

Abstract
Follicle-stimulating hormone (FSH) regulates follicular growth and stimulates estrogen synthesis in the ovaries. FSH is a heterodimer consisting of an α subunit, also present in luteinizing hormone, and a unique β subunit, which is transcriptionally regulated by gonadotropin-releasing hormone 1 (GNRH). Because most FSH is constitutively secreted, tight transcriptional regulation is critical for maintaining FSH levels within a narrow physiological range. Previously, we reported that GNRH induces FSHβ (Fshb) transcription via induction of the AP-1 transcription factor, a heterodimer of c-FOS and c-JUN. Herein, we identify c-JUN-dimerization protein 2 (JDP2) as a novel repressor of GNRH-mediated Fshb induction. JDP2 exhibited high basal expression and bound the Fshb promoter at an AP-1-binding site in a complex with c-JUN. GNRH treatment induced c-FOS to replace JDP2 as a c-JUN binding partner, forming transcriptionally active AP-1. Subsequently, rapid c-FOS degradation enabled reformation of the JDP2 complex. In vivo studies revealed that JDP2 null male mice have normal reproductive function, as expected from a negative regulator of the FSH hormone. Female JDP2 null mice, however, exhibited early puberty, observed as early vaginal opening, larger litters, and early reproductive senescence. JDP2 null females had increased levels of circulating FSH and higher expression of the Fshb subunit in the pituitary, resulting in elevated serum estrogen and higher numbers of large ovarian follicles. Disruption of JDP2 function therefore appears to cause early cessation of reproductive function, a condition that has been associated with elevated FSH in women.
Keywords: AP-1 transcription factor (AP-1), c-FOS, c-JUN transcription factor, endocrinology, follicle-stimulating hormone (FSH), gene expression, gene transcription
Introduction
Gonadotrope cells in the anterior pituitary synthesize and secrete luteinizing hormone (LH)2 and follicle-stimulating hormone (FSH) in response to pulses of hypothalamic gonadotropin-releasing hormone 1 (GNRH), which can be modulated by activin/inhibin and steroid feedback from the gonads. Gonadotropin hormones are heterodimers of a common α subunit and a unique β subunit. Gonadotropin levels fluctuate 3–4-fold throughout the menstrual or estrous cycle, preceded by changes in the β subunit transcription (1, 2). Dysregulation of the tightly controlled gonadotropin levels leads to reproductive pathophysiology. Low levels of gonadotropin hormones lead to infertility; low LH leads to hypogonadism and anovulation, whereas low FSH causes follicular growth arrest (3–5). High levels also lead to reproductive pathologies, particularly in females, such as polycystic ovary syndrome, characterized by an increase specifically in LH (6–8), and premature ovarian failure or primary ovarian insufficiency, which is associated with an increase in FSH (9, 10). Recently, it was determined that increased levels of FSH in the circulation, due to retrafficking of FSH to the GNRH-regulated secretory pathway, resulted in 5-fold more corpora lutea in the ovaries, implicating higher FSH levels in greater follicle recruitment (11). Furthermore, polymorphism in the FSHB promoter in women, which regulates β subunit expression, increases the level of FSH and has been associated with early menarche and menopause (12). Another genome-wide association study reported that polymorphism 5′ of the FSHB gene, which causes a higher level of FSH in the circulation, is also associated with premature puberty, early natural menopause, and dizygotic twinning (13).
Normal reproductive function requires pulsatile secretion of GNRH into the hypophysial portal system, whereas long term tonic exposure to GNRH adversely affects the hypothalamic-pituitary-gonadal axis (14–16). GNRH receptors lack the C-terminal tail that normally mediates desensitization through β-arrestin association and encapsulation into clathrin-coated vesicles, rendering the receptor resistant to this regulatory mechanism. Thus, the receptor does not internalize as rapidly as other GPCRs (17). Given that GNRH receptors do not internalize within the frequency of the pulse, due to the lack of a cytoplasmic tail, other feedback mechanisms must exist within the gonadotrope to shut off the signal and maintain gonadotropin expression within the narrow physiological range. This is especially true for FSH, because once synthesized, the majority of this hormone is constitutively secreted (18). The GNRH receptor primarily couples to Gαq/11 and activates phospholipase Cβ, causing an increase in inositol 1,4,5-triphosphate (IP3), intracellular calcium, and diacylglycerol (19). The degree of desensitization that does occur after receptor occupancy may be due to loss of IP3 receptors necessary for propagation of signaling (17, 20, 21).
In addition to desensitization of signaling molecules, such as IP3 receptors, it is likely that mechanisms exist in the nucleus that prevent further transcription of the gonadotropin subunits. There are several possible mechanisms, including dephosphorylation, degradation, and deactivation of transcription factors and RNA polymerase II; return of chromatin to a closed conformation by histone modification; or induction or reactivation of transcriptional repressors. We identified that GNRH induces Fshb through the activating protein 1 (AP-1) site (22), which is bound by the AP-1 transcription factor, composed of heterodimers of Fos and Jun isoforms. Prototypical members c-FOS and c-JUN are activated rapidly and transiently and exhibit a very short half-life to bring forth very tight temporal regulation of target genes (23). We also determined that GNRH specifically induces c-FOS (24) and c-JUN (25) transcription in the mature gonadotrope cells, whereas other hormones and stimuli that activate these genes in other cell types are not sufficient to induce AP-1 in the gonadotrope. c-FOS is not present without stimulus in quiescent cells and is more strictly regulated, both at the transcriptional and posttranslational level by GNRH, whereas c-JUN has detectable basal expression and is induced 6-fold by GNRH (26). Following c-FOS induction, it dimerizes with c-JUN to bind the AP-1 site, which is currently the only known element to convey GNRH responsiveness of the Fshb promoter. Negative regulators at the AP-1 site or the highly similar CRE element include activating transcription factor 3 (ATF3 (27)), inducible cAMP early repressor (ICER (28)), and c-JUN dimerization protein 2 (JDP2) (29).
JDP2, previously known to form a heterodimer with c-JUN (29), represses transcription of AP-1 target genes. Herein, we determined that JDP2 interacts with c-JUN at the AP-1 site in the Fshb promoter. Consistent with prior reports that JDP2 can also interact with ATF2 (30), which we previously determined conveys GNRH induction of c-Jun in gonadotrope cells (25), JDP2 inhibits induction of c-Jun mRNA by GNRH. The repressor, JDP2, may function before the GNRH signal and after a wave of transcription to shut off the signal. Like many other repressors, JDP2 is known to recruit histone deacetylases (HDACs) to repress transcription through chromatin modification. In particular, JDP2 interacts directly with HDAC3 (31, 32). Previously, it has been shown that HDAC3 dissociates from the Fshb promoter following GNRH treatment to increase Fshb transcription (33). Identification of JDP2 provides a mechanism of histone deacetylation in the absence of the GNRH signal.
In this report, we identify a mechanism of negative regulation of FSHβ transcription in the gonadotrope in vivo and in the LβT2 model cell line. Previous reports identified that Nab family members antagonize Egr1 and negatively affect GNRH induction of Lhb (34), whereas Fshb induction by activin is negatively regulated by Smad pathway antagonists SKIL and TGIF (35). We identified a member of the AP-1 superfamily, JDP2, that serves as a negative regulator of Fshb induction by GNRH, both directly, by repressing AP-1 transcriptional activity, and indirectly, by limiting c-JUN expression. We further demonstrate that a lack of this repressor in vivo leads to early puberty and early cessation of fertility in the null female mice, which may mimic premature ovarian failure in human population. Therefore, identification of the novel function of JDP2 leads to better understanding of the regulation of FSH physiological levels and pathophysiologies where FSH levels are dysregulated.
Results
JDP2 Is a Novel Negative Regulator of FSHβ Induction by GNRH
The physiological range and responsiveness of gene expression to subsequent, specific stimuli is tightly controlled by both positive and negative regulators of transcription. This is critical for gonadotropin hormones, whose increase is associated with polycystic ovary syndrome or premature ovarian failure in females. Although repressors are relatively more difficult to identify, negative regulators of GNRH induction of LHβ and activin induction of FSHβ have been determined (34, 35). We sought to identify a protein that serves as a transcriptional repressor of GNRH induction of Fshb transcription, because Fshb is normally expressed at a low basal level. We first determined the time course of induction of GNRH target genes in LβT2 cells, a model of mature gonadotrope (Fig. 1A). The time course for induction of GNRH target genes is different, but all are biphasic. Similar to the previously reported time course for c-Fos induction (26), the maximal induction of c-Jun of 15.5-fold is at 1 h of treatment, after which the message rapidly declines. -Fold induction diminished to 5.4-fold of control at 2 h and 3.8-fold at 6 h, and induction returned to basal levels at 12 h of treatment. Compared with our previous report analyzing c-Fos, the major difference is that c-Fos is not present at a basal state, without stimuli, whereas c-Jun is present at both mRNA (Fig. 1A) and protein levels (26). Fshb expression is very low at the basal state (Fig. 1A), and maximal induction of 12-fold by GNRH is observed at 2 h of treatment. After the maximum, the message level diminishes and returns to basal level at 12 h of treatment. We performed DNA pull-downs followed by mass spectrometry sequencing and identified JDP2 as a novel protein that interacts with the FSHβ promoter at the AP-1 site, which was previously known to form a heterodimer with the Jun subfamily (29). We then analyzed whether JDP2 is induced by GNRH and determined that Jdp2 exists in the cells at a relatively high basal level and is induced 2-fold at 2 h of GNRH treatment (Fig. 1B). We also analyzed the expression of a close family member, ATF3, which is induced by GNRH (25, 36), and determined that Atf3 mRNA is not found at a basal level and is induced rapidly by GNRH with a maximal induction of 23-fold at 1 h, after which the induction diminishes (Fig. 1B). Therefore, both ATF3 and JDP2, which can serve as negative regulators of transcription at the AP1 and CRE sites, are induced by GNRH. The difference is that Jdp2 is present at the basal level, whereas Atf3 is non-detectable.
FIGURE 1.
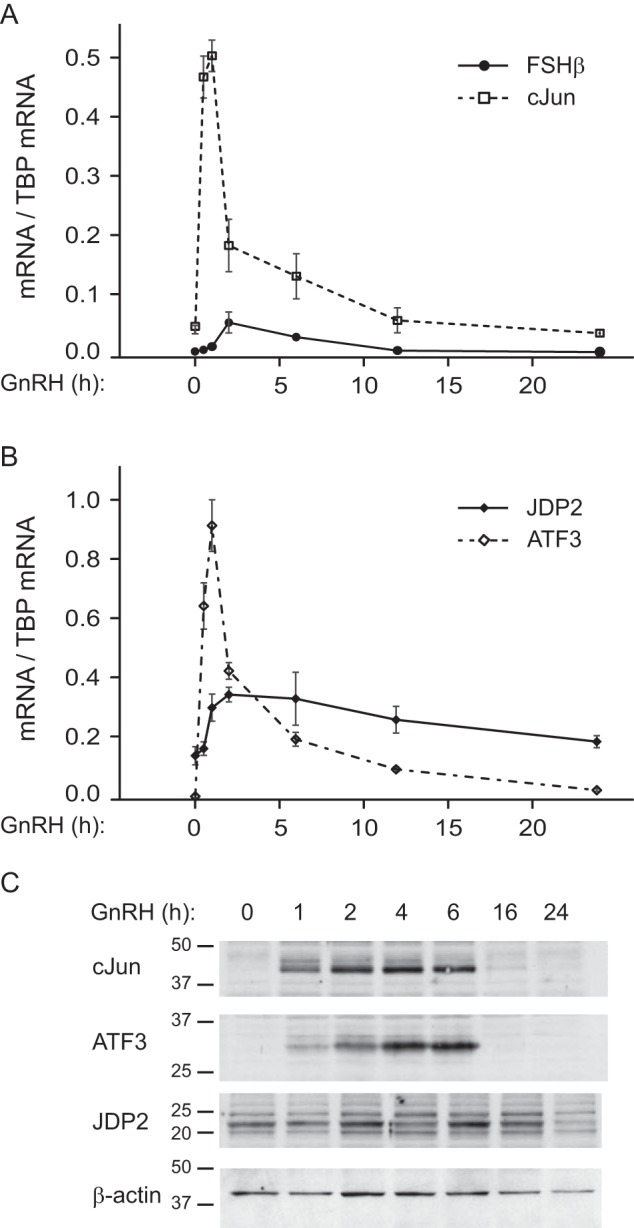
Time line of GNRH induction of gonadotrope genes. A, FSHβ (Fshb; solid line) and c-JUN (c-Jun; dashed line) mRNA expression was determined by qPCR following 10 nm GNRH treatment of LβT2 cells for the times indicated on the x axis and normalized to the Tbp mRNA. B, mRNA of the putative repressors, Jdp2 (solid line) and Atf3 (dashed line), was analyzed in the same samples and also normalized to Tbp mRNA. The experiment was repeated three times, and results are presented as an average ± S.E. (error bars). C, protein levels of transcriptional regulators in the whole cell lysates following GNRH treatment for the times indicated above each lane were analyzed by Western blotting.
Protein levels of c-JUN, ATF3, and JDP2 transcription factors correspond to changes in mRNA levels (Fig. 1C). c-JUN protein is induced after 1 h of GNRH treatment and exhibits a maximum at 2 h, as we determined previously (26). c-JUN protein level returns to basal after 16 h of treatment. ATF3 protein induction, as determined before (25), is observed after 1 h of GNRH, exhibits a maximum at 4 h, and returns to basal level by 16 h. JDP2 protein is present at basal level without GNRH treatment, and the levels change less compared with ATF3 and c-JUN, with an increase at 2 h of treatment and return to basal level by 24 h.
Next, we determined whether JDP2 can interfere with GNRH induction of gonadotrope genes. We determined previously that GNRH phosphorylation of ATF2 activates c-JUN transcription in the gonadotrope (25) and that GNRH induction of c-JUN and c-FOS activates Fshb transcription (22). Because JDP2 in other cells interacts with ATF2 (30) as well as c-JUN (29), we overexpressed JDP2 with c-JUN luciferase (Fig. 2B) and FSHβ luciferase (Fig. 2E) reporters. To determine the specificity of JDP2 function in gene induction by GNRH, we also overexpressed ATF3, which can serve as a repressor and is a close family member of JDP2. JDP2 can bind at either the TRE/AP-1 site (TGAGTCA), where it interacts with c-JUN, or the CRE site (TGACGTCA), where it interacts with ATF2. The TRE/AP-1 half-site conveys GNRH responsiveness of the Fshb promoter (22), whereas the CRE site in the c-Jun promoter is responsible for c-Jun induction by GNRH (25). Thus, in addition to c-JUN luciferase and FSHβ luciferase, we transfected CRE luciferase and TRE luciferase to assay GNRH induction via these sites in the presence of JDP2 or ATF3. These reporters contain four tandem copies of the 8-bp consensus CRE element (Fig. 2A) or of the similar 7-bp consensus TRE multimer, which has four tandem copies of the TRE element (Fig. 2D) ligated to the minimal thymidine kinase promoter driving luciferase expression. GNRH induced the CRE site multimer, as we have shown previously, whereas the addition of either ATF3 or JDP2 diminished both basal expression and GNRH induction, indicated with a pound sign (Fig. 2A). Both ATF3 and JDP2 also reduced -fold induction of the CRE site by GNRH, indicated with an asterisk. -Fold induction was calculated by normalizing samples treated with GNRH to the vehicle-treated samples transfected with the same expression vectors, for easier observation of the induction by GNRH without the effect of basal repression. On the other hand, basal expression and GNRH induction of the c-JUN promoter, which contains other elements in addition to the CRE site, was reduced by overexpression of JDP2 but not ATF3 (Fig. 2B). The CRE site in the c-JUN promoter is necessary for the maximal induction of the c-JUN gene by GNRH, because GNRH induction of c-JUN is diminished when CRE is mutated (Fig. 2C, CRE mut) (25). CRE site mutation prevented further repression of GNRH induction of c-JUN by JDP2, because the CRE site mutant is induced by GNRH to the same level with and without JDP2 (Fig. 2C). This indicates that JDP2 represses c-JUN promoter via the CRE site. JDP2 overexpression diminished both basal and -fold induction of the TRE/AP-1 multimer by GNRH, whereas ATF3 overexpression reduced basal expression and consequently the level of induction by GNRH but had no effect on -fold induction, contrary to the CRE multimer (Fig. 2D). JDP2, but not ATF3, reduced FSHβ -fold induction by GNRH (Fig. 2E). As stated previously, GNRH induces FSHβ through the TRE/AP-1 site, because when this site is mutated, GNRH induction is reduced (Fig. 3F, TRE mut) (22). Similarly to the c-JUN promoter, JDP2 does not repress FSHβ induction by GNRH when the TRE site is mutated, indicating that the TRE site is necessary for JDP2 function (Fig. 2F). Therefore, native promoters that contain elements other than GNRH-responsive CRE and/or TRE sites are resistant to the repression by ATF3. However, JDP2 reduces both c-JUN and FSHβ induction by GNRH via CRE and TRE elements, respectively.
FIGURE 2.

JDP2 diminishes GNRH induction of c-Jun and FSHβ. A, four tandem copies of the CRE element (TGACGTCA) were linked to the minimal heterologous promoter and luciferase reporter in pGL3 backbone and transfected into LβT2 cells with the herpesvirus thymidine kinase-driven β-galactosidase gene as an internal control for transfection efficiency; additionally, cells were co-transfected with empty vector control (ctrl) or expression vector for ATF3 or JDP2. After overnight starvation, cells were treated with vehicle or 10 nm GNRH for 5 h, after which the luciferase and β-galactosidase values were obtained. B and C, c-JUN luciferase plasmid (−1000 bp) was used as a reporter. D, four copies of TRE element (TGAGTCA) served as a reporter. E and F, −1000 bp FSHβ luciferase was transfected as a reporter. *, significant change (p < 0.05) in -fold induction by GNRH, where the GNRH-treated luciferase/β-galactosidase ratio was normalized to vehicle-treated samples transfected with the same expression vector. #, significantly lower luciferase expression from the empty vector control (vehicle-treated samples transfected with overexpression vector compared with vehicle-treated empty vector, and GNRH-treated samples transfected with overexpression compared with GNRH-treated vector control). White bars, vehicle; black bars, GNRH-treated. Error bars, S.E.
FIGURE 3.

JDP2 binds FSHβ (A and B) and c-Jun (C) promoters. Nuclear extract from either LβT2 cells treated with GNRH for the times indicated above the lanes or COS-1 cells transfected with overexpression vectors for proteins indicated above the corresponding lanes was incubated with a 30-bp radiolabeled probe that encompasses the AP-1/TRE site in the FSHβ promoter (A), a 30-bp radiolabeled probe encompassing the AP-1/TRE site in the FSHβ promoter (TTGGTCA) or its mutation (TTGaaaA) compared with AP-1/TRE consensus (TGAGTCA) or its mutation (TGAaaaA) (B), or a 30-bp radiolabeled probe encompassing the CRE site in the c-JUN promoter (C). EMSA was performed three times, and representative gels subjected to autoradiography are shown.
JDP2 Binds c-JUN and FSHβ Promoters in Complex with ATF2 and c-JUN, Respectively
To determine whether JDP2 can bind FSHβ and c-JUN promoters, we performed EMSA using nuclear extracts from LβT2 gonadotrope cells following treatment with GNRH. Due to a lack of specific antibodies for JDP2, we were unable to perform a supershift with LβT2 cell extracts. Instead, to determine whether JDP2 can bind by itself and/or in combination with other factors known to bind the TRE/AP-1 site in the FSHβ promoter and the CRE site in the c-JUN promoter, we overexpressed proteins of interest in COS-1 cells: c-FOS, c-JUN, and JDP2 and their combination for FSHβ promoter analysis (Fig. 3A) and ATF2, ATF3, c-JUN, and JDP2 for c-JUN promoter analysis (Fig. 3C). We monitored not only their binding but also co-migration with complexes from LβT2 cells. Two-hour GNRH treatment of LβT2 gonadotrope cells induced two different complexes that bind FSHβ AP-1 probe (Fig. 3A), consistent with our previous report (22). The identity of the proteins that comprise these complexes was determined previously by antibody supershift; the higher complex is composed of c-JUN and c-FOS, and the lower complex is composed of c-JUN and FosB (22). Here, we show that the higher complex of c-JUN and c-FOS heterodimer comigrates with a c-JUN and c-FOS heterodimer overexpressed in COS-1 cells. These GNRH-induced complexes are not present in the extracts from LβT2 cells at the basal state or following 12 h of treatment, at which times two other complexes bind that are partially obscured by more abundant GNRH-induced complexes at 2 h of treatment. Of the two basal complexes, the higher one was previously identified as the basal factor NFY, and the lower complex was previously unidentified. Here, this lower complex comigrates with a complex composed of c-JUN and JDP2 overexpressed in COS-1 cells, indicating that a dimer of c-JUN and JDP2 can bind the FSHβ TRE/AP-1 probe. JDP2 can bind the AP-1 probe by itself as well; however, the JDP2-only band from COS-1 cells does not comigrate with any complex from LβT2 cell extracts, probably because c-JUN is expressed at a basal level in LβT2 cells, and it can heterodimerize with JDP2. When oligonucleotides with a mutation in the TRE/AP-1 site were used as competitors with the wild-type probe, competitors were not able to compete for c-JUN·c-FOS, c-JUN·FosB, or c-JUN·JDP2 complexes, indicating that the TRE/AP-1 site is necessary for binding of these proteins (Fig. 3A, right).
The TRE site in the FSHβ promoter (TTGGTCA) differs slightly from the TRE consensus sequence (TGAGTCA) (22). Thus, we compared binding to the TRE site in the FSHβ promoter and to the consensus TRE site (Fig. 3B). To more easily observe differences in binding intensity, the film was underexposed compared with Fig. 3A. For consistency, all binding reactions contained the same specific activity of FSHβ or consensus 30-bp probe and the same protein amount. Binding of c-JUN·c-FOS and c-JUN·FosB complexes from LβT2 cells following GNRH treatment was stronger when the TRE consensus was used as a probe than when the FSHβ TRE/AP-1 site was used as a probe. Similar results were observed with overexpressed proteins, and binding intensity was stronger to the consensus probe. The core TRE sequence is necessary for binding because neither c-JUN·c-FOS nor JDP2 bound to either probe when the TRE site was mutated (Fig. 3B, mut).
As determined previously, ATF2 binds the c-JUN promoter at the CRE site in the basal state (25), because its phosphorylation leads to activation and induction of c-Jun transcription (Fig. 3C). As shown in Fig. 1, c-Jun induction diminishes after 1 h of treatment, and previous studies determined that c-JUN can reduce its own expression in a negative feedback loop manner (37). We determined using overexpression in COS-1 cells that, indeed, c-JUN can bind its own promoter, not alone, but in a complex with ATF2 and in the complex with JDP2 (Fig. 3C). We further determined, also with overexpression in COS-1 cells, that JDP2 can dimerize with ATF2, and their complex can bind the c-JUN probe, in addition to the JDP2 and c-JUN complex. JDP2-containing complexes comigrate with complexes from LβT2 extracts after 2- and 5-h GNRH treatment, when c-Jun transcription is diminished. The CRE site mutant used as a competitor in 200-fold excess was unable to compete for GNRH-induced complexes, whereas wild-type competitor successfully competed (Fig. 3C, right). Thus, the CRE site in the c-JUN promoter is necessary for JDP2 and ATF2 binding, because, when mutated, the JDP2 or ATF2 did not bind; however, intensity of binding to the CRE site from the c-JUN promoter and to the CRE consensus was the same (data not shown). Therefore, JDP2 binds both FSHβ and c-JUN promoters at the TRE and CRE sites, respectively, alone and in complex with c-JUN and ATF2.
Activation Site on the JDP2 Protein Is Required for Repressor Function
We sought to determine the mechanism of negative regulation of c-Jun and Fshb gene expression by JDP2 and the specificity of action, because highly homologous family member ATF3 does not reduce GNRH induction of either gene (Fig. 2, B and E). Both the c-JUN promoter and the FSHβ promoter contain the CCAAT element, which binds the NFY basal transcription factor, in close proximity to the GNRH-regulated elements, CRE and TRE/AP-1, respectively (see Fig. 11). To assess whether JDP2 is recruited to the promoters via interaction with NFY, we performed GST pull-down experiments (Fig. 4A). Using GST pull-downs, 35S-labeled JDP2, ATF2, ATF3, and c-JUN are retained in the precipitate with glutathione beads following interaction with GST-NFY, but not with GST control (Fig. 4A). NFY does not interact with c-FOS, whereas it interacts with c-JUN and ATF2, which we have shown previously (22, 25). Both ATF3 and JDP2 can interact with NFY; thus, interaction with NFY is not a reason for the absence of c-JUN and FSHβ promoter repression by ATF3 and for the specificity of JDP2 function.
FIGURE 11.

JDP2 repression of c-Jun and FSHβ gene expression. JDP2 is present in the gonadotrope at the basal level and inhibits c-JUN and FSHβ transcription, indicated with X. JDP2 binds c-JUN and FSHβ promoters at the CRE and TRE elements, respectively, and interacts with basal transcription factor NFY. JDP2 also interacts with ATF2 that binds the c-JUN promoter at the basal state alone and as a heterodimer with JDP2 (top left) and with c-JUN that is expressed at some basal level and binds the FSHβ promoter as a heterodimer with JDP2 (bottom left). GNRH treatment leads to phosphorylation and activation of ATF2 by p38 MAPK, which causes dissociation of JDP2 and induction of c-JUN transcription, indicated with an arrow (top middle). GNRH treatment also leads to induction of c-FOS via calcium calmodulin kinase II (CKII) activation, after which c-FOS displaces JDP2 as a c-JUN binding partner and activates FSHβ transcription (bottom middle). Dephosphorylation of ATF2 (top right) or degradation of c-FOS proteins due to their short half-life (bottom right) leads to recruitment of JDP2 and cessation of transcription until the next pulse of GNRH.
FIGURE 4.

JDP2 interacts with NFY. A, GST pull-down assays demonstrate that NFY can interact directly with JDP2, ATF2, ATF3, and c-JUN, but not c-FOS. 35S-Labeled proteins, indicated above each panel, were used in the binding assay with GST-NFY fusion protein or control GST, labeled above each panel. In the input panel, 10% of the in vitro transcribed and translated labeled proteins that were used in the binding reaction were run on the gel as a control for their expression and labeling. B, cells were transfected with histidine tag-JDP2 (His) or FLAG-tagged-c-FOS, and complexes were precipitated (IP) with antibodies to His tag or FLAG tag, run on the gel, and transferred, and membranes were probed with antibodies to c-JUN, c-FOS, and NFY-A to determine proteins that interact with JDP2. WB, Western blotting.
Additionally, to analyze protein in complex with JDP2, we performed immunoprecipitation following overexpression of histidine-tagged JDP2. FLAG-tagged c-FOS served as a positive control because c-FOS is a known c-JUN-interacting partner. c-JUN was detected in the immunoprecipitate with both His and FLAG tag antibodies, indicating that c-JUN can interact with both JDP2 and c-FOS, as expected (Fig. 4B). JDP2 also precipitated c-FOS and NFY-A protein, which is a part of the NFY complex. Because immunoprecipitation will detect all proteins in complex, whereas GST pull-downs detect direct protein-protein interactions, taken together, these results indicate that JDP2 directly interacts with c-JUN and NFY-A and, via c-JUN, with the c-JUN·c-FOS complex.
It was demonstrated previously that although JDP2 and ATF3 exhibit 61% overall homology and 90% homology in their bZIP domains, a major difference in the structures of these highly homologous proteins is the presence of a putative activation site on Thr-148 in JDP2, which is absent in ATF3 (38). Hence, the activation site was mutated to alanine to inactivate it, and the effect on c-JUN and FSHβ induction by GNRH of the mutant (T148A) was compared with the effect of the wild type (JDP2; Fig. 5, A and B). Consistent with previous results, wild-type JDP2 repressed basal and GNRH induction of c-JUN (Fig. 5A), whereas the T148A mutant increased both basal and GNRH induction of c-JUN, indicating that JDP2 is involved in basal repression of c-JUN without the stimulus. FSHβ induction by GNRH was diminished with overexpression of wild-type JDP2 as before. The T148A mutant not only inhibited the repression observed with wild-type JDP2 but significantly increased the -fold induction (Fig. 5B). Thus, Thr-148 is required for the repressor function of JDP2. Next, we mutated the same threonine residue to Asp, which mimics activation, and determined that this mutant (T148D) repressed GNRH induction of the c-JUN and FSHβ reporters to the same level as the wild type (JDP2; Fig. 5, A and B). This may indicate that in these cells, JDP2 protein is constitutively activated. Mutation of the DNA-binding domain (DBDm) produced the inactive protein that was unable to repress GNRH induction of either c-JUN or FSHβ (Fig. 5, A and B). Thus, the JDP2 DNA-binding domain is necessary for its repressor function.
FIGURE 5.
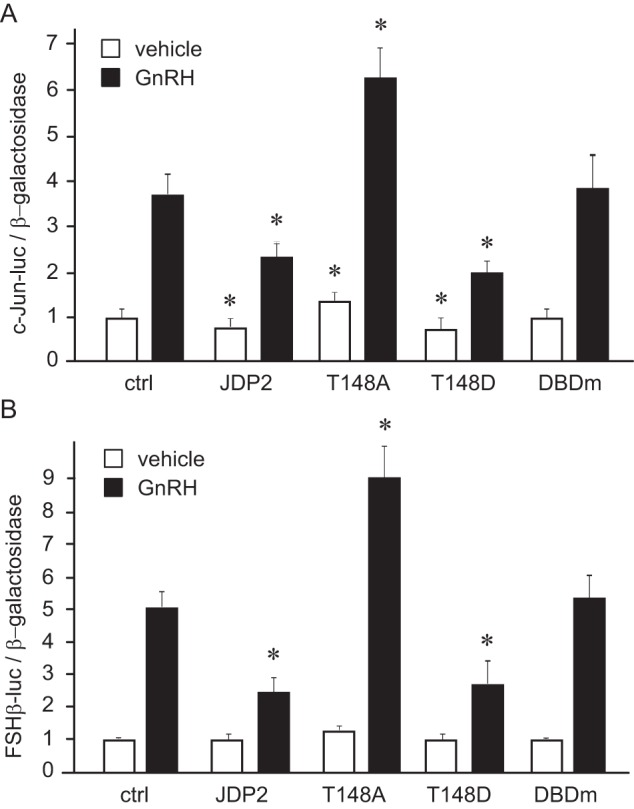
Threonine 148 is necessary for JDP2 repressor activity. c-JUN luciferase (A) or FSHβ luciferase (B) reporter were transfected into LβT2 cells with empty vector control, wild-type JDP2 (JDP2), or JDP2 mutated at the threonine 148 to alanine (T148A) or to glutamic acid (T148D) or with JDP2 with a mutated DNA-binding domain (DBDm) and treated with vehicle or GNRH for 5 h. *, statistically significant difference (p < 0.05) in reporter expression with JDP2 overexpression following vehicle or GNRH treatment, compared with empty vector control following vehicle or GNRH, respectively. White bars, vehicle; black bars, GNRH-treated. Error bars, S.E.
JDP2 directly binds histones and recruits histone deacetylases, resulting in the inhibition of histone acetylation (32, 39, 40). In particular, JDP2 interacts with HDAC3 (41), which is the same HDAC that binds the Fshb promoter at the basal state and is dissociated from the promoter following GNRH treatment (33). Again, due to the lack of specific antibodies to JDP2, we were not able to analyze JDP2 recruitment to the Fshb promoter in vivo. Thus, we analyzed histone acetylation at the Fshb promoter following GNRH treatment using chromatin immunoprecipitation (ChIP) with antibodies to acetylated histone H3, expressed as percentage input, and normalized to the amount of precipitated chromatin with total H3 antibody, also expressed as percentage input based on the standard curve of input chromatin for each time point. Indeed, 1-h GNRH treatment resulted in a higher level of histone acetylation at the Fshb promoter (Fig. 6), which may allow for gene activation following dissociation of the JDP2 from the promoter. This is consistent with previous reports that JDP2 inhibits histone acetylation and our hypothesis that GNRH treatment dissociates JDP2 from the Fshb promoter by inducing c-FOS as a c-JUN binding partner.
FIGURE 6.
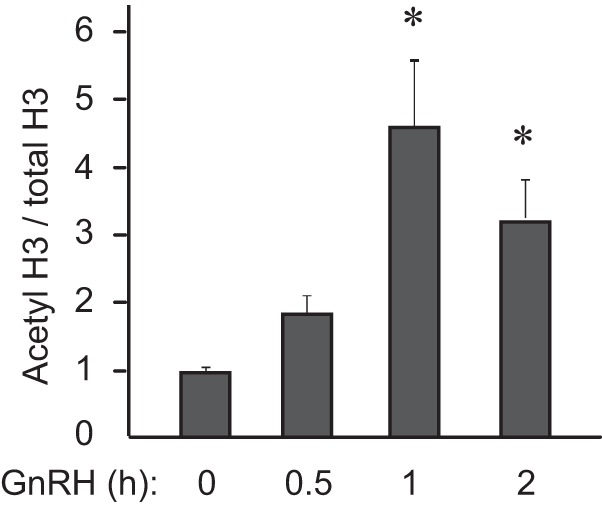
Histone 3 acetylation increases following GNRH treatment. Chromatin immunoprecipitation was performed using antibodies against acetylated histone H3 and total histone H3 following LβT2 cell treatment with GNRH for the times indicated below each bar. The percentage of precipitated chromatin with each antibody at each time point was calculated using serial dilution of total input chromatin, and then acetylated H3 was normalized to total H3. *, statistically significant increase (p < 0.05) in acetylated H3 compared with the zero time point. Error bars, S.E.
JDP2 Regulates Reproductive Function in Vivo
To determine whether JDP2 plays a role in reproduction in vivo, we obtained JDP2 null mice (42) and analyzed their fertility. There is no weight difference between genotypes, and food intake is the same. Male mice had normal sperm count, gonadotropin hormone levels, and reproductive function, as expected from a negative regulator of the FSH hormone. Female mice, on the other hand, exhibited a number of reproductive anomalies (Fig. 7). Female JDP2 null mice exhibited significantly earlier vaginal opening, an external sign of puberty, at postnatal day 24.3 compared with postnatal day 29.4 for wild-type littermates. At 8 weeks of age, females were paired with a wild-type male of proven fertility for a year, and litter birthdates, numbers of litters, and numbers of pups in each litter were recorded. Female null mice delivered their first litter significantly earlier than wild-type mice. In fertility assessment studies, during which we counted the number of pups, the combined number of pups in the first four litters of JDP2 null mice was significantly higher than the number of pups in four litters of wild-type female littermates. We also observed an early cessation of fertility in null mice. JDP2 null mice had their last litter at p268, whereas wild-type mice on average stopped reproducing at postnatal day 338. This difference may stem from early depletion of ovarian reserves due to larger litters in younger animals. Thus, female nulls have early vaginal opening, an earlier first litter, and larger litters, but they stop reproducing at a younger age than wild-type mice. This early cessation of reproductive function corresponds to our hypothesis that JDP2 nulls may mimic premature ovarian failure in women.
FIGURE 7.
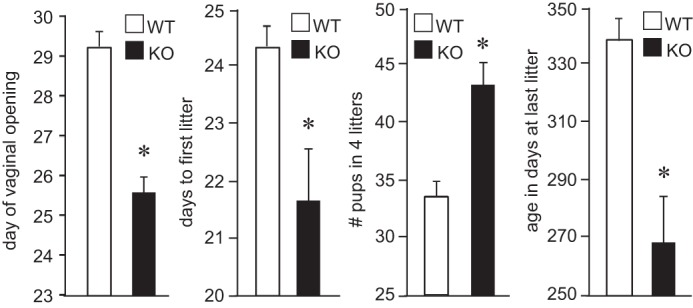
JDP2 null females exhibit reproductive anomalies. White bars, wild type (WT); black bars, JDP2 nulls (KO); 5 mice/group. *, significant difference (p < 0.05) in analyzed reproductive parameters in the female JDP2 null animals from the wild type animals. Error bars, S.E.
Given that in our model cell line, JDP2 negatively regulated Fshb levels, we first assessed gonadotropin hormone levels in the serum and gene expression in the pituitary (Fig. 8). There is no difference in the LH serum levels (Fig. 8A), but FSH was higher in the diestrus of 8-week old null females compared with the wild-type littermates in diestrus (Fig. 8B). Difference in the expression of the Lhb subunit in the pituitary that confers biological specificity to LH did not reach significance (Fig. 8C), whereas Fshb subunit expression was significantly higher in the pituitaries of the null mice (Fig. 8D). On the other hand, the expression of the common Cga, which encodes the α-GSU subunit that heterodimerizes with both LHβ and FSHβ was the same between genotypes (Fig. 8E). Because expression of the Fshb subunit was significantly increased, we assessed whether it stems from an increase in the number of gonadotropes. Pituitary morphology and size and gonadotrope number are the same between wild-type and null females (Fig. 9, A–C). Because hypothalamic neuropeptides GNRH and kisspeptin regulate expression and secretion of gonadotropins in the pituitary, we measured the expression of these neurohormones. Expression of Gnrh and Kiss1 was the same as well between genotypes (Fig. 9, D and E). Therefore, Fshb expression and FSH hormone levels are higher in the JDP2 null females, which is consistent with our findings in the cell model, that JDP2 serves as a negative regulator of FSH synthesis.
FIGURE 8.
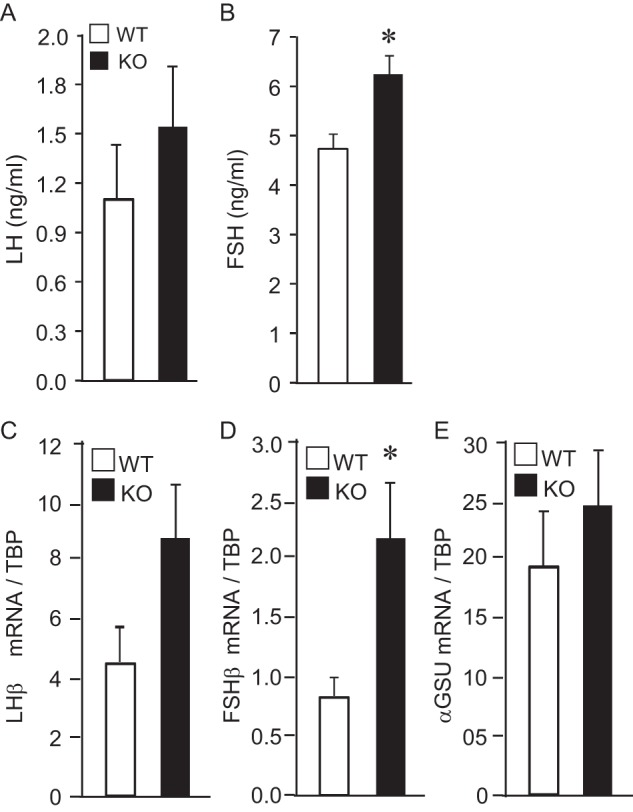
JDP2 null females have higher levels of FSH and higher expression of Fshb. A and B, serum from six 8-week-old females per genotype was analyzed by a radioimmunoassay for LH and FSH. C–E, whole pituitary mRNA was extracted from 6 animals/group, and mRNA levels for Lhb (C), Fshb (D), and common Cga encoding α-GSU (E) were determined by qPCR. White bars, wild type; black bars, JDP2 nulls. *, significant difference in the JDP2 null animals from the wild type animals; p < 0.05. Error bars, S.E.
FIGURE 9.
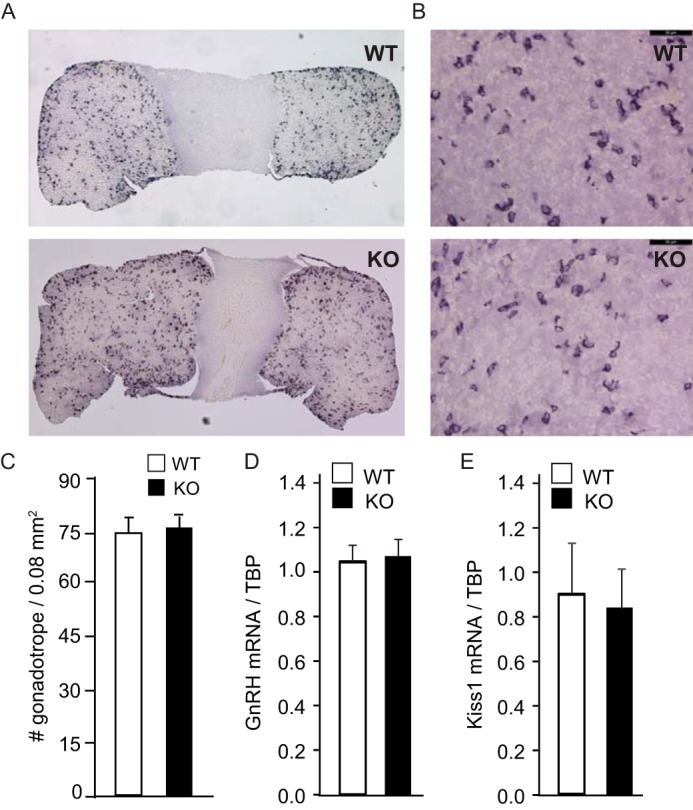
Pituitaries and gonadotrope number are unchanged in JDP2 null females. A and B, pituitaries were stained with antibodies to LH, and representative images are shown at low magnification (A) and higher magnification (B) (×40 objective; bar, 50 μm). C, gonadotropes from three different 8-week-old females per group were counted, and numbers are presented per 0.08 mm2. White bar, wild type; black bar, JDP2 nulls. D and E, hypothalamus mRNA was extracted from 6 females/genotype, and Gnrh and kisspeptin (Kiss1) mRNA were analyzed by qPCR. White bars, wild type (WT); black bars, JDP2 nulls (KO).
Next, we evaluated ovarian function in JDP2 null mice (Fig. 10). In the ovary, anti-Müllerian hormone (AMH) is produced by the granulosa cells of growing follicles to modulate the recruitment of primordial follicles and the FSH-dependent follicular growth (43, 44). Given that AMH levels may correlate with the increased number of pups and early reproductive senescence, we first analyzed the levels of AMH. AMH levels exhibited a trend toward lower levels in null females (p = 0.11), although the difference did not reach significance (Fig. 10A). On the other hand, estradiol levels were significantly higher in JDP2 null proestrus females (Fig. 10B), whereas there was no change in inhibin A concentration (Fig. 10C). On histological sections, we counted a higher number of large follicles with multilayer granulosa cells in the ovaries of JDP2 null mice (Fig. 10, D and E). Therefore, JDP2 null mice demonstrate elevated serum estradiol levels, which may stem from higher FSH levels, and increased follicular recruitment to the growing pool, resulting in the higher number of large follicles in the ovaries and increased number of pups per litter.
FIGURE 10.
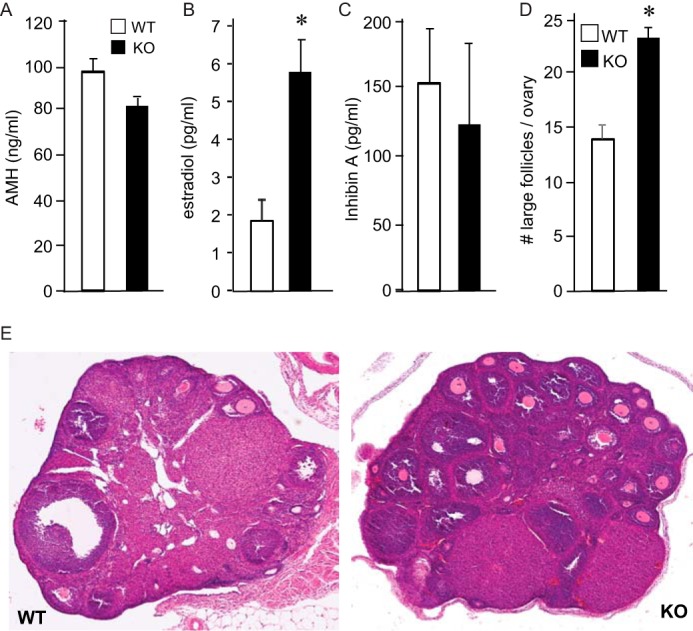
JDP2 null females exhibit elevated estrogen levels and higher number of large follicles in the ovaries. White bars, wild type; black bars, JDP2 nulls. A–C, serum AMH, estradiol, and inhibin A were analyzed in estrus females by ELISA. D, large follicles with multilayer granulosa cells were counted throughout the ovary. *, significant difference in the JDP2 null animals from the wild type animals. E, bottom panels, representative image of H&E stain of the wild-type (WT) and null (KO) ovaries. Error bars, S.E.
Discussion
Notwithstanding extensive studies on the regulation of gonadotropin subunit gene expression and synthesis, little is known about negative regulators that keep the hormone levels within the tight physiological range. Here, we identified a negative regulator of FSHβ synthesis, JDP2, and analyzed its role, first in the model cell line and then in vivo using JDP2 null mice. JDP2 binds the Fshb promoter as a c-JUN binding partner and represses GNRH induction of Fshb transcription. Just as FSHβ null males are not infertile while FSHβ null females are unable to reproduce, JDP2 null males do not exhibit any adverse effects on reproduction, whereas females have several significant changes. JDP2 null females have higher FSH levels, exhibit earlier vaginal opening, deliver their first litter sooner, and have larger litters, but they stop reproducing at a younger age than wild-type mice. The early cessation of reproductive function parallels our hypothesis that JDP2 null mice may mimic premature ovarian failure in women, a condition associated with increased FSH levels (9, 10). Genome-wide association studies in a human population identified polymorphisms in the 5′ region of the FSHB gene, which are associated with elevated FSH levels, premature menarche, early natural menopause, and dizygotic twinning (12, 13), implicating regulation of FSHB transcription in the etiology of these reproductive phenotypes.
JDP2 belongs to the AP-1 superfamily of transcription factors that bind the AP-1/TRE element in the promoter of target genes. Classical AP-1 transcription factor is a heterodimer of Fos and Jun immediate early genes, which are activated rapidly and transiently by various stimuli and growth factors in numerous cell types (23). In turn, AP-1 regulates a wide range of cellular processes, including proliferation, death, survival, and differentiation. However, each member of the family, although induced in several tissues, has a cell- and stimulus-specific function and at times antagonizes other family members (45, 46). We have shown previously that c-FOS has a cell-specific function at each level of the hypothalamic-pituitary-gonadal axis and precise roles in the regulation of reproduction (47). We and others have also shown that c-FOS is rapidly induced by GNRH in the pituitary gonadotrope (48–50), dimerizes with c-JUN to induce Fshb expression, and is degraded shortly thereafter. JDP2 was initially described as a repressor of c-JUN activity, via interaction with c-JUN and binding to the AP-1/TRE site, probably by displacing c-FOS. Later studies expanded these findings to include repression of ATF2 function on the highly similar CRE element.
JDP2 is expressed at the high basal level, and its levels change little with GNRH treatment. GNRH stimulus, however, rapidly increases the transcription of immediate early genes, c-Fos and c-Jun, which leads to induction of Fshb transcription (50). We previously reported that c-FOS is not present without the stimulus and, after induction, diminishes rapidly due to its short half-life of 10 min for mRNA and about 1 h for protein (26). c-JUN, on the other hand, is present in cells at the basal state, is further induced by GNRH, and has a longer half-life. Because both JDP2 and c-JUN are present before the stimuli, they form a complex that binds the FSHβ promoter, as we demonstrate here. The DNA-binding domain of JDP2 is necessary for its function, and JDP2 binds the TRE/AP-1 site in the FSHβ promoter. Following GNRH stimulus, c-FOS is induced at a high level and outcompetes JDP2 as a c-JUN binding partner, which leads to a transcriptionally active AP-1 factor. Due to a short half-life, c-FOS is degraded, allowing JDP2 to repress the transcription again until the next stimulus. In part, JDP2 represses transcription via recruitment of HDAC3 that deacetylates the histones (31, 32, 40). Although it is not clear whether threonine 148 is necessary for chromatin deacetylation, it is an activation site that represents a major difference between JDP2 and highly similar ATF3 (51). We establish that threonine 148 is necessary for repressive function of JDP2 at the c-JUN and FSHβ promoters. We also demonstrate that 1-h GNRH treatment, coinciding with an increase in AP-1 active complex of c-JUN and c-FOS formation that replaced the c-JUN and JDP2 complex, leads to an increase in histone acetylation followed by a subsequent increase in Fshb transcription.
Although the mouse model we used to analyze the in vivo role of JDP2 is a complete null, and we cannot exclude potential effects outside the pituitary, expression analysis of relevant hypothalamic neuropeptides, GNRH and kisspeptin, did not reveal any differences. Previous studies that analyzed JDP2 expression determined that although expressed in variety of tissues, JDP2 is highly expressed in female reproductive tissues, where it interacts specifically with the progesterone receptor (52). Thus, previous studies postulated a role of JDP2 in female reproduction. We determined that, indeed, JDP2 controls female reproductive function via regulation of FSH synthesis. We did not observe changes in progesterone. Female null mice had an increase in Fshb expression in the pituitary and FSH levels in the circulation, which may have caused an increase in estrogen and a higher number of large follicles.
FSH plays a role in follicular growth during the antral stage and stimulates estrogen synthesis in the granulosa cells (53, 54). Thus, in our mouse model, it is difficult to distinguish effects of the elevated FSH and possible effects of the lack of JDP2 in the ovary. Future studies, afforded by creation of the floxed JDP2 mouse model, may elucidate tissue-specific effects of JDP2. Herein, we determined that JDP2 null mice exhibit higher estrogen levels, which may stem from the increase in FSH concentration. Surprisingly, increased estrogen does not exacerbate negative feedback on kisspeptin expression; however, the mix of the persistent positive and negative feedback may cancel each other, resulting in the same level of hypothalamic neuropeptides.
AMH concentration in JDP2 nulls exhibited a trend toward a lower level. The effect on AMH levels may correlate with the increased number of pups and early reproductive senescence. In the ovary, AMH is produced by the granulosa cells of growing ovarian follicles, from the primary follicle stage after follicular growth activation to the small antral stage (43, 55). Interestingly, AMH has been shown to inhibit the recruitment of primordial follicles to the growing pool, thereby avoiding the premature exhaustion of the ovarian reserve. The regulation of AMH production in follicles remains poorly understood. Although it is not believed that AMH exhibits feedback on the pituitary FSH expression as inhibin does, FSH may regulate AMH levels. The role of FSH in AMH expression in the literature is controversial, with one study reporting induction of AMH by FSH (56) and another reporting repression (43). Clinically, AMH serves as a biomarker that accurately reflects ovarian reserve. Although relatively stable throughout the menstrual cycle (57), changes in serum AMH levels are associated with ovarian aging (55, 58). More importantly, serum AMH levels in patients with premature ovarian failure are extremely low (59, 60). Therefore, somewhat lower AMH levels may contribute to a higher number of growing follicles in the ovary of JDP2 null females and premature cessation of reproduction.
Thus, we postulate that JDP2 serves as negative regulator of FSH to keep this hormone within the tight physiological range (Fig. 11). Without GNRH stimulus, Fshb transcription (and c-Jun transcription) is quiescent, due to JDP2 association with the Fshb promoter and its recruitment of HDAC3 that keeps chromatin, via deacetylation of histone 3, in a closed conformation. Following GNRH stimulus, others have shown that HDAC3 dissociates from the Fshb promoter, and herein we provide a mechanism of increased acetylation. Specifically, GNRH signaling leads to phosphorylation of ATF2, which displaces JDP2 and induces c-Jun transcription. Increased expression of c-JUN together with elevated c-FOS, which removes JDP2 as a c-JUN binding partner, induce Fshb transcription. Following a wave of transcription, ATF2 is dephosphorylated, c-FOS is degraded, and JDP2 returns to the c-Jun and Fshb promoters to shut down their transcription until the next pulse of GNRH. In vivo studies strengthen our hypothesis, because JDP2 null females exhibit higher FSH levels and elevated serum estradiol that may result in higher follicular recruitment to the growing pool, a larger number of late secondary and antral follicles in the ovaries leading to larger litters in young animals, and early cessation of reproductive function in older animals. These findings may have provided a candidate gene for premature ovarian failure, a condition associated with increased FSH levels and early loss of ovarian function in women.
Experimental Procedures
JDP2 Null Mice
The JDP2 null mice were obtained from Dr. Shizuo Akira (WPI Immunology Frontier Research Center, Osaka University, Japan (42)). Animals were maintained under a 12-h light, 12-h dark cycle and received food and water ad libitum. All experiments were performed with approval from the University of California (Riverside, CA) Animal Care and Use Committee and in accordance with the National Institutes of Health Animal Care and Use Guidelines. For genotyping, genomic DNA was extracted from toe biopsies and analyzed with PCR. Because male animals did not exhibit differences in hormone levels, sperm count, or reproductive capacity, only female animals were analyzed in detail. At least 5 animals/genotype (WT and null) were analyzed unless otherwise indicated, and differences were compared by Student's t test and Tukey's post hoc test; p < 0.05 was considered a statistically significant difference between wild-type and null mice.
Cell Culture and Transient Transfections
The expression vectors for JDP2 and ATF3 were kindly provided by Dr. Ami Aronheim (Technion-Israel Institute of Technology, Haifa, Israel). The mouse FSHβ luciferase, c-JUN luciferase, 4× CRE, and 4× TRE multimer reporter vectors were published previously (22, 25, 61).
LβT2 cells, kindly provided by Pamela Mellon (University of California San Diego, La Jolla, CA) were cultured at 37 °C in DMEM (Cellgro, Mediatech, Inc., Herndon, VA) containing 10% fetal bovine serum (Omega Scientific Inc., Tarzana, CA) and penicillin. Cells were split into 12-well plates 1 day before transfection and transfected using Fugene 6 reagent (Roche Applied Science) in accordance with the manufacturer's protocol. Wells were transfected with 500 ng of reporter plasmid, 100 ng of the β-galactosidase reporter plasmid driven by the Herpesvirus thymidine kinase promoter to serve as an internal control for transfection efficiency, and 200 ng of expression vectors or control, as indicated in the figure legends. Cells were incubated in serum-free DMEM containing 0.1% BSA and antibiotics overnight before hormone treatment with 10 nm GNRH (Sigma-Aldrich). Subsequent to treatment, cells were lysed with 60 μl of 0.1 m potassium phosphate buffer, pH 7.8, with 0.2% Triton X-100. A 96-well luminometer plate was loaded with 20 μl of each of the lysates, and luciferase activity was measured after injection of a buffer containing 100 mm Tris-HCl, pH 7.8, 15 mm MgSO4, 10 mm ATP, and 65 μm luciferin per well, using a Veritas Microplate Luminometer (Turner Biosystems, Sunnyvale, CA). The Galacto-Light Assay (Tropix, Bedford, MA) was performed according to the manufacturer's protocol to measure galactosidase activity. All experiments were performed a minimum of three times and in triplicates within each experiment. Luciferase values were normalized to β-galactosidase for each sample, and results are presented as an average of three experiments ± S.E. Statistical significance was determined with analysis of variance followed by Tukey's post hoc test, and significance was set at p < 0.05.
EMSA
Nuclear extracts from GNRH or vehicle-treated LβT2 cells or COS-1 cells following overexpression for 48 h were obtained by swelling the cells with hypotonic buffer (20 mm Tris, pH 7.4, 10 mm NaCl, 1 mm MgCl2, 1 mm PMSF, protease inhibitor mixture (Sigma-Aldrich), 10 mm NaF, 0.5 mm EDTA, 0.1 mm EGTA). Cells were broken by passing through a 25⅝-gauge needle three times. Samples were centrifuged at 4000 rpm for 4 min, and the nuclear pellets were resuspended in hypertonic buffer (20 mm Hepes, pH 7.8, 20% glycerol, 420 mm KCl, 1.5 mm MgCl2, 1 mm PMSF, protease inhibitor mixture (Sigma-Aldrich), 10 mm NaF, 0.5 mm EDTA, and 0.1 mm EGTA). Protein determination was performed using the Bradford reagent (Bio-Rad). The oligonucleotides that were used as 30-bp probes, encompassing the sites of interest from FSHβ and c-JUN promoters, were reported previously (22, 25). Oligonucleotides were annealed and labeled with [γ-32P]ATP using T4 polynucleotide kinase (New England Biolabs, Inc., Beverly, MA). Binding reactions contained 2 μg of nuclear proteins in a total volume of 20 μl containing the following: 10 mm Hepes, pH 7.8, 50 mm KCl, 0.5 mm MgCl2, 10% glycerol, 0.1% Nonidet P-40, 0.25 μg of dIdC, 5 mm DTT, and 5 fmol of labeled probe. Reactions were loaded onto a 5% nondenaturing poly(dI-dC) and run in 0.25× Tris borate-EDTA buffer. Gels were run at 250 V/cm2 constant voltage and dried. Autoradiography was performed to identify complexes. The experiment was repeated at least three times, and a representative image is presented.
Western Blotting
Following overnight starvation and hormone treatment, LβT2 cells were rinsed with PBS and lysed with lysis buffer (20 mm Tris-HCl, pH 7.4, 140 mm NaCl, 0.5% Nonidet P-40, 0.5 mm EDTA, with protease inhibitors (aprotinin, pepstatin, and leupeptin at 10 μg/ml each) and 1 mm PMSF). Protein concentrations were determined with Bradford reagent (Bio-Rad), and an equal amount of protein per sample was loaded onto SDS-polyacrylamide gels or first immunoprecipitated with antibodies to protein tags, and the precipitate was loaded. After proteins had been resolved by electrophoresis and transferred to a membrane, they were probed with specific antibodies for c-JUN (sc-1694), ATF3 (sc-188,), or JDP2 (sc-23458) (Santa Cruz Biotechnology, Inc., Dallas, TX). The complexes were detected with secondary antibodies linked to horseradish peroxidase and enhanced chemiluminescence reagent (Amersham Biosciences).
GST Interaction Assay
The GST-NFY in the pGEX vector was reported previously (22). 35S-Labeled proteins were produced using the TnT® T7 coupled reticulocyte lysate system (Promega Corp., Madison, WI). Bacteria transformed with the pGEX vectors were grown to an OD of 0.6, upon which protein expression was induced by the addition of 0.25 mm isopropyl-β-d-thiogalactosidase. Bacterial pellets were sonicated in PBS with 5 mm EDTA and 0.1% Triton X-100 and centrifuged, and the supernatant was bound to glutathione-Sepharose beads (Amersham Biosciences). Beads were washed four times with sonication buffer followed by equilibration in the binding buffer (see below) and split equally between different samples and the control. 35S-Labeled proteins were added to the beads and bound for 1 h at 4 °C in 20 mm Hepes (pH 7.8), with 50 mm NaCl, 10 mg/ml BSA, 0.1% Nonidet P-40, and 5 mm DTT. After extensive washing, samples were eluted and subjected to SDS-PAGE. Afterward, the gels were dried and autoradiographed. The experiment was repeated three times, and representative images are presented.
Immunohistochemistry
Tissues were fixed in 4% paraformaldehyde overnight at 4 °C and dehydrated in ethanol/water washes before embedding in paraffin. Embedded tissues were cut into 14-μm sections with a microtome and floated onto SuperFrost Plus slides (Fisher) and dried overnight at room temperature. Slides were incubated at 60 °C for 30 min, deparaffinized in xylene washes, and rehydrated in ethanol/water washes. The slides were then used for either for hematoxylin-eosin staining (ovary) or immunohistochemistry (pituitary). For immunohistochemistry, antigen unmasking was performed by heating for 10 min in a Tris/EDTA/Tween 20 mixture, and endogenous peroxidase was quenched by incubating for 10 min in 0.3% hydrogen peroxide. After washing in PBS, slides were blocked (PBS, 5% goat serum, 0.3% Triton X-100) for 45 min, incubated with primary antibodies against LH (1:1000; obtained from the National Hormone and Peptide Program, NIDDK, National Institutes of Health) overnight at 4 °C. After washing, slides were incubated with biotinylated goat anti-rabbit IgG (1:300; Vector Laboratories) for 30 min. The Vectastain ABC elite kit (Vector Laboratories) was used per the manufacturer's instructions and incubated for 30 min. After washing, the VIP peroxidase kit was used for colorimetric staining for 3 min. Slides were dehydrated in an ethyl alcohol series and xylene and covered using Vectamount (Vector Laboratories). Images were obtained using a Leica microscope system.
qPCR Analysis
Tissues were dissected, and total RNA was extracted and reverse transcribed using Superscript III (Invitrogen). qPCR was performed using an iQ SYBR Green supermix and an IQ5 real-time PCR machine (Bio-Rad), with primers reported previously (47), under the following conditions: 95 ºC for 15 min, followed by 40 cycles at 95 ºC for 20 s, 56 ºC for 30 s, and 72 ºC for 30 s. A standard curve with dilutions of 10 pg/well, 1 pg/well, 100 fg/well, and 10 fg/well of a plasmid containing LHβ or FSHβ cDNA was generated in each run with the samples. The amount of the gene of interest was calculated by comparing the threshold cycle obtained for each sample with the standard curve generated in the same run. Replicates were averaged and divided by the mean value of the housekeeping gene in the same sample. After each run, a melting curve analysis was performed to confirm that a single amplicon was generated in each reaction. Statistical differences (p < 0.05) in expression between genotypes were determined using Student's t test and Tukey's post hoc test using JMP software (SAS Institute, Cary, NC).
Hormone Analysis
For serum collection, 8-week-old mice were sacrificed by isoflurane inhalation, and blood was obtained from the inferior vena cava. The blood was left to coagulate for 15 min at room temperature and then centrifuged at 2000 relative centrifugal force for 15 min for serum separation. Hormone assays were performed by the University of Virginia Ligand Core. LH was analyzed using a sensitive two-site sandwich immunoassay (62), and mouse LH reference preparation (AFP5306A; provided by Dr. A. F. Parlow and the National Hormone and Peptide program) was used as a standard. FSH was assayed by a radioimmunoassay using reagents provided by Dr. A. F. Parlow and the National Hormone and Peptide Program, as described previously (63). Mouse FSH reference preparation AFP5308D was used for assay standards. Estradiol, AMH, and inhibin levels were analyzed using validated commercially available assays, information for which can be found on the core's website and reported previously (64). Limits of detection were 0.24 ng/ml for LH, 2.4 ng/ml for FSH, and 3 pg/ml for estradiol. Intra- and interassay coefficients of variation were 6.4/8.0%, 6.9/7.5%, 6.0/11.4%, and 4.4/6.4% for the LH, FSH, AMH, and estrogen (E2), respectively. For the assays used for this study, interassay coefficients of variation data are the result of 30 assays. Six animals per group were used for each hormone analysis. Statistical differences (p < 0.05) in hormone levels between wild-type and null group were determined by Student's t test and the Tukey-Kramer post hoc honest significant difference for multiple comparisons using JMP software (SAS Institute).
Author Contributions
C. R. J. performed analysis of JDP2 null animals, including reproductive function, tissue dissection, and histological staining. N. M. L. performed experiments for the revision of the manuscript. L. L. R. conducted experiments in the gonadotrope cell line, including EMSA and transient transfections. A. D. W. performed analysis of gene expression in tissues and cell lines. D. C. conceived and coordinated the study and wrote the paper. All authors reviewed the results, revised the manuscript, and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Aronheim for the JDP2 expression vector. We are especially grateful to Drs. Akira for JDP2 null mice and Mellon for LβT2 cells. The University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core is a fee-for-service core facility and is supported in part by Eunice Kennedy Shriver NICHD/National Institutes of Health (SCCPIR) Grant U54-HD28934.
This work was supported by National Institutes of Health Grant R01 HD057549 (to D. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- LH
- luteinizing hormone
- GNRH
- gonadotropin-releasing hormone
- AMH
- anti-Müllerian hormone
- CRE
- cyclic AMP-response element
- TRE
- TPA response element
- HDAC
- histone deacetylase
- IP3
- inositol 1,4,5-trisphosphate
- qPCR
- quantitative PCR.
References
- 1. Ortolano G. A., Haisenleder D. J., Dalkin A. C., Iliff-Sizemore S. A., Landefeld T. D., Maurer R. A., and Marshall J. C. (1988) Follicle-stimulating hormone β subunit messenger ribonucleic acid concentrations during the rat estrous cycle. Endocrinology 123, 2946–2948 [DOI] [PubMed] [Google Scholar]
- 2. Zmeili S. M., Papavasiliou S. S., Thorner M. O., Evans W. S., Marshall J. C., and Landefeld T. D. (1986) α and luteinizing hormone β subunit messenger ribonucleic acids during the rat estrous cycle. Endocrinology 119, 1867–1869 [DOI] [PubMed] [Google Scholar]
- 3. Huhtaniemi I. (2006) Mutations along the pituitary-gonadal axis affecting sexual maturation: novel information from transgenic and knockout mice. Mol. Cell Endocrinol. 254, 84–90 [DOI] [PubMed] [Google Scholar]
- 4. Huhtaniemi I., Ahtiainen P., Pakarainen T., Rulli S. B., Zhang F. P., and Poutanen M. (2006) Genetically modified mouse models in studies of luteinising hormone action. Mol. Cell Endocrinol. 252, 126–135 [DOI] [PubMed] [Google Scholar]
- 5. Lamminen T., Jokinen P., Jiang M., Pakarinen P., Simonsen H., and Huhtaniemi I. (2005) Human FSH beta subunit gene is highly conserved. Mol. Hum. Reprod. 11, 601–605 [DOI] [PubMed] [Google Scholar]
- 6. Morales A. J., Laughlin G. A., Bützow T., Maheshwari H., Baumann G., and Yen S. S. (1996) Insulin, somatotropic, and luteinizing hormone axes in lean and obese women with polycystic ovary syndrome: common and distinct features. J. Clin. Endocrinol. Metab. 81, 2854–2864 [DOI] [PubMed] [Google Scholar]
- 7. Hall J. E., Taylor A. E., Hayes F. J., and Crowley W. F. Jr. (1998) Insights into hypothalamic-pituitary dysfunction in polycystic ovary syndrome. J. Endocrinol. Invest. 21, 602–6011 [DOI] [PubMed] [Google Scholar]
- 8. Chang R. J. (2007) The reproductive phenotype in polycystic ovary syndrome. Nat. Clin. Pract. Endocrinol. Metab. 3, 688–695 [DOI] [PubMed] [Google Scholar]
- 9. Goswami D., and Conway G. S. (2007) Premature ovarian failure. Horm. Res. 68, 196–202 [DOI] [PubMed] [Google Scholar]
- 10. Chand A. L., Harrison C. A., and Shelling A. N. (2010) Inhibin and premature ovarian failure. Hum. Reprod. Update 16, 39–50 [DOI] [PubMed] [Google Scholar]
- 11. Wang H., Larson M., Jablonka-Shariff A., Pearl C. A., Miller W. L., Conn P. M., Boime I., and Kumar T. R. (2014) Redirecting intracellular trafficking and the secretion pattern of FSH dramatically enhances ovarian function in mice. Proc. Natl. Acad. Sci. U.S.A. 111, 5735–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. He C., Kraft P., Chasman D. I., Buring J. E., Chen C., Hankinson S. E., Paré G., Chanock S., Ridker P. M., and Hunter D. J. (2010) A large-scale candidate gene association study of age at menarche and age at natural menopause. Hum. Genet. 128, 515–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mbarek H., Steinberg S., Nyholt D. R., Gordon S. D., Miller M. B., McRae A. F., Hottenga J. J., Day F. R., Willemsen G., de Geus E. J., Davies G. E., Martin H. C., Penninx B. W., Jansen R., McAloney K., et al. (2016) Identification of common genetic variants influencing spontaneous dizygotic twinning and female fertility. Am. J. Hum. Genet. 98, 898–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burrin J. M., and Jameson J. L. (1989) Regulation of transfected glycoprotein hormone α-gene expression in primary pituitary cell cultures. Mol. Endocrinol. 3, 1643–1651 [DOI] [PubMed] [Google Scholar]
- 15. Shupnik M. A. (1990) Effects of gonadotropin-releasing hormone on rat gonadotropin gene transcription in vitro: requirement for pulsatile administration for luteinizing hormone-β gene stimulation. Mol. Endocrinol. 4, 1444–1450 [DOI] [PubMed] [Google Scholar]
- 16. Belchetz P. E., Plant T. M., Nakai Y., Keogh E. J., and Knobil E. (1978) Hypophysial responses to continuous and intermittent delivery of hypopthalamic gonadotropin-releasing hormone. Science 202, 631–633 [DOI] [PubMed] [Google Scholar]
- 17. McArdle C. A., Franklin J., Green L., and Hislop J. N. (2002) Signalling, cycling and desensitisation of gonadotrophin-releasing hormone receptors. J. Endocrinol. 173, 1–11 [DOI] [PubMed] [Google Scholar]
- 18. Nicol L., McNeilly J. R., Stridsberg M., and McNeilly A. S. (2004) Differential secretion of gonadotrophins: investigation of the role of secretogranin II and chromogranin A in the release of LH and FSH in LβT2 cells. J. Mol. Endocrinol. 32, 467–480 [DOI] [PubMed] [Google Scholar]
- 19. Stojilkovic S. S., Reinhart J., and Catt K. J. (1994) Gonadotropin-releasing hormone receptors: structure and signal transduction pathways. Endocrine Rev. 15, 462–499 [DOI] [PubMed] [Google Scholar]
- 20. McArdle C. A., Davidson J. S., and Willars G. B. (1999) The tail of the gonadotrophin-releasing hormone receptor: desensitization at, and distal to, G protein-coupled receptors. Mol. Cell Endocrinol. 151, 129–136 [DOI] [PubMed] [Google Scholar]
- 21. McArdle C. A., Willars G. B., Fowkes R. C., Nahorski S. R., Davidson J. S., and Forrest-Owen W. (1996) Desensitization of gonadotropin-releasing hormone action in αT3–1 cells due to uncoupling of inositol 1,4,5-trisphosphate generation and Ca2+ mobilization. J. Biol. Chem. 271, 23711–23717 [DOI] [PubMed] [Google Scholar]
- 22. Coss D., Jacobs S. B., Bender C. E., and Mellon P. L. (2004) A novel AP-1 site is critical for maximal induction of the follicle-stimulating hormone β gene by gonadotropin-releasing hormone. J. Biol. Chem. 279, 152–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karin M., Liu Zg., and Zandi E. (1997) AP-1 function and regulation. Curr. Opin. Cell Biol. 9, 240–246 [DOI] [PubMed] [Google Scholar]
- 24. Ely H. A., Mellon P. L., and Coss D. (2011) GnRH Induces the c-Fos gene via phosphorylation of SRF by the calcium/calmodulin kinase II pathway. Mol. Endocrinol. 25, 669–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lindaman L. L., Yeh D. M., Xie C., Breen K. M., and Coss D. (2013) Phosphorylation of ATF2 and interaction with NFY induces c-Jun in the gonadotrope. Mol. Cell Endocrinol. 365, 316–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reddy G. R., Xie C., Lindaman L. L., and Coss D. (2013) GnRH increases c-Fos half-life contributing to higher FSHβ induction. Mol. Endocrinol. 27, 253–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen B. P., Wolfgang C. D., and Hai T. (1996) Analysis of ATF3, a transcription factor induced by physiological stresses and modulated by gadd153/Chop10. Mol. Cell Biol. 16, 1157–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ciccone N. A., Xu S., Lacza C. T., Carroll R. S., and Kaiser U. B. (2010) Frequency-dependent regulation of follicle-stimulating hormone β by pulsatile gonadotropin-releasing hormone is mediated by functional antagonism of bZIP transcription factors. Mol. Cell. Biol. 30, 1028–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aronheim A., Zandi E., Hennemann H., Elledge S. J., and Karin M. (1997) Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol. Cell. Biol. 17, 3094–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jin C., Ugai H., Song J., Murata T., Nili F., Sun K., Horikoshi M., and Yokoyama K. K. (2001) Identification of mouse Jun dimerization protein 2 as a novel repressor of ATF-2. FEBS Lett. 489, 34–41 [DOI] [PubMed] [Google Scholar]
- 31. Jin C., Li H., Murata T., Sun K., Horikoshi M., Chiu R., and Yokoyama K. K. (2002) JDP2, a repressor of AP-1, recruits a histone deacetylase 3 complex to inhibit the retinoic acid-induced differentiation of F9 cells. Mol. Cell. Biol. 22, 4815–4826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jin C., Kato K., Chimura T., Yamasaki T., Nakade K., Murata T., Li H., Pan J., Zhao M., Sun K., Chiu R., Ito T., Nagata K., Horikoshi M., and Yokoyama K. K. (2006) Regulation of histone acetylation and nucleosome assembly by transcription factor JDP2. Nat. Struct. Mol. Biol. 13, 331–338 [DOI] [PubMed] [Google Scholar]
- 33. Lim S., Luo M., Koh M., Yang M., bin Abdul Kadir M. N., Tan J. H., Ye Z., Wang W., and Melamed P. (2007) Distinct mechanisms involving diverse histone deacetylases repress expression of the two gonadotropin β-subunit genes in immature gonadotropes, and their actions are overcome by gonadotropin-releasing hormone. Mol. Cell Biol. 27, 4105–4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lawson M. A., Tsutsumi R., Zhang H., Talukdar I., Butler B. K., Santos S. J., Mellon P. L., and Webster N. J. (2007) Pulse sensitivity of the luteinizing hormone β promoter is determined by a negative feedback loop Involving early growth response-1 and Ngfi-A binding protein 1 and 2. Mol. Endocrinol. 21, 1175–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mistry D. S., Tsutsumi R., Fernandez M., Sharma S., Cardenas S. A., Lawson M. A., and Webster N. J. (2011) Gonadotropin-releasing hormone pulse sensitivity of follicle-stimulating hormone-β gene is mediated by differential expression of positive regulatory activator protein 1 factors and corepressors SKIL and TGIF1. Mol. Endocrinol. 25, 1387–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xie J., Bliss S. P., Nett T. M., Ebersole B. J., Sealfon S. C., and Roberson M. S. (2005) Transcript profiling of immediate early genes reveals a unique role for activating transcription factor 3 in mediating activation of the glycoprotein hormone α-subunit promoter by gonadotropin-releasing hormone. Mol. Endocrinol. 19, 2624–2638 [DOI] [PubMed] [Google Scholar]
- 37. Shaulian E., and Karin M. (2001) AP-1 in cell proliferation and survival. Oncogene 20, 2390–2400 [DOI] [PubMed] [Google Scholar]
- 38. Katz S., Heinrich R., and Aronheim A. (2001) The AP-1 repressor, JDP2, is a bona fide substrate for the c-Jun N-terminal kinase. FEBS Lett. 506, 196–200 [DOI] [PubMed] [Google Scholar]
- 39. Pan J., Jin C., Murata T., and Yokoyama K. K. (2004) Histone modification activities of JDP2 associated with retinoic acid-induced differentiation of F9 cells. Nucleic Acids Symp. Ser. 10.1093/nass/48.1.189 [DOI] [PubMed] [Google Scholar]
- 40. Nakade K., Pan J., Yoshiki A., Ugai H., Kimura M., Liu B., Li H., Obata Y., Iwama M., Itohara S., Murata T., and Yokoyama K. K. (2007) JDP2 suppresses adipocyte differentiation by regulating histone acetylation. Cell Death Differ. 14, 1398–1405 [DOI] [PubMed] [Google Scholar]
- 41. Darlyuk-Saadon I., Weidenfeld-Baranboim K., Yokoyama K. K., Hai T., and Aronheim A. (2012) The bZIP repressor proteins, c-Jun dimerization protein 2 and activating transcription factor 3, recruit multiple HDAC members to the ATF3 promoter. Biochim. Biophys. Acta 1819, 1142–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Maruyama K., Fukasaka M., Vandenbon A., Saitoh T., Kawasaki T., Kondo T., Yokoyama K. K., Kidoya H., Takakura N., Standley D., Takeuchi O., and Akira S. (2012) The transcription factor Jdp2 controls bone homeostasis and antibacterial immunity by regulating osteoclast and neutrophil differentiation. Immunity 37, 1024–1036 [DOI] [PubMed] [Google Scholar]
- 43. Baarends W. M., Uilenbroek J. T., Kramer P., Hoogerbrugge J. W., van Leeuwen E. C., Themmen A. P., and Grootegoed J. A. (1995) Anti-müllerian hormone and anti-müllerian hormone type II receptor messenger ribonucleic acid expression in rat ovaries during postnatal development, the estrous cycle, and gonadotropin-induced follicle growth. Endocrinology 136, 4951–4962 [DOI] [PubMed] [Google Scholar]
- 44. Park M., Suh D.-S., Lee K., and Bae J. (2014) Positive cross talk between FOXL2 and antimüllerian hormone regulates ovarian reserve. Fertil. Steril. 102, 847–855.e1 [DOI] [PubMed] [Google Scholar]
- 45. Shaulian E., and Karin M. (2002) AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–E136 [DOI] [PubMed] [Google Scholar]
- 46. Wagner E. F., and Eferl R. (2005) Fos/AP-1 proteins in bone and the immune system. Immunol. Rev. 208, 126–140 [DOI] [PubMed] [Google Scholar]
- 47. Xie C., Jonak C. R., Kauffman A. S., and Coss D. (2015) Gonadotropin and kisspeptin gene expression, but not GnRH, are impaired in cFOS deficient mice. Mol. Cell Endocrinol. 411, 223–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cesnjaj M., Catt K. J., and Stojilkovic S. S. (1994) Coordinate actions of calcium and protein kinase-C in the expression of primary response genes in pituitary gonadotrophs. Endocrinology 135, 692–701 [DOI] [PubMed] [Google Scholar]
- 49. Padmanabhan V., Dalkin A., Yasin M., Haisenleder D. J., Marshall J. C., and Landefeld T. D. (1995) Are immediate early genes involved in gonadotropin-releasing hormone receptor gene regulation? Characterization of changes in GnRH receptor (GnRH-R), c-fos, and c-jun messenger ribonucleic acids during the ovine estrous cycle. Biol. Reprod. 53, 263–269 [DOI] [PubMed] [Google Scholar]
- 50. Wurmbach E., Yuen T., Ebersole B. J., and Sealfon S. C. (2001) Gonadotropin-releasing hormone receptor-coupled gene network organization. J. Biol. Chem. 276, 47195–47201 [DOI] [PubMed] [Google Scholar]
- 51. Weidenfeld-Baranboim K., Koren L., and Aronheim A. (2011) Phosphorylation of JDP2 on threonine-148 by the c-Jun N-terminal kinase targets it for proteosomal degradation. Biochem. J. 436, 661–669 [DOI] [PubMed] [Google Scholar]
- 52. Wardell S. E., Boonyaratanakornkit V., Adelman J. S., Aronheim A., and Edwards D. P. (2002) Jun dimerization protein 2 functions as a progesterone receptor N-terminal domain coactivator. Mol. Cell Biol. 22, 5451–5466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Howles C. M. (2000) Role of LH and FSH in ovarian function. Mol. Cell Endocrinol. 161, 25–30 [DOI] [PubMed] [Google Scholar]
- 54. Layman L. C. (2000) Mutations in the follicle-stimulating hormone-beta (FSH β) and FSH receptor genes in mice and humans. Semin. Reprod. Med. 18, 5–10 [DOI] [PubMed] [Google Scholar]
- 55. Broekmans F. J., Visser J. A., Laven J. S. E., Broer S. L., Themmen A. P. N., and Fauser B. C. (2008) Anti-Müllerian hormone and ovarian dysfunction. Trends Endocrinol. Metab. 19, 340–347 [DOI] [PubMed] [Google Scholar]
- 56. Taieb J., Grynberg M., Pierre A., Arouche N., Massart P., Belville C., Hesters L., Frydman R., Catteau-Jonard S., Fanchin R., Picard J.-Y., Josso N., Rey R. A., and di Clemente N. (2011) FSH and Its Second Messenger cAMP Stimulate the Transcription of Human Anti-Müllerian Hormone in Cultured Granulosa Cells. Molecular Endocrinology 25, 645–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kissell K. A., Danaher M. R., Schisterman E. F., Wactawski-Wende J., Ahrens K. A., Schliep K., Perkins N. J., Sjaarda L., Weck J., and Mumford S. L. (2014) Biological variability in serum anti-Müllerian hormone throughout the menstrual cycle in ovulatory and sporadic anovulatory cycles in eumenorrheic women. Hum. Reprod. 29, 1764–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. de Vet A., Laven J. S. E., de Jong F. H., Themmen A. P. N., and Fauser B. C. J. M. (2002) Antimüllerian hormone serum levels: a putative marker for ovarian aging. Fertil. Steril. 77, 357–362 [DOI] [PubMed] [Google Scholar]
- 59. Méduri G., Massin N., Guibourdenche J., Bachelot A., Fiori O., Kuttenn F., Misrahi M., and Touraine P. (2007) Serum anti-Müllerian hormone expression in women with premature ovarian failure. Hum. Reprod. 22, 117–123 [DOI] [PubMed] [Google Scholar]
- 60. Visser J. A., Schipper I., Laven J. S. E., and Themmen A. P. N. (2012) Anti-Mullerian hormone: an ovarian reserve marker in primary ovarian insufficiency. Nat. Rev. Endocrinol. 8, 331–341 [DOI] [PubMed] [Google Scholar]
- 61. Coss D., Hand C. M., Yaphockun K. K., Ely H. A., and Mellon P. L. (2007) p38 mitogen-activated kinase is critical for synergistic induction of the FSH β gene by gonadotropin-releasing hormone and activin through augmentation of c-Fos induction and Smad phosphorylation. Mol. Endocrinol. 21, 3071–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Haavisto A. M., Pettersson K., Bergendahl M., Perheentupa A., Roser J. F., and Huhtaniemi I. (1993) A supersensitive immunofluorometric assay for rat luteinizing hormone. Endocrinology 132, 1687–1691 [DOI] [PubMed] [Google Scholar]
- 63. Gay V. L., Midgley A. R. Jr, and Niswender G. D. (1970) Patterns of gonadotrophin secretion associated with ovulation. Fed. Proc. 29, 1880–1887 [PubMed] [Google Scholar]
- 64. Haisenleder D. J., Schoenfelder A. H., Marcinko E. S., Geddis L. M., and Marshall J. C. (2011) Estimation of estradiol in mouse serum samples: evaluation of commercial estradiol immunoassays. Endocrinology 152, 4443–4447 [DOI] [PMC free article] [PubMed] [Google Scholar]