

Abstract
Inflammation is a central pathophysiologic mechanism that contributes to diabetes mellitus and diabetic nephropathy. Recently, we showed that macrophages directly contribute to diabetic renal injury, and that pharmacological blockade or genetic deficiency of chemokine (C-C motif) receptor 2 (CCR2) confers kidney protection in diabetic nephropathy. However, the direct role of CCR2 in kidney-derived cells such as podocytes in diabetic nephropathy remains unclear. To study this, we developed a transgenic mouse model expressing CCR2 specifically in podocytes (Tg(NPHS2-Ccr2)) on a nephropathy prone (DBA/2J) and CCR2 deficient (Ccr2−/−) background with heterozygous Ccr2+/− littermate controls. Diabetes was induced by streptozotocin. As expected, absence of CCR2 conferred kidney protection after nine weeks of diabetes. In contrast, transgenic CCR2 over expression in the podocytes of Ccr2−/− mice resulted in significantly increased albuminuria, blood urea nitrogen, histopathologic changes, kidney fibronectin and type-1 collagen expression, podocyte loss, and glomerular apoptosis after nine weeks of streptozotocin-induced diabetes. Interestingly, there was no concurrent increase in kidney macrophage recruitment or inflammatory cytokine levels in the mice. These findings support a direct role for CCR2 expression in podocytes to mediate diabetic renal injury, independent of monocyte/macrophage recruitment. Thus, targeting the CCR2 signaling cascade in podocytes could be a novel therapeutic approach for treatment of diabetic nephropathy.
Keywords: Podocytes, CCR2, Diabetic Nephropathy
Introduction
Diabetes mellitus (DM) is a global health problem due to its high prevalence and associated risk of end-stage-renal disease (ESRD), accounting for nearly half of incident ESRD cases.1,2 Over the last decade, the incidence of ESRD due to diabetes has doubled. Current therapy, including blood pressure and glucose control and other life style changes, has been only modestly successful in delaying the progression of renal failure. Thus, identifying the molecular mechanisms in the development of diabetic kidney disease may have substantial impact on morbidity and mortality of the diabetic population.
Several lines of evidence have incriminated monocytes/macrophages as a mediator of DN. Specifically, monocytes and/or macrophages and their adherence to endothelial cells and overexpression of proinflammatory cytokines and chemokines has been shown to contribute to the pathogenesis of DN.3–7 Furthermore, in humans, glomerular macrophage accumulation occurs in DN8–10 and correlates strongly with the progression of renal impairment.10 Infiltrating macrophages release lysosomal enzymes, nitric oxide, reactive oxygen species, transforming growth factor-beta (TGF-β), vascular endothelial growth factor and cytokines such as TNF-α, interleukin (IL)-1 and interferon (IFN)-γ,4 which could play pivotal roles in the development and progression of DN. Our previous work11 provides evidence that macrophages directly contribute to kidney injury in DN; perhaps by altering podocyte integrity through the pro-inflammatory M1 subset of macrophages. Furthermore, we recently showed that macrophage-derived TNF-α plays a major role in diabetic renal injury and that blocking TNF-α could be a novel therapeutic approach for treatment of DN.12
Chemokine (C-C motif) receptor 2 (CCR2) and its ligands such as MCP-1 and MCP-5 regulate monocyte/macrophage migration into injured tissues.13,14 CCR2 deficient (Ccr2−/−) and MCP-1 deficient (Ccl2−/−) mice are protected from monocyte/macrophage induced injury in several disease models, including DN.15–19 We have demonstrated previously that CCR2 is necessary for monocyte/macrophage-induced diabetic renal injury in mice.7 Our previous publication suggests that activation of CCR2 may alter podocyte integrity and function, thereby contributing to the glomerular permeability defects associated with DN.7 Unfortunately, all published data have focused on the role of CCR2 in macrophage recruitment. However, CCR2 is also expressed on other kidney cell populations, such as podocytes.20,21 For instance, CCR2 inhibition ameliorated high glucose-induced podocyte apoptosis.20 These findings suggested a possible direct role of CCR2 in the development and progression of DN via expression in podocytes. Therefore, we hypothesize that expression of CCR2 specifically in podocytes will exacerbate renal tissue injury in diabetes. Towards this goal, we have developed a transgenic mouse that expresses CCR2 in podocytes.
Our results confirm our previous report7 that global CCR2 deficiency confers renal tissue protection in DN. Conversely, rescue expression of CCR2 in podocytes exacerbates diabetic renal injury independent of monocyte/macrophage recruitment. Targeting the CCR2 signaling cascade in podocytes could be a novel therapeutic approach for treatment of DN.
Results
Diabetes induces CCR2 expression in mouse podocytes
Our previous publication demonstrated that CCR2 deletion protects kidneys from diabetic injury in a mouse model of type-1 diabetes.22 Additionally, CCR2 is overexpressed in podocytes in human DN patients.23 Here we show that high glucose (HG) media significantly increased CCR2 mRNA expression in immortalized murine podocytes (kindly provided by Dr. John R. Sedor) compared to normal glucose (NG) media (Figure 1A). Similarly, isolated glomeruli derived from diabetic mice are associated with increased CCR2 mRNA expression compared to control (Figure 1B).
Figure 1. Diabetes induces CCR2 expression in podocytes.

A: Immortalized murine podocytes were cultured and treated with high glucose for 10 days. Cells were harvested and RNA was extracted to measure CCR2 mRNA expression. B: Glomeruli were isolated from DBA mice at week 9 after STZ injection. RNA was extracted from isolated glomeruli for the measurement of CCR2 mRNA expression. Data are presented as mean ± SEM. *p<0.05 compared to control.
Generation and characterization of podocin2.5-CCR2 transgenic mice
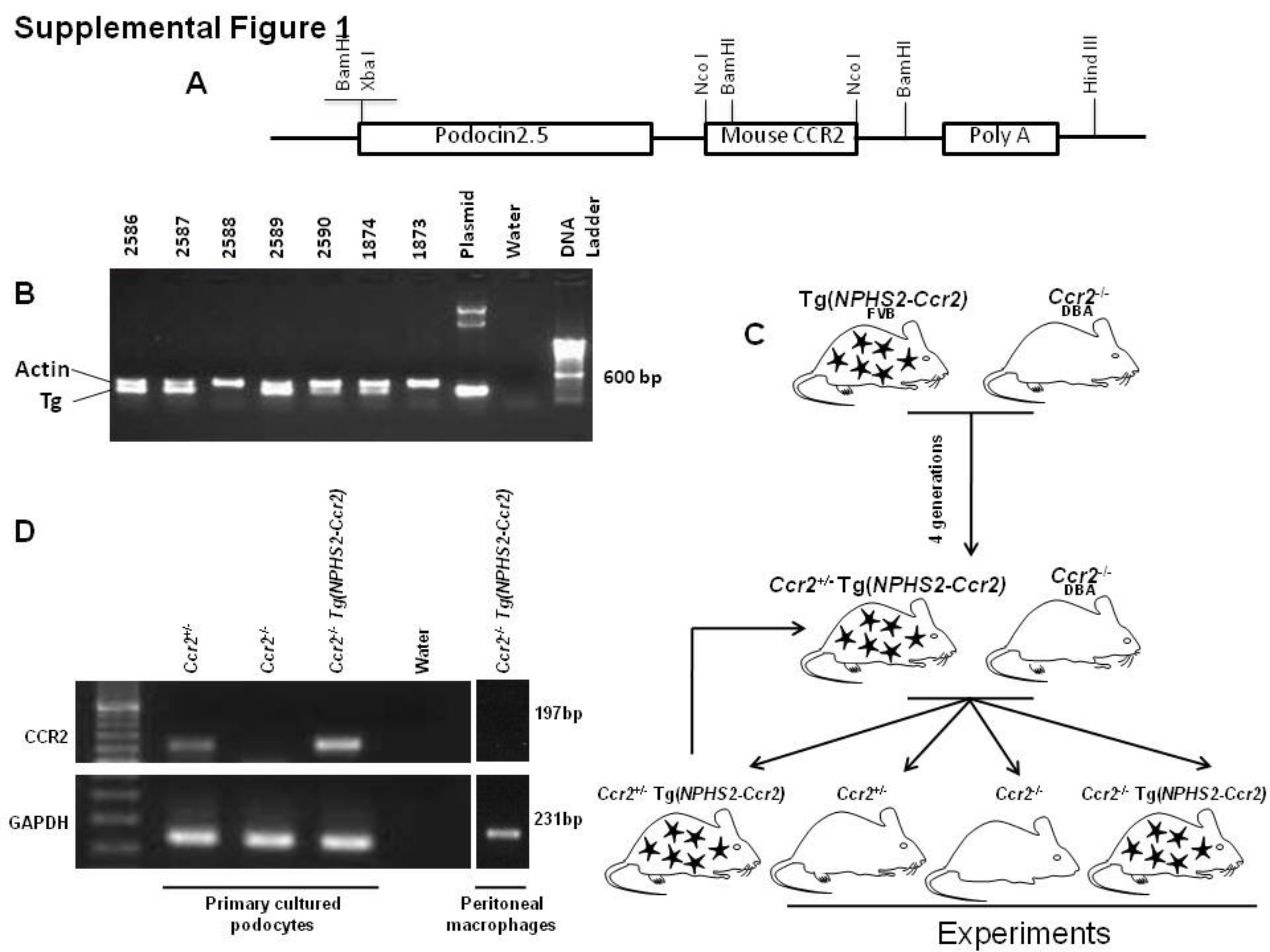
As described in “Materials and Methods” section, we created transgenic mice in which podocytes overexpress mouse CCR2 driven by the human podocin (NPHS2) promoter (Supplemental Figure 1A). Five founders were obtained and identified by PCR amplification of a transgene-specific assay (Supplemental Figure 1B). CCR2 expression in podocytes was verified by RT-PCR of RNA isolated from primary cultures of podocytes isolated from WT and transgenic mice (not shown). Founder mice on the FVB/N background were mated with Ccr2−/− mice on the DBA/2J background (D2;B6-Ccr2tm1Ifc/J) for 4 generations. DBA/2J mice are recommended by the Animal Models of Diabetes Complications Consortium (AMDCC) as a nephropathy prone and an optimal model of DN.24,25 CCR2 transgenic CCR2 heterozygous (Ccr2+/− Tg(NPHS2-Ccr2) mice were then bred with CCR2 deficient (Ccr2−/−) mice to yield the F5 generation. Male Ccr2+/− (positive control), Ccr2−/− (negative control) and Ccr2−/− Tg(NPHS2-Ccr2) mice were used in our experiments (Supplemental Figure 1C). As shown in supplemental Figure 1D, CCR2 mRNA was expressed in podocytes from the transgenic mice, but not CCR2 deficient mice. Importantly, CCR2 mRNA was not expressed in peritoneal macrophages from the transgenic mice.
Effect of CCR2 overexpression in podocytes on general characteristics of diabetic mice
Diabetes was induced by STZ injection as described in “Materials and Methods” section in male Ccr2+/−, Ccr2−/− and Ccr2−/− Tg(NPHS2-Ccr2) mice (Supplemental Figure 1C), and was confirmed by non-fasted blood glucose levels. As shown in table 1, diabetic mice had significantly increased blood glucose levels, kidney weight to body weight ratio, and significantly decreased body weight compared to control mice after 9 weeks of diabetes. There were no significant changes in systolic blood pressure in control or diabetic mice. The blood glucose levels, kidney-to-body weight ratio and body weights were similar in diabetic mice regardless of the genetic background. However, diabetes significantly increased average glomerular size/area in Ccr2+/− and Ccr2−/− Tg(NPHS2-Ccr2); but not in Ccr2−/− mice compared to control mice.
Table 1.
Effect of podocin2.5-CCR2 expression after 9 weeks of STZ-induced diabetes.
| Genotype | CCR2+/− | CCR2−/− | CCR2−/− Tg(NPHS2-Ccr2) | |||
|---|---|---|---|---|---|---|
| Treatment | Control | Diabetes | Control | Diabetes | Control | Diabetes |
| Mice number | 11 | 11 | 8 | 10 | 11 | 13 |
| Body Weight (g) | 30.69±0.71 | 21.61±0.84a | 29.19±0.98 | 21.43±1.02a | 29.28±0.63 | 19.84±0.79a |
| Blood glucose (mg/dL) | 144.7±4.98 | 499.1±0.25a | 149.6±7.85 | 483.6±15.5a | 150.1±4.14 | 497.7±1.40a |
| SBP (mmHg) | 111.4±2.4 | 118.3±2.3 | 121.1±3.9 | 117.5±3.0 | 114.2±2.8 | 116.8±2.2 |
| KW/BW (g/100 g BW) | 0.84±0.04 | 1.18±0.05a | 0.95±0.03 | 1.11±0.05b | 0.86±0.03 | 1.16±0.04a |
| Average Glomerular Size (µm2) | 20211±89 | 2305±81b | 2227±86 | 2350±68 | 2103±90 | 2595±103a |
| BUN (mg/dL) | 23.60±0.89 | 32.24±1.88a | 19.79±0.88 | 25.95±1.38a,c | 20.65±0.88 | 32.21±1.10a |
| Creatinine Clearance (ml/min) | 0.12±0.04 | 0.05±0.02 | 0.12±0.08 | 0.11±0.03 | 0.12±0.05 | 0.04±0.02 |
Data are mean ± SEM.
: p<0.01 compared to control;
: p<0.05 compared to control;
: p<0.05 compared to diabetic Ccr2+/− and diabetic Ccr2−/− Tg(NPHS2-Ccr2) groups.
SBP: systolic blood pressure, KW/BW: kidney weight/body weight.
CCR2 expression in podocytes increases albuminuria and blood urea nitrogen (BUN) in diabetic mice
To determine the role of podocyte-specific CCR2 in diabetic renal function, 24-hour UAER and BUN were determined after 9 weeks of diabetes as indicators of diabetic kidney injury. Diabetes significantly increased UAER (Figure 2) and BUN (Table 1) in Ccr2+/− mice. In contrast, CCR2 deficiency (Ccr2−/−) significantly reduced albuminuria and BUN compared to diabetic Ccr2+/− mice; similar to our previous findings.7 Conversely, podocyte-specific expression of CCR2 (Ccr2−/− Tg(NPHS2-Ccr2)) significantly increased albuminuria and BUN to a level that was not significantly different from diabetic Ccr2+/− mice. Furthermore, there was a trend towards reduced creatinine clearance in diabetic Ccr2+/− and diabetic Ccr2−/− Tg(NPHS2-Ccr2) compared to control mice; although data was not significant (Table 1).
Figure 2. Effect of podocin2.5-CCR2 expression on renal function in diabetic mice.

Mouse 24 hour urine was collected at week 9 after STZ-induced diabetes for measurement of urinary albumin excretion (UAER). Data are presented as mean ± SEM. Open bar, control groups; black-filled bar, diabetic groups. *p<0.01 compared to control. #p<0.05 compared to diabetic Ccr2−/− mice.
CCR2 expression in podocytes increases renal histological changes in diabetic mice
PAS staining of kidney sections (Figure 3) revealed increased glomerular cellularity and mesangial expansion in diabetic Ccr2+/− mice compared to control Ccr2+/− mice after 9 weeks of diabetes (Score: 0.83±0.06 vs. 0.67±0.04; p<0.05). There was no significant difference between diabetic and control Ccr2−/− mice (Score: 0.75±0.02 vs. 0.71±0.03). In contrast, diabetic Ccr2−/− Tg(NPHS2-Ccr2) mice had significantly increased glomerular cellularity and mesangial expansion compared to control Ccr2−/− Tg(NPHS2-Ccr2) mice (0.95±0.05 vs. 0.66±0.03, p<0.01).
Figure 3. Effect of podocin2.5-CCR2 expression on histological changes in diabetic mice.

Sections were stained with PAS and all glomeruli were graded individually at 400× magnification after 9 weeks of STZ-induced diabetes. Images were taken with 100× (oil) objective with a total magnification of 1000×. Scale bar: 10 µm.
Masson's trichrome staining of kidney sections (Figure 4) was also used to evaluate glomerular fibrosis. Diabetes significantly induced glomerular fibrosis in Ccr2+/− mice compared to control Ccr2+/− mice after 9 weeks of diabetes (Score: 0.18±0.03 vs. 0.08±0.02; p<0.05). There was no significant difference between diabetic and control Ccr2−/− mice (Score: 0.09±0.02 vs. 0.06±0.01). In contrast, diabetic Ccr2−/− Tg(NPHS2- Ccr2) mice exhibited significantly increased glomerular fibrosis compared to control Ccr2−/− Tg(NPHS2-Ccr2) mice (Score: 0.26±0.04 vs. 0.09±0.02; p<0.01). Additionally, Masson's trichrome staining did not show any significant differences in tubulointerstitial fibrosis between control and diabetic mice regardless of genetic background (Data not shown).
Figure 4. Effect of podocin2.5-CCR2 expression on glomerular fibrosis in diabetic mice.

Sections were stained with Masson's trichrome and all glomeruli were graded individually at 400× magnification after 9 weeks of STZ-induced diabetes. Images were taken with 100× (oil) objective with a total magnification of 1000×. Scale bar: 10 µm.
CCR2 expression in podocytes increases renal fibronectin and type-1 collagen mRNA expression in diabetic mice
Fibronectin and type-1 collagen are biomarkers of tissue fibrosis. Our results show that both kidney fibronectin (Figure 5A) and type-1 collagen (Figure 5B) mRNA expression were significantly increased in diabetic Ccr2+/− mice, an effect significantly reduced in diabetic Ccr2−/− mice. In contrast, podocyte-specific CCR2 expression in diabetic Ccr2−/− Tg(NPHS2-Ccr2) mice significantly increased kidney fibronectin and type-1 collagen mRNA expression compared to control Ccr2−/− Tg(NPHS2-Ccr2) mice.
Figure 5. Effect of podocin2.5-CCR2 expression on kidney fibronectin and type-1 collagen mRNA expression in diabetic mice.

Quantitative RT-PCR was performed on whole mouse kidney total RNA after 9 weeks following diabetes. Fibronectin (A), and type-1 collagen (B) mRNA expression were normalized with GAPDH mRNA. Open bar, control groups; black-filled bar, diabetic groups. Results are means ± SEM. *p<0.05, **p<0.01 compared to control. #p<0.05, ##p<0.01 compared to diabetic Ccr2−/− mice.
CCR2 expression in podocytes does not increase renal macrophage infiltration in diabetic mice
Since CCR2 induces monocyte/macrophage migration into diabetic kidneys, we questioned whether podocyte-specific CCR2 expression would increase monocyte/macrophage recruitment in diabetic kidneys. Using immunohistochemistry for Mac-2 to detect glomerular macrophages (Figure 6), we found a significant increase in glomerular macrophages in diabetic Ccr2+/− mice, an effect significantly reduced in diabetic Ccr2−/− mice. Interestingly, podocyte-specific CCR2 expression in diabetic Ccr2−/− Tg(NPHS2-Ccr2) mice did not increase glomerular macrophage recruitment and was similar to diabetic Ccr2−/− mice.
Figure 6. Effect of podocin2.5-CCR2 expression on glomerular macrophage infiltration in diabetic mice.

(A) Mac-2-positive macrophages in glomeruli were identified by immunohistochemical staining at 9 weeks following diabetes. B) Summary data for macrophages/glomerulus. Glomerular macrophages were counted in a double-blinded manner. Open bar, control groups; black-filled bar, diabetic groups. Results are means ± SEM. *p<0.05, **p<0.001 compared to control. #p<0.01 compared to diabetic Ccr2−/− and diabetic Ccr2−/− Tg(NPHS2-Ccr2) mice.
These results were confirmed by flow cytometry of total kidney macrophage content (Figure 7). In control mice, both Ccr2−/− and Ccr2−/− Tg(NPHS2-Ccr2) mice had significantly fewer kidney macrophages compared to control Ccr2+/− mice (p<0.01). As expected, diabetes increases kidney macrophage content in diabetic Ccr2+/− mice (p<0.05) compared to control Ccr2+/− mice. However, diabetes was not associated with increased kidney macrophage contents in diabetic Ccr2−/− or Ccr2−/− Tg(NPHS2-Ccr2) mice.
Figure 7. Effect of podocin2.5-CCR2 expression on total kidney macrophage infiltration in diabetic mice.

(A) Kidneys were harvested at 9 weeks following diabetes and processed for fluorescence-activated cell sorting (FACS) analysis as described in the method section. Macrophages were identified as CD11bhighF4/80low. Graphs show representative density contour plots. B) Summary data for macrophages/gram kidney tissues. Open bar, control groups; black-filled bar, diabetic groups. Results are means ± SEM. *p<0.05 compared to control. #p<0.001 compared to diabetic Ccr2−/− and diabetic Ccr2−/− Tg(NPHS2-Ccr2) mice.
CCR2 expression in podocytes does not increase inflammatory cytokines in diabetic mice
Increased proinflammatory cytokines and chemokines contribute to the pathogenesis of DN.3–7 We previously demonstrated that TNF-α produced by macrophages mediates diabetic renal injury.12 Thus, we questioned whether podocyte-specific CCR2 expression would increase proinflammatory cytokines and chemokines in the diabetic kidneys. Our data support our previous results of kidney macrophage recruitment (Figures 6–7) and indicate that podocyte-specific CCR2 expression in diabetic Ccr2−/− Tg(NPHS2-Ccr2) mice did not increase kidney TNF-α (Figure 8A), or NOS-2 (Figure 8B) mRNA expression after 9 weeks of diabetes. Furthermore, podocyte-specific CCR2 expression in diabetic Ccr2−/− Tg(NPHS2-Ccr2) mice did not increase kidney TNF-α (Figure 8C) and IL-2 (Figure 8D) protein expression after 9 weeks of diabetes. There were no significant differences in IL-4 or IL-1β in all groups regardless of genetic background (Data not shown). We also evaluated CCL2, CCL7 and CCL12 mRNA expression as ligands for CCR2 receptor. Our data showed that diabetes up-regulates CCL2, CCL7 and CCL12 mRNA expression regardless of genetic background (Supplemental Figure 2).
Figure 8. Effect of podocin2.5-CCR2 expression on kidney inflammatory cytokines in diabetic mice.

Quantitative RT-PCR was performed on whole mouse kidney total RNA after 9 weeks following diabetes. TNF-α (A), and NOS2 (B) mRNA expression was normalized with GAPDH mRNA. MSD multi-spot assay system was performed to measure TNF-α (C), and IL-2 (D) protein expression in kidney tissues. Open bar, control groups; black-filled bar, diabetic groups. Results are means ± SEM. *p<0.05 compared to control.
CCR2 expression in podocytes induces podocyte depletion
Our data demonstrated that podocyte-specific CCR2 expression increases albuminuria, which could be implicated in podocyte injury/loss. Additionally, podocyte loss is associated DN.26 Our data shows that diabetes is associated with a significant reduction in podocyte number (Figure 9A–B) in Ccr2+/− mice; an effect significantly restored in diabetic Ccr2−/− mice. In contrast, podocyte-specific CCR2 expression in diabetic Ccr2−/− Tg(NPHS2-Ccr2) mice significantly reduced podocyte number compared to control Ccr2−/− Tg(NPHS2-Ccr2) mice.
Figure 9. Effect of podocin2.5-CCR2 expression on podocyte number and appoptosis in diabetic mice.

(A) Wt-1-positive podocytes were identified by immunohistochemical staining at 9 weeks following diabetes. B) Summary data for Wt-1 positive podocytes/10 glomerular sections. Podocyte numbers were counted in a double-blinded manner. C) Summary data for glomerular apoptotic cell numbers using anti cleaved caspase 3/100 glomerular sections. Cleaved caspase 3 positive cells were counted in a double-blinded manner. Open bar, control groups; black-filled bar, diabetic groups. Results are means ± SEM. *p<0.05 compared to control.
CCR2 expression in podocytes induces apoptosis in the diabetic glomeruli
A previous report demonstrated that CCR2 is involved in podocyte apoptosis under diabetic conditions.20 Our current data confirms a significant increases in apoptosis using cleaved caspase 3 in diabetic Ccr2+/− and Ccr2−/− Tg(NPHS2-Ccr2) mice (Figure 9C).
Discussion
Immunologic and inflammatory mechanisms play a cardinal role in the development and progression of DN.27,28 Specifically, monocyte/macrophage recruitment correlates strongly with the progression of renal impairment in DN.3–6 CCR2 receptors are required for monocyte emigration from the bone-marrow (BM), such that Ccr2−/− mice have fewer circulating monocytes.14 Data from our laboratory7 and others29,30 showed that CCR2 inhibitors can block the development of DN in association with a reduction in kidney macrophage infiltration in both type 1 and type 2 diabetic mice. Recently, a multicenter randomized double-blind, placebo-controlled clinical trial was designed to investigate the potential efficacy of CCR2 inhibition in type 2 diabetic patients with nephropathy.31 Patients were followed for 52 weeks, and the results showed renoprotective effects of CCR2 inhibition on top of the current standard of care. Recruitment of monocytes is considered the predominant mechanism by which CCR2 mediates renal tissue damage. However, in addition to being expressed on monocyte subsets,14 CCR2 is also expressed in other cell types, both in vitro32–35 and in vivo.36–38 Taken together, these data suggest a role for CCR2 in cells other than monocytes, such as podocytes. Previous reports confirmed the expression and regulation of CCR2 in podocytes in vitro.20,21,39 The present studies address the direct role of CCR2 expression on podocytes in vivo in DN.
In a murine model of type 1 DM, we confirmed that CCR2 is expressed in podocytes and contributes to diabetic renal injury, independent of kidney monocyte/macrophage recruitment. These results clearly establish a new role for CCR2 in the pathogenesis of DN.
Previous studies have identified a role for CCR2-induced injury in atherosclerosis,15 experimental autoimmune encephalitis,16 bronchiolitis obliterans syndrome,17 hepatotoxicity,18 and ureteral obstruction;19 mainly via monocyte/macrophage infiltration. In these instances, inhibition of CCR2 may serve to block a variety of other inflammatory mediators and account for its beneficial effect in DN. CCR2 is a receptor for multiple ligands, such as CCL2, CCL7, and CCL12. The binding of these ligands to CCR2 may facilitate macrophage migration and infiltration, and act to maintain monocyte homeostasis.13 All these ligands are able to induce monocyte migration through CCR2,13 indicating that targeting CCR2 is potentially more efficient than targeting multiple CCR2 ligands. In the current study, we show that diabetes up-regulates CCL2, CCL7 and CCL12 mRNA expression regardless of genetic background (Supplemental Figure 2). These results provide a possible direct effect of CCR2 in DN independent of its ligands.
Podocytes are important components of the glomerular filtration barrier. They are anchored to the glomerular basement membrane and play a pivotal role in the pathophysiology of DN. In humans, an increase in podocyte foot process width and a reduced podocyte number and density correlate directly with increased urinary albumin excretion rate in both type 1 and type 2 diabetes.40,41 Furthermore, podocyte expression of B7-142 and MCP-143 have been clearly established; suggesting a possible role for podocytes as an immune cells.
To address the possibility that podocyte-specific CCR2 expression might contribute to the pathogenesis of DN, we generated a transgenic mouse model that expresses CCR2 specifically in podocytes. To determine whether podocyte-restricted CCR2 suffices to render Ccr2−/− mice more susceptible to DN, transgenic mice were crossed to the Ccr2−/− background and diabetes was induced via STZ. Our study shows that expression of CCR2 exclusively in podocytes mediates renal tissue injury as evidenced by an increase in albuminuria, BUN, average glomerular size/area, histological changes and kidney fibronectin and type-1 collagen expression.
We also questioned whether the effect of podocyte-specific CCR2 expression to induce renal tissue injury is mediated via increased monocyte/macrophage recruitment. Interestingly, our results showed that podocyte-specific CCR2 expression did not affect renal macrophage infiltration. These data were confirmed by lack of effect of podocyte-specific CCR2 expression to induce kidney inflammatory cytokines; such as TNF-α, Nos2, and IL-2.
The mechanism by which podocyte-specific CCR2 expression induces diabetic kidney damage is not completely clear at this time; but could be mediated via podocyte loss and apoptosis. CCR2 has direct effects on podocyte function. For example, in cultured human podocytes, CCL2/CCR2 binding was able to induce dose-dependent migration of podocytes; an effect blocked by CCR2 inhibition.21 In addition, CCR2 inhibition ameliorated high glucose-induced podocyte apoptosis.20 In mesangial cells, CCR2 induced fibronectin and collagen expression in response to high glucose treatment.44 We saw an increase in fibronectin and collagen in the Ccr2−/− Tg(NPHS2-Ccr2) mice, suggesting that CCR2 stimulation of podocytes may also result in production of fibrosis. In a cultured immortalized murine podocyte model, high-glucose treatment induces CCL2, TGF β1 expression and apoptosis of podocytes. High glucose-induced podocyte apoptosis was inhibited by CCL2/CCR2 and TGFβ1 inhibition without affecting podocyte proliferation; similar data were obtained in a C57BLKS-Leprdb mouse model.20 These findings indicated that activation of the CCL2/CCR2 pathway contributes to not only monocyte/macrophage migration/infiltration, but multiple downstream bio-chemical functions in the different glomerular compartments including podocytes. In our study, CCR2 overexpression induced podocyte loss as evidenced by Wt-1 immuno-staining. In addition, CCR2 overexpression in podocytes increased glomerular cell apoptosis. Therefore, we speculate that the effect of CCR2 overexpression in DN is mediated via podocyte loss and apoptosis.
In summary, we describe a new transgenic mouse model that express CCR2 exclusively in podocytes. We provide strong evidence that podocyte-specific CCR2 expression promotes the pathogenesis of DN, independent of monocyte/macrophage recruitment; possibly via its effect on podocyte loss and apoptosis. Targeting CCR2 signaling could constitute a novel therapeutic approach for diabetic kidney disease.
Materials and Methods
Generation of podocin2.5-CCR2 transgenic mice
We generated a new transgenic mouse model in which murine CCR2 is expressed exclusively in podocytes. Briefly, a 2.5-kb fragment derived from the human podocin gene (NPHS2), in plasmid p2.5P-nlacF, was received from Dr. L. Holzman.45 The mouse Ccr2 full-length cDNA was cloned by PCR amplification using kidney RNA template provided by Thermo Scientific (Waltham, MA). To facilitate introduction into p2.5P, an NcoI site was introduced to both ends of the cDNA as described previously.45 The reporter gene was excised from p2.5P-nlacF and the mouse CCR2 full-length cDNA fragment was inserted. Correct sequences were verified. The podocin2.5-CCR2 construct was sent to Cyagen Biosciences (Santa Clara, CA) for micro-injection to create podocin2.5-CCR2 transgenic mice on the FVB/NCrl background.
Transgene detection and mouse characterization
A PCR assay was used to identify the transgene. The following primers were used in the PCR assay. Transgene: forward 5’ TATCCTCCTTAGCCCATGCAA 3’; reverse 5’ ACATGATGGCAGAAATAATGCTC 3’, product size is 273 bp. Internal control actin: forward 5’ ACTCCAAGGCCACTTATCACC 3’; reverse 5’ ATTGTTACCAACTGGGACGACA 3’, product size is 413 bp. To determine if the CCR2 mRNA was expressed in podocytes, mouse primary podocytes were isolated from mouse kidneys as previously described.46 Briefly, kidney cortex was dissected free and cut into very small pieces in culture medium. The tissue suspension was then passed through a 250 µm-pore screen followed by a 75 µm-pore screen; and glomeruli enriched tissues were retained. Tissue fragments were collected and cultured in RPMI 1640 medium supplemented with 10% FBS for 7–10 days. RNA was isolated from primary cultured podocytes; samples were incubated with DNase-I (Ambion; Grand Island, NY) to remove contaminated genomic DNA. RT-PCR was performed to detect CCR2 mRNA expression using the following primers: CCR2: Forward: 5’ GTG ATT GAC AAG CAC TTA GAC 3’; Reverse: 5’ ACT CGA TCT GCT GTC TCC 3’, product size was 197 bp. GAPDH: Forward: 5’ ACG GCA AAT TCA ACG GCA CAG TCA 3’; Reverse: 5’ TGG GGG CAT CGG CAG AAG G 3’, product size was 231 bp. CCR2 deletion was conducted using genotyping PCR assay according to the protocol provided by The Jackson Laboratory (mouse stock #: 004999).
Generation of a diabetic mouse model
All animal experiments were approved by the Institutional Animal Care and Use Committee of Penn State University College of Medicine. Founder mice were mated with Ccr2−/− mice on the DBA/2J background; generated in our laboratory after breeding Ccr2−/− mice (B6.129S4-Ccr2tm1Ifc/J Jackson laboratory, stock # 004999) with DBA/2J for 7 generations. DBA/2J mice are recommended by the Animal Models of Diabetes Complications Consortium (AMDCC) as a nephropathy prone and an optimal model of DN.24,25 The Ccr2+/− Tg(NPHS2-Ccr2 (transgenic mice on a heterozygous Ccr2+/− background) were mated with Ccr2−/− mice. The male Ccr2+/−, Ccr2−/− and Ccr2−/− Tg(NPHS2-Ccr2) mice were used in all subsequent experiments. Type 1 diabetes was induced by multiple low doses of streptozocin (STZ; Sigma, St. Louis, MO; 50 mg/kg body weight dissolved in lactated Ringers solution intraperitoneally for 5 consecutive days) as recommended by AMDCC24,25 at 6 weeks of age. Diabetes was confirmed one week later by measurement of blood glucose level. Mice with blood glucose levels > 350 mg/dl were considered diabetic. Mice were provided ad lib access to food and water. Urine collections were obtained by housing mice in metabolic cages. At the end of experiments (9 weeks after diabetes), mice were euthanized and kidney and blood samples were collected.
Analytical methodology
As described previously,11,12,47 urine albumin was measured by ELISA using an Albuwell M kit (Exocell, Philadelphia, PA); Blood urea nitrogen (BUN) was determined using QuantiChrom Urea Assay Kit (BioAssay Systems cat#DIUR-500), blood glucose was measured using a ReliOn micro blood glucose monitor (Bentonville, PA). Mouse systolic blood pressure was determined using the Coda blood pressure system (Kent Scientific Corp, Torrington, Connecticut) as previously described.48,49 Mice were habituated to the blood pressure measurement for 5 days before the day of the experiment and then were allowed to rest quietly for 15 minutes at room temperature. All measurements were performed at the same time for all groups to avoid any diurnal variations.
Flow cytometry
Kidney macrophages were identified by fluorescence-activated cell sorting in a LSR-II flow cytometer (BD, Franklin Lakes, NJ) as previously described.11,22 In brief, kidney tissues were minced, digested and then passed through a 40 µm mesh. Samples were pre-incubated with CD16/32 (2.4G) to block non-specific FcR binding site and 7-AAD (Invitrogen, Carlsbad, CA) to exclude dead cells. Kidney macrophages were defined as CD45+CD11bhigh/F4/80low. Counting beads (Product #: PCB100, Invitrogen) were used in the experiments to calculate the total number of CD45+ cell per gram of kidney tissue. Data were analyzed using FlowJo software 9.6.2 (Tree Star, Ashland, OR). All the antibodies were provided by eBioscience.
Histology and immunohistochemistry
Mouse kidney tissues were fixed in 4% neutral buffered formalin and embedded in paraffin. Periodic acid Schiff (PAS) or Masson's trichrome staining was performed on 3 µm kidney tissue sections. All glomeruli (between 50–110 glomeruli) in a single transverse section for each mouse were scored individually at 400× in a blinded manner by a pathologist and then averaged as previously described.50–53 Images were captured with an Olympus BX51 microscope and DP71 digital camera using cellSens Standard 1.12 image software (Olympus America, Center Valley, PA). Semiquantitative scores (0–4+) were assigned based on the masked readings as previously described.11,47,54
Immunohistochemistry was performed using rat anti-mouse Mac-2 antibody (clone M3/38, Cedarlane, Burlington, NC), Wt-1 antibody (Cat #: SC-192, Santa Cruz Biotechnology, Santa Cruz, CA), cleaved caspase 3 antibody (Cat #: 9661, Cell Signaling Technology, Danvers, MA), on paraffin embedded sections. The number of glomerular macrophages, Wt-1 positive cells and apoptotic cells were counted in 15–40 glomeruli per section in blinded fashion under 40× magnification and averaged as described previously.11,47,54
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from kidney tissues using Tri reagent (Molecular Research Center, Cincinnati, OH) according to manufacturer’s protocol. Single-strand cDNA was synthesized using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) for two-step qRT-PCR. Quantitative PCR was performed in a Bio-Rad CFX96 Real-Time system (Bio-Rad, Hercules, CA). Data were processed using Bio-Rad CFX Manager software (version 2.0). Relative expression quantification was calculated using the 2−ΔΔCT equation after normalization to GAPDH. Taqman gene expression assays were used to determine following mRNA expressions: GAPDH: Mm99999915_g1; TNF-α: Mm00443260_g1; Nos2: Mm00440502_m1; Fibronectin: Mm01256744_m1; and type-1 Collagen: Mm00801666_g1; CCL2: Mm00441242; CCL7: Mm00443113_m1; CCL12: Mm01617100_m1 as described previously.11,54
MSD multi-spot assay
Mouse kidney cytokines were measured using a Multi-spot assay system (Mouse proinflammatory panel 1, Meso Scale Diagnostics, Rockville, MD). The assay was performed per manufacturer’s instructions. Ninety µg kidney lysates were used for each assay.
Statistical analysis
Comparisons between groups were analyzed using SPSS (version 19.0, SPSS, Chicago, IL). Data are expressed as mean ± SEM. One-way ANOVA was used when more than two groups were compared, and significance of observed differences among the groups was evaluated with a least significant difference post hoc test. Statistical significance was identified at p< 0.05.
Supplementary Material
(A) Schematic of podocin2.5-CCR2 transgenic mouse creation. p2.5P-nlacF was used as a backbone construct to generate podocin2.5-CCR2 transgenic mice. The reporter gene was replaced by mouse full-length CCR2 cDNA. B) Genotyping PCR of founder mice using PCR assay. C) Schematic of experimental protocol and breeding strategy. Tg: transgene allele. D) Characterization of podocin2.5-CCR2 transgenic mice. Mouse CCR2 mRNA expression was verified by RT-PCR in primary cultured podocytes isolated from indicated mouse lines and peritoneal macrophages from Ccr2−/− Tg(NPHS2-Ccr2) mouse.
{kind=link}
Quantitative RT-PCR was performed on whole mouse kidney total RNA after 9 weeks following diabetes. CCL2 (A), CCL12 (B), and CCL7 (C) mRNA expression was normalized with GAPDH mRNA. Open bar, control groups; black-filled bar, diabetic groups. Results are means ± SEM. *p<0.05, *p<0.01, *p<0.001 compared to control.
{kind=link}
Acknowledgments
This study was supported by NIH Grants DK094930 and DK094930S1. The authors gratefully acknowledge Dr. L. Holzman (University of Pennsylvania) for providing the plasmid p2.5P-nlacF, and Alane Seidel (Penn State College of Medicine) for her help with generation of the transgenic mouse model.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
None
References
- 1.Association AD. Position Statement: Diabetic nephropathy. Diabetes Care. 2001;24:S69–S72. [Google Scholar]
- 2.USRDS TUSRDS. Annual Data Report. Bethesda: The National Institutes of Diabetes and Digestive and Kidney Diseases; 2001. [Google Scholar]
- 3.Parving HHOR, Ritz E. Diabetic nephropathy. In: BM B, editor. The Kidney. Philadelphia: W.B. Saunders Company; 2000. pp. 1731–1773. [Google Scholar]
- 4.Cooper ME. Pathogenesis, prevention, and treatment of diabetic nephropathy. Lancet. 1998;352:213–219. doi: 10.1016/S0140-6736(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 5.Nikolic-Paterson DJ, Atkins RC. The role of macrophages in glomerulonephritis. Nephrol Dial Transplant. 2001;16(Suppl 5):3–7. doi: 10.1093/ndt/16.suppl_5.3. [DOI] [PubMed] [Google Scholar]
- 6.Rovin BH, Doe N, Tan LC. Monocyte chemoattractant protein-1 levels in patients with glomerular disease. Am J Kidney Dis. 1996;27:640–646. doi: 10.1016/s0272-6386(96)90097-9. [DOI] [PubMed] [Google Scholar]
- 7.Awad AS, Kinsey GR, Khutsishvili K, et al. Monocyte/macrophage chemokine receptor CCR2 mediates diabetic renal injury. Am J Physiol Renal Physiol. 2011;301:F1358–F1366. doi: 10.1152/ajprenal.00332.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furuta T, Saito T, Ootaka T, et al. The role of macrophages in diabetic glomerulosclerosis. Am J Kidney Dis. 1993;21:480–485. doi: 10.1016/s0272-6386(12)80393-3. [DOI] [PubMed] [Google Scholar]
- 9.Hirata K, Shikata K, Matsuda M, et al. Increased expression of selectins in kidneys of patients with diabetic nephropathy. Diabetologia. 1998;41:185–192. doi: 10.1007/s001250050888. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen D, Ping F, Mu W, et al. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology (Carlton) 2006;11:226–231. doi: 10.1111/j.1440-1797.2006.00576.x. [DOI] [PubMed] [Google Scholar]
- 11.You H, Gao T, Cooper TK, et al. Macrophages directly mediate diabetic renal injury. Am J Physiol Renal Physiol. 2013;305:F1719–F1727. doi: 10.1152/ajprenal.00141.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Awad AS, You H, Gao T, et al. Macrophage-derived tumor necrosis factor-alpha mediates diabetic renal injury. Kidney Int. 2015;88:722–733. doi: 10.1038/ki.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsou CL, Peters W, Si Y, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 15.Dawson TC, Kuziel WA, Osahar TA, et al. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 1999;143:205–211. doi: 10.1016/s0021-9150(98)00318-9. [DOI] [PubMed] [Google Scholar]
- 16.Izikson L, Klein RS, Charo IF, et al. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belperio JA, Keane MP, Burdick MD, et al. Critical role for the chemokine MCP-1/CCR2 in the pathogenesis of bronchiolitis obliterans syndrome. J Clin Invest. 2001;108:547–556. doi: 10.1172/JCI12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dambach DM, Watson LM, Gray KR, et al. Role of CCR2 in macrophage migration into the liver during acetaminophen-induced hepatotoxicity in the mouse. Hepatology. 2002;35:1093–1103. doi: 10.1053/jhep.2002.33162. [DOI] [PubMed] [Google Scholar]
- 19.Kitagawa K, Wada T, Furuichi K, et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol. 2004;165:237–246. doi: 10.1016/S0002-9440(10)63292-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nam BY, Paeng J, Kim SH, et al. The MCP-1/CCR2 axis in podocytes is involved in apoptosis induced by diabetic conditions. Apoptosis. 2012;17:1–13. doi: 10.1007/s10495-011-0661-6. [DOI] [PubMed] [Google Scholar]
- 21.Burt D, Salvidio G, Tarabra E, et al. The monocyte chemoattractant protein-1/cognate CC chemokine receptor 2 system affects cell motility in cultured human podocytes. Am J Pathol. 2007;171:1789–1799. doi: 10.2353/ajpath.2007.070398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Awad AS, Kinsey GR, Khutsishvili K, et al. Monocyte/macrophage chemokine receptor CCR2 mediates diabetic renal injury. Am J Physiol Renal Physiol. 301:F1358–F1366. doi: 10.1152/ajprenal.00332.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tarabra E, Giunti S, Barutta F, et al. Effect of the monocyte chemoattractant protein-1/CC chemokine receptor 2 system on nephrin expression in streptozotocin-treated mice and human cultured podocytes. Diabetes. 2009;58:2109–2118. doi: 10.2337/db08-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Breyer MD, Bottinger E, Brosius FC, 3rd, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 25.Brosius FC, 3rd, Alpers CE, Bottinger EP, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20:2503–2512. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jefferson JA, Shankland SJ, Pichler RH. Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney Int. 2008;74:22–36. doi: 10.1038/ki.2008.128. [DOI] [PubMed] [Google Scholar]
- 27.Kalantarinia K, Awad AS, Siragy HM. Urinary and renal interstitial concentrations of TNF-alpha increase prior to the rise in albuminuria in diabetic rats. Kidney Int. 2003;64:1208–1213. doi: 10.1046/j.1523-1755.2003.00237.x. [DOI] [PubMed] [Google Scholar]
- 28.Navarro-Gonzalez JF, Jarque A, Muros M, et al. Tumor necrosis factor-alpha as a therapeutic target for diabetic nephropathy. Cytokine Growth Factor Rev. 2009;20:165–173. doi: 10.1016/j.cytogfr.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Kang YS, Lee MH, Song HK, et al. CCR2 antagonism improves insulin resistance, lipid metabolism, and diabetic nephropathy in type 2 diabetic mice. Kidney Int. 2010;78:883–894. doi: 10.1038/ki.2010.263. [DOI] [PubMed] [Google Scholar]
- 30.Sayyed SG, Ryu M, Kulkarni OP, et al. An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int. 2011 doi: 10.1038/ki.2011.102. [DOI] [PubMed] [Google Scholar]
- 31.de Zeeuw D, Bekker P, Henkel E, et al. The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. Lancet Diabetes Endocrinol. 2015;3:687–696. doi: 10.1016/S2213-8587(15)00261-2. [DOI] [PubMed] [Google Scholar]
- 32.Giunti S, Pinach S, Arnaldi L, et al. The MCP-1/CCR2 system has direct proinflammatory effects in human mesangial cells. Kidney Int. 2006;69:856–863. doi: 10.1038/sj.ki.5000197. [DOI] [PubMed] [Google Scholar]
- 33.Christensen PJ, Du M, Moore B, et al. Expression and functional implications of CCR2 expression on murine alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L68–L72. doi: 10.1152/ajplung.00079.2003. [DOI] [PubMed] [Google Scholar]
- 34.Weber KS, Nelson PJ, Grone HJ, et al. Expression of CCR2 by endothelial cells : implications for MCP-1 mediated wound injury repair and In vivo inflammatory activation of endothelium. Arterioscler Thromb Vasc Biol. 1999;19:2085–2093. doi: 10.1161/01.atv.19.9.2085. [DOI] [PubMed] [Google Scholar]
- 35.Lundien MC, Mohammed KA, Nasreen N, et al. Induction of MCP-1 expression in airway epithelial cells: role of CCR2 receptor in airway epithelial injury. J Clin Immunol. 2002;22:144–152. doi: 10.1023/a:1015420029430. [DOI] [PubMed] [Google Scholar]
- 36.Banisadr G, Queraud-Lesaux F, Boutterin MC, et al. Distribution, cellular localization and functional role of CCR2 chemokine receptors in adult rat brain. J Neurochem. 2002;81:257–269. doi: 10.1046/j.1471-4159.2002.00809.x. [DOI] [PubMed] [Google Scholar]
- 37.Warren GL, Hulderman T, Mishra D, et al. Chemokine receptor CCR2 involvement in skeletal muscle regeneration. FASEB J. 2005;19:413–415. doi: 10.1096/fj.04-2421fje. [DOI] [PubMed] [Google Scholar]
- 38.Moore BB, Kolodsick JE, Thannickal VJ, et al. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 2005;166:675–684. doi: 10.1016/S0002-9440(10)62289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rao VH, Meehan DT, Delimont D, et al. Role for macrophage metalloelastase in glomerular basement membrane damage associated with alport syndrome. Am J Pathol. 2006;169:32–46. doi: 10.2353/ajpath.2006.050896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steffes MW, Schmidt D, McCrery R, et al. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001;59:2104–2113. doi: 10.1046/j.1523-1755.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- 41.Pagtalunan ME, Miller PL, Jumping-Eagle S, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99:342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reiser J, von Gersdorff G, Loos M, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004;113:1390–1397. doi: 10.1172/JCI20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu L, Hagiwara S, Fan Q, et al. Role of receptor for advanced glycation endproducts and signalling events in advanced glycation end-product-induced monocyte chemoattractant protein-1 expression in differentiated mouse podocytes. Nephrol Dial Transplant. 2006;21:299–313. doi: 10.1093/ndt/gfi210. [DOI] [PubMed] [Google Scholar]
- 44.Park J, Ryu DR, Li JJ, et al. MCP-1/CCR2 system is involved in high glucose-induced fibronectin and type IV collagen expression in cultured mesangial cells. Am J Physiol Renal Physiol. 2008;295:F749–F757. doi: 10.1152/ajprenal.00547.2007. [DOI] [PubMed] [Google Scholar]
- 45.Moeller MJ, Sanden SK, Soofi A, et al. Two gene fragments that direct podocyte-specific expression in transgenic mice. J Am Soc Nephrol. 2002;13:1561–1567. doi: 10.1097/01.asn.0000015614.68893.0b. [DOI] [PubMed] [Google Scholar]
- 46.Yaoita E, Kurihara H, Sakai T, et al. Phenotypic modulation of parietal epithelial cells of Bowman's capsule in culture. Cell Tissue Res. 2001;304:339–349. doi: 10.1007/s004410100380. [DOI] [PubMed] [Google Scholar]
- 47.Morris SM, Jr, Gao T, Cooper TK, et al. Arginase-2 mediates diabetic renal injury. Diabetes. 60:3015–3022. doi: 10.2337/db11-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Awad AS, Kinsey GR, Khutsishvili K, et al. Monocyte/Macrophage Chemokine Receptor CCR2 Mediates Diabetic Renal Injury. Am J Physiol Renal Physiol. 2011 doi: 10.1152/ajprenal.00332.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morris SM, Jr, Gao T, Cooper TK, et al. Arginase-2 mediates diabetic renal injury. Diabetes. 2011;60:3015–3022. doi: 10.2337/db11-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Awad AS, Rouse MD, Khutsishvili K, et al. Chronic sphingosine 1-phosphate 1 receptor activation attenuates early-stage diabetic nephropathy independent of lymphocytes. Kidney Int. 2011;79:1090–1098. doi: 10.1038/ki.2010.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujita H, Fujishima H, Chida S, et al. Reduction of renal superoxide dismutase in progressive diabetic nephropathy. J Am Soc Nephrol. 2009;20:1303–1313. doi: 10.1681/ASN.2008080844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.You H, Gao T, Cooper TK, et al. Diabetic nephropathy is resistant to oral Larginine or L-citrulline supplementation. Am J Physiol Renal Physiol. 2014;307:F1292–F1301. doi: 10.1152/ajprenal.00176.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.You H, Gao T, Cooper TK, et al. Arginase inhibition: a new treatment for preventing progression of established diabetic nephropathy. Am J Physiol Renal Physiol. 2015;309:F447–F455. doi: 10.1152/ajprenal.00137.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.You H, Gao T, Cooper TK, et al. Arginase inhibition mediates renal tissue protection in diabetic nephropathy by a nitric oxide synthase 3-dependent mechanism. Kidney Int. 2013 doi: 10.1038/ki.2013.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Schematic of podocin2.5-CCR2 transgenic mouse creation. p2.5P-nlacF was used as a backbone construct to generate podocin2.5-CCR2 transgenic mice. The reporter gene was replaced by mouse full-length CCR2 cDNA. B) Genotyping PCR of founder mice using PCR assay. C) Schematic of experimental protocol and breeding strategy. Tg: transgene allele. D) Characterization of podocin2.5-CCR2 transgenic mice. Mouse CCR2 mRNA expression was verified by RT-PCR in primary cultured podocytes isolated from indicated mouse lines and peritoneal macrophages from Ccr2−/− Tg(NPHS2-Ccr2) mouse.
Quantitative RT-PCR was performed on whole mouse kidney total RNA after 9 weeks following diabetes. CCL2 (A), CCL12 (B), and CCL7 (C) mRNA expression was normalized with GAPDH mRNA. Open bar, control groups; black-filled bar, diabetic groups. Results are means ± SEM. *p<0.05, *p<0.01, *p<0.001 compared to control.