

Abstract
The Hypothalamic Pituitary Adrenal (HPA) axis has been implicated in the pathophysiology of a variety of mood and cognitive disorders. Neuroendocrine studies have demonstrated HPA axis overactivity in major depression, a relationship of HPA axis activity to cognitive performance, and a potential role of HPA axis genetic variation in cognition. The present study investigated the simultaneous roles HPA axis activity, clinical symptomatology, and HPA genetic variation play in cognitive performance. Patients with major depression with psychosis (PMD) and without psychosis (NPMD) and healthy controls (HC) were studied. All participants underwent a diagnostic interview and psychiatric ratings, a comprehensive neuropsychological battery, overnight hourly blood sampling for cortisol, and genetic assessment. Cognitive performance differed as a function of depression subtype. Across all subjects, cognitive performance was negatively correlated with higher cortisol, and PMD patients had higher cortisol than did NPMDs and HCs. Cortisol, clinical symptoms, and variation in genes, NR3C1 (glucocorticoid receptor - GR) and NR3C2 (minercorticoid receptor – MR) that encode for glucocorticoid and mineralcorticoid receptors, predicted cognitive performance. Beyond the effects of cortisol, demographics, and clinical symptoms, NR3C1 variation predicted attention and working memory, whereas NR3C2 polymorphisms predicted memory performance. These findings parallel the distribution of GR and MR in primate brain and their putative roles in specific cognitive tasks. HPA axis genetic variation and activity were important predictors of cognition across the entire sample of depressed subjects and healthy controls. GR and MR genetic variation predicted unique cognitive functions, beyond the influence of cortisol and clinical symptoms. GR genetic variation was implicated in attention and working memory, whereas MR was implicated in verbal memory.
Keywords: Major depression, psychosis, cortisol, glucocorticoid genes, HPA axis, cognition
Introduction
The stress responsive hypothalamic pituitary adrenal (HPA) axis has been implicated in the pathophysiology of anxiety and depression as well as cognitive functioning. The axis consists of stimulating forward and feedback inhibition loops involving the brain, pituitary, and adrenal glands, which regulates glucocorticoid production. Cortisol released from the adrenal glands, binds in brain with high affinity to mineralocorticoid receptors (MRs) and with lower affinity to glucocorticoid receptors (GRs). GR is distributed widely throughout the primate brain, whereas MR is heavily localized to the hippocampus (1). In addition, glucorticoid responsive elements are found in the regulatory regions of many genes in brain. Cortisol exerts its tonic influences predominantly via hippocampal MR, whereas feedback actions at the level of the pituitary and activated brain areas such as the amygdala are mediated by GR (2, 3). The development of major depression has been postulated to reflect a dysregulation of MR and/or GR within the hypothalamic-pituitary-adrenocortical system (4, 5).
Upwards of 40-60% of depressed patients experience hypercortisolemia (6) or other disturbances of the HPA system, such as flattened circadian state rhythm (7), or an earlier (8) or elevated nadir (9). However, we and others have found elevated HPA activity is more closely associated with specific depression subtypes, such as psychotic features (PMD). PMD patients demonstrate elevated activity of the HPA axis as compared with nonpsychotic depressives (NPMD) or healthy controls (HC) (10-13). PMD patients have significantly elevated evening (11) and afternoon (1-4 pm) (10) serum cortisol levels. Furthermore, PMD patients demonstrate a blunted response to fludrocortisone, a mixed mineralocorticoid/glucocorticoid agonist (12, 14), high rates of non-suppression on challenge with dexamethasone, with particularly high post-dexamethasone cortisol levels (13).
Disrupted cognition is also a feature of depression with the most consistent deficits being in memory and executive function (15). PMD patients have even greater decrements in cognition than do NPMDs (16). Furthermore, higher cortisol levels have been associated with impaired cognitive functioning in both healthy controls and depressed patients (16, 17).
The relative roles of GR and MR in cognitive dysfunction in depression have been a focus of limited study, although, animal data have long pointed to a role for MR in both cortisol secretion and memory and executive function performance (18). In HCs, blocking MR impairs memory and executive function (19). In contrast, fludrocortisone decreases cortisol in healthy controls and depressives and enhances verbal memory (20); significant correlations between cortisol inhibition and verbal memory (list learning) performance were observed. MR stimulation with fludrocortisone inhibits cortisol secretion (21), but fludrocortisone inhibition of cortisol is attenuated in patients with PMD suggesting impaired MR function in those patients (12). Last, there is evidence for decreased MR expression in the hippocampus and prefrontal cortex in depressed patients (22, 23).
Although mineralocorticoid receptors (MR) mediate neuronal changes required for learning and memory, NR3C2 (MR) genetic variation thus far has not been associated with specific aspects of cognition. NR3C2 genetic variation has been associated with higher serum levels of brain derived neurotrophic factor (sBDNF), where those with intermediate and high exposure to physical neglect showed higher sBDNF levels but only in those with the CC genotype (24). While APOE gene is confirmed as a high risk of Alzheimer's Disease (AD), Sun and colleagues (25)examined four large datasets, looking for other potential risk genes. They concluded that one of 10 potential risk genes was NR3C2 (MR). Since MR is densely distributed in the hippocampal region (26) and involved in hemodynamic centers in brain (27), it may be involved in the progression of AD inresponse to stressful stimuli (28).
Recent studies on specific single nucleotide polymorphisms (SNP's) for GR indicate that some are associated with altered GR sensitivity (29), and in conjunction with COMT genetic variation are associated with poorer working memory in healthy controls (30). In addition, Young's group reported that the GR antagonist mifepristone significantly improves visual memory in bipolar depression (31). Our group has recently reported that GR (but not MR) genetic variation accounted for a significant amount of variance in mean cortisol levels and severity of psychosis (32). However, the relationship of variation in NR3C1 that encodes for GR gene and the directionality of influence on cognition in depression is not yet known.
We have reported previously on potential relationships of cortisol and cognition in limited samples of subjects (16), but neither we nor others have explored the relationships among genetic variation, clinical symptoms, and cognition. The specific aim of this project was to extend on previous findings on cortisol dysregulation to cognitive performance by exploring whether HPA axis genetic variation (see Table 1 for list of SNPs) also contributes to specific cognitive performance beyond that predicted by cortisol or clinical measures.
Table 1.
List of NR3C1 (GR) and NR3C2 (MR) SNPs examined in this study.
| HPA axis Genes Assessed | |||
|---|---|---|---|
| Gene | Function | Brain Location in humans | SNPs Studied |
| NR3C1 (GR) | Feedback inhibition of HPA axis; cognition; immune response | Cortex widely, hypothalamus, amygdala, hippocampus | N=10 * rs56149945 (formerly rs6195) ** rs6198 rs33388 ^ rs2918419 rs10052957 rs10482633 **rs12521436 ** rs12655166 ** rs17209258 rs41423247 |
| NR3C2 (MR) | Inhibitory control of HPA axis; memory; blood pressure | Hypothalamus, hippocampus, amygdala | N=13 ** rs5525 * rs5530 rs1879829 rs2070951 ** rs2272089 rs3910052 rs4835488 rs6535578 rs7658048 ** rs7694064 ** rs10213471 * rs17024360 ** rs17484245 rs2070950** rs5522 |
| FKBP5 | Co-chaperone to heat shock protein for GR; stabilizes GR confirmation | See GR | N=2 rs1360780 rs3800373 |
Little or no variance in the SNPs; SNP not used in analyses
Less than 4 total in the rare homozygous, collapsed into the heterozygous SNP group
Only 2 SNP variations present.
Methods
Participants
Psychiatric participants were recruited through inpatient and outpatients facilities at Stanford University or self-referred from online and print study advertisements. Fifty-nine patients with psychotic major depression (PMD) and 58 patients with nonpsychotic major depression (NPMD) participated in two waves of a larger study on HPA axis in depression.
Depressed patients were required to have a minimum score of 21 on the 21-item Hamilton Depression Rating Scale (HDRS) and a minimum score of 6 on the Thase Core Endogenomorphic Scale, with one exception of a PMD who scored 5. These latter two criteria were designed to ensure inclusion of participants with similar minimum levels of severity of endogenous-type symptoms. PMDs were also required to have a minimum total score of 5 on the positive symptom subscale of the Brief Psychotic Rating Scale (BPRS) which consists of four items: conceptual disorganization, suspiciousness, hallucinations, and unusual thought content. A score of 4 on the PSS indicates no positive psychotic symptoms. NPMD subjects had no history of psychotic symptoms. All patients met the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) criteria for a current major depressive episode, with or without psychotic features.
Healthy control subjects were recruited through online and print study advertisements. Overall, 63 healthy controls participated in the larger HPA study and 29 provided blood samples for genetic analyses. Healthy controls (HC) were assessed for Axis I disorders with the Structured Clinical Interview for DSM-IV. They had a score of less than 6 on the HDRS-21 and no psychotic symptoms as measured by the BPRS positive symptom subscale. Furthermore, they had no current or history of Axis I psychiatric illness.
Participants were allowed to remain on their psychiatric medications but were required to maintain a stable medication dosing regimen for at least 1 week prior to the start of the study. Depressed patients were taking a combination of antidepressants, antipsychotics, anxiolytics, and mood stabilizers (see Table 2).
Table 2.
Subject Demographics.
| PMD (N=46) | NPMD (N=37) | HC (N=46) | Analysis | Post-hoc comparison | |
|---|---|---|---|---|---|
|
| |||||
| Age | 36.67 (12.4) | 42.38 (12.4) | 36.17 (12.6) | F(2,126)=3.01, p=.053 | |
|
| |||||
| Education | 15.04 (2.8) | 14.81 (1.9) | 15.68 (2.2) | F(2,126)=1.58, ns | |
|
| |||||
| WTAR predicted FIQ | N=35 110.57 (10.5) | N=32 109.16 (10.4) | N=42 111.31 (9.0) | F(2,106)=.433, ns | |
|
| |||||
| Gender | χ2=.454, ns | ||||
| Male | 21 | 15 | 22 | ||
| Female | 25 | 22 | 24 | ||
|
| |||||
| Ethnicity | χ2(8)=8.51, ns | ||||
| Caucasian | 31 | 28 | 26 | ||
| African Am. | 4 | 3 | 3 | ||
| Asian Am | 5 | 1 | 10 | ||
| Latino | 2 | 4 | 3 | ||
| Other | 1 | 1 | 1 | ||
|
| |||||
| Daily Psych Medications* | 39/46 | 18/37 | 0/46 | χ2(2)=67.46, p<.001 | |
| • Antidep | 32/46 | 17/37 | |||
| • Antipsychotics | 28/46 | 2/37 | |||
| • Anxiolytics/ benzodiazapines | 19/46 | 4/37 | |||
| • Mood stabilizers | 8/46 | 3/37 | |||
|
| |||||
| Daily Psych Medications - Genetic sample only* | 29/33 | 11/23 | 0/24 | χ2(1)=10.64, p=.001 | |
| • Antidep | 24/33 | 10/23 | χ2(1)=4.96, p=.027 | ||
| • Antipsychotics | 21/33 | 2/23 | χ2(1)=16.90, p<.001 | ||
| • Anxiolytics/ benzodiazapines | 15/33 | 3/23 | χ2(1)=6.53, p=.011 | ||
| • Mood stabilizers | 7/33 | 2/23 | χ2(1)=1.54, ns | ||
|
| |||||
| Comorbidity Diagnoses | |||||
| • Panic Disorder | 9 | 3 | |||
| • Agorophobia | 3 | 2 | |||
| • Social Phobia | 7 | 4 | |||
| • PTSD | 5 | 5 | |||
| • GAD | 5 | 3 | |||
|
| |||||
| HDRS | 30.33 (5.4) | 24.08 (3.2) | .56 (93) | F(2, 126)=811.5, p<.001 | P > (N > HC) |
|
| |||||
| Thase Endog. | 9.32 (1.8) | 8.21 (1.7) | .087 (.28) | F(2, 126)= 576.6, p<.001 | P > (N > HC) |
|
| |||||
| BPRS | 48.04 (7.4) | 33.62 (4.2) | 18.56 (1.1) | F(2, 126)=398.5, p<.001 | P > (N > HC) |
|
| |||||
| Positive Symptom Scale | 12.04 (3.8) | 4.21 (.53) | 4.07 (.33) | F(2, 126)=177.7, p<.001 | P > (N = HC) |
HDRS= Hamilton Depression Rating Scale; BPRS = Brief Psychiatric Rating Scale. Under Post-hoc comparisons, P = Psychotic Major Depression; N= Nonpsychotic Major Depression; HC = Healthy controls. This refers to the significance (p value) of the pairwise comparisons of the three groups. For example, under HDRS, PMD subjects are significantly higher than both NPMDs and HCs, and NPMDs are significantly higher than HCs.
Any daily psychiatric medication includes use of antidepressants, antipsychotics, anxiolytics, or mood stabilizers
Of the 153 original subjects, various subsets of patients are used in different analyses. See Supplemental Figure 1 for a consort chart of usable subjects in each aspect of the omnibus study. Nine patients had unusable or missing neuropsychological assessments, and 15 additional participants were taking estrogens, so the total sample for neuropsychological analyses is 129. Nine patients had the CVLT-1 instead of the CVLT-II, so those patients were dropped only from the CVLT analyses. An additional 6 had missing or unusable cortisol data, and 44 were missing genetic data. Thus, for the genetic analyses the sample was 80 subjects. The sample sizes were based on effect sizes of our previous work on cortisol and neuropsychological testing results.
Procedure
The study was approved by the Stanford University Institutional Review Board, and all subjects gave written informed consent before screening. Eligibility screening procedures included the SCID, HAM-D, BPRS, clinical laboratory tests, comprehensive metabolic panel, urine drug screening, and urine screening for pregnancy in female subjects. If participants met inclusion criteria at eligibility screening, they returned for baseline procedures.
At baseline, participants were re-administered the HAMD-21 and the BPRS to assess for clinical symptomatology. Participants then were assessed for cognitive function. Overnight blood sampling then took place on the Stanford Hospital Clinical and Translational Research Unit. An intravenous line was inserted at 4 PM and blood collected at the top of each hour for cortisol from 6 pm to 9 am the next day. Sixteen blood samples per patient were collected. Part way through the first wave of study, collection of blood samples for genetic analysis began. The majority of available subjects consented to provide blood for genetic analyses.
Data on subsets of participants have been previously reported. Cortisol and cortisol/cognitive data were previously reported (11) on a subset of 73 of 129 subjects reported here. Data on another subset 78 of 80 subjects who had both cortisol and genetic data were also recently reported (32). No published paper from this dataset has examined the relationships of cognition, cortisol, and genetics together.
Cognition
Participants completed a neuropsychological assessment battery that assessed four primary cognitive domains: attention, working memory, executive function, and verbal memory [see (16) for full details and Table 3 for a list of tests].
Table 3.
Means (and Standard Deviations) of neuropsychological measures and cortisol (mcg/dl) by group. All analyses include age as a covariate.
| PMD (N=46) | NPMD (N=37) | HC (N=46) | Analysis & Post-hoc comparisons | |
|---|---|---|---|---|
| Attention | ||||
| Digit Span Forwards | 10.41 (3.0) | 10.51 (2.7) | 11.32 (2.1) | F(2,118)=1.38, ns |
| Trail Making Test A | 34.2 (14.5) | 29.75 (12.8) | 24.54 (8.8) | F(2,123)=7.60, p=.001 P>(N=H) |
| Working Memory | ||||
| Digit Span Backwards | 6.78 (2.3) | 7.71 (2.4) | 8.12 (2.5) | F(2,118)=3.60, p=.03 |
| Letter Number Sequencing* | 9.60 (2.8) | 11.30 (2.6) | 11.63 (2.9) | F(2,124)=7.64, p=.001 P < (N = HC) |
| Executive Function | ||||
| Stroop (Color-Word) | 36.24 (10.2) | 42.11 (9.6) | 46.80 (12.8) | F(2,124)=12.23, p<.001 P < (N = HC) |
| Trail Making Test B | 81.05 (34.8) | 68.89 (25.6) | 60.63 (24.6) | F(2,121)=5.79, p=.004 P > (N = HC) |
| COWA (FAS) | 37.30 (11.7) | 38.14 (10.6) | 45.82 (13.2) | F(2,123)=6.52, p=.002 (P = N) < HC |
| Verbal Memory | ||||
| CVLT-II | N=40 | N=33 | N=41 | |
| Total Learning Trials 1-5 | 45.00 (12.4) | 47.52 (9.3) | 56.61 (9.1) | F(2,110)=12.74, p<.001 (P = N)< HC |
| Short delay free | 9.45 (3.6) | 10.58 (3.5) | ||
| Short delay cued | 10.35 (3.3) | 11.73 (2.6) | 12.68 (2.4) | F(2,110)=7.08, p=.001 P< (N = HC) |
| Long delay free | 9.45 (4.0) | 11.03 (3.2) | 12.56 (2.8) | F(2,110)=8.32, p<.001 P< (N = HC) |
| Long delay cued | 10.30 (3.4) | 11.70 (2.6) | 12.85 (2.5) | F(2,114)=7.94, p=.001 P< (N = HC) |
| Recognition | 14.20 (2.2) | 14.61 (2.1) | 14.85 (1.6) | F(2,114)=1.17, ns |
| Logical Memory | ||||
| Immediate Recall | 21.80 (6.4) | 22.92 (7.6) | 28.75 (8.4) | F(2,122)=10.30, p<.001 (P = N) < HC |
| Delayed Recall | 20.44 (8.2) | 23.00 (8.1) | 29.77 (6.8) | F(2,122)=16.77, p<.001 (P = N)< HC |
| Recognition | 24.36 (2.9) | 25.14 (2.6) | 26.98 (2.3) | F(2,121)=11.25, p<.001 (P = N) < HC |
| Mean Cortisol 1800-0100 | N=434.82 (2.4) | N=373.63 (1.6) | N=433.37 (1.3) | F(2,120)=7.37, p=.001 P > (N = HC) |
| Mean Cortisol 0100-0900 | N=43 9.20 (4.0) | N=37 9.07 (2.4) | N=43 8.70 (2.2) | F(2,120)=.322, ns |
Cortisol Determination
Cortisol assays were conducted by the Brigham Women's Hospital, General Clinical Research Laboratory in Boston. The analytic sensitivity for cortisol was 0.4 micrograms/dl with a coefficient of variation of less than 7.9%. Because of the natural diurnal rhythm of the cortisol slopes, the 15-hour blood collection period was divided into two phases based on the apparent nadir: the evening level from 1800 to 0100 hours and the morning level from 0100 to 0900 hours. These epochs correspond to the natural descending and ascending slopes of the cortisol rhythm and are based on the nadir observed in previous studies (11) of subsamples from this study. Cortisol means were then computed for these two epochs (1800 to 0100 and 0100 to 0900 hrs).
Medication
Patients were taking a variety of psychiatric medications: antidepressants, anxiolytics, antipsychotics, and/or mood stabilizers. The sample size is not large enough to subdivide into specific medications; however, because benzodiazepines in particular can affect cognition (specifically memory) subjects were classified by whether or not they were regularly taking them, and this variable was entered into the regression model.
Genetics
Blood was collected for assessment of HPA axis genetic markers and herein we report on three genes: NR3C1 (Glucocorticoid receptor; GR), NR3C2 (Mineralcorticoid Receptor; MR), and FK506 binding protein 5 (FKBP5). Alleles studied were selected using a standard protocol that utilized 5 specific criteria (see (32) for more details).
DNA was extracted from EDTA-treated whole blood using the Gentra Puregene kit (Qiagen, Valencia, CA). Genotyping was performed using Taqman real-time PCR (Applied Biosystems, Foster City, CA). All genotypes were tested for deviation from Hardy Weinberg equilibrium. SNPs and their frequencies were assayed for each HPA axis gene (32). 27 SNPs in toto were assessed (see Table 1 for list of SNPs; Supplemental Figures 2 and 3 for linkage disequilibrium (LD) maps; Supplemental Table 1 for allelic distribution). SNPs that had no or minimal variability in our sample were excluded from the analysis. If SNPs had fewer than 4 subjects across all 3 groups homozygous for the rare allele, they were collapsed into the heterozygous group.
Statistical Analyses
All analyses were conducted using SPSS statistical software. First, ANOVA and Chi Square analyses were used to examine group differences in demographic variables (age, education, and gender), as well as clinical ratings (HDRS, BPRS). Next, ANCOVAs were utilized to examine differences among the 3 diagnostic groups for each cognitive test. Since age influences neuropsychological functions, it was used as a covariatein the model. Then, ANOVAs were utilized to examine differences in evening and early morning cortisol between the three groups, with age as a covariate since it is also known to influence cortisol. To account for the number of comparisons, the alpha level was set to p < .005 for each omnibus test. On subsequent pairwise comparisons, an alpha level of .05 was used.
Finally linear regression analyses were run predicting cognitive performance using subjects across all three groups. To avoid imposing our own bias, we ran a regression model that allowed the data to guide the outcomes, resulting in the most important predictors in the model to be detected. Thus, a forward regression approach was used for each dependent variable, and variable entry was set at .05 and variable removal was set at .10. The dependent variables were the individual cognitive tests. Independent variables included age, sex, depression severity, psychotic symptom severity, daily benzodiazepines use, mean evening cortisol (6 pm – 1 am), mean early morning cortisol (1 am – 9 am), and all SNPs in a given gene. We analyzed for effects of NR3C1 (GR), NR3C2 (MR), and FKPB5 separately. The final model which was developed based on the forward regression was set to an overall significance level p < .005. This was done to account for the large number of comparisons. Secondary analyses that examined the independent contributions of the chosen variables was set to alpha < .05.
In addition to examining individual SNPs, potential haplotypes were created separately for NR3C1 and NR3C2. NR3C1 potential haplotypes were created using 5 NR3C1 SNPS, including ER22/23EK (rs6189 and rs6190), rs6195, rs41423247, and rs6198, replicating the methods of Kumsta et al. (33). Similar to Kumksta, only Haplotypes 1, 4, and 5 had adequate sample size for further analyses; thus, haplotypes 2 and 3 from the combination of SNPs were not examined. NR3C2 haplotypes were created using rs5522 an rs2070951, which replicates several other studies (34, 35). For all haplotype analyses, if the participant had at least one of each of the target alleles, they were considered a “potential haplotype carrier”. Regressions were performed as outlined above with the exception that instead of adding the individual SNPs into the model, the potential carrier status of each haplotype (yes/no) was used.
Results
Demographics
Across the three groups, there were no differences in age, education, gender, estimated IQ, or ethnicity (see Table 2). As expected, there were significant differences in all psychiatric rating scales (HAMD, Thase Endogenous scale, BPRS, and PSS). PMDs were more severe on all measures compared to NPMD, and both depression groups were higher than the healthy controls. The only exception was the Positive Symptom Subscale, in which NPMD patients and HC did not differ.
Cognitive Function
PMD patients performed significant worse than did both HCs on almost all measures of cognition (all p's < .005; see Table 3), with the exception of digit span and CVLT-II recognition memory and worse than NPMDs on most measures of attention, working memory, and executive function. NPMDs and HCs were generally similar on performance with a few exceptions where NPMDs did more poorly, including COWA, learning on CVLT-II, and Logical Memory. Interestingly, on the verbal list learning measure, total learning over the 5 trials differentiated the three groups, with PMDs performing significantly worse than NPMDs, who performed worse than HCs. On CVLT-II recall, PMD patients had poorer recall than NPMDs, who performed similar to HCs. This pattern held true for both free and cued recall, but no differences were apparent on the recognition trial. In contrast, PMDs and NPMDs both performed worse than HCs on story memory. PMDs performed similarly to NPMDs on immediate recall and recognition but worse on delayed recall. For executive function, PMDs demonstrated greater dysfunction than did both NPMD and HCs. Performance on measures of attention and working memory produced mixed results, with PMDs performing more poorly than HC and NPMDs on some tasks.
Cortisol
Significant relationships were observed between age and evening (r = .253, p=.005) and early morning cortisol (r= .298, p=.001). Thus, age was used as a covariate in cortisol analyses. As expected, PMD patients had a higher evening cortisol than did both HCs and NPMDs, who did not differ from one another (see Table 3). There were no significant differences between the 3 groups on early morning (1 am – 9 am) cortisol. Correlations between cortisol and cognitive variables demonstrated moderate negative correlations but most did not achieve significance cut-off of p<.005 (see Supplemental Table 2). There was a significant correlation between mean evening and early morning cortisol, r = .336 (p<.001); however, the correlations were not so high as to exclude one from the regression model to predict cognition.
Genetics
Results of the Forward Linear Regression with GR and MR individually are displayed in Tables 4 (NR3C1) and 5 (NR3C2). Of the NR3C1 SNP's, rs10052957 and rs41423247 predicted performance on attention tasks, beyond the contributions of cortisol, age, gender, medication status, or clinical measures. Other NR3C1 SNPs sporadically predicted working memory (rs6198) and executive function (rs2918419). However, NR3C1 SNPs did not contribute significantly to verbal memory. Interestingly, cortisol and gender were a strong predictors of verbal memory performance on CVLT list learning, while depression and gender were strong predictors of story memory.
Table 4.
Results of the Forward Regression Models predicting cognitive performance. Age, sex, depression severity, psychosis severity, mean evening cortisol, mean early morning cortisol, daily benzodiazepine medication use, and NR3C1 (GR) were all potential predictors. Statistics for the full model are given, and the predictors that made it into the model are immediately below. Beta and t and p values once the other predictors are accounted for are provided.
| Attention | Full Model | Beta value | t value | p value | %variance |
|---|---|---|---|---|---|
| Digit Span Forward | F(2,72)=6.60, p<.001, 26.8% | ||||
| Mean Evening Cortisol | -0.332 | -3.01 | .004 | 6.2 | |
| Mean Early Morning Cortisol | .348 | 3.24 | .002 | 9.1 | |
| rs10052957 | -0.277 | -2.73 | .008 | 6.8 | |
| Rs41423247 | .228 | 2.18 | .033 | 4.8 | |
| TMT A | F(1,77)=12.29, p=.001, 13.8% | ||||
| Age | 0.371 | 3.5 | 0.001 | 13.8 | |
| Working Memory | |||||
| DS back | F(2,74)=5.93, p=.004, 13.8% | ||||
| Positive symptoms | -0.324 | -2.97 | 0.004 | 8.5 | |
| rs6198 | -0.233 | -2.14 | 0.036 | 5.3 | |
| LNS | F(2,77)=16.49, p<.001, 30.0% | ||||
| Positive symptoms | -0.390 | -3.98 | <.001 | 21.0 | |
| Mean Evening Cortisol | -0.308 | -3.15 | .002 | 9.0 | |
| Executive Function | |||||
| Stroop Color Word | F(3,76)=10.00, p<.001, 28.3% | ||||
| Age | -0.292 | -2.87 | .005 | 14.3 | |
| Positive symptoms | -0.281 | -2.82 | 0.006 | 10.3 | |
| Evening cortisol | -.207 | -1.99 | .05 | 3.7 | |
| TMT B | F(2,75)=5.73, p=.005, 13.3% | ||||
| Evening cortisol | .263 | 2.45 | .017 | 6.4 | |
| Rs2918419 | .262 | 2.43 | .017 | 6.9 | |
| Cowa FAS | F(1,78)=10.24, p=.002, 11.6% | ||||
| Mean Evening Cortisol | -.341 | -3.20 | .002 | 11.6 | |
| Verbal Memory | |||||
| Total Trials 1-5 | F(3,73)=11.14, p<.001, 31.4% | ||||
| HDRS Depression Score | -.375 | -3.87 | <.001 | 15.3 | |
| Mean Evening Cortisol | -.311 | -3.20 | .002 | 10.1 | |
| Sex | -.244 | -2.52 | .014 | 6.0 | |
| SDFR | F(2,74)=7.14, p=.001, 16.2% | ||||
| Benzos | -0.266 | -2.46 | 0.016 | 9.9 | |
| Mean Evening Cortisol | -.255 | -2.34 | .021 | 6.3 | |
| SDCR | F(2,74)=9.05, p<.001, 19.7% | ||||
| Mean Evening Cortisol | -.326 | -3.11 | .003 | 12.3 | |
| Sex | -.273 | -2.61 | .011 | 7.4 | |
| LDFR | F(3,73)=7.21, p<.001, 22.9% | ||||
| Benzos | -.216 | -2.00 | .049 | 10.8 | |
| Early Morning Cortisol | -.264 | -2.52 | .014 | 6.5 | |
| Sex | -.245 | -2.30 | .024 | 5.6 | |
| LDCR | F(2,74)=9.43, p<.001, 20.3% | ||||
| Mean Evening Cortisol | -.358 | -3.43 | .001 | 14.4 | |
| Sex | -.244 | -2.34 | .022 | 5.9 | |
| Recognition | F(3,73)=13.29, p<.001; 35.3% | ||||
| rs17209258 | -.455 | -4.77 | <.001 | 15.0 | |
| Mean Evening Cortisol | -.401 | -4.20 | <.001 | 16.1 | |
| Sex | -.207 | -2.20 | .031 | 4.3 | |
| LM | |||||
| Immediate Memory | F(2,77)=7.22, p=.001, 15.8% | ||||
| HDRS Depression Score | -0.337 | -3.22 | .002 | 10.8 | |
| Sex | -.224 | -2.14 | .035 | 5.0 | |
| Delayed Memory | F(2, 77)=6.05, p=.004, 13.6% | ||||
| HDRS Depression Score | -.300 | -2.83 | .006 | 8.5 | |
| Sex | -.225 | -2.12 | .037 | 5.1 | |
| Recognition | F(2, 76)=7.29, p=.001, 16.1% | ||||
| Positive symptoms | -0.269 | -2.48 | .015 | 10.7 | |
| Mean Evening Cortisol | -0.239 | -2.21 | .030 | 5.4 |
Note: TMT A is the Trail Making Test – Subtest A; DS back = Digit Span Backwards from the Weschler Adult Intelligent Scale – III; LNS = Letter Number Sequencing from the WAIS-III; TMT B = Trail Making Test – Subtest B; COWA FAS = Controlled Oral Word Association Test using the letters F, A, S.;CVLT = California Verbal Learning Test 2nd Edition; SDFR = CVLT short delay free recall score; SDCR = CVLT short delay cued recall score; LDFR = CVLT long delay free recall score; LDCR = CVLT long delay cued recall score; LM = Logical Memory Subtest of the Weschler Memory Scale-III.
In contrast, as seen in Table 5, NR3C2 (MR) did not predict attention or working memory but did predict verbal memory. Variation in four separate SNPs (rs5525, rs4835488, rs 10213471, and rs17484245) predicted verbal memory performance for stories. When NR3C2 was added to the regression equation, gender was no longer a significant predictor of story memory. In addition, variation in rs7694064 was significant for predicting long-delay free recall in list learning measures, and the addition of this SNP usurped the variance that was related to gender.
Table 5.
Results of the Forward Regression Models for predicting cognitive performance. Age, sex, depression severity, psychosis, mean evening cortisol, mean early morning cortisol, daily benzodiazepine use, and NR3C2 (MR) were all potential predictors. Statistics for the full model are given, and the predictors that made it into the model are immediately below. The Beta and t and p values are presented after accounting for the other predictors.
| Attention | Full Model | Beta value | t value | p value | % Variance |
|---|---|---|---|---|---|
| Digit Span Forward | F(2,72)=6.08, p=.004, 14.4% | ||||
| Mean Evening Cortisol | -.351 | -3.04 | .003 | 6.3 | |
| Mean Early Morning Cortisol | .303 | 2.62 | .011 | 8.2 | |
| TMT A | F(1,75)=11.60, p=.001, 13.4% | ||||
| Age | 0.366 | 3.41 | 0.001 | 13.4 | |
| Working Memory | |||||
| DS back | No significant model | ||||
| LNS | F(2,75)=12.89, p<.001, 25.6% | ||||
| Mean Evening Cortisol | -.333 | -3.26 | .002 | 16.0 | |
| Positive symptoms | -.316 | -3.10 | .003 | 9.5 | |
| Executive Function | |||||
| Stroop Color Word | F(3,74)=8.54, p<.001, 25.7% | ||||
| Age | -.281 | -2.68 | .009 | 13.5 | |
| Positive symptoms | -.231 | -2.25 | .027 | 7.5 | |
| Mean Evening Cortisol | -.231 | -2.16 | .034 | 4.7 | |
| TMT B | No significant model | ||||
| Cowa FAS | F(2,75)=7.04, p=.002, 15.8% | ||||
| Mean Evening Cortisol | -.333 | -3.14 | .002 | 11.3 | |
| Rs2070951 | -.211 | -2.00 | .05 | 4.5 | |
| Verbal Memory | |||||
| Total Learning Trials 1-5 | F(3,71)=10.83, p<.001, 31.4% | ||||
| HDRS Depression Score | -.376 | -3.82 | <.001 | 15.7 | |
| Age | -.309 | -3.14 | .002 | 10.1 | |
| Gender | -.238 | -2.42 | .018 | 5.7 | |
| SDFR | F(2,72)=6.95, p=.002, 16.2% | ||||
| Mean evening cortisol | -.262 | -2.28 | .020 | 9.8 | |
| Benzos | -.259 | -2.35 | .022 | 6.4 | |
| SDCR | F(2,72)=8.74, p<.001, 19.5% | ||||
| Mean Evening Cortisol | -0.334 | -3.15 | .002 | 12.9 | |
| Sex | -.258 | -2.43 | .017 | 6.6 | |
| LDFR | F(3,71)=7.99, p<.001, 25.2% | ||||
| Benzos | -.301 | -2.86 | .006 | 10.4 | |
| rs7694064 | .286 | 2.76 | .007 | 7.9 | |
| Mean Early Morning Cortisol | -.270 | -2.58 | .012 | 7.0 | |
| LDCR | F(3,71)=7.79, p<.001, 24.8% | ||||
| Mean Evening Cortisol | -.319 | -3.01 | .004 | 15.1 | |
| Sex | -.227 | -2.19 | .032 | 5.1 | |
| HDRS Depression Score | -.218 | -2.07 | .042 | 4.5 | |
| Recognition | F(1,73)=9.14, p=.003, 11.1% | ||||
| Mean Evening Cortisol | -.334 | -3.02 | .003 | 11.1 | |
| LM | |||||
| Immediate Memory | F(2,75)=11.66, p<.001, 23.7% | ||||
| HDRS Depression Score | -0.373 | -3.70 | <.001 | 12.8 | |
| rs4835488 | 0.331 | 3.28 | 0.002 | 10.9 | |
| Delayed Memory | F(4,73)=7.00, p<.001, 27.7% | ||||
| HDRS Depression Score | -0.313 | -3.06 | 0.003 | 11.2 | |
| rs5525 | -0.531 | -3.44 | 0.001 | 5.2 | |
| Rs10213471 | .398 | 2.59 | .012 | 6.4 | |
| Mean Evening Cortisol | -.227 | -2.23 | .029 | 4.9 | |
| Recognition | F(2, 74)=11.16, p<.001, 23.2% | ||||
| Positive symptoms | -0.366 | -3.59 | .001 | 14.6 | |
| rs17484245 | -.2946 | -2.88 | 0.005 | 8.6 |
Note: TMT A is the Trail Making Test – Subtest A; DS back = Digit Span Backwards from the Weschler Adult Intelligent Scale – III; LNS = Letter Number Sequencing from the WAIS-III; TMT B = Trail Making Test – Subtest B; COWA FAS = Controlled Oral Word Association Test using the letters F, A, S.;CVLT = California Verbal Learning Test 2nd Edition; SDFR = CVLT short delay free recall score; SDCR = CVLT short delay cued recall score; LDFR = CVLT long delay free recall score; LDCR = CVLT long delay cued recall score; LM = Logical Memory Subtest of the Weschler Memory Scale-III.
Finally, FKBP5 appeared to not exert an effect on cognition. FKPB5 SNPs did not predict any measure of cognition.
Since ethnicity has been known to influence genetic results, ethnicity was examined specifically among the participants with genetic data. In this subset, ethnicity did not differ across the three diagnostic (χ2(6)=5.88, ns). Thus, separate analyses were run for the largest subset of patients, European Caucasians (% Caucasians: PMD=72%, NPMD=78%, HC=67%). Results were generally similar between the full sample (N=80) and the subsample with European Caucasians only (N=58; see supplemental Tables 3-5).
In the Caucasian sample, the overall prediction of cognition with NR3C1 SNPs held true for attention and working memory. Rs4142347 predicted attention for both the full and the Caucasian-only samples. However, additional SNPS achieved significance in the Caucasian only sample, including rs33377, rs2918419, and rs12521436. Furthermore, consistent across the two samples, NR3C1 did not predict memory performance.
Similar results were found in the Caucasian only and the overall samples when NR3C2 was put in the model to predict cognition (see Supplemental Table 4). Specifically, the five NR3C2 SNPs found to predict list memory and story memory were identical in the two samples. In the Caucasian-only sample, several additional SNPs emerged. In particular, rs227089 emerged as a relevant factor for list learning and long delay list recall.
Finally, although FKPB5 did not emerge as a factor for any measure of cognition in the full sample, one FKBP5 SNP emerged as an important factor in the Caucasian-only analyses. Rs3800373 predicted delayed memory recall for list learning. Given the different distribution of rs3800373 among African- and Asian-American populations, it is likely that the reduced variability in the Caucasian-only sample led to the emergence of this SNP in predicting memory.
Haplotype Analysis
For the NR3C1 haplotypes, there were no significant effects to potential haplotypes 1 or 5 and few significant effects for haplotype 4. NR3C1 haplotypes 4 and NR3C2 haplotype 2 significantly predicted performance on the Controlled Oral Word Association (FAS) test, accounting for 7.4 and 5.1% of the variance, respectively, after other variables were accounted for (p<.001; see Supplemental material). In addition, NR3C1 Haplotype 4 predicted 4.4% of the variance on the CVLT raw score trials 1-5 (learning over time) after depression level, early morning cortisol, and gender were controlled for (p<.001). No other significant findings emerged from haplotype analyses. Analyses were also performed for those who were confirmed as a carrier for a given Haplotype (i.e., had 2 copies of the target allele on each SNP in the haplotype), and only the FAS test was significant for Haplotype 4. No other haplotype analyses were significant.
Discussion
Data presented confirm, in a larger sample, previous reports of poorer cognitive performance and higher evening cortisol levels in PMD patients as compared with non-psychotic depressed patients and healthy controls. PMD patients performed significantly more poorly than did NPMD's and HC's on most measures of cognition, including working memory, executive function, and verbal memory (list learning). Our data that NPMDs performed more similarly to healthy controls is contrary to some previous findings (36, 37) but not all (38). There is variability in the depression literature regarding what cognitive domains are impaired in depression, with severity (39, 40) often being discussed as a main factor that mediates poor functioning. Our findings suggest that although depression level was an important variable in predicting total learning over the 5 trials on the CLVT-II and memory for stories, severity of depression accounted for little variance on most cognitive measures. Thus, severity of depression, which has been hypothesized to be a key factor in the variability of findings in the neuropsychological literature on depression, was not found to play a key role in a variety of cognitive measures in this study. This lack of association is consistent with several other studies (41, 42). Our study, however, included severely depressed patients (both nonpsychotic and psychotic) and as such we cannot comment on possible effect of depression severity across the spectrum.
Cortisol was a major contributor to cognition in the present study. PMD patients demonstrated higher evening cortisol levels than did both NPMD and HC's, who did not differ from each other. However, early morning cortisol did not differ between the groups. Regression analyses suggest that cortisol is potentially a stronger predictor of poor cognition than are clinical symptoms. These data point to a direct effect of cortisol on cognitive function that is independent of symptoms and medication usage. Cortisol accounted for between 5 to 16% of the variance of a variety of cognitive domains, and it generally accounted for higher levels of variance in memory tests. In the present data set, PMD was associated with both higher levels of depression and cortisol. Thus, the higher levels of depression and psychosis may serve as a proxy for higher levels of cortisol in depression and cognition studies.
Finally, genetic variation for NR3C1 (GR) and NR3C2 (MR) also at times contributed significantly to cognition, after accounting for the effects of cortisol. The degree of SNP contributions appears less than or equivalent to that of cortisol itself. In addition, NR3C1 and NR3C2 genetic variation did not predict cognition in the same manner. GR variation appeared more likely to predict attention and executive function tasks, suggesting involvement of the prefrontal cortex or cingulate; whereas MR variation was more likely to predict encoding and retrieval of longer term memories, functions more dependent on the hippocampus and medial temporal region. These results generally parallel GR and MR receptor distribution in primate brain (1) as well as previous reports on their putative roles in specific cognitive functions (3).
Our results suggest roles for NR3C1 polymorphisms in attentional tasks, including the Digit Span Forward, and tasks that require both attention and working memory, such as Digit Span Backward, consistent with Fortier et al. (43), who reported that a specific haplotype of NR3C1 SNP's (including rs6198) was associated with thought and attentional problems in ADHD. Furthermore, as indicated above, El Hage et al. (30) found significant interactions between NR3C1 rs41423247 and COMT polymorphisms on working memory performance in healthy controls.
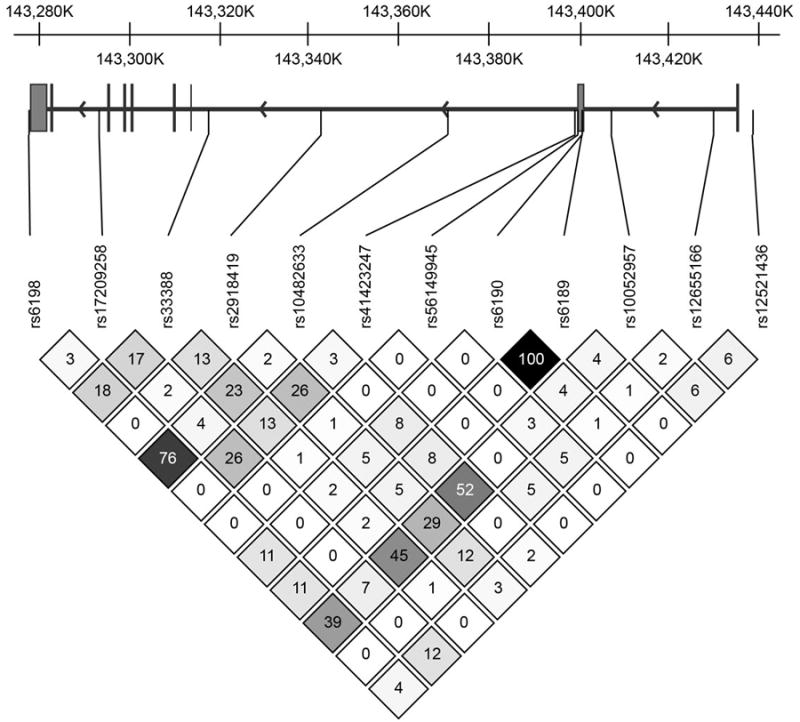
In addition, one of our predictive GR SNP's (i.e., rs10052957) on attentional tasks has been related to metabolic syndrome and metabolic risk factors (44) including higher fasting insulin concentrations and another SNP, rs2918419, contributed to executive function at the p=.001 level. The LD map for NR3C1 (Figure 1) shows that rs10052957 and rs2918419 are in moderate but not complete LD. SNPs rs10052957 and rs2918419 are both intronic, suggesting that they are proxies for one or more functional genetic variants not assessed in our sample.
Figure 1.
Cortisol elevation is well known to cause insulin resistance, and this may explain that metabolic syndrome and Type II diabetes have been found to be more prevalent in patients with recurrent depression as compared to controls (45). Depression may precede Type II diabetes (T2D; (46)), independent of any effects of antidepressant medication (47). T2D is an outcome of a metabolic syndrome; T2D, metabolic syndrome, and depression are all associated with elevated cortisol levels (48). Additionally, elevated cortisol levels are associated with impaired cognition in healthy controls, depressed patients, and T2D (49). T2D may also be associated with hippocampal damage (50). A common genetic pathway underpinning all of these disorders has been hypothesized to stem from genetic variation in the HPA axis (51).
NR3C1 SNP rs6198 in the 3′ UTR region has perhaps been examined more widely than other NR3C1 SNP's, and it has been implicated in risk of major depressive disorder (52), smoking susceptibility (53), binge eating (54) and attentional problems (43). Each of these conditions has a high comorbidity with major depressive disorder, potentially related to shared genetic susceptibility.
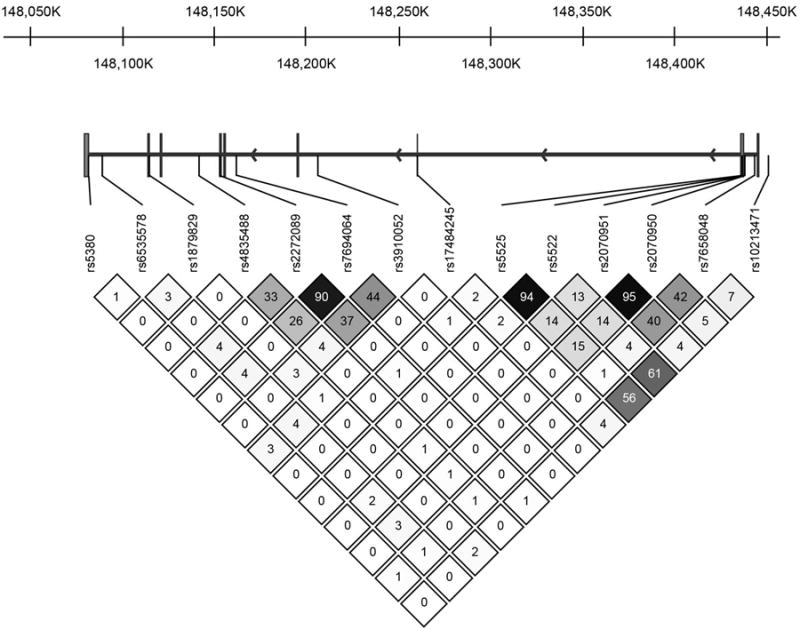
Several NR3C2 SNPs related specifically to verbal memory performance in the present study. Of these, only the rs10213471 promoter SNP might be expected to have a functional effect. The others are hypothesized to be proxies for as yet unknown genetic variants that are in LD. Of the SNP's studied herein, only the rs10212471 promoter SNP and the rs5525 synonymous exon SNP were in moderate in LD. Otherwise the significant NR3C2 SNPs represent largely independent effects (See LD map Figure 2).
Figure 2.
MR has been reported to play a major role in memory consolidation in rodents (3, 18). A number of years ago, we noted the relationships among insomnia, cortisol, and memory impairment in geriatric subjects, suggesting MR involvement since it controls cortisol activity at the nighttime nadir (55). Vogel et al. (56) linked NR3C2 genetic variance in healthy controls to negative memory bias, particularly in those with high life adversity. These findings are consistent with studies that find decreased MR expression in the hippocampus (and prefrontal cortex) in depressed patients (22). Otte and colleagues (20) found that MR stimulation in man with fludrocortisone improved verbal memory and executive functioning compared to placebo. Lastly, Sun and colleagues (25) examined four large datasets on Alzheimer's disease (AD) exploring other genes that can confer risk for AD beyond that of APO-E, and concluded that one of the 10 potential risk genes was NR3C2 (MR).
Beyond cortisol and genes predicting cognitive function, several other seemingly important factors were added to the model, with some influencing cognitive function. For example, gender was a consistent factor in predicting memory performance. In particular, when added to the model with NR3C1, gender predicted between 4.3-7.4% of the variance for memory for lists and stories. However, when in the model with NR3C2, much of the gender effect for story memory appears to be usurped by MR SNPs. Reasons for this finding need to be explored if such a finding is replicable. Medications is another factor which is theorized to play a role in cognition. In this particular case, we used benzodiazepine use because of its likelihood of having an effect on memory specifically. Indeed, benzodiazapine use accounted for about 10% of the variance in list memory, even when NR3C2 was is the model. Thus, medication use and gender factors, if replicable, warrant further understanding in relationship to cortisol and genetics on cognition. Additionally, it is interesting to note differences in findings between the individual SNPs as compared with the haplotype analyses, despite using haplotypes previously observed in the literature (33-35). It is possible that the reductions in sample size seen with using the haplotypes may have contributed to the limited observed significant findings, in part by the ambiguity that comes in inferring or estimating haplotypes (57). Our results are consistent with others who have not found a relationship between haplotypes and clinical features of depression (58) nor with recurrence of major depression (59). The conflicting results speak to the need for enhancement of genetic methods in psychiatric research, given the relatively small sample sizes in these populations.
The limited genetic research on cognition in psychiatric disorders, often point to interactions, particularly with environment, stress, and other genes. This is particularly true for NR3C1 and FKBP5. Although several other important models could be examined, including the interactions of NR3C1 and NR3C2 genes, FKBP5, and COMT genes, the available sample size with complete data (cognition, ratings, cortisol, and genetics), precluded such important analyses, as did our lack of early childhood trauma data. Consequently, we decided to conduct more exploratory, forward regression analyses for each gene (GR and MR) separately. Further testing on larger, independent samples is needed. to confirm the present findings and to extend upon them in understand the inherently complex interactions of genes that encode for specific receptors of the HPA axis as well as those that encode for receptors and enzymes of other neurotransmitter systems.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
Aspects of this study were supported by grants from the Pritzker Foundation, NIH MH50604 to Alan Schatzberg, NIH MH19938 to Alan Schatzberg, and NIH/NCRR CTSA award number UL1 RR025744.The clinical trial registration number was NCT00576095.
Footnotes
Conflict of Interest: Although there is not a conflict of interest, there may appear to be one with Dr. Schatzberg. He has a significant interest in Corcept Therapeutics, Inc., which licensed the use patent for mifepristone in psychotic major depression. Some of the data presented here were part of a larger study examining the effectiveness of mifepristone in psychotic major depression. No data on mifepristone's effectiveness are presented in the submitted report.
Several authors have been named on a use patent related to the SNPs reported in this manuscript, including Alan Schatzberg, Greer Murphy, and Jennifer Keller.
Dr. Greer Murphy has been a consultant for Brain Resource, Ltd.
References
- 1.Patel PD, Lopez JF, Lyons DM, Burke S, Wallace M, Schatzberg AF. Glucocorticoid and mineralocorticoid receptor mRNA expression in squirrel monkey brain. J Psychiatr Res. 2000;34(6):383–92. doi: 10.1016/s0022-3956(00)00035-2. [DOI] [PubMed] [Google Scholar]
- 2.de Kloet ER, Oitzl MS, Joels M. Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci. 1999;22(10):422–6. doi: 10.1016/s0166-2236(99)01438-1. [DOI] [PubMed] [Google Scholar]
- 3.de Kloet ER, Reul JM. Feedback action and tonic influence of corticosteroids on brain function: a concept arising from the heterogeneity of brain receptor systems. Psychoneuroendocrinology. 1987;12(2):83–105. doi: 10.1016/0306-4530(87)90040-0. [DOI] [PubMed] [Google Scholar]
- 4.Young EA, Lopez JF, Murphy-Weinberg V, Watson SJ, Akil H. Mineralocorticoid receptor function in major depression. Arch Gen Psychiatry. 2003;60(1):24–8. doi: 10.1001/archpsyc.60.1.24. [DOI] [PubMed] [Google Scholar]
- 5.Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23(5):477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 6.Murphy BE. Steroids and depression. J Steroid Biochem Mol Biol. 1991 May;38(5):537–59. doi: 10.1016/0960-0760(91)90312-s. 1991;38(5):537-59. [DOI] [PubMed] [Google Scholar]
- 7.Deuschle M, Gotthardt U, Schweiger U, Weber B, Korner A, Schmider J, et al. With aging in humans the activity of the hypothalamus-pituitary-adrenal system increases and its diurnal amplitude flattens. Life Sci. 1997;61(22):2239–46. doi: 10.1016/s0024-3205(97)00926-0. [DOI] [PubMed] [Google Scholar]
- 8.Pfohl B, Sherman B, Schlechte J, Stone R. Pituitary-adrenal axis rhythm disturbances in psychiatric depression. Arch Gen Psychiatry. 1985;42(9):897–903. doi: 10.1001/archpsyc.1985.01790320069009. [DOI] [PubMed] [Google Scholar]
- 9.Yehuda R, Teicher MH, Trestman RL, Levengood RA, Siever LJ. Cortisol regulation in posttraumatic stress disorder and major depression: a chronobiological analysis. Biol Psychiatry. 1996;40(2):79–88. doi: 10.1016/0006-3223(95)00451-3. [DOI] [PubMed] [Google Scholar]
- 10.Belanoff JK, Kalehzan M, Sund B, Fleming Ficek SK, Schatzberg AF. Cortisol activity and cognitive changes in psychotic major depression. Am J Psychiatry. 2001;158(10):1612–6. doi: 10.1176/appi.ajp.158.10.1612. [DOI] [PubMed] [Google Scholar]
- 11.Keller J, Flores B, Gomez RG, Solvason HB, Kenna H, Williams GH, et al. Cortisol circadian rhythm alterations in psychotic major depression. Biol Psychiatry. 2006;60(3):275–81. doi: 10.1016/j.biopsych.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 12.Lembke A, Gomez R, Tenakoon L, Keller J, Cohen G, Williams GH, et al. The mineralocorticoid receptor agonist, fludrocortisone, differentially inhibits pituitary-adrenal activity in humans with psychotic major depression. Psychoneuroendocrinology. 2013;38(1):115–21. doi: 10.1016/j.psyneuen.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson JC, Davis JM. DST studies in psychotic depression: a meta-analysis. Am J Psychiatry. 1997;154(11):1497–503. doi: 10.1176/ajp.154.11.1497. [DOI] [PubMed] [Google Scholar]
- 14.Grossmann C, Scholz T, Rochel M, Bumke-Vogt C, Oelkers W, Pfeiffer AF, et al. Transactivation via the human glucocorticoid and mineralocorticoid receptor by therapeutically used steroids in CV-1 cells: a comparison of their glucocorticoid and mineralocorticoid properties. Eur J Endocrinol. 2004;151(3):397–406. doi: 10.1530/eje.0.1510397. [DOI] [PubMed] [Google Scholar]
- 15.Rock PL, Roiser JP, Riedel WJ, Blackwell AD. Cognitive impairment in depression: a systematic review and meta-analysis. Psychol Med. 2013:1–12. doi: 10.1017/S0033291713002535. [DOI] [PubMed] [Google Scholar]
- 16.Gomez RG, Fleming SH, Keller J, Flores B, Kenna H, DeBattista C, et al. The neuropsychological profile of psychotic major depression and its relation to cortisol. Biol Psychiatry. 2006;60(5):472–8. doi: 10.1016/j.biopsych.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 17.Lupien SJ, Gillin CJ, Hauger RL. Working memory is more sensitive than declarative memory to the acute effects of corticosteroids: a dose-response study in humans. Behav Neurosci. 1999;113(3):420–30. doi: 10.1037//0735-7044.113.3.420. [DOI] [PubMed] [Google Scholar]
- 18.Joels M, Karst H, DeRijk R, de Kloet ER. The coming out of the brain mineralocorticoid receptor. Trends Neurosci. 2008;31(1):1–7. doi: 10.1016/j.tins.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Rimmele U, Besedovsky L, Lange T, Born J. Blocking mineralocorticoid receptors impairs, blocking glucocorticoid receptors enhances memory retrieval in humans. Neuropsychopharmacology. 2013;38(5):884–94. doi: 10.1038/npp.2012.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otte C, Wingenfeld K, Kuehl LK, Kaczmarczyk M, Richter S, Quante A, et al. Mineralocorticoid Receptor Stimulation Improves Cognitive Function and Decreases Cortisol Secretion in Depressed Patients and Healthy Individuals. Neuropsychopharmacology. 2014 doi: 10.1038/npp.2014.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Otte C, Hinkelmann K, Moritz S, Yassouridis A, Jahn H, Wiedemann K, et al. Modulation of the mineralocorticoid receptor as add-on treatment in depression: a randomized, double-blind, placebo-controlled proof-of-concept study. J Psychiatr Res. 2010;44(6):339–46. doi: 10.1016/j.jpsychires.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Medina A, Seasholtz AF, Sharma V, Burke S, Bunney W, Jr, Myers RM, et al. Glucocorticoid and mineralocorticoid receptor expression in the human hippocampus in major depressive disorder. J Psychiatr Res. 2013;47(3):307–14. doi: 10.1016/j.jpsychires.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucassen PJ, Heine VM, Muller MB, van der Beek EM, Wiegant VM, De Kloet ER, et al. Stress, depression and hippocampal apoptosis. CNS Neurol Disord Drug Targets. 2006;5(5):531–46. doi: 10.2174/187152706778559273. [DOI] [PubMed] [Google Scholar]
- 24.Bortoluzzi A, Salum GA, Blaya C, Silveira PP, Grassi-Oliveira R, Dias da Rosa E, et al. J Psychiatr Res. 2014. Mineralocorticoid receptor genotype moderates the association between physical neglect and serum BDNF. [DOI] [PubMed] [Google Scholar]
- 25.Sun J, Song F, Wang J, Han G, Bai Z, Xie B, et al. Hidden risk genes with high-order intragenic epistasis in Alzheimer's disease. J Alzheimers Dis. 2014;41(4):1039–56. doi: 10.3233/JAD-140054. [DOI] [PubMed] [Google Scholar]
- 26.Seckl JR, Dickson KL, Yates C, Fink G. Distribution of glucocorticoid and mineralocorticoid receptor messenger RNA expression in human postmortem hippocampus. Brain Res. 1991;561(2):332–7. doi: 10.1016/0006-8993(91)91612-5. [DOI] [PubMed] [Google Scholar]
- 27.Gomez-Sanchez EP, Gomez-Sanchez CE. Central regulation of blood pressure by the mineralocorticoid receptor. Mol Cell Endocrinol. 2012;350(2):289–98. doi: 10.1016/j.mce.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gil-Bea FJ, Aisa B, Solomon A, Solas M, del Carmen Mugueta M, Winblad B, et al. HPA axis dysregulation associated to apolipoprotein E4 genotype in Alzheimer's disease. J Alzheimers Dis. 2010;22(3):829–38. doi: 10.3233/JAD-2010-100663. [DOI] [PubMed] [Google Scholar]
- 29.van Rossum EF, van den Akker EL. Glucocorticoid resistance. Endocr Dev. 2011;20:127–36. doi: 10.1159/000321234. [DOI] [PubMed] [Google Scholar]
- 30.El-Hage W, Phillips ML, Radua J, Gohier B, Zelaya FO, Collier DA, et al. Genetic modulation of neural response during working memory in healthy individuals: interaction of glucocorticoid receptor and dopaminergic genes. Mol Psychiatry. 2013;18(2):174–82. doi: 10.1038/mp.2011.145. [DOI] [PubMed] [Google Scholar]
- 31.Young AH, Gallagher P, Watson S, Del-Estal D, Owen BM, Ferrier IN. Improvements in neurocognitive function and mood following adjunctive treatment with mifepristone (RU-486) in bipolar disorder. Neuropsychopharmacology. 2004;29(8):1538–45. doi: 10.1038/sj.npp.1300471. [DOI] [PubMed] [Google Scholar]
- 32.Schatzberg AF, Keller J, Tennakoon L, Lembke A, Williams G, Kraemer FB, et al. HPA axis genetic variation, cortisol and psychosis in major depression. Mol Psychiatry. 2014;19(10):1151. doi: 10.1038/mp.2013.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumsta R, Entringer S, Koper JW, van Rossum EF, Hellhammer DH, Wust S. Sex specific associations between common glucocorticoid receptor gene variants and hypothalamus-pituitary-adrenal axis responses to psychosocial stress. Biol Psychiatry. 2007;62(8):863–9. doi: 10.1016/j.biopsych.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 34.de Kloet ER, Otte C, Kumsta R, Kok L, Hillegers MH, Hasselmann H, et al. STRESS and DEPRESSION a crucial role of the mineralocorticoid receptor. J Neuroendocrinol. 2016 doi: 10.1111/jne.12379. [DOI] [PubMed] [Google Scholar]
- 35.Hamstra DA, de Kloet ER, van Hemert AM, de Rijk RH, Van der Does AJ. Mineralocorticoid receptor haplotype, oral contraceptives and emotional information processing. Neuroscience. 2015;286:412–22. doi: 10.1016/j.neuroscience.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 36.Naismith SL, Longley WA, Scott EM, Hickie IB. Disability in major depression related to self-rated and objectively-measured cognitive deficits: a preliminary study. BMC Psychiatry. 2007;7:32. doi: 10.1186/1471-244X-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reppermund S, Ising M, Lucae S, Zihl J. Cognitive impairment in unipolar depression is persistent and non-specific: further evidence for the final common pathway disorder hypothesis. Psychol Med. 2009;39(4):603–14. doi: 10.1017/S003329170800411X. [DOI] [PubMed] [Google Scholar]
- 38.Wang CE, Halvorsen M, Sundet K, Steffensen AL, Holte A, Waterloo K. Verbal memory performance of mildly to moderately depressed outpatient younger adults. J Affect Disord. 2006;92(2-3):283–6. doi: 10.1016/j.jad.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 39.McClintock SM, Husain MM, Greer TL, Cullum CM. Association between depression severity and neurocognitive function in major depressive disorder: a review and synthesis. Neuropsychology. 2010;24(1):9–34. doi: 10.1037/a0017336. [DOI] [PubMed] [Google Scholar]
- 40.McDermott LM, Ebmeier KP. A meta-analysis of depression severity and cognitive function. J Affect Disord. 2009;119(1-3):1–8. doi: 10.1016/j.jad.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 41.Landro NI, Stiles TC, Sletvold H. Neuropsychological function in nonpsychotic unipolar major depression. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(4):233–40. [PubMed] [Google Scholar]
- 42.Murphy FC, Michael A, Robbins TW, Sahakian BJ. Neuropsychological impairment in patients with major depressive disorder: the effects of feedback on task performance. Psychol Med. 2003;33(3):455–67. doi: 10.1017/s0033291702007018. [DOI] [PubMed] [Google Scholar]
- 43.Fortier ME, Sengupta SM, Grizenko N, Choudhry Z, Thakur G, Joober R. Genetic evidence for the association of the hypothalamic-pituitary-adrenal (HPA) axis with ADHD and methylphenidate treatment response. Neuromolecular Med. 2013;15(1):122–32. doi: 10.1007/s12017-012-8202-1. [DOI] [PubMed] [Google Scholar]
- 44.Yan YX, Dong J, Zhang J, Liu F, Wang W, Zhang L, et al. Polymorphisms in NR3C1 gene associated with risk of metabolic syndrome in a Chinese population. Endocrine. 2014 doi: 10.1007/s12020-014-0324-9. [DOI] [PubMed] [Google Scholar]
- 45.Lasic D, Bevanda M, Bosnjak N, Uglesic B, Glavina T, Franic T. Metabolic syndrome and inflammation markers in patients with schizophrenia and recurrent depressive disorder. Psychiatr Danub. 2014;26(3):214–9. [PubMed] [Google Scholar]
- 46.Mezuk B, Eaton WW, Albrecht S, Golden SH. Depression and type 2 diabetes over the lifespan: a meta-analysis. Diabetes Care. 2008;31(12):2383–90. doi: 10.2337/dc08-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pan A, Lucas M, Sun Q, van Dam RM, Franco OH, Manson JE, et al. Bidirectional association between depression and type 2 diabetes mellitus in women. Arch Intern Med. 2010;170(21):1884–91. doi: 10.1001/archinternmed.2010.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vinson GP. Angiotensin II, corticosteroids, type II diabetes and the metabolic syndrome. Med Hypotheses. 2007;68(6):1200–7. doi: 10.1016/j.mehy.2006.09.065. [DOI] [PubMed] [Google Scholar]
- 49.Bruehl H, Rueger M, Dziobek I, Sweat V, Tirsi A, Javier E, et al. Hypothalamic-pituitary-adrenal axis dysregulation and memory impairments in type 2 diabetes. J Clin Endocrinol Metab. 2007;92(7):2439–45. doi: 10.1210/jc.2006-2540. [DOI] [PubMed] [Google Scholar]
- 50.Gold SM, Dziobek I, Sweat V, Tirsi A, Rogers K, Bruehl H, et al. Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia. 2007;50(4):711–9. doi: 10.1007/s00125-007-0602-7. [DOI] [PubMed] [Google Scholar]
- 51.Gragnoli C. Hypothesis of the neuroendocrine cortisol pathway gene role in the comorbidity of depression, type 2 diabetes, and metabolic syndrome. Appl Clin Genet. 2014;7:43–53. doi: 10.2147/TACG.S39993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szczepankiewicz A, Leszczynska-Rodziewicz A, Pawlak J, Rajewska-Rager A, Dmitrzak-Weglarz M, Wilkosc M, et al. Glucocorticoid receptor polymorphism is associated with major depression and predominance of depression in the course of bipolar disorder. J Affect Disord. 2011;134(1-3):138–44. doi: 10.1016/j.jad.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 53.Rovaris DL, Mota NR, de Azeredo LA, Cupertino RB, Bertuzzi GP, Polina ER, et al. MR and GR functional SNPs may modulate tobacco smoking susceptibility. J Neural Transm. 2013;120(10):1499–505. doi: 10.1007/s00702-013-1012-2. [DOI] [PubMed] [Google Scholar]
- 54.Cellini E, Castellini G, Ricca V, Bagnoli S, Tedde A, Rotella CM, et al. Glucocorticoid receptor gene polymorphisms in Italian patients with eating disorders and obesity. Psychiatr Genet. 2010;20(6):282–8. doi: 10.1097/YPG.0b013e32833a2142. [DOI] [PubMed] [Google Scholar]
- 55.Buckley TM, Schatzberg AF. Aging and the role of the HPA axis and rhythm in sleep and memory-consolidation. Am J Geriatr Psychiatry. 2005;13(5):344–52. doi: 10.1176/appi.ajgp.13.5.344. [DOI] [PubMed] [Google Scholar]
- 56.Vogel S, Gerritsen L, van Oostrom I, Arias-Vasquez A, Rijpkema M, Joels M, et al. Linking genetic variants of the mineralocorticoid receptor and negative memory bias: interaction with prior life adversity. Psychoneuroendocrinology. 2014;40:181–90. doi: 10.1016/j.psyneuen.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 57.Liu N, Zhang K, Zhao H. Haplotype-association analysis. Adv Genet. 2008;60:335–405. doi: 10.1016/S0065-2660(07)00414-2. [DOI] [PubMed] [Google Scholar]
- 58.Leszczynska-Rodziewicz A, Szczepankiewicz A, Dmitrzak-Weglarz M, Rajewska-Rager A, Skibinska M, Hauser J. No association between polymorphisms and haplotypes of the AVPR1b, CRHR1 and NR3C1 genes and depression with melancholic features in the course of bipolar disorder. Psychiatry Res. 2013;207(1-2):140–2. doi: 10.1016/j.psychres.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 59.Hardeveld F, Spijker J, Peyrot WJ, de Graaf R, Hendriks SM, Nolen WA, et al. Glucocorticoid and mineralocorticoid receptor polymorphisms and recurrence of major depressive disorder. Psychoneuroendocrinology. 2015;55:154–63. doi: 10.1016/j.psyneuen.2015.02.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.