

Abstract
The architecture of chromatin is governed, in part, by ATP-dependent chromatin remodelers. These multiprotein complexes contain targeting domains that recognize post-translational marks on histones. One such targeting domain is the bromodomain (BD), which recognizes acetyl-lysines and recruits proteins to sites of acetylation across the genome. Polybromo1 (PBRM1), a subunit of the Polybromo-associated BRG1- or hBRM-associated factors (PBAF) chromatin remodeler, contains six tandem BDs and is frequently mutated in clear cell renal cell carcinoma (ccRCC). Mutations in the PBRM1 gene often lead to the loss of protein expression; however, missense mutations in PBRM1 have been identified and tend to cluster in the BDs, particularly BD2 and BD4, suggesting that individual BDs are critical for PBRM1 function. To study the role of these six BDs, we inactivated each of the six BDs of PBRM1 and re-expressed these mutants in Caki2 cells (ccRCC cells with the loss of function mutation in PBRM1). Four of the six BDs abrogated PBRM1 tumor suppressor function, gene regulation, and chromatin affinity with the degree of importance correlating strongly to the rate of missense mutations in patients. Furthermore, we identified BD2 as the most critical for PBRM1 and confirmed BD2-mediated association to histone H3 peptides acetylated at lysine 14 (H3K14Ac), validating the importance of this specific acetylation mark for PBRM1 binding. From these data, we conclude that four of the BDs act together to target PBRM1 to sites on chromatin; when a single BD is mutated, PBRM1 no longer controls gene expression properly, leading to increased cell proliferation.
Keywords: acetylation, cancer, cell growth, chromatin remodeling, epigenetics, histone acetylation, Polybromo-1, bromodomain, bromodomains, renal cancer
Introduction
Chromatin structure in eukaryotic cells is intricately tied to proper gene expression. The structure of chromatin is regulated by multiple factors, most prominently the post-transcriptional modification of histones. Histone methylation, phosphorylation, and acetylation control DNA structure by altering nucleosome interaction with DNA and by recruiting effector proteins to specific regions of the genome (1). Effector proteins are targeted to these regions through chromatin recognition domains that can bind to these modifications (2, 3). One such chromatin recognition domain is the bromodomain (BD),2 a highly conserved 110-amino acid domain composed of four α-helices that form a hydrophobic pocket that preferentially binds acetyl-lysines on histone tails (4, 5). One example of a BD-containing protein is Polybromo-1 (PBRM1), which contains six consecutive BDs, two bromo-adjacent homolog (BAH) domains involved in protein-protein interactions (6), and a high mobility group (HMG), which are typically DNA-interacting domains (7, 8). PBRM1 is a subunit of the Polybromo-associated BRG1- or hBRM-associated factors (PBAF) complex, which is a member of the mammalian SWI/SNF family of chromatin remodeling complexes along with the highly related BRG1-or hBRM-associated factors (BAF) complex (Fig. 1). These complexes use energy from ATP to insert, remove, or slide nucleosomes, effectively controlling DNA accessibility (9, 10). PBAF shares many of the same subunits as the BAF complex, and of the subunits exclusive for PBAF (PBRM1, ARID2, BAF45A, and BRD7), all except PBRM1 have closely related homologs in the BAF complex (11). PBRM1, with a uniquely high number of chromatin-interacting domains, is the defining subunit of the PBAF complex and is likely involved in unique function of PBAF not observed for BAF.
FIGURE 1.
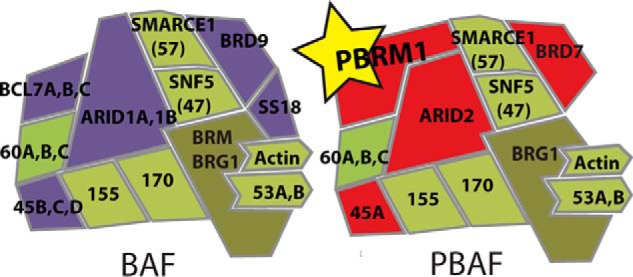
The BAF and PBAF complexes coexist in all cell types. Although BAF and PBAF share many subunits (green), BAF exclusively contains ARID1A/B, BAF45B/C/D, BRD9, SS18, and BLC7A/B/C (purple), whereas PBAF exclusively contains PBRM1, ARID2, BAF45A, and BRD7 (red). PBRM1 is the most distinguishing feature of PBAF with no homolog in BAF.
Recent studies have identified PBRM1 mutations in 40% of clear cell renal cell carcinomas (ccRCC), the most common form of kidney cancer, making PBRM1 the second most frequently mutated gene after von Hippel-Lindau (VHL) (12–15). Studies have indicated that PBRM1 acts as a tumor suppressor in ccRCC, although the mechanism is unclear (12, 16). Due to the presence of the multiple BDs, it is likely that PBRM1, via the BDs, targets PBAF to sites of histone acetylation at genes that need to be expressed for the regulation of proper cell growth. Exome sequencing of patient tumors indicates that PBRM1 mutations most often lead to the complete loss of PBRM1 expression; however, missense mutations are observed in ∼15% of cases, leading to the expression of mutant forms of PBRM1 (14). By mapping the location of these mutations, we noticed a high incident of missense mutations in BD2 and BD4, although mutations have been identified in all the BDs except BD3 (see Fig. 2) (17). Although mutations are observed throughout the domains, many have been predicted to affect acetyl-lysine binding or domain stability (18). Missense mutations that occur outside the domains include sites of post-translational modification such as phosphorylation or acetylation (19), as well as putative LLXXL nuclear hormone receptor binding sites that might contribute to PBRM1 function through other mechanisms.
FIGURE 2.

Missense mutations in PBRM1 identified from ccRCC patient samples indicated with black bars. Data were obtained from the COSMIC database (17).
Our lab recently characterized the effects of re-expressing wild-type PBRM1 in Caki2 cells, a ccRCC cell line with the loss of PBRM1 expression (16). We observed that re-addition of PBRM1 significantly decreased the proliferation rate of these cells and up-regulated genes involved in cell adhesion, apoptosis, and negative cell proliferation. Using the phenotype we characterized in Caki2 cells, we sought to define how each BD contributes to PBRM1 function in ccRCC. By creating PBRM1 mutants with a single inactive BD, we determined that the ability of PBRM1 to regulate growth and gene expression is completely reliant on BD2-mediated binding to chromatin, which appears to be facilitated through association with histone 3 lysine 14 acetylation (H3K14Ac). Additionally, we observed that mutation of BD1, BD4, BD5, and BD6 moderately affected PBRM1 chromatin association and tumor suppressor function, and that mutation of BD3 has no effect on PBRM1 function, perfectly agreeing with the mutational spectrum from patient tumors.
Results
The Contribution of Individual Bromodomains to the Tumor Suppressor Function of PBRM1
As a subunit of the PBAF chromatin remodeling complex, PBRM1 presumably acts as a tumor suppressor by controlling chromatin structure at specific regions of the genome. PBRM1 contains six tandem BDs, which are predicted to bind acetylated lysines to target chromatin remodeling activity to regions of histone acetylation (4). Due to the frequency of missense mutations found in the BDs (Fig. 2), we predict that even a mutation within a single BD can alter the functional properties of PBRM1. To examine how each individual BD contributes to the tumor-suppressive function of PBRM1, we utilized our Caki2 PBRM1 doxycycline-inducible system (16) and systematically mutated each BD to disrupt acetyl-lysine binding. The highly conserved asparagine located at the end of the αB helix forms a hydrogen bond with the acetylated lysine, anchoring it in the binding pocket (4, 5, 20). BD1 is unusual in that it has a tyrosine at this position instead of an asparagine; however, it has been shown to perform the same role (20). To confirm this role in vitro, we performed a thermal shift assay (21) with recombinant BD2 and H3K14Ac peptide, a confirmed ligand for this BD (20). A reproducible dose-dependent shift in the melting temperature (Tm) of the BD was observed upon incubation with H3K14Ac peptide (Fig. 3A). In contrast, the Tm of the asparagine mutant (BD2N263A) did not increase upon incubation with the H3K14Ac peptide (Fig. 3A). It is also important to note that the Tm of the mutant was not significantly shifted when compared with wild type, implying that this conserved asparagine is critical for acetyl-lysine binding but not domain stability, which was noted previously for BDs from BRD4 (22). To study the role of each BD, we created eight cell lines mutating this highly conserved residue in the six BDs of PBRM1: Caki2+Vector, Caki2+PBRM1WT, Caki2+PBRM1mBD1 (Y127A), Caki2+PBRM1mBD2 (N263A), Caki2+PBRM1mBD3 (N463A), Caki2+PBRM1mBD4 (N601A), Caki2+PBRM1mBD5 (N739A), and Caki2+PBRM1mBD6 (N855A). PBRM1 expression was even across the seven lines after treatment with doxycycline (Fig. 3B). In addition, we confirmed that all the mutants were incorporated into the complex with equivalent efficiency by immunoprecipitation of the PBRM1 followed by immunoblotting for BRG1, the ATPase subunit of the PBAF complex (Fig. 3B).
FIGURE 3.

Proliferation and gene expression is altered in PBRM1 BD mutant cell lines. A, thermal shift stability assays indicate that mutation of conserved asparagine 263 to alanine in recombinant BD2 abrogates binding to an H3K14Ac peptide. B, immunoblotting analysis of protein expression level of PBRM1 and BRG1 in the BD mutant (mut) cell lines from lysates (top) and PBRM1 immunoprecipitations (bottom). TBP, TATA-binding protein. C, the proliferation rate of BD mutant cell lines normalized to Caki2+Vector cells. n = 3 independent biological replicates D, transcriptional analysis by quantitative RT-PCR of PBRM1-dependent genes (IGFBP4 and CNTN6) in BD mutant cell lines normalized to Caki2+Vector cells. n = 6 independent biological replicates. A designation of * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001 (paired Student's t test). ns, not significant. Error bars represent S.D.
Previously, we observed that re-expression of wild-type PBRM1 led to a significant decrease in growth of the Caki2 cells (16). To determine whether individual BDs contribute to the tumor-suppressive phenotype of PBRM1, we performed growth curves for Caki2+Vector and Caki2+PBRM1WT, along with the six mutants. Surprisingly, cell proliferation when compared with Caki2+PBRM1WT significantly increased when we mutated a single amino acid in five of the six BDs (Fig. 3C). Mutation of BD1, BD2, BD4, and BD5 almost completely obliterated the tumor-suppressive phenotype, whereas BD6 had moderate effects on the growth rate. Mutation of BD3 had no effect on the growth of cells, and this mutant was indistinguishable from Caki2+PBRM1WT. This dramatic effect on growth signifies the importance of individual BDs in the functionality of PBRM1, while also indicating that the BDs are working in a cooperative manner to suppress tumor growth.
From our previous study, we determined that PBRM1 controls genes involved in pathways that influence cell growth and mobility (16). If PBRM1 controls growth by regulating gene expression, we would expect that the BD mutations that disrupted growth suppression would be crucial in gene expression. To examine how the BD mutants influence gene expression, we quantitated the transcript levels of CNTN6 and IGFBP4, two genes up-regulated upon PBRM1 re-expression in Caki2 cells, which are involved in cell adhesion and metabolism, respectively (16). Mutation of BD2 and BD4 greatly impaired the ability of PBRM1 to up-regulate gene expression, mutation of BD1 and BD5 moderately impaired gene up-regulation, and mutation of BD3 had no effect (Fig. 3D). BD6 mutation significantly inhibited IGFBP4 up-regulation but had no effect on CNTN6. Again these data suggest a cooperative role for multiple BDs at all PBRM1 sites of action; however, results for BD6 suggest that this domain may be required only at a subset of PBRM1 gene targets.
The Effect of PBRM1 on the Affinity of PBAF to Bulk Chromatin
Although we have shown that the individual BDs are important for tumor suppression and gene expression, it is unclear what global binding properties are dependent on the BDs. We first examined whether PBAF has distinguishable chromatin binding characteristics when compared with BAF. Although BAF and PBAF share many of the same subunits, PBRM1 contributes six additional chromatin-interacting domains to the complex, leading us to the hypothesis that it binds more tightly to chromatin. Using a sequential salt extraction (SSE), we quantitated the relative binding strength of the BAF and PBAF complexes to chromatin in Caki1 cells, a ccRCC cell line expressing wild-type PBRM1. After isolating nuclei, we eluted nuclear proteins from the insoluble chromatin fraction using increasing concentrations of salt. The resulting fractions were run on a gel for immunoblotting analysis of ARID1A, a BAF-specific subunit, and PBRM1, the PBAF-specific subunit. The majority of PBRM1 eluted at 300–400 mm NaCl, whereas the peak of elution for ARID1A was at 200–300 mm NaCl, indicating that PBAF inherently binds tighter to chromatin than BAF (Fig. 4A). This was not unique to ccRCC, as the same effect was observed in HeLa cells (Fig. 4B). We next examined whether this increase in affinity of PBAF over BAF was dependent on PBRM1. We repeated this experiment with Caki2+Vector and Caki2+PBRM1 and blotted for ARID2 (Fig. 1) to identify PBAF in the absence of PBRM1. In Caki2+Vector, ARID2 elution peaked at 200–300 mm NaCl fraction, appearing very similar to the ARID1A (BAF) elution pattern. Upon re-expression of PBRM1, ARID2 elution shifts to a higher salt concentration, indicating that in the absence of PBRM1, PBAF still associates with chromatin, but its affinity now appears more similar to BAF, as indicated by ARID1A staining. (Fig. 4C). To validate that this is a general role for PBRM1 in the PBAF complex, and not unique to Caki2 cells, we knocked down PBRM1 in Caki1 and observed a shift in the ARID2 elution to a lower salt concentration, similar to what was observed in Caki2 cells (Fig. 4D). To rule out the possibility that these observations are a result of differential growth rates due to loss or gain of PBRM1, we performed SSE on serum-starved Caki1 cells and confirmed that the changes in chromatin affinity were inherent functions of PBRM1 and not reflective of alterations in cell cycle (Fig. 4E) In total, these results indicate that PBRM1 is responsible for the previously unobserved increased strength of chromatin binding detected for PBAF when compared with BAF.
FIGURE 4.

Chromatin binding properties of BAF and PBAF complexes. A, analysis of binding affinity to chromatin of the BAF (ARID1A) and PBAF (PBRM1) in Caki1 cells by SSE. n = 2 independent biological replicates. B, SSE analysis of BAF and PBAF affinity in HeLa cells. C, analysis of PBAF affinity to chromatin in Caki2+Vector and Caki2+PBRM1WT cells indicated by the elution of ARID2. n = 4 independent biological replicates. D, analysis of PBAF affinity to chromatin Caki1+Vector and Caki1+shPBRM indicated by the elution of ARID2. E, comparison of PBAF elution in Caki1 cells grown under normal conditions or serum-starved conditions for 16 h. A designation of * = p < 0.05 (paired Student's t test). Error bars represent S.D.
The Contribution of Individual Bromodomains to the Affinity of PBAF to Bulk Chromatin
After defining the chromatin binding properties of PBRM1 within the PBAF complex, we wanted to use this readout to determine whether the BD mutations that disrupt PBRM1 function also disrupt the global association of PBAF with chromatin. We performed SSE on our six BD mutant lines and compared them to the SSE profile for wild-type PBRM1 (Fig. 5A). Because we saw the largest differential in elution at 200 mm between ARID1A and PBRM1 in Caki1 (Fig. 4A) or between ARID2 with and without PBRM1 re-expression in Caki2 (Fig. 4C), we compared the amount of PBRM1 eluted from chromatin at this salt concentration across all seven cell lines. In cells containing wild-type PBRM1, ∼18% of the complex elutes in the 200 mm fraction. However, mutation of a single asparagine in BD2 leads to ∼33% of the PBAF complex eluting in 200 mm salt, similar to that observed upon deletion of PBRM1 (Fig. 5B). Although these changes may appear small, they are very consistent between experiments and can be quantified for statistical analysis. Further validating this approach, a similar phenotype was recently observed in live cells for the BD2 mutant using fluorescence recovery after photobleaching using GFP-tagged PBRM1 (23). Mutation of BD1, BD4, and BD5 also significantly increased the amount of PBRM1 eluted at 200 mm, whereas mutation of BD6 slightly increased the amount of PBRM1 eluted, although it was not statistically significant. In contrast, the mutation of BD3 caused no alteration in PBRM1 elution from chromatin, consistent with the cell proliferation, transcriptional profile, and patient mutation data observed for the BD3 mutant.
FIGURE 5.

Chromatin binding properties are altered in PBRM1 BD mutants. A, representative immunoblotting analysis of SSE assays designed to assess relative chromatin binding affinity of BD mutant (m) PBRM1 re-expressed in Caki2 cells. B, analysis of binding affinity to chromatin by SSE of PBRM1 in the BD mutants (mut) indicated by the percentage of PBRM1 eluted at 200 mm NaCl. n = 4 independent biological replicates. A designation of * = p < 0.05, ** = p < 0.01 (paired Student's t test). ns, not significant. Error bars represent S.D.
PBRM1 Binding to Acetylated Histone Peptides
Several studies have attempted to determine the specific targets of individual PBRM1 BDs using recombinantly expressed single BDs with acetylated peptides but have shown very little agreement, except that BD2 binds H3K14Ac (20, 24–26). We decided to pursue a more holistic approach to define the affinity of the full PBRM1-containing complex to acetylated histone peptides. We incubated nuclear lysate from HeLa cells with biotin-labeled acetylated histone peptides bound to streptavidin beads. We found that only H3K14Ac peptides are able to specifically enrich PBRM1 from lysates (Fig. 6A), in agreement with in vitro studies defining a specific association between BD2 and H3K14Ac (20, 24–26). We confirmed this interaction in our PBRM1 re-expression system and determined that wild-type PBRM1 preferentially binds H3K14Ac over unmodified H3 peptides, indicating that PBRM1 binding to histones is at least partly mediated through H3K14 acetylation (Fig. 6B). Using SS18, a BAF-specific subunit (Fig. 1), we determined that binding of H3K14Ac is PBAF-specific as SS18 showed no preference for the H3K14Ac peptide over the H3 peptide (Fig. 6B), although BAF also contains BDs with in vitro binding to H3K14Ac peptides (24). Because BD2 has been shown to bind H3K14Ac in vitro and because mutation of BD2 most dramatically impedes PBRM1 function, we investigated the affinity of PBAF complexes from lysates for this acetylation mark. To determine whether this association is mediated through BD2, we also incubated Caki2+PBRM1mBD2 nuclear lysates with the peptides and found that mutation of BD2 leads to an ∼50% loss in PBRM1 enrichment by the H3K14Ac peptide (Fig. 6C). The loss of binding is not complete, likely due to promiscuous weak binding of the other PBRM1 BDs to acetylated peptides, including H3K14Ac peptides (24). We tested this possibility by measuring the enrichment from lysates of PBRM1 to H3K14Ac peptides with additional acetylation marks. We did not observe a significant increase in enrichment upon incubation with H3 peptides containing acetylation at Lys-9 and Lys-14, but did see a dramatic increase in enrichment using H3 peptides with acetylation at Lys-14, Lys-18, Lys-23, and Lys-27. Further, mutation in any of the BDs, except BD3, reduces the affinity of PBRM1 to histone peptides containing multiple acetylated lysines. Together, this suggests that PBAF is targeted to H3K14Ac via specific association with BD2 and that the association of BD1, BD4, BD5, and potentially BD6 to additional acetylation marks is required for maximal affinity of the PBAF complex to chromatin at sites of gene regulation.
FIGURE 6.

PBRM1 binding to acetylated histone peptides. A, immunoblotting analysis of PBRM1 after peptide pulldowns with H3 or singly acetylated peptide from HeLa nuclear cell lysate. B, immunoblotting analysis after peptide pulldowns with H3 or H3K14Ac peptide from Caki2+PBRM1WT and Caki2+PBRM1mBD2 nuclear lysate. Enrichment of BAF (SS18) and PBAF (PBRM1) was determined by immunoblot analysis. C, immunoblot analysis of PBRM1 after peptide pulldowns with differentially acetylated peptides from HeLa nuclear cell lysate. D, immunoblot analysis of PBRM1 after peptide pulldowns with H3 or multiply acetylated H3 peptide from the nuclear lysates of Caki2 with re-expression of wild-type and BD mutant (mut) PBRM1.
The Role for Individual Bromodomains in PBRM1 Localization to a Discrete Genomic Site
To extend these results to PBRM1 targeting at a discrete genomic locus in vivo, we performed a ChIP-Seq analysis for PBRM1 in Caki2 cells.3 We did not observe PBRM1 enrichment at IGFBP4, indicating that IGFBP4 may be an indirect transcriptional target of PBRM1; however we did identify a site of PBRM1 enrichment 3 kb downstream of the transcription start site (TSS) of CNTN6. We further confirmed both H3K14Ac and PBRM1 enrichment at CNTN6 (Fig. 7A) using ChIP-qPCR, and identified maximal enrichment of both H3K14Ac and PBRM1 binding at 200 bp downstream of the TSS. This is in agreement with published ChIP-Seq data indicating that H3K14Ac is highest in the 1-kb region directly following the TSS (27). Using this primer set, we performed PBRM1 ChIP-qPCR experiments with the BD mutations to define how mutations in the BDs affect PBRM1 enrichment in vivo. We found good agreement with the SSE and peptide pulldown assays, indicating a strong role for BD2 and BD5 in PBRM1 binding, some role for BD1, BD4, and BD6, and no role for BD3 (Fig. 7B).
FIGURE 7.
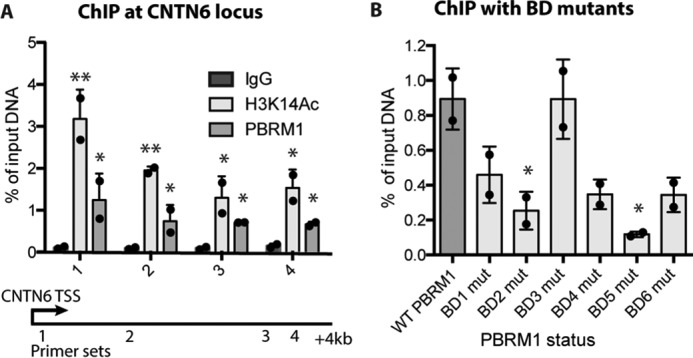
Quantitative ChIP in Caki2 cells. A, ChIP of PBRM1 and H3K14Ac enrichment at four sites across the CNTN6 locus in Caki2 cells. A designation of * = p < 0.05 and ** = p < 0.01 (paired Student's t test) when compared with IgG. Error bars represent S.D. B, PBRM1 ChIP at the CNTN6 locus (primer site 1) from Caki2 cells expressing wild-type or BD mutant (mut) PBRM1. A designation of * = p < 0.05 (paired Student's t test) when compared with WT PBRM1. Error bars represent S.D.
Discussion
PBRM1 is a tumor suppressor frequently mutated in ccRCC (12–15). Mutations in BAF subunits such as ARID1A lead to the total loss of protein expression (28, 29); in contrast, PBRM1 is expressed with missense mutations in about 15% of ccRCC patients with PBRM1 mutations (14). By mapping these missense mutations, we observed that point mutations in PBRM1 cluster in the BDs, suggesting that individual BDs may be critical for PBRM1 to act as a tumor suppressor. In this study, we have demonstrated that four of the six BDs of PBRM1 work in conjunction, and are all individually necessary for full PBRM1 activity. By examining how individual domains contribute to the function of PBRM1, we have found that growth suppression, gene expression, chromatin binding, and acetylation recognition were completely obstructed by the loss of BD2 function, indicating that this BD is the most critical for PBRM1 function. This correlates with data from yeast, in which the second BD of PBRM1 homolog RSC4 binds H3K14ac and is the most essential for viability in yeast (30).
In addition to BD2, patient mutations have been observed in BD1, BD4, BD5, and BD6. In accordance with this, mutation of BD1, BD4, and BD5 in our cell-based system consistently decreased the PBRM1 activity for all of the phenotypes we explored, and BD6 decreased the effectiveness of PBRM1 in some settings. Mutation of the sixth BD moderately impaired growth suppression, but there was no significant change in affinity of the complex. Additionally, the loss of BD6 significantly affected the transcription of IGFBP4, a metabolism gene, but not CNTN6, a gene important in cell adhesion. Intriguingly, the structure of BD6 is the most structurally unique of the six PBRM1 BDs, as it contains an extra helix and has an unusually short ZA loop (24, 31). The length of the ZA loop is implicated to influence BD specificity, and this suggests that BD6 may recognize a unique substrate when compared with the other PBRM1 BDs. The only histone peptides recognized by BD6 include acetylation marks on H2A and H2B core residues, although these interactions are weak (24), raising the possibility that BD6 may be involved in a function separate from acetyl-lysine binding. Finally, mutation of BD3 seemed to have no effect on any of the phenotypes we explored, which was also consistent with patient data, in which no mutations were observed in this domain. Although the hydrophobic pockets of the BDs are conserved, the electrostatic potential of the surface of BD3 has a positively charged surface that may prevent interaction with histone tails (24). Therefore, BD3 may bind to non-histone proteins that are not relevant for PBRM1 function in ccRCC. Alternatively, because only the individual domains have been crystallized and the structure of the whole protein is unknown, it is possible that BD3 is buried in PBRM1 and not involved in acetyl-lysine binding at all.
From the data in this study, it is clear that BD2 is pivotal for PBRM1 function, BD1, BD4, and BD5 all contribute significantly to PBRM1 function, and BD6 and BD3 showed either inconsistent or no effect on PBRM1 activity. Previous studies that attempted to differentiate acetylation marks associated with the individual domains (20, 24, 25) resulted in broad and conflicting results. Certain BDs appear to be promiscuous, binding to almost any acetylated peptide; in particular, BD1, BD3, and BD4 bind a wide array of peptides from all the histones (24). In contrast, the same study only identified three peptides that interacted with BD2, one of which was H3K14ac, an interaction that has been well characterized (20, 24, 26). It is important to note that BDs typically bind weakly to acetylation targets (10–100 μm) and are usually found in proteins or complexes with multiple chromatin binding domains, such as additional BDs, chromodomains, plant homeodomain (PHD) fingers, high mobility group domains, and AT-rich interactive domains (24). This suggests that for proper BD function, they work in conjunction with additional binding domains to obtain optimal affinity and specificity for chromatin targets. With our system, we have demonstrated that PBRM1 association with H3K14ac is dependent on BD2. From these data, we propose that BD2 initiates binding of the complex and anchors the complex to chromatin, allowing BD1, BD4, BD5, and possibly BD6 to bind to nearby acetyl-lysine residues (Fig. 8).
FIGURE 8.
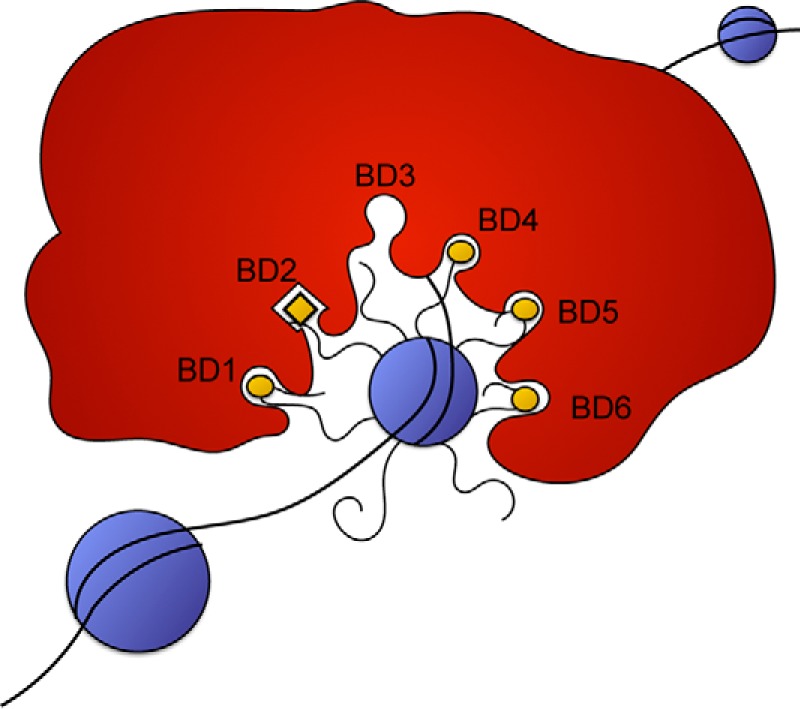
Proposed models for PBRM1 recognition of histone marks. BD2 binds specifically to H3K14ac, and the remaining bromodomains recognize a pattern of acetylation marks based on spatial orientation.
Although many chromatin-modifying complexes display an array of chromatin binding domains, these domains are often expressed on separate proteins. PBRM1, with its six tandem BDs within a single protein, makes a very appealing model for dissecting the mechanism of these highly multivalent systems. We have found that four of these six domains are essential for proper function of the protein, which implies that these domains are acting cooperatively. We have determined that BD2 is the most important BD and that it interacts with H3K14Ac; however, the acetylation targets of the remaining domains are still unknown. The first possibility is that BD2 binds to H3K14Ac and the remaining BDs bind to any acetylation site in the near vicinity and stabilize the interaction of the complex with chromatin. The second possibility is that there is a defined set of acetylation sites that are dictated by the spatial arrangement of acetyl-lysines as opposed to peptide sequence. Recent ChIP-MS studies identify PBRM1 associated with sites of H3K27Ac (32), although peptide studies never indicated any particular specificity for this mark by any of the recombinant BDs (20, 24, 26). Currently, this information is correlative and does not conclusively determine whether H3K27Ac is required for PBRM1 targeting, or whether it correlates with other acetylation marks required for PBRM1 targeting. The next step will be to further elucidate the combination of acetylation marks that define PBRM1 targets in vitro and in vivo, which will allow for further understanding of upstream pathways that regulate PBRM1 function in ccRCC.
Experimental Procedures
Recombinant Protein Expression
Recombinant PBRM1 (His-tagged BD2 construct) was a gift from Nicola Burgess-Brown (Addgene plasmid number 39103). Mutagenesis of BD2N263A was accomplished with the Phusion Site-Directed Mutagenesis kit (Thermo Scientific). Wild-type and mutated BD2 were expressed in BL21(DE3) competent Escherichia coli cells (New England Biolabs Inc., Ipswich, MA), and expression was induced by isopropyl-1-thio-β-d-galactopyranoside (1 mm). Protein purification was performed by lysing cells resuspended in Binding Buffer (20 mm Tris-HCl, 150 mm NaCl, 20 mm imidazole) with a probe sonicator, and debris was pelleted by centrifugation. Supernatant was incubated with His60 Ni Superflow Resin. Resin was washed with Binding Buffer, and protein was eluted in Elution Buffer (20 mm Tris-HCl, 150 mm NaCl, 500 mm imidazole.)
Thermal Shift Assay
Purified protein (5 μg) was incubated with varying concentrations of H3K14Ac peptide (AnaSpec, Fremont, CA) in 10 mm Hepes, pH 7.0, 140 mm NaCl, along with 7.5× SYPRO Orange (Sigma-Aldrich) as described in Vivoli et al (21). Differential scanning fluorometry was performed with Applied Biosystems StepOneTM Real-Time PCR System, and Tm values were calculated using GraphPad Prism 6 graphing software.
Cell Culture
Caki1 and Caki2 cells (American Type Culture Collection, Manassas, VA) were grown in McCoy's 5A medium (Corning Mediatech, Inc., Manassas, VA) supplemented with 10% fetal bovine serum (Omega Scientific Inc., Tarzana, CA), 1% antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin; Corning Mediatech), 1% nonessential amino acids (Corning Mediatech), and 1% l-glutamine (Corning Mediatech) at 37 °C in a humidified atmosphere in a 5% CO2 incubator. Serum starvation was accomplished by growing Caki1 (American Type Culture Collection) in McCoy's 5A medium (Corning Mediatech) supplemented with 0.2% fetal bovine serum (Omega Scientific), 1% antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin; Corning Mediatech), 1% nonessential amino acids (Corning Mediatech), and 1% l-glutamine (Corning Mediatech) at 37 °C in a humidified atmosphere in a 5% CO2 incubator for 16 h.
HEK-293T and HeLa cells (American Type Culture Collection) were grown in DMEM (Corning Mediatech) supplemented with 10% fetal bovine serum (Omega Scientific), 1% antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin; Corning Mediatech), 1% sodium pyruvate (Corning Mediatech), and 1% l-glutamine (Corning Mediatech) at 37 °C in a humidified atmosphere in a 5% CO2 incubator.
Constructs for Mammalian Cell Culture
PBRM1 knockdown was performed by transfection with empty pLKO.1 vector or shPBRM1 pLKO.1 (TRCN0000015994, Thermo Fisher Scientific). PBRM1 mutagenesis was accomplished with In-Fusion®HD cloning kit (Clontech Laboratories, Inc.). Mutagenesis was performed on each BD using pBabepuroBAF180 (a gift from Ramon Parsons, Addgene plasmid number 41078). After confirming proper mutation using Sanger sequencing, full-length PBRM1 containing a single point mutation was cloned from pBabepuroBAF180 into tetracycline-inducible conditional lentiviral vector TetO-FUW (a gift from Rudolf Jaenisch, Addgene plasmid number 20323), which was used with pLenti CMV rtTA3 Hygro (w785-1) (a gift from Eric Campeau, Addgene plasmid number 26730) for tetracycline-inducible expression.
Lentiviral Infection
HEK293T cells were transfected with lentivirus constructs along with packaging vectors pMD2.G and psPAX2. After 48 h, the supernatant was collected and concentrated by ultracentrifugation (17,300 rpm for 2 h) and resuspended in 200 μl of PBS. Cells were infected with concentrated virus using spinfection (1500 rpm in swing bucket centrifuge for 1 h). Fresh medium was added 16 h after infection, and cells were allowed to recover for 24 h before selection. Cells were selected for 2 weeks with puromycin (0.6 μg/ml) (Sigma-Aldrich) and hygromycin (200 μg/ml) (Corning Mediatech). Cells were cultured with 2 μg/ml doxycycline (EMD Chemicals, San Diego, CA) for 72 h prior to experiments to induce protein expression.
Sequential Salt Extractions
Cells (1 × 107 cells) were harvested and washed with PBS for each cell line. Cells were resuspended in 1 ml of Buffer A (0.3 m sucrose, 60 mm KCl, 60 mm Tris, pH 8.0, 2 mm EDTA, 0.5% Nonidet P-40, plus protease inhibitors) and rotated at 4 °C for 10 min. Samples were spun down at 6500 × g for 5 min. The pellet was resuspended in 200 μl of Modified Radioimmunoprecipitation Assay Buffer (50 mm Tris, pH 8.0, 1% Nonidet P-40, 0.25% sodium deoxycholate, plus protease inhibitors) by pipetting up and down 15 times and incubated on ice for 2 min. Then the sample was centrifuged for 3 min at 6500 × g. The supernatant was saved in a separate tube and labeled as the 0 mm fraction. The pellet was sequentially resuspended in 200 μl of Modified Radioimmunoprecipitation Assay Buffer with increasing NaCl concentrations (100, 200, 300, 400, and 500 mm). The process for 0 mm was repeated for each salt concentration. 4× Bolt LDS Sample Buffer (Invitrogen) with 10% β-mercaptoethanol (AMRESCO LLC, Solon, OH) was added to each sample, and 30 μl of each fraction was loaded onto a 4–12% gradient gel (Invitrogen) for immunoblotting analysis of the BAF subunits of interest. ImageJ was employed to quantitate the protein bands.
Growth Curves
Cells (3 × 105) were plated onto a 60-mm plate with 3 ml of medium containing doxycycline (2 μg/ml). Cells were grown for 72 h before the cells were trypsinized and counted with the Moxi cell counter (Orflo, Ketchum, ID). Cells (3 × 105) were replated. This was repeated three times for each cell line, and results were averaged.
Peptide Pulldown
Streptavidin Agarose Ultra Performance Resin (15 μl) (Solulink, San Diego, CA) was washed three times with Binding Buffer 2 (0.5 mm DTT, 150 mm NaCl, 50 mm Tris). The resin was resuspended in 300 μl of Binding Buffer 2 with 2 μg of biotin-labeled peptide (AnaSpec), and samples were rotated at 4 °C for 1 h. The following peptides were used: H3(21–44), H3K9Ac(1–21), H3K14Ac(1–21), H3K18Ac(1–21), H3K23Ac(15–23), H3K27Ac(21–44), H3K9/14Ac (1–21), and H3K14/18/23/27Ac(1–30). Cells (1.5 × 106 were harvested and lysed in 500 μl of Buffer B (25 mm Hepes, pH 7.6, 25 mm KCl, 5 mm MgCl2, 0.05 mm EDTA, 0.1% Nonidet P-40, and 10% glycerol) and centrifuged. The nuclei were then resuspended in 250 μl of IP Buffer (25 mm Tris, pH 8, 300 mm NaCl, 1% Nonidet P-40, 1 mm EDTA, plus protease inhibitors) and rotated at 4 °C for 10 min. The samples were then spun down at 15,000 rpm for 5 min. The 250 μl of lysate was added to the peptide and resin solution and rotated overnight. The samples were washed for 10 min three times in Binding Buffer 2. The resin was resuspended in 1× Bolt LDS Sample Buffer (Invitrogen) and boiled for 5 min. Nuclear lysate input and the samples were processed for immunoblotting analysis.
Real-time Polymerase Chain Reaction (RT-PCR)
Total RNA was converted into cDNA using iScript Reverse Transcription Kit (Bio-Rad Laboratories). Primers used were published in Ref. 16. Real-time PCR was performed using a Bio-Rad CFX Connect Real-Time system and a FastStart Universal SYBR Green Master (Roche Diagnostics). The results were analyzed using the 2(−ΔΔCT) comparative method. Each sample was tested in quadruplicate.
ChIP
Cells were harvested and counted. Cells (6 × 106) were resuspended in 10 ml of PBS with 1% formaldehyde (Thermo Scientific) and allowed to crosslink for 8 min. Crosslinking was quenched by the addition of glycine to a final concentration of 125 mm and incubated for 5 min. Cells were washed three times with PBS and resuspended in 10 ml of Rinse Buffer 1 (50 mm Hepes, pH 8, 1 mm EDTA, 0.5 mm EGTA and 100 mm NaCl) and then incubated on ice for 5 min and pelleted by centrifugation at 600 × g for 3 min. Cells were resuspended in 5 ml of Rinse Buffer 2 (50 mm Hepes, pH 8, 140 mm NaCl, 1 mm EDTA, 10% Glycerol, 0.5% Nonidet P-40, and 0.25% Triton X-100) and incubated on ice for 5 min. Cells were then pelleted by centrifugation (600 × g) at 4 °C. The cells were then resuspended in 1 ml of ChIP IP Buffer (50 mm Hepes, pH 7.5, 300 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% deoxycholate, 0.1% SDS, plus protease inhibitors.) Samples were sonicated with a Branson Sonifier 250 probe sonicator for 7 min in 30-s bursts followed by 30 s on ice at a duty cycle of 35% and an output of 3. After sonication, debris was pelleted by centrifugation at 20,000 × g for 10 min at 4 °C. Supernatant was collected, and 450 μg was precleared with 15 μl of Dynabeads Protein G (Thermo Scientific). Lysate (200 μg) was incubated with 2 μg of antibody for 3 h followed by a 1-h incubation with 15 μl of washed Dynabeads. Following incubation, beads were washed two times for 5 min with ChIP IP Buffer, followed by washes with Deoxycholate Buffer (10 mm Tris, pH 8, 250 mm LiCl, 0.5% Nonidet P-40, 0.5% deoxycholate) and 1× Tris-EDTA Buffer (10 mm Tris-HCl, 1 mm EDTA pH 8.0). Beads were rotated at room temperature for 20 min in 150 μl of Elution Buffer 2 (1% SDS, 100 mm NaHCO3) for 20 min. The elution step was repeated a second time. Following elution, NaCl was added to a final concentration of 200 mm, and samples were uncrosslinked at 65 °C overnight. EDTA (10 mm) and Tris (40 mm) were added to each sample along with 60 μg of RNase (AMRESCO) and 40 μg of Proteinase K (New England Biolabs). Samples were incubated at 37 °C for 30 min followed by 55 °C for 2.5 h. DNA was isolated using phenol chloroform extraction followed by DNA precipitation. The DNA pellets were resuspended in 20 μl of 1× Tris-EDTA, and RT-PCR was performed as described above.
ChIP Primers
The following ChIP primers were used at the CNTN6 locus (chr3:1,134,629–1,445,278): 1, Chr3:1,134830–1134940 (+ 201 from TSS), forward, GCTGTGTCTGCTGCAATGAG, reverse, CAGTGACTTCTCCCCCAACC; 2, Chr3:1,135824–1135958 (+ 1195 from TSS), forward, TTGCTTGTTTTGAGCAATTTTCAT, reverse, AGGAGGGAAAAAGAGTCTGCT; 3, Chr3:1,138124–1138273 (+ 3495 from TSS), forward, TCTTTCTTTTCTTCAGGATCACCA, reverse, GTGGCAAACAAGAGAACAAGT; 4, Chr3:1,138324-113956 (+ 3695 from TSS), forward, GCCTCTTTGTACTTCAGTTTCCC, reverse, GAGTGCTTCTTAATTGCCAGG.
Immunoblot
Protein samples from cell lysates, SSE, IPs, and peptide pulldowns were denatured for 10 min at 100 °C, separated on a 4–12% SDS-polyacrylamide gel (Invitrogen), and transferred to a nitrocellulose membrane (Millipore). The membrane was blocked with 5% bovine serum albumin (VWR International, Batavia, IL) in PBS containing 0.1% Tween 20 (PBST) for 30 min at room temperature and then incubated in primary antibodies overnight at 4 °C. The primary antibodies were detected by incubating the membranes in goat-anti-rabbit or goat-anti-mouse secondary antibodies (LI-COR Biotechnology, Lincoln, NE) conjugated to IRDye 800CW or IRDye 680, respectively, for 1 h at room temperature, and the signals were visualized using Odyssey Clx imager (LI-COR Biotechnology).
Antibodies
Antibodies used include: ARID1A, Santa Cruz Biotechnology sc-32761 (for immunoblot); ARID2, Santa Cruz Biotechnology sc-81050 (for immunoblot); PBRM1, Bethyl A301-591A (for immunoblot), Bethyl A301-590A (for immunoprecipitation and ChIP); SS18, Santa Cruz Biotechnology sc-28698 (for immunoblot); IgG Rabbit, Santa Cruz Biotechnology sc-2027 (for ChIP); and H3K14Ac, Millipore 07-353 (for ChIP).
Author Contributions
E. G. P. and E. C. D. conceived and designed experiments, E. G. P. performed experiments, E. G. P. and E. C. D. analyzed the results, and E. G. P. and E. C. D. wrote the manuscript.
Acknowledgments
Sanger sequencing data were acquired by the Purdue Genomics Facility supported by National Institutes of Health Grant P30 CA023168. We thank Laura Deemer for assistance with cloning and mutagenesis, and Brian Smith from Medical College of Wisconsin for helpful discussion.
This work was supported by a V scholar award (V2014-004) and a V scholar plus award (D2016-030) from the V Foundation for Cancer Research and an American Cancer Society Institutional Research Grant (ACS IRG Grant 58-006-53) to the Purdue University Center for Cancer Research. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
B. Chowdhury, E. G. Porter, and E. C. Dykhuizen, unpublished data.
- BD
- bromodomain
- BAF
- BRG1-or hBRM-associated factors
- hBRM
- human brahma
- BRG1
- brahma related gene 1
- PBAF
- Polybromo-associated BAF
- ccRCC
- clear cell renal cell carcinoma
- SSE
- sequential salt extraction
- TSS
- transcription start site
- ChIP-qPCR
- ChIP quantitative PCR
- ChIP-Seq
- ChIP-sequencing.
References
- 1. Bannister A. J., and Kouzarides T. (2011) Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Musselman C. A., Lalonde M.-E., Côté J., and Kutateladze T. G. (2012) Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yun M., Wu J., Workman J. L., and Li B. (2011) Readers of histone modifications. Cell Res. 21, 564–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dhalluin C., Carlson J. E., Zeng L., He C., Aggarwal A. K., and Zhou M. M. (1999) Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491–496 [DOI] [PubMed] [Google Scholar]
- 5. Owen D. J., Ornaghi P., Yang J. C., Lowe N., Evans P. R., Ballario P., Neuhaus D., Filetici P., and Travers A. A. (2000) The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase Gcn5p. EMBO J. 19, 6141–6149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang N., and Xu R.-M. (2013) Structure and function of the BAH domain in chromatin biology. Crit. Rev. Biochem. Mol. Biol. 48, 211–221 [DOI] [PubMed] [Google Scholar]
- 7. Xue Y., Canman J. C., Lee C. S., Nie Z., Yang D., Moreno G. T., Young M. K., Salmon E. D., and Wang W. (2000) The human SWI/SNF-B chromatin-remodeling complex is related to yeast Rsc and localizes at kinetochores of mitotic chromosomes. Proc. Natl. Acad. Sci. U.S.A. 97, 13015–13020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thompson M. (2009) Polybromo-1: the chromatin targeting subunit of the PBAF complex. Biochimie 91, 309–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Narlikar G. J., Sundaramoorthy R., and Owen-Hughes T. (2013) Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 154, 490–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hargreaves D. C., and Crabtree G. R. (2011) ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 21, 396–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Middeljans E., Wan X., Jansen P. W., Sharma V., Stunnenberg H. G., and Logie C. (2012) SS18 together with animal-specific factors defines human BAF-type SWI/SNF complexes. PLoS ONE 7, e33834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Varela I., Tarpey P., Raine K., Huang D., Ong C. K., Stephens P., Davies H., Jones D., Lin M.-L., Teague J., Bignell G., Butler A., Cho J., Dalgliesh G. L., Galappaththige D., et al. (2011) Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469, 539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peña-Llopis S., Vega-Rubín-de-Celis S., Liao A., Leng N., Pavía-Jiménez A., Wang S., Yamasaki T., Zhrebker L., Sivanand S., Spence P., Kinch L., Hambuch T., Jain S., Lotan Y., Margulis V., et al. (2012) BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 44, 751–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cancer Genome Atlas Research Network (2013) Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gerlinger M., Horswell S., Larkin J., Rowan A. J., Salm M. P., Varela I., Fisher R., McGranahan N., Matthews N., Santos C. R., Martinez P., Phillimore B., Begum S., Rabinowitz A., Spencer-Dene B., et al. (2014) Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 46, 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chowdhury B., Porter E. G., Stewart J. C., Ferreira C. R., Schipma M. J., and Dykhuizen E. C. (2016) PBRM1 regulates the expression of genes involved in metabolism and cell adhesion in renal clear cell carcinoma. PLoS ONE 11, e0153718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Forbes S. A., Bindal N., Bamford S., Cole C., Kok C. Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A., Teague J. W., Campbell P. J., Stratton M. R., and Futreal P. A. (2011) COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 39, D945–D950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brownlee P. M., Chambers A. L., Oliver A. W., and Downs J. A. (2012) Cancer and the bromodomains of BAF180. Biochem. Soc. Trans. 40, 364–369 [DOI] [PubMed] [Google Scholar]
- 19. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., and Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Charlop-Powers Z., Zeng L., Zhang Q., and Zhou M.-M. (2010) Structural insights into selective histone H3 recognition by the human Polybromo bromodomain 2. Cell Res. 20, 529–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vivoli M., Novak H. R., Littlechild J. A., and Harmer N. J. (2014) Determination of protein-ligand interactions using differential scanning fluorimetry. J. Vis. Exp. e51809, 10.3791/51809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jung M., Philpott M., Müller S., Schulze J., Badock V., Eberspächer U., Moosmayer D., Bader B., Schmees N., Fernández-Montalván A., and Haendler B. (2014) Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J. Biol. Chem. 289, 9304–9319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Myrianthopoulos V., Gaboriaud-Kolar N., Tallant C., Hall M. L., Grigoriou S., Brownlee P. M., Fedorov O., Rogers C., Heidenreich D., Wanior M., Drosos N., Mexia N., Savitsky P., Bagratuni T., Kastritis E., et al. (2016) Discovery and optimization of a selective ligand for the switch/sucrose non-fermenting-related bromodomains of Polybromo protein-1 by the use of virtual screening and hydration analysis. J. Med. Chem. 59, 8787–8803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Filippakopoulos P., Picaud S., Mangos M., Keates T., Lambert J.-P., Barsyte-Lovejoy D., Felletar I., Volkmer R., Müller S., Pawson T., Gingras A.-C., Arrowsmith C. H., and Knapp S. (2012) Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chandrasekaran R., and Thompson M. (2007) Polybromo-1-bromodomains bind histone H3 at specific acetyl-lysine positions. Biochem. Biophys. Res. Commun. 355, 661–666 [DOI] [PubMed] [Google Scholar]
- 26. Sanchez O. F., Williamson D., Cai L., and Yuan C. (2015) A sensitive protein-based sensor for quantifying histone acetylation levels. Talanta 140, 212–218 [DOI] [PubMed] [Google Scholar]
- 27. Karmodiya K., Krebs A. R., Oulad-Abdelghani M., Kimura H., and Tora L. (2012) H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics 13, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jones S., Wang T.-L., Shih I.-M., Mao T.-L., Nakayama K., Roden R., Glas R., Slamon D., Diaz L. A. Jr., Vogelstein B., Kinzler K. W., Velculescu V. E., and Papadopoulos N. (2010) Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330, 228–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cancer Genome Atlas Research Network (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615; Correction (2012) Nature490, 298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. VanDemark A. P., Kasten M. M., Ferris E., Heroux A., Hill C. P., and Cairns B. R. (2007) Autoregulation of the Rsc4 tandem bromodomain by Gcn5 acetylation. Mol. Cell 27, 817–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sanchez R., and Zhou M.-M. (2009) The role of human bromodomains in chromatin biology and gene transcription. Curr. Opin. Drug Discov. Devel. 12, 659–665 [PMC free article] [PubMed] [Google Scholar]
- 32. Ji X., Dadon D. B., Abraham B. J., Lee T. I., Jaenisch R., Bradner J. E., and Young R. A. (2015) Chromatin proteomic profiling reveals novel proteins associated with histone-marked genomic regions. Proc. Natl. Acad. Sci. U.S.A. 112, 3841–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]