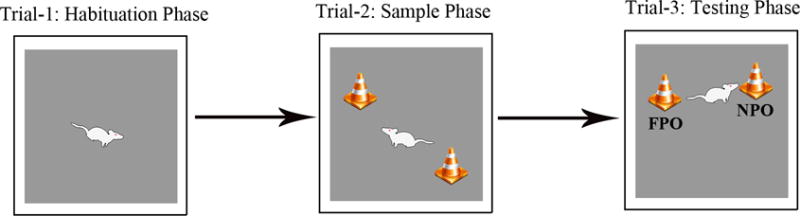
Figure shows a schematic of different trials involved in performing an object location test (OLT). The duration of exploration in each phase is 5 minutes and the delay between trials 2 and 3 varies from 15–60 minutes, which is empirically determined depending on the experimental design employed. Please note that, in trial 3, one of the objects employed in trial 2 was moved to a new location (referred to as novel place object [NPO] in the figure). The percentage of object exploration time spent with NPO, in comparison to the percentage of time spent with the object in the familiar location (referred to as familiar place object [FPO] in the figure) in trial 3 serves as a measure of object location memory.
Handling of CERs prior to test
A day before the test, all animals need to be handled. Additionally, make sure that all chosen CERs explore the open field apparatus individually for 10–15 minutes. This pre-test exploration of the apparatus reduces anxiety in CERs on the day of testing. This was evidenced through increased time spent in exploration of objects by CERS in trials 2 and 3.
Procedures
Trial 1 (Habituation phase): Remove the animal from its home cage and gently place it in the center of an empty open field box. Allow the animal to explore the open field box for 5–15 minutes and then place it back in its home cage.
Trial 2 (Sample phase): After an inter-trial interval of 15–60 minutes (which is determined empirically for different animal models and studies), place the animal again in the center of the open field with two similar objects placed on right and left sides of the box. Allow the animal to freely explore the objects for 5 minutes and then place it back in its home cage. Video-record the entire trial 2 using Noldus-Ethovision or Any Maze video-tracking system to determine whether animals explore both objects in this phase.
-
Trial 3 (Testing phase): After an inter-trial interval of 15–60 minutes (which is determined empirically for different animal models and studies), place the animal again in the center of the same open field box with the left side object placed in its original position and the right side object moved to another corner. Allow the animal to explore for 5 minutes, and video-record the entire trial 3 using Noldus-Ethovision or Any Maze video-tracking system.
NOTE: The open field apparatus needs to be cleaned thoroughly with 70% alcohol and air-dried prior to the commencement of each trial for every rat. This removes odor (and fecal matter or urine when present) of the previously tested animal. Handle and release each rat gently during the test to minimize anxiety.
Interpretation of results
Export data such as times spent with specific objects and the total object exploration time from the software. Calculate percentages of the total object exploration time spent with the object moved to a novel location vis-à-vis the object that remained in its original location. Compare these values within each animal group using two-tailed, unpaired Student’s t-test to determine the ability of animals for place discrimination. The choice to explore the object displaced to a novel location reflects the ability of animal to discern minor changes in the location of objects in its immediate environment. Typically, CERs (not receiving any therapeutic treatment) display a clear impairment in this cognitive function, as they do not show affinity for the object moved to a novel place in trial 3. Rather, they spend either nearly equal amounts of time with the object in the familiar place (FP object) and the object in the novel place (NP object) or spend greater amount of time with the FP object.
BASIC PROTOCOL 4: HARVESTING AND PREPARATION OF MGE-NSC SUSPENSION FOR GRAFTING
Chronic epilepsy is associated with both loss of functional inhibition and a reduction in the number of GABA-ergic interneurons in the hippocampus. Therefore, transplantation of cells that are capable of differentiating into functional GABA-ergic interneurons in the hippocampus may be useful for improving inhibition as well as restraining SRS. In this context, NSCs derived from the embryonic MGE appear ideal as donor cells for grafting because a significant fraction of these cells can differentiate into GABA-ergic interneurons following grafting.
Equipment and Materials
Timed-pregnant (day-14) F344 rats
Isoflurane
Anesthesia chamber for anesthetizing pregnant rats using isoflurane
Proliferation medium (see recipe)
0.4% trypan blue stain
5′-bromodeoxyuridine (BrdU; Sigma-Aldrich)
Differentiation medium (see recipe)
1:200 dilution of 80% (w/v) stock DAPI (Sigma) in distilled water
2% (w/v) paraformaldehyde (see recipe; containing 0.01% glutaraldehyde if performing GABA immunostaining)
0.1 M sodium phosphate buffer, pH 7.4 (Fisher Scientific, cat. no. NC9552713)
2 N HCl
0.1 M borate buffer, pH 8.5 (see recipe)
10% normal goat serum
Mouse anti-BrdU antibody (BD Bioscience)
Tris-buffered saline, pH 7.5 (see recipe)
Slow-fade/anti-fade solution (Invitrogen)
Primary and secondary antibodies for dual immunofluorescence studies (see Table)
Dissection microscope
Fine surgical instruments (Dumont forceps, microdissecting forceps, tissue forceps, spring scissors, microdissecting scissors, operating scissors, scalpel, and scalpel blades) for microdissection of the desired region (e.g., MGE), autoclaved
Sterilized fire-polished Pasteur pipets
Centrifuge
Sterile 25-cm2 canted culture flasks
Humidified 37°C 5% CO2 incubator
Biosafety Cabinet or Laminar flow hood for tissue culture studies
15-ml conical centrifuge tubes
Poly-D-lysine-coated culture dishes (BD Biosciences, cat. no. 354577)
Inverted fluorescence or confocal microscope capable of creating digital images
Table.
Combination of various primary and secondary antibodies used in single and dual immunofluorescence staining
| Name | Blocking Serum | Primary Antibody, Source, and Dilution | Secondary Antibodies |
|---|---|---|---|
| BrdU Staining | NHS | Mouse anti BrdU (BD Biosci.), 1:200 | Biotinylated anti mouse IgG (Vector Labs) followed by ABC reagent and DAB. |
| DCX staining Staining | NHS | Goat anti DCX (SCBT), 1:200 | Biotinylated anti goat IgG (Vector Labs) followed by ABC reagent and VG. |
| BrdU-NeuN Dual IF |
NGS & NDS |
Rat anti BrdU (Serotec), 1:200 Mouse anti-NeuN (Millipore), 1:1000 |
Goat anti-rat IgG with Alexa Fluor 594 Donkey anti-mouse IgG with AF 488 |
| BrdU-GABA Dual IF |
NGS |
Rat anti BrdU (Serotec), 1:200 Rabbit anti GABA (Sigma), 1:5000 |
Goat anti-rat IgG with Alexa Fluor 594 Goat anti-rabbit IgG with Alexa Fluor 488 |
| BrdU-GFAP Dual IF |
NGS |
Rat anti BrdU (Serotec), 1:200 Rabbit anti GFAP (Sigma), 1:1000 |
Goat anti-rat IgG with Alexa Fluor 594 Goat anti-rabbit IgG with Alexa Fluor 488 |
| BrdU-s100β Dual IF |
NGS |
Rat anti BrdU (Serotec), 1:200 Rabbit anti s100β (Millipore), 1:1000 |
Goat anti-rat IgG with Alexa Fluor 594 Goat anti-rabbit IgG with Alexa Fluor 488 |
| BrdU-O1 Dual IF |
NDS |
Rat anti BrdU (Serotec), 1:200 Mouse anti-O1 (Millipore), 1:1000 |
Goat anti-rat IgG with Alexa Fluor 594 Donkey anti-mouse IgG with AF 488 |
| BrdU-NG2 Dual IF |
NGS |
Rat anti BrdU (Serotec), 1:200 Rabbit anti NG2 (Millipore), 1:1000 |
Goat anti-rat IgG with Alexa Fluor 594 Goat anti-rabbit IgG with Alexa Fluor 488 |
| BrdU-Sox2 Dual IF |
NGS |
Rat anti BrdU (Serotec), 1:200 Rabbit anti sox2 (Millipore), 1:1000 |
Goat anti-rat IgG with Alexa Fluor 594 Goat anti-rabbit IgG with Alexa Fluor 488 |
| Tuj1-GABA Dual IF |
NGS |
Mouse anti-Tuj1 (COVANCE), 1:1000 Rabbit anti GABA (Millipore), 1:5000 |
Goat anti-mouse IgG with Alexa Fluor 594 Goat anti-rabbit IgG with Alexa Fluor 488 |
| Tuj1-GFAP Dual IF |
NGS |
Mouse anti-Tuj1 (COVANCE), 1:1000 Rabbit anti GFAP (Millipore), 1:1000 |
Goat anti-mouse IgG with Alexa Fluor 594 Goat anti-rabbit IgG with Alexa Fluor 488 |
| O1-S100β Dual IF |
NGS |
Mouse anti-O1 (R&D), 1:500 Rabbit anti s100β (Millipore), 1:1000 |
Goat anti-mouse IgG with Alexa Fluor 594 Goat anti-rabbit IgG with Alexa Fluor 488 |
| S100β-GDNF Dual IF |
NGS |
Mouse anti s100β (Millipore), 1:1000 Rabbit anti GDNF (SCBT), 1:1000 |
Goat anti-mouse IgG with Alexa Fluor 594 Goat anti-rabbit IgG with Alexa Fluor 488 |
| Human Nuclear Antigen (HNA) IHC |
NHS |
Mouse anti HNA (EMD Millipore), 1:200 | Biotinylated anti mouse IgG (Vector Labs) followed by ABC reagent and DAB. |
| NK×2.1 IF staining |
NDS | Mouse anti NKx2.1 (EMD Millipore) | Donkey anti-mouse Alexa Fluor 488 |
| GABA IF staining |
NDS | Rabbit anti GABA (Sigma) 1:5000 | Donkey anti-Rabbit Alexa Fluor 568 |
| HNA-NeuN Dual IF |
NDS |
Mouse anti HNA (Millipore), 1:200 Rabbit anti-NeuN (Millipore), 1:1000 |
Donkey anti-mouse Alexa Fluor 488 Donkey anti-rabbit Alexa Fluor 568 |
| HNA-GABA Dual |
NDS |
Mouse anti HNA (Millipore), 1:200 Rabbit anti GABA (Sigma), 1:5000 |
Donkey anti-mouse Alexa Fluor 488 Donkey anti-rabbit Alexa Fluor 568 |
| HNA-GFAP Dual IF |
NDS |
Mouse anti HNA (Millipore), 1:200 Rabbit anti GFAP (Sigma), 1:1000 |
Donkey anti-rat Alexa Fluor 594 Donkey anti-rabbit Alexa Fluor 488 |
| HNA-S100β Dual IF |
NDS | Mouse anti HNA (Millipore), 1:200 Rabbit anti s100β (Millipore), 1:1000 |
Donkey anti-rat Alexa Fluor 594 Donkey anti-rabbit Alexa Fluor 488 |
| HNA-O1 Dual IF |
NDS |
Mouse anti HNA (Millipore), 1:200 Rabbit anti-O1 (Millipore), 1: 500 |
Donkey anti-mouse Alexa Fluor 568 Donkey anti-rabbit Alexa Fluor 488 |
| HNA-NG2 Dual IF |
NDS |
Mouse anti HNA (Millipore), 1:200 Rabbit anti NG2 (Millipore), 1:500 |
Donkey anti-mouse Alexa Fluor 594 Donkey anti-rabbit Alexa Fluor 488 |
| HNA-Sox2 Dual IF |
NDS |
Mouse anti HNA (Millipore), 1:200 Rabbit anti sox2 (Millipore), 1:500 |
Donkey anti-mouse Alexa Fluor 568 Donkey anti-rabbit Alexa Fluor 488 |
| HNA-GABA Dual IF |
NDS |
Mouse anti-Tuj1 (COVANCE), 1:1000 Rabbit anti GABA (Millipore), 1:5000 |
Donkey anti-mouse Alexa Fluor 594 Donkey anti-rabbit Alexa Fluor 488 |
| HNA-Tuj1-GFAP Tipple IF |
NDS |
Mouse anti-HNA (Millipore), 1:1000 Rabbit anti Tuj1 (COVANCE), 1:1000 Goat anti GFAP (Millipore), 1:1000 |
Donkey anti-mouse Alexa Fluor 594 Donkey anti-rabbit Alexa Fluor 488 Donkey anti goat Alexa Fluor 405 |
| O1-S100β Dual IF |
NGS |
Mouse anti-O1 (R&D), 1:500 Rabbit anti S100β (Millipore), 1:1000 |
Goat anti-mouse Alexa Fluor 594 Goat anti-rabbit Alexa Fluor 488 |
| S100β-GDNF Dual IF |
NGS |
Mouse anti S100β (Millipore), 1:1000 Rabbit anti GDNF (SCBT), 1:1000 |
Goat anti-mouse Alexa Fluor 594 Goat anti-rabbit Alexa Fluor 488 |
| HNA-NPY Dual IF |
NDS |
Mouse anti HNA (Millipore), 1:200 Rabbit anti NPY (Millipore), 1:10,000 |
Donkey anti-mouse Alexa Fluor 568 Donkey anti-rabbit Alexa Fluor 488 |
| HNA-PV Dual IF |
NDS |
Mouse anti HNA (Millipore), 1:200 Rabbit anti PV (Sigma), 1:1000 |
Donkey anti-mouse Alexa Fluor 568 Donkey anti-rabbit Alexa Fluor 488 |
| HNA-SOM Dual IF |
NDS |
Mouse anti HNA (Millipore), 1:200 Rabbit anti SOM (Millipore), 1:1000 |
Donkey anti-mouse Alexa Fluor 568 Donkey anti-rabbit Alexa Fluor 488 |
| HNA-CBN Dual IF |
NDS |
Mouse anti HNA (Millipore), 1:200 Rabbit anti CBN (Sigma), 1:1000 |
Donkey anti-mouse Alexa Fluor 568 Donkey anti-rabbit Alexa Fluor 488 |
| STEM121 IF | NDS | Mouse anti STEM121 (Stem Cell Inc.), 1:200 | Donkey anti-mouse Alexa Fluor 568 |
| HNA-Ki67 Dual IF |
NDS |
Mouse anti HNA (Millipore), 1:200 Rabbit anti Ki-67 (Vector Labs), 1:10,000 |
Donkey anti-mouse Alexa Fluor 568 Donkey anti-rabbit Alexa Fluor 488 |
| HNA-Oct4 Dual IF |
NDS |
Mouse anti HNA (Millipore), 1:200 Rabbit anti Oct4 (Millipore), 1:10,000 |
Donkey anti-mouse Alexa Fluor 568 Donkey anti-rabbit Alexa Fluor 488 |
Abbreviations: NHS; normal horse serum; NGS, normal goat serum; NDS normal donkey serum; IF, immunofluorescence. AF, Alexa fluor
Secondary antibodies purchased from Invitrogen were used at 1:200 dilution in all staining described above.
Dissect MGE tissues and plate MGE cells for expansion of NSCs
-
1
Deeply anesthetize the timed-pregnant rat (gestation day 14) in isoflurane using a vaporizer. For this, place the pregnant rat in an anesthetic chamber having a direct access to receive 5% isoflurane. Following the cessation of respiration with isoflurane exposure, remove the pregnant rat from the chamber, and surgically open the abdominal cavity using sterile procedures.
-
2
Detach uterine horns from the abdomen cavity by cutting at its trunk, and immerse them in a sterile proliferation medium using a large petriplate.
-
3Using scissors, make cuts through uterine horns to expose fetuses and collect fetuses with amniotic sacs in a fresh petriplate containing sterile proliferation medium. Dissect out fetuses by cutting through amniotic sacs, euthanize them through decapitation and collect their heads in a fresh petriplate containing sterile proliferation medium for dissection of MGEs.
- This procedure should be performed inside a biosafety cabinet.
-
2
Dissect the brain from each fetus and carefully isolate the two cerebral hemispheres from the brain stem using fine micro-scissors under a dissection microscope.
-
3
From each hemisphere, expose the caudal, medial, and lateral ganglionic eminences located in the basal forebrain by making a cut through the superolateral wall of the primitive cerebral cortex (or the cortical plate). Identify the MGE and carefully scoop out the MGE tissue using fine curved micro-scissors, leaving behind both caudal and lateral ganglionic eminences.
-
4
Gently triturate the tissue pieces ~10 times using a fire-polished Pasteur pipet and obtain a homogenous suspension of individual cells with good viability (as determined by trypan blue exclusion; see step 5).
-
5Wash the cell suspension twice, each time by centrifuging 8 min at 800 × g, room temperature, removing the supernatant, resuspending the cells in 1 ml fresh proliferation medium, then centrifuging again as before and removing the supernatant. Resuspend the final pellet in 1 ml of proliferation medium and assess the viability of MGE cells in the suspension using the trypan blue exclusion test.
- Note that a greater percentage of dead cells in the suspension would interfere with the proliferation of stem/progenitor cells in culture. Hence, it is important to have at least 80% viable cells.
-
6
Adjust the density of viable cells to 300,000 live cells per 10 ml proliferation medium, plate the medium into 25-cm2 canted tissue culture flasks at 10 ml/flask, and place in a humidified cell culture incubator maintaining a temperature of 37°C and 5% CO2.
-
7For BrdU labeling, prepare 1 mM stock solution by dissolving 3 mg of BrdU in 10 ml proliferation medium. Add 2.5 μl of this stock solution to each culture flask containing 10 ml of the proliferation medium to obtain a final concentration of 2.5 μM. Allow 5 to 6 days for the neurospheres to grow.
- Note that neurospheres continue to float during the proliferation phase.
-
8
On day 6 or 7, transfer the medium containing the NSC-derived neurospheres from the flasks into 15-ml centrifuge tubes and let the neurospheres settle down. Remove the supernatant medium in each tube, pool neurospheres from all tubes in a single 15-ml centrifuge tube, and gently triturate using a fire-polished Pasteur pipet until a clear cell suspension is obtained.
-
9Wash triturated cells three times to get rid of most of the dead cells in the suspension, each time by centrifuging 8 min at 800 × g, room temperature, removing the supernatant, resuspending the cells in 1 ml fresh proliferation medium, then centrifuging again as before and removing the supernatant. Reconstitute the final pellet in a 30–50μl differentiation medium and count the viable and dead cells using a trypan blue exclusion test (see step 5). Adjust the density of viable cells to 80,000–100,000 cells/μl using a differentiation medium.
- If viability is <75%, wash cells again to obtain the higher ratio of viable cells in the final cell suspension. Typically, the cell suspension exhibiting >80% viability is ideal for transplantation studies.
-
10Use an aliquot of the final cell suspension to determine the BrdU labeling index at the time of grafting. For this:
- Plate cells on poly-D-lysine-coated culture slides/dishes, then incubate in the differentiation medium for 1 hr.
- Fix the cultures by adding 500 μl of 2% paraformaldehyde solution per dish. Following incubation in the fixative for 20 min at room temperature, rinse cultures with PBS three times.
- Perform BrdU immunofluorescence. For this, immerse cultures in 2 N HCl for 2 hr at 37°C and incubate in 0.1 M borate buffer, pH 8.5, for 10 min. Following this, successively treat cultures with 10% normal goat serum for 30 min at room temperature, mouse anti-BrdU antibody solution for 18 hr at 4°C, TBS for 10 to 15 min (with three changes), goat anti–mouse Alexa Fluor 594 for 60 min, and TBS for 10 to 15 min (with three changes). Remove TBS, add 500 μl of the slow-fade/anti-fade solution, and observe under a fluorescence microscope.
- The BrdU+ cells will display red fluorescence (Shetty et al., 1994).
- Counterstain with DAPI by treating with 1:200 dilution of 80% stock DAPI (Sigma) solution in distilled water for 10 to 15 min.
- All nuclei will display blue fluorescence under a fluorescent microscope.
- Calculate the fraction of cells labeled with BrdU by measuring the percentages of BrdU+ nuclei (exhibiting red fluorescence) among all nuclei (exhibiting blue fluorescence) in multiple regions of the culture dish under a fluorescence microscope.
- Typically, expanding neurospheres in a medium containing 2.5 μM of BrdU results in labeling of over 90% of neurosphere cells.
Characterize the differentiation potential of NSCs in vitro
In order to ascertain the differentiation potential of the chosen donor NSCs, it will be useful to examine their differentiation in culture. The protocols we typically use are described below.
-
11
Plate samples of the NSC cell suspension (200,000 cells/dish) into poly-D-lysine-coated 35-mm culture dishes containing differentiation medium.
-
12
Incubate cells for 6 to 8 days at 37°C in a humidified 5% CO2 incubator. Replace one-half of the medium every other day.
-
13
Terminate cultures by treating with 2% paraformaldehyde for 20 min. If GABA immunostaining is planned, fix cultures in 2% paraformaldehyde containing 0.01% glutaraldehyde. Following the above fixation, thoroughly wash cultures with 0.1 M sodium phosphate buffer, pH 7.4.
-
14Process cultures for single or dual immunofluorescence for identifying: (i) neurons expressing beta-III tubulin (TuJ-1); (ii) astrocytes expressing the glial fibrillary acidic protein (GFAP); (iii) oligodendrocytes expressing the protein O1; and (iv) inhibitory interneurons expressing GABA.
- Table illustrates the combination of primary and secondary antibodies that we typically use in our dual immunofluorescence studies.
-
15Counterstain the immunostained cultures with DAPI by treating with a 1:200 dilution of 80% stock DAPI (Sigma) solution in distilled water for 2 to 5 min.
- All nuclei will display blue fluorescence under a fluorescent microscope.
-
16Using a systematic random sampling scheme, obtain digital illustrations of dual/triple labeled samples (at least six fields per each of the four independent samples) using an inverted fluorescence or confocal microscope. Count the total number of cells (i.e., all DAPI+ blue nuclei), as well as cells that are positive for a neural cell marker, to measure the percentages of cells expressing different neural markers.
- Figure 4 shows neural phenotypes derived from MGE-NSCs following 4 and 8 days of incubation in differentiation medium.
Figure 4.
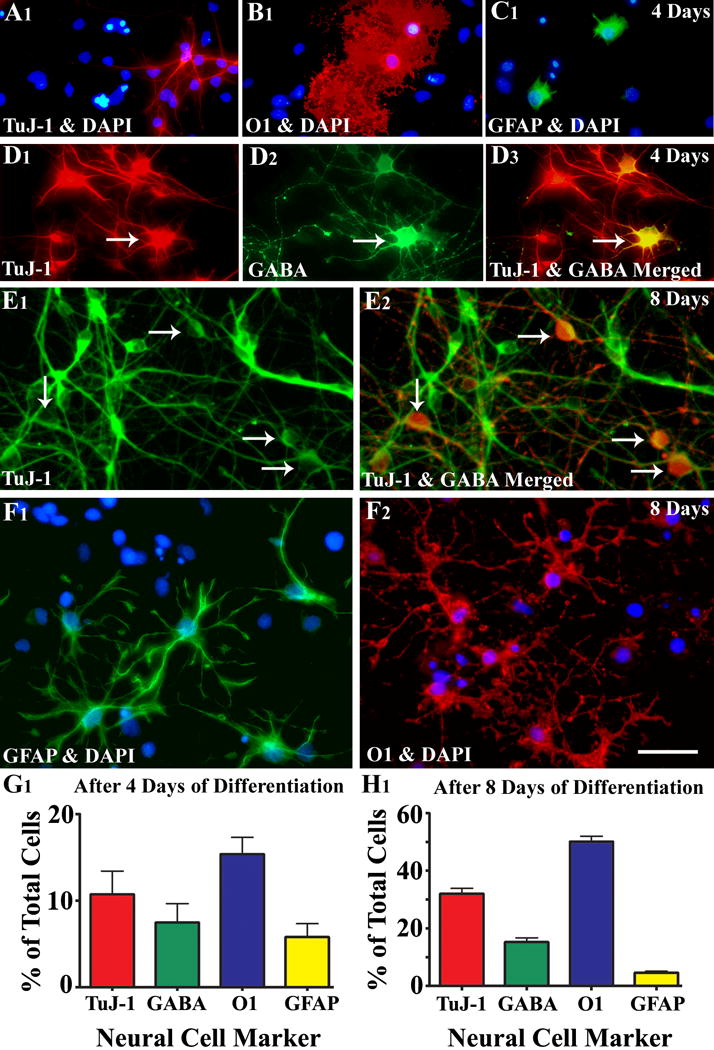
Differentiation of medial ganglionic eminence–neural stem cells (MGE-NSCs) after their dissociation from CldU-labeled neurospheres and incubation in the differentiation medium for 4 days (A1–D3) or eight days (E1–F2). Differentiation of fractions of MGE-NSCs into TuJ-1+ neurons (A1, E1), O1+ oligodendrocytes (B1, F2), and GFAP+ astrocytes (C1, F1) could be seen at both time-points. Furthermore, fractions of MGE-NSCs also differentiate into GABA-ergic neurons (arrows in D1–D3 and E1–E2). Scale bar: 50 μm. The bar charts (G1, H1) illustrate percentages of MGE-NSCs that exhibit differentiation into TuJ-1+ neurons, GABA+ neurons, O1+ oligodendrocytes, and GFAP+ astrocytes after incubation in the differentiation medium for 4 days (G1) or 8 days (H1). Note that the expression of GABA among TuJ-1+ neuronal population is ~70% after 4 days and ~50% after 8 days of incubation in the differentiation medium. Reproduced from Waldau et al. (2010)
BASIC PROTOCOL 5: PREPARATION OF MGE-LIKE GABA-ERGIC PROGENITOR CELLS FROM HUMAN INDUCED PLURIPOTENT STEM CELLS
For the generation of MGE-like GABA-ergic progenitor cells from hiPSCs (i.e. human GPCs), we employ a protocol developed by Su-Chun Zhang laboratory (Liu et al., 2013) with some modifications.
Materials and Reagents
BD Matrigel™ Basement Membrane Matrix Growth Factor Reduced (cat. no. 354230)
B27 supplement, without retinoic acid, 50× (Gibco, cat. no. 12587-010)
Brain-derived neurotrophic factor (BDNF; PeproTech, cat. no. 450-02)
BSA (Sigma-Aldrich, cat. no. A-7906)
Cyclic AMP (cAMP; Sigma-Aldrich, cat. no. D-0260)
Dispase (Gibco, cat. no. 17105-041)
DMSO (Sigma-Aldrich, cat. no. D8418)
DMEM/F-12, medium 1:1 (Gibco, cat. no. 11330)
FBS (Gibco, cat. no. 10437)
Glial cell line–derived neurotrophic factor (GDNF; PeproTech, cat. no. 450-10)
Heparin sodium salt from porcine intestinal mucosa (Sigma-Aldrich, cat. no. H3149)
Insulin-like growth factor (IGF1; PeproTech, cat. no. 100-11)
Human induced pluripotent stem cell lines of interest.
NOTE: We tried a few hiPSC lines for generating GABA-ergic progenitors. However, IMR90-4 (Wisconsin International Stem Cell Bank, cat. no. iPS (IMR90)-4 line gave us consistent and reproducible results.
MEM non-essential amino acids solution, 100×, liquid (NEAA; Gibco, cat. no. 11140)
N-2 supplement, 100×, liquid (Gibco, cat. no. 17502-048)
Natural mouse laminin, 1 mg ml−1 (laminin; Invitrogen, cat. no. 23017-015)
Neurobasal medium, 1×, liquid (Gibco, cat. no. 21103)
Poly-l-ornithine solution (PLO; Sigma-Aldrich, cat. no. P4957)
Purmorphamine (StemGent, cat. no. 04-0009
Recombinant human sonic hedgehog (C24II) N-terminus, CF (SHH; R&D Systems, cat. no. 1845-SH-025/CF)
TeSR™-E6™medium (StemCell Technologies, cat. no. 5946)
TeSR™-E8™ medium (StemCell Technologies, cat. no. 5940)
TGF-β3, human recombinant (Sigma-Aldrich, cat. no. SRP3171)
TrypLE Express Enzyme 1× (Invitrogen, cat. no. 12604-021)
Tissue culture plates (6-well and 24-well)
Centrifuge tubes (15 ml and 50 ml)
Benchtop centrifuge
CO2 incubator
Biosafety Cabinet or Laminar flow hood for tissue culture studies
Phase-contrast inverted microscope
Disposable serological pipettes, 5, 10 and 25 ml
Reconstitution of media and other reagents
Matrigel
Thaw the required amount of BD Matrigel™ on ice (see manufacturer’s instructions for dilution). Dilute the matrigel using cold DMEM/F-12 into a 15 ml tube, mix well. Do not allow formation of a gel inside the tube. Immediately transfer 1 ml of the diluted matrigel into each well of a 6-well plate. Uniformly spread the matrigel into the entire area of the well through slow swirling of the plate. Incubate the plate at room temperature (15 – 25°C) for an hr prior to plating cells. Do not allow the matrigel to dry. Remove the excess matrigel after an hour of incubation and immediately add 2 ml of E8 medium to each well.
Complete TeSR™-E8™ (E8) medium
Thaw all supplements to room temperature. Prepare the complete TeSR™-E8™ medium (Basal Medium + 20× Supplement + 500× Supplement) under sterile conditions. Aliquot the medium into sterile 50 ml tubes and store at −20°C up to six months or at 2 – 8°C for up to 2 weeks.
Complete TeSR™-E6™ (E6) medium
Thaw 20× supplement to room temperature. Prepare the complete TeSR™-E6™ medium (Basal Medium + 20× Supplement) under sterile conditions. Aliquot the medium into sterile 50 ml tubes and store at −20°C up to a month or at 2 – 8°C for up to 2 weeks.
Dispase
Dissolve this enzyme in Dulbecco’s Phosphate-Buffered Saline (DPBS) without calcium and magnesium to 10 mg/ml. Dilute this solution further with DPBS without calcium and magnesium to a final concentration of 1U/mL for the use.
Heparin (20 mg ml−1)
Dissolve 20 mg of heparin in 1 ml of DMEM/F-12 medium. Prepare aliquots and store them at −80 °C for up to 6 months.
Neural induction medium (NIM)
For 500 ml of NIM preparation, combine 490 ml of DMEM/F-12, 5 ml of NEAA, 5 ml of N-2 supplement and 50 μl of heparin inside a sterile biosafety cabinet. Store the medium at 2–8 °C for up to 2 weeks.
Purmorphamine (10 mM)
Completely dissolve 5 mg of purmorphamine in 480 μl of ethanol and 480 μl of DMSO. Aliquot the solution and store them at −80 °C for up to 6 months.
Shh, 500 μg ml−1
Dissolve 500 μg of Shh into 1 ml of sterile DPBS with 0.1% (wt/vol) human serum albumin or BSA for a 500-μg ml−1 stock. Aliquots and store at −80 °C for up to 6 months.
Neuronal differentiation medium (NDM)
For 50 ml of NDM preparation, combine 49 ml of Neurobasal medium, 0.5 ml of NEAA and 0.5 ml of N-2 supplement inside a sterile biosafety cabinet. Store the medium at 2–8 °C for up to 2 weeks.
BDNF, GDNF, IGFI (100 μg ml−1)
Dissolve 100 μg into 1 ml of sterile DPBS with 0.1% (wt/vol) human serum albumin or BSA. Aliquot and store them at −80 °C for up to 6 months.
cAMP (1 mM)
Dissolve 4.914 mg of cAMP in 10 ml of sterilized water. Aliquot and store at −80 °C for up to 6 months.
Plating the hiPSC cells and embryoid body formation
-
1
Dilute freshly derived or frozen hiPSC cells after thawing in E8 medium. Distribute cells equally to each well of a matrigel coated 6 well plate.
-
2
Feed cells on alternate days with a fresh E8 medium. Use cells for experiments once they become 70–80% confluent.
NOTE: We typically use wells containing 70–80% confluent hiPSCs for expansion of GPCs. This stage is designated as day 0 for expansion of GPCs.
-
3
Day 0: Remove E8 medium from each chosen well of a six well plate. Slowly add 1ml of dispase solution, gently rinse cells and aspirate dispase. Add 1ml of fresh dispase solution to each well and incubate at 37 °C with 5% CO2 for 5–10 minutes until hiPSC colonies begin to curl.
-
4
Remove dispase solution and wash cells once with 1 ml of prewarmed E6 medium. Remove E6 medium slowly without disturbing the detaching colonies.
-
5
Add 1 ml of fresh E6 medium on to cells to blow off the colonies.
-
6
Transfer the detached colonies to 50 ml tube. Repeat the step one more time so that majority of colonies get detached. Transfer all detached colonies to 50 ml tube.
-
7
Gently triturate colonies inside the 50 ml tube 2–3 times with a pipette so that slightly smaller fragments of hiPSC colonies are obtained. However, do not triturate extensively, as this can cause single cell suspension of colonies. Let smaller fragments of hiPSC colonies settle down for 5 minutes.
-
8
Remove the supernatant, resuspend smaller fragments of hiPSC colonies in 3 ml of E6 medium supplemented with 2ng/ml TGFβ.
-
9
Distribute fragments of hiPSC colonies equally into wells of two 6-well plates and incubate for 24 hr at 37 °C with 5% CO2 for the formation of embryoid bodies (EBs).
-
10
Day 1: By holding 6-well plates in a slightly slanted position, remove 3/4th of the media using 1ml pipette and add the same amount of fresh E6 medium supplemented with 2ng/ml TGFβ to the EBs formed. Incubate at 37 °C with 5% CO2 for two days.
-
11
Day 3: Repeat step 10 on day 3.
-
12
Day 4: Transfer all EBs to 50 ml tube and allow them to settle down for 5 minutes. Discard the supernatant and resuspend EBs in 3ml of NIM. Distribute EBs to one or two six well plates containing 3 ml of NIM and incubate at 37 °C with 5% CO2.
-
13
Days 5–6: Replace 1 ml of medium with a fresh NIM medium to each well.
Formation of neural rosettes
-
14
Day 7: Transfer all EBs to 50 ml tube and allow them to settle down for 5 minutes. Discard the supernatant and resuspend EBs in NIM containing 5% FBS. Distribute EBs to one or two six well plates containing NIM with 5% FBS.
NOTE: Allow EBs to attach by incubating at 37 °C with 5% CO2 for 6 hours. After 6 hrs, replace the FBS containing NIM with 3 ml of NIM lacking FBS and incubate at 37 °C with 5% CO2.
-
15
Days 8–9: Replace 1 ml of medium with fresh NIM.
Patterning of neural rosettes to MGE progenitors
-
16
Day 10: Remove the medium and add 3 ml of fresh NIM containing 500 ng/ml of SHH and 1.2 uM purmorphamine and incubate at 37 °C with 5% CO2.
-
17
Days 12 and 14: By holding the 6-well plate in a slightly slanted position, remove 3/4th of the medium and add same amount of fresh NIM containing SHH and purmorphamine.
-
18
Day 15: Remove the medium completely and slowly blow off neural rosettes using fresh NIM media in a 1 ml pipette. Transfer all detached rosettes to 50 ml tube. Repeat this step to detach and obtain the additional rosettes.
NOTE: Care should be taken not to detach non-rosette cell types.
Transfer all rosettes to 50 ml tube and triturate gently (by pipetting 2–3 times) to break down the rosettes to slightly smaller fragments. Allow rosettes to settle down for 5 minutes.
NOTE: Avoid extensive trituration, as this would make a single cell suspension.
-
19
Discard the supernatant and resuspend rosettes in 2 ml of NIM with B27 (1:50) containing 500 ng/ml of Shh and 1.2 μM purmorphamine. Distribute rosettes into one or two 6 well plates. Incubate at 37 °C with 5% CO2.
-
20
Day 16: Examine cultures to see the formation of neurospheres from rosettes.
-
21
Days 17 to 24: On alternate days, by holding the 6-well plate in a slightly slanted position, replace 3/4th of the medium with fresh NIM with B27 containing 500 ng/ml of Shh and 1.2 μM concentrations of purmorphamine. By day 24, more than 90% of the cells will be NKX2.1 positive MGE-like GPCs.
Dissociation of neurospheres containing MGE-like GPCs for transplantation
From day 24 onwards, neurospheres containing MGE-like GPCs can be used for characterizing and confirming the presence of NKX2.1+ cells through immunostaining procedures, growing long term cultures to generate mature GABA-ergic interneurons or for transplantation studies. For all these purposes neurospheres are broken down to single cells or into smaller clumps containing <10 cells.
Preparation of single cell suspension
Transfer neurospheres to a 15 ml tube containing the same medium and wait for 5 minutes for them to settle down at the bottom.
Aspirate the supernatant and add 1–2 ml of trypLE, mix well and incubate at 37°C for 10 minutes.
Centrifuge at 500g for 5 minutes and discard the supernatant.
Add 2 ml of NDM media and triturate slowly for a few times to dissociate cells.
Centrifuge at 500g for 5 minutes and discard the supernatant.
Resuspend the cell pellet in required concentrations of fresh NDM.
Preparation of cells for transplantation
Reconstitute the final pellet in a 30–50 μl differentiation medium and count the viable and dead cells using a trypan blue exclusion test. Adjust the density of viable cells to 80,000–100,000 cells/μl.
NOTE: If viability is <75%, wash cells again to obtain the higher ratio of viable cells in the final cell suspension. Typically, the cell suspension exhibiting >80% viability is ideal for transplantation studies.
Characterization of GPCs at different stages or generating mature GABA-ergic neurons from GPCs
Plate cells onto poly-L-ornithine and laminin coated coverslips in a 24 well plate using NDM supplemented with 1 μM cAMP, 10 ng ml−1 BDNF, 10 ng ml−1 GDNF, 10 ng ml−1 IGF1.
Replace 3/4th of the medium with a fresh NDM every other day for 42 days.
Terminate cultures at different time-points for immunocytochemical characterization of cells using markers of: GPCs (NKX2.1), pluripotent stem cells, proliferating cells (Ki-67), GABA-ergic interneurons (GABA or GAD-67) or subtypes of GABA-ergic interneurons such as neuropeptide Y, somatostatin, parvalbumin, calbindin, calretinin for 3–6 weeks.
Figure 5 illustrates the sequence of steps involved in grafting of MGE-like GABA-ergic cells into the hippocampus of rats with chronic TLE.
Figure 5.
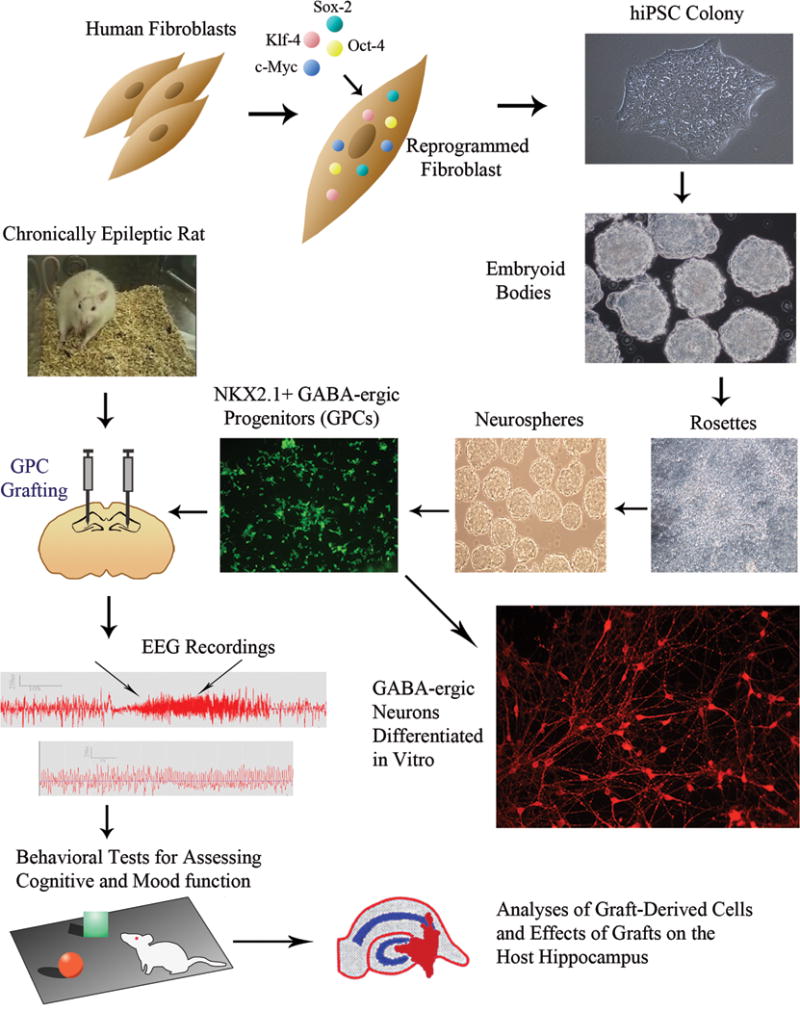
Figure illustrates steps involved in the generation of GABA-ergic progenitor cells (GPCs) from human fibroblasts, their transplantation into hippocampi of chronically epileptic rats and analyses of graft-mediated effects. Human skin fibroblasts are reprogrammed with Oct-4, Sox-2, c-Myc and Klf-4 to generate human induced pluripotent stem cells (hiPSCs). The subsequent steps comprise: expansion of hiPSCs as colonies, culturing of detached colonies in E6 media supplemented with TGFβ to generate embryoid bodies, neural induction in embryoid bodies to generate neuroepithelial cells as neural rosettes, and patterning of neuroepithelial cells towards NKX2.1 positive GPCs through long term treatment with sonic hedgehog and purmorphamine. Virtually all GPCs differentiate into GABA-ergic neurons in culture. The subsequent steps in the figure shows grafting of GPCs into hippocampi of a chronically epileptic rat through stereotaxic survival surgery, EEG recordings and behavioral tests after grafting to examine the effects of grafts on spontaneous recurrent seizures and cognitive and mood function, analyses of survival and differentiation of grafted GPCs and the effects of GPC grafting on the host hippocampus.
BASIC PROTOCOL 6: GRAFTING OF NSCS OR GPCS INTO THE HIPPOCAMPI OF CHRONICALLY EPILEPTIC RATS
Prior to the surgery, choose CERs exhibiting a similar extent of SRS as well as cognitive dysfunction. Choosing rats displaying the above characteristics of TLE would facilitate testing the efficacy of NSC grafts for both restraining seizures and reversing the cognitive dysfunction.
NOTE: Animals chosen for sham-grafting surgery need to undergo a surgical procedure that is identical to what is described here for the transplantation of MGE-NSCs or GPCs. The only difference is that these animals will receive a sterile culture medium (1 μl/site) in place of the cell suspension. Furthermore, animals receiving human GPC grafts need to undergo moderate immune suppression to prevent the rejection of grafts through lymphocytic infiltration. This is typically accomplished through daily subcutaneous injections of cyclosporine (at a dose of 10–12 mg/Kg). Injections need to commence 1–2 days prior to grafting and continue during the entire post-grafting survival period. This strategy prevents the rejection of human cell grafts in the rat brain.
Materials
Chronically epileptic rats (CERs; see Basic Protocols 1 and 2)
Anesthetic cocktail (see recipe)
Artifical tears
70% ethanol
Betadine
cyclosporine
Isopropyl alcohol
3% hydrogen peroxide
NSC suspension, BrdU-labeled (Basic Protocol 4)
Bone wax
Buprenorphine
Sterile saline (0.9% NaCl)
Surgical shaver
Sterile surgical drape
Stereotaxic equipment: any stereotaxic device made for rat neurosurgery can be used; in our laboratory, we use digital stereotaxic equipment purchased from MyNeurolab (http://www.myneurolab.com)
Sterile applicators with cotton tips
- Autoclaved surgical instruments:
- Scalpel blade holders
- Skin retractors
- Blunt forceps of different sizes
- Autoclipping device loaded with 9-mm wound clips (for stapling the skin incision after the grafting surgery)
- Autoclip remover
Sterile scalpel blades
Hand- or foot controlled electrical driller with different sizes of drill bits to make small burr holes in the skull using the desired stereotaxic coordinates
10-μl Hamilton syringes
Heating pads to maintain the body temperature of the rat during and after the surgery
Additional reagents and equipment for injection of the rat (Donovan and Brown, 2006)
NOTE: Personnel protective equipment including sterile surgical latex gloves, disposable lab coats, head cover, booties, surgical masks and approved respirators (for handling human GPCs) should be worn.
NOTE: A glass bead sterilizer should be available to sterilize the stainless steel surgical instruments during the surgery of multiple animals in one surgery session.
(A) Pre-operative procedures
-
1
Ensure that the surgery room has been prepared for BL-2 surgery if human GPC grafting is planned.
-
2
Prepare the aseptic surgical area, arrange all autoclaved surgical instruments and other required supplies around the stereotaxic device, and brightly illuminate the area of the stereotaxic device that holds the head of the rat.
-
3
Anesthetize the animal using an intramuscular injection of the anesthetic cocktail (which is a mixture of ketamine at 50 mg/ml, xylazine at 4.5 mg/ml and Acepromazine at 0.4 mg/ml) at a dose of 0.7ml/Kg body weight. This is equivalent to ketamine at 35mg/Kg, xylazine at 3.2 mg/Kg, and acepromazine at 0.3 mg/Kg. If additional anesthesia is required during the procedure, booster doses (~25% of the initial dose) are given intraperitoneally as required.
-
4
Following anesthetic injection, place the animal in a cage placed over a heating pad maintained at 38 degree C. Five minutes later, check the plane of anesthesia using toe and tail pinches and make sure that animal is well anesthetized but breathing normally.
-
5
Hold the animal gently and take it to a location in the room that is remote from the surgical area. Shave or clip fur on the top of head (i.e. the chosen surgical site) with enough border area to keep hair from contaminating the incision site. Wipe off the cut hair from the shaved area with 70% ethanol and carefully drape the rat with a sterile drape, exposing only the head area. Make sure that animal is still breathing appropriately.
-
7
Carefully fix animal’s head to the stereotaxic device (that has been cleaned prior with bactericidal solution). Place the animal on heating plate (that comes attached to some stereotaxic devices) or heating pad maintaining a temperature of 38 degree C. Examine the breathing pattern and check the plane of anesthesia using tail and toe pinches.
-
8
Give the pre-operative analgesic injection (Buprenorphine at 0.1 mg/Kg, subcutaneous or intraperitoneal).
-
9
Apply artificial tears to animal’s eyes to keep it moist during the surgery.
-
10
Wash hands with a bactericidal scrub (e.g. 70% alcohol). Put on the surgical gown, surgical mask (or approved respirator if handling human cells during the surgery) and a pair of sterile surgical gloves.
(B) Surgical procedures
-
11Wipe the surgical site sequentially with betadine, isopropyl alcohol, and betadine.
- Artificial tears should be applied to the eyes periodically during surgery.
-
12
Make a midline incision in the rat’s head using a sterile scalpel blade, retract the skin flaps with surgical skin retractors, clean the fascia on top of the cranium using 3% hydrogen peroxide, and identify the anatomical landmarks on the cranium such as the bregma (meeting point of the frontal bone and the two parietal bones) and the lambda (meeting point of the two parietal bones and the occipital bone).
-
13Using the stereotaxic device and coordinates and the bregma as a reference point, mark the chosen grafting sites on the skull. Using a hand or a foot-controlled driller, gently make burr holes in these marked sites without damaging the dura mater covering of the brain.
- In our cell grafting studies using CERs, we place grafts into 3 or 4 sites in each hippocampus using the following coordinates. (i) antero-posterior (AP)—3 mm posterior to the bregma, lateral (L)—1.8 mm lateral to the midline, ventral (V)—3.5 mm from the brain surface; (ii) AP—3.6 mm, L–2.5 mm, V–3.5 mm; (iii) AP—4.2 mm, L—3.2 mm, V—3.5 mm; (iv) AP—4.8 mm, L—4 mm, V—4.0 mm.
- These coordinates were chosen using the rat brain atlas (Paxinos and Watson, 2004) especially to place the grafts at the end of the hippocampal fissure (i.e., just above the CA3 pyramidal cell layer and the adjoining the lateral ventricle) in CERs. If placement in other areas of the hippocampus is desired (such as CA1 pyramidal cell layer), adjust the coordinates using the rat brain atlas. Furthermore, as hippocampal shrinkage can vary between different models, determine the appropriate coordinates for the desired location empirically using injections of the dye in a set of pilot studies.
-
14Load a small volume of the cell suspension (2 to 3 μl) into a 10-μl Hamilton syringe, fix it to the stereotaxic instrument, and make sure that no air bubble is trapped between the needle and the cell suspension by ejecting ~0.5 μl of the cell suspension.
- NOTE: Load fresh cell suspension for each injection to minimize inconsistency between grafts placed at different injection sites in a rat or between rats in a group.
-
15
Inject the cell suspension into the hippocampus by first lowering the Hamilton syringe needle to the surface of the brain through the burr hole. Then, using the ventral stereotaxic coordinate, gently lower the needle through meninges and cortex and reach the desired depth; dispense 0.8–1.0 μl of the cell suspension in spurts of ~0.2 μl/min over a period of 5 min.
-
16Following injection of each spurt of 0.2 μl of the cell suspension, gently withdraw the needle by ~0.05 mm to create room for the injected NSC suspension.
- NOTE: The above procedure enhances the probability of placing the graft in the chosen location and minimizes the flow of injected cells into the adjoining lateral ventricle.
-
17Allow the needle to stay in place for 8–10 min following the injection of cell suspension and then slowly withdraw the needle.
- NOTE: This procedure ensures that the injected cells disperse in the hippocampus and minimizes the backflow of the injected fluid along the needle track into the corpus callosum area or the overlying cortex.
-
18
Repeat the above procedure for each of the remaining graft sites (i.e., 3–4 sites/hippocampus in every CER).
-
19Following cell injections to all sites, close the top of the burr holes with a small amount of bone wax, staple the skin flaps using a autoclipping device loaded with 9-mm wound clips, and apply bupivicaine solution to the stapled skin.
- NOTE: Betadine may also be applied at the wound site.
(C) Post-operative Procedures
-
20
Gently remove the animal from the stereotaxic device and place it inside the post-operative cage placed over a hot water circulating pads maintaining 38 degree C temperature. Keep animals warm until they recover from anesthesia or overnight if surgery is performed in the evening.
-
21
Subcutaneously administer 5 ml of Ringer’s lactate or saline immediately after the surgery. Give these injections daily for two days, if needed (once daily, 5 ml/injection).
-
22
Perform the post-operative analgesic administration plan to minimize the post-surgical pain and distress: Since each animal undergoing surgery received a pre-operative analgesic dose (Buprenorphine at 0.1 mg/Kg) just prior to the surgical incision, three injections of buprenorphine (each at 0.1 mg/Kg) need to be administered during the post-operative period. These post-operative analgesic injections can be administered as “twice-daily” basis: one injection in the AM and another injection in the PM until three injections are completed. This plan ensures 48 hours of analgesic support to animals after surgery. Alternatively, give a single injection of longer acting buprenorphine (Buprenorphine SR from SR Veterinary Technologies) at a dose of 0.6 mg/Kg. As per the manufacturer, this single injection (given as a preoperative injection) provides 72 hours of analgesic support.
-
23
Provide soft food/hydrogel/transgel inside the cage for 4 days after surgery.
-
24Observe animals at least twice daily in the first two days after surgery and once daily in the following two days.
- NOTE: The CERs typically recover from the grafting surgery within 2 to 3 days. It is important to carefully observe these rats during this period and ensure that they eat and drink well. If they are not actively eating or drinking, it will be necessary to subcutaneously administer saline (5 ml/day) and place soft rat chow and/or transgel inside the cage.
-
25
Remove wound clips on or before the 14th day after surgery, since wound healing normally takes ~10–14 days.
BASIC PROTOCOL 7
Analyses of rat MGE-NSC or human GPC Grafting-Mediated Changes in Seizures.
Measurement of SRS
Measure changes in the extent of SRS rigorously beginning from a week after the transplantation surgery continuing for at least 3 months after the grafting/sham grafting procedure.
- Quantify SRS after grafting as described earlier for quantification of the pre-grafting SRS [i.e., a minimum of 32 hr of observation/month in eight intermittent sessions or continuous (24/7) video monitoring].
- NOTE: Monitoring of SRS for a longer duration after the grafting (i.e., for 6 to 8 months post-grafting) will be helpful to determine whether any beneficial effects observed on seizure control in CERs at early time-points after the grafting persist at extended periods after the grafting, or are just transient.
From the above measurement of SRS, calculate the average frequencies of all SRS and stage V seizures, the average duration of individual SRS, and the total time spent in seizures for every month.
- Compare the post-grafting seizure scores (for different months post-grafting) with the pre-grafting seizure scores using repeated measures ANOVA and ascertain the extent of seizure suppression mediated by NSC or GPC grafting (Fig. 8 illustrates the efficacy of MGE-NSC grafts for restraining SRS in CERs). For CERs undergoing the sham-grafting surgery, compare the post-sham grafting seizure scores with the pre-sham grafting seizure scores using repeated measures ANOVA and ascertain the sham-grafting mediated changes in the SRS.
- NOTE: Different parameters of SRS can also be compared statistically across different age-matched animal groups (CERs receiving NSC or GPC grafts, CERs receiving sham-grafting surgery, CERs receiving neither grafts nor surgery and CERs receiving immunosuppressant [cyclosporine] treatment alone) using one-way ANOVA, if all three groups of rats had similar extent of SRS at the time of their classification into different groups (i.e., prior to the grafting procedure).
Figure 8.
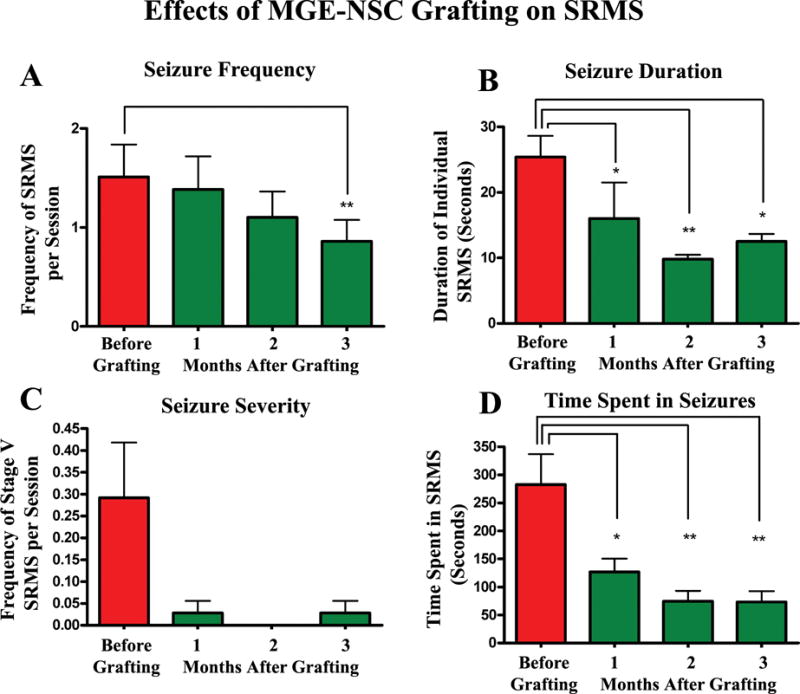
Efficacy of MGE-NSC grafts for restraining spontaneous recurrent motor seizures (SRS) in chronically epileptic rats. The y axis in bar charts A and C denotes the average numbers of seizures per session (4-hr block) of observation. Note that MGE-NSC grafting considerably decreases the seizure frequency (A), the duration of individual seizures (B), the severity of seizures (C), and the total time spent in seizures (D). * = p < 0.05; ** = p <0.01. Reproduced from Waldau et al. (2010)
Measurement of spontaneous recurrent seizures (SRS) via electroencephalographic (EEG) recordings
In addition to the quantification of behavioral SRS as described above, one can also quantify the long-term changes in all SRS (i.e., electrographic seizures with or without a behavioral component) after the grafting using continuous electroencephalographic (EEG) recordings for 2–6 weeks at an extended time-point after grafting.
It is ideal to do EEG recordings following completion of all behavioral measures of SRS and cognitive and mood function.
In our laboratory, we use a time-locked video-digital EEG monitoring system (AS40 from Grass Telefactor) for measuring SRS from CERs.
For EEG recordings from CERs (with or without grafts), we implant sterile metal EEG recording electrodes with mounting screws (Plastics One) epidurally, one over the right fronto-parietal cortex for recording EEG from the cortex, and another over the left cerebellum as a reference electrode. To record EEG directly from the hippocampus, an intracranial stainless steel electrode (Teflon-coated except for the tip) with socket (Plastics One) is also placed into the right DG (Rao et al., 2006a). We also implant a couple of anchoring screws on the skull to secure the EEG electrodes with dental cement. The screws and electrodes are cemented in place, and electrode leads are attached to a microplug, which is then cemented to the animal’s head. The above implantation procedures are done in one surgery session (as detailed in Rao et al., 2006a). In grafting studies, this is typically done at ~2 to 3 months after the grafting.
Two weeks after the implantation surgery, each rat is placed in a Plexiglas cage, and the connector cable of the video-EEG system is fixed into the electrode pedestal on the rat’s head. The video-EEG system monitors simultaneously occurring behavior and EEG activity in awake, freely behaving rats with ad libitum access to food and water.
In our laboratory, the EEG recordings are done with a low-frequency filter (LF) set at 0.3 Hz, a high-frequency (HF) filter set at 35 Hz, and data rate set at 200 Hz. Furthermore, the SZAC detector component of AS40 is turned on to quantify the number of all high-amplitude spikes (first half-wave amplitude ratio set at ≥1.5 and 2nd half-wave amplitude ratio set at ≥2.5), and seizure events (high-frequency multispike complexes at 3 to 20 Hz and 50% faster than background), and/or high-voltage synchronized spike or wave activity (≥45% variation in amplitude, ≥36% variation in duration, amplitude ratio of 3, and lasting for ≥6 sec).
Both frequency and severity of EEG seizures with or without behavioral manifestation is continuously measured for a period of time (e.g. 2–6 weeks). An “EEG seizure with behavior” is a generalized electrographic seizure with accompanying motor seizure activity—e.g., unilateral or bilateral forelimb clonus, and rearing and falling (Stages III to V seizures). The various parameters for comparison across the age-matched animal groups (CERs receiving NSC or GPC grafts, CERs receiving sham-grafting surgery, CERs receiving neither grafts nor surgery and CERs receiving cyclosporine treatment alone) using one-way ANOVA include the following: (1) frequency of all SRS; (2) EEG-SRS with behavior; (3) EEG-SRS without behavior (electrographic SRS); (4) average duration of individual SRS; (5) total time spent in SRS; (6) number of high-amplitude spikes.
The EEG studies are labor intensive and clearly need personnel with expertise in appropriate implantation of electrodes and ability to connect the electrodes on the rat’s head to the video-EEG system through a connector cable without anesthetizing the animal. One pitfall of this procedure is that the number of rats initially implanted with electrodes may be reduced at the time of recordings due to malfunction of electrodes. Alternatively, electrodes may fall off due to accidents in the cage or during daily cyclosporine injections in rats receiving human GPCs. To circumvent these difficulties, it is important to include a larger cohort of CERs than required for statistics in each group. Furthermore, it will be necessary to individually house rats in cages with flat tops, which minimizes the loss/malfunction of electrodes due to accidents in the cage.
BASIC PROTOCOL 8: ANALYSES OF NSC OR GPC GRAFTING MEDIATED EFFECTS ON COGNITIVE AND MOOD FUNCTION
If CERs chosen for different groups (e.g. NSC or GPC graft groups) had undergone examination of cognitive function using one or two hippocampus-dependent cognitive tests (e.g. OLT) prior to the grafting or sham-grafting surgery (as described in an earlier section of this article), their pre-grafting/pre–sham grafting cognitive scores are available. In such scenario, the same tests can be employed at 2 to 4 months post-grafting or post-sham grafting surgery to ascertain changes in the cognitive or memory function with NSC/GPC grafting or sham-grafting surgery
Furthermore, regardless of pre-grafting behavioral tests, it will be important to perform a series of behavioral tests to examine cognitive, memory and mood function at 2–4 months post-grafting. In our laboratory, we employ the following behavioral tests for examining cognitive and mood function:
-
(1)
Object Location Test (OLT): This test examines cognitive ability to detect subtle changes in the environment (Hattiangady et al., 2014). Maintenance of this function depends upon the integrity of the hippocampus circuitry. The procedures involved in OLT are described in the previous section, “Basic Protocol 3”. The choice to explore the object displaced to a novel location reflects the ability of animal to discern minor changes in the location of objects in its immediate environment. Typically, CERs (not receiving any therapeutic treatment) display a clear impairment in this cognitive function, as they do not show affinity for the object moved to a novel place in Trial 3. If NSC or GPC grafting alleviates cognitive dysfunction, then animals receiving NSC or GPC grafts would demonstrate ability for discriminating NP object from the FP object by spending a greater amount of time with the NP object in trial 3.
-
(2)
Novel Object Recognition Test (NORT): This test examines object recognition memory (Hattiangady et al., 2014). Maintenance of this function primarily depends upon the integrity of the perirhinal cortex and partially on the hippocampus.
Apparatus
This test is performed in an open field box measuring 100 cm (L) × 100 cm (W) × 60 cm (H). Each animal is subjected to three successive trials in this test (Fig.6).
Figure 6.
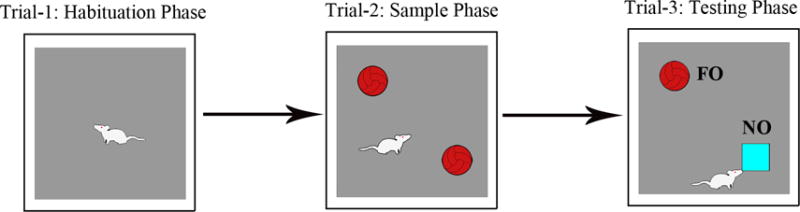
Figure shows a schematic of different trials involved in performing a novel object recognition test (NORT). The duration of exploration in each phase is 5 minutes and the delay between trials 2 and 3 varies from 15–60 minutes, which is empirically determined depending on the experimental design employed. Please note that, in trial 3, one of the objects employed in trial 2 was replaced by a novel object (referred to as NO in the figure). The percentage of object exploration time spent with NO, in comparison to the percentage of time spent with the familiar object (referred to as FO in the figure) in trial 3 serves as a measure of novel object recognition memory.
Handling of CERs prior to test
A day before the test, all animals need to be handled. Additionally, make sure that all chosen CERs explore the open field apparatus individually for 10–15 minutes. This pre-test exploration of the apparatus reduces anxiety in CERs on the day of testing. This was evidenced through increased time spent in exploration of objects by CERS in trials 2 and 3.
Procedures
Trial 1 (Habituation phase): Remove the animal from its home cage and gently place it in the center of an empty open field box. Allow the animal to explore the open field box for 5–15 minutes and then place it back in its home cage.
Trial 2 (Exploration phase): After an inter-trial interval of 15–60 minutes (which is determined empirically for different animal models and studies), place the animal again in the center of the open field with two identical objects placed on right and left sides of the box. Allow the animal to freely explore the objects for 5 minutes and then place it back in its home cage. Video-record this trial using Noldus-Ethovision or Any Maze video-tracking system to determine whether animals explore both objects in this phase.
- Trial 3 (Testing phase): After an inter-trial interval of 15–60 minutes (which is determined empirically for different animal models and studies), place the animal again in the center of the same open field box with objects in the same arena as in the exploration phase but with replacement of one of the objects with a new object. Allow the animal to explore for 5 minutes, and video-record the entire trial 3 using Noldus-Ethovision or Any Maze video-tracking system.
- NOTE: The open field apparatus needs to be cleaned thoroughly with 70% alcohol and air-dried prior to the commencement of each trial for every rat. This removes odor (and fecal matter or urine when present) of the previously tested animal. Handle and release each rat gently during the test to minimize anxiety.
Interpretation of results
Export data such as times spent with novel and familiar objects and the total object exploration time from the software. Calculate percentages of the total object exploration time spent with the novel object vis-à-vis the familiar object. Compare these values within each animal group using two-tailed, unpaired Student’s t-test to determine the ability of animals for novel object recognition. The choice to explore the novel object over the familiar object reflects the ability of animal for recognition memory function. Typically, CERs (not receiving any therapeutic treatment) display a clear impairment in this recognition memory test, as they do not show affinity for the novel object in trial 3. Rather, they spend either nearly equal amounts of time with the familiar and novel objects or spend greater amount of time with the familiar object. If NSC or GPC grafting alleviates recognition memory dysfunction, then animals receiving NSC or GPC grafts would demonstrate ability for discriminating novel object from the familiar object by spending a greater amount of time with the novel object in trial 3.
-
(3)
Pattern Separation Test (PST): Pattern separation function reflects proficiency for discriminating analogous experiences through storage of similar representations in a non-overlapping manner (Leutgeb et al., 2007; Yassa and Stark, 2011). Each animal is subjected to four successive trials in this test (Fig. 7).
Figure 7.
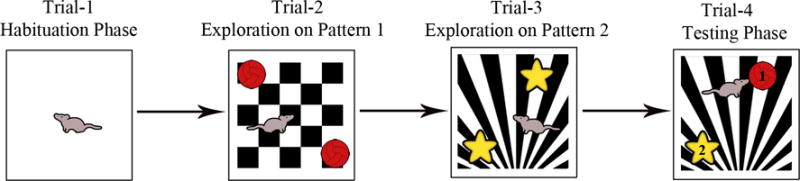
Figure shows a schematic of different trials involved in performing a pattern separation test (PST). In this test, two different floor patterns (P1 in trial 2 and P2 in trials 3 and 4) and two sets of objects are employed. The duration of object exploration in each phase lasts 5 minutes and the delay between trials 2 and 3 and trials 3 and 4 varies from 15–60 minutes, which is empirically determined depending on experimental design employed. Note that, in trial 4, one of the objects used in trial 3 was replaced by an object used in trial 2 (i.e. novel object on P2 for trial 4, indicated by “1” in the figure), and one of the objects using in trial 3 was retained (i.e. familiar object on P2 for trial 4, indicated by “2” in the figure). The percentage of object exploration time spent with novel object (#1 in the figure), in comparison to the percentage of time spent with the familiar object (#2 in the figure) in trial 4 serves as a measure of pattern separation ability.
Apparatus
This test is performed in an open field box measuring 100 cm (L) × 100 cm (W) × 60 cm (H). Each animal is subjected to three successive trials in this test.
Handling of CERs prior to test
A day before the test, all animals need to be handled. Additionally, make sure that all chosen CERs explore the open field apparatus individually for 10–15 minutes. This pre-test exploration of the apparatus reduces anxiety in CERs on the day of testing. This was evidenced through increased time spent in exploration of objects by CERS in trials 2–4.
Procedures
Trial 1 (Habituation phase): Remove the animal from its home cage and gently place it in the center of an empty open field box. Allow the animal to explore the open field box for 5–15 minutes and then place it back in its home cage.
Trial 2 (Exploration of the first set of identical objects placed on floor pattern 1): After an inter-trial interval of 30–90 minutes (which is determined empirically for different animal models and studies), place the animal again in the center of the open field box, now containing the first set of identical objects (object types 1 and 2) placed on a floor pattern 1 (P1). Allow the animal to explore for 5–15 minutes and then place it back in its home cage. Video-record this trial using Noldus-Ethovision or Any Maze video-tracking system to determine whether animals explore both objects in this phase.
Trial 3 (Exploration of the second set of identical objects placed on floor pattern 2): After an inter-trial interval of 30–90 minutes (which is determined empirically for different animal models and studies), place the animal again in the center of the same open field box, now containing the second set of identical objects (object types 1 and 2) placed on a floor pattern 2 (P2). Allow the animal to explore for 5–15 minutes and then place it back in its home cage. Video-record this trial using Noldus-Ethovision or Any Maze video-tracking system to determine whether animals explore both objects in this phase.
- Trial 4 (Testing phase): After an inter-trial interval of 30–90 minutes (which is determined empirically for different animal models and studies), place the animal again in the center of the same open field box, containing an object from trial 2 (which is now a familiar object) and an object from trial-1 (which is now a novel object) placed on the floor pattern employed in trial 2 (P2). Allow the animal to explore for 5–15 minutes and then place it back in its home cage. Video-record the entire trial 4 using Noldus-Ethovision or Any Maze video-tracking system.
- NOTE: The open field apparatus needs to be cleaned thoroughly with 70% alcohol and air-dried prior to the commencement of each trial for every rat. This removes odor (and fecal matter or urine when present) of the previously tested animal. Handle and release each rat gently during the test to minimize anxiety.
Interpretation of results
Export data of trial 3 such as times spent with the object from trial 1 (i.e. novel object on pattern 2, NO on P2]) and the object from trial 2 (familiar object on pattern 2 [FO on P2] and the total object exploration time. Calculate percentages of the total object exploration time spent with the NO on P2 vis-à-vis FO on P2. Compare these values within each animal group using two-tailed, unpaired Student’s t-test to determine the ability of animals for pattern separation. Excellent pattern separation ability (i.e. ability to distinguish between similar experiences) in naïve rats is revealed through greater exploration of the object from trial 1 (i.e. NO on P2) than the object from trial 2 (i.e. FO on P2). Previous studies have shown that this task requires normal levels of dentate neurogenesis (Jain et al., 2012; Oomen et al., 2014; McAvoy et al., 2015). CERs (not receiving any therapeutic treatment) typically display no preference for the NO on P2, as they spend either nearly similar amounts of time with novel and familiar objects on P2 or spend greater amount of time with the familiar object, implying loss of ability for pattern separation, which is not surprising as chronic epilepsy is associated with a great decline in dentate neurogenesis (Hattiangady et al., 2004, 2010; Kuruba et al., 2009). If NSC or GPC grafting alleviates pattern separation dysfunction, then animals receiving NSC or GPC grafts would demonstrate ability for discriminating NO on P2 from FO on P2 by spending a greater amount of time with the NO in trial 4.
-
(4)
Sucrose Preference Test (SPT): For analyzing anhedonia (inability to feel pleasure in activities that offer pleasure in normal conditions), we employ a sucrose preference test (SPT). This test measures mood dysfunction in rodents by examining decreases in the preference for sweet fluids such as sucrose or saccharin solutions (Fawcett et al., 1983; Willner et al., 1987; Muscat and Willner, 1992; Willner et al., 1993). The protocol described here is adapted from a test previously described by Willner and colleagues (Snyder et al., 2011). This test usually takes four days to be completed.
Materials
Water bottles for rats
Drinking water
1% sucrose solution (Sucrose, Sigma-Aldrich)
Graduated measuring cylinder
Procedures
Day 1 – Animals are trained to adapt to only sucrose solution for 24 hours. House animals individually in cages and provide free access to two identical bottles containing 100 mL of 1% sucrose solution. Provide ad libitum access to food.
Day 2 – Animals are trained with 1% sucrose solution and normal water for 24 hours. Provide access to two identical bottles, one bottle containing 100 mL of 1% sucrose solution and another bottle containing 100 mL of regular water. Provide ad libitum access to food.
Day 3 – Animals are deprived of food and fluids for 22 hours. Remove water bottles and food from the cage.
Day 4 – Animals are tested for their preference towards consuming sucrose-containing water or normal water after fasting. Provide free access to two identical bottles: one containing 100 ml of sucrose solution and another containing 100 ml of regular water. Place bottles carefully in the cage so that no leakage occurs from bottles. Do not provide any food.
Two hour later, remove both bottles carefully and measure the consumption of sucrose solution and water by using a graduated measuring cylinder. Place all animals back to their previous housing conditions and provide ad libitum access to water and food.
-
Calculate sucrose preference rate using the formula, sucrose consumption/(water consumption + sucrose consumption) × 100.
Interpretation of results: Naïve control rats show preference for drinking sucrose-containing water over the normal water. On the other hand, CERs typically do not exhibit such preference; they either consume normal and sucrose-containing water approximately in equal proportions or mostly consume normal water. If NSC or GPC grafting alleviates anhedonia, then animals receiving NSC or GPC grafts would consume greater amount of sucrose-containing water than normal water on day 4 of the test.
-
(5)
Eating related depression test (ERDT): This test examines motivation for eating food following 24 hours of food deprivation. This test is adapted from a novelty suppressed feeding test (NSFT, Samuels and Hen, 2011; Powell et al., 2012). NSFT assesses the ability of the animal to resolve a conflict between a context that induces heightened anxiety and a drive to approach an appetitive stimulus. However, this test was found not feasible for CERs because of greater levels of anxiety than typically observed in naïve control rats. This was evidenced by the absence of movement towards the food during the test in a vast majority of CERs, in spite of food deprivation for 24 hours. Hence, we modified this test with a major change that reduced anxiety, i.e. the test is conducted in the home cage instead of a novel open field box. Since anxiety is no longer a significant parameter, this modified test examined the motivation of the animal to eat food following food deprivation for 24 hours.
Materials
Rat cages
Petriplates
Food Pellets
Video camera or video tracking system (Noldus-Ethovision or Any Maze)
Timers (if measurements are made manually)
Procedure
Food deprivation for 24 hours: Remove food from animal’s home cage but provide ad libitum access to water.
Remove animal from its home cage and place temporarily in an identical new cage.
Place pellets of food (regular chow) in one corner of animal’s home cage over a white paper.
Remove animal from the new cage, and place back in its home cage diagonally opposite to the position of the food.
Record movement of the animal until it eats food or for a maximum of five minutes, using a video camera or video-tracking system of Noldus-Ethovision or Any Maze. Using video-recordings, calculate the latency to commencement of eating food pellets. The start of eating is when the rat bites pellets with the use of its forepaws for the first time (i.e. latency to first bite). If the animal fails to eat food in the 5 minutes duration of this test, record the latency value as 300 seconds.
If manual scoring is planned, start the timer as soon as the animal is released into the cage, continuously observe the animal from a distance and stop the timer when the animal picks up and starts eating food pellets. If the animal fails to eat food in the 5 minutes duration of this test, record the latency value as 300 seconds.
-
Once the test is done, provide the animal with ad libitum access to food and water.
Interpretation of results: Naïve control rats typically display shorter latencies to eat food in ERDT, which reflects their higher level of motivation for eating food following deprivation or normal mood function. In contrast, CERs either display much greater latencies to eat food or lack motivation to move towards the food in the 5-minute duration of the test implying impaired motivation to eat food despite being hungry, which is also indicative of depressive-like behavior. If NSC or GPC grafting alleviates depressive-like behavior in CERs, then animals receiving NSC or GPC grafts would exhibit shorter latencies to eat food ERDT.
-
(6)
Forced Swim Test (FST): This test, originally developed by Porsolt and colleagues (Porsolt et al., 1977) measures forced helplessness or depressive-like behavior in rodents.
Materials
Porsolt chambers (Stoelting Inc) – with 18.7 cm inner diameter and 42 cm depth.
Video camera
Dry towels
Procedure
-
Day 1 (Acclimatize the animal to the Porsolt chamber and swimming): Fill the Porsolt chamber with regular water (with a temperature of ~23–25°C) to a depth of 30 cm so that animal’s forepaws do not touch the bottom of the container during the test. Place the animal slowly inside the chamber and let it swim for 10 minutes. Normal rats struggle initially but swim for most of their time in the water and show floating behavior (immobility) for shorter durations when tired. Remove the animal from the cylinder, dry it with towels and place it back in its home age. Place the home cage on a heating pad for 30 minutes.
NOTE: Monitor the animal for drowning. If drowning is seen, remove the animal quickly from the chamber, dry it with towels and exclude this animal for FST since the animal displayed problems with swimming or floating.
-
Day 2 (Testing phase): Fill the Porsolt chamber with fresh water (~23–25°C) to a depth of 30 cm. Twenty-four hours after the pre-test, release the animal slowly into the chamber and let it swim for 10 minutes. Record the behavior of animal for the entire duration of 10 minutes using a video camera. Remove the animal from the cylinder and dry it nicely with towels and place it back in its home age.
NOTE: Make sure that the background of the Porsolt chamber is not reflective during the test, as reflections affect the quality of video and makes scoring immobility times difficult. A contrasting background color (i.e. a dark sheet of card board behind the Porsolt chamber while testing albino rats) gives best quality of videos for scoring immobility.
-
Using video-recordings, score all intermittent durations of time spent in immobility (or floating) during the 10-minute test. Immobility (also termed as floating) is defined as the minimal movement necessary to keep the head above the water level. On the other hand, swimming is identified as a continuous horizontal movement in the swim chamber whereas climbing (also termed as struggling) is defined as a vertically directed movement with forepaws typically above water along the walls of the swim chamber. Use the percentage of time spent in floating as a measure of depressive-like behavior.
NOTE: Identify animals through experimental codes during the test so that investigators scoring video-recordings remain blind to the identity of the animal or animal assignments. Additionally, it is prudent to obtain immobility scores of video-recordings from two independent researchers in the lab.
-
Calculate times spent in immobility during the first and last 5-min segments of the test. The first five minutes of the test is considered important because normal animals actively swim during the first five minutes and factors such as fatigue may cause confounds in the final 5 minutes.
Interpretation of results: Naïve control rats typically spend shorter durations of time in floating, which reflects their normal mood function. In contrast, CERs give up swimming quickly and spend greater amounts of time in floating implying increased depressive-like (or helplessness-like) behavior in these rats. If NSC or GPC grafting alleviates depressive-like behavior in CERs, then animals receiving NSC or GPC grafts would exhibit shorter durations of time in floating.
BASIC PROTOCOL 9: ANALYSES OF THE YIELD AND DIFFERENTIATION OF GRAFT DERIVED CELLS
Following the completion of all behavioral measures described above, animals are perfused with 4% paraformaldehyde for analyses of the yield and differentiation of NSC or GPC graft derived cells. This comprises tissue fixation, tissue processing for cryostat sectioning, immunostaining and/or immunofluorescence procedures, quantification of the yield of graft-derived cells using stereology, and measurement of the percentages of the graft-derived cells that differentiate into NeuN+ neurons, GABA-ergic interneurons, and subclasses of GABA-ergic neurons expressing various markers, astrocytes, astrocytes expressing neurotrophic factors such as the GDNF, oligodendrocyte progenitors, and mature oligodendrocytes.
Equipment and Materials
NSC or GPC grafted chronically epileptic rats (Basic Protocol 5)
Isoflurane
Flat fixation board (e.g., rectangular Thermocol board covered with thin plastic sheet)
Pushpins
Dissection instruments (heavy-duty scissors, tissue forceps, microdissecting scissors, bone cutter, tissue retractors and clamps, scalpel, scalpel blades, and spatula) to open the thoracic cavity and to remove the perfusion-fixed brain from the skull
Perfusion pump and perfusion tube (World Precision Instruments, cat. no. 500328, http://www.wpi-europe.com/en/index.shtml)
20- to 25-G needles
Cryostat (Leica)
24-well plates
37° and 65°C oven
1% (w/v) heparin in 0.9% NaCl, sterile
4% paraformaldehyde (see recipe)
0.1 M sodium phosphate buffer, pH 7.4 (Fisher Scientific, cat. no. NC9552713)
10%, 20%, and 30% (w/v) sucrose in sodium phosphate buffer, pH 7.4
Cryo-embedding gel (Fisher Scientific cat. no. SH75-125D)
0.1 M sodium phosphate buffer, pH 7.4, containing 0.01% sodium azide
0.1 M Tris-buffered saline (TBS; see recipe) containing 20% (v/v) methanol and 2% (v/v) hydrogen peroxide
0.1 M Tris-buffered saline (TBS; see recipe)
50% (v/v) formamide/2× SSC (see SSC recipe in UNIT 1D.5)
2× SSC (UNIT 1D.5)
0.1 M borate buffer, pH 8.5 (see recipe)
Blocking solution: TBS containing 10% (v/v) normal horse serum and 0.01% Triton X-100
Primary antibody solution: mouse anti-BrdU (BD Biosciences, 1:500) or mouse-human nuclear antigen (HNA, Millipore, 1: 200)
Secondary antibody solution: biotinylated anti–mouse IgG (Vector Labs) diluted 1:200 in TBS containing 2% normal horse serum
ABC reagent (from Elite ABC kit; Vector Labs)
DAB kit (Vector Labs)
Light and fluorescent microscopes and a confocal microscope (Nikon, Olympus)
Imaging system for stereological counting of graft-derived cells e.g., the StereoInvestigator system; MicroBrightField (http://www.mbfbioscience.com/)
Additional reagents and equipment for immunohistochemistry (Hofman, 2002)
Harvest tissue and cut sections
For more detail on the techniques described below, see Hofman (2002).
- Deeply anesthetize rats using a lethal dose of isoflurane by using a vaporizer. For this, place each rat in an anesthetic chamber having a direct access to receive 5% isoflurane. Once the animal ceases respiration, secure all four limbs on a flat fixation board using pushpins.
- Perform the entire procedure inside a fume hood.
Cut open the thoracic cavity, open the pericardium, turn on the perfusion pump, and carefully insert a 20- to 25-G syringe needle (attached to a long plastic perfusion tube immersed in 200 ml of normal saline solution containing 1% heparin) into the left ventricle. Immediately following this, make a nick in the right atrium for the blood and the perfusion fluid to flow out of the heart and adjust the speed of the perfusion pump so that 200 ml of saline is perfused in ~10 to 12 min.
Immerse the perfusion tube in a 4% paraformaldehyde solution and perfuse ~500 ml of 4% paraformaldehyde in 30 to 40 min.
Cut open the skull using bone cutters and blunt scissors, carefully remove the entire brain from the skull by cutting the meninges and cranial nerves, immerse the brain in 4% paraformaldehyde solution overnight at 4°C, then transfer it to 0.1 M sodium phosphate buffer, pH 7.4.
- Detach the hindbrain and process the rest of the brain for cryo-protection by successively treating the brain tissue with 10%, 20%, and 30% sucrose at 4°C.
- Note that tissues should be placed in each percentage of the sucrose solution until the entire block of tissue sinks to the bottom.
- Fix the brain tissue to the cryostat chuck using the cryo-embedding gel and let it stay for ~30 min. Following this, fix the cryostat chuck containing the frozen brain tissue into the object holder of the cryostat, adjust the cryostat knife settings and section thickness to desired values, and cut coronal sections through the brain.
- We routinely collect 30-μm-thick serial coronal sections through the entire hippocampus in 24-well plates filled with PB containing 0.01% sodium azide. For long-term storage, we store sections in the antifreeze solution at −20°C.
Perform BrdU immunostaining for identifying rat MGE-NSC graft-derived cells
For more detail on the techniques described below, see Rao and Shetty (2004).
- Choose every 10th or 15th section through the entire hippocampus for analyses of the yield of graft-derived cells. Process these serial sections for BrdU immunostaining in 24-well plates.
- Basic immunohistochemical techniques are described in Hofman et al. (2002).
Etching: Treat sections with 0.1 M Tris-buffered saline (TBS) containing 20% methanol and 3% hydrogen peroxide to remove the endogenous peroxidase.
Wash sections thoroughly in TBS, incubate sections in a 50% formamide solution prepared in 2× saline sodium citrate buffer (2× SSC buffer) for 2 hr at 65°C using an oven.
Wash sections in a cold 2× SSC buffer twice and incubate in 2 N HCl at 37°C for 60 min using an oven. Following this, treat sections for 10 min with a 0.1 M borate buffer (pH 8.5) and wash three times in TBS.
Incubate sections in blocking solution for 30 min at room temperature.
Incubate sections overnight at 4°C in the primary antibody solution.
Wash sections three time in TBS and incubate 1 hr in secondary antibody solution.
Rinse sections three times in TBS and incubate 1 hr in ABC reagent.
- Wash sections three times in TBS and then incubate for 5 to 10 min in DAB solution prepared according to the manufacturer’s instructions (DAB kit, Vector Labs). Give three washes in TBS before mounting on slides for dehydration, clearing, and cover slipping using standard histological methods.
- It is important to standardize the incubation time required for an optimal color reaction using a couple of sections. Terminate the DAB reaction by transferring sections into the distilled water.
- An example of the distribution of BrdU+ NSC graft-derived cells in the chronically epileptic hippocampus is illustrated in Figure 9.
Figure 9.
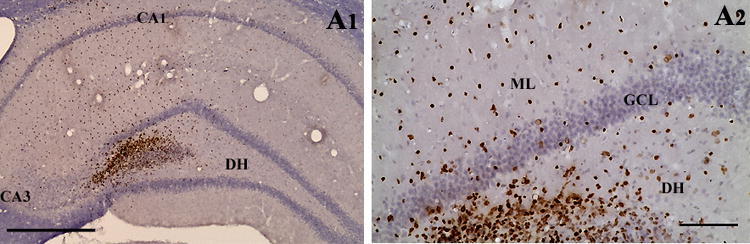
An example showing the distribution of BrdU+ NSC graft-derived cells in the hippocampus of a chronically epileptic rat (A1). A2 is a magnified view of a region from A1 showing the migration of cells into different regions of the host dentate gyrus. Scale bar, A1, 400 μm; A2, 100 μm.
Perform human nuclear antigen immunostaining for identifying human GPC graft-derived cells
Select every 10th or 15th section through the entire hippocampus for analyses of the yield of human GPC graft-derived cells. Process these serial sections for human nuclear antigen (HNA) immunostaining in 24-well plates.
Etching: Treat sections with 0.1 M Tris-buffered saline (PBS) containing 20% methanol and 3% hydrogen peroxide to remove the endogenous peroxidase.
Wash sections thoroughly in PBS and incubate sections in blocking solution for 30 min at room temperature.
Incubate sections overnight at 4°C in the primary antibody solution.
Wash sections three times in PBS and incubate 1 hr in secondary antibody solution.
Rinse sections three times in PBS and incubate 1 hr in ABC reagent.
Wash sections three times in PBS and incubate for 5 to 10 min in DAB solution prepared according to the manufacturer’s instructions (DAB kit, Vector Labs). Give three washes in PBS before mounting on slides for dehydration, clearing, and cover slipping using standard histological methods.
Quantify the yield of graft-derived cells (BrdU+ cells or HNA+ cells) using stereology
For quantifying the yield of graft-derived cells, we typically count all BrdU+ or HNA+ cells in every 10th or 15th section through the entire anterior-posterior extent of the hippocampus using the optical-fractionator counting method in the StereoInvestigator system (MicroBrightField Inc.; http://www.mbfbioscience.com/). The overall yield of graft-derived cells may be expressed as the total number of engrafted cells per hippocampus or as the percentage of the total injected cells per hippocampus (i.e., the total number of BrdU+/HNA+ cells placed at four sites in each hippocampus).
It is important to note that the yield of graft-derived cells does not represent the absolute survival of cells that are originally implanted. However, assessment of the yield does give an idea about the numbers of graft-derived cells that engraft for prolonged periods when a given number of NSCs or GPCs are implanted. Gauging the yield is important for NSC and GPC grafts, as quantification of the absolute graft cell survival is difficult due to the likelihood that both cell proliferation and cell death occur in these grafts. Thus, one can conclude that the overall engraftment is robust if the yield of graft-derived cells is close to or greater than the number of cells originally implanted. A yield greater than the number of cells initially injected can happen if the NSCs or GPCs divide a couple of times after the grafting. The major aspects of the optical fractionator cell counting methodology are described below.
The StereoInvestigator system consists of a color digital video camera (Optronics Inc., http://www.optronics.com/) interfaced with a Nikon E600 microscope. In each hippocampus, the BrdU+ cells are counted from randomly and systematically selected frames (e.g., each measuring 150 × 150 μm, 0.0225 mm2 area) in every 10th section using the 100× oil-immersion objective lens. The numbers and densities of frames are determined by entering the parameter grid size (e.g., 60 × 60 μm) in the optical fractionator component of the StereoInvestigator system (Rao and Shetty, 2004). Although 30-μm-thick sections through the hippocampus are cut using a cryostat, the section thickness at the time of counting reduces to 40% to 50% of the initial thickness because of the shrinkage of the tissue with the BrdU immunostaining. Hence, at the time of data collection, the thickness of sections varies from 12 to 15 μm.
It is very important to check the thickness of each section used for counting before entering the section thickness, as shrinkage can vary between sections.
For commencing cell counting in the each chosen section, open the serial section manager dialog box and enter different parameters such as the number of sections, section evaluation interval, section cut thickness, mounted section thickness, and the starting z level.
Mark the contour of the transplant area (i.e., the area of the hippocampus containing the graft-derived [i.e., BrdU+ or HNA+] cells) using the 4× or 10× objective lens and the tracing function of the StereoInvestigator.
Activate the optical fractionator component of the program and select the numbers and locations of the counting frames (using a systematic random sampling scheme) and the counting depth by entering parameters such as the grid size (e.g., 60 × 60 μm), the thickness of top guard zone (e.g., 4 μm), and the optical dissector height (e.g., 6 μm).
The number of frames per section counted varies, as the overall area comprising the graft-derived cells varies from section to section. This sampling scheme is consistent with the principle of the optical fractionator counting method in that the sample concentration must remain constant for each section, implying that the number of unit volumes to be counted per volume of the structure on a given section needs to be a constant ratio from section to section. Thus, counting of cells from randomly and systematically chosen frames in every 10th or 15th section through the hippocampus guarantees that effectively every BrdU+/HNA+ cell within the transplant area has equal odds of being counted. This is imperative because the scattering of BrdU+/HNA+ cells within the graft area is visibly heterogeneous.
A computer-driven motorized stage then allows the section to be analyzed at each of the counting-frame locations. In every counting-frame location, identify the top of the section, and then move the plane of the focus to a depth of 4 μm (guard zone) to get rid of the problem of uneven section surface. This plane serves as the first point of the counting process. Count all BrdU+/HNA+ cells that come into focus in the next 6-μm section thickness if they are entirely within the counting frame or touching the upper or right side of the counting frame.
Repeat the above protocol for every chosen section in each hippocampus. Following collection of data from all sections, select “display probe run list” to obtain the total number of graft-derived (BrdU+/HNA+) cells. The extent of migration of graft-derived cells can be assessed indirectly by obtaining the total volume of the hippocampal tissue comprising the graft-derived cells from the “display probe run list.” Based on the different parameters that are entered at the beginning of the counting and the number of cells sampled at different counting locations in all sections, the StereoInvestigator program calculates the total number of BrdU+ cells per hippocampus by utilizing the optical fractionator formula, N = 1/ssf.1/asf.1/hsf. EQ− (Dorph-Petersen et al., 2001). The abbreviation “ssf” stands for the section sampling fraction, which is 10 in this example; “asf” symbolizes the area sampling fraction, which is calculated by dividing the total areas sampled by the total area of the transplant (i.e., the sum of transplant areas in every 10th section); “hsf” stands for the height sampling fraction, which is calculated by dividing the height sampled (which is 6 μm in this example) by the section thickness at the time of analysis (i.e., 12 to 15 μm); “EQ− denotes the total count of particles (i.e., BrdU+ or HNA+ cells) sampled for the entire hippocampus. Figure 10 illustrates the distribution of cells derived from NSC grafts along the septo-temporal axis of the hippocampus of a chronically epileptic rat (Waldau et al., 2010).
Figure 10.
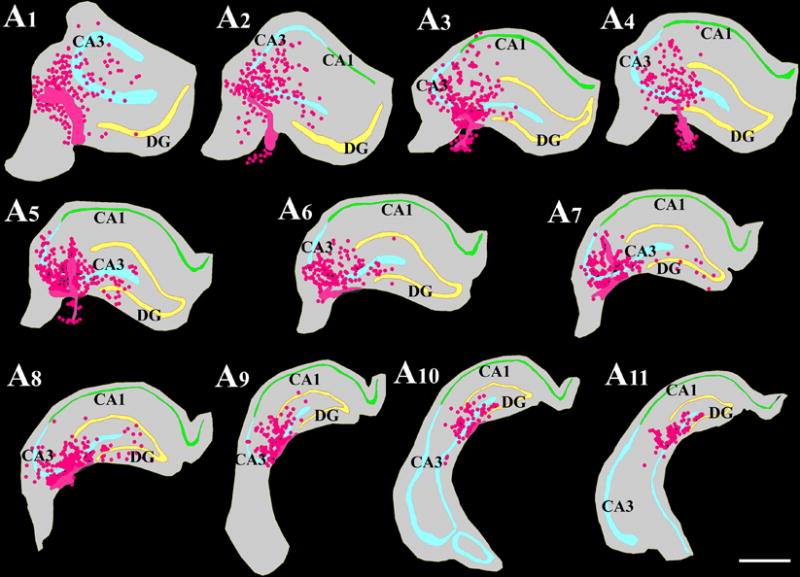
Location of NSC grafts and NSC graft-derived cells (shown in pink color based on chlorodeoxyuridine+ [CldU+] graft-derived cells) with respect to hippocampal cell layers and subfields in a chronically epileptic rat. These tracings, performed using the Neurolucida software (MicroBrightField Inc), represent every tenth 30-μm thick section through a chronically epileptic hippocampus that received four MGE-NSC grafts. Scale bar, 1000 μm. Reproduced from Waldau et al. (2010)
Following collection of data (such as the total number of BrdU+/HNA+ cells per hippocampus), check the Gundersen coefficient of error (CE) for each sample. The CE values that are <0.05 suggest that the counts obtained are valid.
If larger CE values are seen for some samples, repeat the counting process by modifying the counting parameters, which may include increasing the number of sections (e.g., every 5th section instead of the every 10th section), altering the grid size to increase the number of sites per section, and changing the counting-frame dimensions to increase the probability of counting more cells at each counting location.
Perform phenotypic analyses of graft-derived cells in the host brain
Cells derived from the NSCs are typically heterogeneous, and each type of cell derived from NSCs has a unique function. Therefore, it is vital to quantify the fraction of graft-derived cells that become mature neurons (e.g., NeuN+ cells), specialized neurons (e.g., GABA-ergic neurons), mature astrocytes (e.g., S-100β+ cells), oligodendrocyte progenitors (e.g., NG2+ cells), and mature oligodendrocytes (e.g., O1+ and Rip+ cells). In addition, as NSC differentiation might depend on factors in the host tissue or the location of engraftment, it is also important to assess the fraction of graft-derived cells that persist as NSCs (e.g., Sox2+ or nestin+ cells) in the host brain. This will provide clues as to whether cells derived from NSC grafts facilitate repair and regeneration of the host brain area via replacement of lost cells or through mechanisms such as the release of neurotrophic or other beneficial factors.
Cells derived from the human GPCs are expected to be mostly homogeneous, as a vast majority of cells derived from GPCs are expected to differentiate into GABA-ergic interneurons. However, smaller fractions of cells from GPCs may also differentiate into astrocytes, oligodendrocytes or may remain as progenitors expressing NKX2.1. Therefore, it is vital to quantify the fraction of graft-derived cells that become mature neurons (e.g., NeuN+ cells), interneurons (e.g., GABA-ergic neurons), subclasses of interneurons (NPY, SOM, PV, CR, CBN, reelin etc.), mature astrocytes (e.g., S-100β+ cells), oligodendrocyte progenitors (e.g., NG2+ cells), and mature oligodendrocytes (e.g., O1+ and Rip+ cells) and GPCs (NKX2.1). In addition, to rule out the continued proliferation of NKX2.1+ GPCs in the host brain, it will be necessary to quantify fractions of graft-derived cells expressing markers of proliferation (e.g. Ki-67). Furthermore, as GPCs derived from hiPSCs are used for grafting, possible occurrence of pluripotent stem cells need to be investigated among graft-derived cells using appropriate markers (e.g. Oct-4).
The differentiation of graft-derived cells is typically assessed through a standard dual immunofluorescence method and z section analyses in a confocal microscope, by identifying both the graft cell marker (e.g. BrdU or HNA) and the chosen neural cell marker (such as NeuN, GABA, S-100β+, NG2, O1, Rip, Sox2, nestin, NPY, SOM, PV, CR, CBN, reelin, NKX2.1, Ki-67, Oct-4 etc.).
As the dual-immunofluorescence approach can be done using different protocols, we restrict our description mainly to the listing of primary and secondary antibody combinations that we typically use in our studies (see Table for details). We employ free-floating sections in 24-well tissue culture plates and sequentially visualize the two antigens. The first antigen is typically the graft cell marker (i.e., BrdU or HNA), whereas the second antigen is the chosen neural cell/NSC marker. The overall procedure takes ~3 days to complete for each combination.
Following dual-immunofluorescence staining, we perform z section analyses in a confocal microscope within a couple of weeks to prevent the fading of fluorescence over time. We obtain z sections at 1-μm intervals and perform orthogonal analysis to confirm the presence of dual antigens in all graft-derived cells that are selected for quantification. This will facilitate the assessment of the percentages of graft-derived cells that express different neural antigens. Examples of the differentiation of cells derived from MGE-NSC grafts into different neural phenotypes in the chronically epileptic hippocampus are illustrated in Figure 11.
Figure 11.
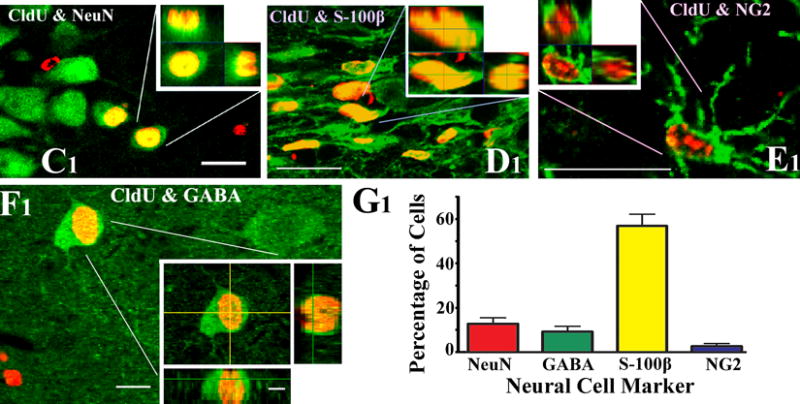
Examples of the differentiation of MGE-NSC graft-derived CldU+ cells into NeuN+ mature neurons (C1), S-100β+ mature astrocytes (D1), NG2+ oligodendrocyte precursors (E1) and GABA+ neurons (F1), visualized via dual immunolabeling for CldU (red) and markers of neurons/glia (green) and z-section analyses under a confocal microscope. Scale bar, C1–E1, 20 μm; F1, 10 μm; orthogonal inset of F1, 5 μm. The bar chart in G1 depicts the percentages of graft-derived cells that differentiate into NeuN+ neurons, GABA+ neurons, S-100β+ astrocytes and NG2+ oligodendrocyte precursors. Reproduced from Waldau et al. (2010).
BASIC PROTOCOL 10: ANALYSES OF EFFECT OF NSC OR GPC GRAFTING ON THE HOST HIPPOCAMPUS
In addition to contributing new neurons synthesizing the inhibitory neurotransmitter GABA and new astrocytes synthesizing the anticonvulsant protein GDNF (Waldau et al., 2010), NSC of GPC grafting into the epileptic hippocampus might also influence the host cells and neurons, as observed in other disease models. For example, NSCs placed into the substantia nigra and caudate nuclei of monkeys exhibiting Parkinson’s disease can induce behavioral recovery with differentiation of only a small fraction of grafted NSCs into dopaminergic neurons (Redmond et al., 2007). As large numbers of grafted NSCs became differentiated into astrocytes and expressed multiple neurotrophic factors including GDNF, it appeared that NSC grafting–mediated changes to the host milieu enhanced the function of the existing dopaminergic neurons (Redmond et al., 2007; Sanberg, 2007). Considering this, it is important to examine the effects of NSC and GPC grafts on the host microenvironment and host cells, in addition to examining the differentiation of graft-derived cells into desired neuronal or glial phenotypes. In our previous study, we utilized a standard dual-immunofluorescence approach to assess the changes in the function of S-100β+ host astrocytes with NSC grafting into the hippocampus of CERs (Waldau et al., 2010). The results showed that NSC grafting into the hippocampus of CERs considerably restored GDNF expression in the S-100β+ host hippocampal astrocytes (Waldau et al., 2010). However, it is likely helpful to use high-throughput genomic (such as microarray and qRT-PCR arrays) and proteomic approaches in the future to understand the effects of NSC or GPC grafting on multiple pathways in the host hippocampus of CERs. The methods for these approaches are not described here, as they are beyond the scope of this article.
REAGENTS AND SOLUTIONS
For culture recipes and steps, use sterile tissue culture–grade water. For other purposes, use deionized, distilled water or equivalent in recipes and protocol steps. For suppliers, see SUPPLIERS APPENDIX.
Anesthetic cocktail
In our laboratory, we use an anesthetic cocktail comprising ketamine (50 mg/ml), xylazine (4.5 mg/ml), and acepromazine (0.4 mg/ml) at a dose of 0.7 ml/kg body weight.
The cocktail may be stored up to 2 weeks at 4°C.
Borate buffer, pH 8.5
Add 6.18 g boric acid (Sigma) to 1000 ml of distilled water and adjust the pH to 8.5.
Proliferation medium for rat MGE-NSCs
For 50 ml:
36 ml DMEM (Invitrogen)
12 ml F12 (Invitrogen)
1 ml B-27 supplement without retinoic acid (Invitrogen)
40 μl 100× antibiotic-antimycotic (Invitrogen)
Epidermal growth factor, 20 ng/ml (Peprotech)
Fibroblast growth factor, 20 ng/ml (Peprotech)
Heparin 5 μM
Prepare fresh
Differentiation medium for rat MGE-NSCs
For 50 ml:
48.335 ml neurobasal medium (Invitrogen, cat. no. 21103-049)
40 μl 100× antibiotic-antimycotic (Invitrogen, cat. no. 15240-062)
1 ml B-27 supplement with retinoic acid (Invitrogen, cat. no. 17504-044)
625 μl L-glutamine (Invitrogen, cat. no. 25030-081)
Prepare fresh
Paraformaldehyde, 2% and 4%
- 8% paraformaldehyde stock:
- Dissolve 16 g paraformaldehyde in 100 ml of distilled water and 100 ml of 0.4 M phosphate buffer, pH 7.2, to obtain 8% paraformaldehyde in 0.2 M phosphate buffer.
- 2% paraformaldehyde:
- For preparation of 2% paraformaldehyde, mix 50 ml of 8% stock paraformaldehyde solution with 150 ml of distilled water and adjust the pH to 7.2.
- 4% paraformaldehyde:
- For preparation of 4% paraformaldehyde, mix 100 ml of 8% stock paraformaldehyde solution with 100 ml of distilled water and adjust the pH to 7.2.
- Proliferation medium (100 ml)
- 74.520 ml Dulbecco’s Modified Eagle Medium (DMEM; Invitrogen, cat. no. 11960-044)
- 24.4 ml F-12 nutrient mixture (Invitrogen, cat. no. 11765-054)
- 1 ml B-27 supplement without retinoic acid (Invitrogen, cat. no. 12587-010)
- 80 μl 100× antibiotic-antimycotic 100× (Invitrogen, cat. no. 15240-062)
- 20 ng/ml FGF-2 (Peprotech, cat. no. AF-100-18B)
- 20 ng/ml EGF (Peprotech, cat. no. AF-100-15)
- 5 μM heparin (Sigma, cat. no. H1027; prepare stock in sterile DMEM)
- Prepare fresh
Tris-buffered saline (TBS), pH 7.5
Add 12.11 g Trizma base (Sigma) and 8.77 g of sodium chloride (Sigma) to 100 ml of distilled water and adjust the pH to 7.5.
COMMENTARY
Background Information
Epilepsy, typified by spontaneous recurrent seizures (SRS) due to hyperexcitability and synchronization of activity within populations of neurons, affects >50 million people (Strine et al., 2005). Temporal lobe epilepsy (TLE), characterized by progressive development of complex partial seizures and hippocampal neurodegeneration, is seen in >30% of epileptic patients (Manford et al., 1992). While the etiology of TLE is unknown in most cases (McNamara, 1999), it is typically seen after an initial precipitating injury such as status epilepticus (SE), brain injury, tumors, meningitis, encephalitis, and febrile seizures (French et al., 1993; Mathern et al., 1995; Lewis, 2005). Seizures in TLE originate from temporal-lobe foci, which are associated with learning and memory impairments, reduced dentate neurogenesis, and depression (Devinsky, 2004; Hattiangady et al., 2004; Detour et al., 2005; Pirttilä et al., 2005; Hattiangady and Shetty, 2010; Hattiangady et al., 2011). Nearly 35% of patients with TLE exhibit seizures that cannot be controlled by antiepileptic drugs (Litt et al., 2001), and memory difficulties are a frequent cognitive complaint in patients with chronic epilepsy (Vannest et al., 2008). Although surgical resection of the epileptic hippocampus gives better seizure control, this option is often associated with significant cognitive impairments (Helmstaedter et al., 2008). Hence, there is a pressing need to develop alternative therapeutic approaches that greatly diminish both frequency and intensity of SRS in patients with chronic TLE on a long-term basis.
Features of KA model of TLE
Pharmacological SE animal models using KA or pilocarpine are popular for studying TLE, as they replicate several features of TLE. Kainic acid (KA) is a widely used excitotoxin in studies of the hippocampus (Nadler et al., 1978; Sperk et al., 1994; Shetty and Turner, 1996, 1999; Rao et al., 2006a,b, 2007; Rao et al., 2008; Shetty et al., 2012). Intraperitoneal (i.p.) administration of KA in rat induces degeneration of dentate hilar neurons and fractions of CA1 and CA3 pyramidal neurons, deafferentation of dentate granule cells due to loss of hilar mossy cells, and reduction in number of GABA-ergic interneurons in the hippocampus. These changes are followed by aberrant sprouting of dentate mossy fibers into the dentate supragranular layer (DSGL), hyperexcitability in the hippocampus, and a chronic epileptic condition, typified by SRS, learning and memory deficits, and depression (Ben-Ari, 1985; Letty et al., 1995; Hellier et al., 1998, 1999; Buckmaster and Dudek, 1999; Wuarin and Dudek, 2001; Rao et al., 2006a). Since the above changes in the adult hippocampus closely resemble human mesial TLE, the i.p. KA model has been widely used for studying TLE. This model has also been found to be useful for studying the effects of cell grafts on chronic seizures (Rao et al., 2007; Waldau et al., 2010). Thus, the intraperitoneal KA model is suitable for testing the efficacy of NSC grafts in alleviating chronic epilepsy characterized by spontaneous seizures and learning and memory impairments.
GABA-ergic cell therapy for TLE
It is believed that an increased excitatory neurotransmission found in the epileptic hippocampus is partly due to a reduced number of GABA-ergic interneurons, loss of functional inhibition, and diminished numbers of GABA-ergic terminals (Ribak et al., 1986; Cornish and Wheal, 1989; During et al., 1995; Shetty and Turner, 2000, 2001; Shetty et al., 2009). From this perspective, the idea of restraining SRS in the epileptic hippocampus via grafting of cells that just release the inhibitory neurotransmitter GABA at the seizure focus has received considerable attention (Löscher et al., 2008). For instance, grafting of GABA-soaked beads, immortalized GABA-ergic cells, cells that are engineered to produce GABA, and fetal GABA-ergic cells derived from medial and lateral ganglionic eminences and GABA-ergic progenitors derived from hESCs into the epileptic foci have been shown to reduce seizures in a variety of animal models (Löscher et al., 1998; Gernert et al., 2002; Thompson, 2005; Castillo et al., 2006; Hattiangady et al., 2008; Hunt et al., 2013, Henderson et al., 2014; Cunningham et al., 2014). Thus, grafting of GABA-producing cells into the epileptic brain has considerable promise for restraining seizures. However, a routine clinical application of human fetal cells for TLE may not be feasible because of the difficulty in obtaining the required quantity of human fetal cells, and ethical issues (Turner and Shetty, 2003). With reference to the other cell types, it is unknown whether immortalized GABA-producing cells have the ability for long-term survival in the chronically epileptic brain, as they are expected to perform as GABA pumps rather than undergo synaptic integration with the host neurons (Löscher et al., 2008). Therefore, there is a need to find types of cells that are capable of providing an unlimited source of donor cells for grafting and have the ability to give rise to large numbers of GABA-ergic interneurons that survive for prolonged periods after grafting into the epileptic brain.
From the above perspective, it appears that NSCs expanded from the embryonic MGE (Waldau et al., 2010) and the postnatal and adult subventricular zone (SVZ) are attractive candidates as donor cells for grafting therapy in TLE. This is because these cells can (i) be expanded for prolonged periods in culture without losing their multipotential property; (ii) give rise substantial numbers of GABA-ergic interneurons; and (iii) release a multitude of neurotrophic factors (Shetty and Hattiangady, 2007; Waldau et al., 2010). Additionally, MGE-like GPCs obtained from sources such as hESCs and iPSCs may also be used once their potential to give rise to full-fledged GABA-ergic neurons and/or to release multiple neurotrophic factors is validated. However, prior to the clinical application of NSCs or GPCs obtained from any source for TLE, it will be necessary to rigorously analyze the ability of the chosen NSCs and GPCs to restrain SRS on a long-term basis using both behavioral and EEG analyses in animal models of TLE. In addition, the efficacy of different NSC or GPC grafts for improving cognitive and mood function in TLE need to be examined. Particularly, detailed anatomical, electrophysiological, and molecular biological analyses of the efficacy of grafting of NSCs and GPCs for easing SRS and cognitive and mood dysfunction in chronic TLE on a long-term basis are needed before considering the clinical application of NSC or GPC therapy for TLE.
Critical Parameters and Troubleshooting
Observation and care of animals during and shortly after SE
Generating CERs via SE induction is a laborious process that needs patience and close attention to every step. Some of the issues and precautions to be taken are described here. (1) The sensitivity to KA may vary among F344 rats purchased from different sources. Hence, it is ideal to use rats obtained from the same source for all experiments in a particular study. (2) It is important to use KA from a single source for the entire study, as KA from different vendors seems to vary in terms of potency to induce acute seizures or SE. It is recommended that the potency of KA from a particular source be checked in a pilot experiment and that the appropriate dose for inducing SE prior to initiating SE studies in a large number of animals be determined. (3) The use of cages having flat tops fitted with foam lining on the inner side is useful for preventing injury to rats in the event of bouncing seizures after the onset of SE. (4) If a rat is having very intense seizures such as bouncing seizures after the onset of SE (which can happen suddenly), carefully handling the rat and inducing hypothermia (e.g., by placing the rat in an empty cage that was kept on a larger container filled with ice) for a few minutes stops such intense seizures and prevents mortality. (5) Injecting diazepam (5 mg/kg body weight) after 2 or 3 hrs of seizure activity considerably reduces the mortality of rats and does not interfere with the occurrences of SRS in the chronic phase after SE. Without diazepam administration, the overnight mortality after SE will be significantly higher, though the rats that survive might exhibit robust chronic epilepsy (characterized by greater frequency and intensity of SRS). On the other hand, if diazepam is administered very early after the onset of SE (e.g., immediately after the first stage V seizure or before the occurrence of stage V seizure), only a small fraction of rats will develop chronic epilepsy at 4 to 6 months post-SE.
Appropriate animal care after SE is another requirement for reducing SE-related mortality. After SE, animals appear weak, lethargic, and anorexic, and do not seem to eat hard pellets or drink from the water bottle. Therefore, preventing dehydration via subcutaneous administration of fluids (such as saline or Ringer’s lactate solution) and providing soft rat chow or transgel within the cage in a dish clearly helps in minimizing mortality in the recovery phase after SE.
Preparation and injection of NSC or GPC suspension
The various methods such as dissection of MGE tissues from the embryonic brain, expansion of NSCs in culture, expansion of NKX2.1+ GPCs from hiPSCs, and preparation of NSC and GPC suspension for grafting can be performed only by personnel with specific expertise in these aspects. Trituration of neurospheres is a highly skilled technique, as too-gentle trituration does not dissociate the neurospheres well and too-harsh trituration will kill a large number of cells. Triturating neurospheres in a proliferation medium minimizes cell death, in comparison to triturating in any other medium. It is important to select the NSC or GPC suspension with a good viability index (e.g., >80%) for grafting studies because excellent viability of donor cells in the suspension at the time of grafting appears to be an important prerequisite for successful engraftment into the host brain and for obtaining higher yields of graft-derived cells at extended time-points after grafting. A cell suspension with a poor overall viability (e.g., <70%) should not be used for grafting, as greater numbers of dead cells in the graft might initiate an inflammatory reaction in the host brain. Higher-volume injections at a single site in the hippocampus should be avoided, as this typically causes tissue damage, opening of the tissue cleavage planes (e.g., the hippocampal fissure), and flow of the injected fluid mostly into the lateral ventricle. Based on our experience, injection of a 1-μl volume comprising 80,000 to 100,000 live cells (per site) into the hippocampus results in good engraftment of graft-derived cells and does not seem to cause tissue damage at the grafted site. Therefore, preparation of NSC or GPC suspension with a high density of cells is required, which clearly requires expansion of NSCs and GPCs in a large number of flasks.
Behavioral studies in CERs
Typically, CERs do not exhibit SRS while handling. However, occasionally, it is possible that a CER can have a seizure in the middle of a behavioral test. In such cases, it is necessary to remove the rat quickly from the testing apparatus to prevent any injury. Such rats should be observed closely for a while and new trials should not be given in the immediate post-SRS confusion period. However, such rats may be re-tested after a delay of 2–3 days, depending upon the type of test. Furthermore, some CERs might show a tendency to drown in pre-trials of FST. Such rats should be excluded from FST.
Anticipated Results
When all guidelines and protocols described in this article are faithfully followed, success of the experiment (i.e., testing the efficacy of NSC of GPC grafting for restraining SRS and improving cognitive and mood function in chronic TLE) in terms of drawing reliable conclusions is ensured. Additionally, the experimental results can be reproduced with sufficiently stringent adherence to the guidelines. However, errors such as inclusion of rats from different sources, the use of KA from different sources, an inappropriate selection of CERs (e.g., a group of rats with widely differing frequency and intensity of SRS) for grafting, grafting donor NSC or GPC suspension with a poor viability of cells, larger-volume cell suspension injections into the hippocampus, and withdrawing the needle immediately after the injection of the cell suspension introduce significant confounds, and the results obtained from such experiments will be unreliable for meaningful conclusions, and non-reproducible.
Time Considerations
The experiment described here for testing the efficacy of NSC or GPC grafting for restraining SRS and improving cognitive function using an animal model of chronic TLE is clearly a long-term experiment. In our experience, animal aspects of the work such as induction of SE, characterization of SRS, and cognitive dysfunction in CERs, as well as expansion and grafting of NSCs and GPCs, post-grafting analyses of SRS (via direct observations/video recordings and video-EEG recordings), and cognitive and mood function (via OLT, NORT, PST, SPT, ERDT and FST), take ~1 year to complete. Following this, processing of tissues via perfusion, serial section cutting of brain tissues, histology, immunostaining, stereological counting of graft-derived cells, dual immunofluorescence and confocal microscopic analyses of the phenotype of graft derived cells, and analyzing the effects of grafts on host cells and microenvironment would need significant additional amounts of time. The overall time required for histological processing, immunostaining, and quantification depends on the number of animals in different groups, numbers of experimental groups, and the type of analysis undertaken. Stereotaxic surgery can be laborious, as injections are made into multiple sites in the hippocampus. However, with practice, transplantation can be performed on six animals in one surgery session if two stereotactic devices are available. Measuring SRS either via direct observation or from the recorded video requires strong observation skills and an ability to distinguish SRS from the normal grooming behavior of rats throughout the observation period. Furthermore, enthusiastic research personnel/scientists who are committed to this type of long-running and labor-intensive (but very interesting and clinically relevant) experiment are needed for successfully completing these studies.
Supplementary Material
Acknowledgments
This work was supported by grants from the Department of Defense (PRMRP award, W81XWH-14-1-0558 to A.K.S.), the Department of Veterans Affairs (VA Merit Award, I01 BX002351 to A.K.S.) and the State of Texas (Emerging Technology Funds to A.K.S.). The authors also acknowledge the previous funding from the National Institute of Neurological Disorders and Stroke (R01 NS054780, R01 NS043507, and R01 NS36742 to A.K.S.) and the Department of Veterans Affairs (Merit Review Awards to A.K.S.) for studies on animal models of temporal lobe epilepsy in SHETTY LAB. The authors wish to thank Vandana Zaman, Muddanna Rao, Ben Waldau, Ramkumar Kuruba, and Bing Shuai for their outstanding contributions to original articles on neural cell grafting in animal models of temporal lobe epilepsy published from SHETTY LAB, and Mr. Luciano Silveira for his contribution to preparation of figures. Dr. A. K. Shetty is also Research Career Scientist at the Olin E. Teague Veterans’ Medical Center, Central Texas Veterans Health Care System Temple, Texas. Dr. Upadhya is an Assistant Professor on study leave from S.D.M. College of Ayurveda, Udupi, India.
Literature Cited
- Astur RS, Taylor LB, Mamelak AN, Philpott L, Sutherland RJ. Humans with hippocampus damage display severe spatial memory impairments in a virtual Morris water task. Behav Brain Res. 2002;132:77–84. doi: 10.1016/s0166-4328(01)00399-0. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Limbic seizures and brain damage produced by kainic acid: Mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. In vivo intracellular analysis of granule cell axon reorganization in epileptic rats. J Neurophysiol. 1999;81:712–721. doi: 10.1152/jn.1999.81.2.712. [DOI] [PubMed] [Google Scholar]
- Castillo CG, Mendoza S, Freed WJ, Giordano M. Intranigral transplants of immortalized GABAergic cells decrease the expression of kainic acid-induced seizures in the rat. Behav Brain Res. 2006;17:109–115. doi: 10.1016/j.bbr.2006.03.025. [DOI] [PubMed] [Google Scholar]
- Coras R, Siebzehnrubl FA, Pauli E, Huttner HB, Njunting M, Kobow K, Villmann C, Hahnen E, Neuhuber W, Weigel D, Buchfelder M, Stefan H, Beck H, Steindler DA, Blümcke I. Low proliferation and differentiation capacities of adult hippocampal stem cells correlate with memory dysfunction in humans. Brain. 2010;133:3359–3372. doi: 10.1093/brain/awq215. [DOI] [PubMed] [Google Scholar]
- Cornish SM, Wheal HV. Long-term loss of paired pulse inhibition in the kainic acid-lesioned hippocampus of the rat. Neuroscience. 1989;28:563–571. doi: 10.1016/0306-4522(89)90005-5. [DOI] [PubMed] [Google Scholar]
- Cunningham M, Cho JH, Leung A, Savvidis G, Ahn S, Moon M, Lee PK, Han JJ, Azimi N, Kim KS, Bolshakov VY, Chung S. hPSC-derived maturing GABAergic interneurons ameliorate seizures and abnormal behavior in epileptic mice. Cell Stem Cell. 2014;15:559–573. doi: 10.1016/j.stem.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detour J, Schroeder H, Desor D, Nehlig A. A 5-month period of epilepsy impairs spatial memory, decreases anxiety, but spares object recognition in the lithium-pilocarpine model in adult rats. Epilepsia. 2005;46:499–508. doi: 10.1111/j.0013-9580.2005.38704.x. [DOI] [PubMed] [Google Scholar]
- Devinsky O. Therapy for neurobehavioral disorders in epilepsy. Epilepsia. 2004;45:34–40. doi: 10.1111/j.0013-9580.2004.452003.x. [DOI] [PubMed] [Google Scholar]
- Donovan J, Brown P. Parenteral injections. Curr Protoc Immunol. 2006;73:1.6.1–1.6.10. doi: 10.1002/0471142735.im0106s73. [DOI] [PubMed] [Google Scholar]
- Dorph-Petersen KA, Nyengaard JR, Gundersen HJ. Tissue shrinkage and unbiased stereological estimation of particle number and size. J Microsc. 2001;204:232–246. doi: 10.1046/j.1365-2818.2001.00958.x. [DOI] [PubMed] [Google Scholar]
- During MJ, Ryder KM, Spencer DD. Hippocampal GABA transporter function in temporal-lobe epilepsy. Nature. 1995;376:174–177. doi: 10.1038/376174a0. [DOI] [PubMed] [Google Scholar]
- Fawcett J, Clark DC, Scheftner WA, Gibbons RD. Assessing anhedonia in psychiatric patients: the pleasure scale. Arch Gen Psychiatry. 1983;40:79–84. doi: 10.1001/archpsyc.1983.01790010081010. [DOI] [PubMed] [Google Scholar]
- Fisher PD, Sperber EF, Moshe SL. Hippocampal sclerosis revisited. Brain Dev. 1998;20:563–573. doi: 10.1016/s0387-7604(98)00069-2. [DOI] [PubMed] [Google Scholar]
- French JA, Williamson PD, Thadani VM, Darcey TM, Mattson RH, Spencer SS, Spencer DD. Characteristics of medial temporal lobe epilepsy: I. Results of history and physical examination. Ann Neurol. 1993;34:774–780. doi: 10.1002/ana.410340604. [DOI] [PubMed] [Google Scholar]
- Gernert M, Thompson KW, Löscher W, Tobin AJ. Genetically engineered GABA-producing cells demonstrate anticonvulsant effects and long-term transgene expression when transplanted into the central piriform cortex of rats. Exp Neurol. 2002;176:183–192. doi: 10.1006/exnr.2002.7914. [DOI] [PubMed] [Google Scholar]
- Henderson KW, Gupta J, Tagliatela S, Litvina E, Zheng X, Van Zandt MA, Woods N, Grund E, Lin D, Royston S, Yanagawa Y, Aaron GB, Naegele JR. Long-term seizure suppression and optogenetic analyses of synaptic connectivity in epileptic mice with hippocampal grafts of GABAergic interneurons. J Neurosci. 2014;34:13492–13504. doi: 10.1523/JNEUROSCI.0005-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattiangady B, Bates A, Shin S, Shuai B, Rao X, Vemuri MC, Shetty AK. Society for Neuroscience meeting abstracts. Chicago, USA: 2015. hiPSC-derived NSC grafting intervention early after hippocampus injury preserves memory and mood function and reduces the occurrence of seizures. [Google Scholar]
- Hattiangady B, Kuruba R, Shetty AK. Acute seizures in old age leads to a greater Loss of CA1 pyramidal neurons, an increased propensity for developing chronic TLE and a severe cognitive dysfunction. Aging and Disease. 2011;2:1–17. [PMC free article] [PubMed] [Google Scholar]
- Hattiangady B, Liu Y, Shuai B, Yao L, Rao X, Zhang SC, Shetty AK. Society for Neuroscience Abstracts. 2013. Survival, differentiation and functional effects of human iPSC-derived MGE progenitor cells in a model of temporal lobe epilepsy. 414.18/C19. [Google Scholar]
- Hattiangady B, Mishra V, Kodali M, Shuai B, Rao X, Shetty AK. Object location and object recognition memory impairments, motivation deficits and depression in a model of Gulf war illness. Front Behav Neurosci. 2014;8:78. doi: 10.3389/fnbeh.2014.00078. eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattiangady B, Shetty AK. Decreased neuronal differentiation of newly generated cells underlies diminished neurogenesis in chronic temporal lobe epilepsy. Hippocampus. 2010;20:97–112. doi: 10.1002/hipo.20594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattiangady B, Rao MS, Shetty AK. Chronic temporal lobe epilepsy is associated with severely diminished dentate neurogenesis in the adult hippocampus. Neurobiol Dis. 2004;17:473–490. doi: 10.1016/j.nbd.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Rao MS, Shetty AK. Grafting of striatal precursor cells into hippocampus shortly after status epilepticus restrains temporal lobe epilepsy. Exp Neurol. 2008;212:468–481. doi: 10.1016/j.expneurol.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattiangady B, Shetty AK. Neural stem cell grafting counteracts hippocampal injury-mediated impairments in mood, memory and neurogenesis. Stem Cells Transl Med. 2012;1:696–708. doi: 10.5966/sctm.2012-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellier JL, Patrylo PR, Buckmaster PS, Dudek FE. Recurrent spontaneous motor seizures after repeated low-dose systemic treatment with kainate: Assessment of a rat model of temporal lobe epilepsy. Epilepsy Res. 1998;31:73–84. doi: 10.1016/s0920-1211(98)00017-5. [DOI] [PubMed] [Google Scholar]
- Hellier JL, Patrylo PR, Dou P, Nett M, Rose GM, Dudek FE. Assessment of inhibition and epileptiform activity in the septal dentate gyrus of freely behaving rats during the first week after kainate treatment. J Neurosci. 1999;19:10053–10064. doi: 10.1523/JNEUROSCI.19-22-10053.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstaedter C, Loer B, Wohlfahrt R, Hammen A, Saar J, Steinhoff BJ, Quiske A, Schulze-Bonhage A. The effects of cognitive rehabilitation on memory outcome after temporal lobe epilepsy surgery. Epilepsy Behav. 2008;12:402–409. doi: 10.1016/j.yebeh.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Hofman F. Immunohistochemistry. Curr Protoc Immunol. 2002:21.4.1–21.4.23. doi: 10.1002/0471142735.im2104s49. [DOI] [PubMed] [Google Scholar]
- Hunt RF, Girskis KM, Rubenstein JL, Alvarez-Buylla A, Baraban SC. GABA progenitors grafted into the adult epileptic brain control seizures and abnormal behavior. Nat Neurosci. 2013;16:692–697. doi: 10.1038/nn.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Yoon SY, Zhu L, Brodbeck J, Dai J, Walker D, Huang Y. Arf4 determines dentate gyrus mediated pattern separation by regulating dendritic spine development. PLoS One. 2012;7:e46340. doi: 10.1371/journal.pone.0046340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruba R, Hattiangady B, Shetty AK. Hippocampal neurogenesis and neural stem cells in chronic temporal lobe epilepsy. Epilepsy & Behavior. 2009;14(Suppl 1):65–73. doi: 10.1016/j.yebeh.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letty S, Lerner-Natoli M, Rondouin G. Differential impairments of spatial memory and social behavior in two models of limbic epilepsy. Epilepsia. 1995;36:973–982. doi: 10.1111/j.1528-1157.1995.tb00955.x. [DOI] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser MB, Moser EI. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science. 2007;315:961–966. doi: 10.1126/science.1135801. [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu H, Sauvey C, Yao L, Zarnowska ED, Zhang SC. Directed differentiation of forebrain GABA interneurons from human pluripotent stem cells. Nat Protoc. 2013a;8:1670–1679. doi: 10.1038/nprot.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DV. Losing neurons: Selective vulnerability and mesial temporal sclerosis. Epilepsia. 2005;46:39–44. doi: 10.1111/j.1528-1167.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- Litt B, Esteller R, Echauz J, D’Alessandro M, Shor R, Henry T, Pennell P, Epstein C, Bakay R, Dichter M, Vachtsevanos G. Epileptic seizures may begin hours in advance of clinical onset: A report of five patients. Neuron. 2001;30:51–64. doi: 10.1016/s0896-6273(01)00262-8. [DOI] [PubMed] [Google Scholar]
- Löscher W, Ebert U, Lehmann H, Rosenthal C, Nikkhah G. Seizure suppression in kindling epilepsy by grafts of fetal GABAergic neurons in rat substantia nigra. J Neurosci Res. 1998;51:196–209. doi: 10.1002/(SICI)1097-4547(19980115)51:2<196::AID-JNR8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Löscher W, Gernert M, Heinemann U. Cell and gene therapies in epilepsy—promising avenues or blind alleys? Trends Neurosci. 2008;31:62–73. doi: 10.1016/j.tins.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Manford M, Hart YM, Sander JW, Shorvon SD. National General Practice Study of Epilepsy (NGPSE): Partial seizure patterns in a general population. Neurology. 1992;42:1911–1917. doi: 10.1212/wnl.42.10.1911. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Pretorius JK, Babb TL. Influence of the type of initial precipitating injury and at what age it occurs on course and outcome in patients with temporal lobe seizures. J Neurosurg. 1995;82:220–227. doi: 10.3171/jns.1995.82.2.0220. [DOI] [PubMed] [Google Scholar]
- McAvoy K, Besnard A, Sahay A. Adult hippocampal neurogenesis and pattern separation in DG: a role for feedback inhibition in modulating sparseness to govern population-based coding. Front Syst Neurosci. 2015;9:120. doi: 10.3389/fnsys.2015.00120. eCollection 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO. Emerging insights into the genesis of epilepsy. Nature. 1999;399:A15–A22. doi: 10.1038/399a015. [DOI] [PubMed] [Google Scholar]
- Muscat R, Willner P. Suppression of sucrose drinking by chronic mild unpredictable stress: a methodological analysis. Neurosci Biobehav Rev. 1992;16:507–517. doi: 10.1016/s0149-7634(05)80192-7. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Cotman CW. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature. 1978;271:676–677. doi: 10.1038/271676a0. [DOI] [PubMed] [Google Scholar]
- Oomen CA, Bekinschtein P, Kent BA, Saksida LM, Bussey TJ. Adult hippocampal neurogenesis and its role in cognition. Wiley Interdiscip Rev Cogn Sci. 2014;5:573–587. doi: 10.1002/wcs.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; San Diego: 2004. [Google Scholar]
- Pirttilä TJ, Manninen A, Jutila L, Nissinen J, Kälviäinen R, Vapalahti M, Immonen A, Paljärvi L, Karkola K, Alafuzoff I, Mervaala E, Pitkänen A. Cystatin C expression is associated with granule cell dispersion in epilepsy. Ann Neurol. 2005;58:211–223. doi: 10.1002/ana.20545. [DOI] [PubMed] [Google Scholar]
- Pirttila TJ, Manninen A, Jutila L, Nissinen J, Kalviainen R, Vapalahti M, Immonen A, Paljarvi L, Karkola K, Alafuzoff I, Mervaala E, Pitkanen A. Cystatin C expression is associated with granule cell dispersion in epilepsy. Ann Neurol. 2005;58:211–223. doi: 10.1002/ana.20545. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Le Pichon M, Jalfre M. Depression: a new animal model sensitive to antidepressant treatments. Nature. 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- Powell TR, Fernandes C, Schalkwyk LC. Depression-related behavioral tests. Curr Protoc Mouse Biol. 2012;2:119–127. doi: 10.1002/9780470942390.mo110176. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rao MS, Shetty AK. Efficacy of doublecortin as a marker to analyze the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci. 2004;19:234–246. doi: 10.1111/j.0953-816x.2003.03123.x. [DOI] [PubMed] [Google Scholar]
- Rao MS, Hattiangady B, Shetty AK. Status Epilepticus during old age is not associated with increased hippocampal neurogenesis. Hippocampus. 2008;18:931–944. doi: 10.1002/hipo.20449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MS, Hattiangady B, Reddy DS, Shetty AK. Hippocampal neurodegeneration, spontaneous seizures, and mossy fiber sprouting in the F344 rat model of temporal lobe epilepsy. J Neurosci Res. 2006;83:1088–1105. doi: 10.1002/jnr.20802. [DOI] [PubMed] [Google Scholar]
- Rao MS, Hattiangady B, Rai KS, Shetty AK. Strategies for promoting anti-seizure effects of hippocampal fetal cells grafted into the hippocampus of rats exhibiting chronic temporal lobe epilepsy. Neurobiol Dis. 2007;27:117–132. doi: 10.1016/j.nbd.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond DE, Jr, Bjugstad KB, Teng YD, Ourednik V, Ourednik J, Wakeman DR, Parsons XH, Gonzalez R, Blanchard BC, Kim SU, Gu Z, Lipton SA, Markakis EA, Roth RH, Elsworth JD, Sladek JR, Jr, Sidman RL, Snyder EY. From the Cover: Behavioral improvement in a primate Parkinson’s model is associated with multiple homeostatic effects of human neural stem cells. Proc Natl Acad Sci USA. 2007;104:12175–12180. doi: 10.1073/pnas.0704091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribak CE, Hunt CA, Bakay RA, Oertel WH. A decrease in the number of GABAergic somata is associated with the preferential loss of GABAergic terminals at epileptic foci. Brain Res. 1986;363:78–90. doi: 10.1016/0006-8993(86)90660-8. [DOI] [PubMed] [Google Scholar]
- Samuels BA, Hen R. Neurogenesis and affective disorders. Eur J Neurosci. 2011;33:1152–1159. doi: 10.1111/j.1460-9568.2011.07614.x. [DOI] [PubMed] [Google Scholar]
- Sanberg PR. Neural stem cells for Parkinson’s disease: To protect and repair. Proc Natl Acad Sci USA. 2007;104:11869–11870. doi: 10.1073/pnas.0704704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK. Progress in Cell Grafting Therapy for Temporal Lobe Epilepsy. Neurotherapeutics. 2011;8:721–735. doi: 10.1007/s13311-011-0064-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK. Hippocampal injury-induced cognitive and mood dysfunction, altered neurogenesis, and epilepsy: Can early neural stem cell grafting intervention provide protection? Epilepsy & Behavior. 2014;38:117–124. doi: 10.1016/j.yebeh.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Bates A. Potential of GABA-ergic cell therapy for schizophrenia, neuropathic pain, and Alzheimer’s and Parkinson’s diseases. Brain Res. 2015 Sep 28; doi: 10.1016/j.brainres.2015.09.019. pii: S0006-8993 (15)00717-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Hattiangady B, Rao MS, Shuai B. Neurogenesis response of middle-aged hippocampus to acute seizure activity. PLoS One. 2012;7:e43286. doi: 10.1371/journal.pone.0043286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Development of fetal hippocampal grafts in intact and lesioned hippocampus. Prog Neurobiol. 1996;50:597–653. doi: 10.1016/s0301-0082(96)00048-2. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Aging impairs axonal sprouting response of dentate granule cells following target loss. J Comp Neurol. 1999;414:238–254. doi: 10.1002/(sici)1096-9861(19991115)414:2<238::aid-cne7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Fetal hippocampal CA3 cell transplants restore host hippocampal GAD-positive interneuron numbers in a rat model of TLE. J Neurosci. 2000;20:8788–8801. doi: 10.1523/JNEUROSCI.20-23-08788.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Glutamic acid decarboxylase positive hippocampal interneurons undergo a permanent reduction in number following kainic acid induced destruction of CA3 pyramidal neurons. Exp Neurol. 2001;176:276–297. doi: 10.1006/exnr.2001.7668. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Hattiangady B. Prospects of stem cell therapy for temporal lobe epilepsy. Stem Cells. 2007;25:2396–407. doi: 10.1634/stemcells.2007-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Madison RD, Turner DA. Quantitative graft integration of fetal hippocampal transplants labeled with 5′ bromodeoxyuridine into normal adult hippocampus. Exp Neurol. 1994;126:205–224. doi: 10.1006/exnr.1994.1059. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Hattiangady B, Rao MS. Vulnerability of hippocampal GABA-ergic interneurons to kainate induced excitotoxic injury during old age. J Cell Mol Med. 2009;13:2408–2423. doi: 10.1111/j.1582-4934.2008.00675.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA. Adult hippocampal neurogenesis buffers stress responses and depressive behavior. Nature. 2011;476:458–61. doi: 10.1038/nature10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperk G, Wieser R, Widmann R, Singer EA. Kainic acid induced seizures: Changes in somatostatin, substance P and neurotensin. Neuroscience. 1994;17:1117–1126. doi: 10.1016/0306-4522(86)90081-3. [DOI] [PubMed] [Google Scholar]
- Strine TW, Kobau R, Chapman DP, Thurman DJ, Price P, Balluz LS. Psychological distress, comorbidities, and health behaviors among U.S. adults with seizures: Results from the 2002 National Health Interview Survey. Epilepsia. 2005;46:1133–1139. doi: 10.1111/j.1528-1167.2005.01605.x. [DOI] [PubMed] [Google Scholar]
- Thompson KW. Genetically engineered cells with regulatable GABA production can affect afterdischarges and behavioral seizures after transplantation into the dentate gyrus. Neuroscience. 2005;133:1029–1037. doi: 10.1016/j.neuroscience.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Turner DA, Shetty AK. Clinical prospects for neural grafting therapy for hippocampal lesions and epilepsy. Neurosurgery. 2003;52:632–644. doi: 10.1227/01.neu.0000047825.91205.e6. [DOI] [PubMed] [Google Scholar]
- Vannest J, Szaflarski JP, Privitera MD, Schefft BK, Holland SK. Medial temporal fMRI activation reflects memory lateralization and memory performance in patients with epilepsy. Epilepsy Behav. 2008;12:410–418. doi: 10.1016/j.yebeh.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Waldau B, Hattiangady B, Kuruba R, Shetty AK. Medial Ganglionic eminence-derived neural stem cell grafts ease spontaneous seizures and restore GDNF expression in a rat model of chronic temporal lobe epilepsy. Stem Cells. 2010;28:1153–1164. doi: 10.1002/stem.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner P, Muscat R, Papp M. Chronic mild stress-induced anhedonia: A realistic animal model of depression. Neurosci Biobehav Rev. 1993;16:525–534. doi: 10.1016/s0149-7634(05)80194-0. [DOI] [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE. Excitatory synaptic input to granule cells increases with time after kainate treatment. J Neurophysiol. 2001;85:1067–1077. doi: 10.1152/jn.2001.85.3.1067. [DOI] [PubMed] [Google Scholar]
- Yassa MA, Stark CE. Pattern separation in the hippocampus. Trends Neurosci. 2011;34:515–525. doi: 10.1016/j.tins.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.