

Abstract
Rhomboid proteases are increasingly being explored as potential drug targets, but their potent and specific inhibitors are not available, and strategies for inhibitor development are hampered by the lack of widely usable and easily modifiable in vitro activity assays. Here we address this bottleneck and report on the development of new fluorogenic transmembrane peptide substrates, which are cleaved by several unrelated rhomboid proteases, can be used both in detergent micelles and in liposomes, and contain red-shifted fluorophores that are suitable for high-throughput screening of compound libraries. We show that nearly the entire transmembrane domain of the substrate is important for efficient cleavage, implying that it extensively interacts with the enzyme. Importantly, we demonstrate that in the detergent micelle system, commonly used for the enzymatic analyses of intramembrane proteolysis, the cleavage rate strongly depends on detergent concentration, because the reaction proceeds only in the micelles. Furthermore, we show that the catalytic efficiency and selectivity toward a rhomboid substrate can be dramatically improved by targeted modification of the sequence of its P5 to P1 region. The fluorogenic substrates that we describe and their sequence variants should find wide use in the detection of activity and development of inhibitors of rhomboid proteases.
Keywords: enzyme kinetics, enzyme mechanism, fluorescence resonance energy transfer (FRET), intramembrane proteolysis, membrane reconstitution, rhomboid protease, substrate specificity, transmembrane domain
Introduction
Rhomboid intramembrane proteases are evolutionarily widespread and regulate important biological processes including growth factor secretion (1, 2), mitochondrial dynamics (3), invasion of the malaria parasite (4), and membrane protein quality control (5). Rhomboid proteases are increasingly being explored as potential drug targets (6–9), but their selective and potent inhibitors are lacking (reviewed in Ref. 10). Rhomboid inhibitor discovery and development are complicated by the lack of widely usable and easily modifiable in vitro activity assays.
Rhomboid activity assays have traditionally relied on recombinant transmembrane protein substrates and gel-based readouts, but such assays are unsuitable for high-throughput screening. A fluorogenic substrate for the Providencia stuartii rhomboid protease AarA lacking most of the transmembrane domain of the parent substrate Gurken is cleaved very poorly by other rhomboids including the main model rhomboid protease GlpG of Escherichia coli (11). Other published variants of fluorogenic substrates can be used only in liposomes (12) or involve large fluorescent protein moieties making them dependent on expression in a biological system and photochemically less variable (13), which may be important for high-throughput screening of compound libraries where bright red-shifted fluorophores are preferred (14). Moreover, each of the described rhomboid substrates has been used only with one or two related rhomboid proteases, and a strategy to design widely usable or specific substrates has been lacking. Other types of activity assays employing MALDI mass spectrometry (15) and fluorescence polarization (16) have been reported, but MALDI is a low-throughput method that requires sophisticated instrumentation, and fluorescence polarization assays are based on competition of small molecular activity probes with inhibitors and are prone to detergent artifacts (16), making both of these methods unfit for routine kinetics measurements or high-throughput screening.
In view of these limitations, we have sought to develop a robust fluorogenic transmembrane peptide substrate platform for continuous activity assays that would capture all the native enzyme-substrate interactions, be applicable to both the detergent micelle system and liposomes, and would be easily adaptable to diverse rhomboid proteases. Because solid phase synthesis of transmembrane peptides and their purification are non-trivial, and their solution behavior often unpredictable, we place emphasis on choosing a robust system and characterizing it thoroughly, and present a generalizable framework for rhomboid substrate design.
Results and Discussion
LacYTM2 Is a Widely Accepted Rhomboid Substrate
To identify a substrate widely accepted by diverse rhomboid proteases, we have measured the efficiency of cleavage of four common model rhomboid substrate transmembrane domains (P. stuartii TatA, Drosophila melanogaster Gurken and Spitz, and E. coli LacYTM2) embedded in a chimeric construct by four unrelated rhomboid proteases (E. coli GlpG, Bacillus subtilis YqgP, P. stuartii AarA, and Bacteroides thetaiotaomicron rhomboid 3 (BtioR3)) (Fig. 1A). Comparison of the efficiencies of cleavage (molar catalytic activities) revealed that the substrate containing the second transmembrane (TM)8 helix of E. coli LacY protein (LacYTM2) (17) was the most “promiscuous” substrate.
FIGURE 1.

Identification of a widely accepted transmembrane substrate for rhomboid proteases. A, comparison of cleavage efficiency of model substrates LacYTM2, Gurken, TatA, and Spitz by bacterial rhomboid proteases GlpG (E. coli), AarA (P. stuartii), YqgP (B. subtilis), and BtioR3 (B. thetaiotaomicron) in vitro. Equal concentrations of purified recombinant substrates were exposed to purified recombinant rhomboid proteases. Cleavage products were separated by SDS-PAGE, stained, and quantified densitometrically to determine initial reaction rates, which were converted to molar catalytic activities to allow comparisons. Displayed values are representative of two independent experiments. B, cleavage of synthetic LacYTM2 transmembrane peptide KSp31 by GlpG. Purified synthetic peptide KSp31 was incubated with purified recombinant GlpG or its inactive mutant S201T in the presence of 0.05% (w/v) DDM, and the reaction mixtures were analyzed by MALDI mass spectrometry. The theoretical molecular masses of the expected cleavage products at the native cleavage site are denoted below the peptide sequence, and unambiguously match those experimentally determined and displayed in the mass spectra. The star-marked peak with molecular mass of 1893.3 is an unidentified minor contaminant in the preparation of KSp31. C, monitoring of cleavage of peptide substrate KSp31 by rhomboid protease GlpG using CE. The N-terminal cleavage product (P) of KSp31 was separated by free-flow CE in the background electrolyte composed of 100 mm H3PO4 and 69 mm Tris, pH 2.5, in bare fused silica capillary at separation voltage +25 kV. Samples for CE were prepared by mixing 20 μl of reaction mixture at selected reaction times (0–90 min) with 2 μl of 2.2 mm tyramine (T) as an internal standard. Samples were injected into the capillary by 20 mbar pressure for 10 s. Quantitative analysis was based on the ratio of corrected (migration time normalized) peak areas of peptides of interest and the internal standard. Analyses were performed in triplicate. P, cleaved N-terminal peptide; X, system peak. D, the importance of the transmembrane domain of the substrate for its recognition and cleavage by rhomboid. A series of synthetic peptides covering LacYTM2 with progressive truncations of its transmembrane domain from the C terminus was exposed to GlpG and initial rates of cleavage were quantified by capillary electrophoresis as denoted in panel C.
Although it is well accepted that the region around the scissile bond, mainly P4 to P2′, is key for the turnover efficiency of rhomboid substrates (12, 18), the role of the TM domain of the substrate for recognition and catalysis by rhomboid is less well understood. We have thus next evaluated the importance of the transmembrane region of LacYTM2 for the recognition by E. coli GlpG, the main model rhomboid protease, by synthesizing a peptide covering the whole transmembrane region and adjacent juxtamembrane segments of LacYTM2, and a series of its C terminally truncated variants. The full-length LacYTM2 transmembrane peptide KSp31 was cleaved by GlpG efficiently and highly specifically at the expected Ser-Asp cleavage site (Fig. 1B). The kinetics of cleavage were monitored by capillary electrophoresis (Fig. 1C). The cleavage rate decreased significantly upon truncating the TM helix of LacYTM2 peptide by more than 5 amino acids from the C terminus (Fig. 1D), suggesting that most of the TM domain of the substrate is important for the interaction with and recognition by rhomboid. Thus, to develop a widely accepted fluorogenic substrate that would faithfully mimic all the relevant enzyme-substrate interactions including the intramembrane ones, we have used the full-length LacYTM2 transmembrane domain peptide KSp31 as a starting point.
Fluorogenic Transmembrane Peptide Substrate Based on LacYTM2, Basic Properties
To generate a fluorogenic variant of the LacYTM2 peptide, we have replaced the P5 and P4′ positions in KSp31 by Glu-EDANS and Lys-DABCYL to yield KSp35 (Fig. 2A). Previously published mutagenic analyses show that these positions are not critical for recognition by rhomboid (18, 19), and they are sufficiently close for Förster resonance energy transfer (FRET) to occur. The KSp35 peptide was soluble up to 500 μm (Fig. 2B) in frequently used detergents at 16 mm decyl maltoside (DM), nonyl glucoside (NG), and dodecyl maltoside (DDM). At a total DDM concentration of 16 mm (0.82%(w/v)), the concentration of micelles is about 110 μm, suggesting a partitioning ratio of more than 1 molecule of the substrate per micelle. When DDM was kept at only 1 mm (0.05% (w/v)) total concentration, which yields about 6–10 μm micelles, the solubility of KSp35 became limited to about 100 μm (Fig. 2B), indicating that the upper limit of the partitioning ratio is about 10–20 molecules of KSp35 per DDM micelle. The solubility of KSp35 in the absence of detergent was negligible (not shown). Circular dichroism of KSp35 in 0.5% (w/v) DDM (Fig. 2C) showed a significant content of α-helical structure (61 ± 18%), which is consistent with the transmembrane character of the peptide and comparable with the helical content of the parent peptide KSp31 (54 ± 15%). Cleavage of KSp35 by GlpG occurred at the expected cleavage site (Fig. 2D), and was accompanied by an increase in fluorescence at 495 nm (Fig. 2E), demonstrating that FRET between the donor and acceptor is occurring in the uncleaved peptide. Collectively, the above results show that KSp35 is a realistic model reflecting all the important interactions between a rhomboid protease and its transmembrane substrate.
FIGURE 2.

Fluorogenic transmembrane peptide substrate based on LacYTM2. A, fluorogenic variant of the LacYTM2 transmembrane helix-derived peptide (KSp31) with the P5 and P4′ positions replaced by Glu-EDANS and Lys-DABCYL, respectively, yielding fluorogenic substrate KSp35. B, solubility of KSp35 in 16 mm detergents DDM, DM, and nonyl glucoside (NG) and at 1 mm DDM. Note that the concentration of DDM micelles is about 100 μm at 16 mm DDM and about 10 μm at 1 mm DDM. The peptide was dissolved to the indicated concentration by dilution from a 10 mm stock solution in DMSO, and after a 2-h incubation at 37 °C the solution was centrifuged at 21,130 × g for 20 min. The absorbance of the supernatant at 455 nm indicated the concentration of the chromophore in solution. C, circular dichroism spectra of LacYTM2-derived transmembrane peptide KSp31 and its fluorogenic variant KSp35 in detergent micelles. Peptides were reconstituted into 0.5% (w/v) DDM to 135 μm (KSp31) and 82 μm (KSp35) concentrations. The spectra show similarly significant helical content for both peptides. D, identification of the cleavage site in KSp35 by GlpG. Purified 95 μm KSp35 was incubated with 26 μm GlpG for 20 h and analyzed by MALDI. The red peak of the mass of 2993.7 corresponds well to the expected size of the C-terminal cleavage product of 2990.690. The second peak lower by 130 Da is visible in both the blue and red traces is probably a deletion product of chemical synthesis lacking a C-terminal lysine. This variant has proven difficult to purify away, but it is cleaved by GlpG and probably does not influence the kinetics properties of the substrate significantly (see Fig. 1D). E, excitation and emission spectra of KSp35 and their change upon cleavage by rhomboid GlpG measured in detergent micelles. The spectra of 10 μm KSp35 substrate in reaction buffer (20 mm HEPES, pH 7.4, 150 mm NaCl, 0.05% (w/v) DDM, 10% (v/v) DMSO) were measured at 37 °C. Excitation wavelengths ranged from 235 to 435 nm with a 10-nm increment and the emission was measured at 493 nm. The emission wavelengths ranged from 365 to 595 nm with a 10-nm increment and excitation at 335 nm.
Kinetic Characterization of the LacYTM2-based Substrate KSp35 in Detergent Micelle System
In the detergent-solubilized state, most commonly used to study the biochemistry of intramembrane proteolysis, the reaction catalyzed by rhomboid protease occurs in detergent micelles due to the hydrophobicity of both enzyme and substrate. The system is thus microheterogeneous, the effective concentrations of the reactants depend on the volume of the micellar milieu and on the partitioning of reaction components between free solution and the micelles. To characterize the kinetic behavior of the new fluorogenic transmembrane substrates in light of these features of the micellar system, steady-state kinetics was measured with 10 μm substrate, 0.4 μm enzyme, and 0.05% (w/v) DDM, always keeping the concentrations of two components constant and varying the third one around the stated values. At 0.05% (w/v) DDM, the concentration of detergent monomers is 980 μm and micelle concentration about 6–10 μm, calculated assuming critical micellar concentration (CMC) of 0.17 mm (20) and aggregation number between 78 and 149 (20). The molar ratio of enzyme:substrate:micelles is thus 4:100:60–100. In these conditions, assuming that all the reaction partners are evenly distributed among micelles, the average number of substrate molecules per micelle is about 1.5, and only up to 4% of micelles carry an enzyme molecule (micelles containing more than one enzyme molecule are strongly improbable).
The cleavage reactions were started by either mixing two preheated solutions containing substrate or enzyme preincubated with detergent, or adding the DMSO-dissolved substrate into the rest of the preheated reaction mixture. In either case, progress curves are linear from the beginning, which implies that the redistribution of the adsorbed molecules among the micelles is significantly faster than substrate cleavage itself. In accordance with this, the reaction rate is proportional to enzyme concentration within the 0–0.6 μm range (Fig. 3A). Within this concentration range, few enzyme molecules are randomly distributed among many more micelles, providing in principle equal conditions for each enzyme molecule. A similar principle can also explain the observation that the dependence of the reaction rate on substrate concentration is linear in the 0–4 μm range (Fig. 3B). At the upper limit of 4 μm substrate, all micelles can be populated by one (or less likely more) substrate molecule, the linear dependence, furthermore, suggests that this substrate concentration is still below the apparent Michaelis constant of this process.
FIGURE 3.

Kinetic characterization of fluorogenic transmembrane peptide substrate KSp35 in the detergent micelle system. A, dependence of the initial reaction rate on enzyme concentration. The fluorogenic substrate KSp35 (10 μm) was incubated with varying concentrations of GlpG in a reaction buffer composed of 20 mm HEPES, pH 7.4, 150 mm NaCl, 0.05% (w/v) DDM, and 10% (v/v) DMSO, and initial reaction rates were measured by following fluorescence at 493 nm. The displayed values are means from duplicate measurements with 2 × S.D. B, dependence of the initial reaction rate on substrate concentration. The rhomboid protease GlpG (0.4 μm) was incubated with varying concentrations of the fluorogenic substrate KSp35 in a reaction buffer composed of 20 mm HEPES, pH 7.4, 150 mm NaCl, 0.05% (w/v) DDM, 10% (v/v) DMSO, and the initial reaction rates were measured by following fluorescence at 493 nm. Representative values from one of three independent experiments are shown. C, dependence of the initial reaction rate on detergent concentration (solid circles, left and lower axes). The fluorogenic substrate KSp35 (10 μm) was incubated with 0.4 μm GlpG at varying concentrations of DDM in a reaction buffer composed of 20 mm HEPES, pH 7.4, 150 mm NaCl, 10% (v/v) DMSO, and initial reaction rates were measured by following fluorescence at 493 nm. Representative values from one of three independent experiments are shown. The open circles (right and upper axes) represent the same plot at the logarithmic scale. When this plot is fitted by second-order polynomial, the equation y = −0.1436x2 − 0.3906x + 2.8852 is obtained, the derivative of which, y′ = −0.2872x − 0.3906, is equal to the power of DDM concentration with which the reaction rate decreases. For high DDM concentrations the derivative tends to −1 (for x = 2, y′ = −0.965), whereas for lower DDM concentrations the absolute value of the power decreases (for x = 0, y′ = −0.3906). D, overall secondary structure of GlpG is not affected by high concentrations of DDM. CD spectra of GlpG at 0.05, 0.5, and 5% (w/v) (98 mm) DDM were recorded and show no variation in the secondary structure content of GlpG depending on DDM concentration. E, the pH dependence of GlpG activity on the LacYTM2-derived chimeric substrate MBP-LacYTM2-Trx. The substrate (2 μm) was incubated with 0.1 μm GlpG in a broad pH range buffer (38) composed of 40 mm H3PO4, 40 mm CH3COOH, and 40 mm H3BO3 adjusted to pH values between 2 and 12, and initial reaction rates were measured by SDS-PAGE and densitometry as described under “Experimental Procedures.” F, the pH dependence of cleavage of the fluorogenic LacYTM2-derived substrate KSp35 by GlpG. The substrate (10 μm) was incubated with 0.4 μm GlpG in a broad pH range buffer (38) composed of 40 mm H3PO4, 40 mm CH3COOH, and 40 mm H3BO3 adjusted to pH values between 2 and 12, and initial reaction rates were measured by recording fluorescence at 493 nm. G, selectivity of the fluorogenic substrate KSp35 for diverse bacterial rhomboid proteases. The purified recombinant rhomboid proteases GlpG, AarA, YqgP (all at 0.4 μm), and BtioR3 (at 0.04 μm) were incubated with 10 μm KSp35 in a reaction buffer composed of 20 mm HEPES, pH 7.4, 150 mm NaCl, 0.05% (w/v) DDM, and 10% (v/v) DMSO, and progress curves were measured by recording the increase in fluorescence at 493 nm.
An important phenomenon is observed when the dependence of the initial rate on detergent concentration is measured. At concentrations above the CMC, the reaction rate rapidly decreases as DDM concentration grows (Fig. 3C), without an obvious impact on the secondary structure content of GlpG (Fig. 3D), suggesting that the effect is caused primarily by the increase in the volume of the micellar phase and consequent decrease of the effective concentrations of both substrate and enzyme. Indeed, mathematical consideration suggests that when substrate and enzyme concentrations are significantly lower than the concentration of micelles (i.e. at high DDM concentrations), the probability of location of a substrate molecule on the same micelle as the enzyme molecule is inversely proportional to the concentration of DDM. Under these conditions, the fraction of substrate-occupied micelles, fSM, is equal to the ratio of the numbers of substrate molecules, n(S), and micelles n(M).
| (Eq. 1) |
The mean number of micelles occupied by both the enzyme and substrate molecules, n(ESM), is then given by this fraction multiplied by the number of enzyme molecules n(E).
| (Eq. 2) |
Hence, when the DDM concentration is increased at constant n(S) and n(E), then n(ESM) reflecting the reaction rate decreases in accord with the growing value of n(M). This causes the proportional decrease of the reaction rate (in other words, the reaction rate is proportional to [DDM]−1). To inspect whether this model is correct, one can conveniently determine the power of the measured rate dependence on DDM concentration by taking a logarithm of the data from Fig. 3C (log an = n × log a). The logarithmic plot (Fig. 3C, open circles, right and upper axes) can be satisfyingly (R2 = 0.9974) fitted by a second-order polynomial, yielding equation: y = −0.1436x2 − 0.3906x + 2.8852, whose derivative y′ = −0.2872x − 0.3906 indicates the power of DDM concentration on which the reaction rate depends. This analysis shows that for high DDM concentrations the derivative indeed tends to −1 (for x = 2, y′ = −0.965; thus rate ∼[DDM]−1), which is in accordance with the above assumption, whereas for the lower end of DDM concentrations the absolute value of the power decreases (for x = 0, y′ = −0.3906; thus rate ∼[DDM]−0.4). This is consistent with a model that upon decreasing the detergent concentration (while still being above the CMC), the density of the adsorbed molecules in the micellar phase increases, whereas total concentration of micelles decreases, which leads to less frequent collisions between them and thus less effective redistribution of the adsorbed molecules among the micelles. Possibly, the redistribution efficiency might also be insufficient because of the higher reaction rate caused by the higher reactant concentrations.
Although the reaction kinetics of intramembrane proteases in liposomes has been described in terms of interfacial kinetics (12, 21), that is, expressing the kinetic constants in relationship to the volume or molar fraction of the lipidic phase, (22, 23), the kinetic effects related to the reaction occurring in detergent micelles have surprisingly not yet been considered in enzyme kinetics studies on rhomboid proteases (12, 13) nor other intramembrane proteases, yet they are evidently important for the interpretation of kinetics measurements. Our data show that for reliable and meaningful measurement of apparent Michaelis-Menten kinetics parameters, the micelle concentration must not be limiting the solubility of the substrate, and the detergent concentration must be kept constant. The latter point also means that having a stock solution of the substrate dissolved in detergent (at a higher concentration than intended in the reaction mixture, which frequently can occur during purification and concentration) may lead to underestimation of reaction rates at high substrate concentrations due to a possibly significant increase of detergent concentration in the final reaction mixture, as shown in Fig. 3C. This could result in pseudo-Michaelis kinetics and yield falsely low Km values. Practical implications are that 1) exact detergent concentrations must be known in any kinetics measurements, and 2) it is advantageous to have the substrate stock solution dissolved in a detergent-free medium or at a detergent concentration lower or equal to that used in the final assay buffer. The transmembrane substrates presented in this article, generated by chemical synthesis, are in principle avoiding this problem, because their stock solutions are detergent-free dissolved in anhydrous dimethyl sulfoxide. Alternatively, they can be reconstituted into a detergent of choice via disaggregation in hexafluoroisopropanol, as described by Deber et al. (24).
The pH dependence of cleavage rate of the unmodified LacYTM2 transmembrane segment in the context of an MBP-thioredoxin fusion protein shows a relatively broad maximum around pH 9, with substantial activity of GlpG between pH 6 and 11 and negligible activity below pH 4 and at pH 12 (Fig. 3E), which is largely in agreement with previous studies (12, 13). The dependence of the cleavage rate of KSp35 on pH also shows that GlpG is completely inactive at pH values below and up to 4, but the initial reaction rate of KSp35 cleavage then appears to grow up to pH 12 (Fig. 3F). This effect cannot be ascribed to the pH-dependent change of EDANS fluorescence (data not shown), and could possibly be due to effects of pH on the conformational dynamics of KSp35. However, this is not a concern because in most cases measurements are performed at a physiologicaly relevant pH near neutral. The apparent catalytic efficiency kcat/Km of GlpG against KSp35 measured at pH 7.4 and 0.05%(w/v) DDM is (2.0 ± 0.5) × 10−3 min−1 μm−1, which is comparable with the values reported for the TatA substrate by Dickey et al. (12) and Arutyunova et al. (13) obtained in similar conditions. Importantly, the LacYTM2-derived fluorogenic peptide substrate KSp35 is cleaved efficiently by unrelated recombinantly purified bacterial rhomboids GlpG, AarA, and BtioR3, and modestly by YqgP (Fig. 3G), which demonstrates its wide usability, surpassing any other currently available rhomboid substrates.
Use of the Transmembrane Peptide Substrate in Liposomes
Because the natural environment of rhomboid proteases is the lipid membrane, we next tested whether the fluorogenic peptide substrate KSp35 can also be used in liposomes. We co-reconstituted KSp35 with GlpG or its inactive mutant S201A/H254A at pH 4 into large unilamellar vesicles (LUVs) formed from E. coli polar lipid extract, and confirmed the composition of the resulting proteoliposomes by SDS-PAGE (Fig. 4A). Negative stain transmission electron microscopy showed that both empty LUVs and proteoliposomes containing KSp35 in the presence or absence of GlpG or its inactive mutant S201A/H254A had similar morphology and size distribution both at pH 7 and 4 (Fig. 4B). The CD spectrum of LUV-reconstituted KSp35 showed helicity of 50 ± 14% (Fig. 4C), which is consistent with its transmembrane helix prediction. GlpG is inactive at pH 4 (Fig. 3, E and F), and, consistently, fluorescence of proteoliposomes containing KSp35 and GlpG at pH 4 was at a constant background level (Fig. 4E). Upon neutralization to pH 7.4, time-dependent increase of fluorescence at 495 nm was observed in the presence of wild type GlpG but not in the presence of its active-site mutant S201A/H254A (Fig. 4D). These results collectively demonstrate that the LacYTM2-based fluorogenic transmembrane substrate KSp35 is widely usable both in detergent micelles or liposomes and with diverse rhomboid proteases.
FIGURE 4.

The use of the transmembrane peptide substrate in liposomes. A, KSp35 was reconstituted into liposomes (LUVs) formed from E. coli polar lipid extract in the presence of GlpG or its inactive mutant S201A at pH 4.0. The resulting large unilamellar vesicles were analyzed by SDS-PAGE. B, the shape, lamellarity, and approximate size distribution of the KSp35+GlpG containing proteoliposomes formed at pH 4.0 were characterized by transmission electron microscopy. C, the integration of KSp35 into liposomes and its secondary structure content were analyzed by electronic CD. The substrate KSp35 (3 μm) was reconstituted with 2 mg/ml of E. coli polar lipid extract yielding an approximate peptide:lipid weight ratio of 1:500. D, activity of GlpG in liposomes detected by the KSp35 fluorogenic substrate. The substrate was co-reconstituted with wild type GlpG or its S201A/H254A mutant in a 30:1 molar ratio into LUVs made of E. coli polar lipid extract at pH 4.0, proteoliposomes were collected by ultracentrifugation and resuspended in 10 mm HEPES, 150 mm NaCl, pH 7.4, to start the cleavage reaction, which was then followed by measuring fluorescence at 493 nm. E, wild type GlpG or its H150A/H254A mutant were co-reconstituted with the substrate KSp35 in a 30:1 molar ratio into LUVs made of E. coli polar lipid extract at pH 4.0, proteoliposomes were collected by ultracentrifugation, resuspended in 50 mm sodium acetate, 150 mm NaCl, pH 4.0, and fluorescence was followed at 493 nm.
A Red-shifted Variant of the Fluorogenic Transmembrane Substrate for Rhomboids
Large compound libraries for high-throughput screening can often contain compounds that absorb in the UV region (14), and fluorogenic substrates operating at red-shifted wavelengths are less affected by such compound interference. Because EDANS is excited in the UV region, and is thus prone to interference in library screening, we have modified the LacYTM2 peptide backbone by instead attaching the red-shifted TAMRA fluorophore to a Lys introduced into the P5 position and a compatible dark quencher QXL610 to a Cys introduced into the P4′ position (Fig. 5A) to yield KSp76. This red-shifted fluorogenic substrate is cleaved by several bacterial rhomboid proteases with efficiencies similar to its UV variant KSp35. The apparent catalytic efficiency kcat/Km of GlpG cleaving KSp76 is (1.6 ± 0.5) × 10−3 min−1 μm−1, which is similar to the EDANS variant KSp35 ((2.0 ± 0.5) × 10−3 min−1 μm−1) under identical reaction conditions within experimental error (Fig. 6C). The utility of this red-shifted variant of the LacYTM2 substrate is demonstrated by measuring the inhibition curves of chloromethylketone ISKA-cmk (19), β-lactam L42 (11), and isocoumarin S037 (25, 26). Using a 60-min enzyme + inhibitor preincubation time, the measurements yielded apparent IC50 values of 370 ± 38, 12.4 ± 1.6, and 0.64 ± 0.08 μm, respectively (Fig. 5B), which are largely in agreement with published values measured in other assay systems and otherwise comparable conditions (11, 15, 19).
FIGURE 5.
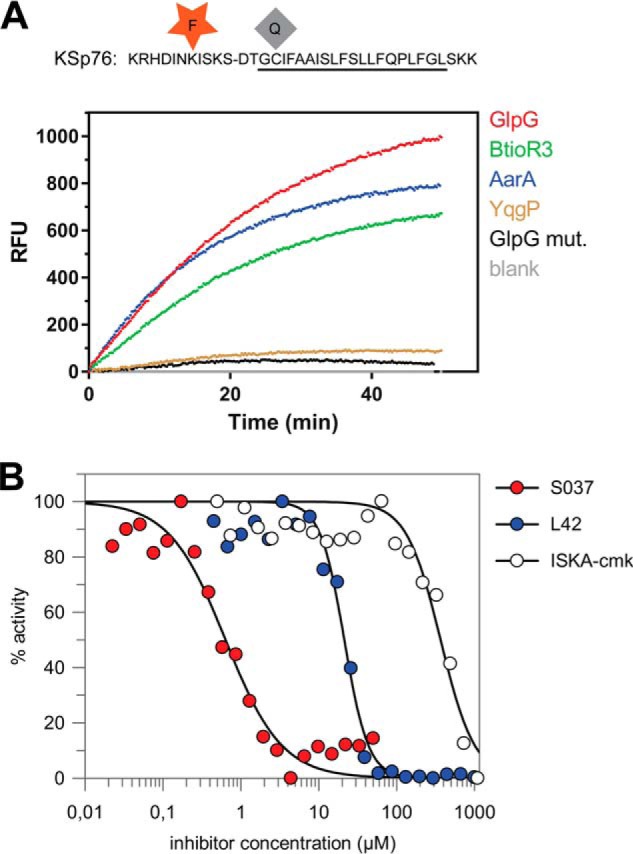
Red-shifted variant of the LacYTM2-based fluorogenic substrate. A, modification of Lys in the P5 position of KSp31 by the red-shifted TAMRA fluorophore and P4′ Cys by a dark quencher QXL610 yields highly fluorogenic substrate KSp76 that is efficiently cleaved by rhomboid proteases GlpG, AarA, YqgP, and BtioR3 at identical concentrations to those used in Fig. 3G. Excitation wavelength was 553 nm, and emission was followed at 583 nm. B, the red-shifted fluorogenic substrate KSp76 allows measurement of inhibition by compounds that absorb in the UV region, such as isocoumarin, and is thus suitable for high-throughput screening. The dose-response curves of the chloromethylketone ISKAcmk, β-lactam L42, and isocoumarin S037 were measured after a 60-min preincubation of enzyme with inhibitor. The curves were fitted in GraFit 7 to yield apparent IC50 values.
FIGURE 6.

The effect of non-prime side substitutions on the catalytic parameters and selectivity of rhomboid substrates. A, preferred amino acids in the P5 to P1 positions of the LacYTM2 transmembrane substrate improve its cleavage by GlpG. The LacYTM2 embedded in the MBP-thioredoxin chimera (18) was point-mutated in the P5 to P1 positions according to the sequence preferences of E. coli GlpG (19). The recombinant substrates were expressed in E. coli ΔglpG, purified, and molar catalytic activity of GlpG in cleaving each of the substrates was determined using gel-based assay (see “Experimental Procedures” for details). The concentration of substrate was always 1.47 μm, concentration of DDM was 0.5%(w/v), the concentration of GlpG was 0.8 μm for wild type substrate (HISKS). and for the RISKS, HVSKS, and HISHS mutants the concentration was 0.08 μm for the HISKA mutant and 0.016 μm for the HIRKS and RVRHA variants (to ensure reliable measurement of the initial reaction rate). Representative values from one of three independent experiments are shown. B, the effects of the preferred amino acids in the P5 to P1 region of LacYTM2 on the steady-state level of cleavage by GlpG in biological membranes in vivo. Plasmids encoding individual mutant versions of the chimeric mutant LacYTM2 substrates described above were transformed into E. coli MC4100 expressing endogenous GlpG, and 2 h after induction of expression of the substrates, the cell lysates were analyzed by immunoblotting using antibody against His tag, located at the C terminus of the constructs. Detection by near-infrared laser scanning, exhibiting linearity over 6 orders of magnitude, enabled reliable quantitation. Integration of product and substrate band intensities yielded steady-state substrate conversion values that are listed below the image. A representative experiment is displayed. C, apparent kinetic parameters of fluorogenic rhomboid substrates derived from LacYTM2. Initial reaction rates at very low substrate concentrations were used to calculate catalytic efficiency values (kcat/Km) of substrates KSp35, KSp64, and Ksp76 cleaved by GlpG at 0.5% (w/v) DDM. The reaction buffer was 20 mm HEPES, pH 7.4, 150 mm NaCl, 10% (v/v) DMSO, enzyme concentration was 0.4 μm, and substrate concentration ranged from 0.5 to 20 μm. Note that a mere optimization of the P5 to P1 region of the substrate increases the catalytic efficiency (kcat/Km) of its cleavage by GlpG by 23-fold. D, influence of the optimization of the P5 to P1 region on the selectivity of a transmembrane substrate for rhomboids. KSp76 underwent cleavage by rhomboid proteases GlpG, AarA, YqgP, and BtioR3 at the same concentrations as described in the legends to Figs. 3G and 5A. Note that optimization of the P5 to P1 region of the substrate increases the selectivity for GlpG dramatically.
Efficiency and Selectivity of the Substrates Can Be Tuned by Varying Their Non-prime Side Amino Acid Sequence
One of the problems with current rhomboid protease assays is that there has been little rationale about how to modify the substrates to improve their kinetic properties and adapt them for different rhomboid proteases. Recent enzymatic analyses (12, 18) have shown that the region between the P4 and P2′ residues determines the kcat of the cleavage reaction, suggesting that selective substrates for rhomboids could be designed by modifying the P4 to P2′ region appropriately. A recent mutagenic study of the TatA substrate and structural analysis of a derived rhomboid·substrate-peptide complex revealed amino acids at the P5 to P1 positions of TatA that are preferred by GlpG (19). We tested the impact of these substitutions in the context of the LacYTM2 substrate.
Although single mutations of the P5 amino acid to the preferred Arg, P4 amino acid to Val, and P2 amino acid to His did not improve the cleavage of the purified recombinant MBP-LacYTM2-Trx substrate in vitro, mutation of the P1 amino acid to Ala improved the cleavage of mutant 7-fold, and mutation of the P3 residue to Arg improved the cleavage of mutant 16-fold (Fig. 6A). Combining all five mutations yielded a mutant substrate (RVRHA) that was cleaved 64-fold better than the wild type substrate (Fig. 6A), which shows that the effects of the preferred substitutions are additive. When analyzed for cleavage in vivo, it turns out that already the wild type MBP-LacYTM2-Trx substrate is such a good substrate of GlpG that it is turned over from 94% (Fig. 6B). The effects of the preferred P5 to P1 mutations thus cannot be assessed in this context as they all exhibit similarly high steady-state turnover (Fig. 6B).
To test this effect in our fluorogenic substrates, we have modified the TAMRA-based LacYTM2-derived fluorogenic substrate by changing the P5 to P1 segment from HISKS to RVRHA to yield KSp64, and compared the kinetic properties of both substrates. The analysis revealed that catalytic efficiency kcat/Km of GlpG cleaving KSp64 is (3.7 ± 0.4) × 10−2 min−1 μm−1, which is 23-fold higher than that of the original red-shifted LacYTM2 substrate KSp76 ((1.6 ± 0.5) × 10−3 min−1 μm−1) (Fig. 6C). The impact of the modifications of the P5 to P1 region on selectivity against other bacterial rhomboid proteases is particularly striking (Fig. 6D), with the initial reaction rate of KSp64 cleavage by GlpG being about 50-fold higher than that of AarA (measured from data displayed in Fig. 6D) and even higher for the other tested rhomboid proteases, revealing a straightforward strategy for designing selective rhomboid substrates.
In summary, we report novel sensitive versatile fluorogenic transmembrane peptide substrates for rhomboid intramembrane proteases that are usable both in detergent micelles and liposomes, are cleaved by diverse rhomboid proteases, and contain a red-shifted fluorophore suitable for high-throughput screening assays. Furthermore, we provide a strategy how to adapt these substrates to individual rhomboid proteases by modifying their P5 to P1 residues, and we demonstrate that controlling the detergent concentration is important for obtaining accurate kinetic data. We expect that the substrates we describe and sequence variants thereof will enable facile detection of activity and development of inhibitors of rhomboid proteases.
Experimental Procedures
General Biochemicals
Lipids were from Avanti Polar Lipids, detergents from Anatrace, buffers and other biochemicals were from Sigma or other suppliers as specified below.
DNA Constructs and Cloning
The expression constructs for rhomboid proteases GlpG, YqgP, and AarA and chimeric MBP-TMD-Trx substrate constructs where TMD = LacYTM2, Gurken, TatA, or Spitz as described previously (18). The expression construct for rhomboid protease BtioR3 was generated by PCR amplification of the entire ORF encoding the Q8A3X2 (Uniprot ID) protein from B. thetaiotaomicron genomic DNA (purchased from ATCC), and its cloning as a C terminally His-tagged construct into pET25b+M as described previously (27). Mutations of the TatA and LacYTM2 recognition motif in the MBP-TMD-Trx construct were generated by overlap assembly PCR (28) and isothermal assembly (29). All constructs were verified by DNA sequencing.
Chemical Synthesis
All reagents were acquired from commercial sources and used without purification. Protected amino acids and amino acid derivatives were purchased from Iris Biotech (Marktredwitz, Germany). Trimellitic anhydride and 3-dimethylaminophenol were from Sigma, QXL610 vinylsulfone was from AnaSpec (Fremont, CA), and N-(9-fluorenyl)methoxycarbonyl (Fmoc)-Glu(EDANS)-OH from Merck KGaA (Darmstadt, Germany). The detailed synthetic procedures, analytical methods, and compound characterization data are included in the supporting information.
Protein Expression and Purification
Bacterial rhomboid proteases AarA, GlpG, BtioR3, and YqgP and the active site mutant GlpG.S201A were overexpressed in E. coli C41(DE3) (30) as full-length, C terminally His-tagged proteins from a modified pET25b+ vector (27). The cultures were grown at 37 °C in LB medium to A600 of 0.4 and induced by 1 mm isopropyl 1-thio-β-d-galactopyranoside. The expression was continued overnight at 20 °C. Cells were harvested, resuspended in buffer A (25 mm HEPES, pH 7.4, 100 mm NaCl, 10%(v/v) glycerol, 1 mm PMSF), and lysed by 2 to 3 passes through Avestin EmulsiFlex-C3. Cell debris was removed by a low-speed centrifugation. Cellular membranes were isolated by a 2-h centrifugation at 100,000 × g and were solubilized in 1.5%(w/v) DDM (solubilization grade, Anatrace) in Buffer B (25 mm HEPES, pH 7.4, 300 mm NaCl, 10%(v/v) glycerol, 10 mm imidazole, EDTA-free Complete Protease Inhibitor mixture (Roche Applied Science)) at room temperature for 1 h. Solubilized proteins were isolated by centrifugation at 100,000 × g for 30 min and loaded onto nickel-nitrilotriacetic acid HiTrap IMAC HP 1-ml columns (GE Healthcare). Nonspecifically bound proteins were washed off with Buffer C (25 mm HEPES, pH 7.4, 300 mm NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM) containing 10, 50, and 125 mm imidazole. The protein was eluted with Buffer C containing 250 to 500 mm imidazole. The peak fractions were buffer exchanged into 25 mm HEPES, pH 7.4, 150 mm NaCl, 10% (v/v) glycerol, and 0.05% (w/v) DDM on a HiPrep 26/10 desalting column (GE Healthcare). If needed, proteins were concentrated using Vivaspin ultrafiltration spin cells with 30-kDa MWCO. Protein concentration was determined from absorbance at 280 nm, and the final concentration of DDM was determined as described (31).
Capillary Electrophoresis (CE)
Analyses of standard peptides and enzymatically cleaved peptide substrates were performed on an Agilent CE 7100 instrument (Agilent, Waldbronn, Germany) equipped with photodiode array UV-visible detector operating in the 190–600 nm range. Electropherograms were acquired at 192, 205, and 214 nm and absorbance data at 192 nm were selected for quantitative evaluation due to the highest signal to noise ratio. CE analyses were carried out in a bare fused silica capillary with polyimide outer coating (internal diameter 50 μm, outer diameter 375 μm, effective length to the detector 40 cm, total length 48.5 cm, supplied by Polymicro Technologies, Phoenix, AZ). Peptides were analyzed as cations in acidic background electrolyte (BGE) composed of 100 mm H3PO4, 69 mm Tris, pH 2.5. For highly hydrophobic peptides, this BGE was modified by the addition of 0.05% (w/v) DDM. The temperature of the air-cooled capillary was set to 20 °C and the sample carousel was kept at the same temperature using a circulating water bath. Prior to each CE run, the capillary was successively washed with 100 mm sodium dodecyl sulfate, ethanol, 1 m NaOH, water, 1 m HCl, and the BGE, to remove any possible carryover of hydrophobic peptides and detergents from the previous run. All washes were done at 8 bar pressure for 30 s. Peptide standards used for identification of cleavage products were solubilized in DMSO at 1 mm concentration and mixed with 50 mm HEPES buffer containing 0.05% (w/v) DDM, resulting in 50 μm peptide concentration.
The enzymatic cleavage reactions were carried out in 20 mm HEPES, pH 7.4, with 0.05%(w/v) DDM and 10% (v/v) DMSO, with 250 μm peptide substrate and 2.6 μm full-length GlpG at 37 °C. To measure the initial reaction rates, fractions were collected every 15 min for up to 2 h and the reaction was terminated by the addition of 10 mm HCl. Samples for CE were prepared by mixing 20 μl of peptide solutions with 2 μl of 2.2 mm tyramine (internal standard for quantitative analysis). Sample solutions were injected into the capillary by 20 mbar pressure for 10 s. Separations were performed at +25 kV (anode at the capillary injection end). The electrode vessels were replenished with fresh BGE after each run. All analyses were performed in triplicate. Quantitative analysis was based on the ratio of corrected (migration time normalized) peak areas of peptides of interest and the internal standard (tyramine) (32).
Mass Spectrometry
The analysis of enzymatic cleavage products of transmembrane peptides was carried out using MALDI-TOF mass spectrometry on an UltrafleXtremeTM MALDI-TOF/TOF mass spectrometer (Bruker Daltonics, Germany) with α-cyano-4-hydroxycinnamic acid matrix using a thin-layer method (33). For routine quality control during peptide synthesis, mass spectra were acquired on a Waters Micromass ZQ ESCi multimode ionization mass spectrometer, and LTQ Orbitrap XL (Thermo Fisher Scientific) for HR-MS experiments, in both cases using ESI(+) ionization.
Gel-based Assay for Rhomboid Activity
For gel-based assays used in Fig. 1, the purified recombinant full-length maltose-binding protein thioredoxin fusion proteins harboring the transmembrane domains of TatA, LacYTM2, Gurken, and Spitz (18) were used as substrates. The reaction was carried out in 50 mm Tris, pH 7.4, 100 mm NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, and 5 μm substrate. Enzyme concentrations varied to ensure adequate conditions for measurement of initial reaction rates for each enzyme-substrate combination. Time courses were measured by withdrawing 10-μl aliquots from the reaction mixture after 10, 20, 30, 40, 50, 60, and 120 min from the start of the reaction, and stopping the reaction by the addition of SDS-PAGE sample buffer. The reaction mixtures were analyzed by SDS-PAGE, Coomassie staining (InstantBlue, Expedeon, UK), and densitometry as described (19), and initial reaction rates were converted to molar catalytic activities defined as the number of substrate molecules converted by a molecule of the enzyme per unit of time (consistent with the definition by IUPAC (34, 35)). Variations in conditions used for measurements in Fig. 6 are denoted in the figure legend.
The in vivo assay of rhomboid activity was carried out essentially as described (19). Cleavage products were detected by SDS-PAGE and Western blotting using primary anti-penta-His mouse monoclonal antibody (Thermo) and IRDye 800CW goat anti-mouse fluorescent secondary antibody (LiCor). Densitometry was done in ImageStudio software (LiCor) and substrate conversion (α) was calculated from band intensities as ατ = [P]/[S] + [P], where [P] and [S] are product and substrate concentrations at time τ, which are proportional to the fluorescence intensity of the product and substrate bands at time τ, because the monoclonal antibody binds to the substrate or product in a constant molar ratio irrespective of their molecular weights.
Fluorescence Assay for Rhomboid Activity
The fluorescence assay of rhomboid activity was performed at 37 °C in 96-well black HTS plates (Greiner Bio-One). The reaction conditions were typically as follows: 20 mm HEPES, pH 7.4, 150 mm NaCl, 0.05% (w/v) DDM, 12% (v/v) DMSO, and 10 μm fluorogenic peptide substrate in a final volume of 50 μl, unless noted otherwise. Concentrations of stock solutions of peptide substrates and inhibitors (if applicable) were determined by quantitative amino acid analysis. Fluorescence was read continuously in a plate reader (Tecan Infinite M1000). Excitation and emission wavelengths were 335 and 493 nm, respectively, for the EDANS-DABCYL substrate, and 553 and 583 nm for the TAMRA-QXL610 substrates. Data were evaluated in i-Control (Tecan), Excel (Microsoft), GraphPad Prism 7 (GraphPad Software, Inc.), and GraFit 7 (Erithacus Software, Ltd.) software.
Inhibition Assays
The inhibition assay was carried out in 20 mm HEPES, pH 7.4, 150 mm NaCl, 12% (v/v) DMSO, 0.05% (w/v) DDM at 37 °C in 96-well black HTS plates (Greiner Bio-one). Purified recombinant full-length GlpG (0.4 μm) was preincubated with each inhibitor at different concentrations for 1 h at 37 °C. The cleavage reaction was started by adding 10 μm KSp76 and fluorescence was read continuously to measure initial reaction rates as described above.
Reconstitution into Liposomes
E. coli polar lipids (20 mg), with optionally 0.1 mg of Lissamine Rhodamine B-labeled phosphatidylethanolamine (16:0) (Avanti Polar Lipids) added for visibility, were dried in a glass test tube by manual rotation under a nitrogen stream. Residual traces of solvent were removed by overnight incubation in a vacuum chamber (Binder). The resulting lipid film was hydrated in 5 ml of 50 mm acetate, 150 mm NaCl, pH 4.0, by 2 min vortexing followed by a 1-h incubation in a horizontal shaker at 200 rpm and 37 °C, and 3 cycles of freezing in liquid nitrogen and thawing in a 37 °C water bath. The lipid suspension was then extruded through a 200-nm pore membrane by 19 strokes in an Avanti Mini Extruder (Avanti Polar Lipids).
For reconstitution of proteins and peptides into liposomes, these unilamellar LUVs were solubilized in DM to a final ratio of 1.5:1 detergent:lipid, and incubated for 1 h at room temperature under gentle rotation. This mixture was diluted to a final lipid concentration of 2 mg/ml in 50 mm acetate, 150 mm NaCl, pH 4.0, and protein (GlpG or its inactive mutant) dissolved in detergent was added to a final concentration of 8 μg/ml; alternatively, the stock solution of substrate peptide KSp35 in DMSO was diluted to 10 μm. The resulting mixture was incubated at room temperature for 1 h under gentle mixing by inversion. Detergent was removed by overnight dialysis against 500-fold excess of 50 mm acetate, 150 mm NaCl, pH 4, followed by 5 h dialysis against 500-fold excess the same buffer, using 10-kDa MWCO dialysis membranes, which allowed reconstitution of proteoliposomes. These were extruded through 200-nm pore filters 9 times to ensure reproducible size distribution and lamellarity. These final proteoliposomes were harvested by ultracentrifugation (250,000 × g for 1 h at 4 °C), and resuspended in 10 mm HEPES, pH 7.4, 150 mm NaCl to a concentration of about 33 mg/ml of lipids. The morphology and size distribution of proteoliposomes was analyzed by electron microscopy.
Transmission Electron Microscopy
Liposome samples were negatively stained with 2% phosphotungstic acid on carbon-coated electron microscopy grids and analyzed with a JEOL JEM-1011 device at 80 kV beam acceleration voltage.
CD Spectroscopy
Protein and peptide samples were dissolved in 50 mm phosphate buffer at the indicated concentrations and in the presence of detergent as indicated, or reconstituted in LUVs made of E. coli polar lipids and extruded by 100-nm filters to minimize light scattering. Electronic circular dichroism spectra were collected by a Jasco 815 spectrometer (Tokyo, Japan) in the spectral 195–280 nm range using a cylindrical 0.02-cm quartz cell with 0.1-nm step resolution, 5 nm/min scanning speed, 16 s response time, and 1 nm spectral band. After baseline correction, the spectra were expressed as molar ellipticity per residue θ (deg cm2 dmol−1). Numerical analysis of the secondary structure and secondary structure assignment were performed using a CDPro software package and CONTIN program (36, 37).
Author Contributions
K. S. conceived and coordinated the study, designed experiments, and wrote the paper with the input of A. T., M. I., S. S., J. B., J. S., M. R., and V. K. S. S. and P. M. designed and S. S. performed all chemical syntheses. M. R. and V. K. designed and performed all capillary eletrophoresis analyses. M. I. analyzed kinetics data, R. H. performed electron microscopy, and L. B. designed and performed all circular dichroism measurements. J. B. performed and evaluated experiments shown in Fig. 6, A and B. JŠ performed and evaluated experiments shown in Fig. 4, A, D, and E. K. Š designed, performed, and analyzed data shown in Fig. 1A. P. R. and E. P. contributed to experiments shown in Fig. 6, A, B, and D. L. P. established the fluorogenic assay and performed and evaluated experiments shown in Fig. 1D. J. Březinová contributed to all mass spectrometry experiments, and A. T. designed, performed, and evaluated all other kinetics and inhibition measurements that are the basis of this manuscript.
Supplementary Material
Acknowledgments
We thank Steven Verhelst (University of Leuven, Belgium) for his kind gift of isocoumarin S037, Matthew Freeman (Oxford University, United Kingdom) for his kind gift of the inhibitor L42, Zdeněk Voburka and Radko Souček for amino acid analyses, Mirka Blechová for peptide synthesis and purification, and Blanka Collis for critical reading of the manuscript.
This work was supported by EMBO Installation Grant 2329, Ministry of Education, Youth and Sports of the Czech Republic Projects LK11206 and LO1302, Marie Curie Career Integration Grant Project 304154 (to K. S.), and National Subvention for Development of Research Organizations RVO: 61388963 to the Institute of Organic Chemistry and Biochemistry. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental information.
- TM
- transmembrane
- DDM
- n-dodecyl-β-d-maltopyranoside
- DM
- n-decyl-β-d-maltopyranoside
- CMC
- critical micellar concentration
- LUV
- large unilamellar vesicles
- MBP
- maltose-binding protein
- TAMRA
- tetramethylrhodamine
- CE
- capillary electrophoresis
- BGE
- background electrolyte.
References
- 1. Urban S., Lee J. R., and Freeman M. (2002) A family of rhomboid intramembrane proteases activates all Drosophila membrane-tethered EGF ligands. EMBO J. 21, 4277–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee J. R., Urban S., Garvey C. F., and Freeman M. (2001) Regulated intracellular ligand transport and proteolysis control EGF signal activation in Drosophila. Cell 107, 161–171 [DOI] [PubMed] [Google Scholar]
- 3. McQuibban G. A., Saurya S., and Freeman M. (2003) Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature 423, 537–541 [DOI] [PubMed] [Google Scholar]
- 4. O'Donnell R. A., Hackett F., Howell S. A., Treeck M., Struck N., Krnajski Z., Withers-Martinez C., Gilberger T. W., and Blackman M. J. (2006) Intramembrane proteolysis mediates shedding of a key adhesin during erythrocyte invasion by the malaria parasite. J. Cell Biol. 174, 1023–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fleig L., Bergbold N., Sahasrabudhe P., Geiger B., Kaltak L., and Lemberg M. K. (2012) Ubiquitin-dependent intramembrane rhomboid protease promotes ERAD of membrane proteins. Mol. Cell 47, 558–569 [DOI] [PubMed] [Google Scholar]
- 6. Riestra A. M., Gandhi S., Sweredoski M. J., Moradian A., Hess S., Urban S., and Johnson P. J. (2015) A Trichomonas vaginalis rhomboid protease and its substrate modulate parasite attachment and cytolysis of host cells. PLoS Pathog. 11, e1005294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Etheridge S. L., Brooke M. A., Kelsell D. P., and Blaydon D. C. (2013) Rhomboid proteins: a role in keratinocyte proliferation and cancer. Cell Tissue Res. 351, 301–307 [DOI] [PubMed] [Google Scholar]
- 8. Chan E. Y., and McQuibban G. A. (2013) The mitochondrial rhomboid protease: its rise from obscurity to the pinnacle of disease-relevant genes. Biochim. Biophys. Acta 1828, 2916–2925 [DOI] [PubMed] [Google Scholar]
- 9. Song W., Liu W., Zhao H., Li S., Guan X., Ying J., Zhang Y., Miao F., Zhang M., Ren X., Li X., Wu F., Zhao Y., Tian Y., Wu W., et al. (2015) Rhomboid domain containing 1 promotes colorectal cancer growth through activation of the EGFR signalling pathway. Nat. Commun. 6, 8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Strisovsky K. (2016) Why cells need intramembrane proteases: a mechanistic perspective. FEBS J. 283, 1837–1845 [DOI] [PubMed] [Google Scholar]
- 11. Pierrat O. A., Strisovsky K., Christova Y., Large J., Ansell K., Bouloc N., Smiljanic E., and Freeman M. (2011) Monocyclic β-lactams are selective, mechanism-based inhibitors of rhomboid intramembrane proteases. ACS Chem. Biol. 6, 325–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dickey S. W., Baker R. P., Cho S., and Urban S. (2013) Proteolysis inside the membrane is a rate-governed reaction not driven by substrate affinity. Cell 155, 1270–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arutyunova E., Panwar P., Skiba P. M., Gale N., Mak M. W., and Lemieux M. J. (2014) Allosteric regulation of rhomboid intramembrane proteolysis. EMBO J. 33, 1869–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simeonov A., Jadhav A., Thomas C. J., Wang Y., Huang R., Southall N. T., Shinn P., Smith J., Austin C. P., Auld D. S., and Inglese J. (2008) Fluorescence spectroscopic profiling of compound libraries. J. Med. Chem. 51, 2363–2371 [DOI] [PubMed] [Google Scholar]
- 15. Vosyka O., Vinothkumar K. R., Wolf E. V., Brouwer A. J., Liskamp R. M., and Verhelst S. H. (2013) Activity-based probes for rhomboid proteases discovered in a mass spectrometry-based assay. Proc. Natl. Acad. Sci. U.S.A. 110, 2472–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wolf E. V., Zeißler A., Vosyka O., Zeiler E., Sieber S., and Verhelst S. H. (2013) A new class of rhomboid protease inhibitors discovered by activity-based fluorescence polarization. PLoS ONE 8, e72307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maegawa S., Ito K., and Akiyama Y. (2005) Proteolytic action of GlpG, a rhomboid protease in the Escherichia coli cytoplasmic membrane. Biochemistry 44, 13543–13552 [DOI] [PubMed] [Google Scholar]
- 18. Strisovsky K., Sharpe H. J., and Freeman M. (2009) Sequence-specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol. Cell 36, 1048–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zoll S., Stanchev S., Began J., Skerle J., Lepšik M., Peclinovská L., Majer P., and Strisovsky K. (2014) Substrate binding and specificity of rhomboid intramembrane protease revealed by substrate-peptide complex structures. EMBO J. 33, 2408–2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. VanAken T., Foxall-VanAken S., Castleman S., and Ferguson-Miller S. (1986) Alkyl glycoside detergents: synthesis and applications to the study of membrane proteins. Methods Enzymol. 125, 27–35 [DOI] [PubMed] [Google Scholar]
- 21. Kamp F., Winkler E., Trambauer J., Ebke A., Fluhrer R., and Steiner H. (2015) Intramembrane proteolysis of β-amyloid precursor protein by γ-secretase is an unusually slow process. Biophys. J. 108, 1229–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scheel G., Acevedo E., Conzelmann E., Nehrkorn H., and Sandhoff K. (1982) Model for the interaction of membrane-bound substrates and enzymes: hydrolysis of ganglioside GD1a by sialidase of neuronal membranes isolated from calf brain. Eur. J. Biochem. 127, 245–253 [DOI] [PubMed] [Google Scholar]
- 23. Parry G., Palmer D. N., and Williams D. J. (1976) Ligand partitioning into membranes: its significance in determining Km and Ks values for cytochrome P-450 and other membrane bound receptors and enzymes. FEBS Lett. 67, 123–129 [DOI] [PubMed] [Google Scholar]
- 24. Rath A., and Deber C. M. (2013) Design of transmembrane peptides: coping with sticky situations. Methods Mol. Biol. 1063, 197–210 [DOI] [PubMed] [Google Scholar]
- 25. Wolf E. V., Zeissler A., and Verhelst S. H. (2015) Inhibitor fingerprinting of rhomboid proteases by activity-based protein profiling reveals inhibitor selectivity and rhomboid autoprocessing. ACS Chem. Biol. 10, 2325–2333 [DOI] [PubMed] [Google Scholar]
- 26. Haedke U., Küttler E. V., Vosyka O., Yang Y., and Verhelst S. H. (2013) Tuning probe selectivity for chemical proteomics applications. Curr. Opin. Chem. Biol. 17, 102–109 [DOI] [PubMed] [Google Scholar]
- 27. Lemberg M. K., Menendez J., Misik A., Garcia M., Koth C. M., and Freeman M. (2005) Mechanism of intramembrane proteolysis investigated with purified rhomboid proteases. EMBO J. 24, 464–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., and Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 29. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A. 3rd, and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 [DOI] [PubMed] [Google Scholar]
- 30. Miroux B., and Walker J. E. (1996) Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260, 289–298 [DOI] [PubMed] [Google Scholar]
- 31. Urbani A., and Warne T. (2005) A colorimetric determination for glycosidic and bile salt-based detergents: applications in membrane protein research. Anal. Biochem. 336, 117–124 [DOI] [PubMed] [Google Scholar]
- 32. Solínová V., Kasicka V., Koval D., Barth T., Ciencialová A., and Záková L. (2004) Analysis of synthetic derivatives of peptide hormones by capillary zone electrophoresis and micellar electrokinetic chromatography with ultraviolet-absorption and laser-induced fluorescence detection. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 808, 75–82 [DOI] [PubMed] [Google Scholar]
- 33. Fenyo D., Wang Q., DeGrasse J. A., Padovan J. C., Cadene M., and Chait B. T. (2007) MALDI sample preparation: the ultra thin layer method. J. Vis. Exp. 192, 10.3791/192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nomenclature Committee of the International Union of Biochemistry (1979) Units of enzyme-activity, recommendations 1978. Eur. J. Biochem. 97, 319–320 [Google Scholar]
- 35. Nomenclature Committee of the International Union of Biochemistry (1983) Symbolism and terminology in enzyme-kinetics, recommendations 1981. Biochem. J. 213, 561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sreerama N., and Woody R. W. (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287, 252–260 [DOI] [PubMed] [Google Scholar]
- 37. Provencher S. W., and Glöckner J. (1981) Estimation of globular protein secondary structure from circular dichroism. Biochemistry 20, 33–37 [DOI] [PubMed] [Google Scholar]
- 38. Mongay C., and Cerda V. (1974) Britton-Robinson buffer of known ionic-strength. Anal. Chim. 64, 409–412 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.