

Abstract
Conventional cytogenetics can categorize patients with acute myeloid leukemia (AML) into favorable, intermediate, and unfavorable‐risk groups; however, patients with intermediate‐risk cytogenetics represent the major population with variable outcomes. Because molecular profiling can assist with AML prognosis and next‐generation sequencing allows simultaneous sequencing of many target genes, we analyzed 260 genes in 112 patients with de novo AML who received standard treatment. Multivariate analysis showed that karyotypes and mutation status of TET2,PHF6,KIT, and NPM1 mutation/FLT3‐ internal tandem duplication (ITD)negative were independent prognostic factors for the entire cohort. Among patients with intermediate‐risk cytogenetics, patients with mutations in CEBPA double mutation, IDH2, and NPM1 in the absence of FLT3‐ITD were associated with improved Overall survival (OS), similar to those with favorable‐risk cytogenetics; patients with mutations in TET2,RUNX1,ASXL1, and DNMT3A were associated with reduced OS, similar to those with unfavorable‐risk cytogenetics. We concluded that integration of cytogenetic and molecular profiling improves prognostic stratification of patients into three groups with more distinct prognoses (P < 0.001) and significantly reduces the number of patients classified as intermediate risk. In addition, our study demonstrates that next‐generation sequencing (NGS)‐based multi‐gene sequencing is clinically applicable in establishing an accurate risk stratification system for guiding therapeutic decisions.
Keywords: Acute myeloid leukemia, gene mutations, next‐generation sequencing, precision medicine, prognosis
Introduction
Acute myeloid leukemia (AML) is a heterogeneous disease that is characterized by impaired differentiation and increased proliferation of immature myeloid cells. For adult AML patients receiving intensive treatment, the 5‐year survival rate is only ~30% 1. The prognosis of AML patients can be classified as favorable, intermediate, and unfavorable groups according to recurrent cytogenetic and genetic abnormalities 2, 3. Patients with a cytogenetic profile that includes the translocation of PML‐RARA [t(15;17) (q24;q21)], RUNX1‐RUNX1T [t(8;21)(q22;q22)], or CBFB‐MYH11 [inv(16) or t(16;16)(p13.1;q22)] are classified as favorable‐risk group and have good outcomes with chemotherapy‐based consolidation treatment 4, 5. Patients with complex cytogenetic changes are classified as unfavorable group and have a poor prognosis 5. Allogeneic hematopoietic stem cell transplantation (HSCT) may be required to improve the outcome of the unfavorable‐risk patients 6. However, half of AML patients belong to an intermediate‐risk group, and most of their leukemia has normal karyotypes 5. Recent translational researches show that mutation profiling of several genes, including FLT3, NPM1, KIT, RAS, CEBPA, IDH1, IDH2, and TET2, provides prognostic prediction and treatment guidance for patients with normal karyotypes 2, 3, 7, 8, 9. For example, patients with NPM1 mutation without FLT3‐internal tandem duplication (ITD) have a favorable prognosis 10, whereas patients with AXSL1 or TET2 mutation have a poor prognosis 11, 12. For accurate risk stratification, the current consensus suggests that cytogenetic and genomic mutation analyses should be integrated for prognostic and therapeutic decisions regarding AML patients 3.
Because next‐generation sequencing (NGS) technology enables parallel analysis of many genes, NGS is used not only in research but also in clinical molecular diagnosis 13, 14. This strategy may solve the challenges of multiple gene screening from conventional platforms. However, the number of genes that should be screened for AML patients is not clear, and it would be beneficial to know whether NGS could define a new genetic mutation profile to serve as a prognostic indicator in AML patients. Previous whole‐genome and exome analyses have demonstrated recurrent mutations in 260 genes in 200 AML patients 15; however, the prognostic impact of these genes remains unclear. Therefore, we used a sequencing panel containing these 260 genes to screen mutations in the 112 patients. First, we demonstrated the clinical feasibility of NGS to the molecular diagnosis of AML. Second, we searched for novel prognostic factors and would like to establish a precise molecular classification based on the integration of cytogenetic and molecular alterations.
Materials and Methods
Patients
The diagnosis of AML was based on the definition of World Health Organization. All of the enrolled patients received standard chemotherapy with or without allogeneic HSCT as previously described. The diagnosis of AML was based on the World Health Organization definition, and all of the enrolled patients received standard chemotherapy with or without allogeneic HSCT as described 16. The mononuclear cells of each bone marrow sample were also collected and cryopreserved in the biobank after the patients had signed informed consent. This study was approved by the Institutional Review Board of China Medical University Hospital (DMR101‐IRB2‐020).
Constructing a shotgun genomic sequencing library
Genomic DNA (gDNA) was isolated from bone marrow mononuclear cells using the QIAGEN Genomic DNA extraction kit. The purities and concentrations of gDNA were confirmed by electrophoresis, Nanodrop 2000 (Thermo Scientific, USA), and a Qubit 2.0 Fluorometer (Life Technologies, USA). Double‐stranded DNA (dsDNA; 2 μg) that passed the quality‐control steps was sheared to ~300 bp with an M220 focused ultrasonicator (Covaris, USA). Size distribution of the fragmented DNA was confirmed using a Bioanalyzer 2100 (Agilent Technologies, USA), and shotgun genomic libraries for use with the MiSeq platform (Illumina, USA) were generated using the KAPA Library Preparation kit (Kapa Biosystems, USA) according to the manufacturer's protocol.
Capture‐based NGS
To test if capture‐based target‐enrichment NGS is applicable, AML genetic testing, the xGen® AML Cancer Panel v1.0 containing 11,743 xGen Lockdown® probes was purchased from Integrated DNA Technologies (USA) which targeted important exons of the AML disease pathway related genes (Table S1) 15. A total of ~1.2 Mbp of gDNA target regions from 6235 exons of genes related to AML were used to design probes. For each capture reaction, multiplex libraries containing 13 libraries pooled equally were used for probe hybridization, and target enrichment was performed according to the Integrated DNA Technologies—optimized xGen 4‐h capture protocol. The libraries were then purified with AMPure XP beads for MiSeq sequencing.
MiSeq high‐throughput sequencing and data processing
The concentrations of captured libraries were determined by real‐time quantitative PCR with Illumina adapter‐specific primers provided with the KAPA Biosystems library quantification kit. Libraries were denatured and sequenced on the MiSeq platform with v2 reagent for paired‐end sequencing (2 × 150 bp). Instrument control, cluster generation, image capture, and base calling were processed with Real Time Analysis software 1.18.54, MiSeq Control software 2.4.1.3, and MiSeq Report software 2.4.60.8. FASTQ files generated by MiSeq Report were used for further analysis.
The post‐NGS bioinformatics was described previously 17. The FASTQ files were aligned to the human reference genome (February 2009, GRCh37/hg19) using the BWA‐MEM algorithm in BWA software (version 0.7.4) 18. Picard tools (version 1.90) were used to perform the necessary data conversion, sorting, and indexing 19. GATK software (version 2.5‐2) was used for variant identification including the UnifiedGenotyper and HaplotypeCaller tools for variant calling and the VariantFiltration tool for variant filtration 20. Gene annotation, amino acid change annotation, SIFT and PolyPhen2 scores, dbSNP identifiers (dbSNP 138), 1000 Genomes Project allele frequencies, and NHLBI‐ESP 6500 exome project allele frequencies of filtered variants were annotated with ANNOVAR (2014‐OCt) 21. In addition to the analysis mentioned above, BAM files were further analyzed by Pindel (version 0.2.4) for FLT3‐ITD identification (Figure S1) 22, 23, 24, 25.
Variant filtration
After annotations, variants were interpreted mainly based on ACMG guideline 26. The frameshift insertion or deletion (indel) variants, nonsense variants, and splice‐site variants with allele frequencies <1% in both the 1000 Genomes Project and NHLBI‐ESP 6500 exome project were included for further analysis. SIFT and PolyPhen2 scores were used to evaluate the effects of specific missense variants on the protein 27, 28; only missense variants with scores >0.95 in PolyPhen2 and SIFT scores <0.05 were included for further analysis. However, due to lack of germline data, the rare germline variants may be falsely considered as the tumor mutations. Therefore, we used Taiwan genomics data (N = 997, https://taiwanview.twbiobank.org.tw/search) to exclude the germline variants which are presented in Taiwan population but rare in Western people.
In addition, previous studies had well established, the definition of pathogenic mutations of NPM1, FLT3, and CEBPA 29, 30, 31. The four nucleotide insertion in exon12 of NPM1 results in dis‐localization of NPM130. Both internal tandem duplication and D835 mutation in FLT3 cause activated transduction signaling 29. The genetic variant of CEBPA is usually a nonframeshift insertion or deletion and the pathogenic mutations are commonly located at transactivation domain (TAD) 1, 2 and basic region mediating DNA binding leucine zipper (bZIP) region. A common benign polymorphism is an in‐frame 6‐bp insertion (ACCCGC) in the transactivation domain 2 (TAD2), resulting in a His‐Pro duplication (HP196–197 insertion)31.
Statistics
Overall survival (OS) was estimated by Kaplan–Meier analysis. The chi‐squared test and Fisher's exact test were used to calculate the significance of variances between each group. Cox proportional hazard regression analysis was used to estimate the hazard ratio (HR) of OS and corresponding 95% confidence interval (CI) for various genetic alterations. All P‐values are two‐sided, and P < 0.05 was considered as significant.
Results
Patient characteristics
The study's 112 patients comprised 45 females and 67 males of median age 42.6 years (range, 11.7–79.0 years). There were 5, 21, 37, 9, 21, 6, 2, 1, and 10 patients diagnosed as AML with M0, M1, M2, M3, M4, M5, M6, M7, and undetermined types, respectively, according to French–American–British classification. Based on cytogenetic data, 22 patients (19.6%) were in the favorable‐risk group, 69 (61.6%) in the intermediate‐risk group, and 21 (18.8%) in the unfavorable‐risk group at initial diagnosis. Nineteen patients received allogeneic HSCT. Table S2 lists the clinical information of the enrolled patients.
Capture enrichment and NGS performance
For all NGS data, an average of 2.65 ± 0.33 million reads that mapped to the reference genome (hg19, GRCh37) were generated per patient, with ~80.1% of reads (range, 74.6–83.3%) mapping to the ~1.2‐Mbp target region. The average mean coverage for the targeted exons was 185.4 ± 23.7 (range, 108.8–263.9), and >0.2 × mean coverage was observed for >96.6% of targets; 94.9 ± 5.9% of the exons had a coverage of ≥50 reads, and the median fragment length was 194 bp (range, 165–216).
Mutation profiling and gene–gene association
Among the 260 gene analyses of the 112 patients, we identified 1926 deleterious mutations, including single‐nucleotide variants (SNVs) and small indels in coding regions, averaging 17.2 mutations per patient (range, 3–185). Forty of the 260 genes can be categorized as DNA methylation, tumor suppressor, activated signaling, myeloid transcriptional factor, chromatin modifier, and spliceosome; these genes and NPM1 are involved in the leukemogenesis and previous studies showed that most of them might be associated with the prognosis of AML patients (Fig. 1)3, 15. Among them, the most commonly mutated gene was FLT3‐ITD (21.4%), followed by ASXL1 (16.1%), NPM1 (15.1%), CEBPA (15.1%), DNMT3A (12.5%), IDH2 (12.5%), WT1 (11.6%), and TET2 (10.7%). The frequency of double allele of CEBPA (CEBPA double mutation) was 6.25%. Other genes had a mutation prevalence of <10%.
Figure 1.

Landscape of mutations in 112 AML patients. (A) A computation plot shows cytogenetic risks and pathogenic mutations in 40 individual genes and sets of genes, grouped into seven categories, as labeled on the left. Mutation frequency for the 112 patients is illustrated in the right bar graph. For cytogenetics, pink boxes indicate favorable‐risk, red boxes indicate intermediate‐risk, and maroon boxes indicate unfavorable‐risk cytogenetics. Single mutations in a gene are labeled as green boxes, and plural mutations in a gene are labeled as yellow boxes. Each column represents data for 1 of the 112 subjects. (B) Relationship between the three cytogenetic groups and the mutation frequency of each gene. CEBPA* represents double mutation of CEBPA (CEBPA double mutation), and CEBPA stands for incidence of all kinds of mutations. AML, acute myeloid leukemia.
Stratified by cytogenetic classification, it was a higher trend to incur mutations of the 260 genes in patients with unfavorable‐risk cytogenetics than in those with favorable‐and intermediate‐risk cytogenetics. The mutation rate of each of TP53, GATA2, and U2AF1 was significantly higher in patients with unfavorable cytogenetics; KIT mutation predominated in patients with favorable‐risk cytogenetics; mutation of CEBPA double mutation was found in patients with intermediate‐risk cytogenetics. Among the 69 patients with intermediate‐risk cytogenetics, the most frequent mutation was FLT3‐ITD (n = 17, 24.6%), followed by mutations in NPM1 (n = 13, 18.8%), DNMT3A (15.9%), and IDH2 (15.9%). The frequency of CEBPA double mutation was 10.1%.
Pairwise mutation analysis was performed in these 40 genes to identify co‐occurring mutations and mutations that occurred exclusively (Table S3). NPM1 mutation was found to significantly co‐occur with FLT3, PTPN11, PRF8, and SF3B1 mutation. IDH2 mutation significantly co‐occurred with mutation of DNMT3A, JAK1, JAK2, JAK3, AXSL1, and U2AF1; IDH2 mutated exclusively with TET2 mutation.
Cytogenetic and genetic alterations affecting complete remission
We assessed the value of cytogenetic and genetic mutations for predicting the remission rate of AML. Patients with favorable‐risk cytogenetics had higher complete remission (CR) rates than those with intermediate‐ and unfavorable‐risk cytogenetics (Fig. 2A, P < 0.001). TP53 (P < 0.001) and U2AF1 (P = 0.025) mutations were identified as unfavorable factors associated with low CR rate (Fig. 2B and C).
Figure 2.
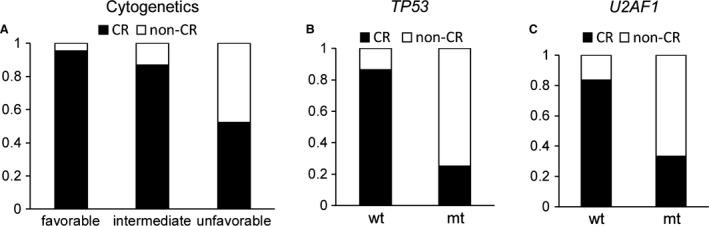
Cytogenetic and genomic lesion associations in response to induction chemotherapy. Complete remission (CR) rate stratified by (A) the three cytogenetic groups (P < 0.001) and (B, C) mutation status of TP53 (P < 0.001) and U2AF1 (P = 0.025). (wt, wild type; mt, mutant.)
Survival analysis according to cytogenetic risk and current known genes
The median follow‐up for the entire cohort was 23.0 months, and 49 surviving patients were followed up for 75.8 months. The 5‐year OS rate was 40.7% [95% CI: 31.6–50.1%]. In the conventional karyotype stratification, the 5‐year OS for patients with favorable‐, intermediate‐, and unfavorable‐risk cytogenetics was 54.5%, 44.7%, and 10.9%, respectively (Fig. 3A, P = 0.004).
Figure 3.
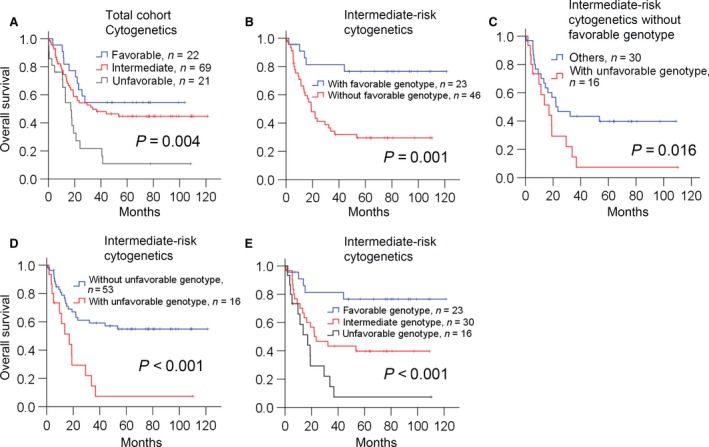
Overall survival estimated by Kaplan–Meier analysis according to (A) cytogenetic risks in the entire cohort. (B) favorable genotype in patients with intermediate‐risk cytogenetics. (C) unfavorable genotype in patients with intermediate‐risk cytogenetics. (D) unfavorable genotype in patients with intermediate‐risk cytogenetics without favorable genotype. (E) revised classification system in patients with intermediate‐risk cytogenetics.
In order to know whether NGS could be a suitable method to identify mutations for serving clinical prognostic indicators in AML patients, we first examined the relationship between survival and mutation profile of the 40 genes, most of which had been reported to be associated with AML prognosis (Fig. 1, Table 1 and Table S4)3. Univariate analysis for all 112 patients revealed that U2AF1 mutation was associated with a significantly worse OS (HR = 4.293, 95% CI: 1.322–13.94, P = 0.015). Mutation of other genes, including KIT, PHF6, TP53, RUNX1, TET2, ASXL1, and FLT3‐ITD, were associated with a nonsignificant trend of reduced OS, whereas mutation of several genes, including CEBPA double mutation, IDH1, IDH2, and NPM1 were trended to correlate with a prolonged survival (Table 1). In the multivariate analysis (Table 1), the independent poor risk factors were karyotypes, mutations in TET2, PHF6, and KIT. The factor of NPM1 mutation in the absence of FLT3‐ITD (NPM1 mutation/FLT3‐ITDnegative) was independently correlated with better prognosis. Mutation of IDH2 or CEBPA double mutation was associated with prolonged OS (Table 1).
Table 1.
Cox regression hazard analysis on the overall survival in the entire cohort
| Variables | N | Univariate | Multivariate | ||
|---|---|---|---|---|---|
| HR (95% CI) | P‐value | HR (95% CI) | P‐value | ||
| Karyotypes | |||||
| Favorable | 22 | Reference | Reference | ||
| Intermediate | 69 | 1.380 (0.685–2.782) | 0.368 | 4.339 (1.559–12.076) | 0.005 |
| Unfavorable | 21 | 3.147 (1.433–6.909) | 0.004 | 7.024 (2.340–21.088) | 0.001 |
| Genetic alterations | |||||
| CEBPA* | 7 | 0.410 (0.100–1.679) | 0.215 | 0.378 (0.089–1.610) | 0.188 |
| DNMT3A | 14 | 0.460 (0.184–1.148) | 0.096 | 0.622 (0.188–2.051) | 0.435 |
| IDH2 | 14 | 0.481 (0.192–1.200) | 0.117 | 0.307 (0.083–1.135) | 0.077 |
| IDH1 | 4 | 0.771 (0.188–3.154) | 0.718 | 3.651 (0.747–18.858) | 0.110 |
| GATA2 | 7 | 0.839 (0.263–2.676) | 0.767 | 0.291 (0.055–1.530) | 0.145 |
| NPM1 | 17 | 0.945 (0.450–1.985) | 0.882 | – | – |
| WT1 | 13 | 0.987 (0.425–2.291) | 0.976 | 1.074 (0.438–2.635) | 0.876 |
| FLT3‐ITD | 24 | 1.223 (0.684–2.186) | 0.497 | – | – |
| ASXL1 | 18 | 1.242 (0.648–2.381) | 0.514 | 1.089 (0.524–2.264) | 0.819 |
| TET2 | 12 | 1.648 (0.784–3.462) | 0.188 | 3.740 (1.598–8.750) | 0.002 |
| RUNX1 | 7 | 1.815 (0.781–4.220) | 0.166 | 2.037 (0.821–5.050) | 0.125 |
| TP53 | 9 | 2.043 (0.816–5.113) | 0.127 | 1.916 (0.628–5.842) | 0.253 |
| PHF6 | 3 | 2.061 (0.645–6.584) | 0.223 | 6.016 (1.255–28.844) | 0.025 |
| KIT | 5 | 2.429 (0.965–6.117) | 0.060 | 12.131 (3.175–46.358) | <0.001 |
| U2AF1 | 3 | 4.293 (1.322–13.94) | 0.015 | 6.575 (0.987–43.815) | 0.052 |
| NPM + /FLT ‐ | 10 | 0.662 (0.240–1.824) | 0.425 | 0.225 (0.059–0.855) | 0.028 |
HR, hazard ratio; ITD, internal tandem duplication.
CEBPA*: CEBPA double mutation; NPM + /FLT ‐: NPM1 mutation/FLT3‐ITDnegative
Analysis of the relationship between other genes and survival
Beyond the 40 genes analyzed, univariate analysis for the other 220 genes showed that C5, GRIK2, MYO5B NMUR2, TOP3B, DOCK2, MAP2 KRT79, and HYDIN might be associated with survival (Table S5). Patient number of other genes was too limited to analyze the survival value (less than 5% of total cohort); only the number of cases with MYO5B, KRT79, and HYDIN were enough. MYO5B (HR = 2.661, 95% CI: 1.064–6.651) was associated with significantly reduced OS, while KRT79 and HYDIN were trended to correlate with a better survival. To avoid incidental statistical significance in our cohort, we then used TCGA dataset to evaluate their potential prognostic impact 15. However, there were only 2, 2, and 1 patients with HYDIN, KRT79 and MYO5B in TCGA dataset, respectively. In addition, the most important value of genetic mutation was to determine the prognosis in patients with intermediate‐risk cytogenetics; these three genes did not affect those patients’ survival. This result indicated that determination of the prognosis was still based on traditional cytogenetics and current known genes.
Prognostic value of genetic mutations in AML with intermediate‐risk cytogenetics
Among the 69 patients with intermediate‐risk cytogenetics, multivariate analysis showed that patients with IDH2 mutation, CEBPA double mutation, or NPM1 mutation/FLT3‐ITDnegative trended to have prolonged OS (all HR<0.5 and P < 0.15). Thus, 23 patients with intermediate‐risk cytogenetics who had at least one of the above genetic alterations had a significantly better survival (Fig. 3B, P = 0.001) as compared with the 46 patients who did not have these mutations.
Subgroup analysis of the 46 patients without favorable genotypes revealed that patients (n = 16) with TET2, RUNX1, ASXL1, or DNMT3A had a trend of reduced OS (Table S7, all HR>1.5). Based on the poor trend of OS and previous studies reporting them as poor prognostic factors 3, we grouped these genetic mutations as an unfavorable subclass. These 16 patients had a significantly inferior OS (Fig. 3C, P = 0.016) among the 46 patients without favorable genotypes, and among the overall 69 patients with intermediate‐risk cytogenetics (Fig. 3D, P < 0.001).
For the 69 patients with intermediate‐risk cytogenetics, we classified them into three groups according to genotype: mutation of IDH2, CEBPA double mutation, or NPM1 mutation in the absence of FLT3‐ITD as a favorable genotype, mutation of TET2, RUNX1, ASXL1, or DNMT3A as an unfavorable genotype, and the remaining was the intermediate genotype (Table 2). The above results indicated that AML patients with intermediate‐risk cytogenetics could be classified into three risk groups according to genotype (Fig. 3E, P < 0.001).
Table 2.
Cox regression hazard analysis of combined gene group on the overall survival in the patients with intermediate‐risk cytogenetics
| Group | N | Univariate | |
|---|---|---|---|
| HR (95% CI) | P‐value | ||
| Mutants NPM1, IDH2, or CEBPA double mutation in the absence of FLT3‐ITD | 23 | Reference | |
| Others | 30 | 3.293 (1.212–8.943) | 0.019 |
| Mutants TET2, RUNX1, AXSL1, or DNMT3A | 16 | 7.735 (2.740–21.833) | <0.001 |
HR, hazard ratio; ITD, internal tandem duplication.
Poor prognostic impact of KIT mutation in favorable‐risk cytogenetic AML
Prior studies reported KIT mutation as a poor factor in favorable‐risk cytogenetic AML 32. In our cohort, five patients had a KIT mutation, and all of them had favorable‐risk cytogenetic AML. Among the 22 patients with favorable‐risk cytogenetic AML, patients with KIT mutation had significantly poorer OS (HR = 7.002, 95% CI: 1.925–25.467, P = 0.003).
Integrated classification of cytogenetic and genetic profiling
Among the 69 patients with intermediate‐risk cytogenetics, 23 patients had the favorable genotype and their 5‐year survival was 76.5% (95% CI: 58.5–94.5%), similar to the prognosis of patients with favorable‐risk cytogenetics (5‐year OS, favorable cytogenetics vs. favorable genotype of intermediate cytogenetics, P = 0.161). For another 16 patients with the unfavorable genotype, the 5‐year OS was also similar to the OS of patients with unfavorable cytogenetics (5‐year OS, unfavorable cytogenetics vs. unfavorable genotype of intermediate cytogenetics, P = 0.674). In addition, five patients with favorable‐risk cytogenetic AML plus KIT mutation had a reduced OS compared with those without KIT mutation, and might be considered as intermediate risk.
The above results allowed us to develop a prognostic classification according to integration of the genetic mutation analysis and cytogenetic data (Fig. 4A). The 5‐year OS rate of patients with the new favorable‐risk, intermediate‐risk, and unfavorable‐risk groups was 73.9%, 35.1%, and 9.1%, respectively (Fig. 4B and C, P < 0.001). The univariate HR of the intermediate‐risk patients was 3.49 (95% CI: 1.64–7.34; intermediate‐risk cytogenetics: 1.38, 95% CI: 0.69–2.78) and HR of the unfavorable‐risk group was 6.77 (95% CI: 3.28–13.98; unfavorable cytogenetics: 3.15, 95% CI: 1.43–6.90), indicating that risk stratification using the integrated system was more clinically informative than that using cytogenetics alone (Figs. 3A vs. and 4C). In addition, integrated risk classification significantly reduces the proportion of intermediate‐risk patients from about 60% to 25%.
Figure 4.
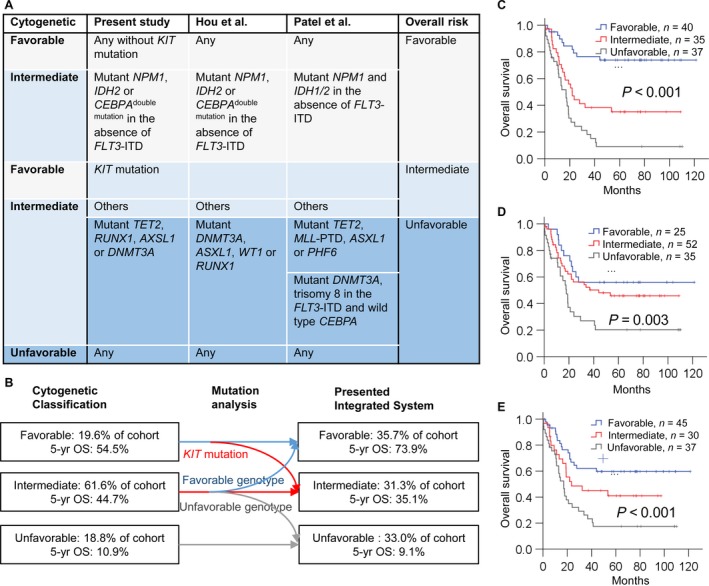
Revised risk classification of acute myeloid leukemia (AML) patients. (A) Mutation profiling of this study, Hou et al.7, and Patel et al.3 for risk stratification of AML patients. (B) Effect of the mutation profiling on the conventional cytogenetic risk. Patients with intermediate‐risk cytogenetic AML who have markedly divergent prognoses are reassigned into the appropriate risk groups according to their mutation profiles. Patients with favorable‐risk cytogenetics and KIT mutation are reclassified as intermediate risk. Overall survival estimated by Kaplan–Meier analysis according to (C) the current integrated stratification system, (D) revised risk system of Patel et al., and (E) risk stratification system of Hou et al.
Discussion
Our results demonstrate that integration of cytogenetic and genetic mutation profiles, with parallel sequencing by NGS, can improve the risk stratification for AML patients, especially for patients with intermediate‐risk cytogenetics.
The ultimate goal of cancer therapy is to establish precision medicine for guiding the best treatment and maximizing patient survival 33. For patients with unfavorable‐risk cytogenetics, treating with allogeneic HSCT in their first CR is currently evident strategy to improve their survival; however, the survival benefit from this strategy is not shown in patients with favorable‐risk cytogenetics 6. For unfavorable genotypes, such as NPM1 negative/FLT3‐ITDpositive or RUNX1 mutation, allogeneic HSCT can prolong patient survival 16, 34, 35. These facts indicate that early identification of poor‐risk patients can guide treatment. Risk classification of AML based on the traditional cytogenetic study is not good enough because most patients belong to intermediate‐risk cytogenetics. Recent studies show that using a list of genetic mutations can establish a prognostic classification to classify AML patients with intermediate‐risk cytogenetics into more definitive prognostic groups 36. European LeukemiaNet first stratified AML patients with normal cytogenetics into two risk groups using the mutation status of NPM1, FLT3, and CEBPA 8. Studies of Patel et al. and Hou et al. utilized 10 and 8 genes to classify intermediated cytogenetics into favorable, intermediate, and unfavorable subgroups 3, 7. In this study, we comprehensively sequenced 260 genes; nine genes were integrated with cytogenetics to develop a revised risk classification (Fig. 4A). For 69 patients with intermediate‐risk cytogenetics, the risk for 23 and 16 patients was revised as favorable and unfavorable, respectively. We also evaluated the two other risk stratification systems on the basis of mutation profiling in our cohort; the survival difference between three risk groups was more significant (Fig. 4D and E) compared with cytogenetic stratification alone (Fig. 3A). These facts suggest that utilizing mutation profiles of multiple genes could classify patients with intermediate‐risk cytogenetics into more accurate risk classification groups so as to significantly reduce the number of AML patients classified as intermediate risk.
In a comparison of our integrated classification and prior two studies, the mutational profiles of each risk classification were similar but not totally the same (Fig. 4A). Patel et al. reported that the IDH1/2 mutation is a favorable prognostic factor for AML with mutated NPM1 without FLT3‐ITD 3; our cohort and Hou et al. did not find IDH1 mutation as a favorable risk factor. IDH1 mutation was reported to be associated with unfavorable risk or did not affect disease outcome in other AML studies 37, 38. These results indicate that the prognostic value of IDH1 mutation is controversial. For IDH2, all three studies revealed that IDH2 mutation is associated with favorable risk only in the absence of FLT3‐ITD (Fig. 4A). However, a survival analysis reported that the IDH2 R140 mutation is associated with favorable prognosis and R172 with poor prognosis 39. Other investigations revealed that AML patients with IDH mutation respond better to treatment with hypomethylating agents 40, 41. These data indicate that risk stratification using IDH mutation may need to consider the therapeutic agents, intrinsic mutation site, and extrinsic genetic modifiers. Another difference in the favorable‐risk genotype is CEBPA double mutation, which was found in our study, Hou et al. and in other studies 7, 42, 43, but the prognostic relevance was not reported by Patel et al. 3. Comparison of the unfavorable genotypes indicated that mutants ASXL1, TET2, and DNMT3A were identified as unfavorable risk factors in the three studies. Mutation of RUNX1 was considered as a poor factor in previous reports and in this study 7, 44. Mutant PHF6 was not seen in our patients with intermediate‐risk cytogenetics, and all three patients carrying this mutation were in the favorable‐ or unfavorable‐risk cytogenetic group and died from the disease. Although the mutation profiles of favorable and unfavorable genotypes across the three studies are not the same, a substantial proportion of the molecular profiles are similar. The differences might be caused by different enrollment criteria and treatment agents, such as the high‐dose daunorubicin used in patients of Patel's study versus the standard‐dose anthracycline used in our patients. In the three studies, a total of 12 genes were analyzed for risk stratification (Fig. 4A). Parallel sequencing using NGS is a good strategy to handle the testing of multiple genes and can provide a rapid and accurate risk classification system for the clinical management of AML patients 14.
This study contained several limitations. This study was retrospective and chemotherapy regimens were not stringently the same, but all patients received Idarubicin and Cytarabine (7 + 3) as induction chemotherapy, followed by high‐dose Cytarabine‐based consolidation. Therefore, we considered this cohort to be appropriate for analyzing genetic values in the prognosis. We also tried to search for new genetic factors associated with patients’ survival, especially for patients with intermediate‐risk cytogenetics; only nine new genes might be related to prognosis (Table S5). However, case numbers of other gene were too limited to analyze their real effect and the prognostic impact cannot be validated in TCGA dataset, indicating no new genetic mutations significantly affecting AML prognosis. Several recent studies found TP53 mutation was associated with a poor survival, especially predicting the worst outcome in patients with unfavorable cytogenetics 45, 46, 47. In our cohort, TP53 mutation was associated with an inferior trend of OS among the whole cohort (Table 1) and usually co‐occurred with unfavorable cytogenetics. TP53 mutation predicted significantly worse OS in patients with unfavorable cytogenetics (P = 0.006), but did not play a role in other cytogenetics. In addition to genetic factors, clinical factors, such as age, were reported to be associated with patients’ survival 48. In this study, age did not significantly affect patients’ outcome and the genetic value in the multivariate analysis (Table S6). These indicated that currently known genes were the most important factors predicting survival.
In conclusion, with early assessment of cytogenetics and mutational profiling, AML patients can be managed by their real risk to reduce the mortality that results from unfavorable cytogenetics or genotypes. Therefore, accurate and rapid molecular diagnosis is important in AML patients. To achieve this goal, our study demonstrates that NGS‐based multi‐gene sequencing is clinically applicable and can be an effective means of establishing an accurate risk stratification system for guiding therapeutic decisions.
Conflicts of Interests
The authors declare that they have no conflicts of interests.
Supporting information
Table S1. Genes analyzed in this study.Table S2. Clinical and laboratory characteristics of AML patients.
Table S3. Pearson chi‐square analysis of pairwise gene–gene associations among the 40 genes involved in leukemogenesis.
Table S4. Univariate analysis of 40 genes with respect to overall survival in the entire cohort.
Table S5. List of genes with positive correlation (P ≤ 0.01) to OS in the entire cohort and their impact on patients with intermediated cytogenetics.Table S6. Cox regression hazard analysis of each gene with age factor on the overall survival in patients with intermediate‐risk cytogenetics.
Table S7. Multivariate analysis of overall survival of patients with intermediate‐risk cytogenetics without favorable genotype (N = 37).
Figure S1. Detection of FLT3 ITDs by GATK (HaplotypeCaller) and Pindel.
Acknowledgments
This work was supported, in part, by research grants from the National Taiwan University Hospital (NTUH. 103‐N2524 and NTUH. 104‐N2901), the Ministry of Science and Technology (I‐RiCE/MOST 104‐2911‐I‐002‐302 and MOST 103‐2314‐B‐002‐005‐MY2), and the Ministry of Health and Welfare (MOHW103‐TDU‐212‐114002). The authors thank the A1 Laboratory of the National Taiwan University Hospital for providing access to the Illumina MiSeq instrument and the National Applied Research Laboratories for providing access to their high‐performance computer for analyzing the post‐NGS data.
Cancer Medicine 2017; 6(2):349–360
Contributor Information
Yen‐Hsuan Ni, Email: yhni@ntu.edu.tw.
Chang‐Fang Chiu, Email: cftl00@gmail.com.
References
- 1. Rowe, J. M. , and Tallman M. S.. 2010. How I treat acute myeloid leukemia. Blood 116:3147–3156. [DOI] [PubMed] [Google Scholar]
- 2. Kihara, R. , Nagata Y., Kiyoi H., Kato T., Yamamoto E., Suzuki K., et al. 2014. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia 28:1586–1595. [DOI] [PubMed] [Google Scholar]
- 3. Patel, J. P. , Gonen M., Figueroa M. E., Fernandez H., Sun Z., Racevskis J., et al. 2012. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 366:1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bloomfield, C. D. , Ruppert A. S., Mrozek K., Kolitz J. E., Moore J. O., Mayer R. J., et al. 2004. Core binding factor acute myeloid leukemia. Cancer and Leukemia Group B (CALGB) Study 8461. Ann. Hematol. 83:S84–S85. [DOI] [PubMed] [Google Scholar]
- 5. Byrd, J. C. , Mrozek K., Dodge R. K., Carroll A. J., Edwards C. G., Arthur D. C., et al. 2002. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood 100:4325–4336. [DOI] [PubMed] [Google Scholar]
- 6. Koreth, J. , Schlenk R., Kopecky K. J., Honda S., Sierra J., Djulbegovic B. J., et al. 2009. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta‐analysis of prospective clinical trials. JAMA 301:2349–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hou, H. A. , Lin C. C., Chou W. C., Liu C. Y., Chen C. Y., Tang J. L., et al. 2014. Integration of cytogenetic and molecular alterations in risk stratification of 318 patients with de novo non‐M3 acute myeloid leukemia. Leukemia 28:50–58. [DOI] [PubMed] [Google Scholar]
- 8. Dohner, H. , Estey E. H., Amadori S., Appelbaum F. R., Buchner T., Burnett A. K., et al. 2010. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115:453–474. [DOI] [PubMed] [Google Scholar]
- 9. Ofran, Y. , and Rowe J. M.. 2013. Genetic profiling in acute myeloid leukaemia–where are we and what is its role in patient management. Br. J. Haematol. 160:303–320. [DOI] [PubMed] [Google Scholar]
- 10. Schlenk, R. F. , Dohner K., Krauter J., Frohling S., Corbacioglu A., Bullinger L., et al. 2008. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N. Engl. J. Med. 358:1909–1918. [DOI] [PubMed] [Google Scholar]
- 11. Liu, W. J. , Tan X. H., Luo X. P., Guo B. P., Wei Z. J., Ke Q., et al. 2014. Prognostic significance of Tet methylcytosine dioxygenase 2 (TET2) gene mutations in adult patients with acute myeloid leukemia: a meta‐analysis. Leuk. Lymphoma 55:2691–2698. [DOI] [PubMed] [Google Scholar]
- 12. Schnittger, S. , Eder C., Jeromin S., Alpermann T., Fasan A., Grossmann V., et al. 2013. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia 27:82–91. [DOI] [PubMed] [Google Scholar]
- 13. Sutton, L. A. , Ljungstrom V., Mansouri L., Young E., Cortese D., Navrkalova V., et al. 2015. Targeted next‐generation sequencing in chronic lymphocytic leukemia: a high‐throughput yet tailored approach will facilitate implementation in a clinical setting. Haematologica 100:370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gagan, J. , and Van Allen E. M.. 2015. Next‐generation sequencing to guide cancer therapy. Genome. Med. 7:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cancer Genome Atlas Research , 2013. N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin, P. H. , Lin C. C., Yang H. I., Li L. Y., Bai L. Y., Chiu C. F., et al. 2013. Prognostic impact of allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia patients with internal tandem duplication of FLT3. Leuk. Res. 37:287–292. [DOI] [PubMed] [Google Scholar]
- 17. Lin, P. H. , Kuo W. H., Huang A. C., Lu Y. S., Lin C. H., Kuo S. H., et al. 2016. Multiple gene sequencing for risk assessment in patients with early‐onset or familial breast cancer. Oncotarget 7:8310–8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li, H. , and Durbin R.. 2009. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li, H. , Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McKenna, A. , Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang, K. , Li M., and Hakonarson H.. 2010. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Spencer, D. H. , Abel H. J., Lockwood C. M., Payton J. E., Szankasi P., Kelley T. W., et al. 2013. Detection of FLT3 internal tandem duplication in targeted, short‐read‐length, next‐generation sequencing data. J. Mol. Diagn. 15:81–93. [DOI] [PubMed] [Google Scholar]
- 23. Ye, K. , Schulz M. H., Long Q., Apweiler R., and Ning Z.. 2009. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired‐end short reads. Bioinformatics 25:2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luthra, R. , Patel K. P., Reddy N. G., Haghshenas V., Routbort M. J., Harmon M. A., et al. 2014. Next‐generation sequencing‐based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring. Haematologica 99:465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mimori, T. , Nariai N., Kojima K., Takahashi M., Ono A., Sato Y., et al. 2013. iSVP: an integrated structural variant calling pipeline from high‐throughput sequencing data. BMC Syst. Biol. 7(Suppl 6):S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Richards, S. , Aziz N., Bale S., Bick D., Das S., Gastier‐Foster J., et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar, P. , Henikoff S., and Ng P. C.. 2009. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 28. Adzhubei, I. A. , Schmidt S., Peshkin L., Ramensky V. E., Gerasimova A., Bork P., et al. 2010. A method and server for predicting damaging missense mutations. Nat. Methods 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gilliland, D. G. , and Griffin J. D.. 2002. The roles of FLT3 in hematopoiesis and leukemia. Blood 100:1532–1542. [DOI] [PubMed] [Google Scholar]
- 30. Falini, B. , Mecucci C., Tiacci E., Alcalay M., Rosati R., Pasqualucci L., et al. 2005. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 352:254–266. [DOI] [PubMed] [Google Scholar]
- 31. Matsuo, H. , Kajihara M., Tomizawa D., Watanabe T., Saito A. M., Fujimoto J., et al. 2014. Prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia: a report from the Japanese Pediatric Leukemia/Lymphoma Study Group. Blood Cancer J. 4:e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Manara, E. , Bisio V., Masetti R., Beqiri V., Rondelli R., Menna G., et al. 2014. Core‐binding factor acute myeloid leukemia in pediatric patients enrolled in the AIEOP AML 2002/01 trial: screening and prognostic impact of c‐KIT mutations. Leukemia 28:1132–1134. [DOI] [PubMed] [Google Scholar]
- 33. Johnson, D. B. , Dahlman K. H., Knol J., Gilbert J., Puzanov I., Means‐Powell J., et al. 2014. Enabling a genetically informed approach to cancer medicine: a retrospective evaluation of the impact of comprehensive tumor profiling using a targeted next‐generation sequencing panel. Oncologist 19:616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Levis, M. 2013. FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Hematology Am. Soc. Hematol. Educ. Program 2013:220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gaidzik, V. I. , Bullinger L., Schlenk R. F., Zimmermann A. S., Rock J., Paschka P., et al. 2011. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J. Clin. Oncol. 29:1364–1372. [DOI] [PubMed] [Google Scholar]
- 36. Cagnetta, A. , Adamia S., Acharya C., Patrone F., Miglino M., Nencioni A., et al. 2014. Role of genotype‐based approach in the clinical management of adult acute myeloid leukemia with normal cytogenetics. Leuk. Res. 38:649–659. [DOI] [PubMed] [Google Scholar]
- 37. Paschka, P. , Schlenk R. F., Gaidzik V. I., Habdank M., Kronke J., Bullinger L., et al. 2010. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 28:3636–3643. [DOI] [PubMed] [Google Scholar]
- 38. Chotirat, S. , Thongnoppakhun W., Promsuwicha O., Boonthimat C., and Auewarakul C. U.. 2012. Molecular alterations of isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) metabolic genes and additional genetic mutations in newly diagnosed acute myeloid leukemia patients. J. Hematol. Oncol. 5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Green, C. L. , Evans C. M., Zhao L., Hills R. K., Burnett A. K., Linch D. C., et al. 2011. The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood 118:409–412. [DOI] [PubMed] [Google Scholar]
- 40. Im, A. P. , Sehgal A. R., Carroll M. P., Smith B. D., Tefferi A., Johnson D. E., et al. 2014. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies. Leukemia 28:1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emadi, A. , Faramand R., Carter‐Cooper B., Tolu S., Ford L. A., Lapidus R. G., et al. 2015. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am. J. Hematol. 90:E77–E79. [DOI] [PubMed] [Google Scholar]
- 42. Taskesen, E. , Bullinger L., Corbacioglu A., Sanders M. A., Erpelinck C. A., Wouters B. J., et al. 2011. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood 117:2469–2475. [DOI] [PubMed] [Google Scholar]
- 43. Dickson, G. J. , Bustraan S., Hills R. K., Ali A., Goldstone A. H., Burnett A. K., et al. 2016. The value of molecular stratification for CEBPA(DM) and NPM1(MUT) FLT3(WT) genotypes in older patients with acute myeloid leukaemia. Br. J. Haematol. 172:573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mendler, J. H. , Maharry K., Radmacher M. D., Mrozek K., Becker H., Metzeler K. H., et al. 2012. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J. Clin. Oncol. 30:3109–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bowen, D. , Groves M. J., Burnett A. K., Patel Y., Allen C., Green C., et al. 2009. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia 23:203–206. [DOI] [PubMed] [Google Scholar]
- 46. Metzeler, K. H. , Herold T., Rothenberg‐Thurley M., Amler S., Sauerland M. C., Gorlich D., et al. 2016. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 128:686–698. [DOI] [PubMed] [Google Scholar]
- 47. Papaemmanuil, E. , Gerstung M., Bullinger L., Gaidzik V. I., Paschka P., Roberts N. D., et al. 2016. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 374:2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mosna, F. , Papayannidis C., Martinelli G., Di Bona E., Bonalumi A., Tecchio C., et al. 2015. Complex karyotype, older age, and reduced first‐line dose intensity determine poor survival in core binding factor acute myeloid leukemia patients with long‐term follow‐up. Am. J. Hematol. 90:515–523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes analyzed in this study.Table S2. Clinical and laboratory characteristics of AML patients.
Table S3. Pearson chi‐square analysis of pairwise gene–gene associations among the 40 genes involved in leukemogenesis.
Table S4. Univariate analysis of 40 genes with respect to overall survival in the entire cohort.
Table S5. List of genes with positive correlation (P ≤ 0.01) to OS in the entire cohort and their impact on patients with intermediated cytogenetics.Table S6. Cox regression hazard analysis of each gene with age factor on the overall survival in patients with intermediate‐risk cytogenetics.
Table S7. Multivariate analysis of overall survival of patients with intermediate‐risk cytogenetics without favorable genotype (N = 37).
Figure S1. Detection of FLT3 ITDs by GATK (HaplotypeCaller) and Pindel.