

Abstract
In eukaryotic cells, two conserved protein kinases, Gcn2 and TOR complex 1 (TORC1), couple amino acid conditions to protein translation. Gcn2 functions as an amino acid sensor and is activated by uncharged tRNAs that accumulate when intracellular amino acids are limited. Activated Gcn2 phosphorylates and inhibits eukaryotic initiation factor-2α (eIF2α), resulting in repression of general protein synthesis. Like Gcn2, TORC1 is also involved in sensing amino acid conditions. However, the underlying mechanism remains unclear. In the present study, we show that TORC1 is a direct target of Gcn2 kinase in the yeast Saccharomyces cerevisiae. In response to amino acid starvation, Gcn2 binds to TORC1 and phosphorylates Kog1, the unique regulatory subunit of TORC1, resulting in down-regulation of TORC1 kinase activity. In the absence of Gcn2, TORC1 signaling activity increases and becomes unresponsive to amino acid starvation. Our findings demonstrate that TORC1 is an effector of Gcn2 in amino acid signaling, hence defining a novel mechanism by which TORC1 senses amino acid starvation.
Keywords: amino acid, autophagy, histidine, TOR complex (TORC), yeast, Gcn2, Kog1, TORC1, amino acid starvation, uncharged tRNA
Introduction
Because amino acids are the building blocks for protein translation, their intracellular availability is tightly coupled to translation rate. Limitation in amino acids triggers prompt responses that lead to reduction in general protein synthesis and selective expression of proteins required for adaptation of the stress condition (1, 2). In eukaryotic cells, two highly conserved mechanisms are involved in sensing amino acid conditions for regulation of protein translation, including the general control nonderepressible 2 (Gcn2) and mechanistic target of rapamycin complex 1 (mTORC1) signaling pathways (3–5).
Gcn2 is a serine/threonine kinase that normally associates with the ribosome 40S subunit in a latent state. It is activated by uncharged tRNAs that are accumulated under amino acid deprivation conditions (6, 7). Activated Gcn2 phosphorylates the α subunit of eukaryotic initiation factor 2 (eIF2α) at serine 51 and converts it into an inhibitor of eIF2B, the guanine nucleotide exchange factor of eIF2. The inactivated eIF2B, in turn, traps eIF2 in the GDP-bound state, leading to repression of general protein translation (8). In mammalian cells, several Gcn2-related kinases are able to phosphorylate eIF2α in response to various stress conditions (9). In Saccharomyces cerevisiae, Gcn2 is the sole eIF2α kinase. In addition to uncharged tRNAs, activation of Gcn2 requires Gcn1 and Gcn20, which form a complex with Gcn2 on ribosome (10, 11). The absence of Gcn1 prevents eIF2α phosphorylation (12, 13), whereas the lack of Gcn20 partially reduces the phosphorylation (14).
In contrast to the negative role of Gcn2 in translation initiation, mTORC1 plays a positive role in translation. In mammalian cells, mTORC1 stimulates phosphorylation of p70 S6 ribosome protein kinase (S6K) and the eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), which leads to activation of ribosomal S6 and eIF-4E, two factors involved in translation initiation (15).
Yeast, like other eukaryotes, contains two TOR complexes termed TOR complex 1 (TORC1) and 2 (TORC2) (16). TORC1 mediates nutrient signaling and is sensitive to rapamycin (17, 18). It consists of four core components. The catalytic subunit is served alternatively by two highly related TOR proteins, Tor1 and Tor2. The three other subunits are Kog1, Lst8, and Tco89, of which Kog1 defines the specificity of TORC1 (16, 19). Under normal growth conditions, TORC1 associates with two small GTPases, Gtr1 and Gtr2, which are counterparts of the Rag GTPases in mammals (20, 21). These two small GTPases, together with several other proteins, form the so-called EGO complex (22–24). This complex resides on the cytosolic surface of the vacuole and is responsible for tethering TORC1 to the vacuolar membrane (23, 25). The membrane association of TORC1 is disrupted when yeast cells are exposed to stress conditions, such as nitrogen starvation or elevated temperatures (26).
Two major targets of TORC1 have been identified in yeast that relay the activity of TORC1 to many cellular processes, including Sch9 and the Tap42-2A phosphatase (27–29). Sch9 controls ribosome biogenesis and translation initiation and is the major effector of TORC1 in yeast translational control. It is phosphorylated directly by TORC1 at multiple sites at its C terminus, which is required for its activity. This TORC1-dependent phosphorylation has been used as a surrogate marker for TORC1 signaling activity in yeast cells (29). The Tap42-2A phosphatase is the effector of TORC1 in transcription of genes involved in nitrogen metabolism, the TCA cycle, and stress response (30, 31). The Tap42-2A phosphatase is associated with membrane-bound TORC1 under normal growth conditions but is released in response to rapamycin treatment and stress conditions (32). Once released, the phosphatase dephosphorylates many factors downstream of the TOR pathway, including Gcn2 (33–35).
Recent studies in both yeast and mammalian cells suggest that amino acids signal TORC1/mTORC1 activation through leucyl-tRNA synthetase (LeuRS).2 It has been shown that the synthetase, upon loading with leucine, interacts with a GTPase complex formed by two Rag GTPases, RagA/B and RagC/D (Gtr1 and Gtr2 in yeast), and stimulates the formation of a RagA/B GTP-RagC/D GDP conformation. This unique GTPase complex then binds and recruits mTORC1 for activation on the cytoplasmic surface of the lysosome. Under leucine deprivation conditions, the switch from the RagA/B GTP-RagC/D GDP conformation to the RagA/B GDP-RagC/D GTP conformation inactivates the GTPase complex and, consequently, mTORC1 (22, 36, 37). Although this model accounts for the role of leucine in stimulating TORC1/mTORC1 activation, it is unclear whether other amino acids utilize the same mechanism for TORC1/mTORC1 regulation. In the present study, we show that Gcn2, the effector kinase of uncharged tRNAs, is the major sensor of TORC1 for amino acid starvation in yeast. In the absence of Gcn2, TORC1 becomes unresponsive to amino acid deprivation. Our findings also suggest that the Gcn2-mediated TORC1 regulation is conserved in mammalian cells.
Results
Gcn2 Is Required for TORC1 Down-regulation in Response to Amino Acid Starvation
The establishment of Sch9 as a direct target of TORC1 in yeast S. cerevisiae allows the use of the TORC1-dependent phosphorylation at its C terminus as a surrogate marker for TORC1 signaling activity (29, 38). Utilizing this feature, we examined several nutrient-mediated kinases for their potential role in regulating TORC1 activity. The results revealed that the absence of several kinases, including Yck1, Yck2, and Vps34, reduced TORC1-dependent phosphorylation of Sch9 (data not shown). Interestingly, in cells deleted for GCN2, we found that TORC1-mediated phosphorylation of Sch9 was increased (Fig. 1A). This observation indicates that Gcn2 may be a negative regulator of TORC1 in yeast cells. Because Gcn2 is a key factor for mediating amino acid starvation response, we further examined whether Gcn2 mediates the down-regulation of TORC1 under amino acid starvation conditions. As shown in Fig. 1B, we found that when wild type cells were deprived of leucine or histidine, two metabolically distinct amino acids, the TORC1-dependent phosphorylation of Sch9 was rapidly reduced. In contrast, the phosphorylation was not affected by the starvation in gcn2 deletion cells, suggesting that Gcn2 is required for amino acid starvation-induced TORC1 down-regulation. Despite being unresponsive to amino acid starvation, TORC1 in gcn2 cells remained sensitive to rapamycin treatment and nitrogen deprivation (Fig. 1B). This finding indicates that Gcn2 is involved in sensing amino acid conditions for TORC1 regulation. To further confirm the role of Gcn2, we examined whether Gcn2 was required for amino acid starvation-induced autophagy, another TORC1-dependent process, which was assayed based on autophagic flux that leads to breakdown of the GFP-Atg8 fusion protein and release of GFP (39). In wild type cells, we found that starvation of leucine or histidine induced the cleavage of GFP-Atg8, as did rapamycin treatment or nitrogen starvation. However, in gcn2 cells, the starvation failed to trigger the cleavage (Fig. 1C). Taken together, these findings demonstrate that Gcn2 acts as the sensor of TORC1 for amino acid starvation.
FIGURE 1.
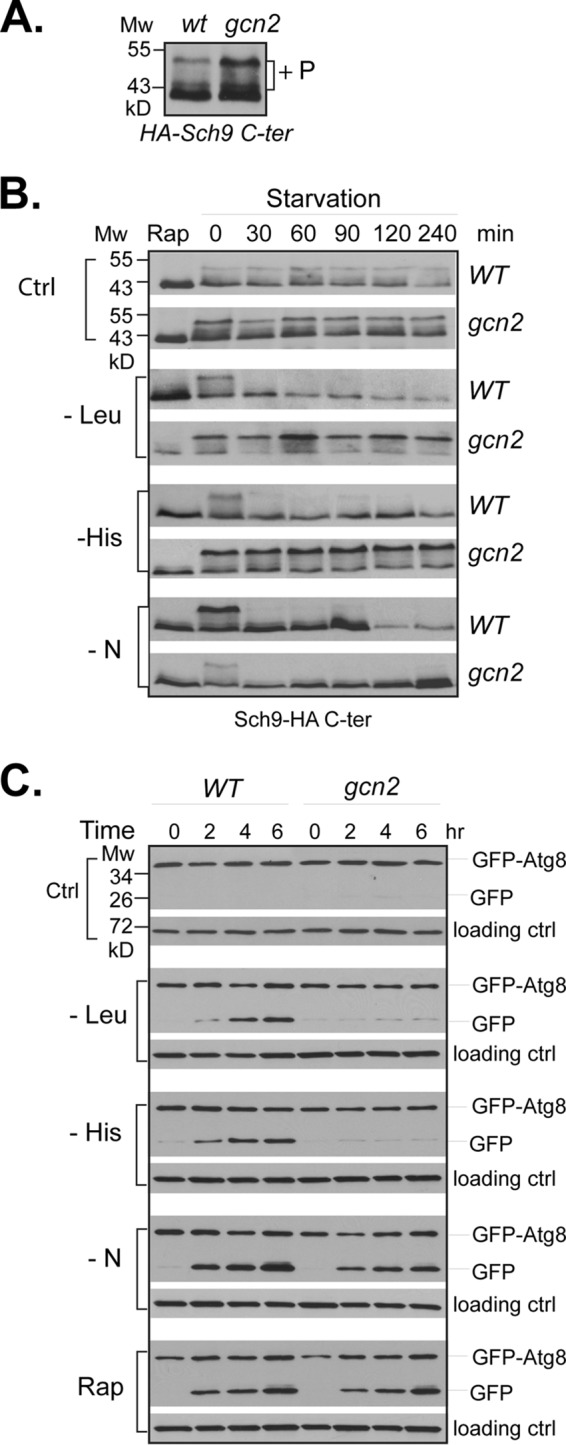
Gcn2 is required for TORC1 down-regulation in response to amino acid starvation. A, wild type (Y280) and gcn2 deletion (Y1527) cells expressing SCH9-HA were grown to early log phase in SC-Ura medium. Levels of TORC1-dependent phosphorylation of Sch9-HA (+P) in the cells were examined by Western blotting with anti-HA antibody. B, the same cells in A were shifted from SC-Ura medium to the same medium for mock treatment (ctrl) or to medium lacking leucine (−Leu), histidine (−His), or nitrogen (−N). Before starvation, an aliquot of wild type and gcn2 deletion cells was treated with rapamycin (Rap). The treated cells were collected at the indicated time points, and the levels of TORC1-dependent phosphorylation of Sch9-HA in the cells were examined by Western blotting. C, wild type (Y280) and gcn2 deletion (Y1527) cells expressing GFP-ATG8 were grown to early log phase in SC-Ura medium and exposed to the indicated treatments. Cells were collected, and autophagic cleavage of GFP-Atg8 in the cells was examined by Western blotting with anti-GFP antibody. The experiments were repeated five times, and representative data are shown.
Gcn1 and Gcn20 Are Involved in Gcn2-mediated TORC1 Down-regulation
Previous studies have shown that, in response to amino acid starvation, Gcn2 functions in a complex that contains Gcn1 and Gcn20 to phosphorylate eIF2α (11). To determine whether Gcn2 acts through the same mechanism for controlling TORC1, we examined the role of Gcn1 and Gcn20 in amino acid starvation-induced down-regulation of TORC1. We found that in gcn1 deletion cells, as in gcn2 deletion cells, TORC1-dependent Sch9 phosphorylation became insensitive to leucine or histidine starvation, whereas it remained sensitive to nitrogen deprivation and rapamycin treatment. In gcn20 deletion cells, the absence of Gcn20 partially blocked the effect of amino acid starvation on the Sch9 phosphorylation, resulting in delayed dephosphorylation (Fig. 2A). Similar results were obtained when the effect of gcn1 on TORC1 activity was assayed using autophagic flux-mediated breakdown of GFP-Atg8. However, in gcn20 deletion cells, amino acid starvation-induced breakdown of GFP-Atg8 was largely unaffected (Fig. 2B). These findings suggest that Gcn1 is required for amino acid starvation-induced TORC1 down-regulation, whereas Gcn20 is partially involved. The effects of gcn1 and gcn20 deletions on TORC1 activity mirror those on eIF2α phosphorylation (Fig. 2C). The partial effect of Gcn20 on TORC1 activity is consistent with its non-essential role for Gcn2 activity (14).
FIGURE 2.
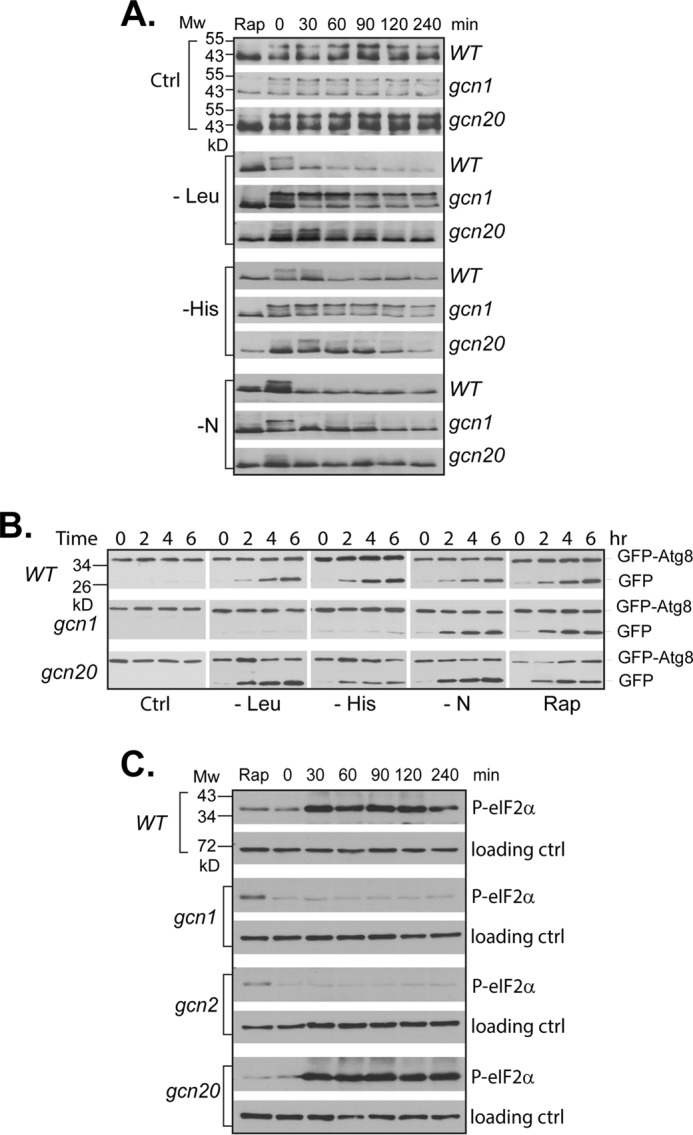
Gcn1 and Gcn20 are involved in Gcn2-mediated TORC1 down-regulation. A, wild type (Y280), gcn1 (Y1622), or gcn20 (Y1629) deletion cells expressing SCH9-HA cells were grown to early log phase in SC-Ura medium and shifted to indicated starvation conditions. Cells were collected at various time points after the shift, and TORC1-dependent phosphorylation of Sch9 was examined by Western blotting. B, wild type (T280), gcn1 (Y1622), or gcn20 (Y1629) deletion cells expressing GFP-ATG8 were shifted from SC-Ura medium to the indicated starvation media. Autophagic cleavage of GFP-Atg8 in the cells collected at different time points after the shift was assayed by Western blotting. C, samples of histidine-starved cells from B were blotted with anti-phospho-eIF2α (Ser-51) antibody for Gcn2-directed phosphorylation of eIF2α and anti-Tpd3 for loading controls. The experiments were repeated four times, and representative data are shown.
Activation of Gcn2 Inhibits TORC1 Activity
To determine whether activation of Gcn2 alone is sufficient to down-regulate TORC1, we examined TORC1-dependent Sch9 phosphorylation and autophagic cleavage of GFP-Atg8 in cells overexpressing an active Gcn2 mutant protein, Gcn2(E803V). The E803V mutation relieves the autoinhibition of Gcn2 kinase, rendering its activation independent of uncharged tRNAs (7, 40). We found that the basal expression of Gcn2(E803V) from a galactose-inducible promoter was sufficient to block the Sch9 phosphorylation in cells grown under non-starving conditions (Fig. 3A). Under the same growth conditions, the autophagic cleavage of GFP-Atg8 was less sensitive to the basal expression of Gcn2(E803V). However, when the expression was increased by galactose induction, the cleavage was enhanced (Fig. 3B). The down-regulation in TORC1 activity was not observed in cells expressing a control vector or a kinase-dead version of Gcn2 (K628R) grown under the same conditions (Fig. 3, A and B). These results demonstrate that activation of Gcn2 alone is able to inhibit TORC1 signaling activity, indicating a direct role for Gcn2 in regulation of TORC1.
FIGURE 3.
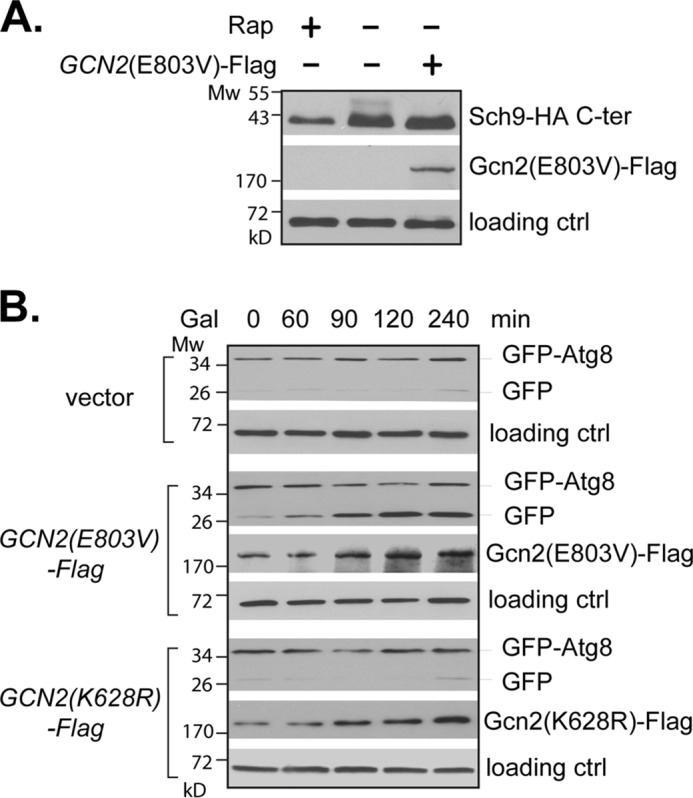
Gcn2 activation inhibits TORC1 activity. A, wild type (Y661) cells expressing SCH9-HA together with a galactose-inducible GCN2(E803V) (+) or control vector (−) grown in medium containing 2% glucose were treated with rapamycin (Rap+) or vehicle control (−). TORC1-dependent phosphorylation of Sch9-HA was examined by Western blotting. B, wild type (Y661) cells expressing GFP-ATG8 together with a control vector or vector expressing a galactose-inducible GCN2(E803V) or gcn2(K628R) were grown in the presence of 2% galactose (Gal) for the indicated times. Autophagic cleavage of GFP-Atg8 was assayed by Western blotting. Tpd3 levels were used as loading controls. The experiments were repeated three times, and representative data are shown.
The Gtr1-Gtr2 Complex Does Not Play a Major Role in Sensing Amino Acid Starvation for TORC1
Previous studies in yeast have shown that amino acids activate TORC1 through two Rag GTPases, Gtr1 and Gtr2. When amino acids are available, the two GTPases form a unique complex with Gtr1 in the GTP-bound state and Gtr2 in the GDP-bound state, which binds and activates TORC1 (22). To determine the role of the Rag GTPase complex in sensing amino acid starvation, we examined whether expressing the active form of the GTPase complex is able to block starvation-induced TORC1 down-regulation. As shown in Fig. 4A, we found that expression of the active Gtr1-Gtr2 complex partially blocked histidine starvation-induced Sch9 dephosphorylation, which is consistent with the findings of a previous study (22). However, the active complex failed to prevent the autophagy cleavage of GFP-Atg8 in response to histidine starvation (Fig. 4B). This finding indicates that Gcn2, but not the Gtr1-Gtr2 complex, plays a major role in sensing amino acid starvation for TORC1 regulation.
FIGURE 4.
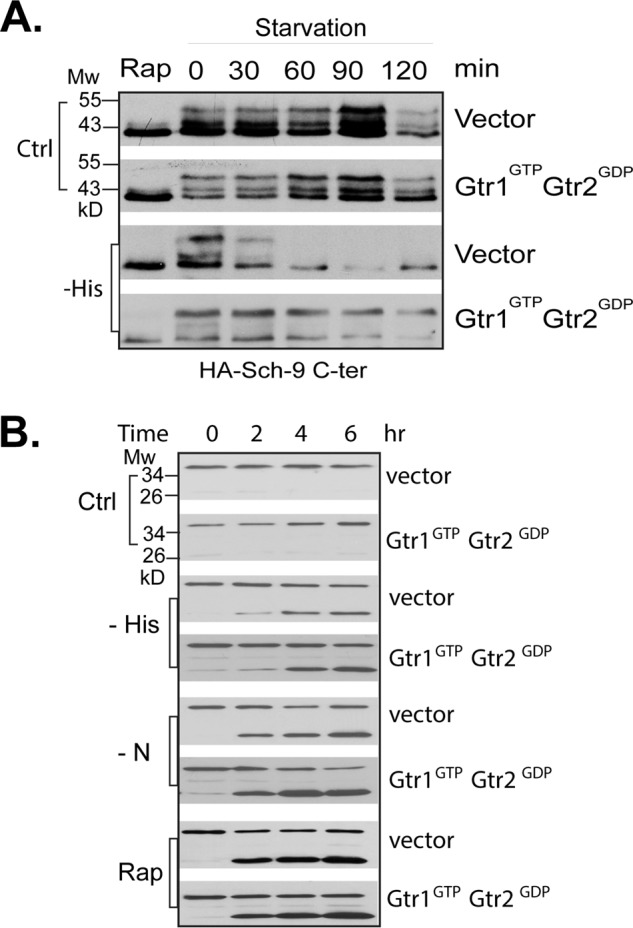
Activation of the Gtr complex does not prevent TORC1 down-regulation under starvation conditions. Wild type cells (Y661) expressing an active form of the Gtr complex containing Gtr1Q65L (Gtr1GTP) and Gtr2S23L (Gtr2GDP) or control vectors were starved for the indicated conditions. TORC1 activity was assayed by the TORC1-dependent phosphorylation of Sch9-HA (A) and autophagy activity by the cleavage of GFP-Atg8 (B). The experiments were repeated five times, and representative data are shown.
Gcn2 Inhibits the Intrinsic Kinase Activity of TORC1
To determine the mechanism by which Gcn2 inhibits TORC1, we examined whether Gcn2 is able to affect the intrinsic kinase activity of TORC1. Accordingly, we assayed TORC1 kinase activity in vitro from wild type and gcn2 deletion cells after the cells were exposed to various starvation conditions. We found that TORC1 immunopurified from wild type cells after leucine or histidine starvation exhibited a reduced activity, as did the TORC1 kinase from cells treated with rapamycin (Fig. 5A). However, the reduction in kinase activity was not observed when TORC1 was immunopurified from gcn2 deletion cells after exposure to leucine or histidine starvation (Fig. 5B). These observations suggest that amino acid starvation inhibits the intrinsic kinase activity of TORC1 and that the inhibitory effect is mediated by Gcn2. Interestingly, although TORC1 signaling activity, as assayed by phosphorylation of Sch9 and autophagic cleavage of GFP-Atg8, was greatly reduced by nitrogen starvation (Fig. 1), its intrinsic kinase activity was unaffected by the starvation (Fig. 5A). This finding indicates that nitrogen and amino acid starvations affect TORC1 function through distinct mechanisms.
FIGURE 5.
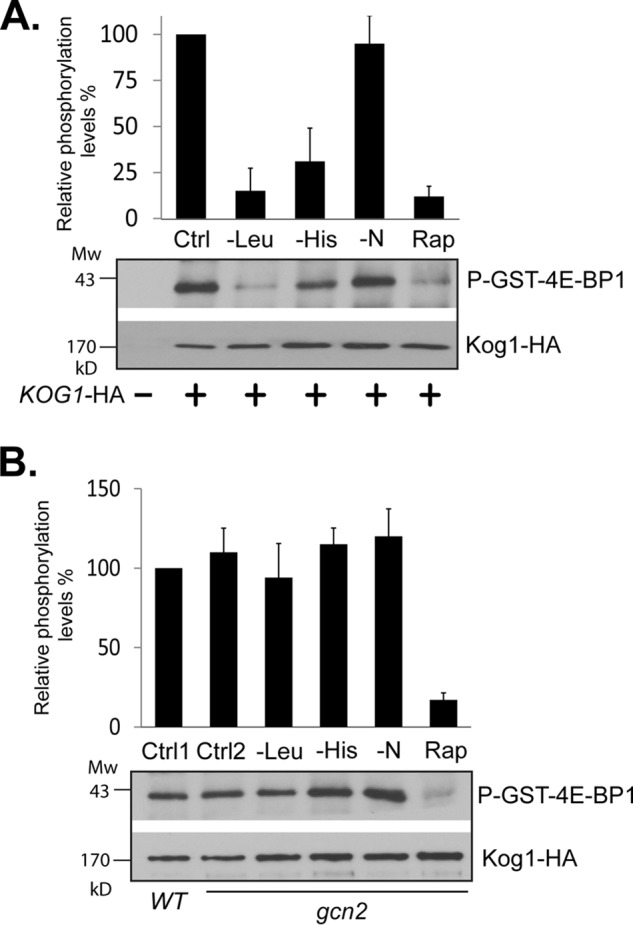
Gcn2 inhibits the intrinsic kinase activity of TORC1. Wild type (Y1599) and gcn2 deletion (Y1600) cells expressing KOG1-HA (+) or empty vector (−) were subjected to the indicated treatments for 15 min. Cells were collected and lysed. TORC1 was immunopurified from cell extracts with HA antibody and assayed for kinase activity toward GST-4E-BP1. A, TORC1 kinase activity from wild type cells. A TORC1 sample from untreated wild type (ctrl) was included as a control. B, TORC1 kinase activity from gcn2 deletion cells. TORC1 samples from untreated wild type (ctrl 1) and gcn2 (ctrl 2) cells were included as controls. TORC1 samples from untreated wild type and gcn2 deletion cells were assayed in the presence of rapamycin and GST-FKBP12 and shown in A and B, respectively, as rapamycin treatment controls (Rap). Bar graphs in A and B are quantitative presentations of TORC1 kinase activities from treated samples that are expressed as percentages of the untreated control. The relative kinase activity was calculated as the ratio between the level of GST-4E-BP1 phosphorylation and that of immunopurified Kog1-HA protein. Data were from five independent experiments and expressed as means ± S.D. (error bars).
Gcn2 Interacts with TORC1 in Response to Amino Acid Starvation
The down-regulation of the intrinsic kinase activity indicates that Gcn2 may act through a direct modification of TORC1. As the first step to test this notion, we examined whether Gcn2 is able to interact with TORC1 using a co-immunoprecipitation assay. Under normal growth conditions, we found that Gcn2 exhibited a weak interaction with Kog1, the unique component of TORC1. Upon starvation for histidine, there was a transient increase in the interaction, which occurred within 5 min after the onset of starvation but became barely detectable after 10 min (Fig. 6A). Similar results were obtained when the association of Gcn2 with Tor1 was assayed, which exists only in TORC1 (Fig. 6B). However, we did not detect any interaction between Gcn2 and Avo1 (Fig. 6C), the unique component of TORC2. These findings demonstrate that amino acid starvation is able to induce a transient interaction of Gcn2 with TORC1 but not TORC2.
FIGURE 6.
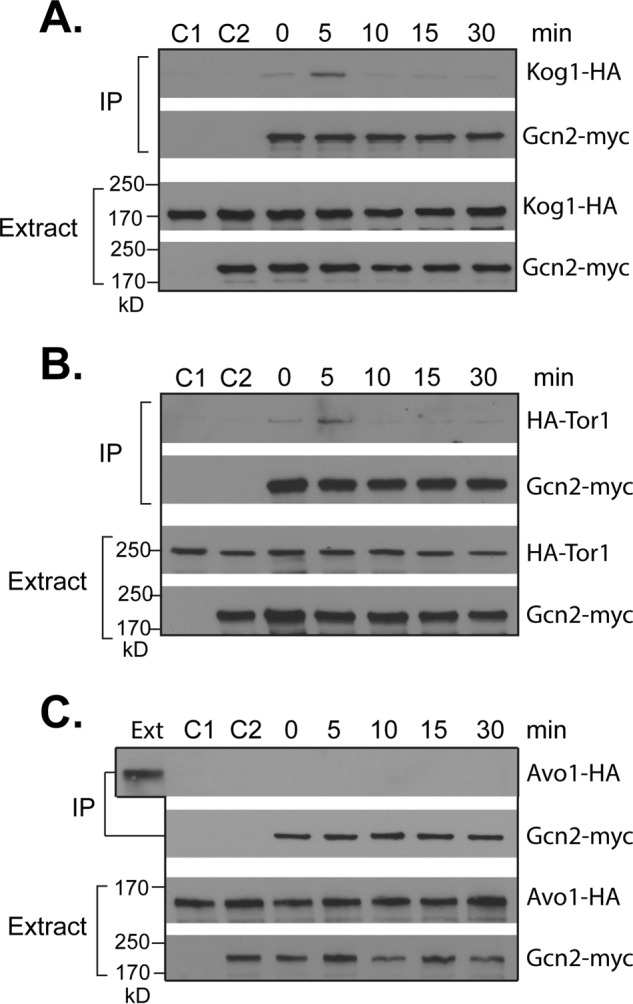
Amino acid starvation induces a transient association of Gcn2 with TORC1. A, wild type cells expressing KOG1-HA (Y1599) alone or together with GCN2-myc (Y1603) were starved for histidine. B, wild type cells expressing TOR1-HA (Y1666) alone or together with GCN2-myc (Y1664) were starved for histidine. C, wild type cells expressing AVO1-HA (Y1659) alone or together with GCN2-myc (Y1658) were starved for histidine. Cells were collected at the indicated times after the onset of starvation. Cell extracts were precipitated with control IgG (C2) or Myc antibody. Levels of the tagged proteins in cell extracts and precipitates (IP) were analyzed by Western blotting. The cells without GCN2-myc expression were used as a negative control (C1). The experiments were repeated three times, and representative data are shown.
Gcn2 Is Able to Phosphorylate Kog1 in Vitro
The physical association between Gcn2 and TORC1 raised a possibility that Gcn2, a serine/threonine kinase, is able to phosphorylate a component of TORC1 and regulates its activity. Because Gcn2 interacts with TORC1 but not TORC2 (Fig. 6), we surmised that the regulatory subunit of TORC1, Kog1, might serve as the target. To test this notion, we used an in vitro kinase assay to analyze the ability of Gcn2 to phosphorylate Kog1. Epitope-tagged wild type, active, and kinase-dead forms of Gcn2 were expressed in yeast cells, immunopurified, and used for phosphorylation of bacterially expressed recombinant Kog1. Due to the large size of Kog1, expressing the full-length protein in bacterial cells proved to be difficult. We thus expressed three fragments of the protein separately in bacterial cells corresponding to N-terminal (aa 1–637), middle (aa 638–1157), and C-terminal (aa 1158–1537) regions. The expressed proteins were purified and used as the substrates for the Gcn2 kinase. When incubated with lysates from cells expressing Gcn2-FLAG, the purified N-terminal region of Kog1 was found to bind with Gcn2-FLAG, indicating that Kog1 of TORC1 is the target of Gcn2 (Fig. 7B). When the kinase activity of Gcn2 was assayed, we found that the immunopurified wild type and active mutant Gcn2 were able to phosphorylate the N-terminal region but not other regions of Kog1 (Fig. 7C). In comparison with wild type Gcn2, the active form of Gcn2 exhibited a higher phosphorylation activity. In contrast, the kinase-dead mutant was incapable of phosphorylating any Kog1 fragments (Fig. 7C). Collectively, these data demonstrate that Gcn2 is capable of binding and phosphorylating Kog1 in vitro, which supports the notion that Kog1 is the direct target of Gcn2.
FIGURE 7.
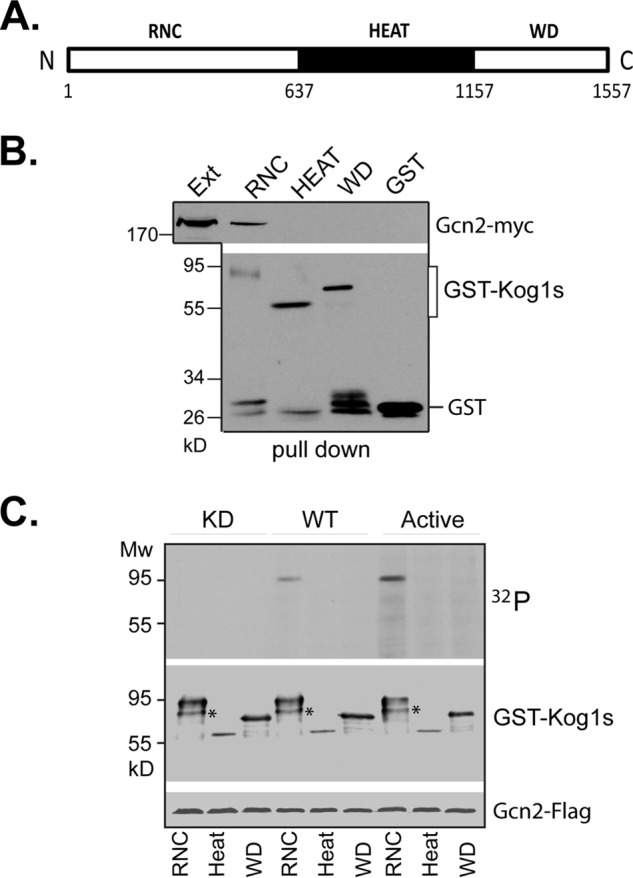
Gcn2 phosphorylates Kog1 directly. A, schematic presentation of Kog1 fragments used for the kinase assay. RNC, raptor N-terminal conserved domain; HEAT, HEAT domains; WD, WD40 domains. B, purified recombinant GST-Kog1 fragments were incubated with lysates from cells expressing GCN2-FLAG. Gcn2-FLAG co-purified with GST-Kog1 fragments was assayed by Western blotting. C, immunopurified FLAG-tagged Gcn2(E803V) (active), wild type, and Gcn2(K628R) (kinase-dead) versions of Gcn2 were assayed for kinase activity against recombinant GST-fused Kog1 fragments. The experiments were repeated three times, and representative data are shown.
Gcn2 Is Involved in Down-regulation of mTORC1 under Amino Acid Starvation Conditions in Mammalian Cells
Because TORC1 and Gcn2 are highly conserved from yeast to humans, we examined the possibility that mammalian Gcn2 is also involved in mTORC1 regulation in response to amino acid starvation. Accordingly, we assayed mTORC1 signaling activity under leucine or histidine starvation condition in wild type and gcn2 null MEF cells. We found that leucine and histidine starvation induced a rapid decrease in mTORC1 activity in wild type MEF cells. However, in gcn2 null MEF cells, the leucine starvation-induced down-regulation in mTORC1 activity was partially prevented, and the effect of histidine starvation on mTORC1 was completely blocked (Fig. 8).
FIGURE 8.
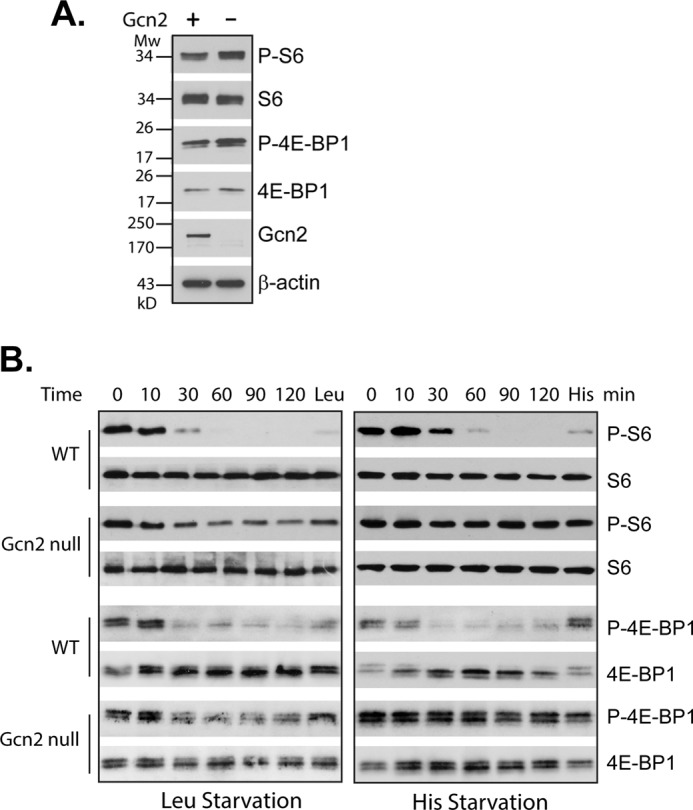
Mammalian Gcn2 is involved in mTORC1 down-regulation in response to amino acid starvation. A, mTORC1-dependent phosphorylation of S6 and 4E-BP1 in wild type (Gcn2 +) and Gcn2 null MEF (Gcn2 −) cells grown under normal conditions. B, WT and Gcn2 null MEF cells were collected after exposure to leucine or histidine starvation for the indicated times. At the end of 2-h starvation, leucine (Leu) or histidine (His) was added back for 15 min. mTORC1 signaling activity in the starved cells and in those after adding back the omitted amino acids was determined by the mTORC1-dependent phosphorylation of S6 and 4E-BP1. The experiments were repeated five times, and representative data are shown.
Discussion
As an energy-consuming process, translation is tightly regulated by amino acid availability. A key mechanism for sensing amino acid starvation involves uncharged tRNA-activated Gcn2. In this Gcn2-mediated mechanism, the only target of Gcn2 identified currently in both yeast and mammalian cells is eIF2α, which is phosphorylated directly by Gcn2 at serine 51 (41, 42). In the present study, we show that TORC1 is another downstream effector of Gcn2 in translation regulation. We find that in the absence of Gcn2, TORC1 signaling activity to Sch9, a key factor that mediates the effect of TORC1 in translation in yeast, becomes unresponsive to amino acid starvation. This finding unveils an important mechanism in both amino acid signaling and TORC1 regulation. It suggests that the down-regulation in general translation under amino acid starvation condition is mediated by Gcn2 through both eIF2α and TORC1. The link between Gcn2 and TORC1 allows uncharged tRNAs to serve as messengers of amino acid starvation for processes mediated by TORC1. In addition to translation and autophagy, this link is expected to impact TORC1-controlled transcription and protein turnover. Such a mechanism is able to orchestrate coordinated cellular responses that are essential for yeast cells to adapt and survive under amino acid starvation conditions.
Our results suggest that Gcn2 controls TORC1 activity through a direct phosphorylation of Kog1, the unique component of TORC1. Three lines of evidence support this notion. First, the kinase activity of Gcn2 is essential for its inhibitory effect on TORC1 (Fig. 3). Second, Gcn2 transiently associates with TORC1 but not TORC2 in response to amino acid starvation (Fig. 6). This transient association is a common characteristic for enzyme-substrate pairing. Third, Gcn2 is able to phosphorylate the N-terminal region of Kog1 in vitro (Fig. 7). The N-terminal region of Kog1 is essential for its function and is highly conserved between yeast and mammals. In a structural analysis of the association between Kog1 and TOR, this region of Kog1 was found to line up with the kinase domain of TOR. This observation has led to the suggestion that the N-terminal region of Kog1 is involved in targeting the catalytic domain of TOR to its substrates (43). It is thus likely that Gcn2, upon activation by uncharged tRNAs, binds to Kog1 and phosphorylates it. This Gcn2-dependent phosphorylation may alter Kog1 conformation and, consequently, affect TORC1 kinase activity. The latter notion is consistent with the observation that the intrinsic kinase activity of TORC1 is reduced from cells subjected to amino acid starvation. However, identification of a Gcn2-directed phosphorylation site in Kog1 has proven to be difficult, due to the large size and labile nature of Kog1, which makes it impractical to purify a sufficient amount of the protein for mapping the site. Analysis of several potential phosphorylation sites located within the N-terminal region of Kog1 that are conserved between yeast and its human counterparts failed to identify a site that affects the phosphorylation (data not shown). It remains possible that Gcn2 phosphorylates Kog1 at multiple sites and that replacing a single one may have no effect. At the present time, we cannot rule out the possibility that Gcn2 may phosphorylate another component of TORC1 or act indirectly on TORC1, although we consider it unlikely, given the evidence listed above. Future study will be directed to identification of Gcn2-directed phosphorylation in Kog1 or another component of TORC1. Despite this shortcoming, our study demonstrates that Gcn2 is the sensor of TORC1 for amino acid starvation.
Although Gcn2 is required for TORC1 down-regulation in response to amino acid starvation, it is not involved in inhibition of TORC1 by nitrogen starvation (Fig. 1). This observation suggests that the two conditions are perceived by TORC1 through distinct mechanisms. Our finding that the intrinsic TORC1 kinase activity is reduced by amino acid starvation but not by nitrogen starvation is consistent with the notion (Fig. 5). Previously, we have shown that nitrogen starvation is sensed as a stress condition that involves activation of Rho1 GTPase. In response to nitrogen starvation and other stress conditions, such as heat shock and cell wall damage, the GTPase binds to TORC1 and disrupts its membrane association, leading to down-regulation of its signaling activity (26). These observations reinforce the notion that Gcn2 controls TORC1 through a unique mechanism that involves a direct inhibition of TORC1 kinase activity, probably through phosphorylation of Kog1.
A cross-talk between Gcn2 and TORC1 has been reported previously. However, in that case, Gcn2 is found to act as a downstream effector of TORC1. It has been shown that upon rapamycin treatment, Gcn2 is dephosphorylated at position Ser-577. The dephosphorylation is mediated by rapamycin-induced activation of the Tap42-PP2A phosphatase, which results in an increase in Gcn2 activity and eIF2α phosphorylation even under amino acid repletion conditions (35). Amino acid starvation per se does not affect the phosphorylation (44). These findings suggest that Gcn2 and TORC1 are regulated by different mechanisms under different adverse conditions.
It has been recently found in yeast that TORC1 is activated by LeuRS. In the presence of leucine, the leucine-bound LeuRS interacts with Gtr1 and promotes its GTP binding. The GTP-bound Gtr1, together with a GDP-bound Gtr2, then activates TORC1 by tethering it to the surface of the vacuole (22). A similar mechanism operates in mammalian cells for mTORC1 activation, although in this case, leucine-bound LeuRS interacts with RagD, the mammalian counterpart of Gtr2, and sequesters the GTPase in a GDP-bound state (36, 37). While this LeuRS-dependent model explains how the availability of leucine and perhaps other amino acids is sensed by TORC1, it falls short in explaining why starvation of any single amino acid is able to down-regulate TORC1. In addition, our finding that expressing an active form of the Gtr complex was ineffective in preventing TORC1 down-regulation in response to amino acid starvation suggests that the LeuRS-dependent mechanism plays a minor role in sensing amino acid starvation for TORC1 regulation.
The Gcn2-mediated TORC1 regulation appears to be conserved in mammalian cells, because we found that in GCN2−/− MEF cells, mTORC1 signaling activity became largely unresponsive to histidine starvation and partially to leucine starvation. These observations are consistent with some early studies that implicated a role for uncharged tRNAs and Gcn2 in mTORC1 regulation in mammalian cells. It was found that inhibition of tRNA synthetases, which resulted in accumulation of uncharged tRNAs, suppressed mTORC1 activity (45). In cultured liver cells, Gcn2 deficiency rendered mTORC1-dependent phosphorylation of S6K1 unresponsive to leucine derivation (46). In an animal study, it was shown that in GCN2−/− mice, the mTORC1 signaling activity became insensitive to a leucine-deficient diet (47). These early findings, together with our results, support a notion that the tRNA-Gcn2 signaling cascade is a conserved mechanism for mTORC1 to sense and respond to amino acid starvation in mammalian cells.
Experimental Procedures
Antibodies and Reagents
Rapamycin was purchased from LC Laboratories (Woburn, MA) and used at a concentration of 100 nm. Anti-4E-BP1 (catalog no. 9452), phospho-4E-BP1 (Thr-37/46) (catalog no. 2855), S6 (catalog no. 2217), phospho-S6(Ser-235/236) (catalog no. 4858), and Gcn2 (catalog no. 3302) antibodies were purchased from Cell Signaling Technology. Anti-phospho-eIF2α (Ser-51) antibody was from Thermo Fisher Scientific (catalog no. 44728G); anti-HA (12CA5) (catalog no. sc-57592), anti-Myc (9E10) (catalog no. sc-40), and anti-GST (catalog no. sc-138) antibodies were from Santa Cruz Biotechnology, Inc.; anti-FLAG antibody (catalog no. F3165) was from Sigma; and anti-GFP (catalog no. 1181446001) antibody was from Roche Diagnostics.
Media, Strains, and Plasmids
Standard YEPD and synthetic complete (SC) dropout media were used. All media contain 2% glucose as the carbon source unless indicated otherwise. Yeast strains used in this study are listed in Table 1, and plasmids are listed in Table 2. Plasmid GFP-ATG8-pRS416 was a gift from Daniel Klionsky (University of Michigan, Ann Arbor, MI). GFP-ATG8-pRS414 was created by cloning the GFP-ATG8 fusion gene into pRS414. Plasmids pDH114, a high copy yeast expressing vector containing a FLAG-tagged GCN2(E803V) gene under control of a galactose-inducible promoter; pDH101, a FLAG-tagged wild type GCN2 in pRS316; and PB115, a kinase-dead gcn2(K628R) in pRS316, were kindly provided by Alan Hinnebusch (National Institutes of Health) (44). pJB1574 was created by replacing the BlpI-SpeI region of GCN2 in pDH101 with the same region from GCN2(E803V) in pDH114. pJB1575 was generated by replacing the BlpI-SpeI region of GCN2 in pDH101 with the same region from gcn2(K628R) in PB115. pJB1421 (gcn2-myc-pRS406) was created by cloning into pRS406 a PCR-generated KpnI-BamHI fragment containing the last 786 bp of the GCN2 gene followed by 13× Myc tags and an ADH1 termination sequence. This plasmid was linearized by digestion with AflII and used for integrating the Myc tags into the genomic copy of GCN2.
TABLE 1.
Strains used in this study
| Strains | Genotypea | Source | Figures |
|---|---|---|---|
| Y280 | MATa, his3-1, leu2-0, met 15-0, ura3 | Laboratory stock | 1A, 1B, 1C, 2A, 2B |
| Y661b | MATa, ura3-1, his3-11, lue2-3,112, trp1-1, ade2-1, can1-100, GAL+ | Laboratory stock | 4A, 4B |
| Y1527 | MATa, his3-1, leu2-0, met 15-0, ura3, lys2, gcn2::KAN | Laboratory stock | 1A, 1B, 1C |
| Y1599 | MATa, his3-1, leu2-0, met 15-0, ura3 kog1::KOG1-HA | This study | 5A, 5B, 6A |
| Y1600 | MATa, his3-1, leu2-0, met 15-0, ura3, lys2, gcn2::KAN, kog1::KOG1-HA | This study | 5A, 5B |
| Y1603 | MATa, his3-1, leu2-0, met 15-0, ura3, kog1::KOG1-HA gcn2::GCN2-myc:URA3 | This study | 6A |
| Y1622 | MATa, his3-1, leu2-0, met 15-0, ura3, gcn1::KAN | Laboratory stock | 2A, 2B |
| Y1629 | MATα, his3-1, leu2-0, met 15-0, ura3, lys2, gcn20::KAN | Laboratory stock | 2A, 2B |
| Y1658 | MATa, his3-1, leu2-0, met 15-0, ura3, gcn2::GCN2-myc, avo1::AVO1-HA:URA3 | This study | 6C |
| Y1659 | MATa, his3-1, leu2-0, met 15-0, ura3, avo1::AVO1-HA:URA3 | This study | 6C |
| Y1664 | MATa, his3-1, leu2-0, met 15-0, ura3, gcn2::GCN2-myc tor1::HA-TOR1:URA3 | This study | 6B |
| Y1666 | MATa, his3-1, leu2-0, met 15-0, ura3, tor1::HA-TOR1:URA3 | This study | 6B |
a All stains are in BY4743 strain background except as indicated.
b Strains in W303 background.
TABLE 2.
Plasmids used in this study
| Plasmids | Characteristics | Source/Reference |
|---|---|---|
| PB115 | gcn2 (K628R) in pRS316 (CEN URA3) | Ref. 44 |
| pDH101 | GCN2-FLAG in pRS316 (CEN URA3) | Ref. 44 |
| pDH109 | pGAL1-gcn2 (K628R)-FLAG in pEMBLyex4 (2μ URA3 leu2-d) | Ref. 7 |
| pDH114 | pGAL1-GCN2 (E803V)-FLAG in pEMBLyex4 (2μ URA3 leu2-d) | Ref. 7 |
| pJB948 | kog1Δ5-3HA in pRS406 (URA3) | Ref. 26 |
| pJB1321 | SCH9-3HA in Ycplace33 (CEN URA3) | Ref. 29 |
| pJB1379 | 3HA-tor1Δ3 in pRS406 (URA3) | Laboratory stock |
| pJB1404 | kog1Δ5-13myc in pRS406 | Ref. 26 |
| pJB1405 | GFP-ATG8-pRS416 (CEN URA3) | Ref. 48 |
| pJB1421 | gcn2Δ5-13myc-ADH1 terminator in pRS406 (CEN URA3) | This study |
| pJB1440 | GFP-ATG8-pRS414 (CEN TRP1) | This study |
| pJB1574 | GCN2 (E803V)-FLAG in pRS316 (CEN URA3) | This study |
| pJB1575 | gcn2 (K628R-FLAG in pRS316 (CEN URA3) | |
| pJB1580 | SCH9-3HA in pRS415 (CEN LEU2) | This study |
| pJM186 | GTR2 (S23L) in pRS414 (CEN TRP1) | This study |
| pJU654 | GTR2 (S23L) in pRS416 (CEN URA3) | Ref. 25 |
| pMB1483 | GTR1 (Q65L) in pRS415 (CEN LEU2) | Ref. 25 |
Starvation Treatments
For amino acid starvation, leucine or histidine auxotrophic cells were grown to early log phase in SC medium with appropriate amino acids omitted for plasmid selection. Cells were collected and resuspended in SC medium without leucine or histidine at a density of ∼3 × 106 cells/ml. For nitrogen starvation, collected cells were resuspended at the same density into nitrogen starvation medium containing 2% glucose and 1× yeast nitrogen base without ammonium sulfate and amino acids. For rapamycin treatment, cells were treated with a 100 nm concentration of the drug. Mock treatment controls were carried out by resuspending cells in SC medium at a density of ∼2 × 106 cells/ml. Treated cells were collected at various time points by centrifugation, and cell pellets were kept on dry ice until all samples were collected.
GFP-Atg8 Autophagic Flux Assay
Cells expressing GFP-ATG8 plasmids were subjected to various treatments as mentioned above. The treated cells were collected, resuspended in SDS sample buffer, and lysed by vortexing with glass beads. Levels of GFP-Atg8 and cleaved GFP in the cell lysates were examined by Western blotting using anti-GFP antibody.
Assay for C-terminal Phosphorylation of Sch9
TORC1-dependent Sch9 phosphorylation was analyzed as described previously (29). An aliquot of cell culture with 2 × 107 cells was collected at each time point during the starvation treatment.
Co-immunoprecipitation
Yeast cells expressing Myc-tagged GCN2 with KOG1-HA, HA-TOR1, or AVO1-HA were grown overnight at 30 °C to early log phase followed by exposure to histidine starvation. At the indicated time points after the onset of the starvation, an aliquot of cells (5 × 108) was transferred to a centrifuge tube filled with ice and collected by centrifugation at 500 × g for 5 min. Cells were resuspended in lysis buffer containing 50 mm Tris-Cl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm PMSF, 1.5× protease inhibitor cocktail (Roche Diagnostics) and lysed by vortexing with glass beads. Triton X-100 was then added to cell extracts to a final concentration of 1% followed by incubation on ice for 15 min. Cell extracts were centrifuged at 10,000 × g for 10 min, and the supernatants were incubated with anti-HA antibody for precipitation as described before (26).
In Vitro Assays
The in vitro assay for TORC1 kinase activity has been described before (26). To assay for Gcn2 kinase activity against Kog1, cells expressing FLAG-tagged wild type, active, and kinase-dead mutants of Gcn2 were grown to mid-log phase at 30 °C. Cells were collected and lysed with glass beads in buffer containing 50 mm HEPES, pH 7.4, 100 mm NaCl, 1 mm DTT, and 1× protease inhibitor cocktail. CHAPS was then added to cell lysates to a final concentration of 1%. After incubation on ice for 15 min, lysates were clarified by centrifugation at 10,000 × g for 10 min. For each sample, the supernatant (5 mg of protein) was incubated with anti-FLAG antibody (2 μg) at 4 °C for 2 h followed by the addition of Protein A beads (20 μl). Beads were washed twice with wash buffer containing 50 mm HEPES, pH 7.4, 300 mm NaCl, 1% CHAPS, and 1 mm DTT and twice with kinase buffer containing 50 mm HEPES, pH 7.4, 50 mm NaCl, 5 mm MgCl2, and 1 mm DTT. The kinase reaction was carried out by incubating the beads at 30 °C for 30 min in 50 μl of kinase buffer containing 1 μg of a purified recombinant GST-fused Kog1 fragment, 200 μm ATP, and 1 μCi of [γ-32P]ATP. The reaction was terminated by adding 15 μl of 5× SDS sample buffer and immediately boiled for 5 min. The reaction mixtures were subjected to SDS-PAGE and transferred to nitromembrane. The incorporation of 32P into the GST-Kog1 fragments was assayed by radioautograph. The bacterial expression and purification of the GST-Kog1 fragments were described before (26).
To assay for Kog1 binding, the clarified lysates (500 μg of protein) were incubated with 1 μg of purified bacterially expressed recombinant GST or GST-Kog1 fragments. After incubation at 4 °C for 1 h, lysates were precipitated with glutathione-conjugated agarose beads, and the precipitates were analyzed by Western blotting.
Mammalian Cell Culture
GCN2 null (catalog no. CRL-2978) and paired wild type MEF (catalog no. CRL-2977) were obtained from ATCC. Cells were normally grown in DMEM medium with high glucose (4.35 g/liter) and 10% FBS. Starvation media were prepared by mixing Hanks' balanced salt solution with bovine serum albumin (1%), glucose (4.35 g/liter), sodium carbonate (3.7 g/liter), 1× vitamin mixture, 1× nonessential amino acids, 1× essential amino acids without leucine or histidine as indicated. Starvation was carried out with cells at 80–90% confluence. All experiments were repeated at least three times, and representative data are shown.
Author Contributions
W. Y. conducted the studies shown in Figs. 1–4 and 8. S. G. did phosphorylation analysis. J. G. performed the studies shown in Figs. 4 and 6. M. Z., G. Y., W. W., and Y. C. assisted with the studies and did other control experiments not shown in the manuscript. W. Y. and Y. J. conceived and planned the experiments and interpreted data. Y. J. performed the experiments shown in Fig. 7 and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Alan Hinnebusch and Daniel Klionsky for providing plasmids and members of our laboratory for comments and stimulating discussion.
This work was supported in part by National Institutes of Health Grant CA169186 (to Y. J.) and National Natural Science Foundation of China Grant 81272269 (to Y. J.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- LeuRS
- leucyl-tRNA synthetase
- aa
- amino acids
- MEF
- mouse embryo fibroblast.
References
- 1. Kimball S. R. (2001) Regulation of translation initiation by amino acids in eukaryotic cells. Prog. Mol. Subcell. Biol. 26, 155–184 [DOI] [PubMed] [Google Scholar]
- 2. Proud C. G. (2014) Control of the translational machinery by amino acids. Am. J. Clin. Nutr. 99, 231S–236S [DOI] [PubMed] [Google Scholar]
- 3. Hinnebusch A. G. (1994) The eIF-2 α kinases: regulators of protein synthesis in starvation and stress. Semin. Cell Biol. 5, 417–426 [DOI] [PubMed] [Google Scholar]
- 4. Kim J., and Guan K. L. (2011) Amino acid signaling in TOR activation. Annu. Rev. Biochem. 80, 1001–1032 [DOI] [PubMed] [Google Scholar]
- 5. Jewell J. L., Russell R. C., and Guan K. L. (2013) Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 14, 133–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhu S., Sobolev A. Y., and Wek R. C. (1996) Histidyl-tRNA synthetase-related sequences in GCN2 protein kinase regulate in vitro phosphorylation of eIF-2. J. Biol. Chem. 271, 24989–24994 [DOI] [PubMed] [Google Scholar]
- 7. Dong J., Qiu H., Garcia-Barrio M., Anderson J., and Hinnebusch A. G. (2000) Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 6, 269–279 [DOI] [PubMed] [Google Scholar]
- 8. Dever T. E., Feng L., Wek R. C., Cigan A. M., Donahue T. F., and Hinnebusch A. G. (1992) Phosphorylation of initiation factor 2 α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68, 585–596 [DOI] [PubMed] [Google Scholar]
- 9. Donnelly N., Gorman A. M., Gupta S., and Samali A. (2013) The eIF2α kinases: their structures and functions. Cell. Mol. Life Sci. 70, 3493–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marton M. J., Vazquez de Aldana C. R., Qiu H., Chakraburtty K., and Hinnebusch A. G. (1997) Evidence that GCN1 and GCN20, translational regulators of GCN4, function on elongating ribosomes in activation of eIF2α kinase GCN2. Mol. Cell. Biol. 17, 4474–4489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garcia-Barrio M., Dong J., Ufano S., and Hinnebusch A. G. (2000) Association of GCN1-GCN20 regulatory complex with the N-terminus of eIF2α kinase GCN2 is required for GCN2 activation. EMBO J. 19, 1887–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marton M. J., Crouch D., and Hinnebusch A. G. (1993) GCN1, a translational activator of GCN4 in Saccharomyces cerevisiae, is required for phosphorylation of eukaryotic translation initiation factor 2 by protein kinase GCN2. Mol. Cell. Biol. 13, 3541–3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kubota H., Ota K., Sakaki Y., and Ito T. (2001) Budding yeast GCN1 binds the GI domain to activate the eIF2α kinase GCN2. J. Biol. Chem. 276, 17591–17596 [DOI] [PubMed] [Google Scholar]
- 14. Vazquez de Aldana C. R., Marton M. J., and Hinnebusch A. G. (1995) GCN20, a novel ATP binding cassette protein, and GCN1 reside in a complex that mediates activation of the eIF-2 α kinase GCN2 in amino acid-starved cells. EMBO J. 14, 3184–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ma X. M., and Blenis J. (2009) Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318 [DOI] [PubMed] [Google Scholar]
- 16. Loewith R., Jacinto E., Wullschleger S., Lorberg A., Crespo J. L., Bonenfant D., Oppliger W., Jenoe P., and Hall M. N. (2002) Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 10, 457–468 [DOI] [PubMed] [Google Scholar]
- 17. Wei Y., and Zheng X. F. (2011) Nutritional control of cell growth via TOR signaling in budding yeast. Methods Mol. Biol. 759, 307–319 [DOI] [PubMed] [Google Scholar]
- 18. Wullschleger S., Loewith R., and Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484 [DOI] [PubMed] [Google Scholar]
- 19. Reinke A., Anderson S., McCaffery J. M., Yates J. 3rd, Aronova S., Chu S., Fairclough S., Iverson C., Wedaman K. P., and Powers T. (2004) TOR complex 1 includes a novel component, Tco89p (YPL180w), and cooperates with Ssd1p to maintain cellular integrity in Saccharomyces cerevisiae. J. Biol. Chem. 279, 14752–14762 [DOI] [PubMed] [Google Scholar]
- 20. Sancak Y., Peterson T. R., Shaul Y. D., Lindquist R. A., Thoreen C. C., Bar-Peled L., and Sabatini D. M. (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shaw R. J. (2008) mTOR signaling: RAG GTPases transmit the amino acid signal. Trends Biochem. Sci. 33, 565–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bonfils G., Jaquenoud M., Bontron S., Ostrowicz C., Ungermann C., and De Virgilio C. (2012) Leucyl-tRNA synthetase controls TORC1 via the EGO complex. Mol. Cell 46, 105–110 [DOI] [PubMed] [Google Scholar]
- 23. Dubouloz F., Deloche O., Wanke V., Cameroni E., and De Virgilio C. (2005) The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol. Cell 19, 15–26 [DOI] [PubMed] [Google Scholar]
- 24. Gao M., and Kaiser C. A. (2006) A conserved GTPase-containing complex is required for intracellular sorting of the general amino-acid permease in yeast. Nat. Cell Biol. 8, 657–667 [DOI] [PubMed] [Google Scholar]
- 25. Binda M., Péli-Gulli M. P., Bonfils G., Panchaud N., Urban J., Sturgill T. W., Loewith R., and De Virgilio C. (2009) The Vam6 GEF controls TORC1 by activating the EGO complex. Mol. Cell 35, 563–573 [DOI] [PubMed] [Google Scholar]
- 26. Yan G., Lai Y., and Jiang Y. (2012) The TOR complex 1 is a direct target of Rho1 GTPase. Mol. Cell 45, 743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Düvel K., and Broach J. R. (2004) The role of phosphatases in TOR signaling in yeast. Curr. Top. Microbiol. Immunol. 279, 19–38 [DOI] [PubMed] [Google Scholar]
- 28. Powers T. (2007) TOR signaling and S6 kinase 1: yeast catches up. Cell Metab. 6, 1–2 [DOI] [PubMed] [Google Scholar]
- 29. Urban J., Soulard A., Huber A., Lippman S., Mukhopadhyay D., Deloche O., Wanke V., Anrather D., Ammerer G., Riezman H., Broach J. R., De Virgilio C., Hall M. N., and Loewith R. (2007) Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 26, 663–674 [DOI] [PubMed] [Google Scholar]
- 30. Düvel K., Santhanam A., Garrett S., Schneper L., and Broach J. R. (2003) Multiple roles of Tap42 in mediating rapamycin-induced transcriptional changes in yeast. Mol. Cell 11, 1467–1478 [DOI] [PubMed] [Google Scholar]
- 31. Shamji A. F., Kuruvilla F. G., and Schreiber S. L. (2000) Partitioning the transcriptional program induced by rapamycin among the effectors of the Tor proteins. Curr. Biol. 10, 1574–1581 [DOI] [PubMed] [Google Scholar]
- 32. Yan G., Shen X., and Jiang Y. (2006) Rapamycin activates Tap42-associated phosphatases by abrogating their association with Tor complex 1. EMBO J. 25, 3546–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beck T., and Hall M. N. (1999) The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402, 689–692 [DOI] [PubMed] [Google Scholar]
- 34. Bertram P. G., Choi J. H., Carvalho J., Ai W., Zeng C., Chan T. F., and Zheng X. F. (2000) Tripartite regulation of Gln3p by TOR, Ure2p, and phosphatases. J. Biol. Chem. 275, 35727–35733 [DOI] [PubMed] [Google Scholar]
- 35. Cherkasova V. A., and Hinnebusch A. G. (2003) Translational control by TOR and TAP42 through dephosphorylation of eIF2α kinase GCN2. Genes Dev. 17, 859–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Han J. M., Jeong S. J., Park M. C., Kim G., Kwon N. H., Kim H. K., Ha S. H., Ryu S. H., and Kim S. (2012) Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 149, 410–424 [DOI] [PubMed] [Google Scholar]
- 37. Durán R. V., and Hall M. N. (2012) Leucyl-tRNA synthetase: double duty in amino acid sensing. Cell Res. 22, 1207–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Panchaud N., Péli-Gulli M. P., and De Virgilio C. (2013) Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Sci. Signal. 6, ra42. [DOI] [PubMed] [Google Scholar]
- 39. Cheong H., and Klionsky D. J. (2008) Biochemical methods to monitor autophagy-related processes in yeast. Methods Enzymol. 451, 1–26 [DOI] [PubMed] [Google Scholar]
- 40. Qiu H., Dong J., Hu C., Francklyn C. S., and Hinnebusch A. G. (2001) The tRNA-binding moiety in GCN2 contains a dimerization domain that interacts with the kinase domain and is required for tRNA binding and kinase activation. EMBO J. 20, 1425–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pain V. M. (1994) Translational control during amino acid starvation. Biochimie 76, 718–728 [DOI] [PubMed] [Google Scholar]
- 42. Cully M., and Downward J. (2009) Translational responses to growth factors and stress. Biochem. Soc. Trans. 37, 284–288 [DOI] [PubMed] [Google Scholar]
- 43. Adami A., García-Alvarez B., Arias-Palomo E., Barford D., and Llorca O. (2007) Structure of TOR and its complex with KOG1. Mol. Cell 27, 509–516 [DOI] [PubMed] [Google Scholar]
- 44. Garcia-Barrio M., Dong J., Cherkasova V. A., Zhang X., Zhang F., Ufano S., Lai R., Qin J., and Hinnebusch A. G. (2002) Serine 577 is phosphorylated and negatively affects the tRNA binding and eIF2α kinase activities of GCN2. J. Biol. Chem. 277, 30675–30683 [DOI] [PubMed] [Google Scholar]
- 45. Iiboshi Y., Papst P. J., Kawasome H., Hosoi H., Abraham R. T., Houghton P. J., and Terada N. (1999) Amino acid-dependent control of p70(s6k). Involvement of tRNA aminoacylation in the regulation. J. Biol. Chem. 274, 1092–1099 [DOI] [PubMed] [Google Scholar]
- 46. Xiao F., Huang Z., Li H., Yu J., Wang C., Chen S., Meng Q., Cheng Y., Gao X., Li J., Liu Y., and Guo F. (2011) Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and AMPK pathways. Diabetes 60, 746–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Anthony T. G., McDaniel B. J., Byerley R. L., McGrath B. C., Cavener D. R., McNurlan M. A., and Wek R. C. (2004) Preservation of liver protein synthesis during dietary leucine deprivation occurs at the expense of skeletal muscle mass in mice deleted for eIF2 kinase GCN2. J. Biol. Chem. 279, 36553–36561 [DOI] [PubMed] [Google Scholar]
- 48. Guan J., Stromhaug P. E., George M. D., Habibzadegah-Tari P., Bevan A., Dunn W. A. Jr., and Klionsky D. J. (2001) Cvt18/Gsa12 is required for cytoplasm-to-vacuole transport, pexophagy, and autophagy in Saccharomyces cerevisiae and Pichia pastoris. Mol. Biol. Cell 12, 3821–3838 [DOI] [PMC free article] [PubMed] [Google Scholar]